

Abstract
Background
Deterioration of ionized calcium (Ca2+) handling in neurons could lead to neurodegenerative disease. Magnesium (Mg) antagonizes Ca during many physiologic activities, including energy metabolism and catalyzation of demethylation from 5-methylcytosine(5-mC) to 5-hydroxymethylcytosine(5-hmC).
Objective
To test the hypothesis that actively reducing the Ca:Mg intake ratio in the diet through Mg supplementation improves cognitive function, and to test whether this effect is partially mediated by modified cytosines in Apolipoprotein E(APOE).
Methods
This study is nested within the Personalized Prevention of Colorectal Cancer Trial (PPCCT), a double-blind 2x2 factorial randomized controlled trial, which enrolled 250 participants from Vanderbilt University Medical Center. Target doses for both Mg and placebo arms were personalized.
Results
Among those aged >65 years old who consumed a high Ca:Mg ratio diet, we found that reducing the Ca:Mg ratio to around 2.3 by personalized Mg supplementation significantly improved cognitive function by 9.1% (p=0.03). We also found that reducing the Ca:Mg ratio significantly reduced 5-mC at the cg13496662 and cg06750524 sites only among those aged >65 years old (p values=0.02 and 0.03, respectively). Furthermore, the beneficial effect of reducing the Ca:Mg ratio on cognitive function in those aged over 65 years was partially mediated by reductions in 5-mC levels (i.e. cg13496662 and cg06750524) in APOE (p for indirect effect=0.05).
Conclusions
Our findings suggest that, among those age 65 and over with a high dietary Ca:Mg ratio, optimal Mg status may improve cognitive function partially through modifications in APOE methylation. These findings, if confirmed, have significant implications for the prevention of cognitive aging and Alzheimer’s disease.
Keywords: magnesium, calcium, ratio, APOE methylation, cognitive function, mediation analysis
INTRODUCTION
Between 2000 and 2015, mortality due to Alzheimer’s disease (AD) increased by 123% [1]. No drugs have yet been approved to stop or slow the progression of AD [1]. A delay of five years in the expression of AD would reduce the incidence rate by half [2;3]. Thus, it is critical to develop novel prevention strategies to delay the onset of this common disease.
The calcium (Ca) hypothesis [4] suggests that deterioration of Ca2+ handling in neurons could lead to Alzheimer’s disease and brain aging through dendrite pruning, synapse loss, aggregated Aβ (amyloid β-peptide), tau, p-tau, inflammation, mitochondrial dysfunction and oxidative stress. Thus, delaying the deterioration of Ca2+ handling in neurons may be a promising strategy to reduce the incidence of cognitive decline and AD. It is known that magnesium (Mg) is a natural physiologic blocker of Ca2+ channels [5;6]. However, no study has examined the Ca:Mg balance in the etiology of cognitive function or AD [4]. Recently, we reported that reducing Ca:Mg intake ratios to around 2.3 in the diet among those with high Ca:Mg ratios optimized vitamin D status [7-10]. Levels of vitamin D, essential for the maintenance of Ca homeostasis [11], have been linked to the risk for AD and dementia [12;13]. We also have shown. in a population with very low dietary Ca:Mg intake ratios (median=1.7), that when Ca:Mg intake ratios were ≤1.7, high dietary Mg intake was significantly associated with increased risks of total mortality and mortality due to cardiovascular disease. Conversely, when Ca:Mg intake ratios were >1.7, intakes of Ca and Mg were associated with reduced risks of total mortality and mortality due to coronary disease [14]. It is known that cardiovascular disease is strongly linked to AD and dementia [15]. In the United States, 76% of the general adult population have a Ca:Mg intake ratio of ≥ 2.6 [7].
Previous studies found that the Apolipoprotein E (APOE)-ε4 allele, the most important genetic factor for sporadic or late-onset AD (LOAD) [16], may contribute to Ca2+ dysregulation [4]. Further, the expression of APOE-ε4 predisposes neurons to Ca2+ dysregulation and, in turn, cell death [17]. However, genotype is as yet unmodifiable. A previous study indicated that increased APOE methylation may serve as a biologic mechanism for the age-related effect of APOE genotype [18]. A recent study found that increased APOE methylation in blood DNA was associated with reduced cognitive function during normal cognitive aging [19]. It is known that DNA methylation changes are inducible by environmental exposures, including nutrients [20;21] and reversible when the exposure disappears [21]. Thus, reducing APOE methylation may serve as a promising strategy for correcting Ca2+ dysregulation and the eventual prevention of AD and cognitive decline.
In addition to being a natural physiologic blocker of Ca2+ channels [5;6], Mg2+ antagonizes Ca2+ in reabsorption [22-25], inflammatory responses [26-29], and many other physiologic activities [5;26;30-40]. Mg also plays an essential role in energy metabolism, including all enzymes utilizing or synthesizing adenosine triphosphate (ATP), and those metabolizing glucose and catalyzing glycolysis [31] and regulating insulin [41-45]. Furthermore, ATP has to bind to a Mg ion to be biologically active [46-48]. Finally, Mg plays a critical role in upregulating insulin-independent glucose transporter (GLUT3) [49] and insulin-responsive GLUT4 [50;51]. Thus, it is not surprising that Mg also affects the metabolism of α-ketoglutarate [52;53], an essential compound for the citric acid cycle. α -ketoglutarate is one key factor for ten-eleven translocations (Tets), [54] which catalyze the oxidation of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC), an active demethylation pathway [55]. Thus, it is possible that improved Mg status may reduce the methylation in APOE. 5-mC is often linked to suppressed expression, [56;57] whereas 5-hmC is specifically enriched in expressed genes, making open chromatin regions. This may play a critical role in activating and/or maintaining gene expression [58;59]. Improved Mg status may therefore reduce 5-mC modification in APOE. However, without differentiating 5-mC from 5-hmC, the results could be contradictory or diluted. In the current study, we examined whether Mg supplementation reduced 5-mC methylation in the APOE gene compared to placebo by using the state-of-the-art TET-assisted bisulfite (TAB)-Array to differentiate 5-mC from 5-hmC signals at base resolution [60].
One randomized trial found that Mg supplementation significantly improved cognitive function among older participants with self-reported cognitive complaints [61]. However, Ca:Mg balance was not considered in this trial. High Ca supplementation consistently increases urinary excretion of Mg in previous randomized trials [22-25]. Kidney reabsorption of Mg plays a major role in maintaining Mg homeostasis, [62] with kidney function declining with age [63]. Thus, reducing the Ca:Mg ratio may improve body Mg status among older individuals who consume high Ca:Mg ratio diets. Furthermore, the incidence of aging-related cognitive decline increases exponentially after age 65 [64;65]. In this regard, we hypothesize that elevated body Mg status caused by reducing the dietary Ca:Mg ratio improves metabolic changes and, in turn, Ca2+ dysregulation. This subsequently improves cognitive function among older people who consume high Ca:Mg ratio diets. Metabolic changes are considered the upstream event of compromised neuronal Ca2+ handling [32] (see Figure 1). Furthermore, in the current study, we focus on testing the hypothesis that one possible pathway by which increased Mg status improves metabolic changes and, in turn, Ca2+ dysregulation is through modifying APOE methylation (i.e. 5-mC and 5-hmC). We also hypothesize that the effect of Mg supplementation on cognitive function may interact with age, being present in older but not younger participants.
Figure 1.
