

Abstract
Failures to produce neutralizing antibodies upon HIV‐1 infection result in part from B‐cell dysfunction due to unspecific B‐cell activation. How HIV‐1 affects antigen‐specific B‐cell functions remains elusive. Using an adoptive transfer mouse model and ex vivo HIV infection of human tonsil tissue, we found that expression of the HIV‐1 pathogenesis factor NEF in CD4 T cells undermines their helper function and impairs cognate B‐cell functions including mounting of efficient specific IgG responses. NEF interfered with T cell help via a specific protein interaction motif that prevents polarized cytokine secretion at the T‐cell–B‐cell immune synapse. This interference reduced B‐cell activation and proliferation and thus disrupted germinal center formation and affinity maturation. These results identify NEF as a key component for HIV‐mediated dysfunction of antigen‐specific B cells. Therapeutic targeting of the identified molecular surface in NEF will facilitate host control of HIV infection.
Keywords: B‐cell dysfunction, HIV‐1 infection, immunological synapse, intravital imaging, NEF
Subject Categories: Immunology
Prevention of polarized cytokine secretion at the immune synapse of infected helper T cells reduces B cell activation, providing a basis for impaired neutralizing antibody responses observed in HIV‐1 patients.
Introduction
Untreated infection with human immunodeficiency virus (HIV) causes a complex pathology that ultimately results in the development of AIDS. In addition to hallmarks of HIV pathogenesis such as progressing depletion of CD4 T cells and chronic immune activation, B‐cell dysfunction is increasingly recognized as a central pathological determinant in HIV‐1 patients (Moir & Fauci, 2009; Moir & Fauci, 2013). Perturbations of B‐cell function in HIV infection prevent the efficient mounting of high‐affinity antibody responses against HIV but also other pathogens or vaccines (Malaspina et al, 2005; Titanji et al, 2006; Frit et al, 2010; Pallikkuth et al, 2012; Kerneis et al, 2014). The lack of efficient broadly neutralizing antibody responses in most HIV‐1‐infected individuals reflects on the one hand that the architecture of the viral glycoprotein Env and the cell entry receptor complex it targets are optimized to reduce accessibility to antibody binding (Mouquet, 2014). On the other hand, the fast dynamics of conformational changes that reveal molecular surfaces susceptible to antibody neutralization as well as the use of cell‐associated modes of virus transmission limit the possibility of antibody neutralization of HIV infection (Malbec et al, 2013; Chen, 2019). In addition to these difficulties to access target structures for antibody neutralization, B‐cell function is generally disturbed in HIV‐infected patients. This B‐cell dysfunction is characterized by a significant enrichment of anergic and exhausted memory B cells paralleled by marked hypergammaglobulinemia (Moir et al, 2008; Moir & Fauci, 2013; Kardava et al, 2014). Such polyclonal, antigen (Ag)‐unspecific exhaustion has been extensively described for Ag‐experienced B cells (Malaspina et al, 2005; Titanji et al, 2006; Fritz et al, 2010; Pallikkuth et al, 2012; Kerneis et al, 2014) and was recently also identified to occur in Ag‐naïve B cells (Liechti et al, 2019).
Whether HIV infection also impacts the functionality of HIV‐specific B cells is much less studied. T follicular helper (TFH) cells, a specialized subset of CD4 T cells that promote B‐cell immune globulin class switching, affinity maturation, and differentiation into plasma cells and long‐lived memory B cells in B‐cell follicles, are preferential targets during chronic HIV‐1 infection (Perreau et al, 2013). Since B‐cell dysfunction is induced in HIV patients despite a significant increase in TFH numbers in early and mid‐stages of disease, preferential infection of TFH by HIV may interfere with their helper function (Wendel et al, 2018). Moreover, altered expression of cytokines and stimulatory receptors have been suggested to result from HIV infection of TFH cells (Graff‐Dubois et al, 2016), but relevance and mechanism of such virus‐induced modification have not been explored.
The viral protein NEF constitutes a major determinant for HIV pathogenesis that drives virus replication (Kestler et al, 1991; Deacon et al, 1995; Kirchhoff et al, 1995) and facilitates evasion of infected cells from CD8 T‐cell recognition and innate immune restriction in the infected host (Schwartz et al, 1996; Rosa et al, 2015; Usami et al, 2015). Acting as a protein interaction adaptor, expression of NEF allows HIV to hijack central host cell vesicular transport and signaling pathways (Geyer et al, 2001; Abraham & Fackler, 2012; Pereira & daSilva, 2016). One result of these activities is the establishment of the activation status of CD4 T cells that supports HIV replication at reduced levels of activation‐induced cell death and thus increases the life span of infected cells (Haller et al, 2006; Schindler et al, 2006; Thoulouze et al, 2006; Arhel et al, 2009; Abraham & Fackler, 2012; Pan et al, 2012). These effects of NEF on T‐cell activation reflect how these cells engage in Ag‐specific cell–cell communication across the immunological synapse (IS). Altered cell surface exposure of receptors and interference with activation‐induced actin polymerization affect the response of infected CD4 T cells to respond to stimulation by antigen presenting cells (APCs). However, these studies relied on the use of superantigen and investigated effects on activation and permissivity for HIV infection in the CD4 T cell. Since superantigen‐mediated T‐cell activation does not trigger Ag‐specific signaling at the APC side of an IS (Fig EV1A and B), these studies did not provide insight into putative effects of HIV‐1 infection in CD4 T cells on APC function.
Figure EV1. Mouse model to study immunomodulatory properties of NEF on antigen‐specific interaction between B cells and T cells.
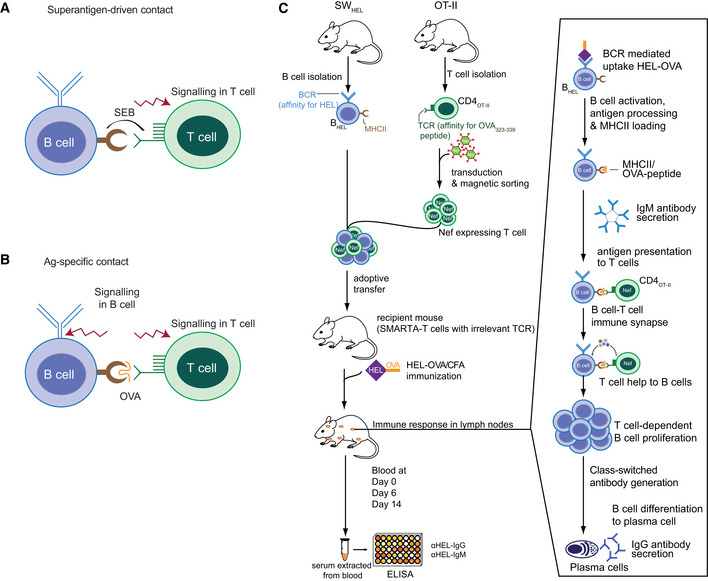
- TCR‐MHC class II crosslinking by superantigen (SEB) initiates signaling exclusively on the T‐cell side, precluding investigation of B‐cell responses upon IS formation.
- Presentation of endogenously processed cognate peptide (OVA323–339) on B‐cell MHC class II to CD4OT‐II cells allows bidirectional signaling in B and T cells enabling analysis of B‐cell downstream functions.
- Retrovirally transduced CD4OT‐II cells were co‐cultured with BHEL cells pulsed with HEL‐OVA in the presence of LPS or adoptively transferred into recipient mice. HEL‐OVA is internalized via BCR‐mediated phagocytosis, processed and presented on MHC class II of B cells. This results in the establishment of an Ag‐specific IS between B cells and transduced/NEF‐expressing CD4OT‐II cells. Upon IS formation, B‐cell signaling is initiated resulting in B‐cell activation, proliferation, and differentiation. This model allows investigation of initial events modulated by NEF during B‐cell activation. For analyzing the effect of NEF on antibody production, SMARTA mice (T cells with TCR specific to irrelevant Ag) were used as recipients for HEL‐OVA/CFA immunization. HEL‐specific IgM and IgG ELISA was performed on serum samples extracted from blood collected on indicated days.
Based on the ability to affect CD4 T‐cell activation, we hypothesized that HIV‐1 NEF may not only modulate the ability of CD4 T cells to respond to Ag‐specific stimulation but also alter their helper function. Using an Ag‐driven adoptive transfer mouse model and infection of human tonsil tissue cultures, we define that NEF potently suppresses T‐cell help to B cells and establish that NEF prevents germinal center (GC) formation by impairing early steps in B‐cell signaling. These findings define NEF as a key viral factor and suitable therapeutic target in HIV‐1 induced dysfunction of HIV‐specific B cells.
Results
CD4 T cells expressing HIV‐1 NEF fail to provide T‐cell help in vivo and are impaired in inducing early B‐cell signaling
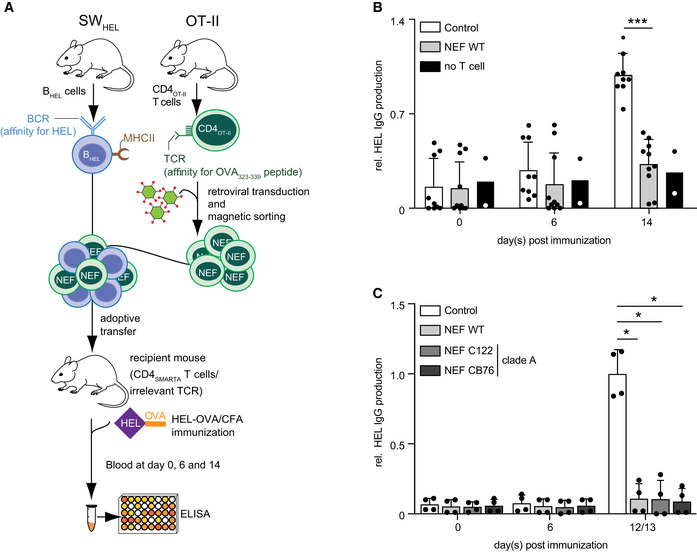
Previous studies demonstrated that HIV‐1 NEF affects T‐cell antigen receptor (TCR) signaling events triggered in the context of a T cell—APC IS (Haller et al, 2006; Thoulouze et al, 2006; Arhel et al, 2009). These experiments were unable to assess potential consequences on B‐cell function due to the use of unidirectionally stimulating SEB superantigen that potently stimulates the TCR without providing sufficient stimulus to the B cell. We therefore sought to test whether expression of HIV‐1 NEF in CD4 T cells affects the ability of these cells to provide help to B cells. To this end, we employed an adoptive transfer mouse model in which CD4+ T cells from OT‐II mice (CD4OT‐II), transgenic for ovalbumin (OVA) peptide (OVA323–339)‐specific TCR, and B cells of SWHEL mice (BHEL), transgenic for hen egg lysozyme (HEL) B‐cell antigen receptor (BCR), are adoptively transferred into recipient SMARTA mice that carry a TCR specific for an unrelated CD4 T‐cell peptide and therefore lack an endogenous T‐cell response toward the exogenous OVA‐HEL antigen (Figs 1A and EV1C). Use of BHEL cells allowed us to study class switch recombination and somatic hypermutation upon immunization in the presence of HIV‐1 NEF which is not possible in most other B‐cell transgenic mice currently available. Immunization with crosslinked HEL‐OVA Ag results in uptake, activation, and processing of HEL‐OVA by BHEL cells and presentation of OVA peptides on their MHC class II molecules to CD4OT‐II cells. This TCR engagement triggers downstream signaling in CD4OT‐II cells inducing their activation and differentiation into T helper cells (Phan et al, 2006; Brink et al, 2015; Tsopoulidis et al, 2019). Vice versa, this interaction provides T‐cell help enabling B cells to undergo proliferation and germinal center reaction (GCR) cycles, eventually resulting in the production of IgM and later class‐switched (IgG) anti‐HEL Abs (De Silva & Klein, 2015; Figs 1A and EV1C). To probe for the effect of HIV‐1 NEF, CD4OT‐II cells were activated in vitro using OVA peptide to facilitate the transduction with a bicistronic retroviral vector expressing HIV‐1SF2 NEF (NEF WT) or an empty vector (Control) together with a truncated version of nerve growth factor receptor (NGFR/CD271) that allows identification and sorting of transduced cells (Fig EV2A; Stolp et al, 2012). After 7 days in culture in the absence of TCR stimuli, cells almost returned to basal activation states (Fig EV2B) and were used to enrich NGFR+ cells (Fig EV2C) for subsequent adoptive transfer. Using this transduction approach in mouse CD4 T cells, NEF is expressed to levels equivalent to those observed in HIV‐1 infection of primary human CD4 T cells (Stolp et al, 2012), does not affect basal cell activation state and viability (Fig EV2D and E), and is biologically active as, e.g., illustrated by the ability to efficiently reduce CD4 cell surface levels (Fig EV2F–H). Analyzing anti‐HEL IgG levels at different time points post‐immunization revealed robust production of HEL‐specific Abs using control transduced CD4OT‐II cells for adoptive transfer, which were significantly above the baseline Ab production observed in animals that were immunized following transfer of BHEL cells without CD4OT‐II cells (no T‐cell control) (Fig 1B). Strikingly, anti‐HEL IgG levels produced in animals that received NEF‐expressing CD4OT‐II cells were almost as low as in no T‐cell control animals. Similar suppression of anti‐HEL IgG production was observed for the primary NEF variants C122 and CB76. Since these NEF variants were derived from clade A HIV‐1 strains with sequences that diverge from clade B HIV‐1SF2, disruption of humoral immunity seems to be largely conserved among HIV‐1 NEF proteins (Fig 1C). NEF WT also impaired the production of early response anti‐HEL IgM over time (Fig EV2I). HIV‐1 NEF therefore strongly suppressed the helper function of transferred CD4OT‐II cells in this model system. Together these results reveal that expression of HIV‐1 NEF potently blocks T‐cell help to drive the production of Ag‐specific Abs.
Figure 1. NEF expression in CD4 T cells blocks their T helper function resulting in a lack of antibody production.
-
AOutline of the experimental setup to assess the immunomodulatory effects of HIV‐1 NEF on the generation of humoral immune responses. Antigen‐specific interactions are established using HEL‐OVA as model antigen with HEL‐specific SWHEL B cells (BHEL) and OVA‐specific OT‐II CD4 T cells (CD4OT‐II).
-
B, CLevels of anti‐HEL IgG in serum as measured by ELISA at the indicated days post‐immunization with HEL‐OVA/CFA following adoptive transfer of BHEL cells without CD4OT‐II cells (no T‐cell control) or with control CD4OT‐II cells or NEF‐expressing CD4OT‐II cells. (B) NEF WT of HIV‐1SF2 (C) patient‐derived NEF variants. Shown are relative levels of HEL IgG production with the mean of IgG levels in mice receiving control cells between days 12–14 post‐immunization arbitrarily set to 1. Each dot represents one animal. Bars represent mean values with SD from 2 to 10 mice analyzed from 3 (B) and 2 (C) independent experiments. Statistical significance was assessed by Mann–Whitney U‐test. ***P ≤ 0.001, *P ≤ 0.05.
