

Mitochondria are dynamic organelles adapting their morphology by cycles of fission and fusion events to control cellular homeostasis. In this issue of The EMBO Journal, Murata and colleagues (2020) show that lack of mitochondrial division leads to safeguard mechanisms, induced by transient mitochondrial membrane depolarization and activation of the metalloprotease OMA1, to prevent extreme mitochondrial fusion and to maintain optimal mitochondrial bioenergetics.
Subject Categories: Membrane & Intracellular Transport,
A new study identifies mitochondrial safeguard mechanisms that compensate unbalanced mitochondrial dynamics and preserve respiratory functions in mammalian cells.

Mitochondria are dynamic organelles that constantly adapt their shape depending on the cellular requirements and metabolic state (Nunnari & Suomalainen, 2012). Therefore, proper organization of the mitochondrial network is essential for cellular homeostasis and survival. Indeed, deregulation of mitochondrial dynamics contributes to the pathogenesis of multiple diseases and abnormal mitochondrial morphology is observed in numerous human diseases (Nunnari & Suomalainen, 2012).
Mitochondrial division is essential not only to ensure the proper segregation of the organelle during cell division but also to facilitate organelle positioning and the degradation of damaged mitochondria. Mitochondrial fission is mainly regulated by the large GTPase dynamin‐related protein‐1 (Drp1), which self‐oligomerize and constrict membranes at mitochondria‐endoplasmic reticulum contact sites (Tilokani et al, 2018). In contrast, mitochondrial fusion allows the mixing of two matrix compartments and is controlled by two specialized fusion machineries that localize either at the outer mitochondrial membrane (OMM) or the inner mitochondrial membrane (IMM). First, OMM fusion is mediated by both the homo‐ or heterotypic in‐trans interaction of the GTPases mitofusin‐1 and mitofusin‐2 (Mfn1 and Mfn2) (Tilokani et al, 2018). IMM fusion is then ensured by the heterotypic interaction between the dynamin‐like GTPase OPA1 and the phospholipid cardiolipin (Ban et al, 2017). Importantly, although both short and long OPA1 isoforms mediate IMM fusion, the excess of the short one inhibits mitochondrial fusion activity promoting a pro‐fission scenario in the cell (Ge et al, 2020). Thus, the regulation of OPA1 cleavage by different mitochondrial proteases is a fundamental process that dictate the shape of the mitochondrial network.
Although these events are regulated by different machineries, these dynamic shape transitions need to be finely spatio‐temporally coordinated to allow optimal mitochondrial functions. Therefore, not only could a fusion event be immediately followed by a division event, but levels of the OMM pro‐fusion factors are also decreased and OPA1 cleavage is enhanced when division is blocked. Importantly, in Drp1‐depleted cells, a localized, repeated, and transient decrease of the inner membrane potential, named flickering, has been observed in individual mitochondria without impacting the overall membrane potential (Lee & Yoon, 2014). This phenomenon is dependent on OPA1 and has been suggested to be linked to mitochondrial functioning (Lee & Yoon, 2014). However, how these events are mechanistically regulated, how they impact mitochondrial fusion, and what is their functional relevance are currently unknown. In this issue of The EMBO Journal, Murata and colleagues (Murata et al, 2020) cast the spotlight on flickering as a safeguard mechanism to prevent deleterious extreme fusion to maintain mitochondrial bioenergetics.
