

Abstract

In addition to its therapeutic value as a chemotherapy drug, gemcitabine is of ongoing interest to the scientific community for its broad-spectrum antiviral activity. Herein the synthesis of 4′-methoxy- and 4′-fluoro-substituted gemcitabine analogues along with their phosphoramidate prodrugs is described. Among these derivatives, 4′-fluorogemcitabine proved to be active against varicella zoster virus (VZV, TK+ strain) with an EC50 of 0.042 μM and produced significant cytotoxicity (CC50 = 0.11 μM). Upon derivatization of this trifluoro nucleoside as its prodrug, decreased anti-VZV activity was observed, but with a concomitantly improved selectivity index (SI = 36). When this prodrug was tested against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), its antiviral activity (EC50 = 0.73 μM) was comparable to or slightly lower than its cytotoxic concentration in measurements of cell growth and cell morphology, respectively.
Keywords: Gemcitabine analogues, phosphoramidate prodrug, varicella zoster virus, severe acute respiratory syndrome coronavirus 2, antiviral activity
Gemcitabine (2′-deoxy-2′,2′-difluorocytidine, dFdC; Figure 1) is a pyrimidine nucleoside analogue that is in clinical use for the treatment of various solid tumors, including nonsmall cell lung, pancreatic, bladder, and breast cancer.1 By acting as a potent antimetabolite, it inhibits two cellular processes that are both required for DNA biosynthesis, i.e., nucleotide reduction and replicative DNA chain elongation. Upon phosphorylation in cells, it is converted to its biologically active metabolites, namely, gemcitabine diphosphate (dFdCDP) and triphosphate (dFdCTP).2 While dFdCDP acts as an inhibitor of ribonucleotide reductase (RNR), thus preventing the biosynthesis of deoxynucleotide building blocks that are required for DNA synthesis, dFdCTP is competitively incorporated into DNA in place of natural 2′-deoxycytosine-5′-triphosphate (dCTP).3
Figure 1.

Gemcitabine and its 4′-substituted derivatives investigated in this study.
Furthermore, gemcitabine was also demonstrated to exert broad-spectrum in vitro activity against a range of RNA viruses. In particular, its antiviral inhibitory effect was assessed against Zika virus (ZIKV) (EC50 = 0.01 μM),4 hepatitis C virus (HCV) (EC50 = 12 nM),5 human immunodeficiency virus 1 (HIV-1) (EC50 = 16.3 nM),6 and poliovirus (IC50 = 0.3 μM)7 as well as respiratory viruses such as influenza A virus (IAV) (EC50 = 0.068 μM),8 human rhinovirus (HRV) (EC50 = 0.81–1.92 μM),9 Middle East respiratory syndrome coronavirus (MERS-CoV) (EC50 = 1.22 μM),10 and severe acute respiratory syndrome coronavirus (SARS-CoV) (EC50 = 4.95 μM).10 In addition, a recent report described the activity of this modified nucleoside against the emerging coronavirus SARS-CoV-2 (EC50 = 1.24 μM) that is responsible for the current worldwide viral pneumonia outbreak.11
The modification of gemcitabine as a means to convert a life-saving antitumoral drug into an active compound for the treatment of severe viral infections represents an attractive research endeavor. In this context, a variety of gemcitabine analogues have been synthesized to increase its selectivity as antiviral agent.12,13 The rationale behind this strategy aims at reducing the inhibitory activity of dFdCDP and dFdCTP against the cellular RNR and polymerase, respectively, while maintaining the viral polymerase inhibitory capacity of the dFdCTP metabolite.
It is well-known that subtle structural modifications of nucleosides can have a profound impact on their biological profiles. In this study, we selected a methoxy group as an electron-withdrawing substituent via the inductive effect and a fluorine atom as a strongly inductive electron-withdrawing substituent to modify the 4′-position of gemcitabine in order to identify compounds that could lead to reduced cell toxicity while retaining activity against selected viruses. Sterically, both substituents are small, as larger groups might be detrimental for an effective interaction with the target enzymes. In addition, the presence of substituents with different electronic properties might influence the reactivity of the primary hydroxyl group, which needs to undergo intracellular phosphorylation to deliver the active metabolite. The synthesis of 4′-methoxy- and 4′-fluorogemcitabine analogues (1a and 1b, respectively; Figure 1) was complemented by that of the corresponding prodrugs (2a and 2b; Figure 1). A comparison of the antiviral activity of a nucleoside itself (which still needs to undergo three consecutive phosphorylations) and its prodrug (bypassing the first phosphorylation to the nucleoside monophosphate) was expected to provide useful information for the design of new gemcitabine analogues.