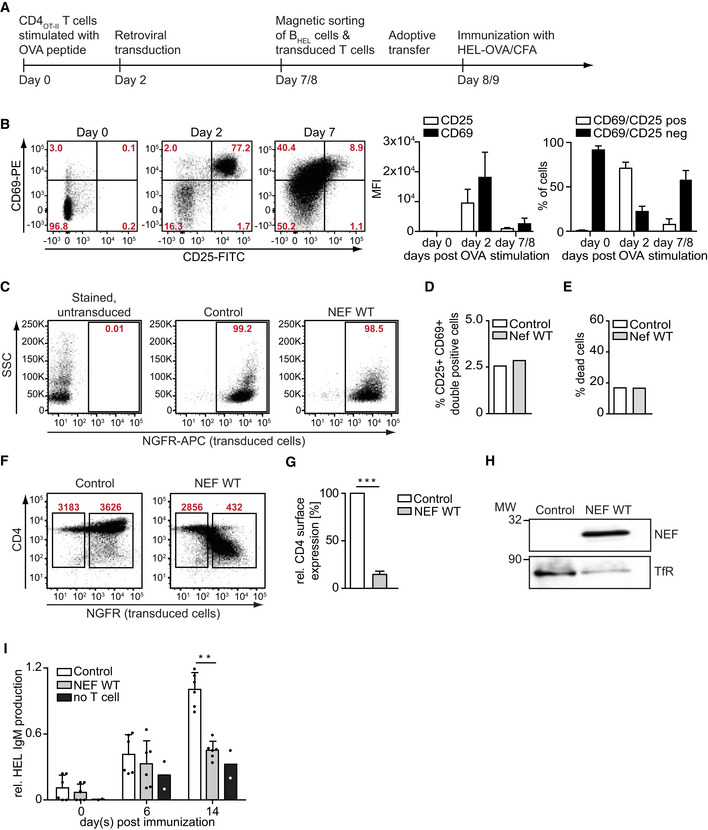
Figure EV2. Activation, transduction, and functionality of NEF in CD4OT‐II cells for adoptive transfer and immunization.
-
AExperimental outline: CD4OT‐II cell stimulation with OVA peptide (d0), retroviral transduction (d2), magnetic enrichment and adoptive transfer (d7/8), and immunization of recipient mice (d8/9).
-
BTo evaluate the activation status, CD4OT‐II cells were stained with anti‐CD25 and anti‐CD69 antibodies and analyzed using flow cytometry. Left: Representative dot plots showing CD25 and CD69 levels on T cells directly after isolation (d0) and d2 and 7 after stimulation of splenocytes with OVA323–339. Shown is the lymphocyte gate. Cells become requiescent by d7 indicating their potential responsiveness to second stimulation. Middle: MFI values for CD25 (white bar) and CD69 (black bar) on CD4OT‐II cells. Right: Percentage of CD69/CD25 double positive (white bar) and double negative (black bar) cells. Shown are mean values with SD from 3 independent experiments.
-
CFlow cytometry dot plots showing percentage of NGFR‐positive cells. Four days after transduction, cells were magnetically enriched, as used for most experiments and stained with anti‐NGFR antibody.
-
D, EActivation (D) and viability (E) of transduced and magnetically sorted CD4OT‐II cells prior to adoptive transfer, measured by the percentage of CD25 and CD69 co‐expressing cells (D) or percentage of dead cells as determined by uptake of viability dye among CD3+ expressing cells. Shown are data from one representative dataset.
-
FRepresentative flow cytometry dot plots showing surface CD4 downmodulation by NEF WT at d7. MFI values for CD4 surface expression in untransduced (left gate: NGFR negative cells) and transduced T cells (right gate: NGFR‐positive cells) are shown above the gate.
-
GQuantification of surface CD4 downregulation as shown in F. Shown is the relative CD4 downmodulation of transduced to untransduced cells with Control cells arbitrarily set to 100%. Shown are mean values with SD from three independent experiment.
-
HNef protein expression in CD4OT‐II cells 2 days after retroviral transduction. Cells were lysed by ultrasonication in the presence of lysis buffer and subjected to Western blotting and antibody detection using anti‐NEF antibody and anti‐transferrin receptor antibody as loading control.
-
IProduction of anti‐HEL IgM at indicated days post‐immunization with HEL‐OVA/CFA, following adoptive transfer of BHEL cells and Control or NEF‐expressing CD4OT‐II cells. Shown are mean values with SD from 2 to 6 mice. Each dot represents one animal.
Data information: Statistical significance was assessed by Mann–Whitney U‐test. ***P ≤ 0.001, *P ≤ 0.05.
Source data are available online for this figure.
HIV‐1 NEF expression in CD4 T cells impairs induction of early B‐cell signaling
To investigate the underlying mechanism of NEF‐mediated suppression of humoral immunity, we assessed the organization and function of ISs formed between HEL‐OVA pulsed BHEL cells with CD4OT‐II cells ex vivo (Fig 2A). Expectedly, BHEL cells and CD4OT‐II cells efficiently formed cell conjugates when cultured together independently of prior pulsing with HEL‐OVA peptide and expression of Nef in CD4OT‐II cells did not impact on the frequency of cell conjugation (Fig 2B and C). However, pulsing of BHEL cells with HEL‐OVA induced the enrichment of polymerized actin (F‐actin) (Fig 2B and D) and phosphorylated tyrosine (p‐Tyr) (Fig 2E and F) at T–BHEL contacts, a hallmark of potent IS formation and signaling. Previous reports on superantigen‐induced ISs established that expression of Nef in CD4 T cells impairs actin polymerization at the T‐cell side of the IS (Haller et al, 2006; Arhel et al, 2009; Rudolph et al, 2009). Consistently, Nef expression in CD4OT‐II potently disrupted this enrichment of F‐actin (Fig 2B and D) and phosphorylated tyrosine (p‐Tyr) (Fig 2E and F) at T–BHEL contacts. The use of specific antigen rather than superantigen allowed us to study actin dynamics also at the B‐cell sides of the IS; however, these stainings did not discriminate between BHEL and CD4OT‐II F‐actin at the IS. To address whether NEF acts exclusively on actin polymerization and signaling in the CD4 T cells or also impacts on related events in B cells, we loaded BHEL cells with silicon rhodamine (SiR)‐actin, a cell permeable fluorescent dye that visualizes actin polymerization, and incubated them with CD4OT‐II cells labeled with a CellTrackerTM dye (Fig 2G and H). When conjugated to control CD4OT‐II cells, BHEL cells rapidly responded by polymerizing actin at cell–cell contacts, eventually resulting in marked actin polymerization at the entire cell periphery (Fig 2G, upper panel, Movie EV1), an established hallmark of early BCR signaling (Harwood & Batista, 2010). Expression of NEF in CD4OT‐II cells engaged by BHEL cells potently abrogated B‐cell actin polymerization (Fig 2G, lower panel, see Fig 2H for quantification) or resulted in small and short‐lived F‐actin puncta in the periphery instead of a strong accumulation of F‐actin at cell–cell contacts (Fig 2G, lower panel, Movie EV2). Together, these results revealed that HIV‐1 NEF, when expressed in CD4OT‐II cells, impairs their ability to provide sufficient stimulation to B cells. This results in a lack of Ag‐specific antibody production by altering proximal steps of BCR signaling triggered by IS engagement.
Figure 2. NEF expression in CD4 T cells impairs early B‐cell signaling and proliferation.
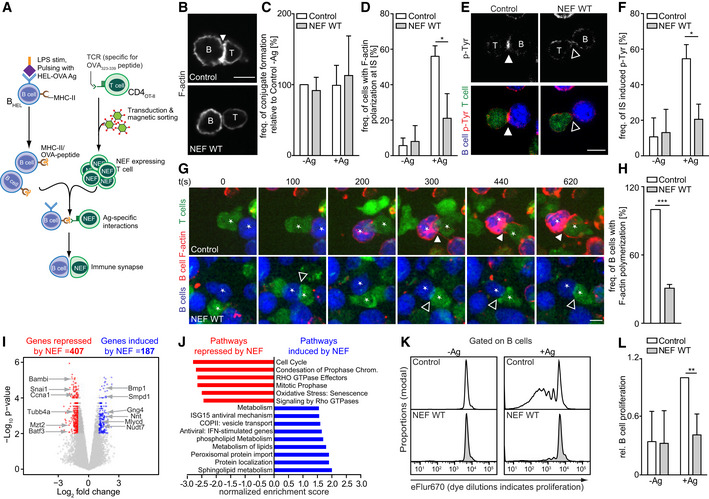
-
ASchematic experimental flow of the analysis of IS formation and function between NEF‐expressing CD4OT‐II cells and antigen‐specific BHEL cells. BHEL cells were stimulated with LPS in the presence (+Ag) or absence (−Ag) of HEL‐OVA antigen. B cells and T cells were labeled with different CellTracker™ dyes before seeding on PLL‐coated cover glasses and analyzed by microscopy.
-
BRepresentative confocal micrographs of F‐actin (gray signal) polarization at B–T‐cell contacts after 1–2 h of co‐culture. Cell conjugates were stained with Phalloidin to visualize F‐actin. Scale bar, 5 µm.
-
C, DQuantification of the frequency of B–T conjugate formation (C) and F‐actin accumulation at the B–T interface (D). Shown are mean values with SD from three independent experiments with at least 50 conjugates analyzed per experiment.
-
ERepresentative confocal micrographs of p‐tyrosine (p‐Tyr) polarization at B–T‐cell contacts after 1–2 h of co‐culture. Cell conjugates were stained with anti‐p‐Tyr antibody. Scale bar, 5 µm. The top panels display only the p‐Tyr signal in gray, the bottom panel depicts p‐Tyr (red), BHEL cells (blue), and CD4OT‐II cells (green).
-
FQuantification of p‐Tyr accumulation at the B–T interface as shown in E. Shown are mean values with SD from three independent experiments with at least 50 conjugates analyzed per experiment.
-
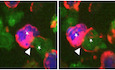
GNEF prevents induction of F‐actin remodeling in BHEL during IS. BHEL cells (blue) and CD4OT‐II cells (green) were placed on PLL‐coated slides and subjected to live‐cell time‐lapse imaging. Shown are representative still images from the indicated time points post‐contact formation (see Movies EV1 and EV2). Red signals indicate actin polymerization specifically in BHEL cells. Scale bars, 5 µm. Filled and empty arrowheads indicate examples of accumulation of polarized actin or p‐Tyr signal at B–T interface during IS formation or lack thereof, respectively. Asterisks mark pairs of interacting T and B cells.
-
HQuantification of B‐cell actin polymerization as shown in G with the frequency of actin polymerization in B cells co‐cultured with control T cells arbitrarily set to 100%. Shown are mean values with SD from three independent experiments with at least 50 conjugates counted per experiment.
-
IVolcano plot of NEF‐induced changes to BHEL cell transcription. NEF‐expressing or control CD4OT‐II cells were co‐cultured with BHEL cells in the presence of HEL‐OVA for 4 h before isolation of BHEL cells and transcriptome analysis by microarray. Shown are mRNAs with log2fold change against −log10 P‐value. Out of around 20,000 genes investigated, 564 genes were induced and 977 genes were suppressed by NEF (see Dataset EV1), with 187 genes induced and 407 suppressed by NEF in a statistically significant manner.
-
JPathways involved in cell proliferation were negatively modulated by NEF, while lipid metabolism pathways were induced by NEF. 1,541 genes (see Dataset EV1) were included for pathway analysis.
-
KEx vivo BHEL proliferation after 3 days of co‐culture with control or NEF‐expressing CD4OT‐II cells. BHEL cells were labeled with the cell proliferation dye eFluor670 and co‐cultured with NGFR‐enriched CD4OT‐II cells expressing NEF in the presence or absence of HEL‐OVA Ag. T cell‐dependent B‐cell proliferation was assessed by quantification of the dilution of eFluor670 using flow cytometry.
-
LRelative B‐cell proliferation (quantified by gating on the last 3–4 generations) is plotted as mean values with SD from four independent experiments. Control T cells + Ag were arbitrarily set to 1.
Data information: Statistical significance was assessed by Students t‐test. ***P ≤ 0.001, **P ≤ 0.01, *P ≤ 0.05.
As this result indicated that NEF expression in T cells impaired early signaling events on the B‐cell side of the IS, we next tested whether this effect of NEF had immediate impact on B‐cell function. Since B‐cell activation driven by T‐cell help is known to induce specific gene expression programs (Bhattacharya et al, 2007), we assessed how expression of the viral protein in the T cell affected the transcriptional changes induced in the B cell after co‐culture in the presence of cognate Ag. To this end, we analyzed the transcriptome of sorted BHEL cells that were pulsed with HEL‐OVA and co‐cultured with CD4OT‐II cells. This microarray analysis investigated approximately 20,000 genes, of which 4,535 were induced and 3,110 repressed when control CD4OT‐II cells were co‐cultured with BHEL cells as compared to naïve BHEL cells. When NEF was expressed in CD4OT‐II cells, 187 genes were induced and 407 repressed in BHEL cells compared to the Control (Fig 2I, Dataset EV1). Gene ontology analysis revealed that NEF expression in CD4OT‐II cells negatively affected expression of genes involved in B‐cell division (e.g., snai, ccna1, tubb4, mzt2) and B‐cell differentiation (e.g., bambi, batf3). B‐cell genes induced by NEF expression in CD4OT‐II included pathways involved in B‐cell metabolism (e.g., bmp1, smpd1, gng4, nnt, mlycd, nudt7) (Fig 2J). These results suggest that B cells are unable to initiate clonal expansion and differentiation when stimulated by NEF‐expressing CD4OT‐II T cells. Consistently, T cell‐dependent proliferation of BHEL cells was potently reduced upon interaction with NEF‐expressing CD4OT‐II cells ex vivo (Fig 2K and L). NEF expression in CD4 T cells thus results in suboptimal stimulation of cognate B cells which limits B‐cell proliferation and differentiation.
