

Abstract
Protein localization is paramount to protein function, and the intracellular movement of proteins underlies the regulation of numerous cellular processes. Given advances in spatial proteomics, the investigation of protein localization at a global scale has become attainable. Also becoming apparent is the need for dedicated analytical frameworks that allow the discovery of global intracellular protein movement events. Here, we describe TRANSPIRE, a computational pipeline that facilitates TRanslocation ANalysis of SPatIal pRotEomics data sets. TRANSPIRE leverages synthetic translocation profiles generated from organelle marker proteins to train a probabilistic Gaussian process classifier that predicts changes in protein distribution. This output is then integrated with information regarding co-translocating proteins and complexes and enriched gene ontology associations to discern the putative regulation and function of movement. We validate TRANSPIRE performance for predicting nuclear-cytoplasmic shuttling events. Analyzing an existing data set of nuclear and cytoplasmic proteomes during Kaposi Sarcoma-associated herpesvirus (KSHV)-induced cellular mRNA decay, we confirm that TRANSPIRE readily discerns expected translocations of RNA binding proteins. We next investigate protein translocations during infection with human cytomegalovirus (HCMV), a β-herpesvirus known to induce global organelle remodeling. We find that HCMV infection induces broad changes in protein localization, with over 800 proteins predicted to translocate during virus replication. Evident are protein movements related to HCMV modulation of host defense, metabolism, cellular trafficking, and Wnt signaling. For example, the low-density lipoprotein receptor (LDLR) translocates to the lysosome early in infection in conjunction with its degradation, which we validate by targeted mass spectrometry. Using microscopy, we also validate the translocation of the multifunctional kinase DAPK3, a movement that may contribute to HCMV activation of Wnt signaling.
Keywords: protein translocation, spatial proteomics, subcellular organelles, machine learning, viral infection
Graphical Abstract
INTRODUCTION
The movement of proteins between organelles lies at the core of essential cellular processes, such as gene expression,1,2 immune signaling,3,4 and apoptosis.5,6 As obligate intracellular parasites, viruses must co-opt these pathways, and consequently, viral infections induce diverse changes in protein localizations that are essential to all phases of the viral lifecycle; from entry, replication, and assembly to egress.7 Of these protein movements, nucleo-cytoplasmic shuttling events are perhaps the most well-characterized and are critical for the ability of viruses to evade immune surveillance8 and to regulate gene expression.9,10 However, it is evident that infection-induced translocations extend to a number of organelles. For example, despite their diverse lifecycles and structures, numerous viruses target the mitochondrial antiviral signaling protein (MAVS) via translocation of viral proteins to the mitochondria to inhibit antiviral signaling and apoptosis.11,12 For human cytomegalovirus (HCMV) and Influenza A this is accomplished via viral protein translocations from the ER and cytoplasm, respectively.13,14 Other proteins frequently observed to undergo virus-induced movements include cell surface-localized immune signaling factors (such as HCMV-targeting of MICA8) or cellular transcription factors.4,15 However, despite individual studies that have elucidated subsets of protein movements during viral infections, our knowledge of the global regulation and functions of protein movements, and their interplay with one another, remains limited.
Fractionation-based spatial proteomics (reviewed in refs 16–18), which combines organelle density fractionation with multiplexed, high-throughput quantitative mass spectrometry19 (MS) and machine learning (ML), has provided the means to investigate protein localization on a proteome-wide scale.20–27 Given the computational complexity of the data analysis process, the continued advancement of quantitative MS and computational and bioinformatics pipelines28,29 has been a critical component of spatial proteomics (reviewed in refs 19 and 30–32). Similarly, the array of ML classifiers for predicting protein localization have been substantial contributors to the growth of this field of research. Among these approaches are support vector machines (SVMs),26,33 neural networks,34,35 K nearest neighbors,36 random forests,37 naïve Bayes,38 partial least-squares discriminate analysis,39 Bayesian mixture models,40 and others (reviewed in16)—each of which require relatively extensive computational implementation. As such, the development of platforms such as Perseus41 and pRoloc42 have helped to provide frameworks for implementation of these classifiers, making this complex data analysis procedure accessible to a broader audience.
Although designed primarily with the goal of assigning proteins to different subcellular compartments, spatial proteomics and its associated tools can also help to predict translocation events when a protein is assigned to different organelles in different conditions (e.g., uninfected vs infected cells). Several studies have taken this concept a step further, developing and implementing a translocation scoring system based on the magnitude and reproducibility of changes in protein spatial profiles between states.25,43 One of the main challenges for the field, however, is predicting the localizations of proteins that reside in multiple compartments,40 and this is estimated to apply to ∼60% of the proteome.16 In the case of protein movement, multiple localization becomes an even more prominent issue given that alterations to protein distribution often occur in a continuous rather than binary manner. These concepts are frequently central to viral infection, where relatively small viral genomes have evolved to produce multifunctional proteins that can be dynamically distributed throughout the cell.
To address the need to broadly understand protein movement during viral infections, here we describe TRANSPIRE, a computational pipeline for TRanslocation ANalysis of SPatIal pRotEomics data. TRANSPIRE provides probabilistic translocation predictions from spatial proteomics data sets and is applicable to diverse biological studies. This pipeline is based on the prediction that by simultaneously analyzing the spatial profiles of a given protein between different states, rather than its static distribution in either state, information about protein movement can be extracted in a manner that is relatively agnostic to whether the protein resides in multiple compartments. In addition to existing translocation scoring methods, TRANSPIRE considers that changes in protein localization may not, necessarily, lead to drastic changes in spatial profiles. For example, even near complete translocation of a protein between organelles with similar spatial profiles (e.g., ER and Golgi) will not exhibit a high magnitude of change in absolute terms. TRANSPIRE, instead, leverages a machine learning classifier to learn how protein translocations are manifested—even if the difference in their profiles may be subtle.
As a proof of concept, we first applied TRANSPIRE to study nuclear-cytoplasmic protein shuttling. We used a data set that investigated cellular mRNA decay-driven shuttling events that were accelerated by a viral endonuclease encoded by Kaposi Sarcoma-associated herpesvirus (KSHV). Our pipeline readily recapitulated the results reported in the original study, capturing broad shuttling of RNA binding proteins. We next applied TRANSPIRE to reveal protein movements during the progression of HCMV infection, a nuclear-replicating β-herpesvirus. Among human pathogens, HCMV induces some of the most prominent remodeling of cellular organelles,34,44 and this is accompanied by numerousestablished protein translocations.34,45–47 We show that our pipeline does not only predict expected (i.e., already reported) protein movements and their temporality during HCMV infection but also uncovers previously unknown translocating proteins. Additionally, the ability of this pipeline to also highlight co-translocating proteins and complexes further aids in understanding the possible function and regulation of these movements. Overall, TRANSPIRE detects global shifts in protein distributions during infection that appear to underlie processes that are critical for HCMV replication. Among these, we discover and further validate by confocal microscopy the translocation of the death-associated protein kinase 3 (DAPK3) from the plasma membrane to the cytoplasm and the nucleus. Considering the functional associations of DAPK3, we propose a role for this kinase in regulating Wnt signaling during infection.
MATERIALS AND METHODS
Cell Culture and Virus Infection.
MRC5 human fibroblasts (ATCC CCL-171) were cultured in complete growth medium (DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin antibiotics) at 37 °C and 5% CO2. The cells were used for the experiments within a maximum of 10 passages. Viral stocks of HCMV strain AD169 were produced from bacterial artificial chromosomes in MRC5 cells, as in Yu et al.48 Virus stocks were stored at −80 °C for no longer than 6 months. Infections for quantitative mass spectrometry and immunofluorescence microscopy experiments were performed at a multiplicity of infection (MOI) of 3.
Sample preparation for MS analysis.
Following collection, cell pellets were washed twice in PBS, pelleted by centrifugation, and stored at −80 °C until ready for analysis. Pellets were lysed in lysis buffer (5% SDS, 50 mM TrisHCl, 0.1 M NaCl, 0.5 mM EDTA, pH8.0) then reduced and alkylated at 70 °C for 20 min using 25 mM TCEP (Thermo Fisher no. 77720) and 50 mM 2-chloroacetamide (MP Biomedicals no ICN15495580). Following reduction and alkylation, proteins were extracted by methanol–chloroform precipitation49 and resuspended in 25 mM HEPES buffer (pH 8.2). Proteins were digested for 16 h at 37 °C using a 1:50 ratio of trypsin to protein (w/w) and then adjusted to 1% trifluoroacetic acid (TFA). Following desalting using the StageTip method50 with C18 material (3 M no. 2215), peptides were washed with 0.5% FA, eluted with 70% acetonitrile (ACN) and 0.5% formic acid (FA), dried via SpeedVac (ThermoFisher), and resuspended in 1% FA and 1% ACN.
Parallel Reaction Monitoring (PRM) Analysis and Quantification.
Samples prepared for PRM analysis were analyzed via LC–MS/MS using a Dionex Ultimate 3000 nanoRSLC coupled to a Q Exactive HF mass spectrometer (Thermo Fisher Scientific), as in ref 51. Peptides were separated by reversed-phase chromatography on a C18 column using a linear gradient (0–35% B) over 60 min. Targeted MS2 scans were performed with the following parameters: resolution of 120000, AGC target of 5 × 105, maximum inject time of 100 ms, isolation window of 0.8, and retention time windows of 5 min. Design and quantification of PRM assays were performed using the Skyline software52 and peptide abundances were calculated using the summed area under the curve for three transition ions per peptide.
Data Processing for TRANSPIRE Analysis.
To demonstrate the utility of TRANSPIRE and simultaneously investigate protein movement during viral infection, we leveraged two data sets—one reported in Gilbertson et al.53 (deposited in ProteomeXchange, PXD009487) and one previously generated in our lab and reported in Jean Beltran et al.34 (PXD003925). To account for changes in protein signal that may occur solely due to changes in protein abundance, protein profiles were normalized such that the sum of values across organelle fractions was equal to 1. In each case, a set of cell-type specific organelle marker proteins were curated from existing literature (i.e., previously reported markers for HEK293T54 and primary human fibroblast cells;34 see Tables S1 and S2). In the case of Gilbertson et al., markers for an array of subcellular components were assigned either a nuclear or cytoplasmic label in accordance with the fractionation scheme of the data set.
To produce synthetic translocation profiles for classifier training, all possible pairwise combinations of organelle marker proteins were generated for combinations of different experimental conditions. For each resulting combination, the spatial profiles for marker proteins of “organelle X” were concatenated with markers for “organelle Y” and the resulting profile was labeled as an “X to Y” translocation. For example, a lysosome to plasma membrane translocation would consist of spatial profiles for a lysosome marker in the uninfected state and a plasma membrane marker in the infected state. Note that this procedure also produces synthetic profiles for combinations of markers for the same organelle, thus allowing TRANSPIRE to distinguish translocating from nontranslocating proteins.
Model Selection and Cross-Validation.
Given the extensive number of samples that the above procedure generates, we required a scalable classification algorithm. We therefore implemented a stochastic variational Gaussian process (SVGP) classifier using the GPFlow package55,56 (which is built upon the TensorFlow ML platform57 in Python). In brief, this model consists of user-defined components that include training data, a kernel function, a likelihood function, n latent variables (traditionally, this number is set equal to the total number of classes), and a subset of the training data to be used as inducing points. Considering the multiclass nature of this classification problem, our choice of likelihood function was limited to two options (robust-max or soft-max).55,58,59 We chose to employ the soft-max likelihood function, as the robust-max can tend to lead to poor confidence calibration.60 We do note that the GPFlow implementation of the soft-max likelihood function can only provide a stochastic estimate of the variational expectations. Consequently, there is some variance in model predictions; however, overall, we found this variance to be quite small (i.e., generally less than 5–10%; see Figure S2A,B).
To address hyperparameter choices regarding kernel type and the number of inducing points, models were built and trained using combinations of five different stationary kernels (squared exponential, rational quadratic, exponential, Matern32, and Matern52) implemented by the GPFlow architecture and an array of numbers of inducing points ranging from 1 to 500 depending on the data set. To evaluate these possible model architectures, the synthetic translocation training data was split into training (50%), validation (25%), and testing (25%) partitions. Throughout the model selection process training data was further stratified into five balanced folds. Each class label was permitted to have at-most three times more samples than the smallest class to help prevent prediction bias due to class imbalance. For each combination of kernel and number of inducing points (n), an SVGP model was built using the training data, the given kernel and likelihood functions, and a set of n inducing points that were determined by KMeans clustering analysis (using Scikit-learn61) of the training data (i.e., n clusters with n cluster centroids leveraged as inducing points). Each kernel was additionally supplemented with a white noise kernel to help prevent overfitting. Internally, the model variational parameters and kernel hyperparameters (i.e., length-scale and variance) were optimized via maximization of the evidence lower bound (ELBO) using the Adam optimizer62 implemented in GPFlow. Externally, the validation set was then used to determine which kernel type and number of inducing points produced the best-performing model. Following determination of kernel type and number of inducing points, both the training and validation sets were combined for final model fitting, and performance was evaluated on the held-out test partition of the synthetic translocation data.
TRANSPIRE Prediction of Protein Translocations.