Herein, varicella zoster virus (VZV) and human cytomegalovirus (HCMV) were chosen as the two most important herpes viruses. Although there are anti-VZV and anti-HCMV drugs on the market, toxicity is an issue for some of these drugs, and the emergence of drug resistance has been described for all of them among immunocompromised patients. Novel nontoxic antiviral chemotherapeutics that are more potent and effective than the currently available drugs are therefore required for the treatment of these viral infections in at-risk populations. At the same time, SARS-CoV-2 was privileged among RNA viruses because of the extremely urgent need to develop new antiviral treatments for this infection. As shown in Scheme 1, commercially available gemcitabine served as a convenient starting point for the introduction of fluoro and methoxy substituents at the 4′-position.
Scheme 1. Synthetic Routes for the Preparation of 4′-Substituted Gemcitabine Analogues.
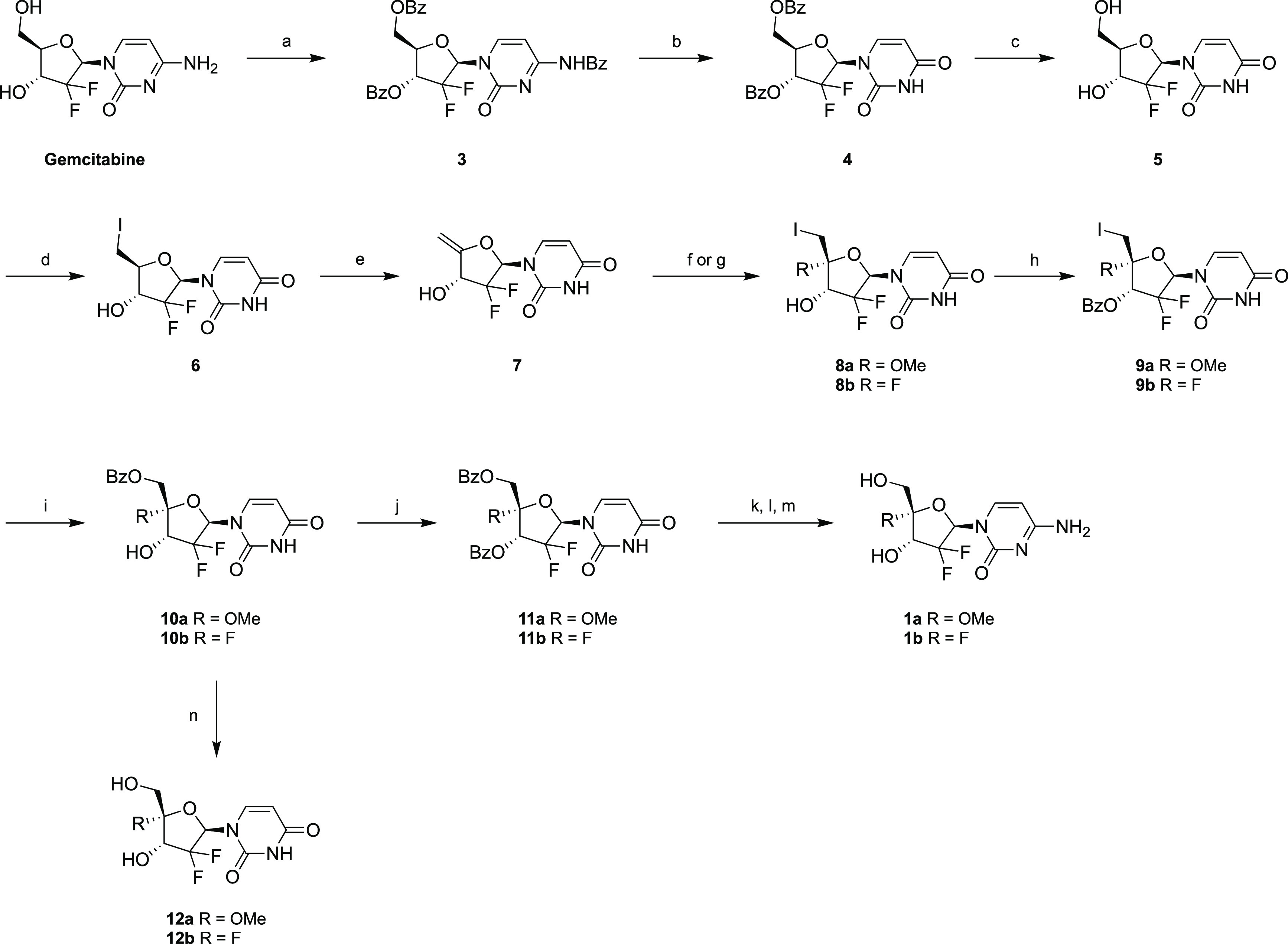
Reagents and conditions: (a) BzCl, pyr, 93%; (b) 80% AcOH, reflux, 90%; (c) 7 M NH3 in MeOH, 85%; (d) imidazole, PPh3, I2, THF, 84%; (e) DBU, THF, 80 °C, 65%; (f) I2, PbCO3, anhydrous MeOH for 8a, 61%; (g) I2, AgF, anhydrous THF for 8b, 35%; (h) BzCl, anhydrous pyr, 0 °C, 94 and 87% for 9a and 9b, respectively; (i) m-CPBA, CH2Cl2/H2O, 40 °C, 72 and 59% for 10a and 10b, respectively; (j) BzCl, triethylamine, DMAP, anhydrous THF, 65 and 75% for 11a and 11b, respectively; (k) POCl3, 1,2,4-triazole, anhydrous MeCN; (l) 25% NH4OH(aq), MeCN; (m) 7 M NH3 in MeOH, 38 and 56% for 1a and 1b, respectively, over three steps; (n) 7 M NH3 in MeOH, 68 and 51% for 12a and 12b, respectively.
First, uridine congener 5 was obtained in an overall yield of 71% via a three-step sequence including complete functional group protection, nucleobase deamination, and O-debenzoylation using standard literature procedures.14 Next, the primary hydroxyl group of compound 5 was substituted with an iodine atom upon treatment with iodine and triphenylphosphine to yield 6 in good yield (84%). When 6 was subjected to an elimination reaction under strongly basic conditions, methylene derivative 7 was obtained in 65% yield and served as a key synthon for the further introduction