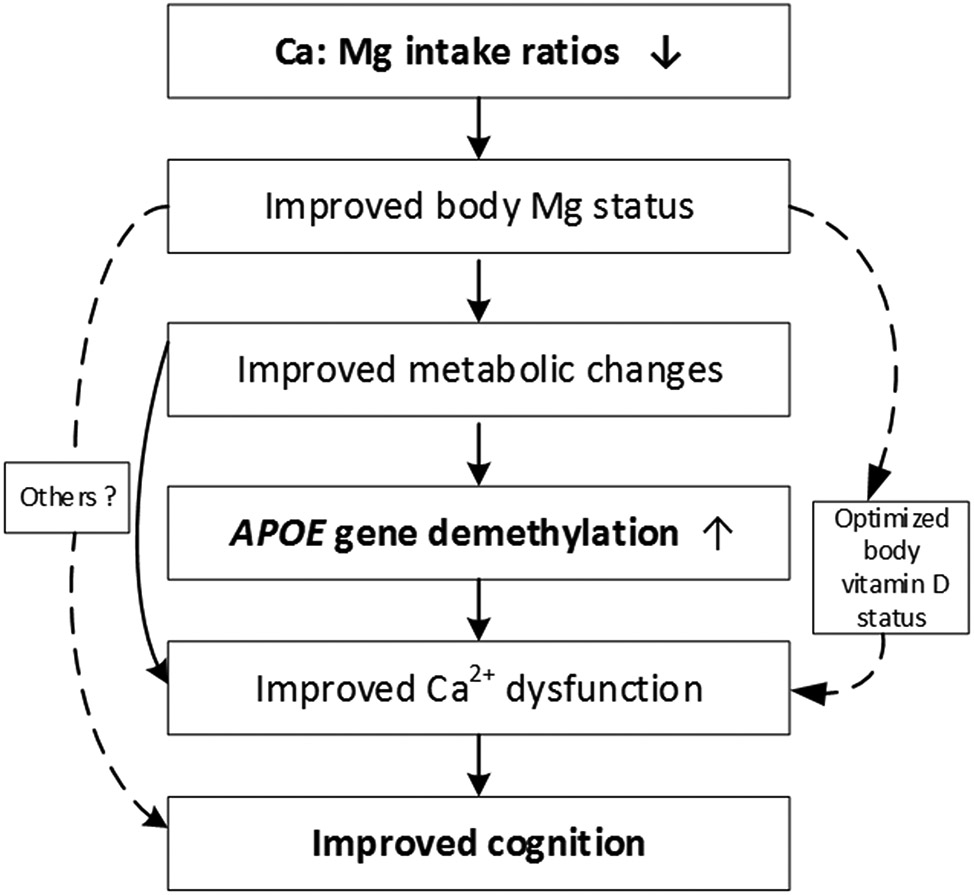
Directed Acyclic Graph To Illustrate Potential Causal Pathways
(Bolded are the focuses to be tested in the current study)
MATERIALS AND METHODS
Participants and randomization.
This is an ancillary study nested in the parent study, the “Personalized Prevention of Colorectal Cancer Trial” (PPCCT, NCT01105169 at ClinicalTrials.gov). The PPCCT is a double-blind 2×2 factorial randomized controlled trial conducted at Vanderbilt University Medical Center, Nashville, TN. The detailed design has been reported previously [7;8]. In brief, participants aged 40 to 85 years were recruited from Vanderbilt patient sources including: 1) 236 individuals with adenomas or hyperplastic polyps diagnosed from 1998 to 2014 or 2) 14 polyp-free individuals with a high risk of colorectal cancer. Detailed inclusion and exclusion criteria have been reported [7;8].
Two 24-hour dietary recalls, which recorded all foods and beverages and their amounts over the 24-hour period, were performed for all participants at the baseline of the PPCCT. We then calculated nutrient intake levels of Ca, Mg and the Ca:Mg intake ratios for each 24-hour dietary recall. The mean intake from these two recalls was used to estimate the baseline intakes of Ca, Mg and the Ca:Mg ratio. For example, an individual had dietary intake levels of Ca and Mg at 1200 mg/day and 300 mg/day, respectively, with a Ca:Mg intake ratio of 4. Three sizes of Mg glycinate capsules were used in this study. Identical-appearing placebo (made from lactose) were made to match these three magnesium capsules. The participant was assigned to a personalized dose of Mg supplementation that would reduce the Ca:Mg intake ratio to around 2.3, suggested by previous studies [6;7;14;66-69]. Identical-appearing placebos were made to match Mg capsules. Mg glycinate and placebo (i.e. microcrystalline cellulose) were provided in capsules. The capsules were filled by the Vanderbilt Investigational Pharmacy personnel following USP 797 conditions according to the compounding instructions. The intervention period was 12 weeks.
To reduce the Ca:Mg ratio from 4 to the target ratio of 2.3, the participant was assigned to a dose (e.g., in this example, 221.7 mg/day) that would reduce the Ca:Mg intake ratio. Each dose corresponds to one or multiple capsules from three different sizes of magnesium supplementation or placebo. Based on a participant’s baseline intakes of Ca and Mg as well as their Ca:Mg intake ratio, eligible participants were those who had a Ca intake ≥ 700 mg/day and < 2000 mg/day and in whom the Ca:Mg intake ratio was ≥ 2.6. Eligible subjects were assigned to Mg treatment or placebo according to the randomization schedule. Participants, study investigators and staff were blinded to the assigned interventions.
The Vanderbilt Clinical Pharmacist in the Investigational Drug Service dispensed the capsules. Blinding was implemented through the Vanderbilt Investigational Drug Service (VIDS). Four additional 24-hour dietary recalls were conducted for all participants during the intervention period with two during weeks 1 to 6 and the other two during weeks 6 to 12. Blood samples were collected and processed at each clinic visit. Anthropometric measurements (weight, height, waist and hip circumference) were measured at least twice at each clinic visit.
265 participants were randomized and allocated to the Mg treatment arm or the placebo arm. 15 participants withdrew consent before taking Mg treatment or placebo. Of these, 250 randomized participants started treatments, and 239 completed the trial, with 11 participants finishing part of the study before withdrawing [7]. Six of the withdrawals were due to self-reported adverse events (four withdrawals in the treatment arm and two in the placebo arm). One of them had donated blood at baseline and at the end of the trial. Therefore, in the current study, 240 participants were included who had blood collected at baseline and at the end of the trial.
Measurement of cognitive function and blood pressure
Cognitive function was measured by the Montreal Cognitive Assessment (MoCA) [70]. The MoCA was developed as a brief but sensitive screening instrument to detect mild cognitive impairment [70]. It measures the following cognitive domains: attention and concentration, executive function, memory, language, visuoconstructional skills, conceptual thinking, calculation, and orientation. IRB approval was obtained to add the MoCA component on Dec.11 2012, which was about 21 months after the trial started. During the clinic visit, participants were asked to complete the MoCA at baseline and the end of the trial, which was administered by trained staff. From Dec. 12, 2012 to Jan 30, 2016, we enrolled 129 participants in the ancillary study of cognitive function (Figure 1). Of these, we measured pre-and post- treatment MoCA for 95.3% (i.e. 123 participants). The mean (standard deviation) days between the first test and the last test was 86.2 (sd=8.8) days. Blood pressures including both systolic blood pressure and diastolic blood pressure were measured for all participants at baseline.
Measure of 5-mC and 5-hmC at single resolution for the APOE gene.
All 240 participants who were enrolled in the PPCCT and had blood DNA samples available at the baseline and the end of the trial were included in the current study to examine the effect of reducing the Ca:Mg ratio on methylation modifications in the APOE gene. In order to minimize potential errors caused by batch effects, samples were randomly organized in treatment-placebo (i.e. one treatment arm with one placebo arm) sets (4 samples in each set: 2 from pre-, and 2 from post-treatment). Lab staff were blinded to sample status (i.e. treatment vs. placebo and pre-treatment vs. post-treatment status).
Genomic DNA was extracted from buffy coat fractions collected using a QIAamp DNA mini-kit (Qiagen Inc, Germantown, MD) according to the manufacturer's protocol [66]. DNA quality was examined using standard molecular biology protocols. We used the TET-assisted bisulfite (TAB)-Array, which combines TET-assisted bisulfite conversion with the Infinium Methylation EPIC array (EPIC array) to differentiate 5-hmC and 5-mC signals at base resolution [60]. Our detailed approach was reported previously [60;71]. Using this technique, we were able to measure conventional bisulfite conversion-based (BS) 5-mC biomarkers as well as both 5-mC from 5-hmC biomarkers simultaneously.
The β-values for 5-mC and 5-hmC were estimated using the Maximum Likelihood Estimate from the paired bisulfite conversion and TAB-treated samples [71]. The following quality control steps were taken: 1) We excluded low-quality probes where the number of beads < 3 or the detection P-value > 0.05 [72]; 2) Exclusion of CpGs with a detection rate <95% and samples with the percentage of low-quality methylation measurements >5% or extremely low signal of BS probes [72]; 3) Exclusion of extreme outliers, as defined by the Tukey’s method [73], based on average total signal value across CpG probes; 4) Remaining samples were preprocessed using the R package ENmix to improve accuracy and reproducibility [72]; 5) Dye bias was corrected using regression on a logarithm of internal control probes [74]; 6) Quantile-normalization of signal for Infinium I or II probes, and 7) Lastly, extreme methylation β value (i.e., the proportion of methylated signal in total signals from 0 to 1) outliers across samples, defined by Tukey’s method, were set as missing. In total, cytosine modification data for 224 participants out of 240 passed the seven quality control steps. In the current study, we kept all 15 CpG sites related to the APOE gene after quality control for 224 participants.
Statistical analyses.
