

SUMMARY
The regulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) trafficking affects multiple brain functions, such as learning and memory. We have previously shown that Thorase plays an important role in the internalization of AMPARs from the synaptic membrane. Here, we show that N-methyl-d-aspartate receptor (NMDAR) activation leads to increased S-nitrosylation of Thorase and N-ethylmaleimide-sensitive factor (NSF). S-nitrosylation of Thorase stabilizes Thorase-AMPAR complexes and enhances the internalization of AMPAR and interaction with protein-interacting C kinase 1 (PICK1). S-nitrosylated NSF is dependent on the S-nitrosylation of Thorase via trans-nitrosylation, which modulates the surface insertion of AMPARs. In the presence of the S-nitrosylation-deficient C137L Thorase mutant, AMPAR trafficking, long-term potentiation, and long-term depression are impaired. Overall, our data suggest that both S-nitrosylation and interactions of Thorase and NSF/PICK1 are required to modulate AMPAR-mediated synaptic plasticity. This study provides critical information that elucidates the mechanism underlying Thorase and NSF-mediated trafficking of AMPAR complexes.
Graphical Abstract
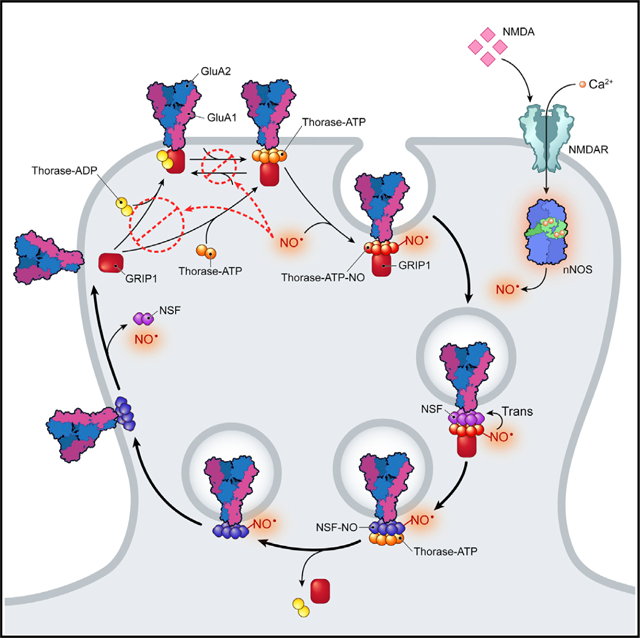
In Brief
Umanah et al. show that the S-nitrosylation of Thorase and the transnitrosylation of NSF are responsible for NMDAR-activated trafficking of AMPARs underlying synaptic plasticity.
INTRODUCTION
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs), which are enriched in the postsynaptic membrane on dendritic spines, are highly dynamic, and cycling in and out of the postsynaptic membrane is thought to regulate synaptic plasticity (Asrar and Jia, 2013; Bassani et al., 2009; Chater and Goda, 2014). Changes in the expression levels of AMPARs, subunit composition, and modifications of their accessory proteins have been shown to mediate synaptic strength. The AMPAR subunit, GluA2, plays critical roles in synaptic plasticity by influencing AMPAR assembly and trafficking, which is fundamentally important for learning, memory formation, and storage (Dong et al., 1997; Tan et al., 2015). Dysregulation of GluA2 on the postsynaptic membrane expression has been implicated in various pathological states and has important con-sequences for several brain disorders, such as epilepsy, Alzheimer disease, Parkinson disease, and schizophrenia (Beneyto and Meador-Woodruff, 2006; Corti et al., 2011; Pøhlsgaard et al., 2011). AMPAR-containing-GluA2s (AMPAR-GluA2) are maintained at the surface of synapses by postsynaptic density (PSD) proteins such as glutamate receptor interacting protein 1 (GRIP1) (Menniti et al., 2013). Other PSD proteins such as N-ethylmaleimide-sensitive factor (NSF) and protein-interacting C kinase 1 (PICK1) have been shown to modulate the trafficking of AMPAR-GluA2 (Arendt et al., 2015; Li et al., 2016). Despite the extensive studies on AMPAR trafficking, the mechanisms that control AMPAR-mediated synaptic plasticity and how NSF regulate recycling of intracellular AMPAR-GluA2 remain unclear.
Thorase, a PSD protein, is a AAA+ ATPase that plays a critical role in synaptic plasticity by modulating the recycling and trafficking of AMPAR-GluA2 (Ahrens-Nicklas et al., 2017; Piard et al., 2018; Prendergast et al., 2014; Umanah et al., 2017; Zhang et al., 2011). Thorase has also been shown to be neuroprotective (Dai et al., 2010; Pignatelli et al., 2017; Umanah et al., 2017; Zhang et al., 2011). The loss of Thorase decreases the internalization of AMPARs, resulting in increased post-synaptic receptor density. The disassembly of the GluA2-GRIP1 complex, which is critical for the internalization of AMPAR-GluA2, is mediated by Thorase (Ahrens-Nicklas et al., 2017; Piard et al., 2018; Prendergast et al., 2014; Umanah et al., 2017; Zhang et al., 2011). Thus, Thorase plays a critical role in modulating AMPAR-mediated neurotransmission. However, the precise mechanisms by which Thorase mediates AMPAR complex disassembly and internalization remain to be elucidated. The intracellular signaling that controls Thorase-mediated internalization of GluA2 in response to N-methyl-d-aspartate receptor (NMDAR) activation is not known. Here, we show that S-nitrosylation of Thorase following NMDAR activation inhibits ATP binding and ATP hydrolysis, resulting in the stabilization of the GluA2/GRIP1 complex and also facilitating AMPAR-GluA2 internalization. Furthermore, S-nitrosylated Thorase transnitrosylates NSF, and in the process, the GluA2/GRIP1 complex disassociates, allowing S-nitrosylated NSF to bind to AMPAR-GluA2 and insert into the postsynaptic membrane. Our study provides insight into the mechanisms by which S-nitrosylation of Thorase and NSF and their interactions with AMPAR-GluA2 complexes modulate AMPAR-mediated neurotransmission and synaptic plasticity.
RESULTS
Thorase Is S-Nitrosylated
To determine whether Thorase is S-nitrosylated, the biotin switch assay (Jaffrey and Snyder, 2001) was conducted on recombinant Thorase. Treatment with the nitric oxide (NO) donor S-nitrosoglutathione (GSNO) S-nitrosylates Thorase, whereas glutathione (GSH) has no effect (Figures 1A and 1B). To ascertain the specific cysteine residues involved in the S-nitrosylation of Thorase, mass spectrometric analysis was performed on Thorase treated with GSNO (Doulias et al., 2010, 2013). The results indicated that Thorase is S-nitrosylated on C137 and C310 residues (Figure S1A). To confirm that C137 and C310 are the residues for S-nitrosylation of Thorase, C137 and C310 residues were mutated to serine. Both wild-type (WT) and C310S recombinant Thorase are S-nitrosylated, whereas S-nitrosylation of C137S Thorase is profoundly reduced (Figures S1B and S1C). These data demonstrate that C137 is the main site for the S-nitrosylation of Thorase. A 3D prediction model of the ATPase domain of Thorase (Figure 1C) suggests that C137 is in close proximity to Walker A residue, K139 (ATP binding) and Walker B residue E193 (ATP hydrolase/ATPase). The multiple alignment of Thorase homologs (Figure 1D) suggests that C137 is conserved in the AAA ATPase domain among the homologs. Substitution of C137 with serine shows defects in ATP hydrolysis, while Cys310 substitution with serine has no effect (Figures S1D and S1E). Therefore, Cys137 was mutated to alanine, leucine, aspartate, and serine and ATP hydrolysis was evaluated (Figures S1F and S1G). The mutation of C137 to alanine (C137A) or leucine (C137L) had no effect on ATP hydrolysis. Since the C137L had similar ATPase activity to WT, it was used for this study. The biotin switch assay confirms that C137L is not S-nitrosylated (Figures 1E and 1F). GSNO S-nitrosylation of WT Thorase inhibits both ATP binding and ATP hydrolysis, whereas GSNO has no effect on C137L Thorase, similar to the effect of GSH on WT Thorase (Figures 1G–1J).
Figure 1. S-Nitrosylation of Thorase at Residue C137 Inhibits ATPase Activity.
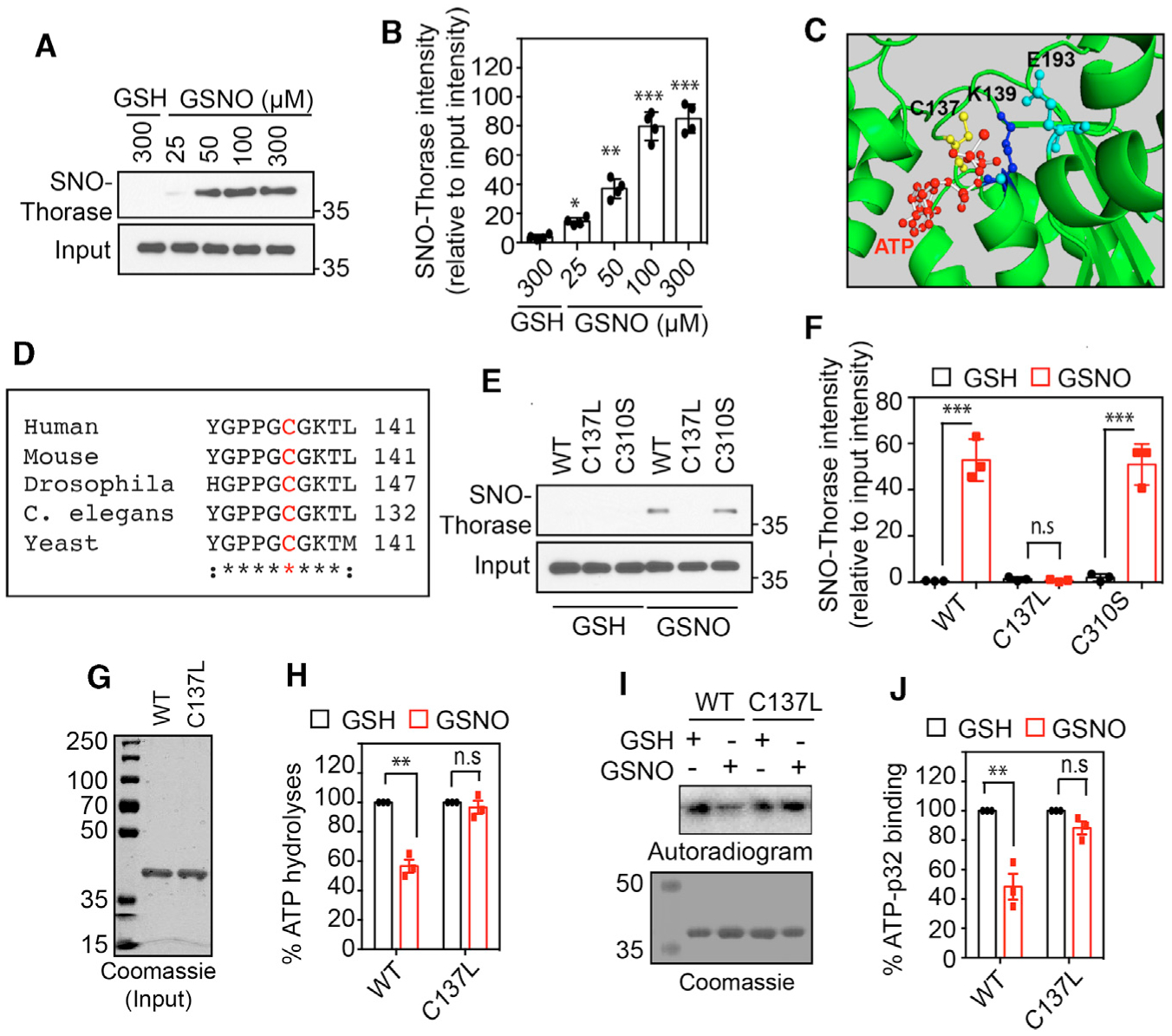
(A) Immunoblot analyses of S-nitrosylation of Thorase with GSNO and control GSH.
(B) Graphical representation of the percentage of S-nitrosylated Thorase (n = 4, p ≤ 0.0001).
(C) A 3D model of Thorase ATPase domain showing C137, K139, and E193 location in the ATP catalytic site.
(D) Multiple alignments of Thorase (ATAD1) homologs showing conserved C137 (red text).
(E) Immunoblot analyses of S-nitrosylation of Thorase wild type (WT), C137L, and C310S.
(F) Graphical representation of the relative amount of S-nitrosylated Thorase compared to control, GSH (n = 3, p ≤ 0.0001).
(G and H) ATPase analyses of Thorase WT and C137L in the presence of GSH and GSNO (n = 4, p = 0.0002). The ATPase activity in the presence of GSH was considered 100% activity.
(I and J) ATP binding activities of Thorase WT and C137L in the presence of GSH and GSNO. GSNO inhibits WT but not C137L ATPase activity (n = 3, p = 0.0036).
Means ± standard errors of the mean (SEMs) of the experiments performed. *p < 0.10, **p < 0.05, ***p < 0.01, n.s., p > 0.10; ANOVA with Holm-Sidak post hoc test compared with WT; power: 1-β error probability = 0.9–1.0.