Studying mouse embryonic fibroblasts (MEF) and using microscopy analysis, Murata et al first confirm previous reports showing that flickering is induced in mitochondria from Drp1‐knock‐out (KO) MEFs, but then also in MEFs depleted of the Drp1 mitochondrial receptor, mitochondrial fission factor (MFF), further demonstrating that this process is enhanced by the hyper‐connectivity of the mitochondrial network and not the loss of Drp1 itself. Mechanistically, the authors demonstrate that these short pulses of membrane depolarization activate the metalloprotease OMA1, which leads to the proteolytic cleavage of OPA1 and accumulation of short OPA1 isoforms (Fig 1). In addition, the authors show that it is the fusion activity of OPA1, rather than its role in cristae maintenance, that is required to induce flickering. Thus, the authors propose that flickering‐induced OMA1 activation drives a safeguard mechanism that Drp1‐deficient cells use to prevent an excessive and deleterious fusion in an already hyperfused mitochondrial network. Drp1‐KO cells also present lower levels of the OMM fusion factors Mfn‐1 and Mfn‐2. All together, these data suggest that Drp1‐KO cells compensate mitochondrial hyperfusion by downregulating the fusion machinery (i.e., accumulating short OPA1 isoforms and diminishing Mfn‐1 and Mfn‐2 levels) (Fig 1). Interestingly, Murata et al show that re‐expression of Mfn1 levels together with inhibition of OMA1‐mediated OPA1 processing increase mitochondrial connectivity in Drp1‐KO cells, resulting in a new mitochondrial phenotype, named extreme fusion (Fig 1). This phenotype is characterized by the complete loss of membrane potential and the decrease of mitochondrial respiration capacity of around 50% of cells, indicating that increased mitochondrial fusion beyond Drp1 deficiency is deleterious. Surprisingly, ectopic expression of Mfn2 in OMA1 knockdown did not lead to the loss of membrane potential in Drp1‐KO cells. These results confirm that OPA1 requires Mfn1 for mitochondrial fusion (Cipolat et al, 2004) and specifically emphasize the new role for the OMA1‐OPA1‐Mfn1 axis as a new mitochondrial stress response that prevent extreme mitochondrial fusion (Fig 1). Finally, the authors highlight the physiological relevance of this process by showing the requirement of OMA1 for OPA1 cleavage in the liver from Drp1‐KO and Drp1‐OMA1‐KO mice.
Figure 1. Safeguard mechanisms preventing extreme mitochondrial fusion to maintain respiratory function.
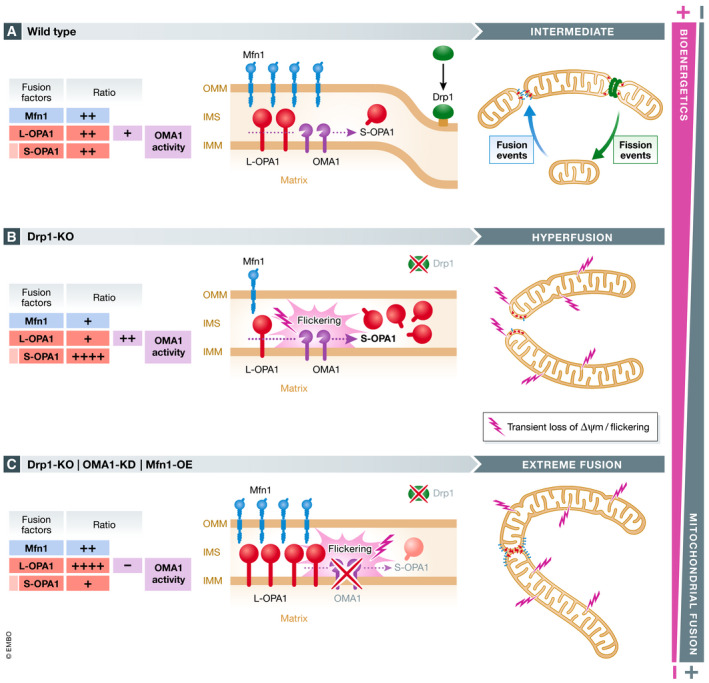
In healthy cells (A), both fission and fusion events are dynamically balanced and mitochondria display an intermediate network distribution, which sustain an optimal respiratory capacity. Inhibition of the fission machinery, by the genetic deletion of Drp1 or its receptor MFF, induces the formation of a hyperfused mitochondrial network (B). Elongated mitochondria in Drp1‐KO cells exhibit localized, repetitive, and transient loss of membrane potential, a phenomenon named flickering, that activate OMA1, which promote OPA1 cleavage, S‐OPA1 accumulation, and, subsequently, the inhibition of excessive mitochondrial fusion. In addition, Drp1‐deficient cells exhibit lower levels of Mfn1, as a second protective mechanism to prevent further and deleterious mitochondrial fusion and thus to maintain the mitochondrial bioenergetic status. Inhibition of OMA1 activity and re‐expression of Mfn1 in Drp1‐KO cells lead to extreme mitochondrial fusion and the complete loss of membrane potential (C), which are correlated to the decrease of mitochondrial bioenergetics. OMM: Outer mitochondrial membrane; IMM: Inner mitochondrial membrane; IMS: Intermembrane space; KO: Knock‐out; WT: Wild‐type; KD: Knockdown; OE: Overexpression.
