

Abstract
Galactose oxidase (GAO) contains a Cu(II)-ligand radical cofactor. The cofactor, which is autocatalytically generated through the oxidation of the copper, consists of a cysteine-tyrosine radical (Cys-Tyr•) as a copper ligand. The formation of the crosslinked thioether bond is accompanied by a C-H bond scission on Tyr272 with few details known thus far. Here, we report the genetic incorporation of 3,5-dichlorotyrosine (Cl2-Tyr) and 3,5-diflurotyrosine (F2-Tyr) to replace Tyr272 in the GAOV previously optimized for expression through directed evolution. The proteins with an unnatural tyrosine residue are catalytically competent. We determined the high-resolution crystal structures of the GAOV, Cl2-Tyr272, and F2-Tyr272 incorporated variants at 1.48, 1.23, and 1.80 Å resolution, respectively. The structural data showed only one halogen remained in the cofactor, indicating that an oxidative carbon-chlorine/fluorine bond scission has occurred during the autocatalytic process of cofactor biogenesis. Using hydroxyurea as a radical scavenger, the spin-coupled hidden Cu(II) was observed by EPR spectroscopy. Thus, the structurally defined catalytic center with genetic unnatural tyrosine substitution is in the radical containing form as in the wild-type, i.e., Cu(II)-(Cl-Tyr•-Cys) or Cu(II)-(F-Tyr•-Cys). These findings illustrate a previously unobserved C−F/C-Cl bond cleavage in biology mediated by a mononuclear copper center.
Keywords: Copper protein, Free radical, Protein-derived cofactor, Unnatural amino acid, Protein engineering, Posttranlational modification, Redox chemistry
Graphical Abstract
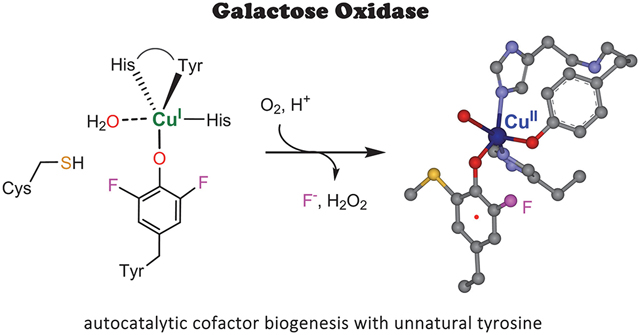
Galactose oxidase (GAO) has a broad spectrum of applications in biosensors, enzymatic synthesis, and diagnostics.1–4 A salient feature of GAO in catalysis is that its active site contains an unusual Cu(II)-bound Cys-Tyr• radical cofactor with its tyrosyl free radical directly coordinating to the type II copper center.5–7 The Cu(II)-radical cofactor performs the regioselective oxidation of primary alcohols, including galactose and other substrates ranging from glycerol, allyl alcohol to galactopyranosides, oligo- and polysaccharides.8–9 Then, the reduced Cu(I)-(Tyr-Cys) is oxidized to the active Cu(II)-(Tyr•-Cys) state through a 2e− oxidation by dioxygen (Scheme 1). A biophysically intriguing aspect is that neither the Cu(II) nor the tyrosyl radical is EPR visible. This spectral silence is due to the antiferromagnetic coupling between the unpaired electron spin of the Cys-Tyr• and the unpaired electron spin of a Cu(II) d electron, resulting in an EPR-invisible status for both paramagnetic centers.5