Continuous demographic variables (mean ± standard deviation) and categorical demographic variables (percent) were compared between treatment and placebo arms (Table 1). In Supplemental Table S1, we compare the demographic variables between all participants (N=240) and those with MoCA scores (n=123). The Wilcoxon rank sum test was conducted to evaluate continuous variables, while Pearson chi-squared tests were conducted to compare categorical variables. Since aging-related reduction in cognitive function most often occurs after age 65 [64;65], we examined the ages from 60 to 70 years old as potential cut-points in the stratified analyses by age. Linear regression models were fitted to examine the effect of Mg treatment on changes of MoCA score and cytosine modifications (BS, 5-mC and 5-hmC) in the APOE gene adjusting for age, sex, education and baseline MoCA score or baseline levels of cytosine modification biomarkers, respectively. Causal Mediation Analysis was used to examine whether the reduction in 5-mC methylation levels in the APOE gene mediated the effect of reducing Ca:Mg ratio on the improvement of MoCA score [75]. The CAUSALMED procedure in SAS was used to implement the regression adjustment method to estimate causal mediation effects. All P values are two sided and statistical significance was determined using an alpha level of 0.05. The data analyses used software SAS Enterprise Guide 7.1.
Table 1.
Descriptive characteristics of treatment vs. placebo at baseline
| Placebo (N=64) |
Treatment (N=59) |
P value | |
|---|---|---|---|
| Age, year | 60.1±8.1 | 60.1±7.6 | 0.731 |
| Age >65 year (%) | 25.0 | 27.1 | 0.79 |
| Sex - male (%) | 43.7 | 44.0 | 0.972 |
| Body mass index (BMI), kg/m2 | 30.9±7.0 | 29.9±6.0 | 0.521 |
| Systolic blood pressure (mmHg) | 127.9±15.2 | 127.4±15.5 | 0.92 |
| Diastolic blood pressure(mmHg) | 75.0±8.5 | 75.3±8.8 | 0.77 |
| eGFR, ml/min/1.73m2 | 79.3±14.9 | 82.7±13.5 | 0.151 |
| Smoking status (%) | 0.902 | ||
| Never | 59.4 | 55.2 | |
| Ever | 34.4 | 37.9 | |
| Current | 6.2 | 6.9 | |
| Drinking status (%) | 0.212 | ||
| Never | 39.0 | 35.6 | |
| Ever | 21.9 | 11.9 | |
| Current | 39.1 | 52.5 | |
| Physically active in ≥ 2 days per week (%) | 87.5 | 84.8 | 0.662 |
| Education under college (%) | 10.9 | 10.2 | 0.892 |
| Race (%) | 0.512 | ||
| White | 98.4 | 96.6 | |
| Daily nutrients intake | |||
| Total energy (kcal) | 2082.4± 663.8 | 1974.3±467.5 | 0.331 |
| Total Ca (mg) | 1283.3±359.0 | 1299.4± 330.7 | 0.681 |
| Total Mg (mg) | 336.8± 100.9 | 350.4±81.0 | 0.231 |
| Ca:Mg intake ratio | 4.0±1.8 | 3.8±1.1 | 0.491 |
| Seasons (%) | |||
| Spring | 20.3 | 20.3 | 0.922 |
| Summer | 43.7 | 47.5 | |
| Fall | 14.1 | 15.2 | |
| Winter | 21.9 | 16.9 | |
| MoCA scores | 27.2±2.2 | 27.0±2.7 | 0.921 |
Continuous variables: X±SD; categorical variables: %
Tests used:
Wilcoxon test
Pearson chi-square test
eGFR: estimated glomerular filtration rate
RESULTS
We compared baseline demographic variables between all 240 participants enrolled in the parent study and 123 participants who completed both pre- and post-treatment MoCA tests (Supplemental Table S1). No significant difference was found for any demographic variables between these two groups.
In Table 1, we compare the baseline characteristics between the treatment and the placebo arm for 123 participants who completed two MoCA tests. Compared to the placebo arm, we did not find that treatment assignment was significantly different on means or distributions for baseline demographic variables, including age, sex, smoking status, alcohol drinking status, physical activity status, educational achievement, race, sample collection season, daily intake of total energy, total Mg, Ca, intake ratio of Ca to Mg and baseline overall MoCA cognitive score as well as factors related to vascular disease, including body mass index (BMI), eGFR and blood pressure (Table 1). However, age, sex and education were still adjusted for in the subsequent analyses due to their potential impact on cognitive function. Furthermore, we found that mean levels were <130 mmHg and <80 mmHg for systolic blood pressure and diastolic blood pressure, respectively. The mean daily dose of personalized Mg supplementation was 216.5 mg with a range from 77.25 mg to 389.55 mg. Compliance with the pill regimen was very high for both the placebo and treatment arms (mean (standard deviation) based on pill counts were 96.1% (8.3) and 95.9% (10.2), respectively, and p=0.37 for difference between the arms). The mean Ca:Mg ratios (standard deviations) for the treatment and placebo arms after administering Mg and placebo supplementation were 2.28 (0.12) and 4.04 (1.80) respectively (p for difference, <0.001), based on the two 24-hour dietary recalls performed at baseline and remained stable at 2.04 (0.64) and 3.57 (1.45), respectively (p for difference, <0.001) based on the four 24-hour dietary recalls conducted over the 12-week period of the trial.
We found that reducing the Ca:Mg ratio by personalized dose of Mg treatment did not significantly change overall MoCA scores compared to the placebo arm (p=0.20) (Table 2). However, based on our hypothesis, we examined whether the effect of reducing Ca:Mg ratio on MoCA scores differed by age. We compared the different ages from 60 to 70 years as an effect modifier on the Mg treatment effect on MoCA (Supplemental Table S2) and found that 65 years old (≤65 vs. >65 years old) provided the best cut-point. Using this cut-point, we found a p-value for interaction between Mg treatment and age of 0.08 for overall MoCA score. In stratified analyses by age, we found that reducing the Ca:Mg ratio significantly improved overall MoCA score compared to the placebo arm (p=0.01) (Table 2) among those aged >65 years, but had no effect among those aged ≤65 years (p=0.90). Reducing the Ca:Mg ratio improved cognitive function by 9.1% in those aged > 65 years old over the 12-week period (Table 2).
Table 2.
Changes in cognitive function by Mg treatment vs. placebo
| Cognitive function | Change from Baseline | P1 | P2 | |||
|---|---|---|---|---|---|---|
| Placebo | Change (%) | Mg Treatment | Change (%) | |||
| Total | ||||||
| Overall MoCA score (n=123) | 0.6 ± 2.6 | 2.1 | 1.1 ± 2.3 | 4.1 | 0.22 | 0.20 |
| Aged >65 years old | ||||||
| Overall MoCA (n=32) | 0.5 ± 1.9 | 1.9 | 2.3 ± 2.7 | 9.1 | 0.03 | 0.01 |
| Age≤ 65 years old | ||||||
| Overall MoCA (n=91) | 0.6 ± 2.8 | 2.4 | 0.7 ± 2.0 | 2.1 | 0.89 | 0.90 |
Mean ± standard deviation
P1: p value for unadjusted GLM model; and P2: p value for GLM model additionally adjusting for age, sex, education and baseline levels
P2: value for interaction between treatment and age: 0.08 for overall MoCA
Mg: magnesium
Shown in Table 3, Supplemental Table S3 and Supplemental Table S4 are the effect of reducing the Ca:Mg ratio on APOE cytosine modifications 5-mC, 5-hmC & BS, respectively. We found that reducing the Ca:Mg ratio did not significantly affect BS or 5-hmC methylation in the APOE gene after false discovery rate (FDR)-adjustment. However, after FDR adjustment, reducing the Ca:Mg ratio significantly reduced 5-mC methylation at the cg13496662 CpG locus, but significantly increased 5-mC methylation at the cg18768621 CpG locus compared to placebo (Table 3). Next, we examined whether age modifies the effect of reducing the Ca:Mg ratio on APOE cytosine modifications. We found that age (>65 vs. ≤65) significantly modified the effect of reducing Ca:Mg ratio on the 5-mC methylation levels at cg06750524 CpG locus (p for interaction, 0.04) (Table 4). In additional analyses, we found that this age cut-point was also the best one for the cg06750524 CpG locus (Supplemental Table S2).
Table 3.