NMDA Induced S-Nitrosylation of Thorase
To determine whether Thorase is endogenously S-nitrosylated, WT or nNOS knockout (KO) primary neurons were stimulated with 20 μM NMDA and 10 μM glycine (Figures 2A and 2B). Thorase is S-nitrosylated within the first minute of NMDA/glycine stimulation and levels are maintained for 5 min, with a slow decrease over the following 60 min in WT neurons. NMDA/glycine stimulation fails to S-nitrosylate Thorase in neuronal NO synthase (nNOS) KO neurons (Figures 2A and 2B). To confirm that endogenous S-nitrosylation of Thorase is NMDAR and nNOS dependent, WT cultures were stimulated with NMDA/glycine in the presence of the NMDA antagonist, MK801, or the NOS inhibitor l-NG-nitroarginine methyl ester (l-NAME). MK801 or l-NAME significantly inhibited the S-nitrosylation of Thorase (Figures 2C–2F).
Figure 2. In Vivo S-Nitrosylation of Thorase Inhibits Oligomer Disassembly.
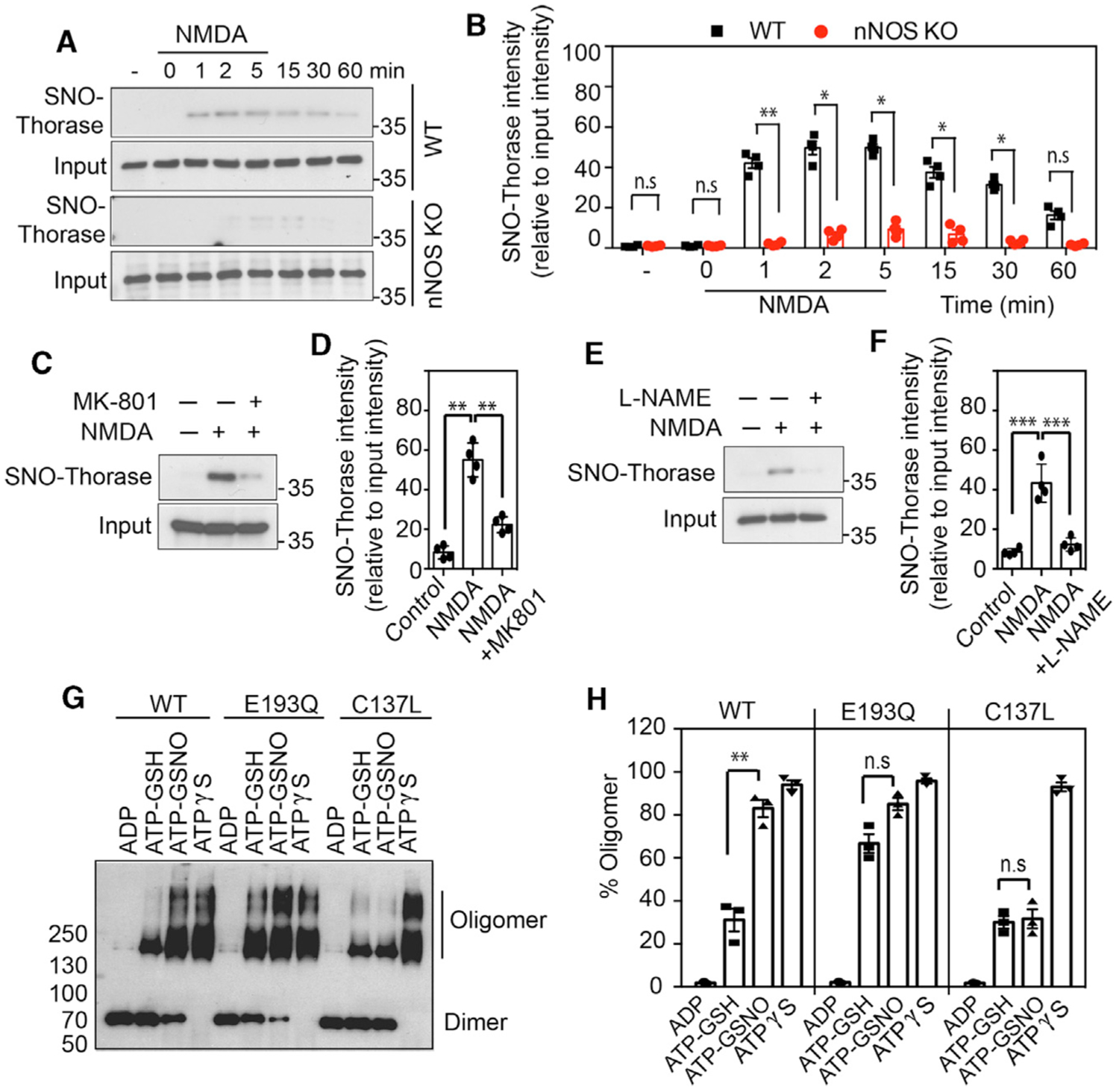
(A) Immunoblot analyses of NMDA-stimulated S-nitrosylation of Thorase in WT and nNOS KO neurons. S-nitrosylation of Thorase was assessed at different times after NMDA treatment of neurons.
(B) Graphical representation of relative amount of S-nitrosylated Thorase (n = 3, p ≤ 0.0001).
(C and D) Immunoblot analyses of NMDA-stimulated (5 min) S-nitrosylation of Thorase in WT primary neurons treated with MK-801 (n = 4, p ≤ 0.0001).
(E and F) Immunoblot analyses of NMDA-stimulated S-nitrosylation of Thorase in WT primary neurons treated with l-NAME (n = 4, p ≤ 0.0001).
(G) Immunoblot analyses of Thorase WT and mutants E193Q and C137L ATP-dependent oligomer formation in the presence of GSH or GSNO with ADP, ATP, or ATPγS.
(H) Graphical representation of percentage of Thorase oligomer in (G) (n = 3, p ≤ 0.0001). Means ± SEMs of the experiments performed. **p < 0.05, ***p < 0.01, n.s. p > 0.10; ANOVA with Holm-Sidak post hoc test compared with WT; power: 1-β error probability = 0.9–1.0.
S-Nitrosylation of Thorase Inhibits Oligomer Disassembly
We evaluated the assembly and disassembly of Thorase in the presence of ATP using chemical crosslinking, as previously described (Babst et al., 1998). The binding of non-hydrolyzable ATP (Figure 2G, ATPγS) to WT Thorase facilitates oligomer assembly and ATP hydrolysis (Figure 2G; ATP-GSH) disassembles the oligomer, as indicated by a change in the ratio of oligomeric to monomeric Thorase (Figure 2H). The ATP hydrolysis-deficient Walker B Thorase mutant (E193Q) is not responsive to ATP hydrolysis-mediated oligomeric disassembly (Figures 2G and 2H). To ascertain whether NO regulates the disassembly of Thorase oligomers, the disassembly assay was performed in the presence of GSNO on recombinant WT or C137L Thorase. GSNO significantly reduces the disassembly of WT from the oligomeric to the monomeric form, but it has no effect on the C137L mutant (Figures 2G and 2H). The results suggest that S-nitrosylation of Thorase impairs the disassembly of the Thorase oligomeric complex.
The Disassembly of the GRIP1/GluA2 Complex Is Inhibited by S-Nitrosylation of Thorase
Thorase disassembles the GluA2/GRIP1 complex in an ATP-dependent manner (Umanah et al., 2017; Zhang et al., 2011). We therefore determined the effects of NO on Thorase-mediated disassembly of the GluA2/GRIP1 complex. As previously reported (Umanah et al., 2017; Zhang et al., 2011), GluA2, GRIP1, and Thorase co-exist as a complex (Figure 3A). The binding of GRIP1 and Thorase to the GluA2 complex is sensitive to ATP hydrolysis since there is less binding in the presence of hydrolyzable ATP (ATP) and elevated in the presence of the non-hydrolyzable form of ATP (ATPγS), compared to ADP-treated samples (Figures 3A–3C). GSNO significantly reduces the disassembly of GRIP1 from GluA2. GSNO has no effect on C137L Thorase-mediated disassembly of GRIP1 from GluA2 (Figures 3B and 3C). Thus, the S-nitrosylation of Thorase inhibits the disassembly of Thorase-GRIP1-GluA2 complex.
Figure 3. S-Nitrosylation of Thorase Is Critical for Endocytosis of GluA2.
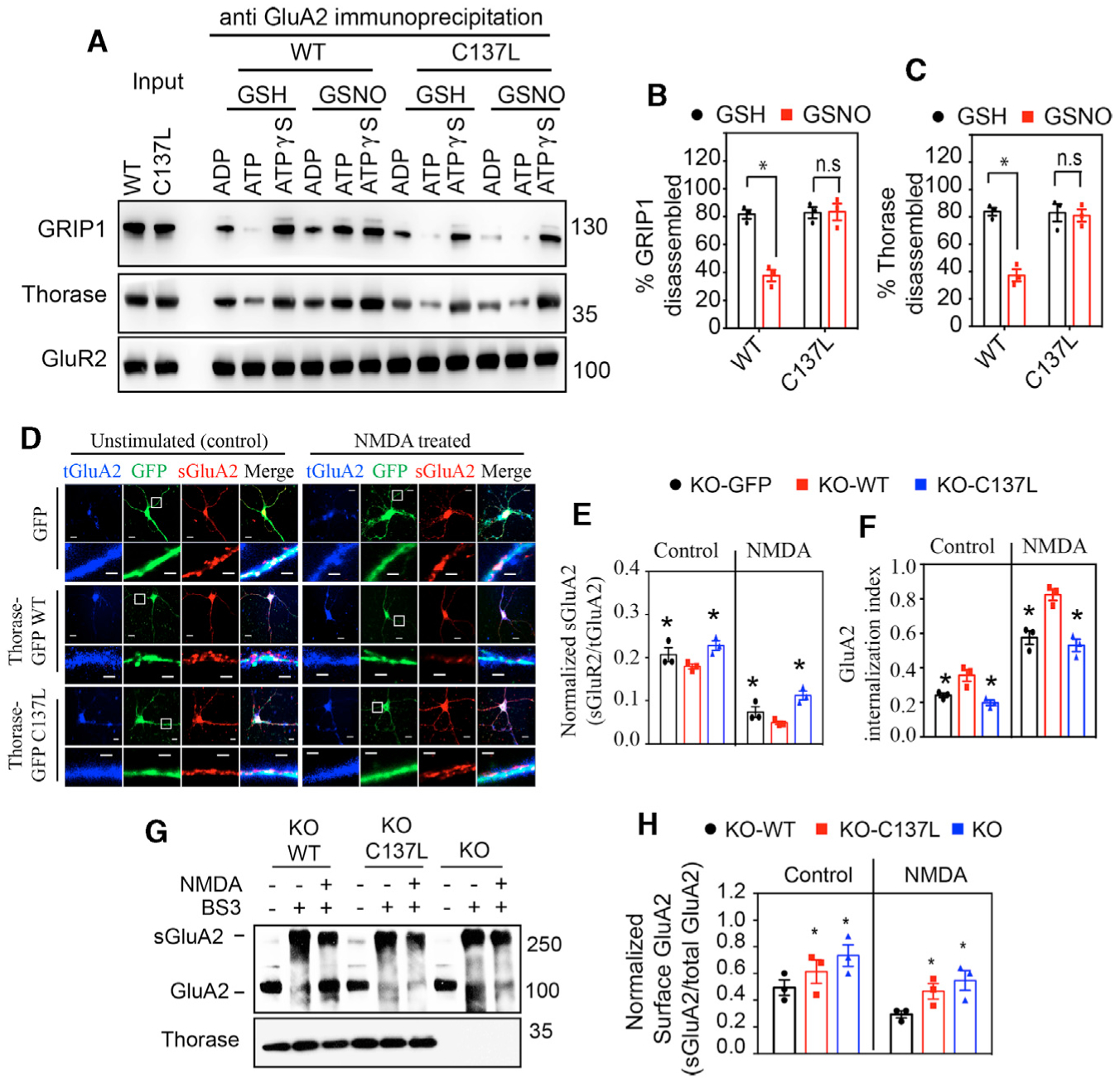
(A) Immunoblot analyses of GluA2 immunoprecipitation (IP) from neurons expressing Thorase WT or C137L in the presence of GSH or GSH with ADP, ATP, or ATPγS.
(B and C) Graphical representation of relative percentage of GRIP1 and Thorase disassembled from GluA2 (n = 3, p = 0.0015).
(D) Representative images of unstimulated or NMDA-induced endocytosis of GluA2 in neurons expressing GFP, Thorase-GFP WT, or C137L mutant. Image scale bar high resolution, 20 μm; low resolution, 2 μm.
(E) Quantification of the ratio of surface GluA2 (sGluA2) to internalized GluA2 (iGluA2) for (n = 3 per replicate, p = 0.0100).
(F) GluA2 internalization index shown in (D) as measured as the ratio of iGluA2 to the total GluA2 (iGluA2 plus sGluA2) fluorescence intensities (n = 3 per replicate, p = 0.0110).
(G) Immunoblot analyses of BS3-crosslinking of sGluA2 in Thorase KO neurons expressing Thorase WT or C137L mutant.
(H) The optical densitometry quantification of sGluA2 for (F) (n = 3, p = 0.0029).
Means ± SEMs of the experiments performed. *p < 0.10, n.s. p > 0.10; ANOVA with Holm-Sidak post hoc test compared with WT; power: 1-β error probability = 0.9–1.0.
S-Nitrosylation of Thorase Is Important for Endocytosis of GluA2
An antibody-feeding internalization assay was used to evaluate the role of S-nitrosylation of Thorase in endocytosis of surface GluA2. Thorase KO primary neurons were transduced with lentiviral WT or C137L mutant GFP-tagged Thorase. Live neurons were incubated with an anti-GluA2 N-terminal antibody followed by treatment with NMDA/glycine to induce GluA2 endocytosis. In control unstimulated cultures, an increased surface expression of GluA2 in neurons expressing control GFP or Thorase-GFP C137L mutant as compared to Thorase-GFP WT is observed (Figures 3D–3F). Moreover, a significant internalization of surface GluA2 in Thorase WT neurons in response to NMDA is detected, while neurons expressing GFP or Thorase C137L mutant display significantly less internalization of surface GluA2 (Figure 3E). In response to NMDA/glycine, the ratio of surface GluA2:intracellular GluA2 is significantly decreased in neurons expressing Thorase WT, while neurons expressing Thorase mutants have a significantly increased ratio of surface GluA2:intracellular GluA2 consistent with a defect in AMPAR endocytosis (Figure 3F).