Following model selection and hyperparameter optimization, the resulting model was used to predict translocations from the actual data set. This consequently yields classifier scores for each sample that range between 0 and 1 for each class of movements (or lack thereof) defined by the synthetic translocation training set. We note that the sum of scores across all classes for each sample is equal to 1, and predictions of class assignment for each sample are then assigned to whichever class has the highest score. We additionally defined an overall translocation score as the summed value of predicted scores for all true translocation classes (eq 1). This allows us to account for scenarios where relatively high classifier scores are split among several translocation classes. Such is the case when multiple classes of translocations have overlapping profiles (e.g., plasma membrane to ER and plasma membrane to Golgi translocations)
| (1) |
where n corresponds to the number of translocation classes and xi corresponds to the given classifier score for the ith tranlocation class.
To facilitate comparison of the agreement between TRANSPIRE predictions and localization assignments reported in Jean Beltran et al., we mapped the original organelle assignment scores (generated by a neural network) to a putative “translocation score” using the following criteria: For proteins assigned to different organelles in different conditions, their score was defined as in eq 2, while translocation scores for proteins assigned to the same compartment in both conditions were defined as in eq 3. In accordance with translocation scores generated by TRANSPIRE, this procedure yields scores that are mapped between 0 and 1. Like for translocation scores generated by TRANSPIRE, a score of 0 therefore represents a prediction of no translocation while 1 represents a translocation prediction. Scores closer to 0.5 represent uncertain predictions
| (2) |
where is the score for organelle assignment i at condition a and is the score for organelle assignment j at condition b
| (3) |
where and are the scores for organelle assignment i at conditions a and b, respecitvely.
Protein Co-translocation Analysis and Scoring.
The extent of similarity between translocation profiles was determined via computation of the Mahalanobis distance metric (eq 4) using the SciPy Python package.63 As this calculation requires calculation of the inverse covariance matrix, we leveraged the minimum covariance determinant method (described in64 and implemented in Scikit-learn61) to determine these values. As a true positive metric, the resulting pairwise distances were compared to distances between profiles of proteins of known protein complexes from the CORUM database.65 To serve as a true negative metric, we additionally compared against distances between marker protein profiles in the synthetic training set for different organelles. Using these two populations, we defined a false-positive rate distance cutoff of 0.05 and proteins with pairwise distances smaller than this cutoff were considered co-translocating
| (4) |
defines the distance between proteins p and q with translocation profiles u and v, respectively, and V is the covariance matrix associated with u and v.
Database Integration and Gene ontology (GO) Enrichment Analysis.
Confident translocations were automatically searched in STRING66 using a custom Python REST API query function (refer to code availability). To be reported in the supporting tables, known interactions were required to have a STRING confidence score of at least 0.4. GO enrichment analysis was performed using the GOATOOLS python package.67 For analysis of the HCMV data set, a custom list of proteins experimentally determined to be expressed in fibroblast cells was used as the background gene list (Table S3). For enrichment analysis, a false discovery rate (FDR)-based resampling approach was used to determine significantly enriched GO terms with an FDR less than 0.05.68
Code Availability and Software Requirements.
The TRANSPIRE Python package can be cloned and/or downloaded directly from GitHub (github.com/cristealab/TRANSPIRE_JASMS2020). Package documentation is available online using Read the Docs (https://transpire.readthedocs.io/en/latest). For the purpose of practicality, we note that the capacities of a standard workstation computer was sufficient for model training and prediction; however, we do caution that model fitting and analysis can be a CPU and RAM-intensive task depending on data set size.
Immunostaining and Immunofluorescence Microscopy.
MRC5 human fibroblasts grown on microscopy coverslips were fixed in 4% paraformaldehyde at room temperature for 15 min, washed with PBS, and permeabilized in 0.2% (v/v) tween in PBS (PBST) for 5 min at room temperature. Blocking was performed in 10% (v/v) goat serum, 5% human serum (Sigma), and 300 mM glycine in PBST for 30 min at room temperature.
Incubation with primary antibody was performed for 2 h at room temperature. The antibody for ZIP kinase immunostain was rabbit monoclonal anti-ZIP kinase (Abcam; ab210528) diluted 1:1000 in blocking solution. The antibody for HCMV protein IE1 immunostain was mouse monoclonal anti-IE1 (clone 1B12 gift from Tom Shenk69) diluted 1:50 in blocking solution. Following primary antibody incubation, samples were washed three times in PBST for 5 min. Secondary incubation was performed with goat secondary antibody conjugated to Alexa Fluor (488 or 568; ThermoFisher Scientific) diluted 1:2000 in blocking solution for 1 h at room temperature. During secondary antibody incubation, the nucleus was stained using 1 μg/mL DAPI (Thermo Fisher; 62248).
Following secondary staining, coverslips were washed three times in PBST, twice in PBS, and once in TBS. Coverslips were then mounted onto slides using 12 μL ProLong Diamond Antifade Mountant (ThermoFisher Scientific; P36970). Confocal images were acquired using an inverted fluorescence confocal microscope (Nikon Ti-E) equipped with a Yokogawa spinning disc (CSU-21), digital CMOS camera (Hamamatsu ORCA-Flash TuCam), and precision microscope stage (Piezo). Z stacks were acquired with 0.2 μm steps throughout the cell depth using a Nikon 100X Plan Apo objective and both Z stacks and maximum projections from each channel were exported as tiff files for quantitative analysis and publication.
Quantitative Image Analysis.
To assess the average localization of DAPK3, line scans were manually acquired in ImageJ70,71 across the middle slice of each cell using a wide (25-pixel), straight line to account for local variation in DAPK3 distribution. The resulting mean gray values for each channel along the given line in each channel were then exported for further analysis. To control for changes in line scan length due to differences in cell size and/or orientation, we normalized each line to its own length (see eq 5) and binned the resulting values into 50 equally sized bins. To make the profiles across different channels comparable, we additionally normalized the profiles of each cell according to eq 6 on a per-channel basis
| (5) |
where x is the set of pixel values for a given line scan and xmax is the length of the line in pixels
| (6) |
where y is the set of mean grey values for a given channel of a given cell, is the arithmetic mean of y, and σ is the standard deviation of y.
RESULTS AND DISCUSSION
Developing TRANSPIRE to Detect and Analyze Protein Translocations.
In general, spatial proteomics studies (Figure 1A) involve separation of organelles into different fractions via density fractionation or differential centrifugation.22–24,26,72 Organelle fractions are then analyzed using quantitative MS, using labeling with isobaric tags (e.g., tandem mass tags; TMT73) or stable isotopes (e.g., SILAC74), yielding spatial profiles for each detected protein across the designated organelle fractions. To ultimately extract information about protein localization, a variety of ML-based approaches have been developed that leverage the spatial profiles of organelle marker proteins to predict unknown protein localizations.22,26,33,40,42 These approaches have so far largely focused on the assignment of proteins to subcellular compartments within a given biological system (e.g., cell type) or stage. With some exceptions,33,43 less focus has been placed on discovering protein movement, i.e., characterizing how these protein localizations may change across different biological conditions.
Figure 1.
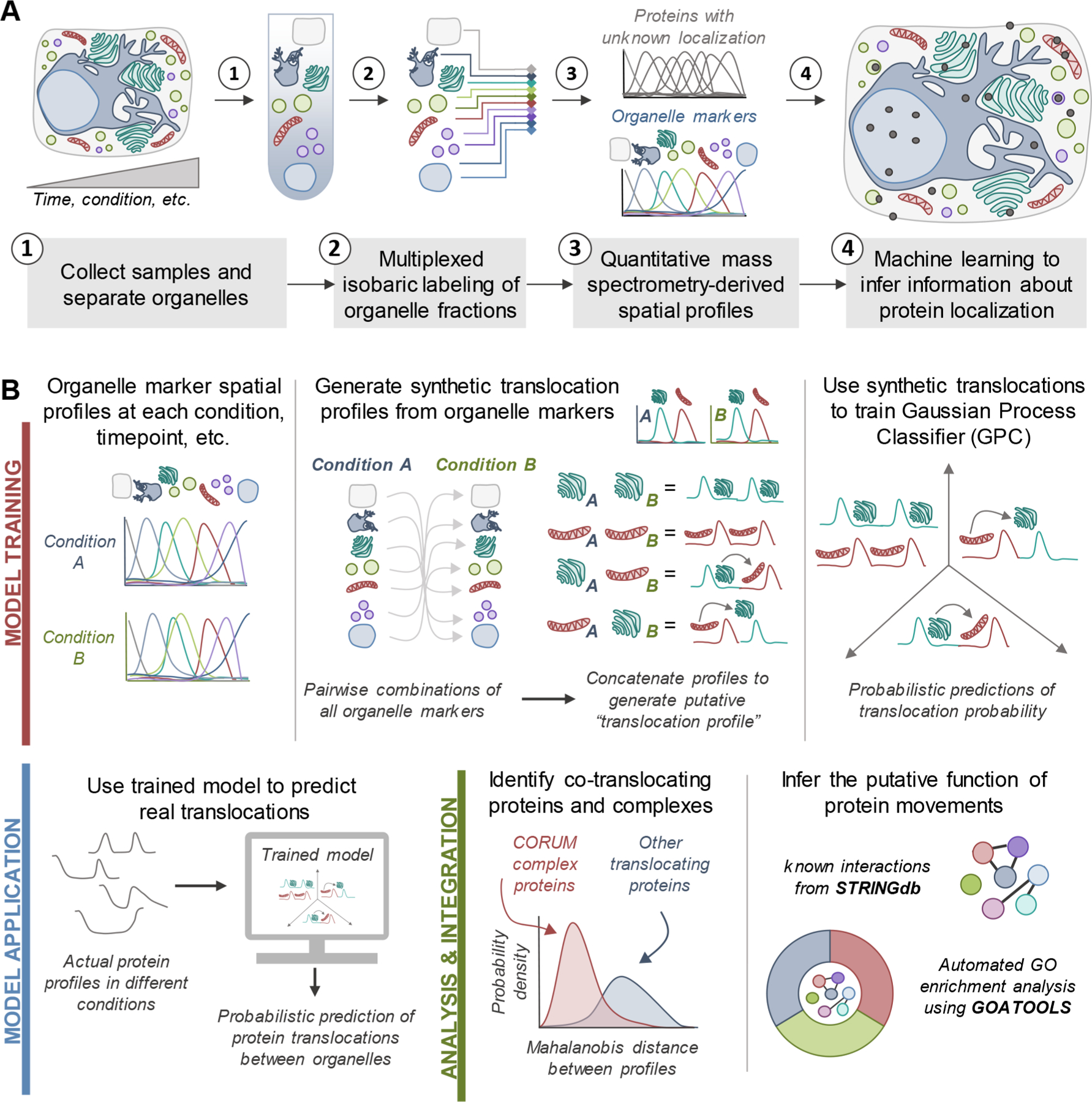
Workflows for spatial proteomics and the TRANPIRE pipeline for studying protein localization and movement. (A) General workflow for assessing the subcellular distribution of cellular proteins using organelle fractionation-based spatial proteomics. Most commonly, these approaches combine multiplexed isobaric labeling and quantitative mass spectrometry with machine learning to discern information regarding protein localization. Organelle marker proteins are used to inform machine learning-enabled classification. (B) Data processing and analysis pipeline for TRANSPIRE, a computational method leveraging a Gaussian process classifier (GPC) to characterize protein movements between organelles. By training the classifier with synthetic translocation profiles derived from different combinations of organelle markers, this classifier can detect and score the probability of protein translocation events. The output of the classifier is further combined with Mahalanobis distance analyses to identify co-translocating proteins and protein complexes. Integration of this analysis with known interactions and gene ontology enrichment can help reveal the putative function of these changes in protein distribution.
To specifically probe for changes in localization between different states in spatial proteomics data sets, we developed an ML-based approach that utilizes custom-generated, “synthetic” translocation profiles to predict changes in protein distributions. Briefly, translocation profiles were generated by concatenating all possible combinations of defined organelle markers at each representative condition (Figure 1B; Model Training). To simulate nontranslocation events, combinations of markers for the same organelle were also included. These profiles were then used for classifier training, and actual translocations in the data set were predicted using the true spatial profiles of a given protein in each state as classifier inputs (Figure 1B; Model Application). Finally, to further extract information about the putative regulation and function of these movements, we identified co-translocating proteins using Mahalanobis distance analysis of translocation profiles, integrated information regarding known protein–protein interactions, and performed gene ontology enrichment analysis on clusters of translocating proteins (Figure 1B; Analysis and Integration).
We predicted that by directly assessing changes across organelle representation between conditions we could extract differences in protein distribution, even if a protein is localized to more than one compartment. One challenge with this approach, however, is that training sets scale exponentially with the number of organelle markers that are available, both in terms of the number of samples and classes. For example, consider a study with markers for 10 compartments and 40 marker proteins for each compartment—this would yield 160000 profiles corresponding to 91 translocation classes. Sample sets of this magnitude are impractical for use with common ML classifiers, such as support SVMs, since fit time scales at least quadratically with sample size. On the other hand, neural networks, k-nearest neighbors, and decision tree classifiers scale better in sample space but frequently suffer from poor calibration (classifier scores tend to not reflect prediction uncertainty75).