Changes in 5-mC methylation (CpG sites) in APOE by Mg treatment vs. placebo
| CpG sites | Changes from baseline | P1 | P2 | FDR | |||
|---|---|---|---|---|---|---|---|
| Placebo | Change (%) |
Treatment | Change (%) |
||||
| cg20051876 | 0.002±0.034 −0.001(−0.020-0.018) | 0.2 | 0.000±0.029 0.002(−0.021-0.018) | 0.0 | 0.63 | 0.63 | 0.67 |
| cg14123992 | −0.002±0.039 −0.003(−0.021-0.024) | −0.2 | 0.001±0.033 0.003(−0.015-0.024) | 0.1 | 0.52 | 0.42 | 0.67 |
| cg04406254 | −0.009±0.059 −0.006(−0.044-0.025) | −1.3 | 0.004±0.047 0.004(−0.034-0.041) | 0.6 | 0.07 | 0.08 | 0.25 |
| cg26190885 | 0.002±0.015 0.000(−0.001-0.005) | −14.6 | 0.004±0.017 0.000(−0.000-0.011) | 46.9 | 0.24 | 0.28 | 0.61 |
| cg12049787 | 0.000±0.008 0.001(−0.004-0.006) | 0.0 | −0.000±0.008 0.000(−0.006-0.005) | 0.0 | 0.41 | 0.45 | 0.67 |
| cg08955609 | −0.001±0.008 −0.000(−0.005-0.004) | −11.5 | 0.001±0.008 0.000(−0.003-0.004) | 12.1 | 0.05 | 0.05 | 0.24 |
| cg18768621 | −0.003±0.025 −0.003(−0.014-0.009) | −6.3 | 0.006±0.025 0.005(−0.010-0.021) | 13.3 | 0.006 | 0.007 | 0.05 |
| cg19514613 | −0.002±0.028 −0.000(−0.015-0.013) | −1.9 | 0.006±0.027 0.007(−0.012-0.025) | 5.7 | 0.05 | 0.07 | 0.25 |
| cg06750524 | 0.006±0.071 0.007(−0.028-0.048) | 3.4 | 0.001±0.056 0.002(−0.030-0.033) | 0.6 | 0.60 | 0.58 | 0.67 |
| cg16471933 | 0.002±0.060 −0.000(−0.040-0.041) | 0.2 | −0.004±0.057 −0.001(−0.039-0.036) | −0.5 | 0.44 | 0.48 | 0.67 |
| cg05501958 | −0.001±0.018 0.001(−0.009-0.009) | −0.1 | −0.000±0.018 0.000(−0.006-0.012) | 0.0 | 0.84 | 0.91 | 0.91 |
| cg18799241 | 0.000±0.020 0.000(−0.012-0.013) | 0.0 | 0.002±0.017 0.002(−0.008-0.013) | 0.2 | 0.57 | 0.54 | 0.67 |
| cg21879725 | 0.002±0.028 0.004(−0.013-0.019) | 0.2 | −0.003±0.030 −0.003(−0.023-0.012) | −0.3 | 0.21 | 0.20 | 0.51 |
| cg17928676 | 0.002±0.027 0.003(−0.014-0.020) | 1.0 | 0.004±0.032 0.004(−0.015-0.024) | 2.0 | 0.55 | 0.59 | 0.67 |
| cg13496662 | 0.006±0.067 0.004(−0.038-0.040) | 1.8 | −0.018±0.060 −0.014(−0.058-0.017) | −5.5 | 0.005 | 0.004 | 0.05 |
Mean ± standard deviation, median (p25, p75)
P1: p value for GLM model; P2: p value for GLM model additionally adjusting for age, sex, education and baseline level
FDR: false discovery rate; Mg: magnesium; APOE: Apolipoprotein E gene
Table 4.
Mg treatment and changes in 5-mC methylation (CpG sites) in APOE stratified by age
| CpG sites | Changes from baseline | P1 | P2 | |||
|---|---|---|---|---|---|---|
| Placebo | Changes (%) | Treatment | Changes (%) | |||
| Aged >65 years old | ||||||
| cg18768621 | −0.001±0.026 −0.003(−0.014-0.015) | −2.1 | 0.006±0.029 0.003(−0.017-0.021) | 13.4 | 0.28 | 0.25 |
| cg06750524 | 0.020±0.055 0.014(−0.020-0.056) | 10.9 | −0.011±0.055 −0.006(−0.047-0.024) | −6.1 | 0.02 | 0.03 |
| cg13496662 | 0.012±0.057 0.026(−0.018-0.043) | 3.9 | −0.021±0.060 −0.023(−0.054-0.011) | −6.8 | 0.02 | 0.02 |
| Age≤ 65 years old | ||||||
| cg18768621 | −0.004±0.024 −0.004(−0.017-0.008) | 0.05 | 0.006±0.023 0.006(−0.008-0.021) | 13.2 | 0.008 | 0.006 |
| cg06750524 | −0.001±0.077 0.002(−0.040-0.043) | 0.17 | 0.006±0.056 0.002(−0.024-0.042) | 3.5 | 0.52 | 0.53 |
| cg13496662 | 0.003±0.071 −0.001(−0.048-0.039) | 0.3 | −0.017±0.061 −0.008(−0.060-0.020) | −5.0 | 0.06 | 0.06 |
Mean ± standard deviation, median (p25, p75)
P1: p value for GLM model; P2: p value for GLM model additionally adjusting for age, sex, education and baseline level P values for interactions: 0.61 for cg18768621, 0.04 for cg06750524, 0.52 for cg13496662
Mg: magnesium; APOE: Apolipoprotein E gene
In stratified analysis by age (Table 4), we found that reducing the Ca:Mg ratio only significantly reduced 5-mC methylation at the cg06750524 CpG locus (by 6.1%) in those aged >65 years old. We also conducted stratified analyses by age for the two CpG sites (cg13496662 cg18768621) that were identified in Table 3. We found that reduction of the Ca:Mg ratio significantly reduced the 5-mC levels at cg13496662 CpG locus (by 6.8%) only among those aged >65 years, and significantly increased the 5-mC levels at the cg18768621 CpG locus (13.2%) among those aged ≤65 years old (Table 4).
Since reducing Ca:Mg ratios significantly reduced both cg13496662 and cg06750524 CpG sites in the APOE gene only among those participants older than 65, we examined whether the reduction in 5-mC methylation at these two CpG sites jointly mediated the effect of reducing Ca:Mg ratio on MoCA scores. Table 5 shows results from the mediation analysis. The analysis showed that a partial mediation exists for reducing the Ca:Mg ratio on MoCA scores through a joint effect of these two CpG sites (p for indirect effect, 0.05). The indirect effect accounted for 86% of the total effect. In the mediation analysis using single 5-mC CpG site (either cg13496662 or cg06750524) in APOE, the indirect effect was not statistically significant (p values=0.28 and 0.46, respectively).
Table 5.
Total, direct and indirect effects of reducing Ca:Mg ratio on MoCA score among those aged older than 65 years
| Estimate | 95% CI* | P value | |
|---|---|---|---|
| Joint effect of cg13496662 and cg06750524 | |||
| Total effect | −1.40 | −3.06, 0.15 | 0.02 |
| Direct effect | −0.19 | −1.94, 2.18 | 0.81 |
| Indirect effect | −1.21 | −3.38, 0.57 | 0.05 |
| cg13496662 CpG site as a mediator | |||
| Total effect | −1.35 | −3.18, −0.07 | 0.02 |
| Direct effect | −1.05 | −2.76, 0.07 | 0.09 |
| Indirect effect | −0.30 | −1.43, 0.17 | 0.28 |
| cg06750524 CpG site as a mediator | |||
| Total effect | −1.48 | −3.06, 0.01 | 0.01 |
| Direct effect | −1.24 | −3.00, 0.19 | 0.06 |
| Indirect effect | −0.23 | −1.06, 0.32 | 0.46 |
Bootstrap bias corrected. Mg: magnesium; APOE: Apolipoprotein E gene
DISCUSSION
In this precision-based randomized trial conducted among participants who consumed high Ca:Mg ratio diets, we found that reducing the Ca:Mg ratio to around 2.3 by personalized Mg supplementation significantly improved cognitive function by 9.1% (p=0.03) compared to placebo only among those participants aged over 65. We also found that reducing the Ca:Mg ratio significantly reduced 5-mC levels at cg13496662 CpG site after FDR adjustment and this effect also existed only in those aged >65 years old. Older age also significantly modified the effect of reducing the Ca:Mg ratio on the 5-mC at the cg06750524 CpG site. Furthermore, we found that the beneficial effect of reducing the Ca:Mg ratio on cognitive function in those aged over 65 years old may be partially mediated by the joint effect of 5-mC modifications from the two CpG sites (i.e. cg13496662 and cg06750524) in the APOE gene in the mediation analysis. To our knowledge, no study has evaluated how to modify APOE cytosine modification, nor examined the effect of Mg treatment on DNA methylation. This is also the first study to examine the effect of modulating the Ca:Mg ratio on cognitive function in those who consumed high Ca:Mg ratio diets.
LOAD comprises 95% of all AD cases [1;76]. In addition to LOAD, the APOE ε4 allele has been linked to an increased risk for atherosclerosis, [77] which contributes to cognitive decline and dementia [78]. One common major risk factor for both LOAD [1;76] and atherosclerosis [79] is advanced age. Aging-related reductions in cognitive function increase exponentially after age 65 [64;65]. Thus, there is biologic plausibility for the effect of reducing the Ca:Mg ratio on cognitive function improvement among older participants. Furthermore, kidney function declines with aging [63]. The loss of Mg in urinary excretion due to a high Ca:Mg ratio may be compensated for by better kidney function among younger participants (≤65 years old) [80].