The effects of Thorase S-nitrosylation on GluA2 surface expression was also assessed by a bis(sulfosuccinimidyl)-sub-erate (BS3) crosslinking assay that allows for the quantification of surface and intracellular receptor pools (Conrad et al., 2008). In unstimulated Thorase KO primary neurons and cultures expressing the Thorase C137L mutant, the ratio of surface GluA2:intracellular GluA2 is increased compared to Thorase WT-expressing cultures (Figures 3G and 3H). In response to NMDA/glycine the ratio of surface GluA2:intracellular GluA2 is significantly decreased in neurons expressing Thorase WT, while Thorase KO neurons and Thorase C137L neurons have a significantly increased ratio of surface GluA2:intracellular GluA2 consistent with a defect in AMPAR endocytosis (Figure 3H).
S-Nitrosylation-Dependent Interaction between Thorase, NSF, and PICK1
The elevated levels of surface GluA2 and defects in endocytosis in the presence of the Thorase C137L mutant suggests that the S-nitrosylation of Thorase may influence the activities of other PSD proteins that are involved in the insertion of GluA2 during AMPAR trafficking. We have suggested that Thorase and NSF may exist in a complex with GluA2 (Prendergast et al., 2014). To further determine the interactions of Thorase-NSF-GluA2 Thorase proteins, recombinant glutathione S-transferase (GST)-tagged Thorase was used to pull down potential Thorase interactors from whole-brain lysates (Figure 4A). When separating protein complexes pulled down by recombinant Thorase from brain lysates according to their molecular size using fast protein liquid chromatography (FPLC), we confirmed via immunoblotting that GluA1, GluA2, NSF, and Thorase co-exist in different complexes of high-molecular-weight (FPLC fractions 2–3 in Figure 4B) Thorase-NSF complex in fraction 4, with the Thorase-PICK1 complex in fractions 5 and 6. The potential interaction of Thorase with NSF and GluA2 was further monitored in the presence of GSH or GSNO. Thorase, GluA2, NSF, and PICK1 were immunoprecipitated from mouse whole-brain lysates (Figure 4C). Thorase exists in a complex with GluA2 and NSF and this complex is dissociated in the presence of ATP-GSH (Figures 4C and 4D). GSNO inhibits the dissociation of the GluA2-Thorase-NSF complex. PICK1 interactions with Thorase were also confirmed in Thorase immunoprecipitated samples and vice versa. The Thorase-PICK1-GluA2 complex dissociates in the presence of ATP (GSH) and is inhibited by GSNO (Figures 4C and 4D).
Figure 4. S-Nitrosylation-Dependent Interaction between Thorase, NSF, and PICK1.
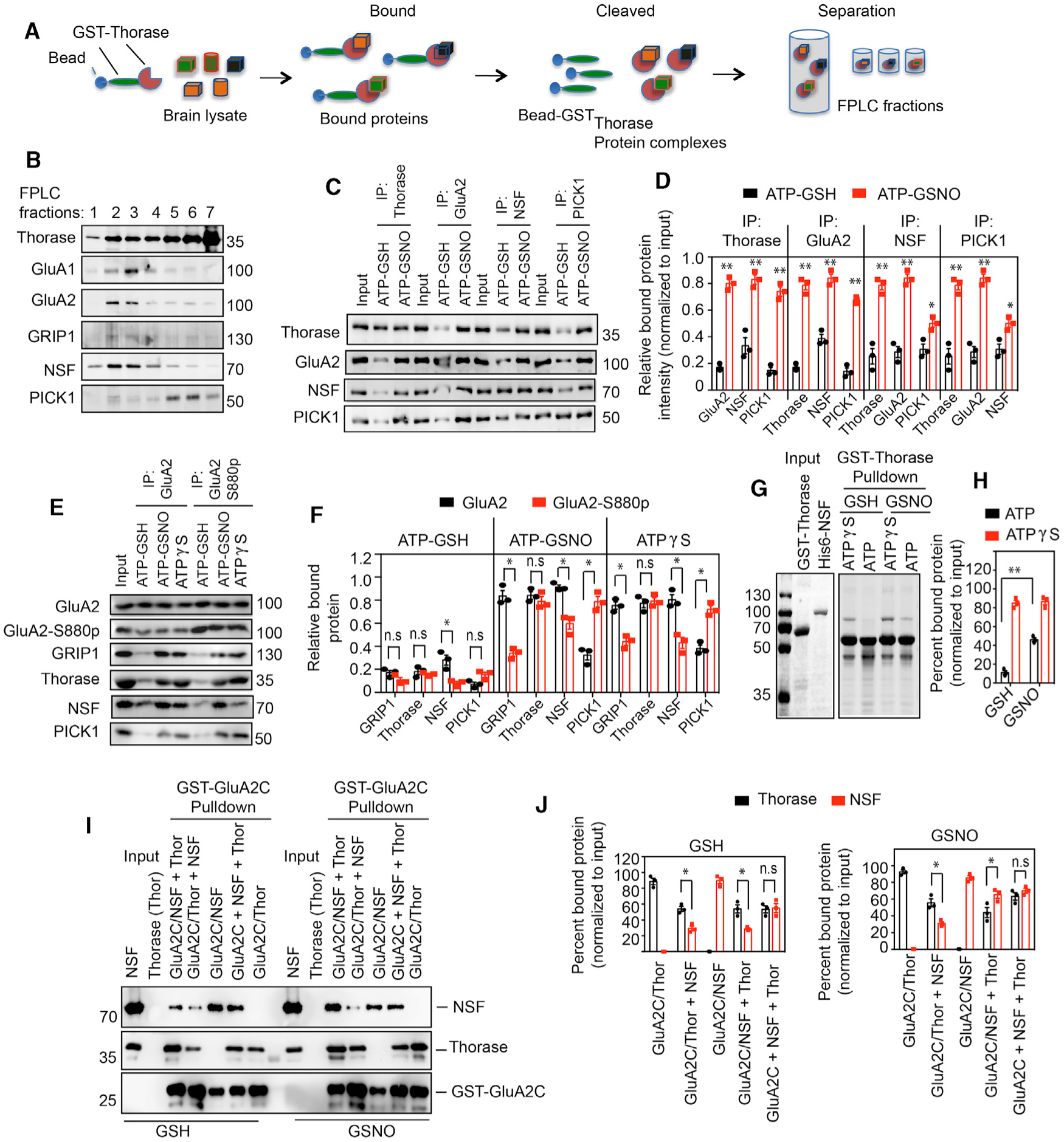
(A) Schematic diagram of recombinant Thorase pull-down from brain lysates to identify binding partners.
(B) Immunoblots of FPLC fractions of Thorase pull-down of protein complexes.
(C) Immunoblot analyses of Thorase, GluA2, NSF, and PICK1 IP from mouse whole-brain lysates in the presence of ATP with GSH or GSNO. The samples were resolved on 10% SDS-PAGE and immunoblotted with anti-Thorase, anti-GluA2, anti-NSF, and anti-PICK1 antibodies.
(D) Normalized relative bound proteins in the IP samples (n = 3, p ≤ 0.0001).
(E) Immunoblot analyses of GluA2 and phosphorylated GluA2 (GluA2 S880p) IP from mouse whole-brain lysates in the presence of ATPγS or ATP with GSH or GSNO.
(F) Normalized relative bound proteins in the IP samples for (C) (n = 3, p ≤ 0.0001).
(G) Coomassie stained SDS-PAGE analyses of GST-Thorase pull-downs of NSF in the presence of GSNO.
(H) Percentage of NSF bound to Thorase in (G) (n = 3, p = 0.0002).
(I) Immunoblot analyses of GST-GluA2C pull-downs of Thorase and NSF in the presence of GSH or GSNO. NSF was added to the prebound GluA2-Thorase complex (GluA2/Thor + NSF), Thorase was added to the prebound GluA2-NSF complex (GluA2/NSF + Thor), or Thorase and NSF were mixed together with GluA2 (GluA2C + NSF + Thor) during the GST-GluA2C pull-down experiments.
(J) Graphical representation of percentage of bound Thorase and NSF bound to GST-GluA2C for (I) (n = 3, p = 0.0139).
Means ± SEMs of the experiments performed. *p < 0.10, **p < 0.05, n.s. p > 0.10; ANOVA with Holm-Sidak post hoc test compared with WT; power: 1-β error probability = 1.0).
The phosphorylation states of GluA2, especially GluA2-S880-phosphorylation (GluA2-S880p), have been shown to influence the interaction of accessory proteins that regulate the trafficking of GluA2 (Chung et al., 2000; Kim et al., 2001). We therefore immunoprecipitated GluA2 (total) and GluA2-S880p from mouse whole-brain lysates in the presence of GSNO to determine the binding preferences of Thorase, NSF, PICK1, and GRIP1. Consistent with a previous study (Lin and Huganir, 2007), PICK1 associates more with GluA2-S880p and GRIP1 (Figures 4E and 4F), and NSF preferentially binds to the non-phosphorylated GluA2 complex. In contrast, Thorase shows no preference for GluA2-S880p complexes or non-phosphorylated GluA2 complexes. The presence of GSNO augments NSF binding to GluA2, but did not influence Thorase binding to GluA2. The presence of GSNO also inhibits the dissociation of Thorase/PICK1 from the GluA2-S880p (Figure 4F).
Thorase Interacts Directly with NSF
To determine whether Thorase directly interacts with NSF, recombinant GST-Thorase and His6-NSF were examined in GST pull-down (Figures 4G, 4H, S1H, and S1I). GST-Thorase pulls down His6-NSF in an ATP-dependent manner (Figures S1H and S1I). GSNO inhibits the ATP-driven dissociation of GST-Thorase from His6-NSF, while GSNO has no effect on ATP-driven dissociation of GST-C137L Thorase from His6-NSF (Figures 4G and 4H). The interaction of GluA2 with Thorase and NSF was also evaluated by GST pull-down assays using the (GST)-tagged C-terminal fragment of GluA2 (GST-GluA2C). In the presence of GSH, both Thorase (GluA2C/Thor) and NSF (GluA2/NSF) strongly bind to GST-GluA2C (Figures 4I, 4J, and S1H–S1J). However, in the presence of equal amounts of Thorase and NSF (GluA2C + NSF + Thor), both display ~50% binding to GluA2C, suggesting that both proteins may bind to the same site on GluA2 C terminus.
We further evaluated the GST-GluA2C pull-down of Thorase and NSF in the presence of GSH or GSNO. Prebound GluA2C-Thorase (GluA2C/Thor) and GluA2C-NSF (GluA2C/NSF) complexes were treated with GSH or GSNO. The addition of excess NSF to GSH and GSNO-pretreated GluA2-Thorase complex (GluA2C/Thor + NSF) displaced 40% and 42% of Thorase, respectively, from GluA2C. However, the addition of excess Thorase to GSH and GSNO-pretreated GluA2-NSF complex (GluA2C/NSF + Thor) displaced 58% and 40% of NSF, respectively, from GluA2C (Figures 4I, 4J, and S1H–S1J). Thus, S-nitrosylation of NSF favors NSF binding to GluA2 over Thorase.
Thorase Transnitrosylates NSF
Previously, we showed that the expression and distribution of synaptic proteins such as PICK1, NSF, and NMDAR are not affected by Thorase expression (Pignatelli et al., 2017; Prendergast et al., 2014; Zhang et al., 2011). We evaluated the expression and colocalization of NSF with GluA2 (Figures S1K–S1N) and did not observe any significant differences between WT and Thorase KO. The data suggest that the absence of Thorase does not alter the localization of NSF. Consistent with a previous study (Huang et al., 2005) NSF is S-nitrosylated by NMDA stimulation in primary neurons (Figures S2A and S2B). However, in contrast to Thorase S-nitrosylation, which decreases after NMDA treatment (see Figures 2A and 2B), NSF remains S-nitrosylated up to 60 min after NMDA treatment (Figures S1H and S1I). In addition, the S-nitrosylation of NSF is significantly reduced in Thorase KO neurons (Figures 5A and 5B). NMDA-dependent S-nitrosylation of NSF is abolished in nNOS KO mice (Figures S2C and S2D), as previously reported (Huang et al., 2005).
Figure 5. Thorase Transnitrosylates NSF In Vivo upon NMDAR Activation.