HIV‐1 NEF interferes with cytokine polarization at the T–B‐cell IS via an N‐terminal protein interaction motif
To gain insight into the molecular mechanism by which NEF impairs CD4 T‐cell help, we analyzed the activity of a series of mutant NEF proteins in the adoptive transfer model (Fig 3A). This included NEF F195A which lacks the ability to associate with the cellular p21‐activated kinase 2 (PAK2) to negatively modulate host cell actin dynamics and motility (O'Neill et al, 2006; Stolp et al, 2009; Stolp et al, 2012), a NEF variant with disrupted di‐leucine motif (NEF LLAA) that lacks the ability to internalize cell surface receptors such as CD4 (Craig et al, 1998; Greenberg et al, 1998), NEF AxxA in which an SH3 domain‐binding PxxP motif is disrupted and fails, e.g., to relocalize the TCR proximal kinase Lck from the plasma membrane to intracellular compartments (Saksela et al, 1995; Pan et al, 2012), as well as NEF variants carrying a deletion of an N‐terminal protein interaction platform (NEF ∆12‐39) or a mutation in an interaction motif within residues 12‐39 (NEF A32‐39) required for CD4 downregulation and SERINC5 antagonism (Ananth et al, 2019). Among these NEF mutants, only deletion/mutation of the motifs in the NEF N‐terminus impaired the ability of the viral protein to disrupt HEL‐specific antibody production (Fig 3B, Appendix Fig S1A). Consistently, NEF Δ12‐39 was also almost entirely defective in preventing actin polymerization in BHEL cells upon IS engagement (Fig 3C and D). Moreover, comparing BHEL cell transcriptomes following interaction with control CD4OT‐II cells or CD4OT‐II cells expressing NEF WT or NEFΔ12‐39 also revealed an involvement of the N‐terminal interaction platform in deregulating BHEL cell gene expression: NEF WT expression affected BHEL cell gene regulation upon interaction with CD4OT‐II for genes that were highly (Fig 3E) or moderately (Fig 3F) induced, or repressed (Fig 3G) by Ag stimulation in control cells. While many genes were similarly affected by expression of NEF and its mutants, for a subset of genes, this NEF‐mediated gene deregulation was strictly dependent on the integrity of the N‐terminal interaction platform. This NEF‐specific deregulation of BHEL cell gene expression associated with disruption of humoral immunity preferentially targeted genes involved in immune activation (Appendix Fig S1B and C), cell survival (Appendix Fig S1D), and cell metabolism (Appendix Fig S1E) and included factors such as Blimp1 (Shaffer et al, 2002), Ezh2 (Guo et al, 2018), and Batf (Ise et al, 2011; Appendix Fig S1C) that are known to be critical for GC formation or plasma cell generation, respectively. Together, these results suggest the deregulation of B‐cell gene expression as an active principle of NEF‐mediated impairment of B‐cell effector functions.
Figure 3. HIV‐1 NEF interferes with B‐cell effector functions via an N‐terminal protein interaction motif.
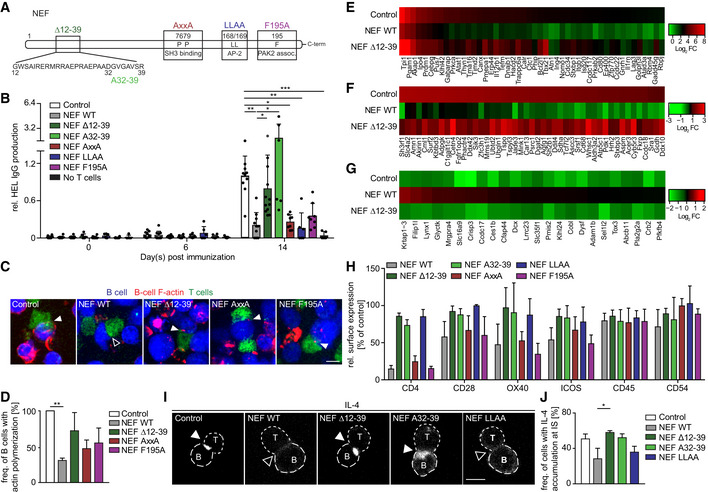
-
ALinear representation of HIV‐1SF2 NEF. To identify a motif of NEF critical for suppression of the humoral immune response, highlighted regions were either deleted or mutated.
-
BProduction of anti‐HEL IgG at the indicated day post‐immunization with HEL‐OVA/CFA following adoptive transfer of BHEL and CD4OT‐II cells expressing NEF or various NEF mutants. Shown are relative levels of HEL IgG production with the median of IgG levels in mice receiving control cells on day 14 post‐immunization arbitrarily set to 1. Each dot represents one animal. Shown are mean values with SD from 5 to 12 mice from 4 experiments.
-
CHEL‐OVA pulsed‐ BHEL cells (blue) and CD4OT‐II cells (green) were placed on PLL‐coated cover glasses and subjected to live‐cell time‐lapse imaging. Shown are representative still images. BHEL cells were loaded with SiRAct to visualize actin polymerization in B cells (F‐actin displayed in red). Filled and empty arrowheads indicate the presence or absence of F‐actin accumulation at the IS, respectively. Scale bars, 5 µm.
-
DQuantification of B‐cell actin polymerization as shown in C with frequency of actin polymerization in B cells co‐cultured with control T cells arbitrarily set to 100%. Shown are mean values with SD from three independent experiments with at least 30 conjugates evaluated per condition.
-
E–GEffects of NEF expression in CD4OT‐II cells on gene expression in BHEL cells. After 24 h of B–T co‐culture, BHEL cells were separated from CD4OT‐II using magnetic beads before RNA isolation for microarray analysis. The top 235 deregulated genes (see Dataset EV2) were grouped into genes that were highly induced (E), moderately induced (F), or repressed (G) by Ag stimulation in control cells and the effects of NEF WT or NEF Δ12–39 displayed as heat maps. Heat map values are plotted as average Log2 FC (n = 3 biological repeats) relative to unstimulated BHEL. Red and green represent higher and lower expression, respectively.
-
HRelative surface expression of CD4, CD28, OX40, ICOS, CD45, and CD54 on T cells expressing NEF WT or the indicated NEF mutants. Analysis of surface expression was performed using flow cytometry 3–7 days post‐transduction of T cells. Depicted are mean values with SD from 3 to 6 independent experiments, with control T cells arbitrarily set to 100% as indicated by the dashed line.
-
IRepresentative confocal images of the subcellular distribution of IL‐4 in CD4OT‐II cells expressing the indicated proteins engaged in an IS with BHEL cells. B cells and T cells were placed on PLL‐coated cover glasses for 60–90 min, fixed, and stained for intracellular IL‐4. Filled and empty arrowheads indicate presence or absence of IL‐4 accumulation at the IS, respectively. Scale bars, 5 µm.
-
JQuantification of IL‐4 accumulation at the IS as depicted in I. Shown are mean values with SD from 3 independent experiments relative to control cells set to 100%.
Data information: Statistical significance of bar graphs was assessed by a Kruskal–Wallis test with Dunn’s multiple comparison test. ***P ≤ 0.001, **P ≤ 0.01, *P ≤ 0.05.
Since supernatants of NEF‐expressing CD4 T cells failed to impact early B‐cell signaling (unpublished observation S. Kaw and O.T. Fackler), we hypothesized that NEF affects B‐cell function by targeting essential processes at the T‐B IS and analyzed the impact of NEF on IS organization at the molecular level. Communication across the IS involves an array of cell surface receptors that engage with their specific ligand on the target cell, which can elicit signal transduction processes (Tangye et al, 2013). Since NEF is known to modulate cell surface exposure of a large array of cell surface receptors (Haller et al, 2014; Matheson et al, 2015), we first screened a series of mouse T‐cell surface receptors known to be involved at the T‐B IS for their cell surface expression in the presence of NEF. While some receptors such as CD45 and CD54 remained unaffected by expression of WT or mutant NEFs, reduced cell surface levels on NEF‐expressing T cells were observed for, e.g., CD4, CD28, OX40, and ICOS (Fig 3H, see Appendix Fig S2A–F for primary data). Cell surface downregulation was in the range of twofold for most receptors but more pronounced for CD4. However, downregulation of cell surface CD4, CD28, OX40, and ICOS by NEF required the di‐leucine motif in NEF, which was dispensable for disruption of IgG production. Moreover, the NEF variant C122 that potently suppressed anti‐HEL Ab production in vivo (Fig 1C) lacked classical NEF activities such as downregulation of cell surface CD4, MHC‐I, inhibition of T‐cell chemotaxis, and interference with CD4 T‐cell actin dynamics (Fig EV3A–E). These activities including receptor downregulation events were thus dispensable for the impairment of humoral immunity by the HIV pathogenesis factor. In search of additional NEF effects that could explain the disruption of humoral immunity, we next assessed whether the viral protein affects the distribution of the essential T helper cytokine IL‐4. In line with a previous report (Kupfer et al, 1994), IL‐4 was markedly polarized toward the IS in conjugates between control CD4OT‐II and BHEL cells (Fig 3I and J, 51 ± 6% of cells with IL‐4 enrichment at the IS). In contrast, expression of NEF WT in CD4OT‐II cells significantly reduced the fraction of cells in which such polarization of IL‐4 toward the IS was observed (28 ± 12% of cells with IL‐4 enrichment at the IS). Similar to disruption of humoral immunity, this effect of NEF was strictly dependent on its N‐terminal interaction motif (see ∆12–39 and A32–39 mutants) but not the di‐leucine motif of NEF. Importantly, although the diffuse cytoplasmic distribution of IL‐4 in cells without IL‐4 polarization toward the IS was difficult to appreciate by microscopy, flow cytometry revealed that the overall cellular levels of IL‐4 were unaffected by NEF expression (Fig EV3F). Moreover, levels of IL‐4 and other cytokines in the supernatant of T‐B co‐cultures were not significantly altered by NEF expression (Appendix Fig S2G–J), which may reflect that NEF specifically affects cytokine polarization but not release or that the amount of NEF‐expressing ISs in these cultures is too low to appreciate effects on overall cytokine production. These results reveal that NEF affects intracellular polarization but not synthesis of IL‐4 and suggest that the viral protein, via its N‐terminal interaction motif, dampens humoral immune responses by impairing polarized secretion of IL‐4 at the T‐B IS.
Figure EV3. Basic characterization of nef alleles and lack of effect of HIV‐1 SF2 NEF on steady state levels of IL‐4.
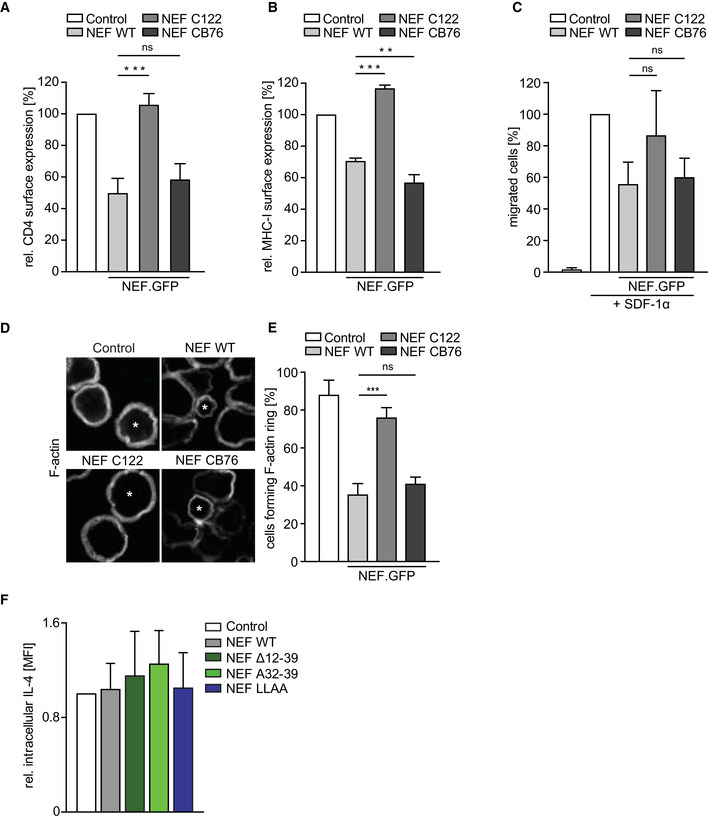
-
A, BFor characterization of nef alleles, standard assays were performed to evaluate the NEF protein function. One day post‐transfection of human T‐cell line (A3.01), cells were either stained with anti‐CD4 (A) or anti‐HLA‐A, B, C (B) antibody to evaluate the effect of nef alleles on receptor cell surface expression. Quantification of receptor expression was carried out as a ratio of mean fluorescence intensity (MFI) values of CD4 or MHC‐I in GFP‐expressing cells to non‐GFP‐expressing cells. The value of control was arbitrarily set to 100%, and all other values were normalized relative to that. Shown are mean values with SD of three independent experiments.
-
CChemotaxis of Jurkat (CCR7) cells toward SDF‐1α was determined by transmigration of cells in a transwell system (5 μm pore size) for 2 h after starvation in 0.5% FCS containing hunger medium. Percentage of migrated cells was calculated as ratio of % GFP‐expressing cells after migration to % GFP‐expressing cells in input. The value of control was arbitrarily set to 100%, and all other values were normalized relative to that. Shown are mean values with SD from at least three independent experiments.
-
DRepresentative confocal images of Jurkat (TAg) T cells on anti‐CD3 coated cover glasses 24 h post‐transfection, fixed, permeabilized, and stained for F‐actin. Asterisks (*) indicate cells expressing either GFP or NEF.GFP. Shown are mean values with SD from at least three independent experiments. Scale bar 10 µm.
-
EQuantification of samples as analyzed in D forming peripheral F‐actin ring upon activation by counting 50–100 GFP‐expressing cells per condition. Shown are mean values with SD from three independent experiments.
-
FIntracellular expression of IL‐4 in T cells expressing NEF or indicated mutants. Transduced T cells were treated with Golgi transport inhibitor for 4–5 h and intracellular IL‐4 was analyzed by flow cytometry. Shown are mean values with SD of the MFI of IL‐4 signal normalized to Control T cells (set to 1) in 3 independent experiments.
Data information: Statistical significance was assessed by one‐way ANOVA with Dunnett’s multiple comparisons test. ***P ≤ 0.001, **P ≤ 0.01.
HIV‐1 NEF‐mediated impairment of T‐cell help prevents B‐cell proliferation and GC formation
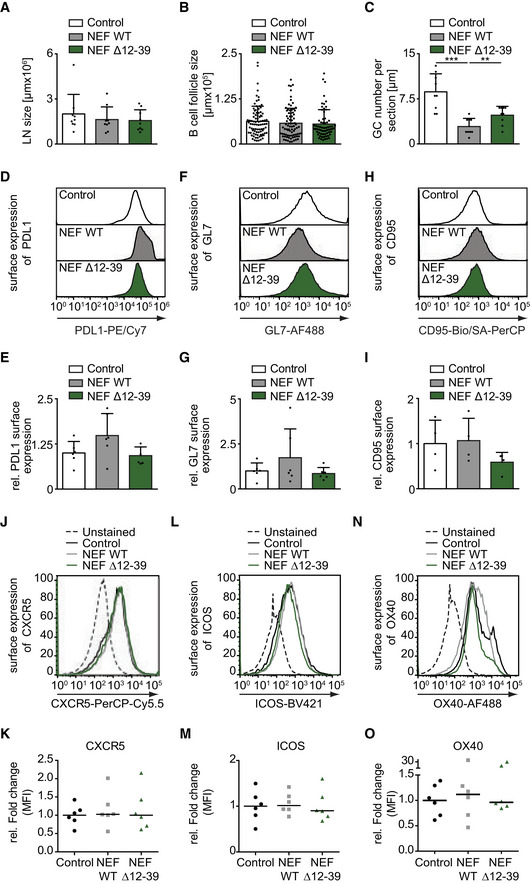
The above finding that HIV‐1 NEF interferes with Ag‐specific T‐B communication via its N‐terminal interaction motif allowed us to more specifically assess which step in mounting of humoral immunity was affected by expression of the viral protein in CD4 T cells. To this end, lymph nodes (LNs) of adoptively transferred, immunized mice were analyzed for abundance and organization of GCs as areas where constant B–T interaction mediate affinity maturation and antibody class switching. Overall, the size of LNs (Fig EV4A) and B‐cell follicles (Fig EV4B) was not affected by expression of NEF in CD4 T cells. In contrast, the number of GCs as assessed by the number of area that were stained with the GC marker GL‐7 was significantly reduced in mice receiving NEF‐expressing CD4 T cells compared to LNs from control mice (Figs EV4, 4) and the GCs formed in these animals were significantly smaller (Fig 4C). Consistently, the quantification of absolute number of BHEL cells and CD4OT‐II cells in draining lymph nodes by flow cytometry revealed that NEF expression compromised B‐cell expansion (Fig 4D and E). The few BHEL cells present in mice receiving NEF WT‐expressing CD4OT‐II cells, however, were phenotypically only mildly altered as judged by their surface expression of PDL‐1, GL7, and CD95 (Fig EV4D–I). Notably, expansion of CD4OT‐II cells, expression of T‐cell activation markers (ICOS and OX40), and expression of the B‐cell follicle localization marker CXCR5 in LNs were also unaffected by expression of NEF (Figs 4F and G, and EV4J–O).