Scheme 1.
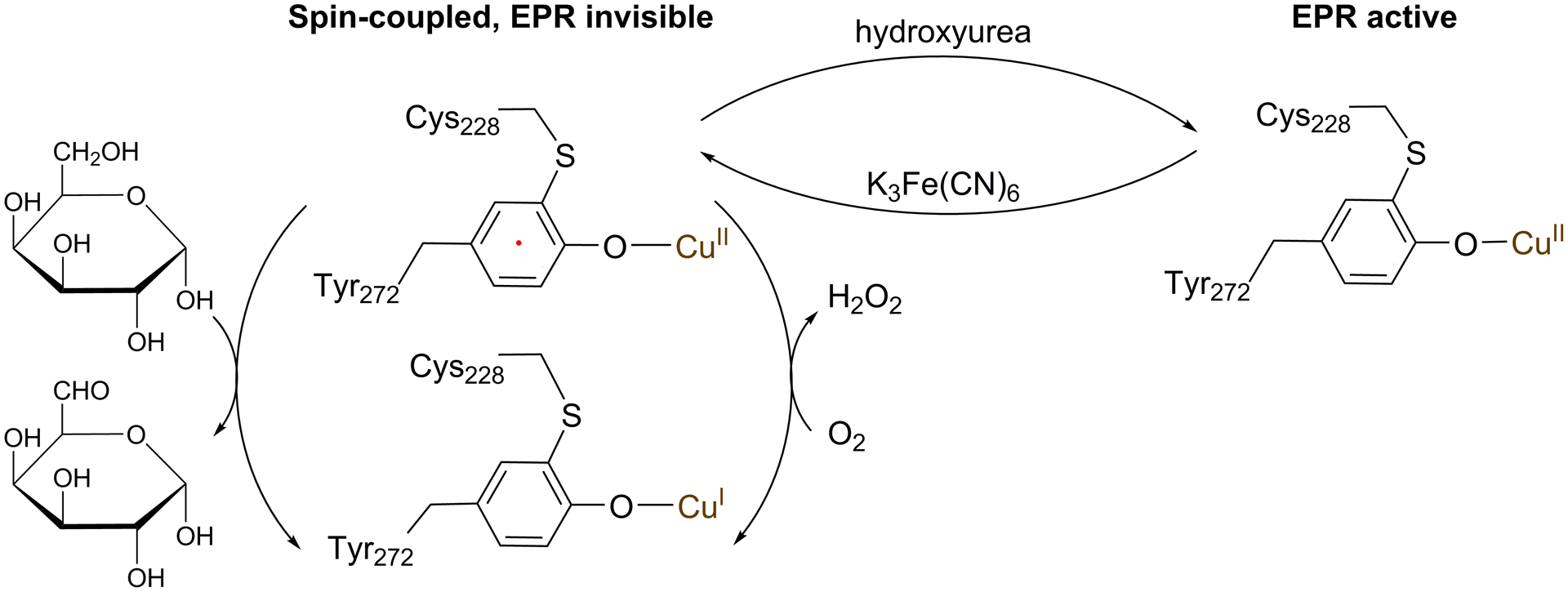
The catalytic reaction of GAO and three oxidation states of the Cu(II)-(Tyr•-Cys) center
The Cu(II)-(Tyr•-Cys) cofactor is also present in glyoxal oxidase.10–11 Although a protein-derived Cys-Tyr cofactor has been found in a growing number of metalloproteins, the cofactor in oxidases is rather unique, as it is a one-electron more oxidized than other known Cys-Tyr cofactors. Moreover, the free radical in oxidases is long-lived at ambient temperature. It carries one of the two oxidizing equivalents as the catalytic driving force to initiate oxidation chemistry on a carbohydrate substrate. In contrast, the radical-free form of Cys-Tyr cofactor found in other enzymes typically plays a role in catalytic amplification, as observed in thiol dioxygenases.12
In all cases, the Cys-Tyr crosslinkage is a result of the redox action of the metal center through an autocatalytic, irreversible, post-translational modification reaction. A major unanswered question is how these oxidases generate a specific tyrosyl free radical during the crosslinking process, and how it is stabilized. This type of information is necessary for a deeper understanding of the bioinorganic chemistry of regenerating the spin-coupled catalytic center after each turnover, and the knowledge would inspire protein design and protein-based inorganic chemistry. In the past decades, a significant effort has been invested in the study of the biogenesis of the catalytic assembly of GAO.13–18 Although notable progress has been made, much of the chemical details are still missing. The self-processing,14 single-turnover nature of the reaction brings a substantial challenge in studying the biogenesis of those Cys-Tyr cofactors.
Recently, we began to employ the genetic code expansion strategy to specifically incorporate unnatural amino acids into cofactor bearing sites. This strategy has resulted in several lines of findings for Cys-Tyr cofactor biogenesis in non-heme iron-dependent thiol dioxygenases and produced otherwise inaccessible mechanistic insights.19–21 A notable finding is that the non-heme iron(II) center cleaves a C-F bond of 3,5-difluoro-L-tyrosine (F2-Tyr) during cofactor formation.19–21 Since the C-F bond is one of the strongest single bonds,22 one fundamental question that arises is whether a type II copper center in GAO would be able to conduct a similar C-F bond scission. If so, would the resulting Cys-Tyr be a radical-containing or radical-free form, and would it be a metal ligand?