of a methoxy or fluorine substituent at the 4′-position to afford intermediate compounds 8a and 8b in 61 and 35% yield, respectively. The stereoselectivity of this step was most likely due to the participation of the C2 carbonyl of the uracil nucleobase via formation of a 4′-anhydro intermediate, as previously postulated.14−16 Subsequent protection of the 3′-hydroxy group of 8a and 8b as a benzoyl moiety furnished compounds 9a and 9b in 94 and 87% yield, respectively; these were further converted to 10a and 10b by transfer of the 3′-benzoyl group to the 5′-position upon treatment with m-CPBA in a solvent mixture (CH2Cl2/H2O, 4/1). Then the benzoyl group of 10a and 10b was deprotected to afford 4′-methoxy- and 4′-fluorouridine analogues 12a and 12b in 68 and 51% yield, respectively. Alternatively, the 3′-hydroxy group of compounds 10a and 10b was reprotected using benzoyl chloride in pyridine followed by nucleobase conversion via a triazole intermediate and final deprotection under basic conditions to afford compounds 1a and 1b in 38 and 56% yield, respectively, over three steps.15,17
The aryloxy triester phosphoramidate prodrug or ProTide technology is a strategy that has proven to be very effective in bypassing the first rate-limiting phosphorylation step in the nucleoside activation cascade to their 5′-triphosphate forms and facilitate intracellular delivery.18 For instance, the application of this approach to gemcitabine led to enhanced biological activities and decreased drug resistance.19 Herein, phosphoramidate prodrugs of compounds 1a and 1b along with that of gemcitabine were synthesized as shown in Scheme 2. First, 1a, 1b, and gemcitabine were selectively protected at the 3′-position by treatment with di-tert-butyl dicarbonate (DBDC) and sodium carbonate in a 4:1 mixture of dioxane and water as the solvent to afford compounds 13a–c, respectively. In parallel, l-alanine benzyl ester hydrochloride (14) was converted to its phenyl aminoacyl phosphorochloridate 15 upon reaction with phenyl phosphorochloridate. Then compounds 13a–c were reacted with compound 15 to furnish compounds 16a–c in moderate yields. Finally, the Boc protecting group was cleaved under acidic conditions to afford the desired compounds 2a–c.
