

Abstract
In the current situation, the importance of vaccines for viral diseases has become the need of the hour. The scientific community in the field of virology has taken it upon themselves to develop vaccines for viral infections before an epidemic or pandemic situation arises. Human herpes virus-5 is an emerging situation that has alarming cases with major health concerns, including congenital impairments and infections leading to cancer states. Vaccination is the route most likely to succeed in the battleground with viral infections and consequences. Hence in the present manuscript, we have formulated the multiepitope subunit vaccine and optimized it with the advanced computational immunological framework. As a result, we report the subunit vaccine for HHV-5, comprised of promiscuous cytotoxic T-lymphocytes epitopes, helper T-lymphocytes, and B-cell epitopes engineered with putative adjuvants to ensure the strong immune response. The formulated subunit vaccine depicted high antigenicity and immunogenicity along with sustainable physicochemical characteristics. Molecular dynamics simulation analyses revealed the strong binding of the vaccine with MHC receptors (MHC-1 and MHC-2) and the virus progression specific membrane receptor TLR2 for a 100 ns MD simulation run. The interacting trajectory analysis of the vaccine showed stable binding with minimal deviations through RMSD, RMSF, and secondary structure confinement plot analyses for a long span of 100 ns. Interestingly, the vaccine showed robust immune response statistics for a prolonged time with evoking T-cell and B-cell populations with other vital players of the immune system, through the machine learning-based immune simulation approach. This study paved the way to a multiepitope vaccine for HHV-5 employing the immunoinformatics networks.
Keywords: multiepitope vaccine, human herpes virus-5, molecular dynamics simulation, immunoinformatics
The world is currently battling one of the most severe health concerns, and it has emerged that preparedness is a key defense against viruses. The scientific community in the field of virology should take it upon themselves to develop vaccines for viral infections before an epidemic or pandemic situation arises. Human herpes virus-5 (HHV-5), also known as cytomegalovirus, is an infectious virus that transmits to a developing fetus. HHV-5 infections are reported to have spread to all geographic locations and infected 60%–70% adults in developed countries.1 Herpes virus mostly harbors the regulation of innate immunity and adaptive immunity, which may affect the immune system and prolonged T-cell surveillance.2 HHV-5 infections cause congenital disabilities that lead to deafness, learning disabilities, and encephalitis and in various cases are reported to develop tumors.3,4 The traditional and conventional therapies have continued to prove inefficient in combating viruses targeting humans, and evoking the body’s immune system is the most effective and faster arsenal against such infections. Though there are many antiviral therapies that have been reported, they are said to be associated with issues like target in-specificity, laborious experimentation with random experimentation, high cost, etc.5,6 Overcoming the problems, the scientists have used the combinatorial methods by adjoining immunological methods with computational analysis to identify the potential vaccine candidates for many viral diseases like acute encephalitis, human immunodeficiency virus, dengue virus, etc.7−10 Hence in the present study, advanced immunoinformatics approaches were employed to design the multiepitope subunit vaccine against HHV-5.
HHV-5 virus consists of double-stranded DNA and is surrounded by a tegument layer and lipoprotein complex envelope studded by glycoproteins.11 HHV-5 is reported to infect mainly with glycoprotein-B (gB), which is responsible for viral entry into the host, and directly involved in tethering and stable attachment to the host cells.12 The virus infection, fusion, and processing to the host are reported to be caused by envelope protein glycoprotein-B. The glycoproteins are mechanistically known to interact with MHC alleles and membrane toll-like receptors (TLR2) and suggested to be involved in therapeutics design and developments.13 TLR2 interacts specifically with gB and plays the central role in identifying the cytomegalovirus infection with a cluster of differentiations and virus infection by activation of transcription factors.14 The glycoprotein-B, along with integrin proteins, increases the progression and entry of viruses and leads to the illness regulatory networks in the host. In many reports, it has also been shown the virion with a lack of gB was not able to attach to the host cell.15 Hence in the present study, HHV-5 envelope glycoprotein-B was employed to design the multiepitope subunit vaccine using the immunological frameworks. The multiepitope subunit vaccine consisted of putative cytotoxic T-lymphocytes (CTL), helper T-lymphocytes (HTL), and B-cell epitopes, which are adjoined with adjuvants to enhance the immune responses.16 Multiple epitopes make the approach highly specific and have the potential to evoke strong humoral and adaptive immune response against the virus.17,18
Moreover, advancements of immunological methods along with molecular docking and molecular dynamics simulations have been widely employed to design the vaccine candidates and other therapeutics agents.19−21 Molecular docking and molecular dynamics simulation assays have been immensely used to assess the molecular binding specificity and further to illustrate the binding mechanism of ligand molecules with the target.22−25 By employing these significant combinatorial computational approaches, we have designed the multiepitope vaccine and optimized it with molecular dynamics simulation for their prolonged interaction with herpes virus receptors MHCs and TLR2. Top-scored CTLs, HTLs, and B-cell epitopes were engineered with robust adjuvants. In addition, we have also demonstrated the antigenicity, immunogenicity, and physicochemical profiles of the vaccine, along with vaccine capability to elicit the immune responses.
Results and Discussion
Infectious Virus Sequence Retrieval and Alignment Assessment
The herpes virus infection, fusion, and progression in the host are reported to be caused by the envelope protein. Hence in this study, the envelope glycoprotein of HHV-5 was employed to design the multiepitope subunit vaccine. The envelope glycoprotein-B of human herpes virus-5 with accession number GenBank: ADE88063.1 was retrieved from the NCBI-Protein database. To assess its homology and conservancy, it was analyzed through the BLAST. Protein–protein BLAST stated the envelope protein is highly conserved with more than 90% sequence similarity to more than 100 viral proteins hits. The length of the envelope protein was of size 907 amino acids present in the herpes virus. Proteomic analysis of HHV-5 suggested envelope glycoprotein as a potential target for vaccine designing.