Figure EV4. NEF interferes with antibody affinity maturation, while GC B‐cell numbers or B‐cell follicle size is not altered by NEF.
-
A–CMice were immunized with HEL‐OVA/CFA following adoptive transfer of BHEL and CD4OT‐II cells expressing WT or mutant NEF. Draining lymph nodes (dLN) were harvested at d7 post‐immunization, cryosectioned, and stained with fluorophore‐coupled antibodies to visualize GCs (GL‐7), follicular dendritic cells (CD21/CD35), and nuclei (Hoechst). Quantification of dLN size (A), B‐cell follicle size (B), and GC numbers (C) per section are quantified from confocal micrographs as shown in Fig 4A. Shown are mean values with SD from three mice in two independent experiments. P‐values were calculated by one‐way ANOVA with Tukey’s post‐test. ***< 0.001, **< 0.01.
-
D–ORepresentative flow cytometry histogram plots showing surface expression of PD‐L1 (D), GL‐7 (F) and CD95 (H) on B cells and CXCR5 (J), ICOS (L), and OX40 (N) on T cells harvested from dLN of immunized mice. Receptor expression was quantified on cells that were positive for CD45.2 (donor cell) and either B220 (for B cells) or CD3 (for T cells). Panels (E, G, I) and (K, M, O) show mean values of the data shown in (D, F, H) and (J–N, respectively. Data shown are derived from 2‐3 independent experiments. Shown are mean values with (E, G, I) and without (K, M, O) SD, each dot represents one animal.
Figure 4. HIV‐1 NEF prevents GC formation upon immunization.
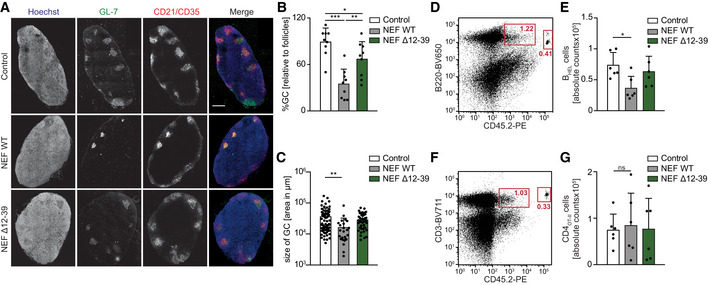
NEF impairs GC formation in immunized mice via the 12–39 region.
-
ARepresentative cross‐sections of draining LNs (dLNs) from mice 7 days post‐immunization with HEL‐OVA/CFA following adoptive transfer of BHEL and transduced CD4OT‐II cells. dLN were cryosectioned and stained with fluorophore‐conjugated antibodies against GL‐7 to reveal GCs (green), CD21/35 to stain for follicular dendritic cells (FDC, red), and Hoechst to stain for cell nuclei (blue). Scale bar, 250 µm.
-
B, CQuantification of the number of GCs relative to B‐cell follicles (B) and size of GC (C) on confocal micrographs as shown in A. Shown are mean values with SD from dLNs from three mice in two independent experiments. Each dot represents one LN (3 LNs/mouse) in B and one GC in C.
-
D–GFlow cytometry dot plots of cells in dLNs from mice 7 days post‐immunization that received the indicated transduced CD4OT‐II and BHEL cells. Adoptively transferred cells were identified and distinguished from endogenous cells of recipient SMARTA mice (CD45.1+) by staining for CD45.2. For quantification of absolute cells numbers, precision beads were used and cells double positive for B220 and CD45.2 (D, BHEL cells) or CD3 and CD45.2 (F, CD4OT‐II cells) analyzed. (E, G) Quantification of absolute BHEL cells (E) as shown in D and CD4OT‐II cells (G) as shown in F. Shown are mean values with SD of 6 mice from 3 independent experiments. Each dot represents one animal.
Data information: Statistical significance was assessed by one‐way ANOVA test with Holm–Sidak’s multiple comparison test. ***P ≤ 0.001, **P ≤ 0.01, *P ≤ 0.05.
We next performed intravital imaging of draining popliteal lymph nodes to assess the impact of NEF WT expression on the in vivo interaction of membrane Tomato (mT) CD4OT‐II cells with cognate BHEL cells at two different time points post‐immunization (Fig 5). Twenty‐four hours post‐immunization, early cognate interactions prior to T‐cell and B‐cell differentiation into TFH and GC B cells, respectively, mainly occur at the T–B border (Cyster & Allen, 2019). We observed abundant control CD4OT‐II cells in this anatomical side which underwent frequent, repeated, and often prolonged (yellow arrows) interactions with BHEL cells (Fig 5A, upper panel, B, Movie EV3). In contrast, only few NEF‐expressing CD4OT‐II cells could be found at the T–B border that mostly underwent transient interactions with BHEL cells (orange arrows, Fig 5A, lower panel, B, Movie EV4). Most NEF‐expressing cells were found in the deep T‐cell area, which was devoid of BHEL cells, suggesting that the recruitment of these cells to the T–B border was impaired (unpublished observation B. Stolp and O.T. Fackler). At 6–8 days post‐immunization, GCs have been induced and prolonged cognate interactions of control CD4OT‐II cells with BHEL cells could be observed in these structures (highlighted by subcutaneous injection of a follicular dendritic cell (FDC) marker) (Fig 5C, white). ity of this T–B interaction was demonstrated by immunization with CFA only in the absence of antigen which did not trigger CD4OT‐II cells and BHEL cells to migrate within the same lymph node areas and to undergo prolonged interactions (maximum interaction observed was less than 4 min, while cells were passing each other) (Appendix Fig S3, Movie EV7). Upon immunization with HEL‐OVA, control CD4OT‐II cells underwent frequent, prolonged interactions with BHEL cells inside GCs (Fig 5C, upper panel, yellow arrows, D, Movie EV5). In contrast, BHEL cells could only be detected in a subset of mice that received NEF‐expressing CD4OT‐II cells (five out of nine mice). In case sufficient BHEL cells were induced be NEF‐expressing CD4OT‐II cells to allow interaction analyses, they only underwent brief, transient interactions (Fig 5C, lower panel, orange arrows, D, Movie EV6). Notably, tracking of migration speeds 4–8 days post‐CFA only immunization revealed the expected drop in migration speed and meandering index of NEF‐expressing CD4OT‐II cells (Murooka et al, 2012; Stolp et al, 2012), illustrating that functional NEF expression was maintained in vivo at this late time point post‐adoptive transfer (Appendix Fig S3 and Movie EV7). Together, these results reveal that NEF expression in CD4OT‐II cells restricts Ag‐specific interactions with B cells in vivo.
Figure 5. NEF restricts T–B cell interactions upon immunization.
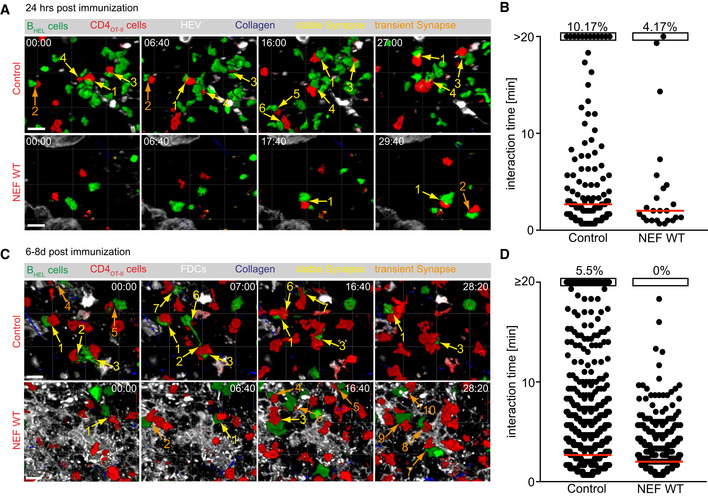
NEF impairs duration of Ag‐induced interaction of CD4OT‐II cells with BHEL cells in vivo.
- Still images of the Movie EV3 (Control, upper panel) and Movie EV4 (NEF, lower panel), obtained by intravital time‐lapse 2PM imaging of draining popliteal LNs 24 h post‐immunization with HEL‐OVA/CFA, following adoptive transfer of GFP BHEL cells (green) and mTomato CD4OT‐II cells (red) expressing empty vector (Control; upper panel) or NEF WT (lower panel), respectively. High endothelial venules (HEVs) (white) were labeled by intravenous injection of Alexa660‐labeled Meca‐79 antibody and collagen (blue) is visualized by second harmonic generation (SHG). Yellow and orange arrows indicate stable and transient ISs, respectively. Numbering of ISs corresponds to that in the respective Supplemental movies. Scale bar 20 µm.
- Quantification of the duration of interactions as indicated by the arrows in A. Touching T and B cells were manually quantification using virtual reality (InViewR, Arivis). Each dot represents one T‐B interaction pair. Red bar shows median. Data are derived from 5 to 6 movies, acquired from 4 mice in 2 independent experiments. Percentages on top indicate frequency of interactions that lasted 20 min or longer.
- Still images of the Movie EV5 (Control, upper panel) and Movie EV6 (NEF, lower panel), obtained by intravital time‐lapse 2PM imaging of draining popliteal LNs 8 days post‐immunization with HEL‐OVA/CFA, following adoptive transfer of GFP BHEL cells (green) and mTomato CD4OT‐II cells (red) expressing empty vector (Control; upper panel) or NEF WT (lower panel). Follicular dendritic cells (FDCs) (white) were labeled by subcutaneous injection of Alexa647‐labeled anti‐CD21/CD35 antibody and collagen (blue) is visualized by SHG. Yellow and orange arrows indicate stable and transient ISs, respectively. Numbering of ISs corresponds to that in the respective Supplemental movies. Scale bar 20 µm.
- Quantification of the duration of interactions as indicated by the arrows in C. Touching T and B cells were manually quantification using virtual reality (InViewR, Arivis). Each dot represents one T‐B interaction pair. Red bar shows median. Shown is one representative dataset out of two, derived from 5 to 8 movies, acquired from 4 mice. Percentages on top indicate frequency of interactions that lasted 20 min or longer.
Since affinity maturation of antibodies relies on iterative cycles of GC reactions governing tightly regulated B‐cell proliferation (Victora & Nussenzweig, 2012), we tested if the observed reduction of GC formation in the presence of NEF‐expressing T cells was associated with an impairment of the ability of B cells to undergo somatic hypermutation. For this, we used HEL3X‐OVA containing three amino acid substitutions in HEL, against which BHEL cells have a 10,000‐fold lower affinity than to HEL‐OVA (Paus et al, 2006). While BCR‐Ag matching supports the induction of IgG production in mice that received BHEL cells in response to HEL‐OVA immunization without the need for somatic hypermutation, HEL3X‐OVA exerts antigenic selection pressure for B cells that acquire high‐affinity BCRs during somatic hypermutation (Fig EV5A). When HEL3X‐OVA was used for immunization following adoptive transfer, NEF moderately but significantly reduced the proportion of B cells expressing high‐affinity BCR for HEL3X to the level comparable with mice that did not receive cognate T cells (Fig EV5B and C). This result indicated that BHEL cells that were in contact with CD4 T cells expressing NEF do not undergo somatic BCR hypermutation. In line with this scenario, adoptive transfer of NEF‐expressing CD4OT‐II cells markedly reduced the production of HEL3X‐specific antibodies (Fig EV5D). Together, these results suggest that the impairment of early B‐cell activation by NEF expression in CD4 T cells translates into reduced GC formation, B‐cell expansion, and somatic hypermutation contributing to disruption of efficient antibody generation.
Figure EV5. NEF interferes with somatic hypermutation.
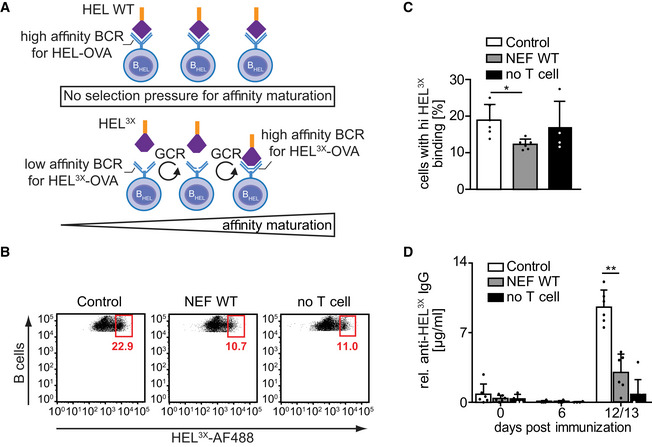
- Schematic indicating process of somatic hypermutation of BCR resulting in affinity maturation of class‐switched antibodies as occurring during germinal center reaction (GCR). Top: BHEL cells have intrinsic high affinity for HEL Ag and do not undergo additional BCR mutation. Bottom: Since HEL‐specific B cells have 10,000‐fold lower affinity for HEL3X Ag as compared to HEL, they undergo a selection process during GCR, based on their relative antigen affinity following somatic hypermutation. This process strictly depends on T‐cell help during GCR cycles.
- To analyze the effect of NEF on T‐cell help, mice were immunized with HEL3X‐OVA/CFA in the presence of Control or NEF‐expressing T cells. Cells were isolated from dLN 7 days after immunization. Single‐cell suspensions were stained with fluorophore‐conjugated anti‐B220 antibody and HEL3X protein. The proportion of B cells with high‐affinity BCR for HEL3X was assessed by the ability of in vivo proliferated and selected BHEL cells to bind to the AlexaFluor 488‐coupled HEL3X by flow cytometry. Shown are representative flow cytometry dot plots gated on B220 (B cell) and CD45.2 (donor population) double positive cells. To estimate how many B cells underwent somatic hypermutation without any T‐cell help, immunization was carried out in the absence of CD4OT‐II cells (no T cell). The gated population indicates the percentage of B cells bearing BCRs with high affinity for HEL3X.