Scheme 2. Conversion of Gemcitabine and Its 4′-Substituted Analogues to Their Phosphoramidate Prodrugs.
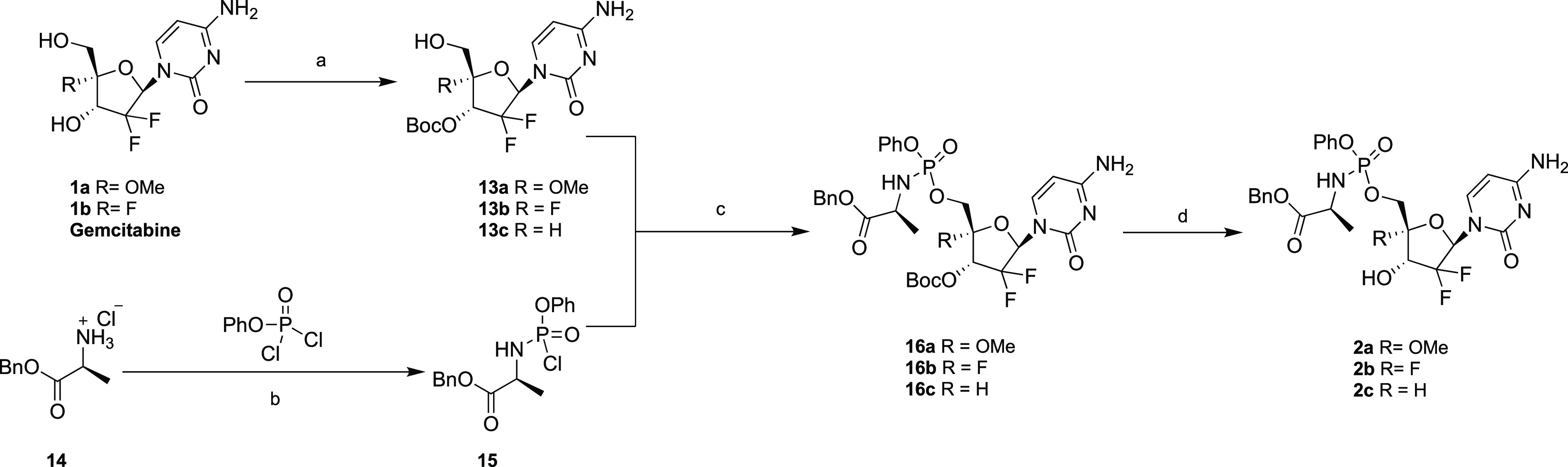
Reagents and conditions: (a) DBDC, Na2CO3, dioxane/H2O, 35% for 13a, 23% for 13b, and 59% for 13c; (b) phenyl dichlorophosphate, triethylamine, CH2Cl2, −78 °C, quantitative yield; (c) tert-butylmagnesium chloride, anhydrous THF; (d) trifluoroacetic acid/CH2Cl2, 0 °C, 49% for 16a, 54% for 16b, and 32% for 16c.
All of the novel compounds (1a, 1b, and 2a–c) were evaluated for their antiviral activity (expressed as EC50) against VZV (strains TK+, thymidine kinase-competent, and TK–, thymidine kinase-deficient) and HCMV (strains AD-169 and Davis), with concomitant determination of cytotoxicity and cytostatic effects (CC50) (Table 1). Gemcitabine was included as a reference compound, and acyclovir, brivudine, ganciclovir, and cidofovir were included as positive controls.