Thus, we probed the autocatalytic cofactor biogenesis by forcing the copper-bound oxidant to oxidize a C-F bond. An efficient E. coli expression system for producing an active form of the enzyme, known as GAO variant A3.E7 (GAOV), was employed in this study. This system was previously developed by Arnold et al. using directed evolution and achieved much-improved expression.23 We determined the crystal structure of GAOV, refined to 1.48 Å resolution (Figure 1, Table S1), which shows the structural outcome of GAO after the directed evolution.
Figure 1.
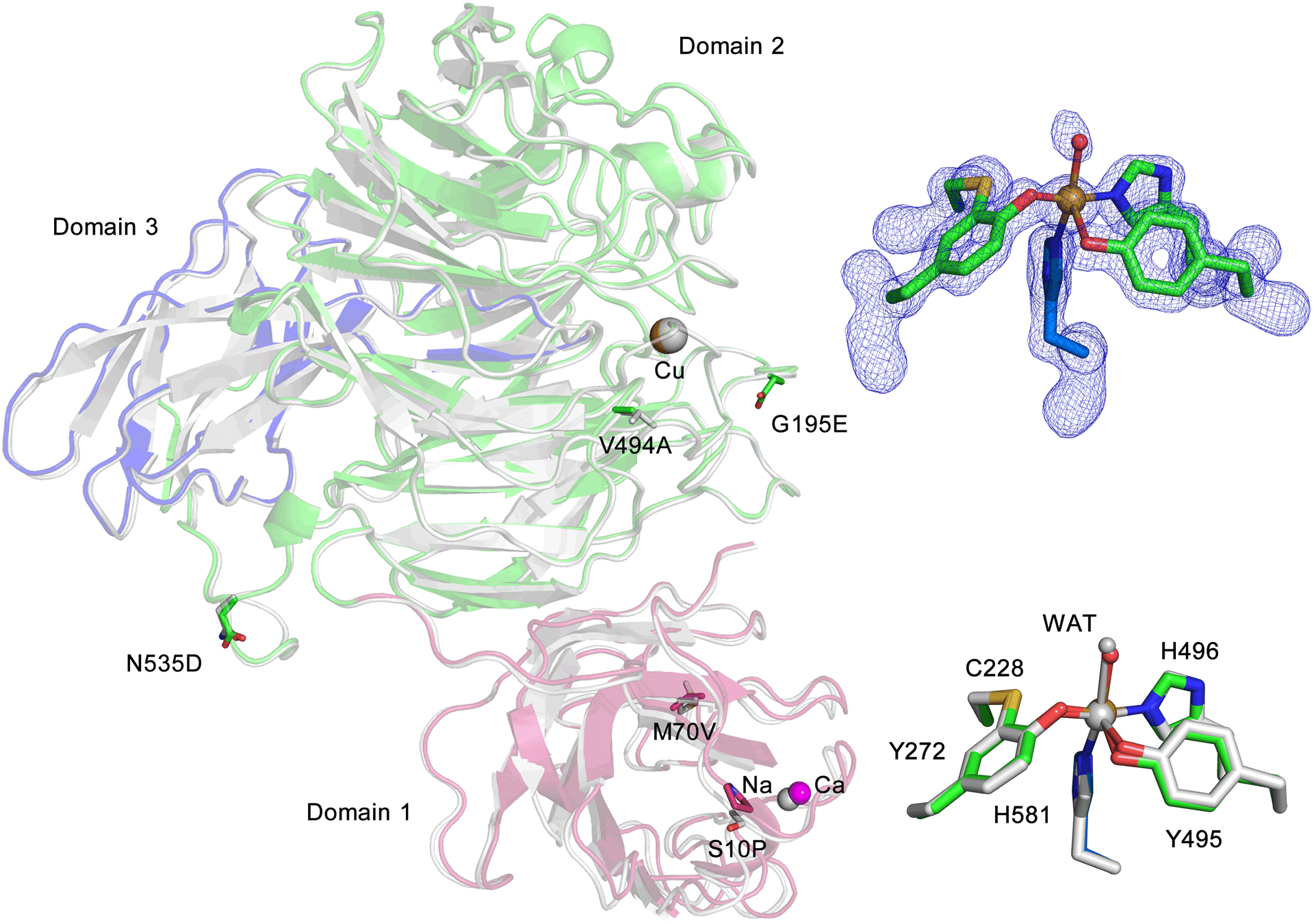
Crystal structure of GAOV and superposition with WT GAO (1GOG). The three domains of GAOV are presented in red, blue, and green with WT GAO in grey. Residues of directed evolution mutations are illustrated in stick form. The inset on the top right shows the active site of the cross-linked form GAOV, and the omit Fo-Fc electron densities (blue) is contoured at 4.0 σ. The inset on the bottom right shows the comparison of the active sites of both structures.
Compared to the wild-type enzyme, GAOV contains six mutations in its three-domain architecture, S10P, M70V, G195E, V494A, N535D, and Pro136 as a silent mutation at the DNA level. No significant changes in the overall structure (RMSD of 0.28 Å for 527 Cα carbons) were observed in comparison to the wild-type structure of GAO (1GOG).6–7 The copper ion is ligated by Tyr272, Tyr495, His496, His581, and one water ligand. Cys228 and Tyr272 are crosslinked through a thioether bond (Figure 1).
Next, we genetically substituted Tyr272 with 3,5-dichloro-L-tyrosine (Cl2-Tyr) and F2-Tyr (SI Experimental). The specific aminoacyl-tRNA synthetase and tRNA pair led to a clean, 100% substitution at the 272 position of GAOV, whereas tyrosine residues in other positions were not altered. The as-isolated proteins showed two bands in SDS-PAGE corresponding to crosslinked and uncrosslinked forms (Figure 2). The rough estimates of the percentage of crosslink formed in the as-isolated samples were approximately 95%, 31%, and 64% for GAOV, F2-Tyr272 GAOV, and Cl2-Tyr272 GAOV, respectively. Further processing of halogen-substituted GAOV with Cu(I) and oxygen-saturated buffer led to a higher ratio of crosslinked form (Figure S1).
Figure 2.
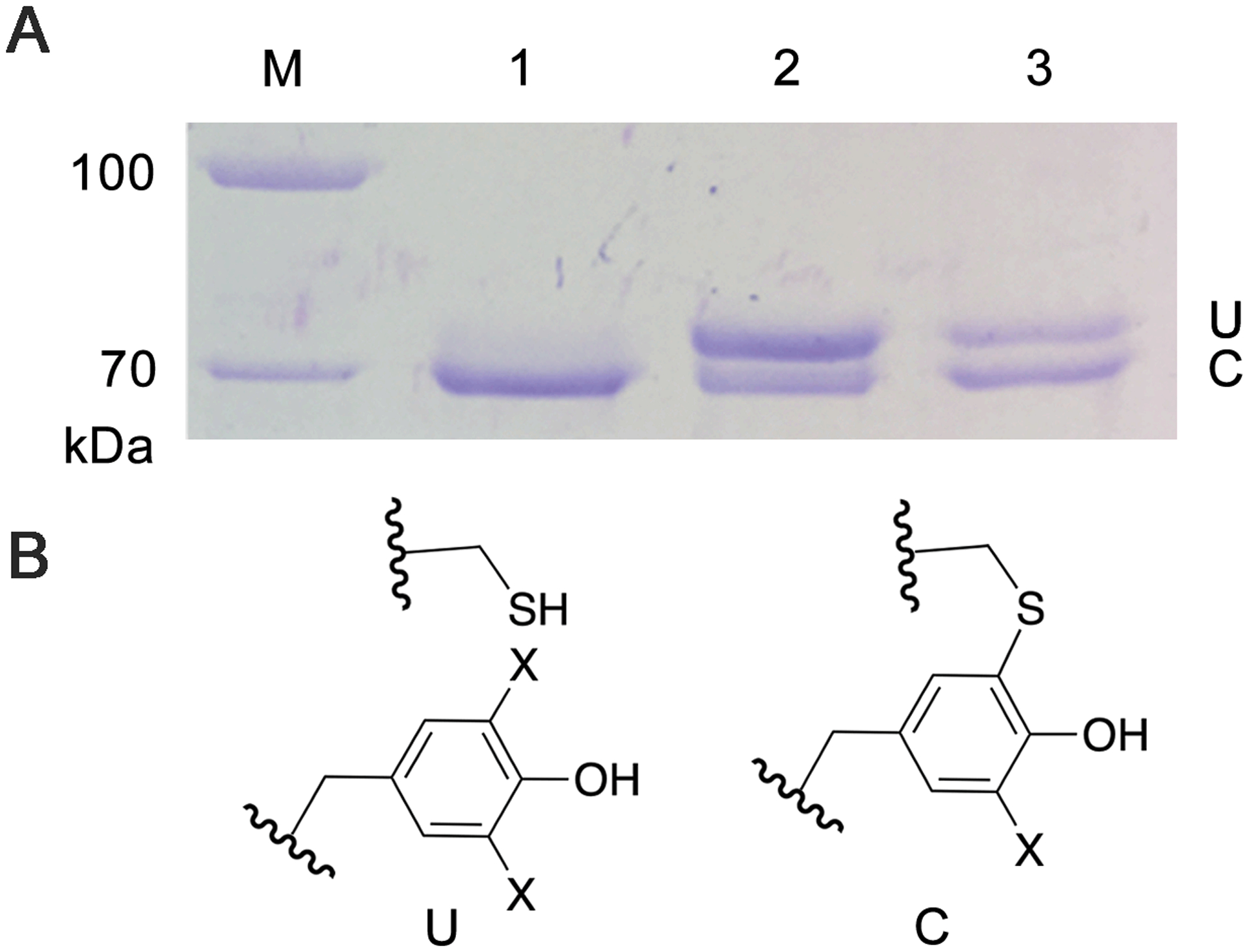
The genetic incorporation of an unnatural tyrosine into GAOV. (A) The slower moving band corresponding to uncrosslinked protein (U) and faster moving band corresponding to the mature GAOV (C). The lanes are molecular marker (M), GAOV (1), F2-Tyr272 GAOV (2), and Cl2-Tyr272 GAOV (3), respectively. (B) The crosslinked and uncrosslinked chemical structures (X=H/F/Cl).