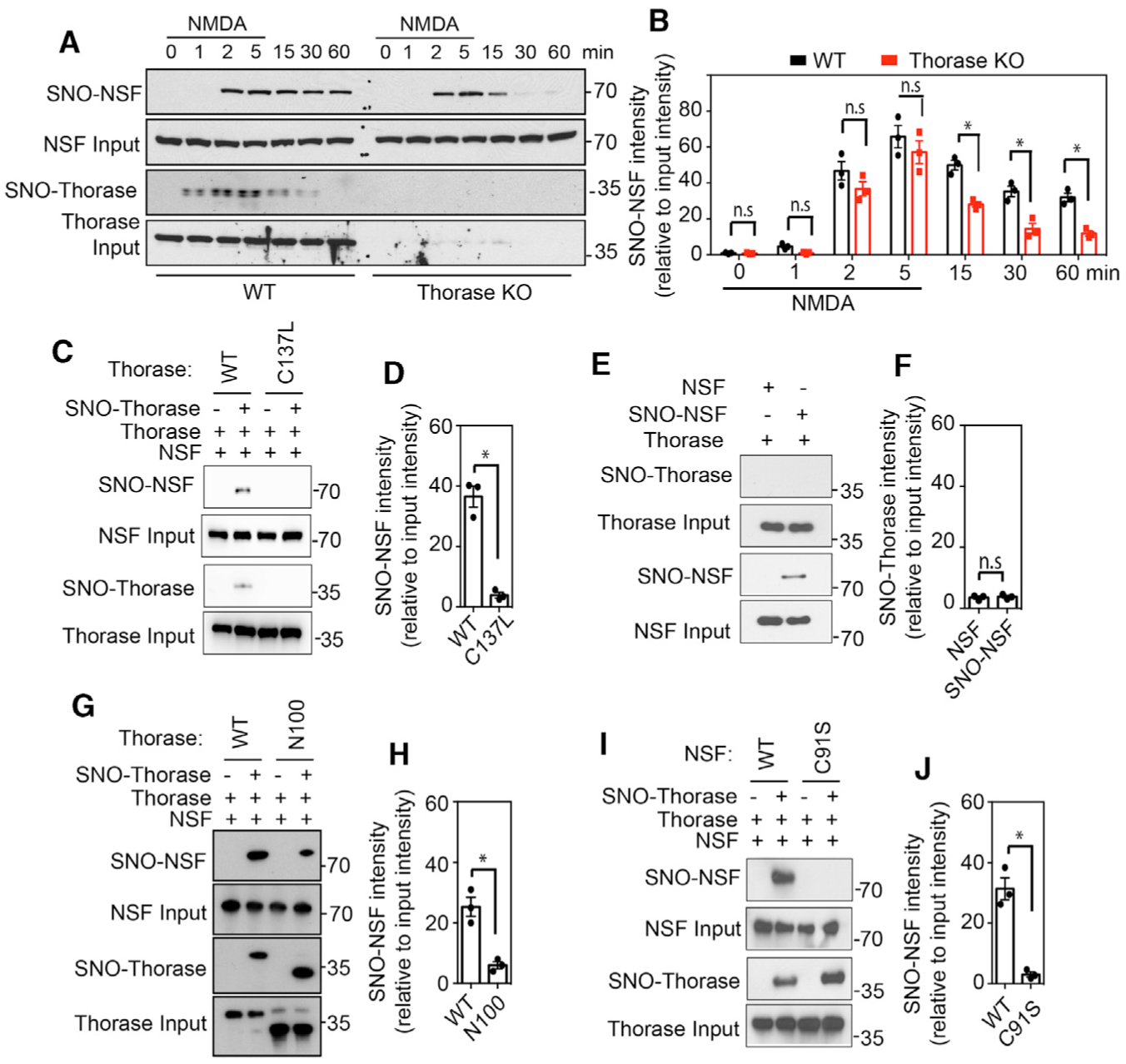
(A) Immunoblot analyses of NMDA-stimulated S-nitrosylation of NSF in WT and Thorase KO neurons. S-nitrosylation of NSF was assessed at different times after NMDA treatment of neurons.
(B) Graphical representation of relative amount of S-nitrosylated NSF (n = 3, p = 0.0111).
(C and D) Immunoblot analyses of trans-S-nitrosylation of NSF by S-nitrosylated Thorase WT (n = 3, p = 0.0106).
(E and F) Immunoblot analyses of trans S-nitrosylation of Thorase by S-nitrosylated NSF (n = 3, p = 0.6964).
(G and H) Immunoblot analyses of trans S-nitrosylation of NSF by S-nitrosylated Thorase WT and Thorase N-terminal deleted mutant, N100 (n = 3, p = 0.0210).
(I and J) Immunoblot analyses of trans-S-nitrosylation of WT NSF but not C91S mutant by Thorase (n = 3, p = 0.0208).
Means ± SEMs of the experiments performed, *p < 0.10, n.s. p > 0.10; ANOVA with Holm-Sidak post hoc test compared with WT; power: 1-β error probability = 0.85–1.0).
To determine whether S-nitrosylated Thorase can transfer its NO group to NSF, we performed an in vitro S-transnitrosylation assay. Purified WT Thorase and C137L Thorase were exposed to GSNO to induce S-nitrosylation, with GSH treatment as a control. After removing excess GSNO from the sample, the proteins were incubated with purified NSF, followed by a biotin switch assay to determine the extent of NSF S-nitrosylation. Incubation with WT Thorase, but not C137L Thorase, leads to the S-nitrosylation of NSF (Figures 5C and 5D), reflecting the transfer of the NO group from the C137 residue of S-nitrosylated Thorase to NSF. However, S-nitrosylated NSF is not able to S-transnitrosylate Thorase (Figures 5E and 5F). Furthermore, a binding interaction between the two proteins is required since an N-terminal deletion of the first 100 amino acids (Thorase N100), which is still S-nitrosylated, significantly reduces the interaction of Thorase with NSF (Prendergast et al., 2014) and it abrogates the S-transnitrosylation of NSF (Figures 5G and 5H).
We observe that adding ATPγS to purified WT, but not C137L Thorase, causes a significant S-transnitrosylation of purified NSF (Figures S2E and S2F). C91 is the S-nitrosylated residue in NSF (Huang et al., 2005), and a mutation of C91 to serine (C91S) mutation abolishes the S-transnitrosylation, indicating that NO group can transfer from C137 of Thorase to C91 of NSF during the S-transnitrosylation process (Figures 5I, 5J, S2G, and S2H). To directly examine Thorase S-transnitrosylation of NSF in cells, we overexpressed NSF in HEK293 cells with or without co-overexpression of WT Thorase and C137L Thorase. The overexpression of WT Thorase leads to markedly elevated levels of NSF S-nitrosylation after GSNO (50 μM) treatment (Figures S2I and S2J). The mutation of C137 also abrogates the effect of Thorase on the S-nitrosylation of NSF. The results also suggest that C137L mutant is not nitrosylated in intact cells. These results indicate that S-nitrosylated Thorase S-transnitrosylates NSF.
S-Nitrosylation of Thorase and Transnitrosylation of NSF Are Important for AMPAR-GluA2 Trafficking
To further evaluate the effects of S-nitrosylation of Thorase on AMPAR trafficking, pH-sensitive GFP (pH-luorin) fused at the N-terminal extracellular domain of GluA2 was used to examine AMPAR distribution at the extracellular surface, as previously described (Umanah et al., 2017; Zhang et al., 2011). Thorase KO neurons were co-transfected with pH-GluA2 and mCherry Ires or mCherry Ires Thorase WT or C137L mutant (Figures 6A and S3). In WT neurons expressing pH-GluA2, NMDA-stimulated internalization of GluA2 results in the loss of the fluorescent signal for pH-GluA2, and washout of NMDA leads to the recovery of fluorescence (Figures 6B, 6C, and S3). NMDA induced a significantly smaller reduction in pH-GluA2 fluorescence and fluorescence recovery that is significantly faster in Thorase KO control (vector) than in WT neurons (Figures 6B and 6C), as previously described (Zhang et al., 2011). However, in Thorase C137L mutant neurons, NMDA stimulation leads to a significantly reduced internalization of GluA2, and recovery is significantly slower than in WT neurons. These results suggest that Thorase S-nitrosylation is important for AMPAR-GluA2 internalization and insertion into the synaptic membrane. Interestingly, NSF S-nitrosylation is significantly decreased in Thorase KO neurons following NMDAR stimulation (see Figures 5A and 5B). These results suggest that the absence of Thorase S-transnitrosylation of NSF is reduced, resulting in a slower rate of AMPAR insertion to the synaptic membrane. In nNOS KO neurons, NMDA stimulation leads to a significantly reduced internalization of GluA2, and recovery is significantly slower than in WT cultures (Figures S4A–S4D), whereas in the presence of the NMDA antagonist MK801 or the NOS inhibitor l-NAME, GluA2 internalization is significantly reduced (Figures S4E–S4H). These results suggest that endogenous S-nitrosylation of Thorase and NSF is NMDAR and nNOS dependent and is critical for GluA2 trafficking.
Figure 6. S-Nitrosylation of Thorase and Transnitrosylation of NSF Are Important for AMPAR-GluA2 Trafficking.
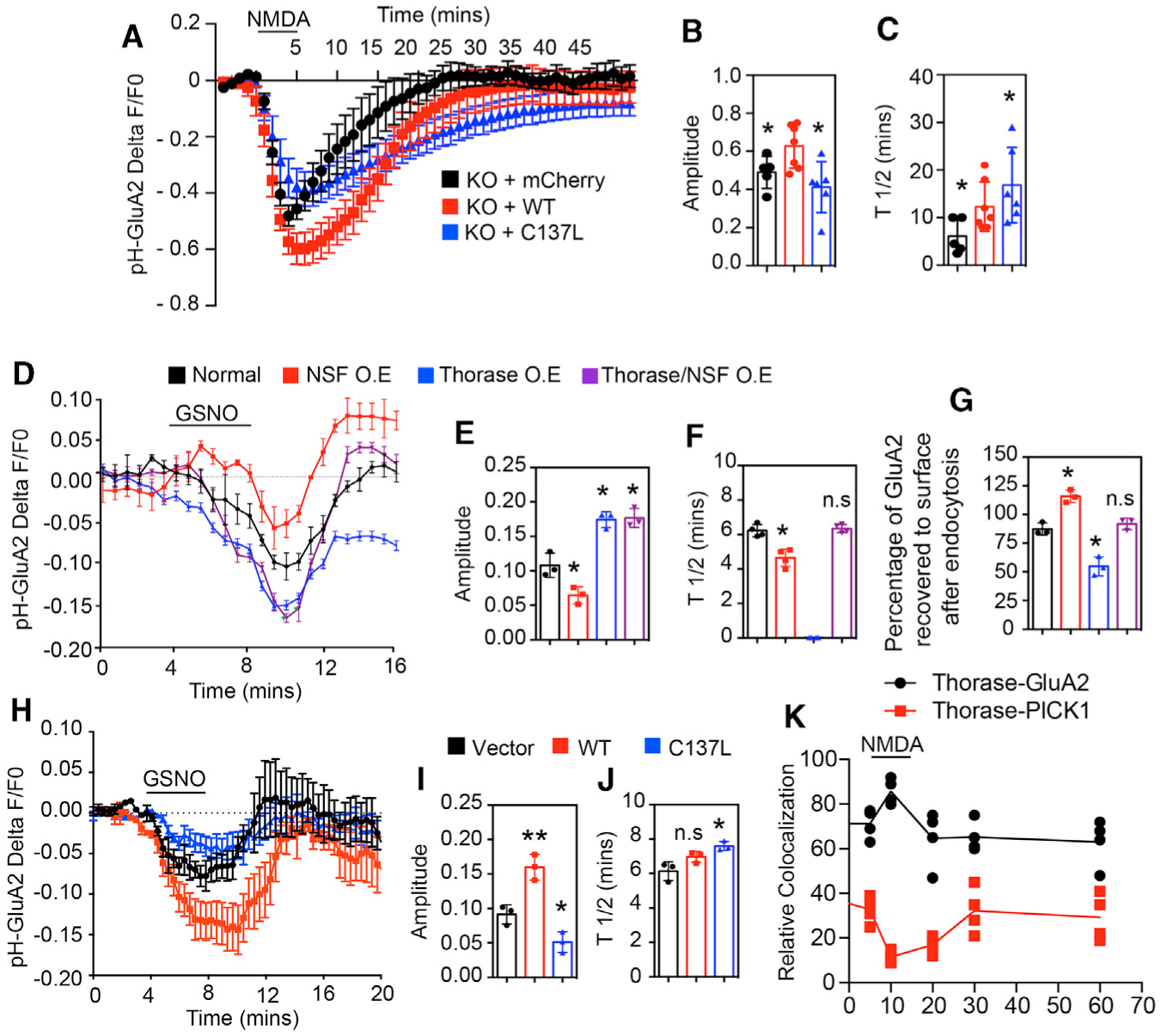
(A) Time trace of pH-GluA2 fluorescence change in response to NMDA in neurons expressing mCherry empty vector or Thorase (WT or C137L mutant).
(B) Amplitudes of fluorescence intensity changes (n = 5–7, p = 0.0367).
(C) Average recycling half-time (T1/2), the time taken from maximum endocytosis to 50% recycling (n = 5–7, p = 0.0299).
(D) Time trace of pH-GluA2 fluorescence change in response to GSNO in HEK293 cells expressing Thorase, NSF, and GRIP1.
(E) Amplitudes of fluorescence intensity changes (n = 3–4, p ≤ 0.0001).
(F) Average T1/2 (n = 3–4, p ≤ 0.0001).
(G) Percentage of pH-GluA2 fluorescence recovered after GSNO washout (n = 3–4, p ≤ 0.0001).
(H) Time trace of pH-GluA2 fluorescence change in response to GSNO in HEK293 cells expressing Thorase WT or C137L.
(I) Amplitudes of fluorescence intensity changes (n = 3, p = 0.0005).
(J) Average T1/2 (n = 3, p ≤ 0.0115).
(K) Time trace of Thorase interactions (colocalization) with GluA2 and PICK1 change in response to NMDA in neurons (n = 4, p = 0.0001).
Means ± SEMs of the experiments performed. *p < 0.10, **p < 0.05, n.s. p > 0.10; ANOVA with Holm-Sidak post hoc test compared with WT; power: 1-β error probability = 0.85–1.0).