Table 1. Antiviral Activities and Cytotoxicities of 4′-Substituted Gemcitabine Analogues and Their Prodrugs against VZV and HCMV.
| antiviral
activity, EC50 (μM)a |
|
|
||||||
|---|---|---|---|---|---|---|---|---|
| VZV |
HCMV |
HEL
cytotoxicity (μM) |
VZV
SIe |
|||||
| compound | TK+ strain | TK– strain | AD-169 strain | Davis strain | MCCb | CC50c | TK+ strain | TK– strain |
| gemcitabine | 0.028 ± 0.020 | 0.054 ± 0.012 | 0.074 ± 0.004 | 0.053 ± 0.049 | 20 | 0.0036 ± 0.0003 | <1 | <1 |
| 1a | >100 | >100 | >100 | >100 | >100 | NDd | – | – |
| 1b | 0.042 ± 0.014 | 0.166 ± 0.13 | 0.815 ± 0.64 | 0.93 ± 0.75 | 20 | 0.11 | 2 | <1 |
| 12a | >100 | >100 | >100 | >100 | >100 | NDd | – | – |
| 12b | >100 | >100 | >100 | >100 | >100 | NDd | – | – |
| 2a | >100 | >100 | >100 | >100 | >100 | NDd | – | – |
| 2b | 2.32 ± 0.43 | 2.84 ± 0.021 | 15.47 ± 6.40 | 4.00 ± 0.00 | >100 | 84.35 ± 22.1 | 36 | 29 |
| 2c | 0.074 ± 0.006 | 0.089 ± 0.011 | 0.066 ± 0.007 | 0.032 ± 0.00 | >100 | 0.34 ± 0.25 | 4 | 3 |
| acyclovir | 1.6 | 22.15 ± 11.6 | NDd | NDd | >440 | 424 ± 21.7 | 265 | 19 |
| brivudin | 0.036 ± 0.004 | 6.04 | NDd | NDd | >300 | >300 | >8333 | >49 |
| ganciclovir | NDd | NDd | 2.77 ± 0.53 | 1.67 ± 0.48 | 350 | 231.22 ± 49.2 | – | – |
| cidofovir | NDd | NDd | 0.65 ± 0.38 | 0.34 ± 0.056 | 300 | 150.02 ± 29.1 | – | – |
Effective concentration required to reduce virus plaque formation by 50%. Virus input was 100 plaque forming units (PFU).
Minimum cytotoxic concentration that causes a microscopically detectable alteration of cell morphology.
Cytotoxic concentration required to reduce the cell growth by 50%.
Not determined.
SI = CC50/EC50.
The potent cytostatic activity of gemcitabine in human embryonic lung (HEL) fibroblasts (CC50 = 0.0036 μM) leads to a complete absence of selective antiviral activity. The phosphoramidate prodrug of gemcitabine (compound 2c) was 100-fold less inhibitory toward HEL cell growth (CC50 = 0.34 μM), which is in agreement with what was previously observed for other ProTides of gemcitabine. The introduction of a fluorine at the 4′-position of gemcitabine yielded compound 1b, which was in general less active against VZV and HCMV. Remarkably, an EC50 of 0.042 μM was observed against the TK+ strain of VZV, making it only 2-fold less active than gemcitabine. Moreover, 1b displayed a 30-fold lower cytostatic effect than gemcitabine (CC50 = 0.11 μM vs 0.0036 μM for gemcitabine).
Interestingly, phosphoramidate prodrug 2b is less active than the parent nucleoside 1b, showing moderate activity against the TK+ and TK– strains of VZV with EC50 values in the 2–3 μM range. However, a 700-fold lower cytostatic activity was observed for ProTide 2b in comparison with the parent compound 1b. Consequently, compound 2b showed a selectivity index (SI) of 36, which was higher than those of 1b (SI = 2) and 2c (SI = 4). On the other hand, the 4′-methoxy gemcitabine analogue 1a and the 4′-substituted uridine analogues 12a and 12b were completely devoid of activity against VZV and HCMV.