To address these issues of scalability and calibration, we took advantage of a modified Gaussian process classifier (GPC) framework that has been implemented in Python via the GPFlow package.76 Briefly, Gaussian process (GP) models constitute a flexible, nonparametric approach to supervised learning that benefit from well-calibrated uncertainty estimates due to their Bayesian treatment of uncertainty.77 Applications of GPC have been primarily limited to contexts where sample number is relatively small due to memory and computational restrictions that scale quadratically and cubically, respectively, with sample size. The introduction of stochastic variational inference for GP models has rendered GPC tenable for large data sets.55,78 Although GPs have been applied across the field of biology to address the prediction of properties such as transcription factor binding targets,79 cell growth rates,80 and epigenetic regulation of cholesterol homeostasis,81 GPC has not yet been used for the purpose of analyzing spatial proteomics data.
Our pipeline for TRanslocation ANalysis of SPatIal pRotEomics data (TRANSPIRE) leverages the GPC framework to detect and provide confidence scores for translocating proteins and their localization dynamics. We specifically leverage the stochastic variational Gaussian process (SVGP) classifier implemented in GPFlow, which has been shown to produce well-calibrated predictions with large training data sets.55 Our pipeline additionally integrates the output of this classifier with bioinformatic metrics to help discern the functional implications of such movements. We provide TRANSPIRE as an installable Python package via GitHub (https://github.com/cristealab/TRANSPIRE_JASMS2020), and provide documentation and example workflows for data analysis. The TRANSPIRE pipeline is applicable to spatial proteomics studies of varying experimental design and biological context, and here, we demonstrate its utility for predicting protein movements during viral infection in the context of two different experimental studies.
Reliable Prediction of Protein Translocation during Virus Infection.
As previously discussed, spatial proteomics studies can take on a variety of experimental designs, with one varied aspect being the type and extent of organelle fractionation. On one end of the spectrum is nuclear-cytoplasmic fractionation, while at the other end are studies that perform consecutive fractionation steps to provide increased organellar resolution. In considering this, we aimed to test the applicability of TRANSPIRE to studies using these different approaches. To start, we analyzed a multiplexed nuclear-cytoplasmic fractionation data set (Figure 2A) reported in Gilbertson et al.53 By using transfection with muSOX, a viral endonuclease encoded by Kaposi Sarcoma-associated herpesvirus (KSHV), this study uncovered nuclear-cytoplasmic shuttling events linked to KSHV-induced cellular mRNA decay. After integrating this data set with a set of known organelle markers for the cells used in this study (HEK293T),54 we leveraged TRANSPIRE to generate synthetic translocation profiles, optimize model parameters and hyperparameters, and finally predict muSOX-induced shuttling events. Our results demonstrate that the classifier generally performs well across the experimental conditions (Figure 2B and Table S4). Moreover, our analysis of muSOX-induced translocation events demonstrated an enrichment for proteins involved in RNA binding (Figure 2C and Table S5), in agreement with the observations reported in the original study. For example, among the top scoring predicted translocations were PABPC1, PABPC4, and LARP4 (Figure 2D and Table S6)—three of the essential factors that were validated and functionally investigated in the original study. We additionally note that TRANSPIRE does not predict translocations of these proteins in cells devoid of the cellular exonuclease Xrn1, further replicating the Xrn1-dependent nature of these movements as reported in the original study.
Figure 2.
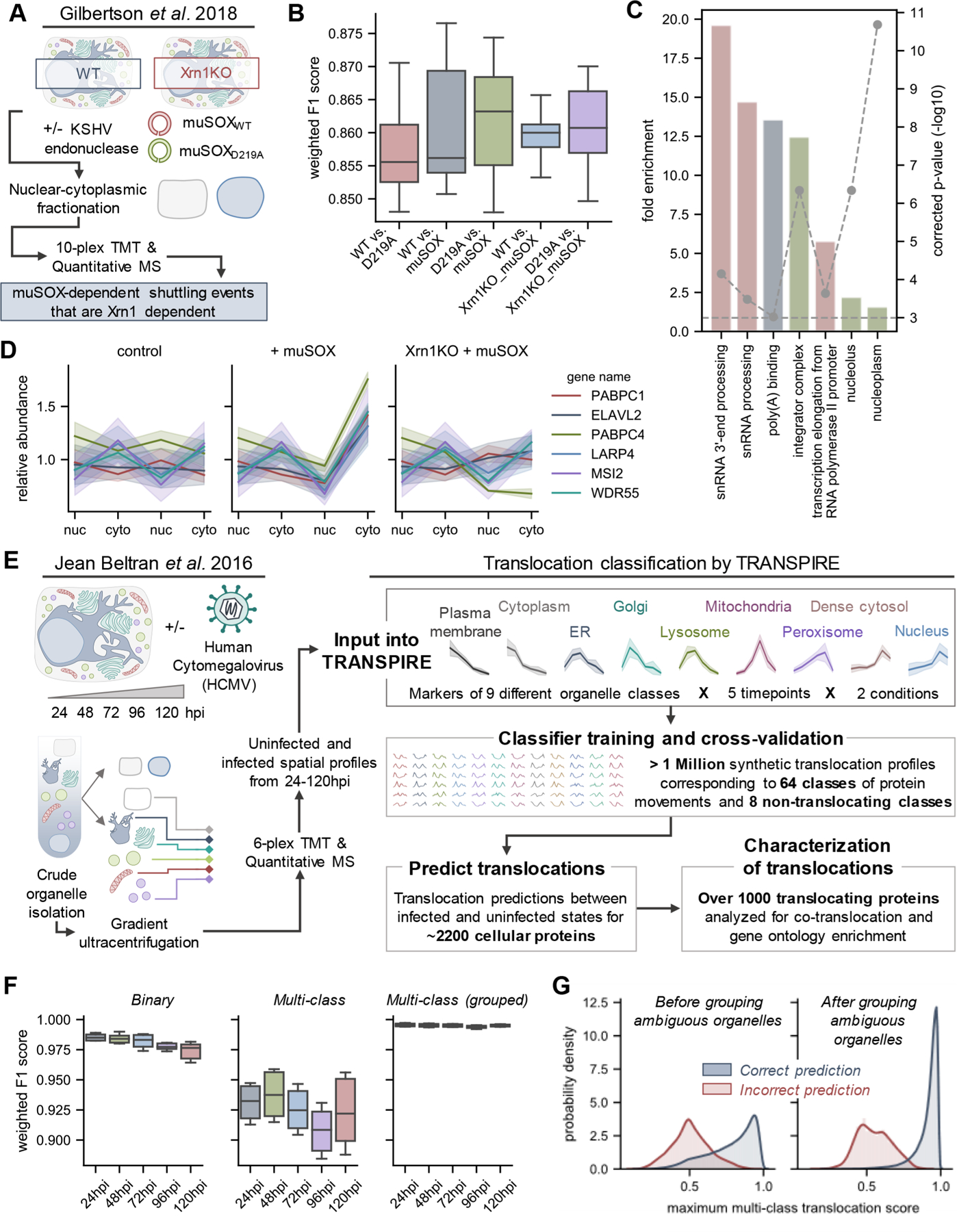
Assessing the reliability of TRASPIRE classification for predicting protein translocations in the context of viral infection. (A) Experimental workflow from Gilbertson et al.,53 a study focused on understanding nuclear-cytoplasmic shuttling events upon KSHV-induced cellular mRNA decay (induced by transfection of the KSHV endonuclease muSOX or its catalytically inactive counterpart muSOX D219A). (B) Boxplots of weighted F1 scores describing classifier performance across five balanced training folds and three biological replicates for each experimental condition. (C) Gene ontology enrichment on proteins predicted to translocate by TRANSPIRE point to RNA binding proteins, in agreement with the results of the original manuscript. (D) Selected profiles of proteins predicted to translocate by TRANSPIRE in an Xrn1-dependent manner. Solid lines and shaded areas represent the mean and standard deviation, respectively, of protein profiles across the three biological replicates reported in the study. (E) Experimental workflow for the HCMV spatial proteomics study that was subsequently analyzed by TRANSPIRE. Using 6-plex TMT labeling, Jean Beltran et al. generated spatial profiles of proteins in uninfected and (HCMV)-infected cells at 24, 48, 72, 96, and 120 hpi. The curated organelle markers defined in the original study were used to generate synthetic translocation profiles from all pairwise combinations of organelle markers. Equal subsets of profiles corresponding to each combination of markers were then used to train the classifier and performance was validated on a held-out subset of test data. Following training, the classifier was then applied to predict translocations within the data set and high confidence predictions were further characterized by integrating information regarding known protein interactions and gene ontology enrichment analysis. (F) Boxplots of weighted F1 scores describing performance on the held-out test data set across all time points of infection at both binary (e.g., translocating versus not translocating) and multiclass levels, as well as for classifier predictions after grouping ambiguous organelles. Ambiguous organelle groups were: plasma membrane/cytoplasm, ER/Golgi/lysosome, and dense cytosol/nucleus. (G) Classifier score distributions for correct and incorrect classifier predictions before and after grouping ambiguous organelles. Note that these scores refer to multiclass translocation assignments rather than the binary translocation scores discussed later in the manuscript.
Upon validation of TRANSPIRE’s ability to predict protein movement in the context of a nuclear-cytoplasmic fractionation workflow, we next tested its performance on a more complex data set that investigated protein localization throughout the course of HCMV replication. Infection with HCMV is known to result in radical alterations of the host proteome,34,82,83 in conjunction with broad remodeling of organelle shape and functions.44,84–86 Our previous finding of temporal changes in protein abundances within a wide range of subcellular organelles34 has led us to propose that HCMV infection induces protein translocations on a global scale. We therefore applied TRANSPIRE to study protein movement throughout the cycle of HCMV replication. We predicted that we could repurpose the data set collected by Jean Beltran et al.,34 which focused on assigning protein localization during infection, to uncover temporal protein movements between organelles. In brief, the study consisted of spatial profiles across six organelle fractions for paired uninfected and infected samples throughout the HCMV replication cycle (24, 48, 72, 96, and 120 h post infection (hpi)) (Figure 2E). To maintain consistency, we retained organelle markers from the original study, and by using this information, TRANSPIRE generated over 1 million synthetic translocation profiles representing 64 classes of movements and eight not-translocating classes (Table S7). Following hyperparameter optimization (see the Materials and Methods), these profiles were then used to train SVGP models for each infected vs uninfected time point comparison (Figure 2E).
To validate classifier performance in the context of the synthetic translocation data, we utilized a stratified cross-validation strategy (detailed in the Materials and Methods, and the results are provided in Table S8). Of note, we observed a level of overlap between organelle marker classes (Figure S1A), which resulted in overlap between synthetic translocation classes (Figure S1B). This is an inherent challenge of spatial proteomics and the experimental workflows leveraged for separating organelles. Despite this overlap, we still obtained relatively high F1 scores across all infection time points at both the level of binary (e.g., translocating vs not translocating) and multiclass assignment (Figure 2F). We further show that by grouping ambiguous organelles (plasma membrane/cytoplasm, ER/Golgi/lysosome, and dense cytosol/nucleus) these scores are further increased (Figure 2F; multiclass (grouped)). This not only demonstrates the extent of classifier accuracy, precision, and recall, but it shows that, even if the model does not predict a single translocation class with high certainty, it retains the ability to predict translocations between groups of organelles. Finally, when the classifier does predict a label incorrectly in either of these scenarios, it generally does so with a much lower score than when it predicts a label correctly, demonstrating that prediction scores scale accordingly with classifier uncertainty (Figure 2G).
If we look more closely at which classes of translocations are incorrectly predicted (Figure S2C), we observe that, in general, the most-frequently misclassified labels include those for translocations involving either the nucleus or the Golgi compartments. This was, perhaps, not surprising given that these two sets of markers overlapped with other marker classes and had much lower representation in the marker set (approximately 10 and 20 marker proteins, respectively). Furthermore, as expected, we observe that the classifier has some difficulty differentiating between sets of organelles that are relatively poorly separated (e.g., components of the secretory system). Again, however, we note that we can rescue many of these lower-scoring classes by grouping together organelles that we observe to be poorly separated (Figure S2D). Additionally, we see that the classifier generally performs well in the prediction of nontranslocating classes (Figure S2C; red boxes), overall, giving us confidence in the classifier’s ability to distinguish translocating from nontranslocating proteins.
HCMV Infection Induces Protein Translocations at a Global Scale.
To minimize the likelihood of false positives, we leveraged the model performance on the synthetic translocation data to calculate false-positive rates corresponding to given translocation score thresholds on a per-condition basis (Figure S3A). We settled on a stringent cutoff of 0.3%, as this cutoff retained proteins already known to translocate during infection. Upon input of the actual infected and uninfected spatial profiles into the trained TRANSPIRE classifier, we found that although most proteins are predicted to remain static, over 800 proteins were projected to translocate with scores greater than our confidence cutoff (Figure 3A and Table S9). Something to consider is that changes in protein distribution may occur as a result of physical movement of a protein between compartments, as well as targeted protein synthesis or degradation. Therefore, we next assessed whether TRANSPIRE-predicted translocations exhibited changes in abundance at the whole proteome level. Performing this additional analysis revealed that, on average, the proteins predicted to translocate exhibit relatively little change in abundance between infected and uninfected states (Figure S3B) and that, globally, translocation scores are not correlated with changes in protein abundance (Figure S3C). As expected, however, several proteins predicted to translocate also have relatively large changes in protein abundance. For example, the immune response factors STAT1 and ISG15 were identified to translocate from the cytoplasm to the dense cytosol, and their abundances increased upon infection by approximately 7-fold and 97-fold, respectively.82 We additionally noted that protein movements were primarily predicted to occur between well-connected organelles, for example, between the plasma membrane and cytoplasm or between secretory system components. The most abundant number of translocations occurred between the plasma membrane/cytoplasm and the dense cytosol/nucleus (Figure 3A). This was encouraging given the number of nucleocytoplasmic shuttling events already reported during HCMV infection and the prominent rearrangements made to secretory organelles upon formation of the HCMV viral assembly complex (vAC).84,87–90
Figure 3.