Putative Cytotoxic T-Lymphocytes Epitope Prediction
Potential cytotoxic T-lymphocytes (CTLs) were predicted for envelope glycoprotein of human herpes virus. CTLs are present on the antigen-presenting cell surface and bind with MHC to evoke the immune system. High binder CTLs (MHC-1 restricted) were identified by using the CTLpred program. The CTLpred application employed two algorithms, artificial neural network and support vector machine-based approach, to identify the high binder epitopes. To identify high binders, the threshold was set to 0.51 for ANN and 0.36 for SVM with high scores, sensitivity, and specificity. The obtained results were shown to cover more than 20 MHC class-1 supertypes that included more than 90% of the world’s population. High scored CTL epitopes for HHV-5 envelope protein are shown in Table 1.
Table 1. List of Cytotoxic T-Lymphocytes Epitopes for Human Herpes Virus-5.
| CTL rank | start position | epitope sequence | ANN score | SVM score | prediction types |
|---|---|---|---|---|---|
| 1 | 643 | STVDSMIAL | 0.77 | 1.3160073 | epitope |
| 2 | 132 | KRNIVAHTF | 0.91 | 0.92765306 | epitope |
| 3 | 524 | NPSAILSAI | 1.00 | 0.70374509 | epitope |
| 4 | 840 | YTNEQAYQM | 0.87 | 0.79583574 | epitope |
| 5 | 689 | SYKQRVKYV | 0.52 | 1.1372672 | epitope |
| 6 | 489 | DTLRNYINR | 0.46 | 1.1909782 | epitope |
| 7 | 439 | KSLLELERL | 0.90 | 0.6890118 | epitope |
| 8 | 756 | TIILVAIAV | 0.82 | 0.76847868 | epitope |
| 9 | 844 | QAYQMLLAL | 0.70 | 0.87768109 | epitope |
| 10 | 701 | VVDPLPPYL | 0.84 | 0.72834914 | epitope |
Helper T-Lymphocytes Epitopes Prediction
Helper T-lymphocyte (MHC-2 restricted) epitopes play a potential role in the generation of adaptive immunity. HTLs activate the antibodies and macrophages to destroy the foreign antigens and simultaneously induce the B-cells to produce a high number of antibodies. Potential HTL epitopes were exploited by the MHCpred program. The MHCpred server performed the additive approaches to determine the MHC-2 confined binder and movement specialized transporter associated with antigen processing (TAP) epitopes. For HTL epitope prediction, a large class of MHC-2 alleles was employed: DRB0101, DRB0701, DRB0401, HLA-DRB1101, and HLA0301 using the partial least-squares approach. Resulting promiscuous HTLs with high binder IC50 score and rank position were short-listed in Table 2.
Table 2. Helper T-Lymphocyte (MHC-2 Restricted) Epitopes and Putative Transporter Associated with Antigen Processing (TAP) Epitopes.
| rank | antigenic sequence | calcd log IC50 (M) | calcd IC50 value (nM) | confidence of prediction (max = 1) | |
|---|---|---|---|---|---|
| HTLs epitopes | 1 | NLFPYLVSA | 7.914 | 12.19 | 1.00 |
| 2 | YIYSTYLLG | 7.913 | 12.22 | 0.89 | |
| 3 | YEYVDYLFK | 7.738 | 18.28 | 0.78 | |
| 4 | YEKYGNVSV | 7.693 | 20.28 | 0.89 | |
| 5 | YLKGLDDLM | 7.691 | 20.37 | 1.00 | |
| TAP epitopes | 1 | RVYQKVLTF | 10.815 | 0.02 | 1.00 |
| 2 | KVVDPLPPY | 10.383 | 0.04 | 1.00 | |
| 3 | RLRHRKNGY | 10.32 | 0.05 | 1.00 | |
| 4 | ARSKYPYHF | 10.241 | 0.06 | 1.00 | |
| 5 | NSYKQRVKY | 10.167 | 0.07 | 1.00 |
Moreover, to evoke a strong immune response and smooth transportation of the designed vaccine, HTLs for tetanus toxin fragment-C (TTFrC) were created. Tetanus toxin fragment-C regulates retrograde transport, internalization, and binding of target molecules and also helps in activation and proliferation of CD4 associated immune cells. High binder TTFrC epitopes were predicted by employing the position-specific scoring matrices using the Rankpep tool (Table 3).
Table 3. High Binder HTLs for Tetanus Toxin Fragment-C (TTFrC).
| rank | epitope sequence | percentile score | starting residue position | end residue position |
|---|---|---|---|---|
| 1 | NDIISDISGFNSSVITYPDAQLVPGINGKAIHLVNNE | 22 | 40 | 66 |
| 2 | IEYNDMFNNFTVSFWLRVPKVSASHLEQYGT | 28.12 | 78 | 108 |
| 3 | LRVGYNAPGIPLYKKMEAVKLRDLK | 17.4 | 362 | 386 |
Putative Transporter Associated with Antigen Processing Epitopes Prediction
Putative transporter associated with antigen processing (TAP) epitopes were predicted by the MHCpred tool. TAP peptides are known to be responsible for transportation and regulatory networks, internalization, and interaction with MHC receptors. TAP epitopes were predicted, and high binder epitopes were short-listed by high binding values, percentile ranks, and IC50 scores. Nanomer size TAP epitopes were identified for efficient translocation to the different locations (Table 2).
Flexible B-Cell Epitope Determination Employing the Karplus and Kolaskar Method
B-cell epitopes were predicted by using the Bepred tool through the stringent data sets of physicochemical parameters. As a result, the top three potential flexible B-cell epitopes were VGVAIGAVGGAV, RKGPGPPSSDAS, and KRSTNNTTTLSL, among the list of predicted epitopes. Notably, the Bcepred tool is reported to possess experimental conversion accuracy of 52.97% to 57.53% on the basis of physicochemical parameters including the flexibility, antigenic propensity, exposed surface, hydrophilicity, polarity, and accessibility with threshold values of 1.9, 1.8, 2.4, 2, 2.3, and 2, respectively, as per algorithm. Resulting B-cell epitopes were found to be in an optimal range for the above-mentioned physicochemical properties lying in the range of −3 to +3 (Table 4).