The protein ESI mass spectra obtained at various charge states showed that both F2-Tyr272 GAOV and Cl2-Tyr272 GAOV are mixtures containing both crosslinked and uncrosslinked forms (Table S2). At the +53 charge state, the uncrosslinked form under denaturing conditions showed experimentally determined masses of 70,010 and 70,047 Da for the GAOV and F2-Tyr272 GAOV, respectively. These data indicated that the uncrosslinked F2-Tyr272 GAOV contains the two expected fluorine atoms (Figure S2).
We set out to determine if crosslink formation is realized by breaking the newly introduced C-F or C-Cl bond. We determined the crystal structures of Cl2-Tyr272 GAOV and F2-Tyr272 GAOV, refined to 1.23 Å and 1.80 Å resolution, respectively (Figure 3). Both structures of the unnatural tyrosine variants are nearly identical to the structure of GAOV, with RMSD of 0.06 Å for 565 Cα carbons of GAOV and Cl2-Tyr272 GAOV and 0.08 Å for 590 Cα carbons of GAOV and F2-Tyr272 GAOV.
Figure 3.
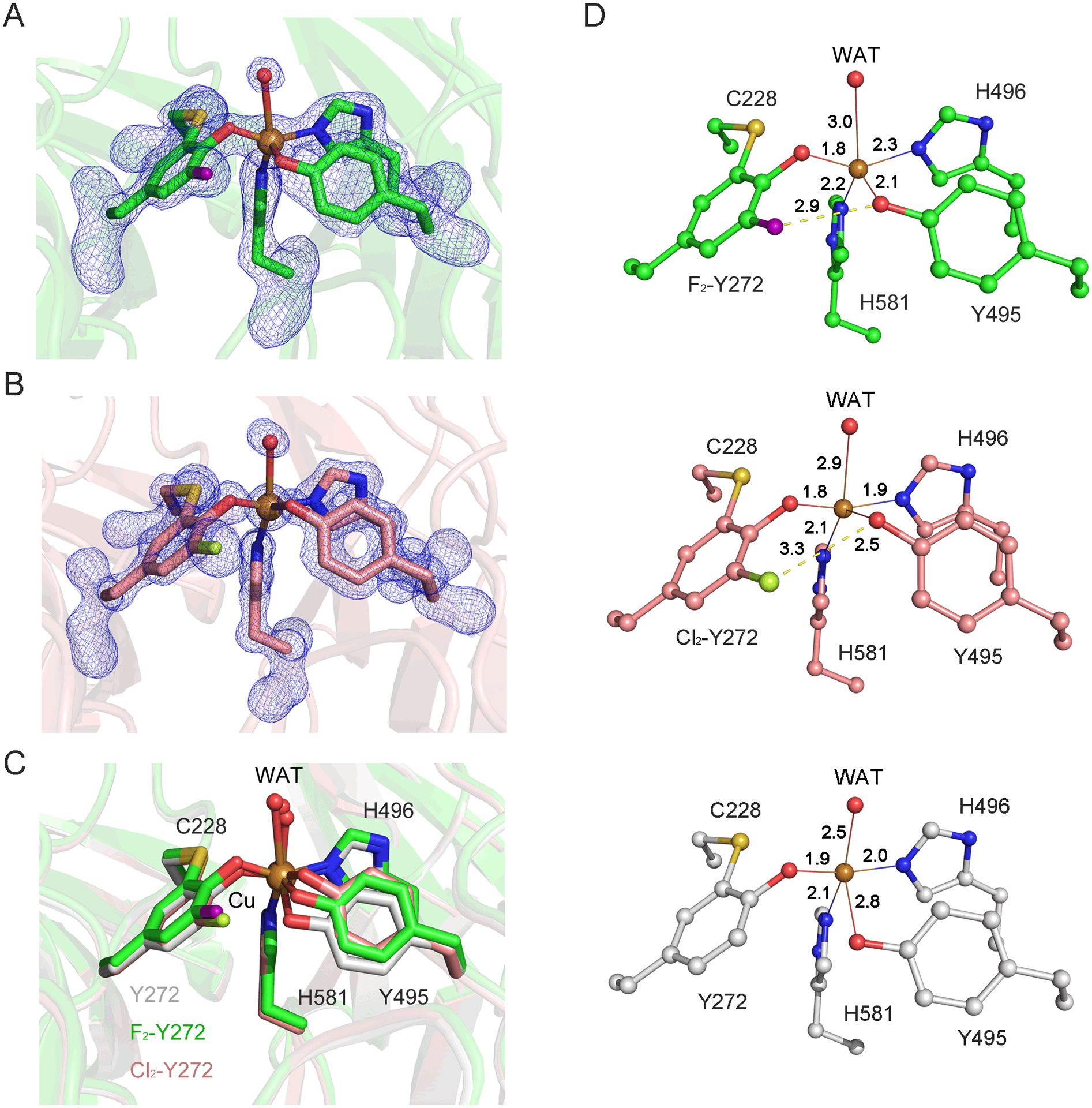
The crystal structures of F2-Tyr272 and Cl2-Tyr272 GAOV reveal C-F and C-Cl bond cleavages in the Cu-bound crosslinked cofactor. (A) The omit Fo-Fc electron densities (blue, 4.0 σ) and the cross-linked form F2-Tyr272 GAOV. The fluorine atom was highlighted in purple. (B) The omit Fo-Fc electron densities (blue, 4.0 σ) and the cross-linked form of Cl2-Tyr272 GAOV active site. The chlorine atom was colored in mint. (C) Comparison of the active sites of all three structures. The active site of F2-Tyr272 GAOV, Cl2-Tyr272 GAOV, GAOV are shown in green, pink, and grey, respectively. WAT represents the water ligand bound at the Cu(II) center. (D) The active site interaction diagrams of F2-Tyr272 GAOV, and Cl2-Tyr272 GAOV, and GAOV.