- Quantification of BHEL cells with high affinity for HEL3X as shown in B. Shown are mean values with SD from 4 to 6 mice from two independent experiments. Each dot represents one animal.
- Production of anti‐HEL3X IgG at the indicated days post‐immunization with HEL3X‐OVA/CFA, following adoptive transfer of BHEL cells and Control or NEF‐expressing CD4OT‐II cells. Shown are mean values with SD from 4 to 10 mice analyzed from 2 independent experiments. Each dot represents one animal.
Data information: P‐values in C and D were calculated by a Kruskal–Wallis with Dunn’s multiple comparison test. **P ≤ 0.01, *P ≤ 0.05.
HIV‐1 NEF disrupts antibody production in HIV‐1 infected human tonsil explant cultures
Finally, we sought to test whether the ability of NEF to impair B‐cell function, observed in ex vivo and in vivo mouse models, is also exerted in the context of HIV‐1 infection in human cells. As current humanized mouse models fail to effectively mount humoral immune responses and were thus unsuitable for such analyses (Villaudy et al, 2014), we employed ex vivo cultures of human tonsil tissue. This model system is readily permissive to HIV‐1 infection and the inhibition of production of antibodies against recall Ags like tetanus toxin (TT) by some but not all HIV‐1 strains has been reported (Grivel & Margolis, 2009) (Glushakova et al, 1995). Blocks of tonsil tissue obtained from routine tonsillectomy from HIV‐negative donors were cultured, infected with equal infectious units of HIV‐1 WT, ΔNEF, or HIV‐1 encoding for the NEF mutant Δ12–39 and subsequently challenged with TT (Fig 6A). The normalization to infectious units overrode the effect of NEF on virion infectivity which provides a replication advantage in ex vivo tonsil cultures (Glushakova et al, 1999; Glushakova et al, 2001; Homann et al, 2009). Under these conditions, NEF did thus not markedly affect HIV replication kinetics as determined by the release of virus particles (determined by measuring reverse transcriptase activity in the cell culture supernatant) (Fig 6B), thus allowing the comparison between similar numbers of infected cells in cultures inoculated with the different HIV‐1 variants. Responsiveness to addition of TT by robust production of anti‐TT IgG was observed in tonsils from half of the donors tested and, in these cases, infection with HIV‐1 WT reduced anti‐TT IgG production almost to background levels observed in the absence of TT (Fig 6C). Of note, lack of NEF expression or deletion of NEF’s N‐terminal interaction motif abrogated this impairment of antibody production. HIV‐1 infection thus disrupts the mounting of humoral immune responses in organotypic cultures of human tonsil tissue via the N‐terminal interaction motif of the pathogenesis factor NEF.
Figure 6. NEF impairs antibody production in HIV‐1 infected human tonsil histocultures.
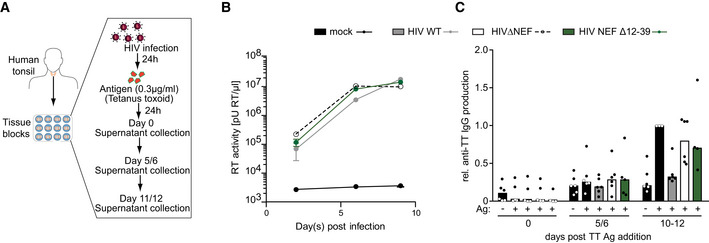
Analysis of the impact of NEF on anti‐TT IgG production in HIV‐1 infected tonsillar blocks.
- Schematic overview of HIV‐1 infection and Tetanus toxoid (TT) stimulation of tonsillar histocultures.
- Virus replication as assessed by quantification of RT activity in the cell culture supernatant. Shown are mean values with SD from 6 donors.
- Production of IgG antibodies against TT from infected blocks at indicated time point. Each dot represents one donor, bar indicates mean value. Tonsillar blocks were infected with indicated virus for 24 h. After washing off the virus, blocks were stimulated with TT antigen for 24 h. Samples were collected at different time intervals to estimate viral replication and antibody production.
Discussion
The clinical outcome of untreated virus infection reflects the relative efficacies of host immune responses and viral evasion strategies. In the case of HIV‐1, humoral immune responses are of particularly low efficacy that only rarely lead to potent neutralizing antibody responses (Burton & Hangartner, 2016; Subbaraman et al, 2018). Broadly neutralizing antibodies as produced by elite neutralizer patients have thus proven to be valuable additions to current therapy and prevention measures (Balazs et al, 2011; Caskey et al, 2015; Gautam et al, 2016; Niessl et al, 2020; Schommers et al, 2020). In addition to the specific topological challenges toward neutralizing the fusion activity of the HIV‐1 glycoprotein Env, the immune system of HIV patients is generally impaired in mounting humoral immune responses. Studies on the mechanisms underlying this B‐cell dysfunction thus far mostly focused on polyclonal B‐cell exhaustion in the context of generalized immune activation. We report here that HIV‐1 infection of CD4 T cells results in a potent impairment of their helper function and thus disruption of Ag‐specific humoral immunity. Expression of the viral factor NEF, known to optimize virus replication and accelerate disease progression in HIV patients, was necessary and sufficient for this impairment of T‐cell help, which was observed in an adoptive transfer mouse model as well as upon experimental infection of human tonsil tissue. These results define (i) an additional mechanism by which HIV‐1 undermines humoral immune responses of its host and (ii) a previously unrecognized activity of NEF that likely contributes to its cardinal role in AIDS pathogenesis.
A key finding of this study was that expression of NEF in CD4 T cells disrupts functional communication with cognate B cells in the context of an IS, which in turn results in a substantial reduction of GC formation (Fig 4B). The previous observations that NEF transgenic mice in which lymphoid architecture is compromised display fewer GCs (Poudrier et al, 2001) thus likely reflect this suppression of B‐cell function by NEF‐expressing CD4 T cells. Our further characterization revealed that NEF prevents GC formation and somatic BCR hypermutation (Fig EV5), which is critical for the subsequent generation of high‐affinity Abs. By blocking these steps, HIV‐1 prevents efficient mounting of humoral immune responses and thus provides the virus with the possibility to escape antibody neutralization. Since in our murine assays, NEF is only expressed in CD4 cells, we were able to distinguish NEF’s effect on Ag‐specific B‐cell activation from polyclonal B‐cell activation, which is largely induced by sustained systemic inflammation (Moir & Fauci, 2009). This novel mechanism of NEF‐mediated disruption of humoral immune responses by suppression of T‐cell help during T–B interactions was also observed for patient‐derived NEF variants and NEF was critical for HIV‐1 mediated reduction of antibody production in tonsillar histocultures. While disruption of antibody production by NEF in mice was observed in all animals analyzed, NEF did not exert this inhibitory effect in HIV‐1 infection of tonsil tissue from all donors. This indicates that, e.g., the genetic constitution or basal immune activation state of the host may determine how efficiently NEF can exert this activity. These effects of NEF in CD4 T cells likely synergize with effects of the viral protein in infected macrophages (Swingler et al, 2008; Xu et al, 2009), suggesting NEF as the central viral determinant of B‐cell dysfunction in HIV‐1 infection. HIV‐1 preferentially replicates in HIV‐specific CD4 cells (Douek et al, 2002). Our results suggest that beyond these cells representing good substrates for virus replication, virus replication in HIV‐specific CD4 cells also ensures specific delivery of the cardinal viral immunosuppressant to the cells that are most needed to support mounting of HIV‐specific B‐cell responses. NEF‐mediated disruption of T‐cell help thus likely contributes to, e.g., the reduced generation of high‐affinity antibodies in HIV patients (Cubas et al, 2013) and their poor response to vaccination (Pallikkuth et al, 2012). In this scenario, the effect of NEF would be similar to the reduction of humoral immune memory induced by infection with Measles virus (Mina et al, 2019; Petrova et al, 2019). These parallel studies suggest that direct suppression of T‐cell help by T cell‐tropic viruses is of high benefit for virus pathogenesis and spread.
Our results also provided insight into the molecular mechanism by which HIV‐1 NEF disrupts T‐B communication to prevent T‐cell helper function (Fig 7). While NEF had no impact on the frequency of interactions of BHEL cells with CD4OT‐II cells ex vivo, NEF‐expressing cells were not efficiently recruited to the T–B border and cells reaching this anatomical site engaged in fewer, less stable and mainly transient interactions with cognate B cells in vivo. Consistently, marked qualitative differences in signal transmission across the T–B IS were observed as NEF impaired B‐cell actin remodeling and deregulated the induction of B‐cell gene expression. Since the NEF‐induced deregulation preferentially affected genes involved in B‐cell activation/differentiation, metabolism, and survival, it is conceivable that the sum of these alterations explains the functional deficits of these B cells in vivo. B‐cell actin remodeling is required for coordinated B‐cell activation including induction of the respective gene expression programs (Song et al, 2014). The inability of NEF+ CD4OT‐II cells to induce B‐cell actin dynamics thus likely represents a key event in the disruption of T‐cell help by the viral protein. Surprisingly, the analysis of Nef mutants and alleles revealed that functions known to be exerted by NEF in CD4 T cells such as reduction of cell surface receptor densities or impairment of actin remodeling were dispensable for this effect. Instead, we identified prevention of CD4OT‐II cell IL‐4 polarization toward the IS as a novel NEF activity mediated by the N‐terminal interaction surface required to disrupt humoral immunity. Since (i) engagement of the IL‐4 receptor triggers coupling to RAC1 and PAK‐dependent actin remodeling (Kurgonaite et al, 2015) and may thus drive the observed actin dynamics and (ii) IL‐4R signaling acts as master switch for survival and differentiation of activated B cells (Reiter & Pfeffer, 2002; Wurster et al, 2002), we propose prevention of polarized IL‐4 secretion as an active principle of NEF for the disruption of T‐B communication. Since in preliminary experiments, we observed that NEF can also alter the recruitment of cell surface receptors in to the IS, effects of the viral protein may be pleiotropic and include activities in addition to alteration of IL‐4 secretion. Very limited information is available on the biosynthetic transport pathways of IL‐4. Future studies will thus focus on using the viral protein as tool to dissect mechanism and regulation of polarized IL‐4 transport, on defining the molecular interactions that NEF employs to interfere with this process and on assessing the relative contribution of interference with IL‐4 polarization to the disruption of T‐cell help. The low frequency of HIV‐1‐infected cells (2–3%) and TT‐specific B cells (below 1%) prevented us from analyzing the effects of NEF on IL‐4 polarization in infected tonsil cultures. A future focus will therefore be to develop experimental approaches that allow to study these events in the context of HIV‐1 infection of human cells.
Figure 7. Schematic model of the disruption of T‐cell help by HIV‐1 NEF.
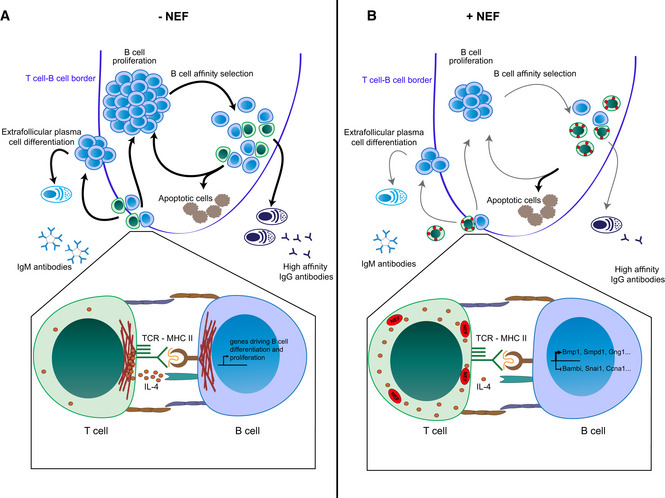
Overview of the effects of HIV‐1 NEF at the level of the GC (upper panels) and the T‐B IS (lower panels).
- Undisturbed physiological reactions in the absence of Nef.
- Effects in the context of HIV‐1 infection due to expression of NEF. See text for details.
Together, these results define disruption of T‐cell help and thus impairment of B‐cell function by the HIV‐1 pathogenesis factor NEF as a previously unrecognized viral evasion mechanism from humoral immune responses of the host. The N‐terminal molecular interaction surface in NEF required for this activity and its ligands represent attractive targets for future therapeutic intervention strategies aimed at boosting humoral immunity in HIV patients.
Materials and Methods
Mice
All mice used in the study were of C57/BL6N genetic background. OT‐II.2 (transgenic TCR that recognizes chicken ovalbumin323‐339 peptide in the context of MHC class II I‐Ab), SWHEL –HL (transgenic for HEL‐specific BCR light chain, knock‐in for HEL‐specific BCR heavy chain), SMARTA/CD45.1 (transgenic TCR that recognizes LCMV GP61–80 in the context of MHC class II I‐Ab, used as recipients in adoptive transfer studies, being unreactive to OVA peptide), CAG‐eCFP, mT/mG, and UBC‐GFP mice were kind gifts from Jens Stein (Bern, Switzerland), Robert Brink (Sydney, Australia), and Inka Zörnig/Anette Oxenius (Zürich, Switzerland), Anette Oxenius, Marc Freichel, and Stephan Herzig, respectively. All mice, despite the SMARTA line, are bred on a CD45.2 background, to be able to distinguish adoptively transferred cells by flow cytometry. OT‐II.2 mice were backcrossed to CAG‐eCFP and mT/mG mouse lines to generate ubiquitously eCFP and membrane Tomato expressing mice as OT‐II T‐cell donors, respectively. SWHEL –HL mice were backcrossed to UBC‐GFP mice to generate ubiquitously GFP‐expressing mice as donors for SWHEL –HL B cells. Six‐ to 10‐week‐old male mice were used for in vivo experiments, as the OT‐II TCR transgene is encoded on the Y chromosome. All experiments with mice were carried out in accordance with the standards approved by central animal facility of the University of Heidelberg (G252/14, G300/19, T45/14, T30/17, and T39/19).
Preparation of HEL‐OVA323‐339 chemical conjugates
OVA323‐339 was maleimide‐activated with 25‐fold molar excess of succinimidyl 4‐(N‐maleimidomethyl)cyclohexane‐1‐carboxylate (SMCC; Thermo Fisher) for 30 min at RT. Desalting columns (SpinOUT GT‐100, G Biosciences) equilibrated with conjugation buffer were used to remove excess cross‐linker. HEL protein (SIGMA) was incubated with a 10‐fold molar excess of N‐succinimidyl S‐acetylthioacetate (SATA; Thermo Fisher) for 30 min, and HEL‐conjugated SATA was deacetylated using 0.5 M hydroxylamine (25 mM EDTA in PBS) for 2 h at RT. Unreacted reagents were removed by using equilibrated desalting columns (Thermo Fisher Scientific). Eluted fractions were selected by measuring the absorbance at 280 nm. Maleimide‐activated OVA323–339 peptides were then conjugated with a fourfold molar excess of sulfhydrylated‐HEL for 30 min at RT. New batches of prepared HEL‐OVA were titrated in an immunization assay with three conditions, i.e., Control, Nef WT, and negative (No T cell) control. The dose of HEL‐OVA at which appreciable difference in IgG level was observed between Control and Nef WT with minimum background from negative control was selected to be used for all the assays.