Table 4. Top Scored B-Cell Epitopes.
| rank | position | epitope | score |
|---|---|---|---|
| 1. | 726 | VGVAIGAVGGAV | 1 |
| 2. | 823 | RKGPGPPSSDAS | 1 |
| 3. | 460 | KRSTNNTTTLSL | 0.997 |
| 4. | 896 | RHLKDSDEEENV | 0.976 |
| 5. | 280 | PFYNGTNRNTSY | 0.96 |
| 6. | 747 | FLKNPFGAFTII | 0.937 |
| 7. | 258 | AARSKYPYHFFA | 0.924 |
| 8. | 165 | GSNTEYVAPPMW | 0.92 |
| 9. | 379 | SKKQEVNMSDPV | 0.912 |
| 10. | 75 | NTTLKYGDVVGV | 0.91 |
Multiepitope Subunit Vaccine Formulation
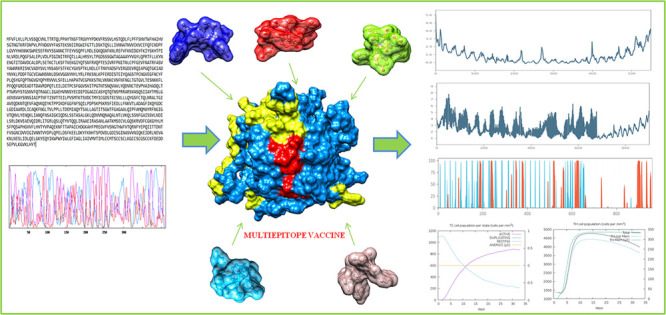
The multiepitope vaccine was formulated by promiscuous CTL, HTL, TAP, tetanus toxin fragment-C, and B-cell epitopes with a total size of 411 amino acids (Figure 1). The vaccine construct was engineered by adjoining epitopes with adjuvants using the linkers. Adjuvants were adjoined for their vital properties to elicit a robust immune response. Cholera toxin subunit-B (CTB), a well-known adjuvant, was also an ingredient in the vaccine construct. CTB belongs to the strain of cholera toxin with no toxic effects and importantly also has been used in many vaccines to enhance the immune responses. Top scored CTL, HTL, TAP, tetanus toxin fragment-C, and B-cell epitopes were employed using the linkers. CTL epitopes were joined together using the AAY linker, HTLs and TTFrC epitopes were joined through the GPGPG linker, B-cell epitopes were united by the KK linker, and CTB adjuvant was linked by EAAAK linkers. These linkers also played an essential role in providing stability, flexibility, and extended conformation of the formulated vaccine. The overall formulated multiepitope vaccine was comprised of adjuvant CTB at the N terminal position: the first domain consisted of top-scored three TTFrC epitopes, the second domain consisted of high binder three CTLs (9mers) and top three HTLs, and the third domain consisted of three top-scored B-cell epitopes and three TAP epitopes at the C terminal. Further, the designed subunit vaccine was optimized.
Figure 1.

Formulation scheme of a multiepitope subunit vaccine.
Secondary and Tertiary Structural Analyses of a Formulated Vaccine
The formulated vaccine construct was assessed for secondary structural and tertiary structural conformations. The secondary fundamental analysis of the vaccine construct depicted the composition of alpha-helices 40.88%, extended beta strands 16.79%, and random coils 42.34% by using the GOR4 server and Psipred server.26,27 The strong constitution with high flexibility stated the globular structure of the vaccine construct (Figure 2). Moreover, the 3D structure of the construct was designed by the de novo method using the I-Tasser module. The basic local sequence alignment (BLAST) tool was used to identify the templates. Five templates (1LTR, 5FYP, 4I6T, 3FFZ, 5ZZM, and 5ELB) were used for the threading approach to predict the 3D structure of the vaccine. Among the designed top five models, the top-scored energy minimized structure was considered for the study. The top structural model-3 of the vaccine construct showed the Tm score of 0.54 ± 0.14 and RMSD of 9.1 ± 4.6 Å, which showed the stability of the structure with high sequence alignment. Also, the C-score was computed to −2.43, which showed the optimal structural quality of residues with a threshold value of (−5 to 2) by structure simulations. The 3D structure of the vaccine construct was also optimized and energy minimized using the Gromacs-minimizer. The optimized structure was further assessed through the stereochemical and physicochemical properties. The Ramachandran plot assessment showed that 92.0% of residues lay in the favored region, 4.3% residues lay in the generously allowed region, and only 3.7% residues lay in the outlier region for the vaccine construct. Moreover, we also performed the intrinsically disordered protein sequence using IUPred2A. IUPred2A, a combined web interface, identified no disordered protein regions and binding regions in the vaccine construct and showed the stable structure of designed vaccine constructs based on the redox state of its environment.28
Figure 2.

(A) Three-dimensional crystal structure of formulated vaccine. (B) Ramachandran plot for the formulated vaccine. (C) The secondary structure analysis and depiction of globularity and stability.
Molecular Binding Analyses of the Vaccine with MHC Receptors and Optimization with the TLR2 Receptor
The molecular binding analyses of the vaccine were performed with MHC-1 and MHC-2 by employing the Cluspro server. Before delivering the docking, the MHC-1 (PDB ID 1I1Y) and MHC-2 (1KG0) receptors were retrieved from the Protein Data Bank and evaluated through the Ramachandran plot (Figure S1). The Cluspro module used the piper rigid body algorithm with a grid point in X, Y, and Z coordinates with 70000 rotations with a receptor spacing of 1 Å. From the molecular docking analyses, we obtained the ten different ligand binding conformation models with a target receptor. Through the molecular docking, ten models with ligand binding conformations were obtained. Among these models, the top-scored model showed the most durable binding of the vaccine to MHC-1 with an energy score of −1275.1 kcal/mol (lowest energy) and center interaction score of −1283.8 kcal/mol. Similarly, the vaccine also showed the optimal binding with the MHC-2 receptor with the lowest energy score of −897.8 kcal/mol and center energy score of −1094.2 kcal/mol.