The high-resolution crystal structures unambiguously demonstrate that the specific substitution of halogen atoms at the 3,5 position of Tyr272 does not halt cofactor biogenesis. These structures also showed that only one halogen atom remains in the crosslinked cofactor, indicating a C-F or C-Cl bond transformation has taken place during a copper-mediated oxidative process. It became evident that the halogen substitution of Tyr272 does not affect the overall geometry of the cofactor, though it does affect the position of Tyr495. Figure 3D shows metal-ligand interaction diagrams of the active site. No significant difference in the distances from the copper center to Tyr272, His581, and His496 in these three structures was observed. However, the lengths of Tyr495 ligand and the water to copper center vary.
The halogen-bearing GAOV proteins were catalytically active, but their activity is lower than that of GAOV (Figure S3). The determined kinetic parameters are summarized in Table 1. The reduced activity in F2-Tyr272 GAOV is likely due to the electron-withdrawing nature of the fluorine atom. In Cl2-Tyr272 GAOV, an additional geometric disturbance likely contributes to the further reduction of catalytic activity because of the elevated KM value (Table 1).
Table 1.
Catalytic parameters of GAOV proteins
| Enzyme | kcat (s−1) | KM (mM) |
kcat/KM (M−1s−1) |
|---|---|---|---|
| GAOV | 544.9 ± 16.1 | 28.5 ± 1.3 | 19,115 |
| F2-Tyr272 | 62.7 ± 5.2 | 14.8 ± 2.5 | 4,265 |
| Cl2-Tyr272 | 16.9 ± 3.3 | 203.5 ± 50.7 | 83 |
An EPR study was performed to examine if the halogen-substituted, crosslinked cofactor contains a free radical and to detect if a significant perturbation is present in the electronic structure and geometry of the copper ion in the radical-free form. The GAOV protein after reconstitution with Cu(II) showed g = 2 EPR resonances arising from a characteristic low-spin (S = 1/2) Cu(II) signal, which is consistent with previous reports.5, 24 This copper signal was derived from the copper center without a free radical. The treatment of GAOV with an oxidant K3Fe(CN)6 caused a decrease in signal intensity (Figure 4A, red trace). The decreased Cu(II) EPR signal indicates the formation of a ligand radical that is spin-coupled with the Cu(II) ion. In reconstituted F2-Tyr272 GAOV, oxidation caused a similar decrease of the Cu(II) EPR signal (Figure 4B), indicating a portion of the copper center becoming hidden due to spin coupling with a ligand radical. A summary of the EPR signal change is included in Table S3.
Figure 4.
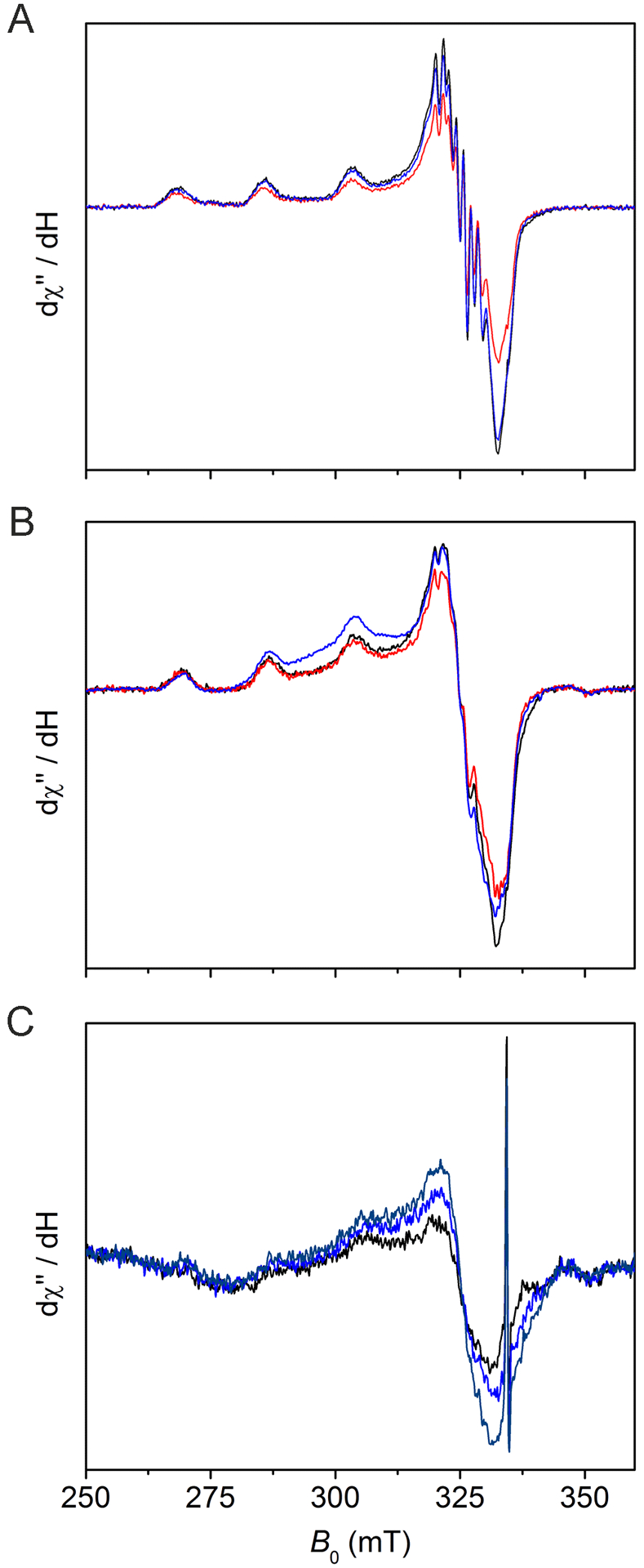
Investigation of the GAOV and variant active site radical content by EPR spectroscopy. Black traces in each panel: (A) Cu(II)-reconstituted GAOV(100) μM, (B) Cu(II)-reconstituted F2-Tyr272 GAOV(50 μM), and (C) as-isolated F2-Tyr272 GAOV (100 μM). Red traces: after K3Fe(CN)6 oxidation. Blue traces: after hydroxyurea incubation for 1 min. Navy trace in panel C: after a prolonged hydroxyurea treatment (2 h). The spectra were collected at 30 K, 0.05 mW microwave power, 0.6 mT modulation amplitude, and 100 kHz modulation frequency.