In a second screening assay, the cytosine-containing compounds were tested against two different clinical isolates of SARS-CoV-2 in Vero cells (Table 2). Remdesivir and hydroxychloroquine were included as positive controls. As expected, the high cytotoxicity of gemcitabine in Vero cells precluded selective antiviral activity. The application of the ProTide technology to gemcitabine (affording compound 2c) led to decreased cytotoxicity but also lack of activity against SARS-CoV-2. The introduction of a 4′-methoxy group afforded compound 1a and the corresponding prodrug 2a, both of which were devoid of antiviral activity and cytotoxicity. The presence of a 4′-fluorine substituent (compound 1b) reduced the cytotoxicity of gemcitabine, although no selective antiviral activity was observed. The 4′-fluorophosphoroamidate prodrug 2b was less cytostatic (CC50 = 1.44 μM) than the parent nucleoside 1b and displayed minimal antiviral activity. Interestingly, in this cellular test system, compounds 1b and 2b were 20 and 10 times more active than remdesivir, respectively.
Table 2. Antiviral Activities and Cytotoxicities of 4′-Substituted Gemcitabine Analogues and Their Prodrugs against SARS-CoV-2 in Vero Cells.
| antiviral
activity, EC50 (μM)a |
cytotoxicity
(μM) |
|||
|---|---|---|---|---|
| compound | UC-1074 strain | UC-1075 strain | MCCb | CC50c |
| gemcitabine | >0.0032 ± 0 | >0.0016 ± 0 | 0.008 | 0.0043 ± 0.0008 |
| 1a | NDd | ≥86.2 ± 20.4 | >100 | >100 ± 0 |
| 1b | 0.36 | 0.096 ± 0.034 | 0.16 | 0.26 ± 0.13 |
| 2a | >100 | ≥55.1 ± 42.8 | ≥100 | ≥93.6 ± 11.1 |
| 2b | ≥2.4 ± 2.3 | 0.73 ± 0.15 | 4 ± 0 | 1.44 ± 0.62 |
| 2c | >0.032 ± 0 | >0.0128 ± 0 | 0.048 ± 0.018 | 0.020 ± 0.009 |
| remdesivir | 5.8 ± 3.1 | 1.52 ± 1.60 | >40 | >40 ± 0 |
| hydroxychloroquine | 8.1 ± 2.4 | 1.74 ± 0.68 | 100 | 36.9 ± 7.5 |
Effective concentration required to reduce virus plaque formation by 50%. Virus input was 100 PFU.
Minimum cytotoxic concentration that causes a microscopically detectable alteration of cell morphology.
Cytotoxic concentration required to reduce cell growth by 50%.
Not determined.
In summary, the synthesis of 4′-methoxy- and 4′-fluorogemcitabine analogues and their phosphoramidate prodrugs and an evaluation of their antiviral activities against VZV, HCMV, and SARS-CoV-2 have been described. The introduction of a fluorine atom at the 4′-position led to a trifluorinated gemcitabine analogue that exhibited potent activity but no selectivity against these three viruses. A phosphoramidate prodrug of the 4′-fluoro congener displayed reduced activity against VZV but less pronounced cytotoxicity with an enhanced selectivity index. A similar although somewhat reduced effect was observed against HCMV. Unfortunately, this effect was not determined upon testing against SARS-CoV-2.
Acknowledgments
Z.Z. thanks the China Scholarship Council (CSC) for funding (Grant 201704910837). The authors are grateful to Mr. Brecht Dirix for excellent technical assistance in the antiviral assays.
Glossary
Abbreviations
- HCMV
human cytomegalovirus
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- VZV
varicella zoster virus
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00485.
Experimental details and characterization data for the reported compounds, NMR spectra, and biological assays (PDF)
Author Contributions
§ P.H. and G.A. contributed equally to this work. All of the authors approved the final version of the manuscript.
The authors declare no competing financial interest.
This article is made available via the ACS COVID-19 subset for unrestricted RESEARCH re-use and analyses in any form or by any means with acknowledgement of the original source. These permissions are granted for the duration of the World Health Organization (WHO) declaration of COVID-19 as a global pandemic.