A promising finding from this study is that the beneficial effect of reducing the Ca:Mg ratio on cognitive function in those aged >65 years old may be partially mediated by the joint effect of 5-mC modifications on two CpG sites (i.e. cg13496662 and cg06750524) in the APOE gene in the mediation analysis. Besides age, the APOE ε4 allele is the most important risk factor for LOAD [16]. Although APOE genotype cannot be modified, not everyone carrying the ε4 allele develops LOAD [81]. Thus, modifying the APOE phenotype is a very promising strategy for the prevention of cognitive decline and LOAD. Interestingly, a recent study found that an increased APOE methylation at multiple CpG sites in blood DNA was associated with reduced cognitive function in African Americans during normal cognitive aging [19]. Furthermore, a large study found that APOE methylation was inversely correlated with gene expression [18] while APOE methylation determined in blood DNA was positively correlated with age and there was an interaction between increasing age and APOE genotype on levels of APOE methylation [18]. In particular, the cg06750524 CpG site was significantly linked to APOE variants (ε2 carriers < ε3/ ε3 < ε4 carriers, p=3.51×10−5). Further, the study observed that APOE methylation patterns were similar across multiple cell types, including brain and blood. Strikingly, the study was consistent with the fact that two single nucleotide polymorphisms (SNPs) determining APOE ε4 variants were CpG-related SNPs and that ε4 carriers have the largest number of CpG sites, including the cg06750524 CpG site [18]. The study found additionally that methylation in cg06750524 was the only CpG site positively associated with low-density lipoprotein (LDL)-cholesterol (c) (p=0.01) [18]. Interestingly, we found that the level of 5-mC at the cg06750524 CpG site was significantly reduced in those aged >65 years old with lowered Ca:Mg ratio compared to placebo, and that the reduction in 5-mC at the cg06750524 CpG site also contributed to cognitive improvement. These findings suggest that we may be able to modify the phenotype of APOE-ε4 variants through modification of 5-mC methylation. In addition to cg06750524, in our study, we found that Mg treatment significantly reduced the 5-mC level at the cg13496662 CpG site compared to the placebo arm. cg13496662 is contained in the EPIC array we used. However, the published study used the Infinium HumanMethylation450 BeadChip array, which was the earlier version of the EPIC array, which does not contain cg13496662 [18]. Furthermore, the published study measured overall methylation, which does not differentiate 5-mC from 5-hmC [18]. Thus, future studies measuring both 5-mC and 5-hmC and covering all CpG sites in the APOE gene are warranted to further understand the functional significance of our findings.
Unlike the aforementioned studies conducted in non-LOAD patients, four previous case-control studies were conducted to compare the methylation in the APOE gene between LOAD cases and controls. One study found elevated methylation levels in APOE in blood DNA in LOAD cases compared to their twin controls [82]. The other three studies, which used both brain tissue DNA and blood DNA, found differences in methylation in the APOE gene between cases and controls [83-85]. However, these studies also found that there were differences in methylation in the APOE gene between brain DNA and blood DNA. Due to the use of the case-control study design, the temporal sequence is unclear in these studies because the difference in methylation in the APOE gene between brain and blood DNA could be caused by AD pathogenesis-related pathology in brain.
Ca2+ signaling regulates neuronal gene expression, energy production, membrane excitability, synaptogenesis, synaptic transmission, and processes supporting learning and memory as well as cell survival [86]. Due to the important role of APOE in Ca2+ signaling [4;17], reducing Ca:Mg ratios may reduce 5-mC in the APOE gene, and in turn, lead to improvements in Ca2+ dysregulation [4;17] and cognitive function. However, in the mediation analysis, we found that the effect of reducing the Ca:Mg ratio was partially mediated by the reduction in 5-mC in the APOE gene. This is consistent with the fact that as an antagonist of Ca2+, elevated Mg status improved energy metabolism. In addition, many physiologic activities related to Ca2+ [5;22-40] may be affected through not only the methylation of the APOE gene, but also through vitamin D status [7-10] or directly on Ca2+ signaling. This subsequently leads to cognitive improvement [4].
Several animal studies have found that elevated levels of Mg in brain through Mg supplementation may protect or even reverse cognitive function and synaptic loss in AD mouse models [35;38;87]. Furthermore, one human trial of Mg supplementation conducted in older adults with self-reported cognitive complaints (memory and concentration) [61] found a 19.8% improvement in overall cognitive score (p=0.047). Unlike these previous animal and human studies conducted in an AD model or at a high risk for AD, our randomized trial was conducted in individuals who were generally healthy and consumed high Ca:Mg ratio diets, a feature of Western diets compared to East Asian diets [6;14;66-69]. Thus, it is not surprising that the improvement in cognitive function (9.1%) observed in our trial is not as strong as that found in previous studies. Additionally, the effect of reducing the Ca:Mg ratio was significant only among those aged >65 years old. Our results may also provide a possible explanation for the inconsistent finding on the associations between Mg intake or serum Mg with risk of dementia or cognitive impairment observed in a few cohort studies [88] because none of those studies have considered Ca:Mg balance. Two cohort studies have been conducted specifically on AD, and both indicated possible U-shaped associations. The Rotterdam Study, which measured serum Mg [89], found that both low serum Mg (≤0.79 mmol/L) or high serum Mg (≥0.90 mmol/L) were associated with an increased risk for dementia and AD. The Hisayama Study in Japan evaluated dietary intake of Mg [90] and found that only the middle two quartiles of Mg intake, but not the highest quartile, were associated with more than a 40% reduced risk for AD compared with the lowest intake quartile. It is possible that the U-shaped associations between Mg and AD risk are due to the modifying effect of the Ca:Mg ratio.
The current study has several strengths, including the double-blinded randomized and placebo-controlled design. Furthermore, a precision-based design is utilized. Thus, all the background intakes of Mg and Ca from both diet and supplements were measured twice before and four times during the treatment and a personalized dosing strategy of Mg supplementation was administered to each participant. We found that the Ca:Mg ratios remained stable over the 12-week study period. In addition, we had a high compliance with the study medication and the dropout rate was very low. The study has some weaknesses though. The primary concern is that in this ancillary study, cognitive function was only measured in a little over half of participants from the parent study. We found no significant differences for any demographic variables between all participants enrolled in the parent study and the participants who completed both pre- and post-treatment MoCA tests. However, our study may be underpowered. For example, the effects of reducing the Ca:Mg ratio on single cognitive domains were not significant. Also, we found that Mg treatment significantly reduced the 5-mC at both cg06750524 and cg13496662 CpG sites compared to the placebo arm in all participants. However, in the mediation analysis in the subset with cognitive function measures, we found that the single CpG site did not significantly contribute to the indirect effect. The non-significant effects could be due to small sample size. Thus, future larger studies are required to confirm these findings. Also, our findings might be explained by regression to the mean. However, we adjusted baseline levels of cognitive function and APOE cytosine modification levels in regression models and the baseline cognitive scores are not different between treatment and placebo arms. In this ancillary study, we only included participants who completed the trial and provided DNA samples at baseline and at the end of the trial, but not those who enrolled and withdrew because DNA samples were required for the assay APOE cytosine modification. Thus, the analyses were not carried out on an intention-to-treat sample. There is a possible training effect that could account for improvements in cognitive performance. However, this effect would exist in the both treatment and placebo arms and all statistical analyses were based on the comparison between the treatment arm and the placebo arm. In addition to intake of Mg, other factors such as intakes of other minerals (e.g. calcium) [23-25;27-29;91;92], alcohol drinking [93;94], absorption and reabsorption rates [28;62], and use of medications (e.g. proton pump inhibitors, and thiazide diuretics) [95-101] affect body Mg status. However, serum Mg, which is used to clinically diagnose Mg deficiency, is a poor measurement of body Mg status [101-103]. Since our study is an RCT, those factors affecting Mg and Ca absorption and reabsorption are likely balanced in the treatment and placebo arms through the randomization process. However, our findings need to be confirmed in future studies by accurately measuring body Mg status. Another weakness of the current study is that we do not have genotyping data for the APOE gene. Future studies are needed to examine whether the effect of reducing Ca:Mg intake ratios differs by APOE genotype. In the parent study, a majority of participants were previously diagnosed with colorectal adenomas or hyperplastic polyps. Although the prevalence of colorectal polyps is very high (almost 2/3 in the U.S. screening population [104]) and polyp(s) and adenoma(s) were removed when they participated in the trial, cautious interpretation of our results is warranted, particularly regarding generalization of our findings to individuals who are not diagnosed with colorectal polyp. No patients diagnosed with Alzheimer’s disease were included in our randomized trial. Thus, the effect of reducing Ca:Mg ratio on Alzheimer’s disease and the demethylation of APOE gene remains to be determined. Future longitudinal studies are required to determine whether the beneficial effect of reducing Ca:Mg intake ratios on cognitive function has a lasting effect beyond three months. Future studies are also required to specifically examine how Mg supplementation affects neuronal calcium homeostasis.