Subsequently, we employed a well-known protein radical scavenger, hydroxyurea (HU), to quench the Cys-Tyr• radical in order to break spin coupling and reveal the Cu(II) center by EPR spectroscopy. We first tested it with GAOV before applying it to F2-Tyr272 GAOV. The addition of HU led to increased Cu(II) EPR signal in all cases (Figure 4, blue traces), indicating conversion of a radical-containing cofactor to the radical-free form (Scheme 1). A sharp spark in panel C was a background signal. A prolonged HU incubation led to a further increase of the Cu(II) EPR signal in the as-isolated sample that had not been reconstituted or oxidized by K3Fe(CN)6 (Figure 4C, navy trace). These results indicate that there is a (Cys-F-Tyr)• radical cofactor ligated to the copper center in the as-isolated and reconstituted F2-Tyr272 GAOV.
Compared to the GAOV, the percentage of the crosslinked cofactor is lower in proteins with halogen-substituted Tyr272. The less complete conversion in halogen-substituted GAOV could be due to a combination effects of the energetics of the C-H/C-F/C-Cl bonds and the size of the halogen atoms. In our previous study, we estimated that the dissociation energy of the aromatic C−F bond (132.66 kcal/mol) is 10.12 kcal/mol greater than that of the corresponding aromatic C−H bond.21 The structural data reported here would enable computational study for further energetics analysis as for cysteine dioxygenase.25 When further processing F2-Tyr272 GAOV with Cu(I) and O2, we observed increased crosslink formation (Figure S1), but far from the level found in GAOV (Figure 2A) even after incubation for 20 h. The observation that the posttranslational modification still occurs, but with reduced efficiency, in the fluorine substituted tyrosine indicates that the C-F bond dissociation energy is near the limit of the oxidizing power of the oxidant. Hence, this fluorine-substitution gauged the oxidizing power of the copper-bound oxidant and the presumed thiyl radical during cofactor biogenesis.
The mononuclear copper-mediated C-F bond cleavage is unprecedented in biology. The only other known system is a multicopper-dependent laccase-mediator system, by which a synthetic mediator is oxidized to the radical form and subsequently performs an oxidative decomposition of fluorinated hydrocarbons through radical propagation and rearrangement.22, 26 The copper-mediated C-F bond transformation in GAO is a result of oxidation on a copper-bound ligand.
Using EPR spectroscopy, the cofactor is shown to be a ligand radical in the unnatural tyrosine substituted variants. However, with an electron-withdrawing substituent in the cofactor, such as F or Cl, the Cu(II)-bound radical cofactor F-Tyr•-Cys or Cl-Tyr•-Cys is catalytically less active. This is consistent with the expectation that the presence of an F or Cl reduces the efficiency of the radical transfer from the cofactor to the monosaccharide substrate during catalysis.
These results led us to propose a mechanistic model for the formation of the Cu(II)-(F-Tyr•-Cys) cofactor (Figure 5). Dioxygen binds to the Cu(I) ion of the enzyme, producing a Cu-bound superoxide anion radical. Subsequent oxidation of Cys228 though hydrogen atom abstraction by the Cu(II)-superoxide generates a thiyl radical at Cys228 and a Cu-bound hydroperoxo species. Oxidative attack on the copper ligand F2-Tyr272 by the thiyl radical creates the crosslink with the formation of a thioether bond and a tyrosyl-like radical. As previously demonstrated in thiol dioxygenases, F− is a leaving group, which carries away two electrons.19, 21
Figure 5.