Supplementary Material
References
- Hertel L. W.; Boder G. B.; Kroin J. S.; Rinzel S. M.; Poore G. A.; Todd G. C.; Grindey G. B. Evaluation of the Antitumor-Activity of Gemcitabine (2′,2′-Difluoro-2′-deoxycytidine). Cancer Res. 1990, 50, 4417–4422. [PubMed] [Google Scholar]
- van der Donk W. A.; Yu G. X.; Perez L.; Sanchez R. J.; Stubbe J.; Samano V.; Robins M. J. Detection of a New Substrate-Derived Radical During Inactivation of Ribonucleotide Reductase from Escherichia coli by Gemcitabine 5′-Diphosphate. Biochemistry 1998, 37, 6419–6426. 10.1021/bi9729357. [DOI] [PubMed] [Google Scholar]
- Wang J.; Lohman G. J. S.; Stubbe J. Enhanced Subunit Interactions with Gemcitabine-5′-Diphosphate Inhibit Ribonucleotide Reductases. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 14324–14329. 10.1073/pnas.0706803104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuivanen S.; Bespalov M. M.; Nandania J.; Ianevski A.; Velagapudi V.; De Brabander J. K.; Kainov D. E.; Vapalahti O. Obatoclax, Saliphenylhalamide and Gemcitabine Inhibit Zika Virus Infection in Vitro and Differentially Affect Cellular Signaling, Transcription and Metabolism. Antiviral Res. 2017, 139, 117–128. 10.1016/j.antiviral.2016.12.022. [DOI] [PubMed] [Google Scholar]
- Beran R. K. F.; Sharma R.; Corsa A. C.; Tian Y.; Golde J.; Lundgaard G.; Delaney W. E.; Zhong W. D.; Greenstein A. E. Cellular Growth Kinetics Distinguish a Cyclophilin Inhibitor from an HSP90 Inhibitor as a Selective Inhibitor of Hepatitis C Virus. PLoS One 2012, 7, e30286. 10.1371/journal.pone.0030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouser C. L.; Holtz C. M.; Mullett M.; Crankshaw D. L.; Briggs J. E.; O’Sullivan M. G.; Patterson S. E.; Mansky L. M. Activity of a Novel Combined Antiretroviral Therapy of Gemcitabine and Decitabine in a Mouse Model for HIV-1. Antimicrob. Agents Chemother. 2012, 56, 1942–1948. 10.1128/AAC.06161-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. R.; Yang E. Z.; Hu C. M.; Cheng H.; Chen C. Y.; Huang D.; Wang R.; Zhao Y.; Rong L. J.; Vignuzzi M.; Shen H. B.; Shen L.; Chen Z. W. Cell-Based High-Throughput Screening Assay Identifies 2′,2′-Difluoro-2′-deoxycytidine Gemcitabine as a Potential Antipoliovirus Agent. ACS Infect. Dis. 2017, 3, 45–53. 10.1021/acsinfecdis.6b00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisova O. V.; Kakkola L.; Feng L.; Stenman J.; Nagaraj A.; Lampe J.; Yadav B.; Aittokallio T.; Kaukinen P.; Ahola T.; Kuivanen S.; Vapalahti O.; Kantele A.; Tynell J.; Julkunen I.; Kallio-Kokko H.; Paavilainen H.; Hukkanen V.; Elliott R. M.; De Brabander J. K.; Saelens X.; Kainov D. E. Obatoclax, Saliphenylhalamide, and Gemcitabine Inhibit Influenza A Virus Infection. J. Biol. Chem. 2012, 287, 35324–35332. 10.1074/jbc.M112.392142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J. H.; Kim S. R.; Heo E. Y.; Lee J. Y.; Kim D. E.; Cho S.; Chang S. Y.; Yoon B. I.; Seong J.; Ko H. J. Antiviral Activity of Gemcitabine Against Human Rhinovirus in Vitro and in Vivo. Antiviral Res. 2017, 145, 6–13. 10.1016/j.antiviral.2017.07.003. [DOI] [PubMed] [Google Scholar]
- Dyall J.; Coleman C. M.; Hart B. J.; Venkataraman T.; Holbrook M. R.; Kindrachuk J.; Johnson R. F.; Olinger G. G.; Jahrling P. B.; Laidlaw M.; Johansen L. M.; Lear-Rooney C. M.; Glass P. J.; Hensley L. E.; Frieman M. B. Repurposing of Clinically Developed Drugs for Treatment of Middle East Respiratory Syndrome Coronavirus Infection. Antimicrob. Agents Chemother. 2014, 58, 4885–4893. 10.1128/AAC.03036-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.-N.; Zhang Q.-Y.; Li X.-D.; Xiong J.; Xiao S.-Q.; Wang Z.; Zhang Z.-R.; Deng C.-L.; Yang X.-L.; Wei H.-P.; Yuan Z.-M.; Ye H.-Q.; Zhang B. Gemcitabine, Lycorine and Oxysophoridine Inhibit Novel Coronavirus (SARS-CoV-2) in Cell Culture. Emerging Microbes Infect. 2020, 9, 1170–1173. 10.1080/22221751.2020.1772676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patent application WO 2014209979 A1.