NO Directly Influences Thorase-Mediated GluA2 Endocytosis
To evaluate the direct effect of the nitrosylation of Thorase on GluA2 surface expression, we designed a live imaging assay using pH-GluA2 trafficking in HEK293 cells expressing functional AMPARs. The addition of GSNO did not significantly affect the intracellular pH as determined by the intracellular pH indicator pHrodo Red AM, a fluorogenic probe (Figure S5A). HEK293 cells were transfected with pH-GluA2, GRIP1, and WT Thorase or C137L Thorase. At baseline, WT Thorase-transfected HEK293 cells show significantly lower levels of pH-GluA2 signal intensity than vector control-transfected cells (Figures S5B and S5C). Treatment with GSNO causes a further reduction in pH-GluA2 signal intensity in the setting of WT Thorase, but has a minimal effect on pH-GluA2 signal intensity in cells transfected with C137L Thorase.
Thorase and NSF Required for Proper GluA2 Surface Expression
To assess the direct effects of Thorase and NSF nitrosylation on GluA2 trafficking, we monitored the surface GluA2 expression in real time in live HEK293 cells. HEK293 cells were co-transfected with pH-GluA2, WT Thorase (mCherry), NSF, and GRIP1. The addition of GSNO during live imaging stimulated the internalization of pH-GluA2, resulting in the loss of fluorescent signal, and the washout of GSNO leads to the recovery of the fluorescence (Figures 6D and S6A). Interestingly, we observed different patterns of the fluorescent signal for pH-GluA2 from different cells in the same cultures treated with GSNO. The cells were therefore immunostained to examine the expression of Thorase, NSF, and GRIP1 (Figures S6A and S6B). The results suggest that control cells that are not overexpressing Thorase and NSF have a significant loss of the fluorescent signal for pH-GluA2, and the washout of GSNO leads to the recovery of fluorescence to baseline (Figures 6D–6G). Cells overexpressing only Thorase (Thorase OE) have loss of the fluorescent signal for pH-GluA2 more than control cells; however, the washout of GSNO leads to less recovery of fluorescence. Cells overexpressing NSF (NSF OE) have loss of the fluorescent signal for pH-GluA2 lower than control cells, and the washout of GSNO leads to the recovery of fluorescence higher than baseline. Moreover, cells overexpressing both Thorase and NSF (Thorase/NSF OE) have loss of the fluorescent signal for pH-GluA2 more than control cells, and the washout of GSNO leads to the recovery of fluorescence to baseline. Thus, both Thorase and NSF are required for proper trafficking of GluA2.
S-Nitrosylation of Thorase Is Important for AMPAR-Mediated Synaptic Plasticity
The surface GluA2 expression was also monitored in live HEK293 cells co-expressing pH-GluA2 and NSF with WT Thorase or the C137L mutant. In WT cells, GSNO-stimulated internalization of GluA2 results in the loss of the fluorescent signal for pH-GluA2, and the washout of GSNO leads to the recovery of fluorescence (Figures 6H–6J and S6C). GSNO induced a significantly smaller reduction in pH-GluA2 fluorescence and recovery that is faster in control cells. However, in Thorase C137L mutant cells, GSNO treatment leads to a significantly reduced internalization of GluA2, and recovery is significantly slower than in WT cells. Thorase interactions with GluA2 increase during NMDA-induced endocytosis of GluA2 as expected; however, Thorase-PICK1 interactions decrease during this process (Figures 6K and S6D). These results suggest that direct NMDA-mediated NO modification on Thorase is important for modulating GluA2 trafficking.
We next addressed whether long-term potentiation (LTP) and long-term depression (LTD) are affected by the S-nitrosylation of Thorase. Adeno-associated viruses (AAVs) expressing GFP-tagged WT or C137L Thorase-GFP were generated and injected into the cortex of Thorase KO mice at postnatal day 2 (P2) (KO-WT and KO-C137L), as described in our previous studies (Umanah et al., 2017). AAVs expressing GFP alone were introduced into Thorase WT and KO mouse brains at P2 as control groups (WT-GFP and KO-GFP) (Figure S7). Two weeks after infection, we prepared acute brain slices from injected mice and performed extracellular field recordings from medial prefrontal cortex (mPFC) (Figure 7A).
Figure 7. Thorase S-Nitrosylation-Modulated GluA2 Trafficking Is Important for Synaptic Plasticity.
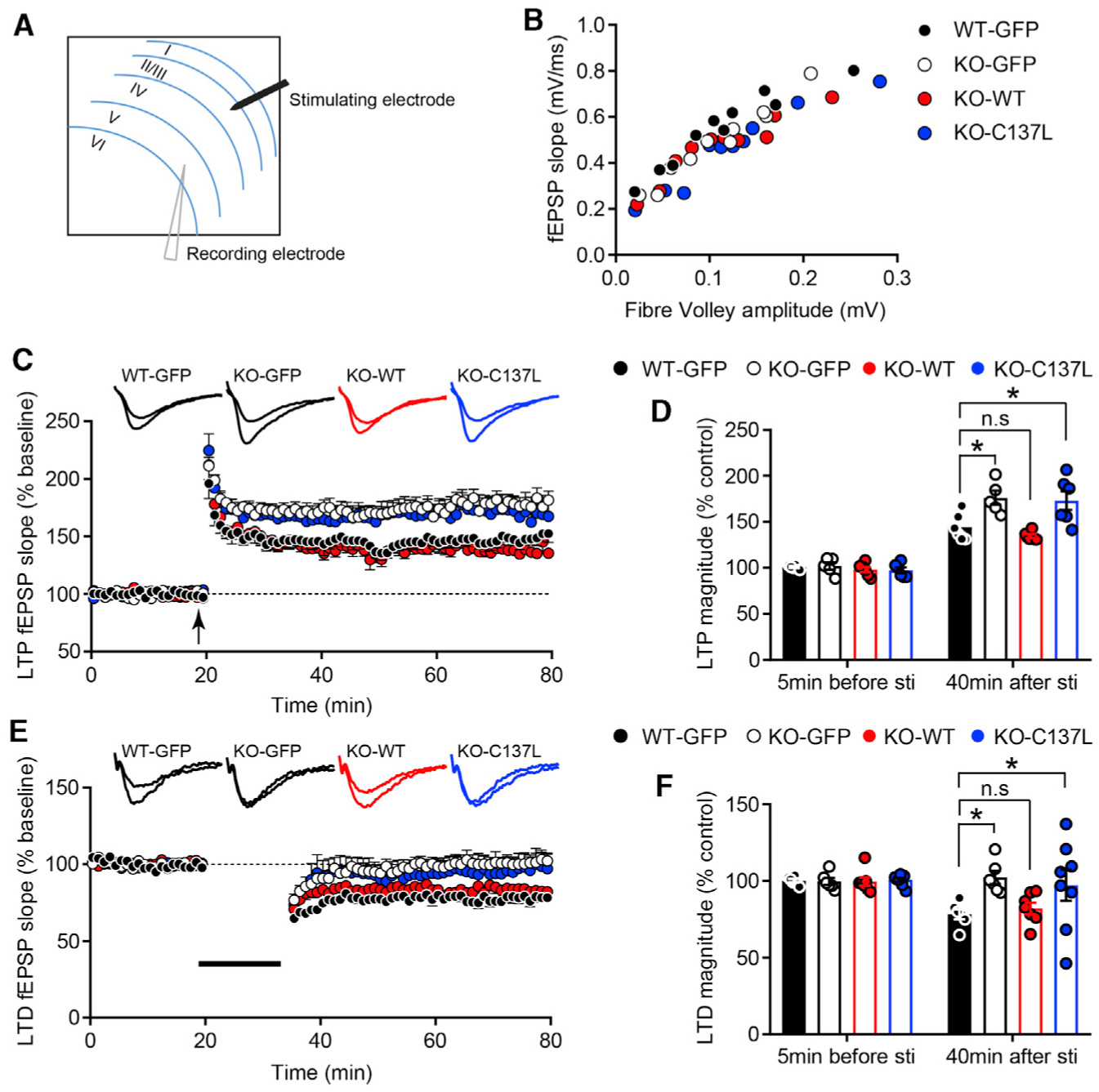
(A) Schematic representation of the prelimbic (PrL) area in a coronal mPFC slice.
(B) Input-output relationships (I/O) of fEPSPs in WT-GFP, KO-GFP, KO-WT, and KO-C137L slices. Traces show representative fEPSPs obtained with different stimulus intensities.
(C) LTP expression was dramatically increased in KO-GFP and KO-C137L slices. Representative traces show fEPSPs recorded at 5 min before stimulation and 40 min after LTP induction (n = 6 slices from 6 mice; KO-GFP, n = 5 slices from 5 mice; KO-WT, n = 6 slices from 6 mice; KO-C137L, n = 6 slices from 6 mice).
(D) LTP magnitude was calculated at 5 min before and 40 min after LTP induction as a percentage of baseline responses (*p < 0.05).
(E) LTD was absent in KO-GFP and KO-C137L mouse brain slices. Representative traces show fEPSPs recorded at 5 min before stimulation and 40 min after LTD induction (n = 6 slices from 6 mice; KO-GFP, n = 6 slices from 5 mice; KO-WT, n = 7 slices from 6 mice; KO-C137L, n = 8 slices from 6 mice).
(F) LTD magnitude was quantified 40 min after low-frequency stimulation as a percentage of baseline responses.
Values are means ± SEMs, *p < 0.05, ANOVA with Holm-Sidak post hoc test compared with WT-GFP.
The slope of the input-output (I/O) relationship was not different among WT-GFP, KO-GFP, KO-WT, and KO-C137L mouse slices (Figure 7B). LTP was induced by a theta-burst stimulation (TBS) protocol consisting of 5 trains of burst with 4 pulses at 100 Hz, repeated 4 times at intervals of 10 s. Consistent with previous results (Zhang et al., 2011), LTP was increased by the deletion of Thorase (Figure 7C). Expression of Thorase WT in Thorase KO mice rescued LTP (Figure 7C). Importantly, a significant increase in LTP was observed in KO-C137L slices compared with WT-GFP slices (LTP magnitude at 40 min, WT-GFP: 144.30% ± 5.92%, KO-GFP: 176.10% ± 7.75%, KO-WT: 134.50% ± 1.98%, KO-C137L: 173.10% ± 10.24%) (Figures 7C and 7D). Next, we compared the levels of LTD, which was induced by a low-frequency train of 1 Hz for 15 min. LTD was eliminated in KO-GFP as well as in KO-C137L slices (field excitatory post-synaptic potential [fEPSP] slope at 40 min after stimulation, WT-GFP: 78.30% ± 3.30%, KO-GFP: 102.30% ± 4.29%, KO-WT: 82.22% ± 3.75%, KO-C137L: 97.22% ± 10.20%) (Figures 7E and 7F). These results imply that GluA2 trafficking modulated by Thorase S-nitrosylation is essential for LTP and LTD.
DISCUSSION
The major findings of this article are the observations that Thorase is S-nitrosylated and that S-nitrosylated Thorase S-transnitrosylates NSF to regulate GluA2 receptor trafficking. Importantly, our data elucidate a mechanism underlying the regulation of the internalization and insertion of AMPARs in the postsynaptic membrane following NMDAR activation. The activation of NMDAR has been shown to enhance NO levels and induce an increase in the S-nitrosylation of proteins (Choi et al., 2000; Huang et al., 2005; Qu et al., 2012; Zorumski and Izumi, 1998). The S-nitrosylation of Thorase at residue C137 inhibits ATP binding and ATPase activities, resulting in a stable Thorase oligomeric complex. Thorase C137 is located within the ATP hydrolysis motif next to the conserved Walker B residue, E193. It is therefore not surprising that NO modification of the ATP hydrolysis motif inhibits ATP hydrolysis, which augments a stable oligomer formation similar to the ATP hydrolysis-deficient Walker B mutant E193Q. The residue C137 is well conserved among Thorase homologs, implying a broader effect of S-nitrosylation on this family of enzymes. Oligomer formation and stability play important roles in how AAA+ proteins assemble and disassemble protein complexes (Kedzierska, 2006; Mogk et al., 2003; Sauer and Baker, 2011). Consistent with our previous study (Zhang et al., 2011), Thorase disassembles the GluA2/GRIP1 complex in an ATP-dependent manner. However, the disassembly of the GluA2/GRIP1 complex is markedly impaired in the presence of NO. The formation of a stable Thorase oligomer in the presence of NO accounts for the stable Thorase/GluA2/GRIP1 complex. These findings suggest that the S-nitrosylation of Thorase is critical in stabilizing Thorase-AMPAR complexes.
Although several studies have shown that NSF participates in the insertion of AMPARs, especially GluA2/GluA3, into the synaptic compartment (Beretta et al., 2005; Huang et al., 2005; Katano et al., 2008; Steinberg et al., 2004) the mechanism of this process is not completely understood. Previously, we showed that NSF and Thorase exist together in the GluA2-containing AMPAR complexes (Prendergast et al., 2014), and the present study shows that NSF directly binds Thorase, suggesting that these two proteins may function together to regulate AMPAR trafficking. Both Thorase and NSF binding to GluA2 complexes is influenced by S-nitrosylation. Thorase binding to GluA2 complexes is augmented by S-nitrosylation via the inhibition of ATP hydrolysis, whereas, S-nitrosylation-mediated increased NSF binding affinity for GluA2 is not dependent on ATPase activity (Huang et al., 2005). Our data show that increased S-nitrosylation of NSF upon NMDAR activation is dependent on interaction with Thorase S-nitrosylation. Thorase S-nitrosylation occurs earlier than S-nitrosylation of NSF upon NMDAR activation. The S-nitrosylation of Thorase augments the Thorase interaction with NSF, allowing Thorase to trans-nitrosylate NSF, thereby increasing NSF S-nitrosylation and enhancing the NSF-mediated insertion of GluA2 at the synapses. However, in the presence of Thorase-deficient S-nitrosylation mutant (C137L), NSF S-nitrosylation is significantly reduced, resulting in the delayed insertion of GluA2. Thus, C137L Thorase may act as a negative regulator of NSF by delaying NSF S-nitrosylation and binding to GluA2, thereby delaying the insertion of GluA2 at the synapses. The data suggest that Thorase interaction and trans-nitrosylation of NSF play a critical role in NSF-mediated insertion of GluA2 at the synapses.