Moreover to validate our results, we performed the binding analysis of the vaccine construct with membrane specific receptor TLR-2. The 3D structure of TLR-2 (PDB ID 2Z7X) was availed from the Protein Data Bank and evaluated with the Ramachandran plot. Thereafter, molecular docking was performed and showed the proximal close binding of the vaccine construct with TLR-2 with the lowest energy score of −940.2 kcal/mol and score −1014.6 kcal/mol from the center of the receptor (Figure 3). These results signified a potent binding of the vaccine construct with MHC receptors and a membrane specific TLR-2 receptor.
Figure 3.

Molecular interaction of the vaccine construct depicted in ribbon view in gray color, with (A) MHC class-1 (surface view in blue color), (B) MHC class-2 (surface view in blue color), and (C) the TLR-2 receptor (surface view in blue color) in dark blue color.
Molecular Dynamics Simulation Binding Analyses of the Vaccine Construct with TLR2
Lead vaccine candidate binding analysis was studied through molecular dynamics simulation. It was performed to determine the stable binding and conformational change during interaction within the epitope-TLR2 complex. Importantly, to get insight into the binding mechanism of the vaccine construct with TLR2, comparative analysis of their complex with TLR2 and the vaccine, individually in globular states, was performed. Molecular dynamics simulation studies were performed for a long time (100 ns run) in order to assess the prolonged stable binding of the vaccine construct, which can be expected to elicit a robust immune response for a long time. The Desmond suite was used to perform the simulation employing the OPLS-2005 force field, CHARMM energy, to get the most confined trajectories. The trajectory systems were immersed in an orthorhombic box of a water association system, that functions as a buffering solvent with charged particles to process and equilibrate the solvation molecular entities. Trajectories were taken with the energy minimization step with the iteration of 5000 steepest descent steps and followed by equilibration steps of NVT (restrained Brownian dynamics) and the NPT phase (1 atmospheric pressure). Obtained trajectories were subjected to run for 100 ns, and atomic coordinates were optimized with recording the simulation calculations for every two ps frames. The resulting estimates were processed to determine the RMSD, RMSF, and secondary structure elements plots. The vaccine construct was found to bind strongly in the binding groove of the target TLR2 receptor with stabilized ranges of RMSD and RMSF.
Obtained results showed the stable binding of the vaccine and made the conformational changes to the TLR2 receptor. The analyses revealed that the subunit vaccine binds to TLR2 in close vicinity with minimal deviations of RMSD in the range of 3 Å–5.2 Å, which signified the stable binding and globular binding complex. The membrane receptor (TLR2) in the singlet state depicted the stabilized structural properties with minimal RMSD fluctuations in the range of 1.4 Å–4.6 Å, and the vaccine construct (singlet state) showed the RMSD deviations in the range from 4.8 to 8.4 Å, which were stabilized by binding to the TLR2 in the comparative simulation analysis (Figure 4).
Figure 4.

Root mean square deviation (RMSD) plots through molecular dynamics simulation analysis of (A) the TLR2 receptor, (B) vaccine construct, and (C) vaccine construct-TLR2 complex.
Thereafter, structural fluctuations of trajectories were performed by analyzing the RMSF plots. RMSF plots also demonstrated the stabilized binding of the vaccine construct with TLR2, for C-alpha atoms mobility with deviations in the range of 0.9 Å–6.4 Å for 100 ns. Though TLR2 in globular form has reduced fluctuations in the range of 0.8–4.2 Å and after binding with the vaccine, the insertion of conformational changes to receptor TLR2 is depicted (Figure 5).
Figure 5.

Root mean square fluctuation (RMSF) structural analysis by molecular dynamics simulation analysis of (A) the TLR2 receptor, (B) vaccine construct, and (C) vaccine construct-TLR2 complex.
Also, throughout the simulation to depth insight into the residue stability and globularity of the interacting complex of the vaccine with the receptor, the secondary structure elements (SSE) of three systems were monitored (Figure S2). The SSE plot analysis showed stable confinement and compactness in the composition of the trajectory frame of the binding complex (vaccine-TLR2) with total SSE content of 25.34% that included the alpha-helices 12.28% and beta strands 13.07% (Figure 6) and lower than with the individual TLR2 receptor and vaccine construct (Figures S3, S4, and S5). The SSE results demonstrated the compactness of the binding complex with fewer fluctuations to alpha helices, beta helices, and residues at N-terminal and C-terminal tails. These results showed the significant binding of the vaccine to the target membrane receptor TLR-2 (Figure 7). Importantly, the stable prolonged interaction of the vaccine candidate with the TLR2 receptor is expected to elicit a robust immune response against the herpes virus.
Figure 6.

(A) Secondary structure element analysis of the vaccine construct and TLR2 complex and (B) the residue content fluctuation plot of secondary structural elements of the vaccine-TLR2 complex system.
Figure 7.

Residue content fluctuation of the secondary structure element analysis of the vaccine construct and TLR2 complex for a 100 ns molecular dynamics simulation run.
Immune Response Elicitation Studies of Vaccine
The immune response generation capability of the designed vaccine against the target virus was performed by an in silico immunization experiment using the C-ImmSimm server. It employed the combining mesoscopic simulation with the machine learning method and was allowed to run for 100 simulation steps and 480 time steps for the immune response. As a result, we found the designed vaccine evokes the strong primary and secondary immune response by the high level of key players of the immune system (helper T-cells, cytotoxic T-cells, and B-cells). The vaccine administration showed the elevated level of IgM, IgG1, and IgG2 immunoglobulins and other immune cells for a long span. The high concentration of IgM and IgG antibodies suggested the robust primary immune response by a vaccine candidate. Moreover, the immunization experiment result also showed a high level of cytotoxic T-cells with the highest level of 1157 cells per mm3 on day 14 and gradually decreased with an optimal level of 1125 cells per mm3. T-cell population (helper T-cells) elevated to the level of 4300 cells per mm3 on days 7–8 of the vaccine induction and maintained until 35 days. Both immune T-cell types remained in high concentration during both active and resting states and evoked the memory cells which would help in boosting strong adaptive immunity against an encounter from the pathogenic virus (Figure 8). As an innate immunity player, B-cell immune cells were reported to increase 6.5 cells per mm3 for IgG and IgM immunoglobulins. Also, the elevated level of other potential players of the immune system, cytokines, interferons, interleukin, and natural killer cells, was found (Figure S6). The obtained results suggested the significant role of the designed vaccine construct elicits a robust immune response against the herpes virus. Moreover, this work further needs to be validated through the in vitro and in vivo experiments.