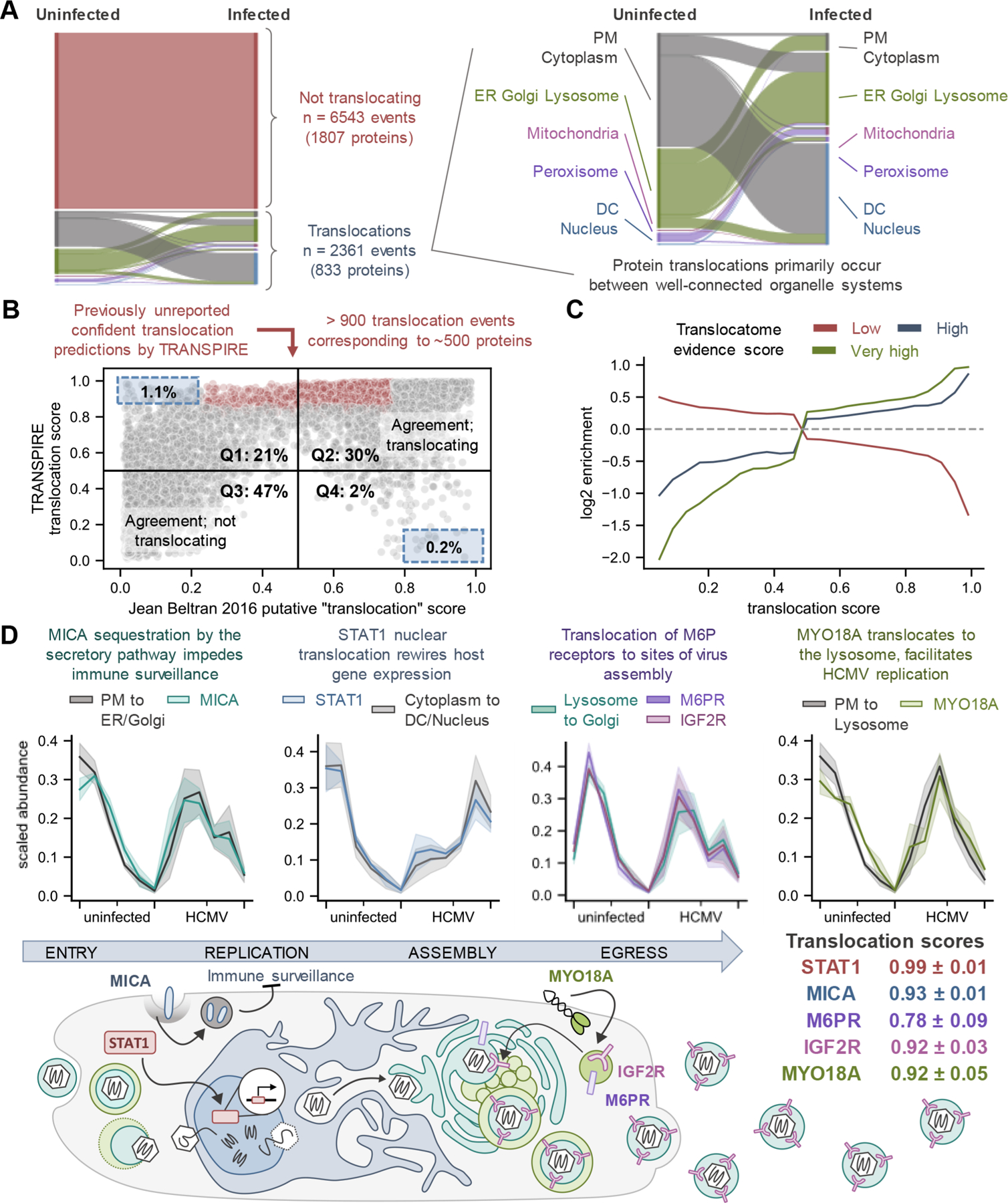
HCMV infection globally induces protein movements. (A) Sankey diagram depicting all high confidence predictions made by TRANSPIRE across all infection conditions. Each color (besides red) represents a different organelle, while the width of the strip represents the number of proteins that correspond to that organelle in each state. (B) Agreement between TRANSPIRE translocation predictions and the original study. To make this comparison, we generated a putative “translocation score” from the organelle assignments described in the original study (see eqs 5 and 6). Scores closer to 0 or closer to 1 indicate less uncertainty, while scores closer to 0.5 represent high uncertainty. (C) Translocation scores plotted against the relative enrichment for proteins with low, high, and very high translocation evidence scores in the Translocatome database.91 Cutoffs were defined as per the Translocatome publication (e.g., low ≤ 0.4487, high > 0.4487 ≤ 0.6167, and very high > 0.6167), and the enrichment baseline was determined by the translocation evidence scores for all proteins detected in the study. D) TRANSPIRE identification of proteins known to translocate upon HCMV infection. (Top) Protein translocation profiles compared to synthetic translocation profiles for the corresponding predicted translocation class. Solid lines and shaded areas represent the mean and standard deviation, respectively, of protein profiles across all time points for uninfected and infected cells. (Bottom) schematic overview of the role of these movements during HCMV infection. Mean translocation scores and their standard deviations across time points are reported. Abbreviations: PM, plasma membrane; DC, dense cytosol.
In general, the translocation predictions made by TRANSPIRE agreed with the original study, particularly for the high confidence predictions (Figure 3B, Q2 and Q3). Of the predictions that were not in agreement with the original study (Figure 3B Q1 and Q4), only 1.1% (Q1) and 0.2% (Q4) of these predictions passed our selection criteria (i.e., few of these disagreements were made with high confidence). Of this small proportion of possible disagreements, it is possible that those in Q1 arise as a result of increased sensitivity of the TRANSPIRE classifier, while those in Q4 may correspond to a small (but expected) proportion of false negatives. However, it is also possible that these proteins may be incorrectly classified by both methods, for example for proteins with ambiguous localizations or for those undergoing localization-dependent changes in abundance (as stated above). Among proteins passing our selection criteria, we observed more than 900 previously unreported confident translocation events corresponding to approximately 500 proteins that were either not identified or identified with low confidence in the original study (Figure 3B, points highlighted in red). Additionally, we show that higher TRANSPIRE translocation scores tend to enrich for proteins known to have the capacity to traffic between compartments, i.e., proteins with high or very high translocation evidence scores from the Translocatome database91 (Figure 3C). On the other hand, lower TRANSPIRE translocation scores tend to be negatively correlated with high Translocatome evidence scores, and instead enrich for proteins with low evidence scores (Figure 3C).
It was reassuring that TRANSPIRE pointed to known protein movements with established functions at various stages during the HCMV replication cycle. For example, early in infection we detected translocations that contribute to immune signaling and HCMV evasion of immune surveillance, while late in infection we identified movements that contribute to virus assembly (Figure 3D). Specifically, we detected the movement of major histocompatibility complex (MHC) class I-related chain A (MICA) away from the cell membrane, which inhibits NK cell-mediated immune surveillance.8,92 Additionally, TRANSPIRE was able to detect the shuttling of STAT1, a critical regulator of interferon-stimulated gene (ISG) expression, from the cytoplasm to the nucleus. This shuttling has been shown to be important for HCMV-induced rewiring of ISG signaling.93,94 By 24 hpi, TRANSPIRE had also identified translocation of mannose-6-phosphate receptors M6PR and IGF2R from the lysosome to the Golgi apparatus. These proteins have been reported to colocalize with HCMV envelope glycoprotein gH at the vAC,95 the formation of which involves the reorganization of Golgi membranes.90 Finally, TRANSPIRE also identified the plasma membrane to lysosome translocation of the unconventional myosin MYO18A, a protein that we have previously reported to be important for efficient HCMV replication.34
Protein Translocations Are Prevalent in Processes Contributing to Virus Replication and Host Defense.
Given the extended time frame of the HCMV replication cycle and its apparent induction of global changes in protein distributions, we wanted to further examine the spatiotemporal dynamics of these movements. At a global level, TRANSPIRE predicted that the vast majority (over 85%) of translocating proteins undergo these movements by or before 72 hpi (Figure 4A). This is in agreement with the understanding that most HCMV-induced cellular dysregulation events are evident (or becoming evident) by this stage of infection. We also observed that translocations to the dense cytosol occurred early in infection (by 24 hpi), while translocations to or from secretory organelles primarily occurred later (Figure 4B), which correlates with the formation of the vAC.
Figure 4.
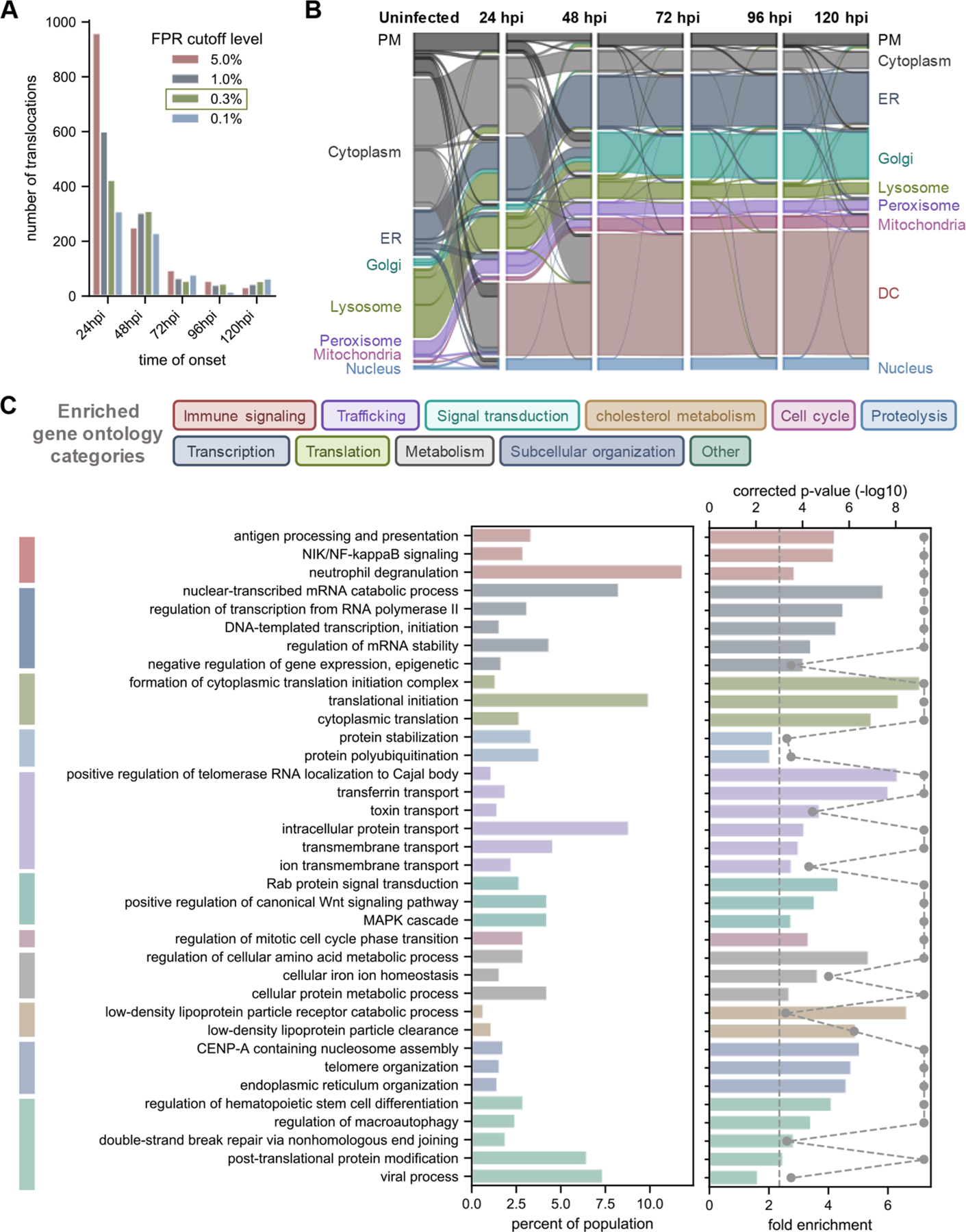
Temporal, spatial, and functional nature of protein movements reflect the HCMV modulation of cellular pathways. (A) Onset times for protein movement events identified by TRANSPIRE. Onset was defined as the first time point with a translocation score above the determined cutoff. (B) Spatiotemporal dynamics of protein movements during HCMV infection illustrated as a Sankey diagram. (C) Results of gene ontology (GO) analysis of all translocating proteins. GO terms are color-coded by the general category that they belong to. Gray data points on the right-most graph represent the −log10(p-value), and the dashed line represents a significance cutoff of 0.05. GO terms that returned a p-value of 0.0 based on FDR resampling are represented with a p-value of 0.0001 for visualization purposes. PM, plasma membrane; DC, dense cytosol.