In summary, among individuals with the Ca:Mg intake ratios equal to or over 2.6, reducing Ca:Mg ratios to around 2.3 improved cognitive function among those aged >65 years old and the effect was partially affected by the reduction in 5-mC modifications (i.e. cg13496662 and cg06750524) in the APOE gene. These findings, if confirmed, have significant implications for the prevention of cognitive decline and LOAD by modifying the phenotypes of the APOE genotype.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by R01 CA149633 (to Qi Dai & Chang Yu) and R01 CA202936 (to Qi Dai & Lifang Hou) from the National Cancer Institute, Department of Health and Human Services as well as the Ingram Cancer Center Endowment Fund. Data collection, sample storage and processing for this study were partially conducted by the Survey and Biospecimen Shared Resource, which is supported in part by P30CA68485. Clinical visits to the Vanderbilt Clinical Research Center were supported in part by the Vanderbilt CTSA grant UL1 RR024975 from NCRR/NIH. The parent study data were stored in Research Electronic Data Capture (REDCap) and data analyses (VR12960) were supported in part by the Vanderbilt Institute for Clinical and Translational Research (UL1TR000445).
Abbreviations
- AD
Alzheimer’s disease
- APOE
Apolipoprotein E
- BMI
body mass index
- Ca
calcium
- CD
cognitive decline
- CI
confidence interval
- CV
coefficients of variation
- MoCA
Montreal Cognitive Assessment
- Mg
magnesium
- NCI
National Cancer Institute
- PPCCT
Personalized Prevention of Colorectal Cancer Trial
- QC
quality control
Footnotes
Clinical Trial Registry number and website: #100106 https://clinicaltrials.gov/ct2/show/NCT03265483
Author disclosures: All authors have no conflicts of interest
Reference
- (1).Alzheimer's Association (2018) 2018 Alzheimer's disease facts and figures. Alzheimers Dement 14, 367–429. [Google Scholar]
- (2).Brookmeyer R, Gray S, Kawas C (1998) Projections of Alzheimer's disease in the United States and the public health impact of delaying disease onset. Am J Public Health 88, 1337–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hake AM, Schere P (2000) On the brink of the pandemic: Epidemiology and risk factors for Alzheimer's Summaries from the World Alzheimer's Congress 2000, Washington, DC. [Google Scholar]
- (4).Alzheimer's Association Calcium Hypothesis Workgroup (2017) Calcium Hypothesis of Alzheimer's disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement 13, 178–182. [DOI] [PubMed] [Google Scholar]
- (5).Iseri LT, French JH (1984) Magnesium: nature's physiologic calcium blocker. Am Heart J 108, 188–193. [DOI] [PubMed] [Google Scholar]
- (6).Rosanoff A, Dai Q, Shapses SA (2016) Essential Nutrient Interactions: Does Low or Suboptimal Magnesium Status Interact with Vitamin D and/or Calcium Status? Adv Nutr 7, 25–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Dai Q, Zhu X, Manson JE, Song Y, Li X, Franke AA, Costello RB, Rosanoff A, Nian H, Fan L, Murff H, Ness RM, Seidner DL, Yu C, Shrubsole MJ (2018) Magnesium status and supplementation influence vitamin D status and metabolism: results from a randomized trial. Am J Clin Nutr 108, 1249–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Liu S, Liu Q (2018) Personalized magnesium intervention to improve vitamin D metabolism: applying a systems approach for precision nutrition in large randomized trials of diverse populations. Am J Clin Nutr 108, 1159–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Deng X, Song Y, Manson JE, Signorello LB, Zhang SM, Shrubsole MJ, Ness RM, Seidner DL, Dai Q (2013) Magnesium, vitamin D status and mortality: results from US National Health and Nutrition Examination Survey (NHANES) 2001 to 2006 and NHANES III. BMC Medicine 11, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zittermann A (2013) Magnesium deficit ? overlooked cause of low vitamin D status? BMC Medicine 11, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Uwitonze AM, Razzaque MS (2018) Role of Magnesium in Vitamin D Activation and Function. J Am Osteopath Assoc 118, 181–189. [DOI] [PubMed] [Google Scholar]
- (12).Littlejohns TJ, Henley WE, Lang IA, Annweiler C, Beauchet O, Chaves PH, Fried L, Kestenbaum BR, Kuller LH, Langa KM, Lopez OL, Kos K, Soni M, Llewellyn DJ (2014) Vitamin D and the risk of dementia and Alzheimer disease. Neurology 83, 920–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jayedi A, Rashidy-Pour A, Shab-Bidar S (2019) Vitamin D status and risk of dementia and Alzheimer's disease: A meta-analysis of dose-response (dagger). Nutr Neurosci 22, 750–759. [DOI] [PubMed] [Google Scholar]
- (14).Dai Q, Shu XO, Deng X, Xiang YB, Li H, Yang G, Shrubsole MJ, Ji B, Cai H, Chow WH, Gao YT, Zheng W (2013) Modifying effect of calcium/magnesium intake ratio and mortality: a population-based cohort study. BMJ Open 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Stampfer MJ (2006) Cardiovascular disease and Alzheimer's disease: common links. J Intern Med 260, 211–223. [DOI] [PubMed] [Google Scholar]
- (16).Michaelson DM (2014) APOE epsilon4: the most prevalent yet understudied risk factor for Alzheimer's disease. Alzheimers Dement 10, 861–868. [DOI] [PubMed] [Google Scholar]
- (17).Wadhwani AR, Affaneh A, Van GS, Kessler JA (2019) Neuronal apolipoprotein E4 increases cell death and phosphorylated tau release in alzheimer disease. Ann Neurol 85, 726–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ma Y, Smith CE, Lai CQ, Irvin MR, Parnell LD, Lee YC, Pham L, Aslibekyan S, Claas SA, Tsai MY, Borecki IB, Kabagambe EK, Berciano S, Ordovas JM, Absher DM et al. (2015) Genetic variants modify the effect of age on APOE methylation in the Genetics of Lipid Lowering Drugs and Diet Network study. Aging Cell 14, 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Liu J, Zhao W, Ware EB, Turner ST, Mosley TH, Smith JA (2018) DNA methylation in the APOE genomic region is associated with cognitive function in African Americans. BMC Med Genomics 11, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ryu MS, Langkamp-Henken B, Chang SM, Shankar MN, Cousins RJ (2011) Genomic analysis, cytokine expression, and microRNA profiling reveal biomarkers of human dietary zinc depletion and homeostasis. Proc Natl Acad Sci U S A 108, 20970–20975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Bishop KS, Ferguson LR (2015) The Interaction between Epigenetics, Nutrition and the Development of Cancer. Nutrients 7, 922–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Institute of Medicine (IOM).Food and Nutrition Board (1997) Dietary Reference Intakes: Calcium, Phosphorus, Magnesium, Vitamin D and Fluoride . Washington, DC: National Academy Press. [Google Scholar]
- (23).Green JH, Booth C, Bunning R (2003) Acute effect of high-calcium milk with or without additional magnesium, or calcium phosphate on parathyroid hormone and biochemical markers of bone resorption. Eur J Clin Nutr 57, 61–68. [DOI] [PubMed] [Google Scholar]
- (24).Domrongkitchaiporn S, Ongphiphadhanakul B, Stitchantrakul W, Piaseu N, Chansirikam S, Puavilai G, Rajatanavin R (2000) Risk of calcium oxalate nephrolithiasis after calcium or combined calcium and calcitriol supplementation in postmenopausal women. Osteoporos Int 11, 486–492. [DOI] [PubMed] [Google Scholar]
- (25).Karkkainen MU, Wiersma JW, Lamberg-Allardt CJ (1997) Postprandial parathyroid hormone response to four calcium-rich foodstuffs. Am J Clin Nutr 65, 1726–1730. [DOI] [PubMed] [Google Scholar]
- (26).Bussiere FI, Gueux E, Rock E, Mazur A, Rayssiguier Y (2002) Protective effect of calcium deficiency on the inflammatory response in magnesium-deficient rats. Eur J Nutr 41, 197–202. [DOI] [PubMed] [Google Scholar]