Figure 8.

Designed vaccine immune response: (A) elevation of immunoglobulins at different concentrations of antigen, (B) cytotoxic T-cell count, (C) T-cytotoxic cell populations at different states; resting and active state (D) helper T-cell population, (E) helper T-cell count at resting and active state, and (F) B-cell population (IgM, IgG1, and IgG2) in response to the designed vaccine.
Antigenicity, Physicochemical, and Allergenicity Characterization of Formulated Vaccine
The antigenicity profile of the designed vaccine construct was assessed by using the Vaxijen and AntigenPro servers. Obtained results depicted the high antigenicity character of the vaccine construct with a score of 0.840463 and 0.530 with threshold 0.4, respectively. Moreover, the allergenicity analysis showed the nonallergenic nature of the vaccine by the Allertop server. We have also assessed the sequence similarity analysis of epitopes sequence leading to the vaccine with the human genome. As a result, we did not find any hits (sequence similarity) against the human genome and were not associated with any risk for autoimmune disease. We have predicted the cellular localization of the lead epitope using the Cello Predictor software, which worked with a multiclass SVM system. Results showed the lead vaccine construct is majorly localized in the nucleus with a score of 0.717 and in the mitochondrial region with a score of 1.95, and fewer ratios localize in cytoplasm with a score of 0.874, extracellular plasma membrane, and other regions of the cell.29 These results signified the high potency of the vaccine. The physicochemical studies showed the stabilizing characteristics of the vaccine. The formulated vaccine of size 411 amino acids and molecular weight 44677.95 Da consisted of theoretical isoelectric point (pI) 9.28. The instability index depicted the stability confinements of the vaccine construct with a score of 25.99 (a score < 40 was considered to be stable by the protparam algorithm). The aliphatic profile was computed to be 81.31, which implied the enhanced thermostability of the globular vaccine. Half-life was defined through the three experimentation models, which showed the half-life values for 1 h in mammalian reticulocytes in in vitro analyses: >10 h in E. coli and >30 min in yeast. Also, the grand average of the hydropathy index showed a score of −0.281 that indicated the optimally hydrophilic nature. The computed physicochemical properties of the vaccine showed the stability and suitability of the vaccine for further in vitro and in vivo analyses.
In Silico Cloning Expression Analysis of the Vaccine Construct
The vaccine construct was accessed to identify the optimized DNA sequence using the JCAT server. Obtained results showed the high sequence expression of a chimeric chain of the vaccine construct in E. coli (k12 strain) with 0.8, elevated more than the codon adaption index (CAI). The CAI for the sequence was calculated to be 1.0, with a high GC content of 50.12% of the improved DNA sequence of the vaccine construct and 50.73% for E. coli. The results indicated the optimal expression of the vaccine construct for restriction enzyme BamHI and XhoI in the pET28a vector (Figure 9).
Figure 9.

In silico cloning expression analysis of vaccine in the plasmid vector.
Conclusion
Human herpes virus-5 infection is associated with multiple diseases leading to cancer and other severe conditions. This breakthrough study provides a new treatment modality for human herpes virus-5 viral infection prevention. We report the multiepitope subunit vaccine against HHV-5 employing the combinatorial methods using the immunological and bioinformatics approaches. The reported vaccine construct was formulated with putative top-scored T-cell and B-cell epitopes and as ingredients with the adjuvants to strengthen the capability of the vaccine to elicit a strong immune response. The vaccine was well optimized, and the obtained results depicted the high antigenicity and immunogenicity along with optimal physicochemical characteristics. The vaccine was also found to bind strongly in the binding groove of MHC-1, MHC-2, and the membrane specific TLR-2 receptor with high molecular docking scores. Molecular dynamics simulation studies confirmed the stable binding of the vaccine with specific receptors with RMSD, RMSF, and secondary structure analysis throughout the MD simulation run (100 ns). Moreover, the vaccine also showed an ability to elicit the strong immune response by generation of a high population of the key players’ immune system by an in silico simulation approach. The present study reports the formulation of the subunit vaccine candidate against HHV-5 and successful optimization with advanced immunoinformatics approaches, which further needs to be validated with in vitro and in vivo studies.