To determine which pathways are targeted by these movements, we performed a gene ontology (GO) association enrichment analysis for all translocating proteins throughout infection (Figure 4C and Table S10). To assess whether groups of proteins exhibit coordinated co-translocation, we additionally calculated the Mahalanobis distance between all translocating proteins (Figure S3D). Comparison of these values with distances between proteins in the CORUM database allowed us to establish a metric for evaluating the extent of co-translocation for given sets of proteins (Figure S3E). We noted that pathways involved in cellular signaling processes (particularly immune signaling), cell cycle regulation, metabolism, and trafficking were significantly enriched for protein movements as well as co-translocations (Figure S4A–K and Table S11). While many of these categories include proteins that have already been identified to translocate during infection, we also observed numerous previously unappreciated protein movements.
A significantly enriched functional category included proteins with roles in cholesterol metabolism, such as the low-density lipoprotein receptor (LDLR), scavenger receptor SCARB1, and members of the clathrin-mediated adaptor protein 2 complex (AP-2) (Figure 5A). For example, LDLR was predicted to translocate from the plasma membrane to the ER/Golgi/Lysosome early during the replication process (Figure 5B). HCMV infection is well-known to dysregulate cellular lipid synthesis96 and, more specifically, upregulate intracellular cholesterol levels.97 Given that cholesterol content of the HCMV viral envelope has been shown to positively correlate with the infectivity of newly formed virions,97 we asked whether LDLR may be degraded at the lysosome, possibly as an antiviral host response to infection. Indeed, referencing one of our previous studies,82 we found that LDLR protein abundance levels decrease throughout HCMV infection (Figure 5C). We further validated this decrease by performing parallel reaction monitoring (PRM) analysis of two unique LDLR peptides during infection (Figures 5D and S5A). However, it remains possible that this decrease in protein abundance is driven by mechanisms other than lysosomal degradation. We therefore consulted a study that monitored alterations to the viral and cellular transcriptome during HCMV infection98 and noted that LDLR mRNA levels are also decreased (Figure S5B). This was concomitant with an upregulation of the mRNA levels of PCSK9, the protein responsible for targeting LDLR to the lysosome for degradation (reviewed in ref 99) (Figure S5B). Given the extended half-life of the LDLR protein (14–24 h100), it is, perhaps, not surprising that a cellular response aiming to rapidly decrease LDLR levels would target its regulation at both the transcriptional and post-translational levels. In support of the importance of LDLR family member proteins, the infection-induced upregulation of LRP1 has also been implicated as a host antiviral response to HCMV infection.97 Given our results, it is possible that LDLR translocation and putative degradation at the lysosome may also contribute to host-mediated cholesterol restriction.
Figure 5.
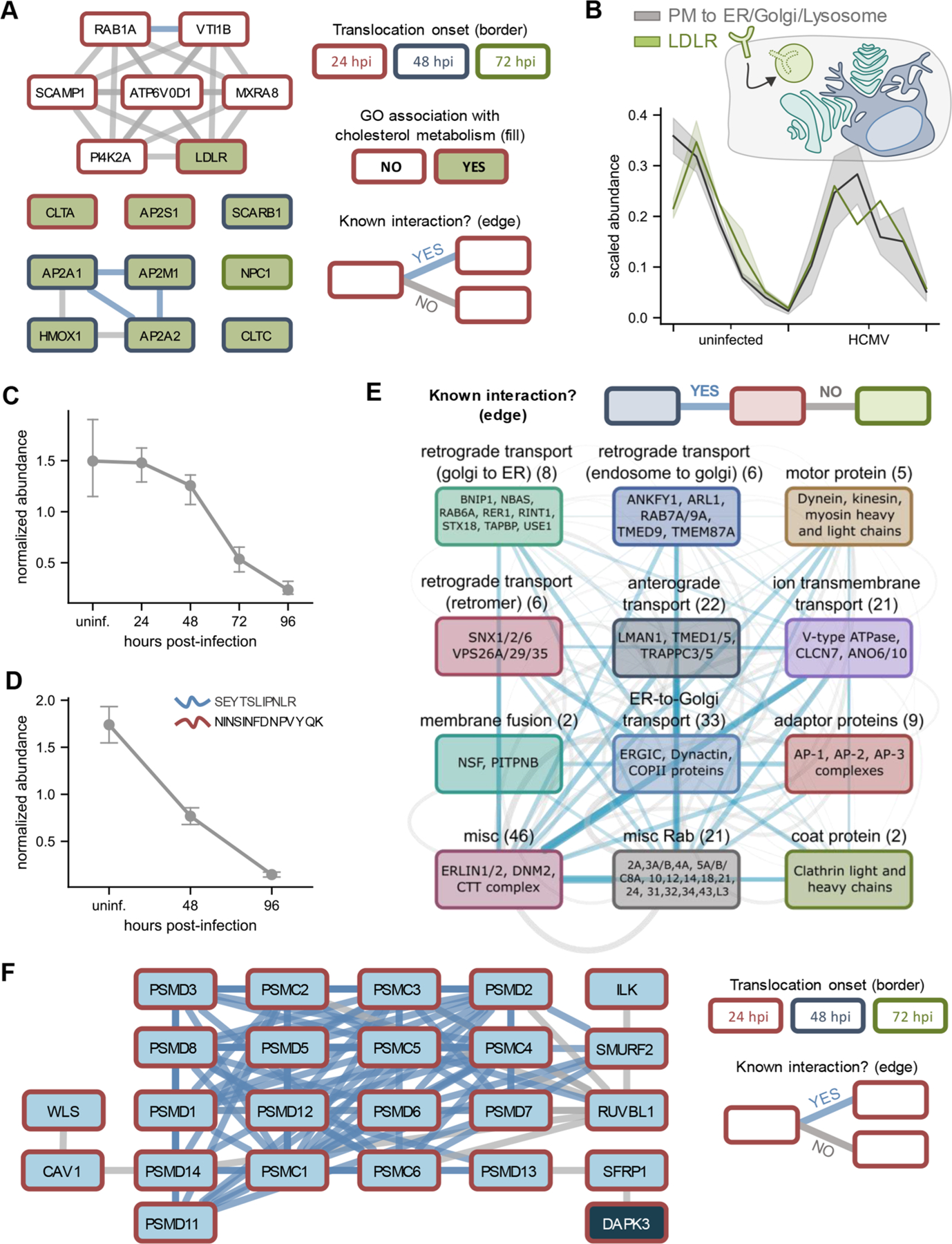
HCMV infection targets cholesterol metabolism, cellular trafficking factors, and Wnt signaling via protein translocations. (A) Translocating and co-translocating proteins involved in cholesterol metabolism. Co-translocations that correspond to a known interaction are shown in blue, while other co-translocations are shown in gray. Node border color denotes the time of translocation onset. (B) Translocation profiles of LDLR relative to the synthetic translocation profiles generated for plasma membrane (PM) to ER/Golgi/Lysosome movements. (C) LDLR protein levels decrease throughout HCMV infection. Error bars represent the standard deviation of three biological replicates. (D) Validation of decrease in LDLR levels by targeted mass spectrometry. Error bars represent the standard deviation across two unique LDLR peptides quantified by parallel reaction monitoring (PRM). (E) Translocating and co-translocating protein categories involved in intracellular trafficking. Edge width scales with the number of TRANSPIRE-identified co-translocations that represent known (blue) or unknown (gray) associations. (F) Translocating and co-translocating proteins involved in Wnt signaling.
Perhaps not surprising, another functional category significantly enriched among the translocating proteins was cellular trafficking. Transport factors are not only necessary for facilitating protein movements, but also for virus trafficking and assembly.90,101 These factors include members of the dynein motor complex and the ER-to-Golgi intermediate compartment (ERGIC) (Figure 5D), both of which have been shown to exhibit reorganization upon infection.90,102 However, some of the other factors that we observed to translocate have not previously been reported to undergo HCMV-induced changes in localization. Among these are members of both clathrin-independent and clathrin-dependent trafficking mechanisms, including coatomer subunits involved in ER-to-Golgi transport and clathrin adaptor protein complexes AP-1, AP-2, and AP-3 (Figure 5E). In uninfected cells, AP-1, AP-2, and AP-3 are generally responsible for trafficking endosomes between the trans-Golgi network (TGN), plasma membrane, and lysosomes, respectively. Intriguingly, each of these complexes exhibited translocation profiles during infection that were distinct from one another—both spatially and temporally (Figure S5C). For example, TRANSPIRE predicted AP-1 to translocate from the plasma membrane/cytoplasm to the ER/Golgi/lysosome at around 72 hpi, which is supported by the recent report of its localization to the vAC.103 On the other hand, AP-3 was predicted to translocate from the cytoplasm to the dense cytosol by 24–48 hpi, while at 48 hpi AP-2 was predicted to undergo movements between components of the secretory system. Of these complexes, all three have been implicated in the replication cycles of a number of other viruses, including human immunodeficiency virus (HIV), dengue virus, and West Nile virus.104,105 However, with the exception of AP-1, the contribution of the redistribution of these factors to HCMV infection is largely unknown. Broadly, clathrin-mediated processes have been shown to contribute to HCMV virion maturation and trafficking, and numerous HCMV viral proteins have been shown to engage with clathrin-associated factors.106–108 Overall, these changes in distribution of clathrin-associated proteins appear to not only reflect HCMV-induced changes in subcellular organization, but also the necessity of HCMV modulation of diverse cellular trafficking pathways to achieve proper replication.
HCMV Infection Induces the Redistribution of DAPK3 as a Possible Link to Activation of Wnt Signaling.
In addition to cholesterol metabolism and trafficking, we also discovered an enrichment of translocating factors involved in the regulation of Wnt signaling—another pathway known to be perturbed by HCMV infection.109,110 Among these proteins are subunits of the proteasome, which are responsible for regulating the cytoplasmic levels of the transcription factor β-catenin, as well as DAPK3 (also known as ZIPK), a multifunctional kinase involved in several cellular processes including Wnt signaling (Figure 5F).111 DAPK3 was predicted to translocate with high confidence (score >0.95), moving from the plasma membrane/cytoplasm to the dense cytosol/nucleus (Figure S5D).
To investigate this further, we assessed DAPK3 distributions in uninfected and infected cells using confocal microscopy (Figure 6A). To better capture its movement during the early stages of infection, we assessed DAPK3 localization at 6, 12, and 24 hpi, using the immediate-early HCMV protein IE1 as a marker of infected cells. We found that in uninfected cells DAPK3 predominantly localizes to the plasma membrane and cytoplasm, with very few puncta found in the nucleus. However, upon infection, the presence of DAPK3 at the plasma membrane sharply decreases. This is particularly evident when looking at infected and uninfected cells directly next to one another in the same image (white and red arrows, respectively, in Figure 6A). We quantitatively confirmed this decrease by acquiring line scans across uninfected and infected cells (Figure 6B). Additionally, this decrease at the plasma membrane generally correlates with little change in the amount of DAPK3 observed in the cytoplasm (Figure 6C), which suggests that the relative ratio of cytoplasmic to plasma membrane associated DAPK3 is significantly higher in infected cells. We also saw that infected nuclei exhibited increased DAPK3 signal, which appeared to manifest as puncta and peak at 12 hpi (Figure 6A and 6C). Thus, we confirmed HCMV induces translocations of DAPK3 between multiple sets of cellular compartments.
Figure 6.