- (27).Norman DA, Fordtran JS, Brinkley LJ, Zerwekh JE, Nicar MJ, Strowig SM, Pak CY (1981) Jejunal and ileal adaptation to alterations in dietary calcium: changes in calcium and magnesium absorption and pathogenetic role of parathyroid hormone and 1,25-dihydroxyvitamin D. J Clin Invest 67, 1599–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hardwick LL, Jones MR, Brautbar N, Lee DB (1991) Magnesium absorption: mechanisms and the influence of vitamin D, calcium and phosphate. J Nutr 121, 13–23. [DOI] [PubMed] [Google Scholar]
- (29).Nielsen FH, Milne DB, Gallagher S, Johnson L, Hoverson B (2007) Moderate magnesium deprivation results in calcium retention and altered potassium and phosphorus excretion by postmenopausal women. Magnes Res 20, 19–31. [PubMed] [Google Scholar]
- (30).SEELIG MS (1990) Increased need for magnesium with the use of combined oestrogen and calcium for osteoporosis treatment. Magnes Res 3, 197–215. [PubMed] [Google Scholar]
- (31).Flatman PW (1991) Mechanisms of magnesium transport. Annu Rev Physiol 53, 259–271. [DOI] [PubMed] [Google Scholar]
- (32).Toffa DH, Magnerou MA, Kassab A, Hassane DF, Sow AD (2019) Can magnesium reduce central neurodegeneration in Alzheimer's disease? Basic evidences and research needs. Neurochem Int 126, 195–202. [DOI] [PubMed] [Google Scholar]
- (33).Yu X, Guan PP, Guo JW, Wang Y, Cao LL, Xu GB, Konstantopoulos K, Wang ZY, Wang P (2015) By suppressing the expression of anterior pharynx-defective-1alpha and −1beta and inhibiting the aggregation of beta-amyloid protein, magnesium ions inhibit the cognitive decline of amyloid precursor protein/presenilin 1 transgenic mice. FASEB J 29, 5044–5058. [DOI] [PubMed] [Google Scholar]
- (34).Yu J, Sun M, Chen Z, Lu J, Liu Y, Zhou L, Xu X, Fan D, Chui D (2010) Magnesium modulates amyloid-beta protein precursor trafficking and processing. J Alzheimers Dis 20, 1091–1106. [DOI] [PubMed] [Google Scholar]
- (35).Li W, Yu J, Liu Y, Huang X, Abumaria N, Zhu Y, Huang X, Xiong W, Ren C, Liu XG, Chui D, Liu G (2014) Elevation of brain magnesium prevents synaptic loss and reverses cognitive deficits in Alzheimer's disease mouse model. Mol Brain 7, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Jia S, Liu Y, Shi Y, Ma Y, Hu Y, Wang M, Li X (2016) Elevation of Brain Magnesium Potentiates Neural Stem Cell Proliferation in the Hippocampus of Young and Aged Mice. J Cell Physiol 231, 1903–1912. [DOI] [PubMed] [Google Scholar]
- (37).Wang P, Yu X, Guan PP, Guo JW, Wang Y, Zhang Y, Zhao H, Wang ZY (2017) Magnesium ion influx reduces neuroinflammation in Abeta precursor protein/Presenilin 1 transgenic mice by suppressing the expression of interleukin-1beta. Cell Mol Immunol 14, 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Xu ZP, Li L, Bao J, Wang ZH, Zeng J, Liu EJ, Li XG, Huang RX, Gao D, Li MZ, Zhang Y, Liu GP, Wang JZ (2014) Magnesium protects cognitive functions and synaptic plasticity in streptozotocin-induced sporadic Alzheimer's model. PLoS One 9, e108645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zhang JY, Liu SJ, Li HL, Wang JZ (2005) Microtubule-associated protein tau is a substrate of ATP/Mg(2+)-dependent proteasome protease system. J Neural Transm (Vienna ) 112, 547–555. [DOI] [PubMed] [Google Scholar]
- (40).Slutsky I, Abumaria N, Wu LJ, Huang C, Zhang L, Li B, Zhao X, Govindarajan A, Zhao MG, Zhuo M, Tonegawa S, Liu G (2010) Enhancement of learning and memory by elevating brain magnesium. Neuron 65, 165–177. [DOI] [PubMed] [Google Scholar]
- (41).Wester PO (1987) Magnesium. Am J Clin Nutr 45, 1305–1312. [DOI] [PubMed] [Google Scholar]
- (42).Saris NE, Mervaala E, Karppanen H, Khawaja JA, Lewenstam A (2000) Magnesium. An update on physiological, clinical and analytical aspects. Clin Chim Acta 294, 1–26. [DOI] [PubMed] [Google Scholar]
- (43).Hartwig A (2001) Role of magnesium in genomic stability. Mutat Res 475, 113–121. [DOI] [PubMed] [Google Scholar]
- (44).Gueux E, Azais-Braesco V, Bussiere L, Grolier P, Mazur A, Rayssiguier Y (1995) Effect of magnesium deficiency on triacylglycerol-rich lipoprotein and tissue susceptibility to peroxidation in relation to vitamin E content. Br J Nutr 74, 849–856. [PubMed] [Google Scholar]
- (45).Hans CP, Chaudhary DP, Bansal DD (2003) Effect of magnesium supplementation on oxidative stress in alloxanic diabetic rats. Magnes Res 16, 13–19. [PubMed] [Google Scholar]
- (46).Paolisso G, Scheen A, D'Onofrio F, Lefebvre P (1990) Magnesium and glucose homeostasis. Diabetologia 33, 511–514. [DOI] [PubMed] [Google Scholar]
- (47).Chaudhary DP, Sharma R, Bansal DD (2010) Implications of magnesium deficiency in type 2 diabetes: a review. Biol Trace Elem Res 134, 119–129. [DOI] [PubMed] [Google Scholar]
- (48).Mooren FC (2015) Magnesium and disturbances in carbohydrate metabolism. Diabetes Obes Metab 17, 813–823. [DOI] [PubMed] [Google Scholar]
- (49).Huang CY, Liou YF, Chung SY, Pai PY, Kan CB, Kuo CH, Tsai CH, Tsai FJ, Chen JL, Lin JY (2010) Increased expression of glucose transporter 3 in gerbil brains following magnesium sulfate treatment and focal cerebral ischemic injury. Cell Biochem Funct 28, 313–320. [DOI] [PubMed] [Google Scholar]
- (50).Morakinyo AO, Samuel TA, Adekunbi DA (2018) Magnesium upregulates insulin receptor and glucose transporter-4 in streptozotocin-nicotinamide-induced type-2 diabetic rats. Endocr Regul 52, 6–16. [DOI] [PubMed] [Google Scholar]
- (51).Solaimani H, Soltani N, Malekzadeh K, Sohrabipour S, Zhang N, Nasri S, Wang Q (2014) Modulation of GLUT4 expression by oral administration of Mg(2+) to control sugar levels in STZ-induced diabetic rats. Can J Physiol Pharmacol 92, 438–444. [DOI] [PubMed] [Google Scholar]
- (52).Mithieux G, Vega FV, Riou JP (1990) The liver glucose-6-phosphatase of intact microsomes is inhibited and displays sigmoid kinetics in the presence of alpha-ketoglutarate-magnesium and oxaloacetate-magnesium chelates. J Biol Chem 265, 20364–20368. [PubMed] [Google Scholar]
- (53).Panov A, Scarpa A (1996) Independent modulation of the activity of alpha-ketoglutarate dehydrogenase complex by Ca2+ and Mg2+. Biochemistry 35, 427–432. [DOI] [PubMed] [Google Scholar]
- (54).Lu X, Zhao BS, He C (2015) TET Family Proteins: Oxidation Activity, Interacting Molecules, and Functions in Diseases. Chem Rev 115, 2225–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Klungland A, Robertson AB (2017) Oxidized C5-methyl cytosine bases in DNA: 5-Hydroxymethylcytosine; 5-formylcytosine; and 5-carboxycytosine. Free Radic Biol Med 107, 62–68. [DOI] [PubMed] [Google Scholar]
- (56).Robertson KD, Wolffe AP (2000) DNA methylation in health and disease. Nat Rev Genet 1, 11–19. [DOI] [PubMed] [Google Scholar]
- (57).Dunn BK (2003) Hypomethylation: one side of a larger picture. Ann N Y Acad Sci 983, 28–42. [DOI] [PubMed] [Google Scholar]
- (58).Mariani CJ, Madzo J, Moen EL, Yesilkanal A, Godley LA (2013) Alterations of 5-hydroxymethylcytosine in human cancers. Cancers (Basel) 5, 786–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Jin SG, Wu X, Li AX, Pfeifer GP (2011) Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res 39, 5015–5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Nazor KL, Boland MJ, Bibikova M, Klotzle B, Yu M, Glenn-Pratola VL, Schell JP, Coleman RL, Cabral-da-Silva MC, Schmidt U, Peterson SE, He C, Loring JF, Fan JB (2014) Application of a low cost array-based technique - TAB-Array - for quantifying and mapping both 5mC and 5hmC at single base resolution in human pluripotent stem cells. Genomics 104, 358–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Liu G, Weinger JG, Lu ZL, Xue F, Sadeghpour S (2016) Efficacy and Safety of MMFS-01, a Synapse Density Enhancer, for Treating Cognitive Impairment in Older Adults: A Randomized, Double-Blind, Placebo-Controlled Trial. J Alzheimers Dis 49, 971–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Blaine J, Chonchol M, Levi M (2015) Renal control of calcium, phosphate, and magnesium homeostasis. Clin J Am Soc Nephrol 10, 1257–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Weinstein JR, Anderson S (2010) The aging kidney: physiological changes. Adv Chronic Kidney Dis 17, 302–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Salthouse TA (2009) When does age-related cognitive decline begin? Neurobiol Aging 30, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Borenstein AR, Mortimer JA (2016) Alzheimer's Disease: Life Course Perspectives on Risk Reduction. Amsterdam: : Elsevier Academic Press. [Google Scholar]