Materials and Methods
HHV-5 Infections Sequence Retrieval and Assessment
The infectious glycoprotein-B sequence of HHV-5 is reported to the primary causative vehicle for viral attachment, infection, and replication. The complete glycoprotein-B sequence was retrieved from the NCBI protein database. The retrieved sequence was evaluated for its conservancy and homology to relative infectious sequences using the protein–protein BLAST (blastp).30 Hits with >90% homology were considered for the study. The physicochemical analysis of the sequence was conducted using the Protparam server.31
Putative Cytotoxic T-Lymphocytes Epitopes Prediction
Cytotoxic T-lymphocytes (CTLs) are present on antigen-presenting cells which bind with a major histocompatibility complex to evoke the immune response. CTL epitopes were predicted by artificial neural networks and the support vector machine-based CTLpred tool. CTLpred used the direct algorithm to identify the promiscuous high binder MHC-1 epitopes.32 Since the optimal size of epitopes is nanomer and also the MHC-1 receptor binds to a ligand of size 8–10 amino acids, hence epitopes of 9 mer size were studied.33
Helper T-Lymphocytes Epitopes Prediction and Transporter Associated with Antigen Processing Epitope Identification
The putative helper T-lymphocyte (HTL) epitopes are known to play a vital role in the generation of adaptive immunity. HTL epitopes were determined by the MHCpred server. MHCpred worked based on an additive approach to define the potential MHC-2 binders.34 Moreover, transporter associated with antigen processing (TAP) epitopes were also identified using the MHCpred server. TAP epitopes are directly involved in signaling regulation of cytosolic antigens to the endoplasmic reticulum and transportation to the surface membrane to activate the cascade of immune response.35 In addition, to derive the enhanced immune responses along with these epitopes, promiscuous HTLs for adjuvant tetanus toxin fragment-C (TTFrC) were designed and adjoined with the vaccine construct. Tetanus toxin fragment-C is known to enhance the MHC-2 assisted robust immune response in the host.36 TTFrC epitopes were predicted by the position-specific scoring matrices using the Rankpep server.37
Flexible B-Cell Epitope Determination Employing the Karplus and Kolaskar Method
Flexible B-cell epitopes were predicted through the Karplus and Kolaskar method by using the Bcepred tool. This algorithm used the large scale flexibility of epitopes based on the movement of peptidic residues by temperature, atomics fluctuations, and alpha carbons of well-known protein structures.38
Formulation of Multiepitope Subunit Vaccine and 3D Structural Assessment
The multiepitope subunit vaccine for HHV-5 was constructed by linking the putative top-scored CTL, HTL, and TAP epitopes, which were ingredients with the potential adjuvants to design the robust and complete vaccine. Various linkers and a motif were used to adjoin the multiple epitopes. To boost the immune response by the vaccine candidate, the adjuvants choler toxin subunit-B and tetanus toxin fragment-C were added to the whole vaccine using the suitable linkers.39 Besides the formulation of the vaccine construct, the 3D structure of the vaccine construct was designed by employing the de novo threading approach using the I-Tasser server.40 Relevant templates for structure prediction were identified using the protein BLAST. Moreover, the created 3D structure of the vaccine was optimized and energy minimized through the Gromacs minimizer and further analyzed for its stereochemical and physicochemical properties through the Ramachandran plot.41−43
Molecular Binding Analysis of Vaccine with MHC-1 and MHC-2 and Membrane Specific TLR2
The binding analysis of the vaccine construct was performed with the MHC-1 and MHC-2 receptors using the molecular docking assays. Notably, to optimize the specificity of the vaccine construct, it was docked with virus-specific membrane receptor TLR2 using the Cluspro docking server.44 TLR2 is the important membrane receptor for virus signaling molecules and plays a significant role as the host sensor in the derivation of innate and adaptive immune responses. TLR2 activation by ligand triggers the initiation and generation of antiviral responses including the interferons with an increased level of IL-6 and TNF- α, which strengthen the immune system to fight against the virus.45 After that, resulting trajectories were energy minimized and analyzed using the PyMOL and Chimera modeling suites.46
Molecular Dynamics Simulation Analysis of Vaccine Binding
Molecular dynamics simulation for 100 ns was performed for detailed analysis of molecular binding of the vaccine construct with the TLR2 receptor. The MD simulation was performed for the docked complex using the Desmond server by Deshaw. A long MD simulation run was conducted to evaluate the binding of the vaccine for a long span. The resulting trajectories were energy minimized and analyzed for root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), a radius of gyration, and secondary structure assessment for the whole MD run to determine the stable conformational binding of the vaccine.47
Immune Response Elicitation Studies of the Vaccine Construct
An in silico immunization experiment was performed to assess the immune response derivation capability of a designed vaccine against the target. The C-ImmSimm server was used to perform the immune response study of the vaccine. It used the position-specific scoring matrix and machine learning methods to demonstrate the antigen responsible for the evoking of the immune cell. The C-ImmSimm server employed the Miyazawa and Jernigan protein–protein analyzing method to define the molecular interaction of immune regulators T-cells, B-cells, and linked regulators.48
Antigenicity, Physicochemical, Allergenicity, and in Silico Cloning Expression Analysis of the Formulated Vaccine
The antigenicity characteristic of the vaccine was determined using the Vaxijen server and Antiegpro server.49 The allergenicity of the vaccine was assessed to determine its nature using the Allertop server.50 Also, physicochemical parameters were analyzed to determine the feasibility and confined nature of the vaccine construct. Moreover, the designed vaccine was investigated for the in silico chimerical cloning expression in E. coli strain through the JCAT server.51
Acknowledgments
Prof. Ramesh Chandra is grateful to the Council of Scientific and Industrial Research (CSIR) and DST-SERB (EEQ/2016/ 000489) for providing financial assistance. Dr. Neeraj Kumar particularly thanks CSIR for the Research Associate fellowship (09/045(1706) 2019 EMR-1) and the University of Delhi for research support. Dr. Damini Sood would like to thank UGC for financial assistance. We would also like to thank Vinod Devaraji from Schrodinger Bangalore India for in silico support and suggestions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00139.
Protein sequence of multiepitope vaccine construct, relevant linkers used for constriction of vaccine construct, three-dimensional structure assessment of MHC-1, MHC-2, and TLR2 (Figure S1), molecular dynamics simulation analyses results for secondary structure element analysis (Figures S2–S5), and in silico immune response results for vaccine (Figure S6) (PDF)
Author Contributions
N.K. and R.C. designed and performed the spectroscopic studies. N.K. and D.S. carried out the in silico experiments. The manuscript was written by N.K. and D.S.
The authors declare no competing financial interest.