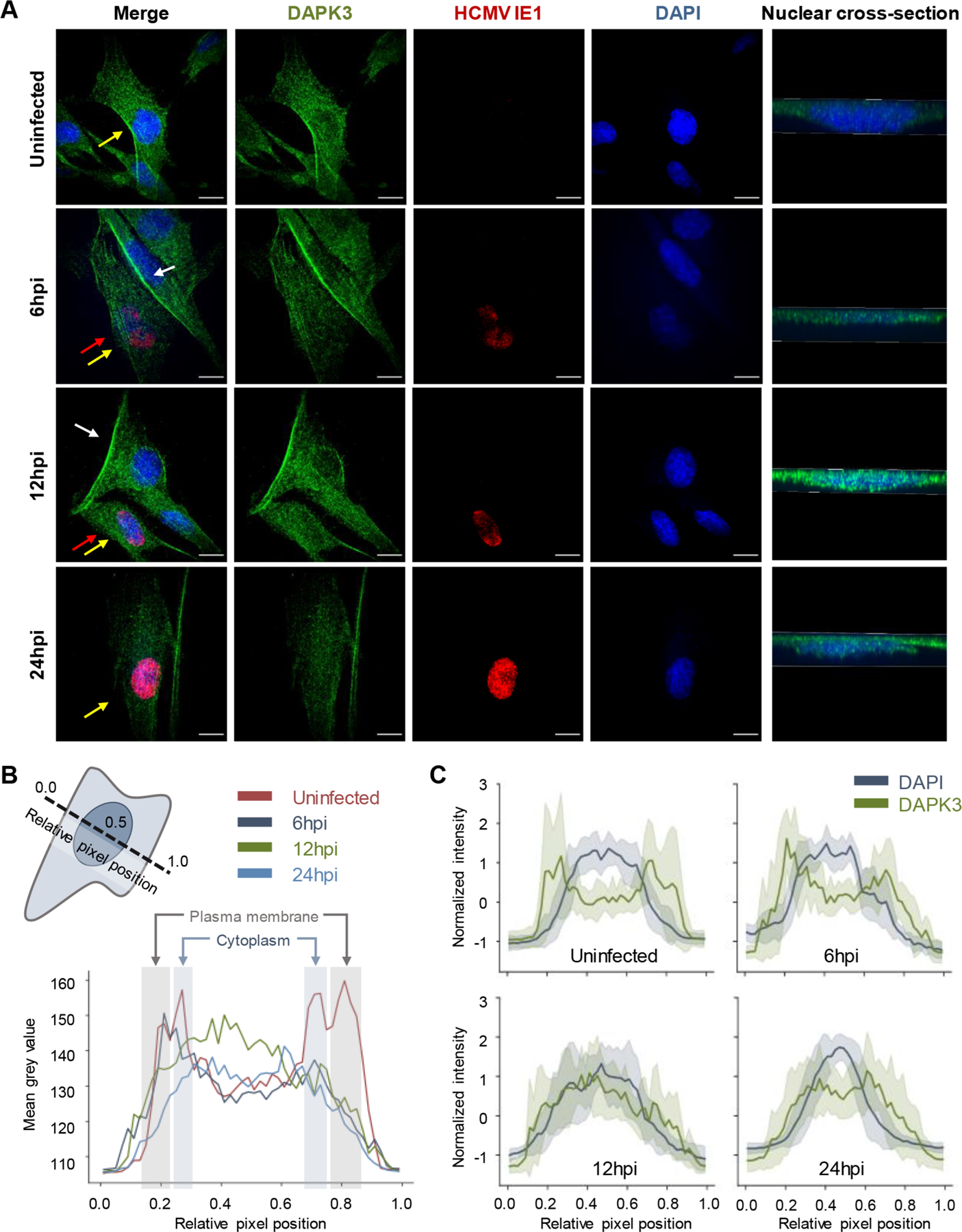
HCMV stimulates DAPK3 translocation between multiple subcellular compartments. (A) Immunofluorescence microscopy images (maximum projections) of DAPK3 distributions in uninfected and HCMV-infected cells early during HCMV infection (6, 12, and 24 hpi). The immediate-early HCMV protein IE1 is provided as marker of infected cells. Yellow arrows denote the nucleus that is cross-sectioned in the right-most panel. For emphasis, white arrows highlight plasma membrane accumulations of DAPK3 in uninfected cells, while red arrows point to infected cells that have lost this phenotype. (B) Line scan analysis of DAPK3 distributions (shaded to highlight the plasma membrane and cytoplasm, with the nucleus between the cytoplasm shadings) in uninfected and infected cells at 6, 12, and 24 hpi. The schematic in the upper-left corner is a representation of the orientation of the line scans relative to the cell body. (C) Overlay of the distribution of DAPK3 relative to the nucleus in uninfected and infected cells at 6, 12, and 24 hpi. Solid lines and shading represent the mean and standard deviation, respectively, across line scans from all cells analyzed at a given condition.
Like other viruses,112,113 HCMV infection has been observed to activate Wnt signaling.110 However, it appears to accomplish this in a noncanonical fashion, relying, in part, on expression of the HCMV protein pUS28.110 pUS28 is a virally encoded chemokine receptor, and its regulation of the Rho-associated kinase (ROCK) signaling axis has been implicated in HCMV-induced Wnt signaling.110 Nevertheless, the downstream effectors of this cascade remain unknown. Given that DAPK3 is a downstream substrate of ROCK114 that positively regulates Wnt signaling,111 it is tempting to speculate that nuclear translocation of DAPK3 contributes to this HCMV-induced increase in Wnt signaling. In support of this notion, ROCK activation has been shown to restrict HCMV propagation,115 and ROCK is known to phosphorylate DAPK3 at a site that inhibits its nuclear localization.114 Given our findings, it is possible that the plasma membrane to cytoplasm and nucleus translocation of DAPK3 represents a missing link in HCMV induction of Wnt signaling.
CONCLUDING REMARKS
Here, we introduce the TRANSPIRE computational pipeline and demonstrate its ability to predict the spatiotemporal dynamics of protein movements from spatial proteomics data sets. This pipeline illustrates the value of using a Gaussian process classifier framework for organelle proteomics studies. We show the applicability of this pipeline for studying nuclear-cytoplasmic protein shuttling, as well as protein movement between diverse subcellular compartments during infection. Our application of this pipeline to studying HCMV infection revealed global, infection-induced protein translocations that were temporally regulated. These protein translocations reflected processes important for virus replication and host defense, such as cholesterol metabolism, subcellular trafficking, and Wnt signaling. Using microscopy, we validated the HCMV-induced translocation of the death associated protein kinase 3 (DAPK3), a movement that may contribute to the ability of the virus to modulate Wnt signaling. Given the ubiquitous importance of protein movement within cellular processes, the methodology employed by TRANSPIRE can be readily applied, as well as expanded to study translocations in the context of a wide variety of experimental designs and biological questions.
Supplementary Material
Supplemental figures and figure legends as referenced in the text (PDF)
Table S10, results of gene ontology analysis of translocating proteins identified in the analysis of Jean Beltran et al.34(XLSX)
Table S9, classifier prediction results for Jean Beltran et al.34 (XLSX)
Table S5, results of gene ontology analysis of translocating proteins identified in the Gilbertson et al.53 analysis (XLSX)
Table S11, poteins identified to co-translocate by TRANSPIRE (including STRINGdb interaction scores if applicable) (XLSX)
Table S6, classifier prediction results for Gilbertson et al.53 (XLSX)
Table S3, experimentally generated list of genes expressed in MRC5 human fibroblast cells used as the background proteome for gene ontology analysis in HCMV-infected cells (XLSX)
Table S2, Gilbertson et al.53 data set formatted for direct pipeline analysis with associated organelle markers for HEK293T cells (XLSX)
Table S1, Jean Beltran et al.34 data set formatted for direct pipeline analysis with associated organelle markers for human fibroblast cells (XLSX)
Table S8, model performance on held-out test data for analysis of data reported in Jean Beltran et al.34 (XLSX)
Table S4, model performance on held-out test data for analysis of data reported in Gilbertson et al.53 (XLSX)
Table S7, synthetic translocation profiles generated using organelle marker proteins used to assess model performance on of data reported in Jean Beltran et al. 201634 (XLSX)
ACKNOWLEDGMENTS
We are grateful for funding from the NIH (GM114141), Mallinckrodt Scholar Award to I.M.C., as well as an NIH training grant from NIGMS (T32GM007388). This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-1656466 (awarded to M.A.K.).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.0c00033.
The authors declare no competing financial interest.
REFERENCES
- (1).Hetz C; Papa FR The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [DOI] [PubMed] [Google Scholar]
- (2).Alchini R; Sato H; Matsumoto N; Shimogori T; Sugo N; Yamamoto N Nucleocytoplasmic Shuttling of Histone Deacetylase 9 Controls Activity-Dependent Thalamocortical Axon Branching. Sci. Rep 2017, 7 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Diner BA; Lum KK; Toettcher JE; Cristea IM Viral DNA Sensors IFI16 and Cyclic GMP-600 AMP Synthase Possess Distinct Functions in Regulating Viral Gene Expression. mBio 2016, DOI: 10.1128/mBio.01553-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lin R; Heylbroeck C; Pitha PM; Hiscott J Virus-Dependent Phosphorylation of the IRF-3 Transcription Factor Regulates Nuclear Translocation, Transactivation Potential, and Proteasome-Mediated Degradation. Mol. Cell. Biol 1998, 18 (5), 2986–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Erster S; Mihara M; Kim RH; Petrenko O; Moll UM In Vivo Mitochondrial P53 Translocation Triggers a Rapid First Wave of Cell Death in Response to DNA Damage That Can Precede P53 Target Gene Activation. Mol. Cell. Biol 2004, 24 (15), 6728–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Prokhorova EA; Kopeina GS; Lavrik IN; Zhivotovsky B Apoptosis Regulation by Subcellular Relocation of Caspases. Sci. Rep 2018, 8 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cook KC; Cristea IM Location Is Everything: Protein Translocations as a Viral Infection Strategy. Curr. Opin. Chem. Biol 2019, 34–43, DOI: 10.1016/j.cbpa.2018.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Fielding CA; Aicheler R; Stanton RJ; Wang ECY; Han S; Seirafian S; Davies J; McSharry BP; Weekes MP; Antrobus PR; Prod’homme V; Blanchet FP; Sugrue D; Cuff S; Roberts D; Davison AJ; Lehner PJ; Wilkinson GWG; Tomasec P Two Novel Human Cytomegalovirus NK Cell Evasion Functions Target MICA for Lysosomal Degradation. PLoS Pathog 2014, 10 (5), No. e1004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kowalik TF; Wing B; Haskill JS; Azizkhan JC; Baldwin AS; Huang E-S Multiple Mechanisms Are Implicated in the Regulation of NF-KB Activity during Human Cytomegalovirus Infection. Proc. Natl. Acad. Sci. U. S. A 1993, 90, 1107–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kumar GR; Glaunsinger BA Nuclear Import of Cytoplasmic Poly(A) Binding Protein Restricts Gene Expression via Hyperadenylation and Nuclear Retention of MRNA. Mol. Cell. Biol 2010, 30 (21), 4996–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Li XD; Sun L; Seth RB; Pineda G; Chen ZJ Hepatitis C Virus Protease NS3/4A Cleaves Mitochondrial Antiviral Signaling Protein off the Mitochondria to Evade Innate Immunity. Proc. Natl. Acad. Sci. U. S. A 2005, 102 (49), 17717–17722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mukherjee A; Morosky SA; Delorme-Axford E; Dybdahl-Sissoko N; Oberste MS; Wang T; Coyne CB The Coxsackievirus B 3Cpro Protease Cleaves MAVS and TRIF to Attenuate Host Type I Interferon and Apoptotic Signaling. PLoS Pathog 2011, 7 (3), No. e1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bozidis P; Williamson CD; Wong DS; Colberg-Poley AM Trafficking of UL37 Proteins into Mitochondrion-Associated Membranes during Permissive Human Cytomegalovirus Infection. J. Virol 2010, 84 (15), 7898–7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Öhman T; Rintahaka J; Kalkkinen N; Matikainen S; Nyman TA Actin and RIG-I/MAVS Signaling Components Translocate to Mitochondria upon Influenza A Virus Infection of Human Primary Macrophages. J. Immunol 2009, 182 (9), 5682–5692. [DOI] [PubMed] [Google Scholar]
- (15).Du Y; Bi J; Liu J; Liu X; Wu X; Jiang P; Yoo D; Zhang Y; Wu J; Wan R; Zhao X; Guo L; Sun W; Cong X; Chen L; Wang J 3Cpro of Foot-and-Mouth Disease Virus Antagonizes the Interferon Signaling Pathway by Blocking STAT1/STAT2 Nuclear Translocation. J. Virol 2014, 88 (9), 4908–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Gatto L; Breckels LM; Burger T; Nightingale DJH; Groen AJ; Campbell C; Nikolovski N; Mulvey CM; Christoforou A; Ferro M; Lilley KS A Foundation for Reliable Spatial Proteomics Data Analysis. Mol. Cell. Proteomics 2014, 13 (8), 1937–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tharkeshwar AK; Gevaert K; Annaert W Organellar Omics-A Reviving Strategy to Untangle the Biomolecular Complexity of the Cell. Proteomics 2018, 18 (5–6), 1700113. [DOI] [PubMed] [Google Scholar]
- (18).Lundberg E; Borner GHH Spatial Proteomics: A Powerful Discovery Tool for Cell Biology. Nat. Rev. Mol. Cell Biol 2019, 20, 285–302. [DOI] [PubMed] [Google Scholar]
- (19).Rauniyar N; Yates JR Isobaric Labeling-Based Relative Quantification in Shotgun Proteomics. J. Proteome Rese 2014, 13, 5293–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Pankow S; Martínez-Bartolomé S; Bamberger C; Yates JR Understanding Molecular Mechanisms of Disease through Spatial Proteomics. Curr. Opin. Chem. Biol 2019, 48, 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Savas JN; Stein BD; Wu CC; Yates JR Mass Spectrometry Accelerates Membrane Protein Analysis. Trends Biochem. Sci 2011, DOI: 10.1016/j.tibs.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Dunkley TPJ; Watson R; Griffin JL; Dupree P; Lilley KS Localization of Organelle Proteins by Isotope Tagging (LOPIT). Mol. Cell. Proteomics 2004, 3 (11), 1128–1134. [DOI] [PubMed] [Google Scholar]
- (23).Mulvey CM; Breckels LM; Geladaki A; Britovsěk NK; Nightingale DJH; Christoforou A; Elzek M; Deery MJ; Gatto L; Lilley KS Using HyperLOPIT to Perform High-Resolution Mapping of the Spatial Proteome. Nat. Protoc 2017, 12 (6), 1110–1135. [DOI] [PubMed] [Google Scholar]
- (24).Foster LJ; de Hoog CL; Zhang YY; Zhang YY; Xie X; Mootha VK; Mann M A Mammalian Organelle Map by Protein Correlation Profiling. Cell 2006, 125 (1), 187–199. [DOI] [PubMed] [Google Scholar]
- (25).Itzhak DN; Davies C; Tyanova S; Mishra A; Williamson J; Antrobus R; Cox J; Weekes MP; Borner GHH A Mass Spectrometry-Based Approach for Mapping Protein Subcellular Localization Reveals the Spatial Proteome of Mouse Primary Neurons. Cell Rep 2017, 20 (11), 2706–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Christoforou A; Mulvey CM; Breckels LM; Geladaki A; Hurrell T; Hayward PC; Naake T; Gatto L; Viner R; Arias AM; Lilley KS A Draft Map of the Mouse Pluripotent Stem Cell Spatial Proteome. Nat. Commun 2016, 7, 9992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yan W; Hwang D; Aebersold R Quantitative Proteomic Analysis to Profile Dynamic Changes in the Spatial Distribution of Cellular Proteins. Methods Mol. Biol 2008, 432, 389–401. [DOI] [PubMed] [Google Scholar]