Nitrosylation has been shown to directly or indirectly influence other proteins that regulate AMPARs. Our data further suggest that S-nitrosylation of Thorase not only enhances endocytosis of GluA2 but also influences the stability of the GluA2-PICK1 complex. Thus, S-nitrosylated Thorase may mediate GluA2-PICK1 complex stability that is critical in retaining a pool of intracellular AMPARs as well as delaying the recycling of AMPARs during endocytosis. NSF regulates GluA2 recycling by binding to the GluA2-PICK1 complex and dissociating PICK1 from GluA2 (Hanley et al., 2002). In the absence of Thorase, the unstable GluA2-PICK1 in the intracellular pool allows NSF to bind the free GluA2-containing AMPARs, thereby accelerating AMPAR recycling and enhancing surface expression of AMPARs.
Although phosphorylation of GluA2 at S880 has been shown to not be required for GluA2 endocytosis, it plays an important role in the recycling of GluA2 during endocytosis (Lin and Huganir, 2007). GRIP1 is observed to associate more with the non-phosphorylated GluA2 complexes, whereas PICK1 associates more with S880-phosphorylated GluA2 complexes, consistent with previous reports (Lin and Huganir, 2007; Chung et al., 2000; Kim et al., 2001). We also observed that NSF preferentially binds to the non-phosphorylated GluA2 complex, whereas Thorase shows no preference for S880-phosphorylated GluA2 complexes or non-phosphorylated GluA2 complexes. The fact that Thorase has similar binding affinity for both the phosphorylated and non-phosphorylated GluA2 allows it to influence the binding of GRIP1, NSF, and PICK1 to different states of GluA2 complexes. The differential binding of these proteins to GluA2 complexes is critical for modulating synaptic plasticity mediated by AMPAR trafficking. We propose that Thorase may play an important role in the differential binding of proteins that interact with the GluA2-containing AMPAR complexes.
Our data suggest that Thorase may mediate GluA2-containing AMPAR trafficking by (1) binding and internalizing GRIP1 with non-phosphorylated GluA2 complexes, (2) retaining intracellular GluA2 by stabilizing phosphorylated GluA2-PICK1 complexes, and (3) inserting the GluA2 surface by enhancing NSF binding to non-phosphorylated GluA2 complexes via trans-nitrosylation. The present study and a previous study (Zhang et al., 2011) suggest that Thorase plays important roles in both LTD and LTP. We propose that decreased synaptic strength during LTD due to a decrease in postsynaptic AMPAR surface expression is mediated by Thorase-GluA2-GRIP1 endocytosis and the stability of the retained intracellular Thorase-GluA2-PICK1 complex. Conversely, increased synaptic strength during LTP due to enhanced AMPAR surface expression is mediated by the increased binding of NSF to GluA2-containing AMPARs as a result of the interaction of NSF with Thorase.
Here, we provide evidence that NO modification of Thorase and its association with AMPAR complexes play an important role in AMPAR trafficking and synaptic plasticity. Overall, the data presented in this study provide important information on how NSF and PICK1 may be influenced by Thorase in regulating the trafficking of AMPAR.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Contact Valina L. Dawson (vdawson@jhmi.edu) for reagent and resource sharing.
Materials Availability
Plasmids and other materials generated in this study will be available upon request. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact.
Data and Code Availability
Datasets is available to readers and there are no restrictions on any data or materials presented in this paper.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All experimental procedures were according to the guidelines of Laboratory Animal Manual of the National Institute of Health Guide to the Care and Use of Animals and were approved by the Johns Hopkins Medical Institute Animal Care and Use Committee. Thorase-heterozygous C57BL/6J mice (ATAD1+/−) males and females generated in our previous studies (Umanah et al., 2017; Zhang et al., 2011). For primary neuron cultures, three months old males and females Thorase-heterozygous C57BL/6J mice (ATAD1+/−) were mated and the females were sacrificed at day 15–17 of pregnancy (E15–E17) to obtain Thorase-knockout (KO) C57BL/6J mice (ATAD1−/−) and litter mates Thorase-wild-type C57BL/6J mice (ATAD1+/+) pups. Genotyping of mice was carried out as we previously described (Umanah et al., 2017; Zhang et al., 2011).
Cell Culture
HEK293 cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) plus 10% (v/v) bovine serum (FBS) and 1% Penicillin-Streptomycin, at 37°C with a 5% CO2 atmosphere in a humidified incubator. Primary hippocampal and cortical neuron cultures were prepared from embryonic day 15 (E15) and 17 (E17) mouse pups, respectively in neurobasal media with B27 supplement (GIBCO) and 1% Penicillin-Streptomycin as previously described (Umanah et al., 2017). Transfections were performed with Lipofectamine 2000 Reagent (Invitrogen) according to the manufacturer’s instruction.
Recombinant proteins
All recombinant proteins were expressed in E. coli BL21(DE3) codon plus and induced by 1.0 mM isopropyl thiogalactoside (IPTG) at 16°C overnight. The bacterial pellets were lysed using a microfluidizer (Microfluidics) in binding/ATPase buffer (100 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM MgCl2, and 5% Glycerol) with protease inhibitors (Sigma) and centrifuged at 15,000 × g for 30 min. Histagged and GST-tagged proteins were purified by using Nickel-NTA and glutathione beads, respectively following manufacturer’s instructions. Protein samples eluted from the beads were further purified by size exclusive chromatography (GE Healthcare) using the ATPase buffer. The purity of the recombinant proteins was checked by Coomassie staining and western immunoblotting.
METHOD DETAILS
ATPase Activities
The ATPase activities of all Thorase mutants generated in this study along side wild-type were examined by ATP binding and hydrolysis assays. The ATP binding was evaluated by photolabeling technique as described by Babst et al. (Babst et al., 1998) with some few modifications. Briefly, 20 μL of purified Thorase protein samples (5 μg) in ATPase buffer were incubated at 4°C with 20 μCi of [α−32P]ATP (2000 Ci/mmol)]. The mixtures were then illuminated by UV light to cross-link the bound [α−32P]ATP to the bound proteins. The protein samples were then precipitated by adding trichloroacetic acid (TCA), washed with acetone and resuspended in 1× laemmli sample buffer (Bio-Rad). The samples were resolved on SDS-PAGE, stained with Coomassie blue to check protein input and exposed to a phosphor screen (Perkin Elmer, USA). Quantitation of [α−32P] ATP labeled protein was performed by scanning the radioautograph with a soft laser scanning densitometer.
The ATP hydrolysis activity was determined by using the ADP Colorimetric Assay Kit (Sigma) following manufacturer’s instruction with some modifications. Briefly, the recombinant proteins (2.0 ug) were incubated with 100 mM ATP for 30 min at 37°C for ATP hydrolysis to ADP. To check ATP degradation to ADP that were not due to enzymatic hydrolyses the ATPase buffer (without protein) containing 100 mM ATP was included as control. The samples were incubated with ADP reaction mix buffer for 30 min at room temperature in the dark and the absorbances at 570 nm for each sample (in triplicates) were measured to determine amount of ADP present in the samples. The rate of ATP hydrolysis was determined by the rate of ADP formation.
The 3D structure modeling of Thorase full length and AAA ATPase domain were generated by the SWISS-MODEL (Arnold et al., 2006) and confirmed by Phyre2 (Kelley et al., 2015) protein modeling. All images obtained were viewed and labeled with Pymol pdb viewer.
Biotin Switch Method (S-nitrosylation and site identification)
The biotin switch method was performed as previously described (Jaffrey et al., 2001; Jaffrey and Snyder, 2001) with some modifications. Briefly, cell lysates or recombinant proteins were prepared in NP40/HEN buffer (250 mM HEPES, 1 mM EDTA, 0.1 mM Neocuproine, 0.5% Nondiet P-40). Blocking buffer (2.5% SDS, 10 mM methyl methane thiosulfonate [MMTS] (Thermo Scientific) in HEN buffer [250 mM HEPES, 1 mM EDTA, 0.1 mM Neocuproine]) was mixed with the samples and incubated for 20 min at 50°C with frequent vortexing to block free thiol groups. After removing excess MMTS by spin columns, newly formed thiols were linked with sulfhydryl-specific biotinylating reagent through incubation with 5 mM ascrobate and 0.4 mM N-[6-(biotinamide)hexyl]-3′-(2′-pyridyldithio)propionamide (Biotin-HPDP, Thermo Scientific) for 1 h at room temperature with rotation. Unreacted Biotin-HPDP was removed by acetone precipitation and centrifugation, and the pellet was resuspended in 100 μl HENS buffer (HEN buffer plus 1.0% SDS). Two volumes of neutralization buffer (250 mM HEPES, 100 mM NaCl, 1 mM EDTA, and 0.5% Triton X-100) were added, and the solution was cleared by centrifugation in order to pellet the undissolved debris. The biotinylated proteins were pulled-down with Neutravidin-agarose beads (Thermo Scientific) from the remaining supernatant. Pellets were then washed 5 times with neutralization buffer plus 0.6 M NaCl and eluted by SDS sample buffer and subjected to western blot analysis.
S-Nitrosylation of either Thorase or NSF in cultured primary cortical neurons was assayed as described above, except that neurons were treated with NMDA (20 μM) and glycine (10 μM) in Tyrodes’ solution (119 mM NaCl, 5 mM KCl, 25 mM HEPES, 2 mM CaCl2, 300 μM MgCl2, and 1 μM TTX) for 5 min at 37°C in the dark in the presence or absence of MK801 (10 μM) or L-NAME (300 μM) for 30 min before NMDA stimulation.
For the S-transnitrosylation assay, purified recombinant Thorase (0.1 μg/μl) or NSF (0.1 μg/μl) bound to beads was incubated with GSNO (100 μM) for 1 h at room temperature in the dark. The beads were then washed with HEN buffer (without GSNO) and the SNO-proteins were then used as NO donors for 1 h at room temperature in the dark in order to test for potential S-transnitrosylation of recombinant Thorase or NSF. The protein samples were then subjected to the biotin switch assay as described above to monitor S-transnitrosylation of each protein.
Detailed protocols for the identification of site-specific S-nitrosylation using organic mercury resin assisted capture approach have been published (Doulias et al., 2010, 2013). In brief, free thiols on recombinant S-nitrosylated Thorase were blocked with incubation with 35 mM methyl methane thiosulfonate [MMTS] (Thermo Scientific) for 30 min at 55°C with frequent vortexing. The Thorase protein suspension was then loaded onto pre-activated organic mercury resin-containing columns for 60 min at room temperature. Upon the completion of the reaction, the column was extensively washed to eliminate nonspecific interactions. Bound Thorase was subjected to on column trypsin digestion overnight at room temperature. Unbound peptides were removed by extensive washes and cysteine-containing peptides were eluted with performic acid for 45 min according to published protocols. The performic acid released peptides were analyzed by mass spectrometry (Doulias et al., 2010; Doulias et al., 2013).
Thorase mediated disassembly of GluA2/GRIP1 assay
The effect of nitrosylation on Thorase disassembling of GluA2-GRIP1 complex was examined by immunoprecipitating GluA2 from isolated mouse hippocampi lysate as previously described with some modification (Umanah et al., 2017; Zhang et al., 2011). Briefly, freshly mouse hippocampi were homogenized in binding buffer (25 mM HEPES-KOH (pH 7.8), 150 mM NaCl, 3 mM MgCl2, 1 mM DTT) containing protease inhibitors. Triton X-100 was added to a final concentration of 1% and incubated at 4°C (with mixing by end-to-end rotation) for 1 hour. Samples were centrifuged at 15,000 × g for 30 min and supernatant was incubated with Dynabead (Invitrogen) pre-bound with anti-GluA2 antibodies plus 2 mM ADP or ATP or ATPγS at 4°C (with mixing by end-to-end rotation) for 1 hour. The samples were then transferred to room temperature and incubated with 100 μM GSH or GSNO (with mixing by end-to-end rotation) for 1 hour. Beads were washed 3 times with binding buffer plus 100 μM GSH or GSNO (with mixing by end-to-end rotation) for 2 minutes per wash. After the last wash beads were resuspended in 1× laemmli sample buffer, resolved on 10% SDS-PAGE and immunoblotted using anti-GluA2, anti-Thorase and anti-NSF antibodies.
Oligomer formation assays
To evaluate the oligomer formation of Thorase, purified recombinant proteins (5 μg in 100 μl) were treated with 5 mM ADP or ATP in modified HEN buffer (250 mM HEPES, 2 mM MgCl, 0.1 mM Neocuproine, 0.5% Nondiet P-40) containing 100 μM GSH or GSNO at 4°C to minimize ATP hydrolysis for 1 hour. Oligomer formation upon ADP or ATP binding was examined by chemical cross-linking with 0.02% glutaraldehyde at room temperature for 5 minutes as previously described (Babst et al., 1998). The cross-linked products were TCA-precipitated, washed with acetone and analyzed by SDS-PAGE and Immunoblotted with anti-Thorase antibody.