Expression plasmids
The expression plasmids for pSTITCH‐GFP, pHit60, pHit123, and MLV‐A were kindly provided by Reno Debets (Department of Medical Oncology, Erasmus MC, Rotterdam, The Netherlands). The cloning strategy to insert NEFSF2 and various mutants in pSTITCH has been described (Stolp et al, 2012; Lamas‐Murua et al, 2018). The coding sequences for NEFSF2A32‐39, NEFSF2LLAA, or patient‐derived nef genes C122 and CB76 were cloned into pSTITCH using the restriction sites AgeI and PacI in a multiple‐cloning site that was previously subcloned along with IRES∆NGFR from pQCXIX backbone. The proviral constructs used are based on HIV‐1NL4‐3 and either lack NEF expression (HIV‐1 ∆NEF), or encode for wild‐type NEF from HIV‐1SF2 (HIV‐1 WT) or specific NEF mutants (Fackler et al, 2006; Ananth et al, 2019).
Preparation of CD4OT‐II cells
Single‐cell suspensions were obtained from spleen, peripheral (axillary, brachial, inguinal, cervical), and mesenteric lymph nodes of OT‐II mice by mechanical disruption (70 µm cell strainer, Falcon), and erythrocyte depletion using ammonium‐chloride‐potassium lysis buffer. Isolated cells were cultured for 2 days in RPMI‐1640 supplemented with 10% FCS, 100 U/ml penicillin, 100 µg/ml streptomycin, 2 mM Glutamine, 50 µM 2‐mercaptoethanol, 0.1 mM non‐essential amino acids, and 0.1 mM sodium pyruvate (all from GIBCO) in the presence of 1 µg/ml OVA323‐339 peptide (InvivoGen). Transduction of murine CD4+ T lymphocytes was carried out as described below.
MLV‐based transduction of murine CD4OT‐II cells
HEK 293T or PlatE cells were used as producer cells and were maintained in DMEM with high glucose, supplemented with 10% FCS, 100 U/ml penicillin, and 100 µg/ml streptomycin. Twenty‐four hours before transfection, 5 × 106 cells were seeded in T‐175 culture flasks. Transfection was performed using JetPEI transfection reagent (Peqlab) according to manufacturer’s instructions using: 20 µg of a pSTITCH vector, 20 µg pHit60 (expressing Gag and Pol), 5 µg pHit123 (expressing ecotrophic Env), and 5 µg MLV‐A (expressing amphotropic Env) in case of HEK 293T cells, or 20 µg of a pSTITCH vector and 5 µg MLV‐A in case of PlatE cells. Viral supernatant was collected after 48 h, filtered (0.45 µm pore‐size filter; Roth), and used immediately for transduction of CD4OT‐II. For transduction, CD4OT‐II were spin‐infected 48 h post‐activation with virus supernatant for 90 min at 2,300 rpm, 32°C in 24‐well plates coated with 16 µg/ml Retronectin (TaKaRa BioTech). Following transduction, cells were cultured with the viral supernatant for 3–5 h followed by a medium change. Transduced cells were transferred to T‐25 culture flasks after 16 h and kept in culture for 4–5 days. During this time, cells return to a resting state and regained responsiveness to activation by OVA presenting B‐cell interactions (Fig EV2B). Transduced cells (NGFR positive) were enriched by magnetic sorting (EasySep™ Human CD271 Positive Selection Kit II; STEMCELL Technologies) and used for ex vivo or in vivo assays.
Cell tracker dyes used for imaging and proliferation assays
B cells or CD4OT‐II were labeled with either CellTrace™ CFSE (5 µM), CellTracker Green CMFDA (2.5 µM), CellTracker Blue CMAC (10 µM), CellTrace™ Violet Cell proliferation dye (5 µM) (all from Thermo Fisher Scientific), or Cell proliferation Dye eFluor™670 (eBiosciences), according to manufacturer’s instructions. Briefly, cells were resuspended in 1× PBS at the cell density of 8–10 × 106 cells/ml and labeled at 37°C for 15–20 min. Unbound dye was washed away using complete medium in the centrifugation step, and labeled cells were immediately analyzed or used for experiments.
Immune synapse
NGFR‐enriched CD4OT‐II (0.25 × 106 cells) were either not labeled or labeled with CFSE (Thermo Fisher Scientific). The cells were then mixed with CMAC‐labeled B cells (0.5 × 106 cells) and incubated on PLL (0.1%, Sigma‐Aldrich)‐coated cover glasses in each well of a 24‐well plate for 1–2 h. Before incubation, the plate was spun at 1,000 rpm for 60 s to reduce the cell settling time. Sample was fixed using 1–3% PFA in PBS and incubated for another 10 min at RT. Cells were then permeabilized with 0.1% Triton X‐100 for 2–3 min and blocked in 1% BSA in PBS (v/v) for 30 min at RT. Following washing with 1× PBS, TRITC‐conjugated Phalloidin (Thermo Fisher), mouse anti‐p‐Tyr 100 (Cell Signaling Technology), and rat anti‐mouse IL‐4 (R&D Systems) was used for staining. Cover glasses were washed three times with 1× PBS and incubation with secondary anti‐mouse Alexa‐568 or anti‐Rat Alexa‐568 (1:2,000, in 1× PBS with 1% BSA, Invitrogen) was added and incubated for 1 h at RT. After washing with 1× PBS, cover glasses were mounted using Mowiol (Calbiochem), dried over night at RT protected from light and stored at 4°C until analyzed by epifluorescence (IX81 Olympus), confocal (TCS SP5), and spinning disc confocal (Nikon Ti PerkinElmer UltraVIEW) microscopy. Images were processed using ImageJ.
Visualization of B‐cell F‐actin polymerization during IS
Magnetically sorted BHEL cells (B‐cell enrichment kit (untouched); STEM CELL TECHNOLIGIES) were cultured in the presence of LPS (Sigma) and HEL‐OVA (both; 3 µg/ml) for 16–20 h. Activated B cells were labeled with CMAC in 1× PBS and then with 0.5 µM of SirAct (Spirochrome) in complete medium for 20 min at 37°C. After washing away excess dye, cells were immediately used for imaging. B cells (4 × 104) were added in PLL‐coated wells of µ‐Slide Angiogenesis (iBidi), 2–3 min later labeled CD4OT‐II were added slowly from the side of the well and imaging was started within 2–3 min. Live microscopy of IS was performed using spinning disc confocal microscope (Nikon Ti PerkinElmer UltraVIEW), with a 60× oil objective and an environmental control chamber (37°C, 5% CO2). Time‐lapse images were taken for 30–60 min with 10 Z planes, each stack spacing 0.5 µm, and laser power between 3 and 6%.
Ex vivo T cell‐dependent B‐cell proliferation
For analyzing the effect of NEF on T cell‐dependent B‐cell proliferation, 2 × 105 labeled CD4OT‐II cells were co‐cultured with labeled 6 × 105 BHEL with titrating amounts of HEL‐OVA (3, 1, 0.3, 0.1, and 0 µg/ml). Three days later, B‐cell proliferation was estimated by assessing dye dilution of B cells using flow cytometry. For analysis, the proportion of B cells in the last 3–4 generations (highly dividing cells) in control (+Ag) was arbitrarily set as 1 and relative proportions were calculated for other samples. Peak B‐cell proliferation was observed with different doses of HEL‐OVA in different experiments. Thus, to minimize the variability the dose of HEL‐OVA showing the highest proliferation was included in the analysis. Different Cell Tracker dye combinations were used to assess B‐cell proliferation in different experiments to rule out the effect of cell labeling on the proliferation quantification. To assess effects of NEF on cytokine production, cell culture supernatants were collected 3 days after T–B‐cell co‐culture and cytokines quantified using a Mouse Cytokine/Chemokine Array 31‐Plex (Eve Technologies, Calgary, Canada).
Antibody production in mice following adoptive transfer and immunization
To study the effect of NEF on antibody production, 2 × 104 transduced CD4TOT‐II cells and 6 × 104 BHEL cells (B‐cell enrichment kit (untouched); STEM CELL TECHNOLIGIES) were adoptively transferred into SMARTA mice. After 24 h, mice were immunized subcutaneously with 10–40 µg of HEL‐OVA323–339 emulsified in Freund’s complete adjuvant (CFA; Santa Cruz) at five positions (cranial, axillary left and right, inguinal left and right). Blood was collected from mice before and after immunization via facial vein puncture and production of IgM and IgG antibodies was tested by ELISA. Briefly, 96‐well plates were coated with 1–2 µg/ml of HEL (SIGMA) overnight. Next day, the plates were washed with PBS (with 0.1% Tween 20). Extracted serum was then added, and plates were incubated for 2 h. Plates were then washed and incubated with biotinylated anti‐IgM and anti‐IgG antibodies (Bio‐Rad) followed by HRP‐conjugated SA (Thermo Fisher Scientific). OPD substrate (Thermo Fisher Scientific or SIGMA) was used to develop the reaction which was then stopped using 3 M H2SO4. To detect antibodies generated against HEL3X, plates were coated with 4 µg/ml of HEL3X protein generated in the laboratory (see below).
Generation of recombinant HEL3X protein to study somatic hypermutation
Mutant protein with a COOH‐terminal His(6) tag was cloned (MWG Eurofins) in yeast expression vector pPIC9K (Invitrogen). Transformation of 1.2 OD competent yeast (Pichia pastoris) was done by electroporation (Bio‐Rad Gene Pulser, 25 μF, 200 ohm, 200 V) in the presence 15 µg of linearized DNA. Cells were immediately mixed with 1 ml of cold 1 M sorbitol and spread on Regenerative Dextrose Medium (RDB) agar plates and kept at 30°C for 4 days. Around 1,000 colonies were picked and mixed with Yeast Extract Peptone Dextrose (YPD) medium in 96‐well plate (around 10 plates) and incubated for 5 days at 30°C. Clones were grown to similar density by successive inoculation to ensure that equivalent number of cells are spotted on Geneticin (GIBCO) plate. Yeast culture (10 µl) was spotted on Geneticin (0, 0.25, 0.5, 0.75, 1, 1.5, 1.75, 1, 3, and 4 mg/ml) containing YPD plates in 96‐well format and incubated for 4 days at 30°C. For protein induction, PCR‐confirmed positive colonies were grown in buffered glycerol‐complex medium and scaled up in buffered methanol‐complex medium at absorbance of 1OD and were further grown for 48 h at 30°C at 250 rpm. After every 24 h, 100% methanol was added to a final concentration of 0.25%. For protein purification, filtered supernatant was incubated with Ni‐NTA agarose beads, spun at 770 g (Beckman Allegra centrifuge: 2,000 rpm) for 5 min (3× washing 150 mM NaCl; 30 mM Tris pH 6.8). The bound protein was eluted using 200 mM Imidazole. The purified protein was concentrated using VIVASPIN 3000 centricons (Satorius) at 3,900 g. Concentrated HEL3X protein was coupled with OVA peptide as mentioned above and used for immunization assay (10–20 µg). To analyze the proportion of B cells with rearranged high‐affinity BCR for HEL3X by flow cytometry, HEL3X was coupled with AlexaFluor‐488 using protein labeling kit (Invitrogen).
Microarray analysis
BHEL were co‐cultured with CD4OT‐II cells expressing NEF WT or NEF ∆12‐39 for 4 and 24 h. BHEL were separated using magnetic isolation (B‐cell enrichment kit; STEM CELL TECHNOLIGIES) and used for RNA isolation (NucleoSpin®; Macherey‐Nagel). RNA samples were sent to DKFZ microarray facility and the transcriptome determined by the Clariom™ S Pico assay. For volcano plots, expression of genes was compared between BHEL cells co‐cultured with either control or Nef‐expressing CD4OT‐II cells for 4 h. Data from two independent experiments were used for the analysis. For the generation of heat maps, gene expression in each sample was compared to naïve BHEL cells and data from three independent experiment were used for the analysis. For pathways analysis, WEB‐based GEne SeT AnaLysis Toolkit and Gene Ontology enRIchment anaLysis and visuaLizAtion tool was used.
Immunohistology
For immunohistology, draining lymph nodes were harvested 7 days after immunization, immediately frozen, and stored at −80°C. LNs were embedded into FSC 22 clear frozen section medium (Leica Biosystems), sectioned to slices of 6–10 µm thickness using a cryostat (Leica CM1950), and fixed using 1% PFA for 20 min. Subsequently, sections were treated with blocking buffer (5% skimmed milk powder, 0.6% Triton X‐100, 0.06% sodium azide) in the presence of 1 µg/ml of Fc‐blocker (anti‐mouse CD16/CD32, BioLegend) for 20 min at RT. After 3× washing, sections were incubated with anti‐mouse IgD‐Biotin (11–26c.2a), anti‐mouse GL7‐Alexa488 (GL7), and anti‐mouse CD21/35‐Alexa647 (7E9; all from BioLegend) for 1 h. Streptavidin‐Alexa568 (Thermo Fisher Scientific) was used for second step staining. Sections were washed 3× and staining with 1:100,000 Hoechst 33342 (Thermo Fisher Scientific) for 3–4 min. Fluoromount‐G® (Thermo Fisher Scientific) was used as mounting solution. Leica TCS SP8 confocal microscope was used for imaging of LN sections with a 20× oil objective using mosaic scanning.
2‐photon intravital imaging of popliteal lymph nodes
For visualization of early and late cognate B cell–T cell interactions, 3–4 × 106 or 104 NGFR‐sorted, transduced CD4OT‐II ‐eCFP or CD4OT‐II ‐mT cells and 4 × 106 or 3 × 104 BHEL GFP, respectively, were adoptively transferred into WT mice. After 24 h, mice were immunized subcutaneously into the hock with 10 µg of HEL‐OVA323‐339 emulsified in Freund’s complete adjuvant (CFA; Santa Cruz) or CFA only for the no antigen control movies. 2PM intravital imaging of the popliteal lymph node was performed after 16–24 h (early) or 6–8 days (late) post‐immunization, as described previously (Stolp et al, 2012). In brief, mice were anesthetized with isoflurane (5% for induction and 1‐1.5% for maintenance), under Buprenorphine (0.05–0.1 mg/kg) analgesia. The right popliteal lymph node was surgically exposed. One day prior to imaging, 5 µg Alexa 647‐conjugated anti‐mouse CD21/35 antibody (Biolegend) was injected s.c. into the footpad to label the follicular dendritic cells (FDCs). 2PM was performed with a Nikon Eclipse FN‐1 upright microscope equipped with a 25× Nikon CFI‐Apo (NA 1.1) objective and a TrimScope II 2PM system controlled by ImSpector software (LaVision BioTec) combined with an automated system for real‐time correction of tissue drift (Vladymyrov et al, 2016). For 2‐photon excitation, a Ti:sapphire laser with an optical parametric oscillator (OPO, Coherent MPX Package) were tuned to 840 and 1,100 nm, respectively. For 4‐dimensional analysis of cell migration, 16 x–y sections with z‐spacing of 4 µm (60 µm depth) were acquired every 20 s for 30–40 min; the field of view was 300 × 300 µm. Emitted light and second harmonic signals were detected through 447/60‐nm, 525/50‐nm, 595/50‐nm, and 690/50‐nm bandpass filters using non‐descanned detectors. Time‐lapse movies were reconstructed and analyzed using Arivis Vision 4D software (Arivis AG). Cell interactions were manually tracked and quantified using InViewR (ArivisAG), and cells were tracked using Imaris (Bitplane).