- (28).Eng JK; McCormack AL; Yates JR An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J. Am. Soc. Mass Spectrom 1994, 5 (11), 976–989. [DOI] [PubMed] [Google Scholar]
- (29).Washburn MP; Wolters D; Yates JR Large-Scale Analysis of the Yeast Proteome by Multidimensional Protein Identification Technology. Nat. Biotechnol 2001, 19 (3), 242–247. [DOI] [PubMed] [Google Scholar]
- (30).Yates JR Recent Technical Advances in Proteomics. F1000Research 2019, DOI: 10.12688/f1000research.16987.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Aebersold R; Mann M Mass-Spectrometric Exploration of Proteome Structure and Function. Nature 2016, 537, 347–355. [DOI] [PubMed] [Google Scholar]
- (32).Jean Beltran PM; Federspiel JD; Sheng X; Cristea IM Proteomics and Integrative Omic Approaches for Understanding Host–Pathogen Interactions and Infectious Diseases. Mol. Syst. Biol 2017, 13 (3), 922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Itzhak DN; Tyanova S; Cox J; Borner GH Global, Quantitative and Dynamic Mapping of Protein Subcellular Localization. eLife 2016, DOI: 10.7554/eLife.16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Jean Beltran PM; Mathias RA; Cristea IM A Portrait of the Human Organelle Proteome In Space and Time during Cytomegalovirus Infection. Cell Syst 2016, 3 (4), 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Tardif M; Atteia A; Specht M; Cogne G; Rolland N; Brugière S; Hippler M; Ferro M; Bruley C; Peltier G; Vallon O; Cournac L PredAlgo: A New Subcellular Localization Prediction Tool Dedicated to Green Algae. Mol. Biol. Evol 2012, 29 (12), 3625–3639. [DOI] [PubMed] [Google Scholar]
- (36).Groen AJ; Sancho-Andreś G; Breckels LM; Gatto L; Aniento F; Lilley KS Identification of Trans-Golgi Network Proteins in Arabidopsis Thaliana Root Tissue. J. Proteome Res 2014, 13 (2), 763–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ohta S; Bukowski-Wills JC; Sanchez-Pulido L; Alves F. de L.; Wood L; Chen ZA; Platani M; Fischer L; Hudson DF; Ponting CP; Fukagawa T; Earnshaw WC; Rappsilber J The Protein Composition of Mitotic Chromosomes Determined Using Multiclassifier Combinatorial Proteomics. Cell 2010, 142 (5), 810–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Nikolovski N; Rubtsov D; Segura MP; Miles GP; Stevens TJ; Dunkley TPJ; Munro S; Lilley KS; Dupree P Putative Glycosyltransferases and Other Plant Golgi Apparatus Proteins Are Revealed by LOPIT Proteomics. Plant Physiol 2012, 160 (2), 1037–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Dunkley TPJ; Hester S; Shadforth IP; Runions J; Weimar T; Hanton SL; Griffin JL; Bessant C; Brandizzi F; Hawes C; Watson RB; Dupree P; Lilley KS Mapping the Arabidopsis Organelle Proteome. Proc. Natl. Acad. Sci. U. S. A 2006, 103 (17), 6518–6523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Crook OM; Mulvey CM; Kirk PDW; Lilley KS; Gatto L A Bayesian Mixture Modelling Approach for Spatial Proteomics. PLoS Comput. Biol 2018, 14 (11), e1006516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Tyanova S; Temu T; Sinitcyn P; Carlson A; Hein MY; Geiger T; Mann M; Cox J The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [DOI] [PubMed] [Google Scholar]
- (42).Gatto L; Breckels LM; Wieczorek S; Burger T; Lilley KS Mass-Spectrometry-Based Spatial Proteomics Data Analysis Using PRoloc and PRolocdata. Bioinformatics 2014, 30 (9), 1322–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Hirst J; Itzhak DN; Antrobus R; Borner GHH; Robinson MS Role of the AP-5 Adaptor Protein Complex in Late Endosome-to-Golgi Retrieval. PLoS Biol 2018, 16 (1), No. e2004411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Jean Beltran PM; Cook KC; Hashimoto Y; Galitzine C; Murray LA; Vitek O; Cristea IM Infection-Induced Peroxisome Biogenesis Is a Metabolic Strategy for Herpesvirus Replication. Cell Host Microbe 2018, 24 (4), 526–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Clippinger AJ; Alwine JC Dynein Mediates the Localization and Activation of MTOR in Normal and Human Cytomegalovirus-Infected Cells. Genes Dev 2012, 26 (18), 2015–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Reitsma JM; Sato H; Nevels M; Terhune SS; Paulus C Human Cytomegalovirus IE1 Protein Disrupts Interleukin-6 Signaling by Sequestering STAT3 in the Nucleus. J. Virol 2013, 87 (19), 10763–10776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Sharon-Friling R; Shenk T Human Cytomegalovirus PUL37 × 1-Induced Calcium Flux Activates PKC, Inducing Altered Cell Shape and Accumulation of Cytoplasmic Vesicles. Proc. Natl. Acad. Sci. U. S. A 2014, 111 (12), E1140–E1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Yu D; Smith GA; Enquist LW; Shenk T Construction of a Self-Excisable Bacterial Artificial Chromosome Containing the Human Cytomegalovirus Genome and Mutagenesis of the Diploid TRL/IRL13 Gene. J. Virol 2002, 76 (5), 2316–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Wessel D; Flügge UI A Method for the Quantitative Recovery of Protein in Dilute Solution in the Presence of Detergents and Lipids. Anal. Biochem 1984, 138 (1), 141–143. [DOI] [PubMed] [Google Scholar]
- (50).Rappsilber J; Mann M; Ishihama Y Protocol for Micro-Purification, Enrichment, Pre-Fractionation and Storage of Peptides for Proteomics Using StageTips. Nat. Protoc 2007, 2 (8), 1896–1906. [DOI] [PubMed] [Google Scholar]
- (51).Lum KK; Song B; Federspiel JD; Diner BA; Howard T; Cristea IM Interactome and Proteome Dynamics Uncover Immune Modulatory Associations of the Pathogen Sensing Factor CGAS. Cell Syst 2018, 7 (6), 627–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Skyline: An Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. Bioinformatics 2010, 26 (7), 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Gilbertson S; Federspiel JD; Hartenian E; Cristea IM; Glaunsinger B Changes in MRNA Abundance Drive Shuttling of RNA Binding Proteins, Linking Cytoplasmic RNA Degradation to Transcription. eLife 2018, DOI: 10.7554/eLife.37663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Breckels LM; Gatto L; Christoforou A; Groen AJ; Lilley KS; Trotter MWB The Effect of Organelle Discovery upon Sub-Cellular Protein Localisation. J. Proteomics 2013, 88, 129–140. [DOI] [PubMed] [Google Scholar]
- (55).Hensman J; Matthews A; Ghahramani Z Scalable Variational Gaussian Process Classification. In Proceedings of the Eighteenth International Conference on Artificial Intelligence and Statistics; Lebanon G, Vishwanathan SVN, Eds.; Proceedings of Machine Learning Research; PMLR: San Diego, 2015; Vol. 38, pp 351–360. [Google Scholar]
- (56).De AG; Matthews G; Nickson T; Fujii K; Boukouvalas A; León-Villagrá P; Ghahramani Z; Hensman J GPflow: A Gaussian Process Library Using TensorFlow Mark van Der Wilk; Gaussian, 2017; Vol. 18. [Google Scholar]
- (57).Abadi M; Agarwal A; Barham P; Brevdo E; Chen Z; Citro C; Corrado GS; Davis A; Dean J; Devin M; Ghemawat S; Goodfellow I; Harp A; Irving G; Isard M; Jia Y; Jozefowicz R; Kaiser L; Kudlur M; Levenberg J; Mane D; Monga R; Moore S; Murray D; Olah C; Schuster M; Shlens J; Steiner B; Sutskever I; Talwar K; Tucker P; Vanhoucke V; Vasudevan V; Viegas F; Vinyals O; Warden P; Wattenberg M; Wicke M; Yu Y; Zheng X TensorFlow: Large-Scale Machine Learning on Heterogeneous Distributed Systems; TensorFlow, 2016. [Google Scholar]
- (58).Hernández-Lobato D; Hernández-Lobato JM; Dupont P Robust Multi-Class Gaussian Process Classification. Proceedings of the 24th International Conference on Neural Information Processing Systems; NIPS’11; Curran Associates, Inc.: Red Hook, NY, 2011; pp 280–288. [Google Scholar]
- (59).Salimbeni H; Eleftheriadis S; Hensman J Natural Gradients in Practice: Non-Conjugate Variational Inference in Gaussian Process Models. Int. Conf. Artif. Intell. Stat. AISTATS 2018, 2018, 689–697. [Google Scholar]
- (60).Galy-Fajou T; Wenzel F; Donner C; Opper M Multi-Class Gaussian Process Classification Made Conjugate: Efficient Inference via Data Augmentation. 35th Conf. Uncertain. Artif. Intell. UAI 2019 2019. [Google Scholar]
- (61).Pedregosa F; Varoquaux G; Gramfort A; Michel V; Thirion B; Grisel O; Blondel M; Prettenhofer P; Weiss R; Dubourg V; Vanderplas J; Passos A; Cournapeau D; Brucher M; Perrot M; Duchesnay E Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res 2011, 12, 2825–2830. [Google Scholar]
- (62).Kingma DP; Ba JL Adam: A Method for Stochastic Optimization. In 3rd International Conference on Learning Representations, ICLR 2015 - Conference Track Proceedings; International Conference on Learning Representations; ICLR, 2015. [Google Scholar]
- (63).Virtanen P; Gommers R; Oliphant TE; Haberland M; Reddy T; Cournapeau D; Burovski E; Peterson P; Weckesser W; Bright J; van der Walt SJ; Brett M; Wilson J; Jarrod Millman K; Mayorov N; Nelson ARJ; Jones E; Kern R; Larson E; Carey CJ; Polat I; Feng Y; Moore EW; Vand erPlas J; Laxalde D; Perktold J; Cimrman R; Henriksen I; Quintero EA; Harris CR; Archibald AM; Ribeiro AH; Pedregosa F; van Mulbregt P; Contributors S 1. 0. SciPy 1.0--Fundamental Algorithms for Scientific Computing in Python. arXiv e-prints 2019, arXiv:1907.10121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Rousseeuw PJ; Van Driessen K A Fast Algorithm for the Minimum Covariance Determinant Estimator. Technometrics 1999, 41 (3), 212–223. [Google Scholar]
- (65).Ruepp A; Brauner B; Dunger-Kaltenbach I; Frishman G; Montrone C; Stransky M; Waegele B; Schmidt T; Doudieu ON; Stümpflen V; Mewes HW CORUM: The Comprehensive Resource of Mammalian Protein Complexes. Nucleic Acids Res 2007, 36 (Suppl 1), D646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Szklarczyk D; Gable AL; Lyon D; Junge A; Wyder S; Huerta-Cepas J; Simonovic M; Doncheva NT; Morris JH; Bork P; Jensen LJ; Von Mering C STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res 2019, 47 (D1), D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Klopfenstein DV; Zhang L; Pedersen BS; Ramírez F; Vesztrocy AW; Naldi A; Mungall CJ; Yunes JM; Botvinnik O; Weigel M; Dampier W; Dessimoz C; Flick P; Tang H GOATOOLS: A Python Library for Gene Ontology Analyses. Sci. Rep 2018, 8 (1), 10872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Zeeberg BR; Qin H; Narasimhan S; Sunshine M; Cao H; Kane DW; Reimers M; Stephens RM; Bryant D; Burt SK; Elnekave E; Hari DM; Wynn TA; Cunningham-Rundles C; Stewart DM; Nelson D; Weinstein JN High-Throughput GoMiner, an “industrial-Strength” Integrative Gene Ontology Tool for Interpretation of Multiple-Microarray Experiments, with Application to Studies of Common Variable Immune Deficiency (CVID). BMC Bioinf 2005, 6 (1), 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Zhu H; Shen Y; Shenk T Human Cytomegalovirus IE1 and IE2 Proteins Block Apoptosis. J. Virol 1995, 69 (12), 7960–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Schneider CA; Rasband WS; Eliceiri KW NIH Image to ImageJ: 25 Years of Image Analysis. Nature Methods July 2012; pp 671–675. DOI: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Schindelin J; Arganda-Carreras I; Frise E; Kaynig V; Longair M; Pietzsch T; Preibisch S; Rueden C; Saalfeld S; Schmid B; Tinevez JY; White DJ; Hartenstein V; Eliceiri K; Tomancak P; Cardona A Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Geladaki A; Kočevar Britovšek N; Breckels LM; Smith TS; Vennard OL; Mulvey CM; Crook OM; Gatto L; Lilley KS Combining LOPIT with Differential Ultracentrifugation for High-Resolution Spatial Proteomics. Nat. Commun 2019, 10 (1), x DOI: 10.1038/s41467-018-08191-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Thompson A; Schäfer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Hamon C Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem 2003, 75 (8), 1895–1904. [DOI] [PubMed] [Google Scholar]
- (74).Ong SE; Blagoev B; Kratchmarova I; Kristensen DB; Steen H; Pandey A; Mann M Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteomics 2002, 1 (5), 376–386. [DOI] [PubMed] [Google Scholar]