Supplementary Material
References
- Ibanez C. E.; Schrier R.; Ghazal P.; Wiley C.; Nelson J. A. (1991) Human cytomegalovirus productively infects primary differentiated macrophages. J. Virol. 65, 6581–6588. 10.1128/JVI.65.12.6581-6588.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinzger C.; Kahl M.; Laib K.; Klingel K.; Rieger P.; Plachter B.; Jahn G. (2000) Tropism of human cytomegalovirus for endothelial cells is determined by a post-entry step dependent on efficient translocation to the nucleus. J. Gen. Virol. 81, 3021–3035. 10.1099/0022-1317-81-12-3021. [DOI] [PubMed] [Google Scholar]
- Isaacson M. K.; Compton T. (2009) Human cytomegalovirus glycoprotein B is required for virus entry and cell-to-cell spread but not for virion attachment, assembly, or egress. J. Virol. 83, 3891–3903. 10.1128/JVI.01251-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schottstedt V.; Blümel J.; Burger R.; Drosten C.; Gröner A.; Gürtler L.; Heiden M.; Hildebrandt M.; Jansen B.; Montag-Lessing T.; Offergeld R.; Pauli G.; Seitz R.; Schlenkrich U.; Strobel J.; Willkommen H.; von König C. H. (2010) Human Cytomegalovirus (HCMV) - Revised. Transfus. Med. Hemoth. 37, 365–375. 10.1159/000322141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oli A. N.; Obialor W. O.; Ifeanyichukwu M. O.; Odimegwu D. C.; Okoyeh J. N.; Emechebe G. O.; Adejumo S. A.; Ibeanu G. C. (2020) Immunoinformatics and Vaccine Development: An Overview. ImmunoTargets Ther. 9, 13–30. 10.2147/ITT.S241064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahrami A. A.; Payandeh Z.; Khalili S.; Zakeri A.; Bandehpour M. (2019) Immunoinformatics: In Silico Approaches and Computational Design of a Multi-epitope, Immunogenic Protein. Int. Rev. Immunol. 38, 307–322. 10.1080/08830185.2019.1657426. [DOI] [PubMed] [Google Scholar]
- Kumar N.; Sood D.; Sharma N.; Chandra R. (2020) Multiepitope Subunit Vaccine to Evoke Immune Response against Acute Encephalitis. J. Chem. Inf. Model. 60, 421–433. 10.1021/acs.jcim.9b01051. [DOI] [PubMed] [Google Scholar]
- Abd El-Baky N.; Uversky V. N.; Redwan E. M. (2018) Virucidal activity of cell-penetrating peptides of viral origin. J. Biomol. Struct. Dyn. 36, 1739–1746. 10.1080/07391102.2017.1333459. [DOI] [PubMed] [Google Scholar]
- Anusuya S.; Gromiha M. M. (2017) Quercetin derivatives as nonnucleoside inhibitors for dengue polymerase: Molecular docking, molecular dynamics simulation, and binding free energy calculation. J. Biomol. Struct. Dyn. 35, 2895–2909. 10.1080/07391102.2016.1234416. [DOI] [PubMed] [Google Scholar]
- Vora J.; Patel S.; Sinha S.; Sharma S.; Srivastava A.; Chhabria M.; Shrivastava N. (2019) Molecular docking, QSAR and ADMET based mining of natural compounds against prime targets of HIV. J. Biomol. Struct. Dyn. 37, 131–146. 10.1080/07391102.2017.1420489. [DOI] [PubMed] [Google Scholar]
- Phillips S. L.; Bresnahan W. A. (2011) Identification of binary interactions between human cytomegalovirus virion proteins. J. Virol. 85, 440–447. 10.1128/JVI.01551-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton T. (2004) Receptors and immune sensors: The complex entry path of human cytomegalovirus. Trends Cell Biol. 14, 5–8. 10.1016/j.tcb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Chakravarti A.; Kashyap B.; Matlani M. (2009) Cytomegalovirus infection: An Indian perspective. Indian J. Med. Microbiol. 27, 3. [PubMed] [Google Scholar]
- Compton T.; Kurt-Jones E. A.; Boehme K. W.; Belko J.; Latz E.; Golenbock D. T.; Finberg R. W. (2003) Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J. Virol. 77, 4588–4596. 10.1128/JVI.77.8.4588-4596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni V.; Gianni T.; Salvioli S.; Campadelli-Fiume G. (2012) Herpes simplex virus glycoproteins gH/gL and gB bind Toll-like receptor 2, and soluble gH/gL is sufficient to activate NF-κB. J. Virol. 86, 6555–6562. 10.1128/JVI.00295-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. (2018) Multi-epitope vaccines: a promising strategy against tumors and viral infections. Cell. Mol. Immunol. 15, 182–184. 10.1038/cmi.2017.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M.; Khan S.; Ali A.; Akbar H.; Sayaf A. M.; Khan A.; Wei D. Q. (2019) Immunoinformatics approaches to explore Helicobacter Pylori proteome (Virulence Factors) to design B and T cell multi-epitope subunit vaccine. Sci. Rep. 9 (1), 13321. 10.1038/s41598-019-49354-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Y.; Zhao F.; Shao J.; Li Y.; Li S.; Chang H.; Zhang Y. (2019) Application of built-in adjuvants for epitope-based vaccines. PeerJ 6, e6185 10.7717/peerj.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N.; Singh A.; Grover S.; Kumari A.; Dhar P. K.; Chandra R.; Grover A. (2019) HHV-5 epitope: A potential vaccine candidate with high antigenicity and large coverage. J. Biomol. Struct. Dyn. 37, 2098–2109. 10.1080/07391102.2018.1477620. [DOI] [PubMed] [Google Scholar]
- Singh V. K.; Kumar N.; Kalsan M.; Saini A.; Chandra R. (2016) A Novel Peptide Thrombopoietin Mimetic Designing and Optimization Using Computational Approach. Front. Bioeng. Biotechnol. 4, 69. 10.3389/fbioe.2016.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N.; Sood D.; Tomar R.; Chandra R. (2019) Antimicrobial Peptide Designing and Optimization Employing Large-Scale Flexibility Analysis of Protein-Peptide Fragments. ACS Omega 4, 21370–21380. 10.1021/acsomega.9b03035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood D.; Kumar N.; Rathee G.; Singh A.; Tomar V.; Chandra R. (2018) Mechanistic Interaction Study of Bromo-Noscapine with Bovine Serum Albumin employing Spectroscopic and Chemoinformatics Approaches. Sci. Rep. 8, 16964. 10.1038/s41598-018-35384-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N.; Kumar N.; Rathee G.; Sood D.; Singh A.; Tomar V.; Dass S. K.; Chandra R. (2020) Privileged Scaffold Chalcone: Synthesis, Characterization and Its Mechanistic Interaction Studies with BSA Employing Spectroscopic and Chemoinformatics Approaches. ACS Omega 5, 2267–2279. 10.1021/acsomega.9b03479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N.; Tomar R.; Pandey A.; Tomar V.; Singh V. K.; Chandra R. (2018) Preclinical evaluation and molecular docking of 1,3-benzodioxole propargyl ether derivatives as novel inhibitor for combating the histone deacetylase enzyme in cancer. Artif. Cells, Nanomed., Biotechnol. 46, 1288–1299. 10.1080/21691401.2017.1369423. [DOI] [PubMed] [Google Scholar]