The findings of Murata and colleagues significantly increase our understanding of how mitochondrial fission and fusion events are interconnected and how excessive mitochondrial connectivity can be detrimental, but also raise important questions. Do membrane flickering and this safeguard mechanism occur in wild‐type (WT) cells? While the authors do not detect flickering in unstimulated WT MEFs, they show that ATP synthesis inhibition by oligomycin treatment or artificial flickering induced by repeated addition and removal of FCCP leads to limited‐OMA1 activation and OPA1 cleavage, similar to what they observe in Drp1‐KO cells. These results suggest that this mechanism is not limited to cells with a block in division, confirming previous reports describing flickering in multiple WT cell lines (Duchen et al, 1998) (Chalmers et al, 2015) (Loew et al, 1993). If these events happen in WT cells, what is their functional relevance? Flickering has been associated to the contraction of the mitochondrial matrix (Lee & Yoon, 2014), a process similar to the constriction of the inner membrane compartment (COMIC), which is also up‐regulated in Drp1‐deficient cells and require OMA1‐mediated OPA1 cleavage to initiate mitochondrial division (Cho et al, 2017). However, COMIC is triggered by mitochondrial calcium influx, whereas membrane flickering has been proposed to be independent of calcium signaling. Therefore, it can be hypothesized that this mechanism could exert an anti‐fusion activity in WT cells to facilitate adequate mitochondrial morphology adaptation. How these specific flickering events are triggered remain an open question. While it has been shown that they are independent of calcium, reactive oxygen species and mitochondrial permeability transition, it has been proposed that proton leak through the IMM could dissipate membrane potential and induce flickering in Drp1‐KO cells. This hypothesis is reinforced by the present study, which show that oligomycin treatment can induce flickering and the safeguard mechanism in WT cells.
Together, Murata et al have described a new safeguard mechanism to prevent an extreme and harmful mitochondrial fusion in cells already harboring a mitochondrial hyperfused network. Future studies will shed light on the exact mechanism of how flickering is triggered and the physiological relevance of this safeguard mechanism in health and pathological conditions.
The EMBO Journal (2020) 39: e107326.
See also: D Murata et al (December 2020)
References
- Ban T, Ishihara T, Kohno H, Saita S, Ichimura A, Maenaka K, Oka T, Mihara K, Ishihara N (2017) Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat Cell Biol 19: 856–863 [DOI] [PubMed] [Google Scholar]
- Chalmers S, Saunter CD, Girkin JM, McCarron JG (2015) Flicker‐assisted localization microscopy reveals altered mitochondrial architecture in hypertension. Sci Rep 5: 16875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho B, Cho HM, Jo Y, Kim HD, Song M, Moon C, Kim H, Kim K, Sesaki H, Rhyu IJ et al (2017) Constriction of the mitochondrial inner compartment is a priming event for mitochondrial division. Nat Commun 8: 15754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L (2004) OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA 101: 15927–15932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Leyssens A, Crompton M (1998) Transient mitochondrial depolarizations reflect focal sarcoplasmic reticular calcium release in single rat cardiomyocytes. J Cell Biol 142: 975–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Shi X, Boopathy S, McDonald J, Smith AW, Chao LH (2020) Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner‐membrane. Elife 9: e50973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Yoon Y (2014) Transient contraction of mitochondria induces depolarization through the inner membrane dynamin OPA1 protein. J Biol Chem 289: 11862–11872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loew LM, Tuft RA, Carrington W, Fay FS (1993) Imaging in five dimensions: time‐dependent membrane potentials in individual mitochondria. Biophys J 65: 2396–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata D, Yamada T, Tokuyama T, Arai K, Quirós PM, López‐Otín C, Iijima M, Sesaki H (2020) 'Mitochondrial Safeguard': a stress response that offsets extreme fusion and protects respiratory function via flickering‐induced Oma1 activation. EMBO J 39: e105074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A (2012) Mitochondria: in sickness and in health. Cell 148: 1145–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilokani L, Nagashima S, Paupe V, Prudent J (2018) Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem 62: 341–360 [DOI] [PMC free article] [PubMed] [Google Scholar]