Characterization of nef alleles
Patient‐derived nef alleles were isolated from plasma samples obtained from the New York HIV Study 2010 cohort as described previously with minor modifications (Salazar‐Gonzalez et al, 2008). From a starting volume of at least 500 µl of plasma, RNA was extracted using QIAamp Viral RNA Mini Kit (Qiagen) according to manufacturer’s instructions. This was followed by cDNA synthesis using an oligo dT primer and Superscript III Reverse Transcriptase (Invitrogen). Full length nef gene was amplified from bulk plasma‐derived viral cDNA using two‐step nested PCR with primers that overlap env and 3ʹLTR regions. Bulk PCR products were then cloned into pGEM®‐T TA vector backbone (Promega), and a minimum of 10 nef sequences with intact open reading frame was analyzed per patient. A predominant sequence that resembles the bulk population was then cloned into pEGFP‐N1 vector as Nef.GFP fusion protein (Galaski et al, 2016) or pSTITCH.IRES.∆NGFR vector (Stolp et al, 2012) for further in vitro or in vivo characterization, respectively. These patient‐derived Nefs were assessed for downregulation of cell surface CD4 and MHC‐I, inhibition of chemotaxis toward stromal cell‐derived factor‐1 alpha (SDF‐1α), and inhibition of anti‐CD3 induced peripheral F‐actin ring formation as described in detail previously (Stolp et al, 2009; Imle et al, 2015; Ananth et al, 2019). The isolated nef sequences are deposited in GenBank (NCBI, NIH) under the following accession numbers: MT223941 and MT223942.
Expansion of adoptively transferred B cell and T cells
For comparing the expansion of B cells and T cells after immunization with HEL‐OVA/CFA, draining LNs (dLNs) were harvested from d7‐immunized mice and single‐cell suspensions stained with antibody combinations. Antibodies (Abs) and reagents used were as follows: anti‐mouse CD45.2‐PE (104), anti‐mouse B220‐BV650 (RA3‐6B2), anti‐mouse GL7‐AlexaFluor‐488 (GL7), anti‐mouse PD‐L1‐PE/Cy7 (10F.9G2), anti‐mouse CXCR5‐Bio (L138D7), anti‐mouse ICOS‐BV420 (C398.4A), anti‐mouse OX40‐FITC (Ber‐ACT‐35), Streptavidin‐PerCP (all BioLegend), anti‐mouse CD95‐Biotin (Jo2), and anti‐mouse CD3‐BV711 (500A2), (all from BD Biosciences). Precision count beads (BioLegend) were added in the samples to estimate the absolute number of BHEL and CD4OT‐II recovered from the dLNs using flow cytometry.
HIV‐1 production
Six million 293T cells were seeded on a 15 cm culture dish and 1 day later cells were transfected (PEI) with various proviral HIV‐1 plasmid DNA (25 µg). Virus supernatant was harvested 48 and 72 h after transfection and filtered using 0.45 µm pore size. Virus was concentrated using 20% sucrose cushion (24,000 rpm, 1.5 h), titers were determined by SYBR Green I‐based PERT (SG‐PERT) or p24 ELISA, and infectivity was evaluated by infection of Tzm‐bl reporter cells (Ananth et al, 2019).
Sybr Green‐based Product Enhanced Reverse Transcriptase (SG‐PERT) assay
For determination of HIV‐1 titers, supernatant was collected every 2–3 days and analyzed by SG‐PERT as described (Ananth et al, 2019). Five µl of supernatant were lysed with 5 µl of 2× Lysis buffer containing 40 U/µl Ribolock for 10 min at RT. Samples were diluted by adding 90 µl of PCR buffer. A serial dilution of the standard ranging from around 109–103 pUnits RT/µl was generated by log dilution with PCR buffer, lysed. Ten µl of this diluted sample and lysed standard was mixed with 10 µl of 2× Reaction buffer containing 5 U/µl Go Taq Hotstart polymerase. Probes were analyzed with Bio‐Rad CFX96.
Preparation and infection of tonsillar histocultures
Tonsillar histocultures were conducted essentially as described (Homann et al, 2009) using RPMI1640 GlutaMAX™ supplemented with 15% (v/v) heat inactivated FBS, 1% (v/v) Sodium pyruvate, non‐essential amino acid 1% (v/v), Gentamycin 1% (v/v), Fungizone 1% (v/v), and 100 µg/ml ampicillin. Surgically removed tonsils (during standard tonsillectomy of anonymous donors with informed consent in accordance with the ethics vote S‐123/2014 from the Heidelberg University Hospital ethics committee) obtained from Heidelberg University hospital were processed by removing burnt areas (black and dark brown colored) and connective tissues (yellow colored). Small tissue blocks of 2–3 mm thickness were generated using scalpel and fine scissors (Introini et al, 2014). Each Gelfoam® (Pfizer) was cut in four parts, and one part was placed in one well on a 12‐well plate. Four to six blocks were placed separately on medium‐soaked Gelfoam®, and 1 ml of medium was added in each well. For infection, 6–10 µl of medium containing 2–3 × 105 infectious units as titered on Tzm‐bl reporter cell was added on top of each block. The virus was washed off after 24 h (three washes with fresh medium) followed by addition of 0.3 mg/ml Tetanus toxoid (Calbiochem) for 24 h. Toxoid was removed by 4–5 times washing 24 h later.
For determination of HIV replication, supernatant was collected every 3–4 days for SG‐PERT assay. For determination of antibodies generated against Tetanus toxoid, ELISA kit (Siemens; TETG5043DB) was used. Production of anti‐tetanus IgG in infected blocks was calculated relative to Mock‐infected blocks stimulated with Tetanus toxoid antigen. Out of 19 donors used in the assay, only nine donors showed IgG production in mock‐stimulated group. Out of these nine donors, three donors did not show inhibition in antibody production in the presence of HIV WT. Thus, only six remaining donors were included in the assay.
Statistical analysis
Statistical analysis of datasets was carried out using Prism version 5.0 (GraphPad). Statistical significance was calculated using the tests indicated in the respective figure legends. n.s., not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
Author contributions
Study design, Experiment plan: OTF, BS, SK; Experiments: SK, SA, NT, KM, BMC, BS; Patient material: RH, OCB, FK; Data analysis and result interpretation: SK, SA, KM, BS, OTF; Manuscript writing: OTF, SK, BS; Manuscript reading and comments: All authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Source Data for Expanded View
Review Process File
Acknowledgements
We are grateful to Jens Stein for OT‐II.2 mice, Robert Brink for SWHEL –HL mice, Inka Zörnig for SMARTA/CD45.1 mice, Anette Oxenius for SMARTA/CD45.1 and CAG‐eCFP mice, Marc Freichel for mT/mG mice, and Stephan Herzig for UBC‐GFP mice. We also thank Nadine Tibroni and Ina Ambiel for technical assistance, Clara Bourgeon for her help in analyzing samples prepared for Fig 3I, Tabea Kramer and Vivien Throm for help in preparing samples analyzed for Fig 4A, Klaus Meese and Frauke Melchior for help with the purification of HEL3x antigen, and Kathrin Bajak for help with preparing manuscript and figures. This project is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) (Projektnummer 240245660—SFB 1129, Projekt 8 TRR83), TTU HIV of German Centre for Infection Research (DZIF) (TTU 04.810—An integrated approach for HIV‐cure “Shock and Kill” strategies to OTF. OTF and FM are members of the CellNetworks cluster of excellence (EXC81). Imaging was supported by the IDIP imaging facility of the CIID with support from DZIF. Open access funding enabled and organized by ProjektDEAL.
The EMBO Journal (2020) 39: e105594.
See also: C Vanpouille & L Margolis (December 2020)
Data availability
The microarray data from this publication have been deposited to NCBI Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE156100) and assigned the identifier GSE156100.
References
- Abraham L, Fackler OT (2012) HIV‐1 Nef: a multifaceted modulator of T cell receptor signaling. Cell Commun Signal 10: 39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananth S, Morath K, Trautz B, Tibroni N, Shytaj IL, Obermaier B, Stolp B, Lusic M, Fackler OT (2019) Multi‐functional roles of the N‐terminal region of HIV‐1SF2Nef are mediated by three independent protein interaction sites. J Virol 94: e01398‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arhel N, Lehmann M, Clauss K, Nienhaus GU, Piguet V, Kirchhoff F (2009) The inability to disrupt the immunological synapse between infected human T cells and APCs distinguishes HIV‐1 from most other primate lentiviruses. J Clin Invest 119: 2965–2975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balazs AB, Chen J, Hong CM, Rao DS, Yang L, Baltimore D (2011) Antibody‐based protection against HIV infection by vectored immunoprophylaxis. Nature 481: 81–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya D, Cheah MT, Franco CB, Hosen N, Pin CL, Sha WC, Weissman IL (2007) Transcriptional profiling of antigen‐dependent murine B cell differentiation and memory formation. J Immunol 179: 6808–6819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brink R, Paus D, Bourne K, Hermes JR, Gardam S, Phan TG, Chan TD (2015) The SW(HEL) system for high‐resolution analysis of in vivo antigen‐specific T‐dependent B cell responses. Methods Mol Biol 1291: 103–123 [DOI] [PubMed] [Google Scholar]
- Burton DR, Hangartner L (2016) Broadly neutralizing antibodies to HIV and their role in vaccine design. Annu Rev Immunol 34: 635–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caskey M, Klein F, Lorenzi JC, Seaman MS, West AP Jr, Buckley N, Kremer G, Nogueira L, Braunschweig M, Scheid JF et al (2015) Viraemia suppressed in HIV‐1‐infected humans by broadly neutralizing antibody 3BNC117. Nature 522: 487–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B (2019) Molecular mechanism of HIV‐1 entry. Trends Microbiol 27: 878–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig HM, Pandori MW, Guatelli JC (1998) Interaction of HIV‐1 Nef with the cellular dileucine‐based sorting pathway is required for CD4 down‐regulation and optimal viral infectivity. Proc Natl Acad Sci USA 95: 11229–11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubas RA, Mudd JC, Savoye AL, Perreau M, van Grevenynghe J, Metcalf T, Connick E, Meditz A, Freeman GJ, Abesada‐Terk G et al (2013) Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med 19: 494–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyster JG, Allen CDC (2019) B cell responses: cell interaction dynamics and decisions. Cell 177: 524–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva NS, Klein U (2015) Dynamics of B cells in germinal centres. Nat Rev Immunol 15: 137–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon NJ, Tsykin A, Solomon A, Smith K, Ludford‐Menting M, Hooker DJ, McPhee DA, Greenway AL, Ellett A, Chatfield C et al (1995) Genomic structure of an attenuated quasi species of HIV‐1 from a blood transfusion donor and recipients. Science 270: 988–991 [DOI] [PubMed] [Google Scholar]
- Douek DC, Brenchley JM, Betts MR, Ambrozak DR, Hill BJ, Okamoto Y, Casazza JP, Kuruppu J, Kunstman K, Wolinsky S et al (2002) HIV preferentially infects HIV‐specific CD4+ T cells. Nature 417: 95–98 [DOI] [PubMed] [Google Scholar]
- Fackler OT, Moris A, Tibroni N, Giese SI, Glass B, Schwartz O, Krausslich HG (2006) Functional characterization of HIV‐1 Nef mutants in the context of viral infection. Virology 351: 322–339 [DOI] [PubMed] [Google Scholar]
- Fritz S, Mossdorf E, Durovic B, Zenhaeusern G, Conen A, Steffen I, Battegay M, Nuesch R, Hess C (2010) Virosomal influenza‐vaccine induced immunity in HIV‐infected individuals with high versus low CD4+ T‐cell counts: clues towards a rational vaccination strategy. AIDS (London, England) 24: 2287–2289 [DOI] [PubMed] [Google Scholar]
- Galaski J, Ahmad F, Tibroni N, Pujol FM, Muller B, Schmidt RE, Fackler OT (2016) Cell surface downregulation of NK cell ligands by patient‐derived HIV‐1 Vpu and Nef alleles. J Acquir Immune Defic Syndr 72: 1–10 [DOI] [PubMed] [Google Scholar]
- Gautam R, Nishimura Y, Pegu A, Nason MC, Klein F, Gazumyan A, Golijanin J, Buckler‐White A, Sadjadpour R, Wang K et al (2016) A single injection of anti‐HIV‐1 antibodies protects against repeated SHIV challenges. Nature 533: 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer M, Fackler OT, Peterlin BM (2001) Structure–function relationships in HIV‐1 Nef. EMBO Rep 2: 580–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glushakova S, Baibakov B, Margolis LB, Zimmerberg J (1995) Infection of human tonsil histocultures: a model for HIV pathogenesis. Nat Med 1: 1320–1322 [DOI] [PubMed] [Google Scholar]
- Glushakova S, Grivel JC, Suryanarayana K, Meylan P, Lifson JD, Desrosiers R, Margolis L (1999) Nef enhances human immunodeficiency virus replication and responsiveness to interleukin‐2 in human lymphoid tissue ex vivo . J Virol 73: 3968–3974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glushakova S, Munch J, Carl S, Greenough TC, Sullivan JL, Margolis L, Kirchhoff F (2001) CD4 down‐modulation by human immunodeficiency virus type 1 Nef correlates with the efficiency of viral replication and with CD4(+) T‐cell depletion in human lymphoid tissue ex vivo . J Virol 75: 10113–10117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff‐Dubois S, Rouers A, Moris A (2016) Impact of chronic HIV/SIV infection on T follicular helper cell subsets and germinal center homeostasis. Front Immunol 7: 501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg ME, Iafrate AJ, Skowronski J (1998) The SH3 domain‐binding surface and an acidic motif in HIV‐1 Nef regulate trafficking of class I MHC complexes. EMBO J 17: 2777–2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivel JC, Margolis L (2009) Use of human tissue explants to study human infectious agents. Nat Protoc 4: 256–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Price MJ, Patterson DG, Barwick BG, Haines RR, Kania AK, Bradley JE, Randall TD, Boss JM, Scharer CD (2018) EZH2 represses the B cell transcriptional program and regulates antibody‐secreting cell metabolism and antibody production. J Immunol 200: 1039–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller C, Rauch S, Michel N, Hannemann S, Lehmann MJ, Keppler OT, Fackler OT (2006) The HIV‐1 pathogenicity factor Nef interferes with maturation of stimulatory T‐lymphocyte contacts by modulation of N‐Wasp activity. J Biol Chem 281: 19618–19630 [DOI] [PubMed] [Google Scholar]