Coimmunoprecipitation and GST and His6 Pull-Down Assays
Whole mouse brains homogenized in ATPase buffer containing protease inhibitors plus 1% Triton X-100 were centrifuged at 15,000 × g for 30 minutes. Approximately 2 mM of ATPγS with 100 μM GSH or GSNO was added to the supernatant and incubated with magnetic-Dyna-beads (Invitrogen) pre-bound to anti-Thorase, anti-GluA2, anti-NSF or anti-PICK1 antibodies at 4°C for 3 hours with end-over-end mixing. The beads were washed 3 times with ATPase buffer plus ATPγS with GSH or GSNO and resuspended in 1× laemmli sample buffer. Proteins bound to the beads were detected by immunoblotting using anti-Thorase, anti-GluA2, anti-NSF and anti-PICK1 antibodies.
For all GST and His6 pull-down experiments purified recombinant GST or His6 fusion proteins bound to glutathione (GST) or His-Nickel affinity beads, respectively were mixed with corresponding binding protein partners in ATPase buffer containing 2 mM ADP or ATP or ATPγS and incubated at 4°C for 2 hours and then additional 1 hour incubation at room temperature with 100 μM GSH or GSNO. The GST or His-Nickel beads were washed 4 times ATPase buffer with GSH or GSNO and samples resolved on 10% SDS-PAGE. The bound proteins were detected by Immunoblotting using corresponding antibodies.
GluA2 recycling assays
GluA2 surface expression recycling assay was carried out as previously described (Lin and Huganir, 2007; Zhang et al., 2011). Hippocampal neurons were cultured from postnatal day 0 mice obtained by mating heterozygote parents (Thorase KO mice). Mice were genotyped by PCR. Neurons from homozygous Thorase knock-out, homozygous knock-out nNOS and wild-type mice littermates were transfected with plasmids on day in vitro (DIV) 15 and imaging experiments were performed on DIV 18–20 using a Zeiss LSM 5 Duo confocal microscope as described previously (Lin and Huganir, 2007; Zhang et al., 2011). Briefly, pHluorin-GluA2 (GluA2-GFP) and mCherry were imaged with 488 nm and 560 nm excitation, respectively. Neurons were continuously perfused at a rate of 1 ml/min in recording buffer (25 mM HEPES, pH = 7.4, 120 mM NaCl, 5 mM KCl, 2.5 mM CaCl2, 1.5 mM MgCl2, 30 mM glucose, and 1 μM TTX). Prior to perfusion with NMDA, images were collected for 5 min (1 image/min) to establish a baseline (F0) in the recording buffer with or without MK801 or L-NAME. The NMDA solution contained 300 μM MgCl2, 10 μM glycine, and 20 μM NMDA. After 5 min perfusion with the NMDA solution, neurons were continuously perfused for 60 min with recording buffer to monitor fluorescence changes. Images were analyzed using NIH ImageJ software (Rasband, W.S., NIH, https://imagej.nih.gov/ij/, 1997–2007).
Nitrosylation mediated recycling of GluA2 surface expression in HEK293 cells
Real-Time Imaging of surface expression of pH-luorin-GluA2 (GluA2-GFP) in HEK293 cells was carried out as previous described by Huang et al. (Huang et al., 2005) with some modifications. To determine if whether treatment of GSNO affects intracellular pH, intracellular pH indicator, pHrodo™Red AM (Life Technologies, USA) was incubated in the HEK293 cells for 30min in Live-cell imaging buffer, Hank’s balanced salt solution (HBSS) without phenol red (Life technologies). Following dye loading, the cells were subjected to live cell imaging using. The fluorescence signals were monitored by time-lapse recording with perfusion at a rate of 1 ml/min in HBSS for 10 minute to establish a baseline followed by perfusion with 0.5 mM GSNO in HBSS for 4 minutes and then continuously perfused with HBSS for another 30 minutes.
For real-time imaging of surface expression of pHluorin-GluA2, HEK293 cells were plated on 35 mm dishes with a glass coverslip and co-transfected with pHluorin-GluA2 (GluA2-GFP), mCherry ires Thorase (wild-type or C137L), GRIP1 and NSF. Prior to imaging, culture medium was replaced with HBSS. Cells expressing pHluorin-GluA2 (GluA2-GFP) and mCherry were imaged with 488 nm and 560 nm excitation, respectively. The Fluorescence signals were monitored by time-lapse recording with an interval of 1 minute for 4 minutes using a Zeiss LSM 5 Duo confocal microscope. Cells were perfused at a rate of 1 ml/min in HBSS for 4 minutes to establish a baseline followed by perfusion with 0.5 mM GSNO in HBSS for 4 minutes and then continuously perfused with HBSS for another 16 minutes. Images were analyzed using NIH ImageJ software (Rasband, W.S., NIH, https://imagej.nih.gov/ij//, 1997–2007).
For Immunostaining of HEK293 cells co-epressing pHluorin-GluA2, Thorase (Ires mCherry), NSF and GRIP1, cells were treated with 0.5 mM GSH or GSNO in HBSS for 2 minutes and then fixed with 4% PFA in PBS (pH 7.5) plus 0.1 mM GSH or GSNO for 15 minutes. The cells were immunostained with mouse monoclonal anti-NSF and rabbit monoclonal anti-GRIP1. Stained NSF and GRIP1 were visualized by using anti-mouse Alexa 350-conjugated and anti-rabbit Alexa 647-conjugated secondary antibodies (Invitrogen), respectively. Imaging of cells was performed using a Zeiss LSM 5 Duo confocal microscope. All images were analyzed using NIH ImageJ software (Rasband, W.S., NIH, https://imagej.nih.gov/ij/, 1997–2007).
Electrophysiology
To express Thorase-C137L in mouse brain approximately 2 μL AAV2 GFP or Thorase-GFP wild-type, or C137L (1X1013 GC/ml, Vector BioLabs) were injected into both sides of intracerebroventricular of the newborn (postnatal day 1) Thorase wild-type of knockout mice as previously described (Umanah et al., 2017). The expressions of GFP or Thorase-GFP were checked by immunohistochemistry and after 14 days.
Mice expressing GFP or Thorase-GFP were sacrificed at P16–18 and brains were rapidly placed into cold artificial cerebrospinal fluid (ACSF) containing (in mM): NaCl 125, KCl 2.5, MgSO4 1.0, NaH2PO4 1.2, NaHCO3 26, CaCl2 2.0, and D-glucose 10. Coronal cortical slices (350 μm) were prepared and incubated in ACSF infused with 95% O2/5% CO2 for recovery at 32°C for 30 min. After another 30 min of equilibration at room temperature, slices were transferred into a submerged recording chamber and perfused continuously with 28–30°C oxygenated ACSF at a rate of 2 ml/min. All experiments were conducted in accordance with the National Institutes of Health guidelines for the care and use of animals, and all procedures were approved by Harvard Medical Area Standing Committee on Animals.
Extracellular recordings of field excitatory postsynaptic potentials (fEPSPs) were performed using HEKA EPC10 amplifier (HEKA Elektronik, Lambrech, Germany). Picrotoxin (100 μM) was routinely added to the perfusion solution to block GABAA receptor-mediated inhibitory synaptic responses. Presynaptic stimuli (0.1 Hz, 100 μs) were delivered at the border of layer I and layer II/III with a concentric electrode (FHC), and fEPSPs were obtained using a glass microelectrode filled with 1 M NaCl placed at Layer V. After 20 min of stable baseline recording, LTP was triggered using a theta-burst stimulation (TBS) protocol consisting of five trains of burst with four pulses at 100 Hz, at 200-ms interval, repeated four times at intervals of 10 s. For LTD studies, 1 Hz stimulation was applied for 15 min. The magnitude of LTP or LTD was calculated at 40 min after stimulation as a percentage of baseline responses (Umanah et al., 2017). Data were acquired by PatchMaster software (HEKA Elektronik, Lambrech, Germany), sampled at 10 kHz, filtered at 2.9 kHz, and analyzed using Clampfit 10.5 software (Molecular devices, Palo Alto, CA, USA).
QUANTIFICATION AND STATISTICAL ANALYSIS
Western blots were visualized using either the SuperSignal West ECL system from Pierce followed by film exposure or the LI-COR Odyssey infrared imaging system. The images were quantified using NIH-ImageJ software. Data are expressed as mean ± SEM from the indicated number of experiments. All experiments were repeated at least three times and quantitative data are presented as the mean ± standard error of the mean (SEM) performed by GraphPad prism6 software (Instat, GraphPad Software). Statistical significance was assessed by one-way or two-way ANOVA or Two-tailed paired t-Test. The significant differences were identified by post hoc analysis using the Holm-Sidak post hoc method for multiple comparisons. Assessments were considered significant with a p < 0.05 and non-significant with a p > 0.05. Power analysis and sample size calculation for all experiments were determined using G*Power 3.1 statistics software (Table S1).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal anti-Thorase | NeuroMab | Cat#75-157; RRID:AB_2290002 |
| Extracellular anti-GluA2 | NeuroMab | Cat# 73-002; RRID:AB_10674575 |
| Intracellular anti-GluA2 | Abcam | Cat# 3520-1 RRID:AB_10703317 |
| anti phoshpo-S880 GluA2 | Abcam | Cat# ab52180; RRID:AB_880227 |
| Monoclonal mouse anti-NSF | Abcam | Cat# ab16681; RRID:AB_2155806 |
| Rabbit anti-PICK1 | Abcam | Cat# 3367-1; RRID:AB_10645748 |
| Rabbit anti-GRIP1 | Abcam | Cat# ab44970; RRID:AB_732721 |
| Rabbit anti-Biotin | Abcam | Cat# ab79111; RRID:AB_1603712 |
| Goat polyclonal Biotin antibody conjugated to HRP | Abcam | Abcam Cat# ab53468 RRID:AB_867864) |
| Anti-rabbit IgG-HRP conjugate secondary | Abcam | Cat# ab98467; RRID:AB_10674445 |
| Anti-mouse IgG-HRP conjugate secondary | Abcam | Cat# ab6823 RRID:AB_955395 |
| Mouse anti-β-actin-HRP | Sigma | Cat#A3854; RRID: AB_262011 |
| Donkey Anti-Rabbit IgG, Whole Ab ECL Antibody, HRP Conjugated | GE Healthcare | Cat#NA934; RRID: AB_772206 |
| Sheep Anti-Mouse IgG, Whole Ab ECL Antibody, HRP Conjugated | GE Healthcare | Cat#NA931; RRID: AB_772210 |
| Donkey Anti-Rabbit Alexa Fluor® Plus 647 | ThermoFisher (Invitrogen) | Cat# A32795TR, RRID:AB_2866496 |
| Donkey Anti-mouse Alexa Fluor® Plus 350 | ThermoFisher (Invitrogen) | Cat# A10035, RRID:AB_2534011 |
| Chemicals and Assay kits | ||
| Glutathione (GSH,) | Millipore-Sigma | Cat#PHR1359 |
| S-nitrosoglutathione (GSNO,), | Millipore-Sigma | Cat#N4148 |
| Nω-Nitro-L-arginine methyl ester (L-NAME,) | Millipore-Sigma | Cat#N5751 |
| Dizocilpine (MK-801,) | Millipore-Sigma | Cat#M107 |
| Glutaraldehyde solution | Millipore-Sigma | Cat# G7651 |
| N-Methyl-D-aspartic acid (NMDA) | Millipore-Sigma | Cat# M3262 |
| Adenosine 5′-triphosphate (ATP) disodium salt hydrate | Millipore-Sigma | Cat#A2383 |
| Adenosine 5′-diphosphate (ADP) sodium salt | Millipore-Sigma | Cat#A2754 |
| Adenosine 5′-[γ-thio]triphosphate (ATPgS) tetralithium salt | Millipore-Sigma | Cat#A1388 |
| Neocuproine | Millipore-Sigma | Cat#N1501 |
| EZ-Link HPDP-Biotin | ThermoFisher Scientific | Cat# 21341 |
| 1X PBS pH 7.4 | Quality Biologicals | Cat#114-058-101 |
| 10X TBS pH 7.4 | Quality Biologicals | Cat#351-086-101 |
| Phosphatase inhibitor cocktail 2 | Millipore-Sigma | Cat#P5726 |
| Phosphatase inhibitor cocktail 3 | Millipore-Sigma | Cat#P0044 |
| Paraformaldehyde (PFA) reagent grade | Millipore-Sigma | Cat#P6148 |
| Normal donkey serum | Jackson ImmunoResearch | Cat#017-000-121 |
| pHrodo Red AM Intracellular pH Indicator | ThermoFisher Scientific | Cat# P35372 |
| ADP Colorimetric Assay Kit | ThermoFisher Scientific | Cat#MAK081 |
| PIERCE BCA PROTEIN ASSAY | ThermoFisher Scientific | Cat#23227 |
| DreamTaq Green PCR Master Mix (2X) | ThermoFisher Scientific | Cat#K1082 |
| Cell culture reagents | ||
| DMEM - Dulbecco’s Modified Eagle Medium | ThermoFisher Scientific | Cat#11965118 |
| DMEM, no phenol red | ThermoFisher Scientific | Cat#31053028 |
| FBS - fetal bovine serum | ThermoFisher Scientific | Cat#16000044 |
| Penicillin-Streptomycin | ThermoFisher Scientific | Cat#15140122 |
| Neurobasal Medium | ThermoFisher Scientific | Cat#21103049 |
| B-27 Plus Supplement (50X) | ThermoFisher Scientific | Cat#A3582801 |
| Neurobasal Medium, minus phenol red | ThermoFisher Scientific | Cat#12348017 |
| CTS TrypLE Select Enzyme | ThermoFisher Scientific | Cat#A1285901 |
| Lipofectamine 2000 Reagent | ThermoFisher Scientific | Cat#11668019 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6;129X1-ATAD11+/− |
Zhang et al., 2011. Umanah et al., 2017 |
N/A |
| Mouse: C57BL/6J | Jackson Laboratory | JAX:000664 |
| Oligonucleotides | ||
| Thorase C137S: 5′primer: CCGGGCTCTGGGAAAACATTAATTGCCAAAGC | Integrated DNA Technologies, Inc. (IDT) | Oligo synthesis |