- Sood D.; Kumar N.; Singh A.; Sakharkar M. K.; Tomar V.; Chandra R. (2018) Antibacterial and Pharmacological Evaluation of Fluoroquinolones: A Chemoinformatics Approach. Genomics Inform. 16, 44–51. 10.5808/GI.2018.16.3.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T. Z.; Jernigan R. L.; Garnier J.; Kloczkowski A. (2005) GOR V server for protein secondary structure prediction. Bioinformatics 21 (11), 2787–2788. 10.1093/bioinformatics/bti408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D. T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292 (2), 195–202. 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- Meszaros B.; Erdos G.; Dosztanyi Z. (2018) IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 46 (W1), W329–W337. 10.1093/nar/gky384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C. S.; Chen Y. C.; Lu C. H.; Hwang J. K. (2006) Prediction of protein subcellular localization. Proteins: Struct., Funct., Genet. 64 (3), 643–651. 10.1002/prot.21018. [DOI] [PubMed] [Google Scholar]
- Altschul S. F.; Gish W.; Miller W.; Myers E. W.; Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Gasteiger E., Hoogland C., Gattiker A., Wilkins M. R., Appel R. D., and Bairoch A. (2005) Protein identification and analysis tools on the ExPASy server. In The proteomics protocols handbook, pp 571–607, Humana Press, 10.1385/1-59259-890-0:571. [DOI] [Google Scholar]
- Bhasin M.; Raghava G. P. (2004) Prediction of CTL epitopes using QM, SVM and ANN techniques. Vaccine 22, 3195–204. 10.1016/j.vaccine.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Madden D. R. (1995) The three-dimensional structure of peptide-MHC complexes. Annu. Rev. Immunol. 13, 587–622. 10.1146/annurev.iy.13.040195.003103. [DOI] [PubMed] [Google Scholar]
- Guan P.; Doytchinova I. A.; Zygouri C.; Flower D. R. (2003) MHCPred: a server for quantitative prediction of peptide-MHC binding. Nucleic Acids Res. 31, 3621–3624. 10.1093/nar/gkg510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palatnik-De-Sousa C. B.; Soares I. D. S.; Rosa D. S. (2018) Editorial: Epitope Discovery and Synthetic Vaccine Design. Front. Immunol. 9, 826. 10.3389/fimmu.2018.00826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo A. C.; Oliván S.; Manzano R.; Zaragoza P.; Aguilera J.; Osta R. (2012) Fragment C of Tetanus Toxin: New Insights into Its Neuronal Signaling Pathway. Int. J. Mol. Sci. 13, 6883–6901. 10.3390/ijms13066883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reche P. A.; Glutting J. P.; Zhang H.; Reinherz E. L. (2004) Enhancement to the RANKPEP resource for the prediction of peptide binding to MHC molecules using profiles. Immunogenetics 56, 405–19. 10.1007/s00251-004-0709-7. [DOI] [PubMed] [Google Scholar]
- Chen J.; Liu H.; Yang J.; Chou K. C. (2007) Prediction of linear Bcell epitopes using amino acid pair antigenicity scale. Amino Acids 33, 423–428. 10.1007/s00726-006-0485-9. [DOI] [PubMed] [Google Scholar]
- Stratmann T. (2015) Cholera Toxin Subunit B as Adjuvant-An Accelerator in Protective Immunity and a Break in Autoimmunity. Vaccines 3, 579–596. 10.3390/vaccines3030579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Yan R.; Roy A.; Xu D.; Poisson J.; Zhang Y. (2015) The ITASSER Suite: Protein structure and function prediction. Nat. Methods 12, 7. 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S., and Raghava G. P. S. (2004) BcePred: prediction of continuous B-cell epitopes in antigenic sequences using physico-chemical properties. In International Conference on Artificial Immune Systems, pp 197–204, Springer, Berlin, Heidelberg, 10.1007/978-3-540-30220-9_16. [DOI]
- Laskowski R. A.; Rullmann J. A. C.; MacArthur M. W.; Kaptein R.; Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486. 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Lindahl E.; Hess B.; van der Spoel D. (2001) GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 7, 306–317. 10.1007/s008940100045. [DOI] [Google Scholar]
- Kozakov D.; Hall D. R.; Xia B.; Porter K. A.; Padhorny D.; Yueh C.; Dmitri Beglov D.; Vajda S. (2017) The ClusPro web server for protein-protein docking. Nat. Protoc. 12 (2), 255–278. 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira-Nascimento L.; Massari P.; Wetzler L. M. (2012) The Role of TLR2 in Infection and Immunity. Front. Immunol. 3, 79. 10.3389/fimmu.2012.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. (2004) UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 25 (13), 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Bowers K. J., Chow D. E., Xu H., Dror R. O., Eastwood M. P., Gregersen B. A., Klepeis J. L., Kolossvary I., Moraes M. A., Sacerdoti F. D., Salmon J. K., Shan Y., and Shaw D. E. (2006) Scalable algorithms for molecular dynamics simulations on commodity clusters. In SC’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, pp 43–43, IEEE.
- Rapin N.; Lund O.; Bernaschi M.; Castiglione F. (2010) Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS One 5, e9862 10.1371/journal.pone.0009862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova I. A.; Flower D. R. (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinf. 8, 4. 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov I.; Flower D. R.; Doytchinova I. (2013) AllerTOP--a server for in silico prediction of allergens. BMC Bioinf. 14, S4. 10.1186/1471-2105-14-S6-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote A.; Hiller K.; Scheer M.; Münch R.; Nörtemann B.; Hempel D. C.; Jahn D. (2005) JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 33, W526–31. 10.1093/nar/gki376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.