- Haller C, Muller B, Fritz JV, Lamas‐Murua M, Stolp B, Pujol FM, Keppler OT, Fackler OT (2014) HIV‐1 Nef and Vpu are functionally redundant broad‐spectrum modulators of cell surface receptors, including tetraspanins. J Virol 88: 14241–14257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood NE, Batista FD (2010) Early events in B cell activation. Annu Rev Immunol 28: 185–210 [DOI] [PubMed] [Google Scholar]
- Homann S, Tibroni N, Baumann I, Sertel S, Keppler OT, Fackler OT (2009) Determinants in HIV‐1 Nef for enhancement of virus replication and depletion of CD4+ T lymphocytes in human lymphoid tissue ex vivo . Retrovirology 6: 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imle A, Abraham L, Tsopoulidis N, Hoflack B, Saksela K, Fackler OT (2015) Association with PAK2 enables functional interactions of lentiviral Nef proteins with the Exocyst complex. MBio 6: e01309–e1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Introini A, Vanpouille C, Grivel JC, Margolis L (2014) An ex vivo model of HIV‐1 infection in human lymphoid tissue and Cervico‐vaginal tissue. Bio Protoc 4: e1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ise W, Kohyama M, Schraml BU, Zhang T, Schwer B, Basu U, Alt FW, Tang J, Oltz EM, Murphy TL et al (2011) The transcription factor BATF controls the global regulators of class‐switch recombination in both B cells and T cells. Nat Immunol 12: 536–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardava L, Moir S, Shah N, Wang W, Wilson R, Buckner CM, Santich BH, Kim LJ, Spurlin EE, Nelson AK et al (2014) Abnormal B cell memory subsets dominate HIV‐specific responses in infected individuals. J Clin Invest 124: 3252–3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerneis S, Launay O, Turbelin C, Batteux F, Hanslik T, Boelle PY (2014) Long‐term immune responses to vaccination in HIV‐infected patients: a systematic review and meta‐analysis. Clin Infect Dis 58: 1130–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kestler HW 3rd, Ringler DJ, Mori K, Panicali DL, Sehgal PK, Daniel MD, Desrosiers RC (1991) Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell 65: 651–662 [DOI] [PubMed] [Google Scholar]
- Kirchhoff F, Greenough TC, Brettler DB, Sullivan JL, Desrosiers RC (1995) Brief report: absence of intact nef sequences in a long‐term survivor with nonprogressive HIV‐1 infection. N Engl J Med 332: 228–232 [DOI] [PubMed] [Google Scholar]
- Kupfer H, Monks CR, Kupfer A (1994) Small splenic B cells that bind to antigen‐specific T helper (Th) cells and face the site of cytokine production in the Th cells selectively proliferate: immunofluorescence microscopic studies of Th‐B antigen‐presenting cell interactions. J Exp Med 179: 1507–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurgonaite K, Gandhi H, Kurth T, Pautot S, Schwille P, Weidemann T, Bökel C (2015) Essential role of endocytosis for interleukin‐4‐receptor‐mediated JAK/STAT signalling. J Cell Sci 128: 3781–3795 [DOI] [PubMed] [Google Scholar]
- Lamas‐Murua M, Stolp B, Kaw S, Thoma J, Tsopoulidis N, Trautz B, Ambiel I, Reif T, Arora S, Imle A et al (2018) HIV‐1 Nef disrupts CD4(+) T lymphocyte polarity, extravasation, and homing to lymph nodes via its Nef‐associated kinase complex interface. J Immunol 201: 2731–2743 [DOI] [PubMed] [Google Scholar]
- Liechti T, Kadelka C, Braun DL, Kuster H, Boni J, Robbiani M, Gunthard HF, Trkola A (2019) Widespread B cell perturbations in HIV‐1 infection afflict naive and marginal zone B cells. J Exp Med 216: 2071–2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaspina A, Moir S, Orsega SM, Vasquez J, Miller NJ, Donoghue ET, Kottilil S, Gezmu M, Follmann D, Vodeiko GM et al (2005) Compromised B cell responses to influenza vaccination in HIV‐infected individuals. J Infect Dis 191: 1442–1450 [DOI] [PubMed] [Google Scholar]
- Malbec M, Porrot F, Rua R, Horwitz J, Klein F, Halper‐Stromberg A, Scheid JF, Eden C, Mouquet H, Nussenzweig MC et al (2013) Broadly neutralizing antibodies that inhibit HIV‐1 cell to cell transmission. J Exp Med 210: 2813–2821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheson NJ, Sumner J, Wals K, Rapiteanu R, Weekes MP, Vigan R, Weinelt J, Schindler M, Antrobus R, Costa AS et al (2015) Cell surface proteomic Map of HIV infection reveals antagonism of amino acid metabolism by Vpu and Nef. Cell Host Microbe 18: 409–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mina MJ, Kula T, Leng Y, Li M, de Vries RD, Knip M, Siljander H, Rewers M, Choy DF, Wilson MS et al (2019) Measles virus infection diminishes preexisting antibodies that offer protection from other pathogens. Science 366: 599–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir S, Ho J, Malaspina A, Wang W, DiPoto AC, O'Shea MA, Roby G, Kottilil S, Arthos J, Proschan MA et al (2008) Evidence for HIV‐associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV‐infected viremic individuals. J Exp Med 205: 1797–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir S, Fauci AS (2009) B cells in HIV infection and disease. Nat Rev Immunol 9: 235–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir S, Fauci AS (2013) Insights into B cells and HIV‐specific B‐cell responses in HIV‐infected individuals. Immunol Rev 254: 207–224 [DOI] [PubMed] [Google Scholar]
- Mouquet H (2014) Antibody B cell responses in HIV‐1 infection. Trends Immunol 35: 549–561 [DOI] [PubMed] [Google Scholar]
- Murooka TT, Deruaz M, Marangoni F, Vrbanac VD, Seung E, von Andrian UH, Tager AM, Luster AD, Mempel TR (2012) HIV‐infected T cells are migratory vehicles for viral dissemination. Nature 490: 283–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessl J, Baxter AE, Mendoza P, Jankovic M, Cohen YZ, Butler AL, Lu CL, Dubé M, Shimeliovich I, Gruell H et al (2020) Combination anti‐HIV‐1 antibody therapy is associated with increased virus‐specific T cell immunity. Nat Med 26: 222–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill E, Baugh LL, Novitsky VA, Essex ME, Garcia JV (2006) Intra‐ and intersubtype alternative Pak2‐activating structural motifs of human immunodeficiency virus type 1 Nef. J Virol 80: 8824–8829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallikkuth S, Parmigiani A, Silva SY, George VK, Fischl M, Pahwa R, Pahwa S (2012) Impaired peripheral blood T‐follicular helper cell function in HIV‐infected nonresponders to the 2009 H1N1/09 vaccine. Blood 120: 985–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Rudolph JM, Abraham L, Habermann A, Haller C, Krijnse‐Locker J, Fackler OT (2012) HIV‐1 Nef compensates for disorganization of the immunological synapse by inducing trans‐Golgi network‐associated Lck signaling. Blood 119: 786–797 [DOI] [PubMed] [Google Scholar]
- Paus D, Phan TG, Chan TD, Gardam S, Basten A, Brink R (2006) Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J Exp Med 203: 1081–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira EA, daSilva LL (2016) HIV‐1 Nef: taking control of protein trafficking. Traffic 17: 976–996 [DOI] [PubMed] [Google Scholar]
- Perreau M, Savoye AL, De Crignis E, Corpataux JM, Cubas R, Haddad EK, De Leval L, Graziosi C, Pantaleo G (2013) Follicular helper T cells serve as the major CD4 T cell compartment for HIV‐1 infection, replication, and production. J Exp Med 210: 143–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova VN, Sawatsky B, Han AX, Laksono BM, Walz L, Parker E, Pieper K, Anderson CA, de Vries RD, Lanzavecchia A et al (2019) Incomplete genetic reconstitution of B cell pools contributes to prolonged immunosuppression after measles. Sci Immunol 4: eaay6125 [DOI] [PubMed] [Google Scholar]
- Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, Brink R (2006) High affinity germinal center B cells are actively selected into the plasma cell compartment. J Exp Med 203: 2419–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poudrier J, Weng X, Kay DG, Pare G, Calvo EL, Hanna Z, Kosco‐Vilbois MH, Jolicoeur P (2001) The AIDS disease of CD4C/HIV transgenic mice shows impaired germinal centers and autoantibodies and develops in the absence of IFN‐gamma and IL‐6. Immunity 15: 173–185 [DOI] [PubMed] [Google Scholar]
- Reiter R, Pfeffer K (2002) Impaired germinal centre formation and humoral immune response in the absence of CD28 and interleukin‐4. Immunology 106: 222–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa A, Chande A, Ziglio S, De Sanctis V, Bertorelli R, Goh SL, McCauley SM, Nowosielska A, Antonarakis SE, Luban J et al (2015) HIV‐1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 526: 212–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph JM, Eickel N, Haller C, Schindler M, Fackler OT (2009) Inhibition of T‐cell receptor‐induced actin remodeling and relocalization of Lck are evolutionarily conserved activities of lentiviral Nef proteins. J Virol 83: 11528–11539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saksela K, Cheng G, Baltimore D (1995) Proline‐rich (PxxP) motifs in HIV‐1 Nef bind to SH3 domains of a subset of Src kinases and are required for the enhanced growth of Nef+ viruses but not for down‐regulation of CD4. EMBO J 14: 484–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar‐Gonzalez JF, Bailes E, Pham KT, Salazar MG, Guffey MB, Keele BF, Derdeyn CA, Farmer P, Hunter E, Allen S et al (2008) Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single‐genome amplification and sequencing. J Virol 82: 3952–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler M, Munch J, Kutsch O, Li H, Santiago ML, Bibollet‐Ruche F, Muller‐Trutwin MC, Novembre FJ, Peeters M, Courgnaud V et al (2006) Nef‐mediated suppression of T cell activation was lost in a lentiviral lineage that gave rise to HIV‐1. Cell 125: 1055–1067 [DOI] [PubMed] [Google Scholar]
- Schommers P, Gruell H, Abernathy ME, Tran MK, Dingens AS, Gristick HB, Barnes CO, Schoofs T, Schlotz M, Vanshylla K et al (2020) Restriction of HIV‐1 escape by a highly broad and potent neutralizing antibody. Cell 180: 471–489.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard JM (1996) Endocytosis of major histocompatibility complex class I molecules is induced by the HIV‐1 Nef protein. Nat Med 2: 338–342 [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, Giltnane JM, Yang L, Zhao H, Calame K et al (2002) Blimp‐1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 17: 51–62 [DOI] [PubMed] [Google Scholar]
- Song W, Liu C, Upadhyaya A (2014) The pivotal position of the actin cytoskeleton in the initiation and regulation of B cell receptor activation. Biochem Biophys Acta 1838: 569–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolp B, Reichman‐Fried M, Abraham L, Pan X, Giese SI, Hannemann S, Goulimari P, Raz E, Grosse R, Fackler OT (2009) HIV‐1 Nef interferes with host cell motility by deregulation of Cofilin. Cell Host Microbe 6: 174–186 [DOI] [PubMed] [Google Scholar]
- Stolp B, Imle A, Coelho FM, Hons M, Gorina R, Lyck R, Stein JV, Fackler OT (2012) HIV‐1 Nef interferes with T‐lymphocyte circulation through confined environments in vivo . Proc Natl Acad Sci USA 109: 18541–18546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbaraman H, Schanz M, Trkola A (2018) Broadly neutralizing antibodies: what is needed to move from a rare event in HIV‐1 infection to vaccine efficacy? Retrovirology 15: 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swingler S, Zhou J, Swingler C, Dauphin A, Greenough T, Jolicoeur P, Stevenson M (2008) Evidence for a pathogenic determinant in HIV‐1 Nef involved in B cell dysfunction in HIV/AIDS. Cell Host Microbe 4: 63–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangye SG, Ma CS, Brink R, Deenick EK (2013) The good, the bad and the ugly – TFH cells in human health and disease. Nat Rev Immunol 13: 412–426 [DOI] [PubMed] [Google Scholar]
- Thoulouze MI, Sol‐Foulon N, Blanchet F, Dautry‐Varsat A, Schwartz O, Alcover A (2006) Human immunodeficiency virus type‐1 infection impairs the formation of the immunological synapse. Immunity 24: 547–561 [DOI] [PubMed] [Google Scholar]
- Titanji K, De Milito A, Cagigi A, Thorstensson R, Grutzmeier S, Atlas A, Hejdeman B, Kroon FP, Lopalco L, Nilsson A et al (2006) Loss of memory B cells impairs maintenance of long‐term serologic memory during HIV‐1 infection. Blood 108: 1580–1587 [DOI] [PubMed] [Google Scholar]
- Tsopoulidis N, Kaw S, Laketa V, Kutscheidt S, Baarlink C, Stolp B, Grosse R, Fackler OT (2019) T cell receptor‐triggered nuclear actin network formation drives CD4(+) T cell effector functions. Sci Immunol 4: eaav1987 [DOI] [PubMed] [Google Scholar]
- Usami Y, Wu Y, Gottlinger HG (2015) SERINC3 and SERINC5 restrict HIV‐1 infectivity and are counteracted by Nef. Nature 526: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victora GD, Nussenzweig MC (2012) Germinal centers. Annu Rev Immunol 30: 429–457 [DOI] [PubMed] [Google Scholar]
- Villaudy J, Schotte R, Legrand N, Spits H (2014) Critical assessment of human antibody generation in humanized mouse models. J Immunol Methods 410: 18–27 [DOI] [PubMed] [Google Scholar]
- Vladymyrov M, Abe J, Moalli F, Stein JV, Ariga A (2016) Real‐time tissue offset correction system for intravital multiphoton microscopy. J Immunol Methods 438: 35–41 [DOI] [PubMed] [Google Scholar]
- Wendel BS, Del Alcazar D, He C, Del Río‐Estrada PM, Aiamkitsumrit B, Ablanedo‐Terrazas Y, Hernandez SM, Ma KY, Betts MR, Pulido L et al (2018) The receptor repertoire and functional profile of follicular T cells in HIV‐infected lymph nodes. Sci Immunol 3: eaan8884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurster AL, Rodgers VL, White MF, Rothstein TL, Grusby MJ (2002) Interleukin‐4‐mediated protection of primary B cells from apoptosis through Stat6‐dependent up‐regulation of Bcl‐xL. J Biol Chem 277: 27169–27175 [DOI] [PubMed] [Google Scholar]
- Xu W, Santini PA, Sullivan JS, He B, Shan M, Ball SC, Dyer WB, Ketas TJ, Chadburn A, Cohen‐Gould L et al (2009) HIV‐1 evades virus‐specific IgG2 and IgA responses by targeting systemic and intestinal B cells via long‐range intercellular conduits. Nat Immunol 10: 1008–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Source Data for Expanded View
Review Process File
Data Availability Statement
The microarray data from this publication have been deposited to NCBI Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE156100) and assigned the identifier GSE156100.