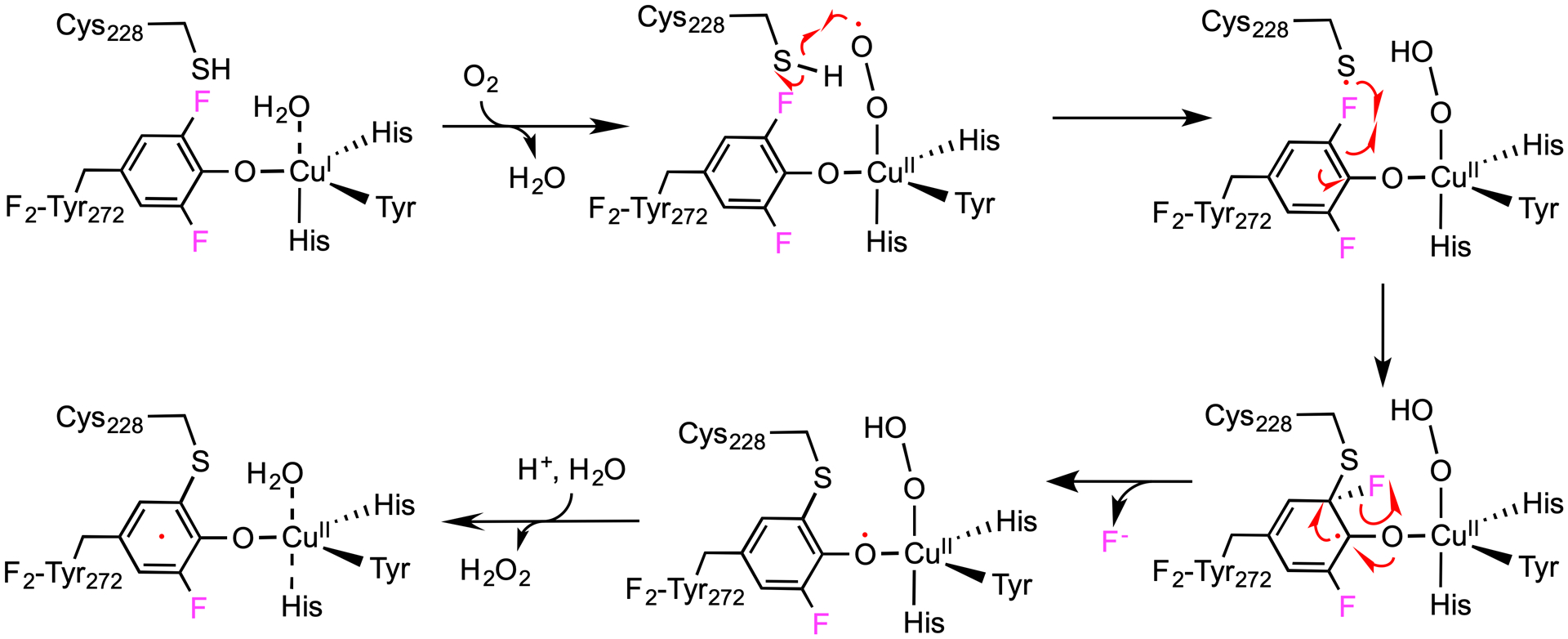
Proposed model for C-F bond session and cofactor biogenesis in F2-Tyr272 GAOV. A copper-bound superoxide radical oxidizes Cys228 and produces a copper-bound hydroperoxide and a thiol radical. Oxidation of F2-Tyr272 by the thiol radical leads to the formation of a thioether bond and Cu(II)-(F-Tyr•-Cys).
The above mechanistic proposal lends support to one of the working models for cofactor biogenesis in the copper-dependent oxidases (Figure S4).18 The difference in the outcome of C-H bond cleavage leads to variations in the subsequent steps. The Cu-bound hydroperoxo species obtains one proton from the phenol of Tyr272 through the water-mediated H-bond network and promotes the formation of a hydrogen peroxide side product. Finally, a second oxygen molecule abstracts two electrons from the Cu(I) center and cofactor, leading to the production of the copper(II)/(Cys-Tyr)• and second hydrogen peroxide molecule.
In conclusion, the highly selective, chemical alteration of Tyr272 through genetic incorporation of F2-Tyr and Cl2-Tyr led to the observation of carbon-halogen bond scission chemistry performed by the copper center in the oxidase and the formation of a mono-halogenated, cross-linked, radical cofactor in the form of Cu(II)-(F-Tyr•-Cys), which is catalytically active with reduced efficiency. This study has revealed new insights into the power of the copper-dependent oxidase, and it is of particular chemical interest for its bioinorganic, biophysical, and environmental chemistries.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Jiangyun Wang for providing the GAOV expression plasmid and tRNA TyrRS system for the TAG codon used in this and our previous studies.19 We thank the staff scientists for assistance remote data collections at the beamline 9-2 of the Stanford Synchrotron Radiation Lightsource (SSRL) and beamlines 19-BM, 19-ID, 22-ID, and 22-BM in the Argonne National Laboratory of Advanced Photon Source (APS). Use of the SSRL and APS is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515 and No. DE-AC02-06CH11357, respectively. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393).
Funding Sources
This work was partially supported by NSF grant CHE-1808637, NIH grant GM108988, and the Lutcher Brown Endowment Fund (to A.L.). The mass spectrometry facility used in this work was sponsored by the National Institutes of Health Grant G12MD007591.
Footnotes
Supporting Information
Experimental procedure, Table S1-S3, and Figures S1-S4. This material is available free of charge via the Internet at http://pubs.acs.org.”
Data Availability
The structural coordinates are available at PDB with the accession codes 6XLR, 6XLS, and 6XLT.