| Thorase C137S: 3′ primer: GTTTTCCCAGAGCCCGGAGGTCCATACAGGAG | IDT | Oligo synthesis |
| Thorase C310S: 5′ primer: CTCCTCTCTGTCAGGGAATATGTCAATTCTAC | IDT | Oligo synthesis |
| Thorase C310S: 3′primer: CCCTGACAGAGAGGAGGGCAGCATCTCGAC | IDT | Oligo synthesis |
| Thorase C137A: 5′ primer: CCGGGCGCTGGGAAAACATTAATTGCCAAAGC | IDT | Oligo synthesis |
| Thorase C137A: 3′primer: GTTTTCCCAGCGCCCGGAGGTCCATACAGGAG | IDT | Oligo synthesis |
| Thorase C137L: 5′ primer: CCGGGCCTCGGGAAAACATTAATTGCCAAAGC | IDT | Oligo synthesis |
| Thorase C137L: 3′primer: GTTTTCCCGAGGCCCGGAGGTCCATACAGGAG | IDT | Oligo synthesis |
| Thorase C137V: 5′ primer: CCGGGCGTCGGGAAAACATTAATTGCCAAAGC | IDT | Oligo synthesis |
| Thorase C137V: 3′primer: GTTTTCCCGACGCCCGGAGGTCCATACAGGAG | IDT | Oligo synthesis |
| Thorase C137N: 5′ primer: CCGGGCAACGGGAAAACATTAATTGCCAAAGC | IDT | Oligo synthesis |
| Thorase C137N: 3′primer: GTTTTCCCGTTGCCCGGAGGTCCATACAGGAG | IDT | Oligo synthesis |
| Recombinant Plasmid DNA | ||
| AAV-CAG-GFP | Addgene | Cat#28014 |
| pET32a | EMD Biosciences | Cat#69015-3 |
| pGEX6P1 | Amersham | Cat#27-4597-01 |
| pThorase-AAV-GFP | This Manuscript | N/A |
| pThorase- His6 | This Manuscript | N/A |
| pThorase-pLVX-IRES-mCherry |
Zhang et al., 2011. Umanah et al., 2017 |
N/A |
| pThorase-Lenti-GFP |
Zhang et al., 2011. Umanah et al., 2017 |
N/A |
| pCDNA-GRIP1 |
Zhang et al., 2011. Umanah et al., 2017 |
N/A |
| pHluorin-GluA2 |
Zhang et al., 2011. Umanah et al., 2017 |
N/A |
| pCDNA-NSF | Huang et al., 2005 | N/A |
| pET-NSF- His6 | Huang et al., 2005 | N/A |
| Deposited Data | ||
| Raw data used for this study | This Manuscript | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Prism 6 | GraphPad software | https://www.graphpad.com/scientific-software/prism/ |
| ZEN lite | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| PyMOL 3D structure viewer | Schrödinger | http://pymolwiki.org |
| Other | ||
| RIPA Lysis and Extraction Buffer | Thermo Scientific | Cat#89901 |
| E. coli BL21(DE3) RIL cells | Agilent Technologies | Cat#230240 |
| 2X Lammli Sample Buffer | Bio-Rad | Cat#1610737 |
| Novex WedgeWell 8-16% Tris-Glycine Mini Gels | Thermo Scientific | Cat#XP08165BOX |
| SUPERSIGNAL WEST PICO PLUS | Thermo Scientific | Cat#34580 |
| Blotting-Grade Blocker, nonfat dry milk | Bio-Rad | Cat#1706404 |
| Restore western blot stripping buffer | Thermo Scientific | Cat#21059 |
| Nitrocellulose Membrane | Bio-Rad | Cat#1620115 |
| X-ray film | RPI | Cat#248300 |
| Hamilton® GASTIGHT® syringe, 1700 series | Sigma | Cat#20972 |
| Pierce High Capacity Endotoxin Removal Spin Column | Thermo Scientific | Cat#88276 |
| Superdex 200 Increase 10/300 G | GE Healthcare | Cat#45-002-570 |
| Ni Sepharose 6 Fast Flow | GE Healthcare | Cat#17-5318-06 |
Highlights.
The activation of NMDARs leads to increased S-nitrosylation of Thorase and NSF
S-nitrosylation of Thorase inhibits ATPase activity and enhances AMPAR internalization
S-nitrosylated Thorase transnitrosylates NSF, thereby modulating AMPAR trafficking
S-nitrosylation of Thorase is required for long-term potentiation and depression
ACKNOWLEDGMENTS
The authors thank Noelle Burgess for creating and assisting with illustrations. This work was supported by grants from the NIH/NIDA DA000266 and DA044123 to T.M.D. and V.L.D. and NIH/NINDS NS099362 to G.K.E.U. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108329.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahrens-Nicklas RC, Umanah GK, Sondheimer N, Deardorff MA, Wilkens AB, Conlin LK, Santani AB, Nesbitt A, Juulsola J, Ma E, et al. (2017). Precision therapy for a new disorder of AMPA receptor recycling due to mutations in ATAD1. Neurol. Genet 3, e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt KL, Zhang Y, Jurado S, Malenka RC, Südhof TC, and Chen L (2015). Retinoic Acid and LTP Recruit Postsynaptic AMPA Receptors Using Distinct SNARE-Dependent Mechanisms. Neuron 86, 442–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, and Schwede T (2006). The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201. [DOI] [PubMed] [Google Scholar]
- Asrar S, and Jia Z (2013). Molecular mechanisms coordinating functional and morphological plasticity at the synapse: role of GluA2/N-cadherin interaction-mediated actin signaling in mGluR-dependent LTD. Cell. Signal 25, 397–402. [DOI] [PubMed] [Google Scholar]
- Babst M, Wendland B, Estepa EJ, and Emr SD (1998). The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J. 17, 2982–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani S, Valnegri P, Beretta F, and Passafaro M (2009). The GLUR2 subunit of AMPA receptors: synaptic role. Neuroscience 158, 55–61. [DOI] [PubMed] [Google Scholar]
- Beneyto M, and Meador-Woodruff JH (2006). Lamina-specific abnormalities of AMPA receptor trafficking and signaling molecule transcripts in the prefrontal cortex in schizophrenia. Synapse 60, 585–598. [DOI] [PubMed] [Google Scholar]
- Beretta F, Sala C, Saglietti L, Hirling H, Sheng M, and Passafaro M (2005). NSF interaction is important for direct insertion of GluR2 at synaptic sites. Mol. Cell. Neurosci 28, 650–660. [DOI] [PubMed] [Google Scholar]
- Chater TE, and Goda Y (2014). The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front. Cell. Neurosci 8, 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YB, Tenneti L, Le DA, Ortiz J, Bai G, Chen HS, and Lipton SA (2000). Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nat. Neurosci 3, 15–21. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Xia J, Scannevin RH, Zhang X, and Huganir RL (2000). Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J. Neurosci 20, 7258–7267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, and Wolf ME (2008). Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454, 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti C, Xuereb JH, Crepaldi L, Corsi M, Michielin F, and Ferraguti F (2011). Altered levels of glutamatergic receptors and Na+/K+ ATPase-α1 in the prefrontal cortex of subjects with schizophrenia. Schizophr. Res 128, 7–14. [DOI] [PubMed] [Google Scholar]
- Dai C, Liang D, Li H, Sasaki M, Dawson TM, and Dawson VL (2010). Functional identification of neuroprotective molecules. PLOS ONE 5, e15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, O’Brien RJ, Fung ET, Lanahan AA, Worley PF, and Huganir RL (1997). GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature 386, 279–284. [DOI] [PubMed] [Google Scholar]
- Doulias PT, Greene JL, Greco TM, Tenopoulou M, Seeholzer SH, Dunbrack RL, and Ischiropoulos H (2010). Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc. Natl. Acad. Sci. USA 107, 16958–16963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulias PT, Tenopoulou M, Raju K, Spruce LA, Seeholzer SH, and Ischiropoulos H (2013). Site specific identification of endogenous S-nitrosocysteine proteomes. J. Proteomics 92, 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley JG, Khatri L, Hanson PI, and Ziff EB (2002). NSF ATPase and alpha-/beta-SNAPs disassemble the AMPA receptor-PICK1 complex. Neuron 34, 53–67. [DOI] [PubMed] [Google Scholar]
- Huang Y, Man HY, Sekine-Aizawa Y, Han Y, Juluri K, Luo H, Cheah J, Lowenstein C, Huganir RL, and Snyder SH (2005). S-nitrosylation of N-ethylmaleimide sensitive factor mediates surface expression of AMPA receptors. Neuron 46, 533–540. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, and Snyder SH (2001). The biotin switch method for the detection of S-nitrosylated proteins. Sci. STKE 2001, pl1. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, and Snyder SH (2001). Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell Biol 3, 193–197. [DOI] [PubMed] [Google Scholar]
- Katano T, Furue H, Okuda-Ashitaka E, Tagaya M, Watanabe M, Yoshimura M, and Ito S (2008). N-ethylmaleimide-sensitive fusion protein (NSF) is involved in central sensitization in the spinal cord through GluR2 subunit composition switch after inflammation. Eur. J. Neurosci 27, 3161–3170. [DOI] [PubMed] [Google Scholar]
- Kedzierska S (2006). [Structure, function and mechanisms of action of ATPases from the AAA superfamily of proteins]. Postepy Biochem. 52, 330–338. [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJ (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Chung HJ, Lee HK, and Huganir RL (2001). Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc. Natl. Acad. Sci. USA 98, 11725–11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YH, Zhang N, Wang YN, Shen Y, and Wang Y (2016). Multiple faces of protein interacting with C kinase 1 (PICK1): structure, function, and diseases. Neurochem. Int 98, 115–121. [DOI] [PubMed] [Google Scholar]
- Lin DT, and Huganir RL (2007). PICK1 and phosphorylation of the glutamate receptor 2 (GluR2) AMPA receptor subunit regulates GluR2 recycling after NMDA receptor-induced internalization. J. Neurosci 27, 13903–13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menniti FS, Lindsley CW, Conn PJ, Pandit J, Zagouras P, and Volkmann RA (2013). Allosteric modulators for the treatment of schizophrenia: targeting glutamatergic networks. Curr. Top. Med. Chem 13, 26–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogk A, Schlieker C, Strub C, Rist W, Weibezahn J, and Bukau B (2003). Roles of individual domains and conserved motifs of the AAA+ chaperone ClpB in oligomerization, ATP hydrolysis, and chaperone activity. J. Biol. Chem 278, 17615–17624. [DOI] [PubMed] [Google Scholar]
- Piard J, Umanah GKE, Harms FL, Abalde-Atristain L, Amram D, Chang M, Chen R, Alawi M, Salpietro V, Rees MI, et al. (2018). A homozygous ATAD1 mutation impairs postsynaptic AMPA receptor trafficking and causes a lethal encephalopathy. Brain 141, 651–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignatelli M, Umanah GKE, Ribeiro SP, Chen R, Karuppagounder SS, Yau HJ, Eacker S, Dawson VL, Dawson TM, and Bonci A (2017). Synaptic Plasticity onto Dopamine Neurons Shapes Fear Learning. Neuron 93, 425–440. [DOI] [PubMed] [Google Scholar]
- Pøhlsgaard J, Frydenvang K, Madsen U, and Kastrup JS (2011). Lessons from more than 80 structures of the GluA2 ligand-binding domain in complex with agonists, antagonists and allosteric modulators. Neuropharmacology 60, 135–150. [DOI] [PubMed] [Google Scholar]
- Prendergast J, Umanah GK, Yoo SW, Lagerlöf O, Motari MG, Cole RN, Huganir RL, Dawson TM, Dawson VL, and Schnaar RL (2014). Ganglioside regulation of AMPA receptor trafficking. J. Neurosci 34, 13246–13258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu ZW, Miao WY, Hu SQ, Li C, Zhuo XL, Zong YY, Wu YP, and Zhang GY (2012). N-methyl-D-aspartate receptor-dependent denitrosylation of neuronal nitric oxide synthase increase the enzyme activity. PLOS ONE 7, e52788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer RT, and Baker TA (2011). AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem 80, 587–612. [DOI] [PubMed] [Google Scholar]
- Steinberg JP, Huganir RL, and Linden DJ (2004). N-ethylmaleimide-sensitive factor is required for the synaptic incorporation and removal of AMPA receptors during cerebellar long-term depression. Proc. Natl. Acad. Sci. USA 101, 18212–18216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HL, Queenan BN, and Huganir RL (2015). GRIP1 is required for homeostatic regulation of AMPAR trafficking. Proc. Natl. Acad. Sci. USA 112, 10026–10031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umanah GKE, Pignatelli M, Yin X, Chen R, Crawford J, Neifert S, Scarffe L, Behensky AA, Guiberson N, Chang M, et al. (2017). Thorase variants are associated with defects in glutamatergic neurotransmission that can be rescued by Perampanel. Sci. Transl. Med 9, eaah4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wang Y, Chi Z, Keuss MJ, Pai YM, Kang HC, Shin JH, Bugayenko A, Wang H, Xiong Y, et al. (2011). The AAA+ ATPase Thorase regulates AMPA receptor-dependent synaptic plasticity and behavior. Cell 145, 284–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorumski CF, and Izumi Y (1998). Modulation of LTP induction by NMDA receptor activation and nitric oxide release. Prog. Brain Res 118, 173–182. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets is available to readers and there are no restrictions on any data or materials presented in this paper.