- Patent application WO 2015054465 A1.
- Wang G. Y.; Deval J.; Hong J.; Dyatkina N.; Prhavc M.; Taylor J.; Fung A.; Jin Z. N.; Stevens S. K.; Serebryany V.; Liu J. W.; Zhang Q. L.; Tam Y. E.; Chanda S. M.; Smith D. B.; Symons J. A.; Blatt L. M.; Beigelman L. Discovery of 4′-Chloromethyl-2′-deoxy-3′,5′-di-O-isobutyryl-2′-fluorocytidine (ALS-8176), A First-in-Class RSV Polymerase Inhibitor for Treatment of Human Respiratory Syncytial Virus Infection. J. Med. Chem. 2015, 58, 1862–1878. 10.1021/jm5017279. [DOI] [PubMed] [Google Scholar]
- Martinez-Montero S.; Deleavey G. F.; Kulkarni A.; Martin-Pintado N.; Lindovska P.; Thomson M.; Gonzalez C.; Gotte M.; Damha M. J. Rigid 2′,4′-Difluororibonucleosides: Synthesis, Conformational Analysis, and Incorporation into Nascent RNA by HCV Polymerase. J. Org. Chem. 2014, 79, 5627–5635. 10.1021/jo500794v. [DOI] [PubMed] [Google Scholar]
- Martinez-Montero S.; Deleavey G. F.; Dierker-Viik A.; Lindovska P.; Ilina T.; Portella G.; Orozco M.; Parniak M. A.; Gonzalez C.; Damha M. J. Synthesis and Properties of 2′-Deoxy-2′,4′-difluoroarabinose-Modified Nucleic Acids. J. Org. Chem. 2015, 80, 3083–3091. 10.1021/jo502948t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakar K. J.; Reese C. B. 4-(1,2,4-Triazol-1-yl) and 4-(3-Nitro-1,2,4-triazol-1-yl)-1-(β-d-2,3,5-tri-O-acetylarabinofuranosyl)pyrimidin-2(1H)-ones—Valuable Intermediates in the Synthesis of Derivatives of 1-(β-d-Arabinofuranosyl)Cytosine (Ara-C). J. Chem. Soc., Perkin Trans. 1 1982, 1171–1176. 10.1039/P19820001171. [DOI] [Google Scholar]
- Mehellou Y.; Balzarini J.; McGuigan C. Aryloxy Phosphoramidate Triesters: A Technology for Delivering Monophosphorylated Nucleosides and Sugars into Cells. ChemMedChem 2009, 4, 1779–1791. 10.1002/cmdc.200900289. [DOI] [PubMed] [Google Scholar]
- Slusarczyk M.; Lopez M. H.; Balzarini J.; Mason M.; Jiang W. G.; Blagden S.; Thompson E.; Ghazaly E.; McGuigan C. Application of ProTide Technology to Gemcitabine: A Successful Approach to Overcome the Key Cancer Resistance Mechanisms Leads to a New Agent (NUC-1031) in Clinical Development. J. Med. Chem. 2014, 57, 1531–1542. 10.1021/jm401853a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.