REFERENCES
- (1).Whittaker JW Free radical catalysis by galactose oxidase. Chem. Rev 2003, 103, 2347–2363. [DOI] [PubMed] [Google Scholar]
- (2).Liu XC; Dordick JS Sugar-containing polyamines prepared using galactose oxidase coupled with chemical reduction. J. Am. Chem. Soc 1999, 121, 466–467. [Google Scholar]
- (3).Kanyong P; Krampa FD; Aniweh Y; Awandare GA Enzyme-based amperometric galactose biosensors: a review. Microchimica Acta 2017, 184, 3663–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Nie YX; Liu Y; Zhang Q; Su XG; Ma Q Novel coreactant modifier-based amplified electrochemiluminescence sensing method for point-of-care diagnostics of galactose. Biosensors & Bioelectronics 2019, 138, 111318. [DOI] [PubMed] [Google Scholar]
- (5).Whittaker MM; Whittaker JW The active site of galactose oxidase. J. Biol. Chem 1988, 263, 6074–6080. [PubMed] [Google Scholar]
- (6).Ito N; Phillips SEV; Stevens C; Ogel ZB; Mcpherson MJ; Keen JN; Yadav KDS; Knowles PF Novel thioether bond revealed by a 1.7 Å crystal structure of galactose oxidase. Nature 1991, 350, 87–90. [DOI] [PubMed] [Google Scholar]
- (7).Ito N; Phillips SE; Yadav KD; Knowles PF Crystal structure of a free radical enzyme, galactose oxidase. J. Mol. Biol 1994, 238, 794–814. [DOI] [PubMed] [Google Scholar]
- (8).Schlegel RA; Gerbeck CM; Montgomery R Substrate specificity of D-galactose oxidase. Carbohyd. Res 1968, 7, 193–199. [Google Scholar]
- (9).Avigad G Oxidation rates of some desialylated glycoproteins by galactose oxidase. Arch. Biochem. Biophys 1985, 239, 531–537. [DOI] [PubMed] [Google Scholar]
- (10).Whittaker MM; Kersten PJ; Cullen D; Whittaker JW Identification of catalytic residues in glyoxal oxidase by targeted mutagenesis. J. Biol. Chem 1999, 274, 36226–36232. [DOI] [PubMed] [Google Scholar]
- (11).Yin DL; Urresti S; Lafond M; Johnston EM; Derikvand F; Ciano L; Berrin JG; Henrissat B; Walton PH; Davies GJ; Brumer H Structure-function characterization reveals new catalytic diversity in the galactose oxidase and glyoxal oxidase family. Nat. Commun 2015, 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Dominy JE Jr.; Hwang J; Guo S; Hirschberger LL; Zhang S; Stipanuk MH Synthesis of amino acid cofactor in cysteine dioxygenase is regulated by substrate and represents a novel post-translational regulation of activity. J. Biol. Chem 2008, 283, 12188–12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Rogers MS; Hurtado-Guerrero R; Firbank SJ; Halcrow MA; Dooley DM; Phillips SE; Knowles PF; McPherson MJ Cross-link formation of the cysteine 228-tyrosine 272 catalytic cofactor of galactose oxidase does not require dioxygen. Biochemistry 2008, 47, 10428–10439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Rogers MS; Baron AJ; McPherson MJ; Knowles PF; Dooley DM Galactose oxidase pro-sequence cleavage and cofactor assembly are self-processing reactions. J. Am. Chem. Soc 2000, 122, 990–991. [Google Scholar]
- (15).Whittaker MM; Whittaker JW Cu(I)-dependent biogenesis of the galactose oxidase redox cofactor. J. Biol. Chem 2003, 278, 22090–22101. [DOI] [PubMed] [Google Scholar]
- (16).Firbank SJ; Rogers MS; Wilmot CM; Dooley DM; Halcrow MA; Knowles PF; McPherson MJ; Phillips SE Crystal structure of the precursor of galactose oxidase: an unusual self-processing enzyme. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 12932–12937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Rokhsana D; Dooley DM; Szilagyi RK Structure of the oxidized active site of galactose oxidase from realistic in silico models. J. Am. Chem. Soc 2006, 128, 15550–15551. [DOI] [PubMed] [Google Scholar]
- (18).Cowley RE; Cirera J; Qayyum MF; Rokhsana D; Hedman B; Hodgson KO; Dooley DM; Solomon EI Structure of the reduced copper active site in preprocessed galactose oxidase: Ligand tuning for one-electron O2 activation in cofactor biogenesis. J. Am. Chem. Soc 2016, 138, 13219–13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Li J; Griffith WP; Davis I; Shin I; Wang J; Li F; Wang Y; Wherritt DJ; Liu A Cleavage of a carbon-fluorine bond by an engineered cysteine dioxygenase. Nat. Chem. Biol 2018, 14, 853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Li J; Koto T; Davis I; Liu A Probing the Cys-Tyr cofactor biogenesis in cysteine dioxygenase by the genetic incorporation of fluorotyrosine. Biochemistry 2019, 58, 2218–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wang Y; Griffith WP; Li J; Koto T; Wherritt DJ; Fritz E; Liu A Cofactor biogenesis in cysteamine dioxygenase: C-F bond cleavage with genetically incorporated unnatural tyrosine. Angew. Chem. Int. Ed 2018, 57, 8149–8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wang Y; Liu A Carbon-fluorine bond cleavage mediated by metalloenzymes. Chem. Soc. Rev 2020, 49, 4906–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sun L; Petrounia IP; Yagasaki M; Bandara G; Arnold FH Expression and stabilization of galactose oxidase in Escherichia coli by directed evolution. Protein Eng 2001, 14, 699–704. [DOI] [PubMed] [Google Scholar]
- (24).Cleveland L; Coffman RE; Coon P; Davis L An investigation of the role of the copper in galactose oxidase. Biochemistry 1975, 14, 1108–1115. [DOI] [PubMed] [Google Scholar]
- (25).Song Z; Yue Y; Feng S; Sun H; Li Y; Xu F; Zhang Q; Wang W Cysteine dioxygenase catalyzed C-F bond cleavage: An in silico approach. Chem. Phys. Lett 2020, 750, article #137449. [Google Scholar]
- (26).Luo Q; Yan X; Lu J; Huang Q Perfluorooctanesulfonate degrades in a laccase-mediator system. Environ. Sci. Technol 2018, 52, 10617–10626. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.