- (75).Zadrozny B; Elkan C Transforming Classifier Scores into Accurate Multiclass Probability Estimates. KDD ‘02: Proceedings of the eighth ACM SIGKDD international conference on Knowledge discovery and data mining, 2002; DOI: 10.1145/775047.775151. [DOI] [Google Scholar]
- (76).Matthews AG; van der Wilk M; Nickson T; Fujii K; Boukouvalas A; León-Villagrá P; Ghahramani Z; Hensman J GPflow: A Gaussian Process Library Using TensorFlow. J. Mach. Learn. Res 2017, 18 (40), 1–6. [Google Scholar]
- (77).Rasmussen CE; Williams CKI Gaussian Processes for Machine Learning; Gaussian, 2006; Vol. 14 DOI: 10.1142/S0129065704001899. [DOI] [Google Scholar]
- (78).Hensman J; Fusi NON; Lawrence ND Gaussian Processes for Big Data. In Proceedings of the Twenty-Ninth Conference on Uncertainty in Artificial Intelligence UAI’13; AUAI Press: Arlington, VA, 2013; pp 282–290. [Google Scholar]
- (79).Penfold CA; Sybirna A; Reid JE; Huang Y; Wernisch L; Ghahramani Z; Grant M; Surani MA Branch-Recombinant Gaussian Processes for Analysis of Perturbations in Biological Time Series. Bioinformatics 2018, 34 (17), i1005–i1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Swain PS; Stevenson K; Leary A; Montano-Gutierrez LF; Clark IBN; Vogel J; Pilizota T Inferring Time Derivatives Including Cell Growth Rates Using Gaussian Processes. Nat. Commun 2016, 7 (1), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Wang C; Scott SM; Subramanian K; Loguercio S; Zhao P; Hutt DM; Farhat NY; Porter FD; Balch WE Quantitating the Epigenetic Transformation Contributing to Cholesterol Homeostasis Using Gaussian Process. Nat. Commun 2019, 10 (1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Murray LA; Sheng X; Cristea IM Orchestration of Protein Acetylation as a Toggle for Cellular Defense and Virus Replication. Nat. Commun 2018, DOI: 10.1038/s41467-018-07179-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Weekes MP; Tomasec P; Huttlin EL; Fielding CA; Nusinow D; Stanton RJ; Wang ECY; Aicheler R; Murrell I; Wilkinson GWG; Lehner PJ; Gygi SP Quantitative Temporal Viromics: An Approach to Investigate Host-Pathogen Interaction. Cell 2014, 157 (6), 1460–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Das S; Vasanji A; Pellett PE Three-Dimensional Structure of the Human Cytomegalovirus Cytoplasmic Virion Assembly Complex Includes a Reoriented Secretory Apparatus. J. Virol 2007, 81, 11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Das S; Pellett PE Spatial Relationships between Markers for Secretory and Endosomal Machinery in Human Cytomegalovirus-Infected Cells versus Those in Uninfected Cells. J. Virol 2011, 85 (12), 5864–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Procter DJ; Banerjee A; Nukui M; Kruse K; Gaponenko V; Murphy EA; Komarova Y; Walsh D The HCMV Assembly Compartment Is a Dynamic Golgi-Derived MTOC That Controls Nuclear Rotation and Virus Spread. Dev. Cell 2018, 45 (1), 83–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Carter DM; Westdorp K; Noon KR; Terhune SS Proteomic Identification of Nuclear Processes Manipulated by Cytomegalovirus Early during Infection. Proteomics 2015, 15 (12), 1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Choi HJ; Park A; Kang S; Lee E; Lee TA; Ra EA; Lee J; Lee S; Park B Human Cytomegalovirus-Encoded US9 Targets MAVS and STING Signaling to Evade Type i Interferon Immune Responses. Nat. Commun 2018, DOI: 10.1038/s41467-017-02624-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Gao Y; Kagele D; Smallenberg K; Pari GS Nucleocytoplasmic Shuttling of Human Cytomegalovirus UL84 Is Essential for Virus Growth. J. Virol 2010, 84 (17), 8484–8494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Alwine JC The Human Cytomegalovirus Assembly Compartment: A Masterpiece of Viral Manipulation of Cellular Processes That Facilitates Assembly and Egress. PLoS Pathog 2012, 8 (9), No. e1002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Mendik P; Dobronyi L; Hári F; Kerepesi C; Maia-Moço L; Buszlai D; Csermely P; Veres DV Translocatome: A Novel Resource for the Analysis of Protein Translocation between Cellular Organelles. Nucleic Acids Res 2019, 47 (D1), D495–D505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Dassa L; Seidel E; Oiknine-Djian E; Yamin R; Wolf DG; Le-Trilling VTK; Mandelboim O The Human Cytomegalovirus Protein UL148A Downregulates the NK Cell-Activating Ligand MICA To Avoid NK Cell Attack. J. Virol 2018, 92 (17), e00162–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Paulus C; Krauss S; Nevels M A Human Cytomegalovirus Antagonist of Type I IFN-Dependent Signal Transducer and Activator of Transcription Signaling. Proc. Natl. Acad. Sci. U. S. A 2006, 103 (10), 3840–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Collins-McMillen D; Stevenson EV; Kim JH; Lee B-J; Cieply SJ; Nogalski MT; Chan GC; Frost RW; Spohn CR; Yurochko AD Human Cytomegalovirus Utilizes a Nontraditional Signal Transducer and Activator of Transcription 1 Activation Cascade via Signaling through Epidermal Growth Factor Receptor and Integrins To Efficiently Promote the Motility, Differentiation, and Polarization of Infected Monocytes. J. Virol 2017, DOI: 10.1128/JVI.00622-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Cepeda V; Esteban M; Fraile-Ramos A Human Cytomegalovirus Final Envelopment on Membranes Containing Both Trans -Golgi Network and Endosomal Markers. Cell. Microbiol 2010, 12 (3), 386–404. [DOI] [PubMed] [Google Scholar]
- (96).Yu Y; Maguire TG; Alwine JC Human Cytomegalovirus Infection Induces Adipocyte-Like Lipogenesis through Activation of Sterol Regulatory Element Binding Protein 1. J. Virol 2012, 86 (6), 2942–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Gudleski-O’Regan N; Greco TM; Cristea IM; Shenk T Increased Expression of LDL Receptor-Related Protein 1 during Human Cytomegalovirus Infection Reduces Virion Cholesterol and Infectivity. Cell Host Microbe 2012, 12 (1), 86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Oberstein A; Shenk T Cellular Responses to Human Cytomegalovirus Infection: Induction of a Mesenchymal-to-Epithelial Transition (MET) Phenotype. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (39), E8244–E8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Luo J; Yang H; Song BL Mechanisms and Regulation of Cholesterol Homeostasis. Nat. Rev. Mol. Cell Biol 2020, 21, 225–245. [DOI] [PubMed] [Google Scholar]
- (100).Hare JF Compartmentation and Turnover of the Low Density Lipoprotein Receptor in Skin Fibroblasts. J. Biol. Chem 1990, 265 (35), 21758–21763. [PubMed] [Google Scholar]
- (101).Indran SV; Ballestas ME; Britt WJ Bicaudal D1-Dependent Trafficking of Human Cytomegalovirus Tegument Protein Pp150 in Virus-Infected Cells. J. Virol 2010, 84 (7), 3162–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Sanchez V; Sztul E; Britt WJ Human Cytomegalovirus Pp28 (UL99) Localizes to a Cytoplasmic Compartment Which Overlaps the Endoplasmic Reticulum-Golgi-Intermediate Compartment. J. Virol 2000, 74 (8), 3842–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Strazic Geljic I; Kucan Brlic P; Angulo G; Brizic I; Lisnic B; Jenus T; Juranic Lisnic V; Pietri G. Pietro; Engel P; Kaynan N; Zeleznjak J; Schu P; Mandelboim O; Krmpotic A; Angulo A; Jonjic S; Lenac Rovis T Cytomegalovirus Protein M154 Perturbs the Adaptor Protein-1 Compartment Mediating Broad-Spectrum Immune Evasion. eLife 2020, DOI: 10.7554/eLife.50803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Noble B; Abada P; Nunez-Iglesias J; Cannon PM Recruitment of the Adaptor Protein 2 Complex by the Human Immunodeficiency Virus Type 2 Envelope Protein Is Necessary for High Levels of Virus Release. J. Virol 2006, 80 (6), 2924–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Agrawal T; Schu P; Medigeshi GR Adaptor Protein Complexes-1 and 3 Are Involved at Distinct Stages of Flavivirus Life-Cycle. Sci. Rep 2013, DOI: 10.1038/srep01813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Eggers M; Bogner LE; Agricola B; Kern HF; Radsak K Inhibition of Human Cytomegalovirus Maturation by Brefeldin A. J. Gen. Virol 1992, 73, 2679. [DOI] [PubMed] [Google Scholar]
- (107).Archer MA; Brechtel TM; Davis LE; Parmar RC; Hasan MH; Tandon R Inhibition of Endocytic Pathways Impacts Cytomegalovirus Maturation. Sci. Rep 2017, DOI: 10.1038/srep46069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Moorman NJ; Sharon-Friling R; Shenk T; Cristea IM A Targeted Spatial-Temporal Proteomics Approach Implicates Multiple Cellular Trafficking Pathways in Human Cytomegalovirus Virion Maturation. Mol. Cell. Proteomics 2010, 9 (5), 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Angelova M; Zwezdaryk K; Ferris M; Shan B; Morris CA; Sullivan DE Human Cytomegalovirus Infection Dysregulates the Canonical Wnt/β-Catenin Signaling Pathway. PLoS Pathog 2012, 8 (10), No. e1002959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Langemeijer EV; Slinger E; de Munnik S; Schreiber A; Maussang D; Vischer H; Verkaar F; Leurs R; Siderius M; Smit MJ Constitutive SS-Catenin Signaling by the Viral Chemokine Receptor US28. PLoS One 2012, 7 (11), No. e48935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Togi S; Ikeda O; Kamitani S; Nakasuji M; Sekine Y; Muromoto R; Nanbo A; Oritani K; Kawai T; Akira S; Matsuda T Zipper-Interacting Protein Kinase (ZIPK) Modulates Canonical Wnt/β-Catenin Signaling through Interaction with Nemo-like Kinase and T-Cell Factor 4 (NLK/TCF4). J. Biol. Chem 2011, 286 (21), 19170–19177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Drayman N; Patel P; Vistain L HSV-1 Single-Cell Analysis Reveals the Activation of Anti-Viral and Developmental Programs in Distinct Sub-Populations. eLife 2019, DOI: 10.7554/eLife.46339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).More S; Yang X; Zhu Z; Bamunuarachchi G; Guo Y; Huang C; Bailey K; Metcalf JP; Liu L Regulation of Influenza Virus Replication by Wnt/β-Catenin Signaling. PLoS One 2018, 13 (1), No. e0191010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Hagerty L; Weitzel DH; Chambers J; Fortner CN; Brush MH; Loiselle D; Hosoya H; Haystead TAJ ROCK1 Phosphorylates and Activates Zipper-Interacting Protein Kinase. J. Biol. Chem 2007, 282 (7), 4884–4893. [DOI] [PubMed] [Google Scholar]
- (115).Eliyahu E; Tirosh O; Dobesova M; Nachshon A; Schwartz M; Stern-Ginossar N Rho-Associated Coiled-Coil Kinase 1 Translocates to the Nucleus and Inhibits Human Cytomegalovirus Propagation. J. Virol 2019, x DOI: 10.1128/JVI.00453-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figures and figure legends as referenced in the text (PDF)
Table S10, results of gene ontology analysis of translocating proteins identified in the analysis of Jean Beltran et al.34(XLSX)
Table S9, classifier prediction results for Jean Beltran et al.34 (XLSX)
Table S5, results of gene ontology analysis of translocating proteins identified in the Gilbertson et al.53 analysis (XLSX)
Table S11, poteins identified to co-translocate by TRANSPIRE (including STRINGdb interaction scores if applicable) (XLSX)
Table S6, classifier prediction results for Gilbertson et al.53 (XLSX)
Table S3, experimentally generated list of genes expressed in MRC5 human fibroblast cells used as the background proteome for gene ontology analysis in HCMV-infected cells (XLSX)
Table S2, Gilbertson et al.53 data set formatted for direct pipeline analysis with associated organelle markers for HEK293T cells (XLSX)
Table S1, Jean Beltran et al.34 data set formatted for direct pipeline analysis with associated organelle markers for human fibroblast cells (XLSX)
Table S8, model performance on held-out test data for analysis of data reported in Jean Beltran et al.34 (XLSX)
Table S4, model performance on held-out test data for analysis of data reported in Gilbertson et al.53 (XLSX)
Table S7, synthetic translocation profiles generated using organelle marker proteins used to assess model performance on of data reported in Jean Beltran et al. 201634 (XLSX)