- (66).Dai Q, Shrubsole MJ, Ness RM, Schlundt D, Cai Q, Smalley WE, Li M, Shyr Y, Zheng W (2007) The relation of magnesium and calcium intakes and a genetic polymorphism in the magnesium transporter to colorectal neoplasia risk. Am J Clin Nutr 86, 743–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Dai Q, Sandler R, Barry E, Summers R, Grau M, Baron J (2012) Calcium, magnesium, and colorectal cancer. Epidemiology 23, 504–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Dai Q, Cantwell MM, Murray LJ, Zheng W, Anderson LA, Coleman HG (2016) Dietary magnesium, calcium:magnesium ratio and risk of reflux oesophagitis, Barrett's oesophagus and oesophageal adenocarcinoma: a population-based case-control study. Br J Nutr 115, 342–350. [DOI] [PubMed] [Google Scholar]
- (69).Zhao J, Giri A, Zhu X, Shrubsole MJ, Jiang Y, Guo X, Ness R, Seidner DL, Giovannucci E, Edwards TL, Dai Q (2019) Calcium: magnesium intake ratio and colorectal carcinogenesis, results from the prostate, lung, colorectal, and ovarian cancer screening trial. Br J Cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Nasreddine ZS, Phillips NA, Bedirian V, Charbonneau S, Whitehead V, Collin I, Cummings JL, Chertkow H (2005) The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 53, 695–699. [DOI] [PubMed] [Google Scholar]
- (71).Zeng C, Zhang Z, Wang J, Chiu BC, Hou L, Zhang W (2019) Application of the High-throughput TAB-Array for the Discovery of Novel 5-Hydroxymethylcytosine Biomarkers in Pancreatic Ductal Adenocarcinoma. Epigenomes 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Xu Z, Niu L, Li L, Taylor JA (2016) ENmix: a novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res 44, e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Tukey J (1977) Exploratory Data Analysis. Pearson. [Google Scholar]
- (74).Xu Z, Langie SA, De BP, Taylor JA, Niu L (2017) RELIC: a novel dye-bias correction method for Illumina Methylation BeadChip. BMC Genomics 18, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).VanderWeele TJ, Vansteelandt S (2014) Mediation Analysis with Multiple Mediators. Epidemiol Methods 2, 95–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Neth BJ, Craft S (2017) Insulin Resistance and Alzheimer's Disease: Bioenergetic Linkages. Front Aging Neurosci 9, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Marais AD (2019) Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology 51, 165–176. [DOI] [PubMed] [Google Scholar]
- (78).Lin YF, Smith AV, Aspelund T, Betensky RA, Smoller JW, Gudnason V, Launer LJ, Blacker D (2019) Genetic overlap between vascular pathologies and Alzheimer's dementia and potential causal mechanisms. Alzheimers Dement 15, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Head T, Daunert S, Goldschmidt-Clermont PJ (2017) The Aging Risk and Atherosclerosis: A Fresh Look at Arterial Homeostasis. Front Genet 8, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Dominguez LJ, Barbagallo M, Lauretani F, Bandinelli S, Bos A, Corsi AM, Simonsick EM, Ferrucci L (2006) Magnesium and muscle performance in older persons: the InCHIANTI study. Am J Clin Nutr 84, 419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Huq AJ, Fransquet P, Laws SM, Ryan J, Sebra R, Masters CL, Winship IM, James PA, Lacaze P (2019) Genetic resilience to Alzheimer's disease in APOE epsilon4 homozygotes: A systematic review. Alzheimers Dement 15, 1612–1623. [DOI] [PubMed] [Google Scholar]
- (82).Karlsson IK, Ploner A, Wang Y, Gatz M, Pedersen NL, Hagg S (2018) Apolipoprotein E DNA methylation and late-life disease. Int J Epidemiol 47, 899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Wang SC, Oelze B, Schumacher A (2008) Age-specific epigenetic drift in late-onset Alzheimer's disease. PLoS One 3, e2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Shao Y, Shaw M, Todd K, Khrestian M, D'Aleo G, Barnard PJ, Zahratka J, Pillai J, Yu CE, Keene CD, Leverenz JB, Bekris LM (2018) DNA methylation of TOMM40-APOE-APOC2 in Alzheimer's disease. J Hum Genet 63, 459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Foraker J, Millard SP, Leong L, Thomson Z, Chen S, Keene CD, Bekris LM, Yu CE (2015) The APOE Gene is Differentially Methylated in Alzheimer's Disease. J Alzheimers Dis 48, 745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Kawamoto EM, Vivar C, Camandola S (2012) Physiology and pathology of calcium signaling in the brain. Front Pharmacol 3, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Huang Y, Huang X, Zhang L, Han F, Pang KL, Li X, Shen JY (2018) Magnesium boosts the memory restorative effect of environmental enrichment in Alzheimer's disease mice. CNS Neurosci Ther 24, 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Kirkland AE, Sarlo GL, Holton KF (2018) The Role of Magnesium in Neurological Disorders. Nutrients 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Kieboom BCT, Licher S, Wolters FJ, Ikram MK, Hoorn EJ, Zietse R, Stricker BH, Ikram MA (2017) Serum magnesium is associated with the risk of dementia. Neurology 89, 1716–1722. [DOI] [PubMed] [Google Scholar]
- (90).Ozawa M, Ninomiya T, Ohara T, Hirakawa Y, Doi Y, Hata J, Uchida K, Shirota T, Kitazono T, Kiyohara Y (2012) Self-reported dietary intake of potassium, calcium, and magnesium and risk of dementia in the Japanese: the Hisayama Study. J Am Geriatr Soc 60, 1515–1520. [DOI] [PubMed] [Google Scholar]
- (91).Brown EM, MacLeod RJ (2001) Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev 81, 239–297. [DOI] [PubMed] [Google Scholar]
- (92).Hoenderop JG, Bindels RJ (2005) Epithelial Ca2+ and Mg2+ channels in health and disease. J Am Soc Nephrol 16, 15–26. [DOI] [PubMed] [Google Scholar]
- (93).Abbott L, Nadler J, Rude RK (1994) Magnesium deficiency in alcoholism: possible contribution to osteoporosis and cardiovascular disease in alcoholics. Alcohol Clin Exp Res 18, 1076–1082. [DOI] [PubMed] [Google Scholar]
- (94).Poikolainen K, Alho H (2008) Magnesium treatment in alcoholics: a randomized clinical trial. Subst Abuse Treat Prev Policy 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Alexandropoulou K, van VJ, Reid F, Poullis A, Kang JY (2013) Temporal trends of Barrett's oesophagus and gastro-oesophageal reflux and related oesophageal cancer over a 10-year period in England and Wales and associated proton pump inhibitor and H2RA prescriptions: a GPRD study. Eur J Gastroenterol Hepatol 25, 15–21. [DOI] [PubMed] [Google Scholar]
- (96).Bai JP, Hausman E, Lionberger R, Zhang X (2012) Modeling and simulation of the effect of proton pump inhibitors on magnesium homeostasis. 1. Oral absorption of magnesium. Mol Pharm 9, 3495–3505. [DOI] [PubMed] [Google Scholar]
- (97).Luk CP, Parsons R, Lee YP, Hughes JD (2013) Proton pump inhibitor-associated hypomagnesemia: what do FDA data tell us? Ann Pharmacother 47, 773–780. [DOI] [PubMed] [Google Scholar]
- (98).Markovits N, Loebstein R, Halkin H, Bialik M, Landes-Westerman J, Lomnicky J, Kurnik D (2014) The association of proton pump inhibitors and hypomagnesemia in the community setting. J Clin Pharmacol 54, 889–895. [DOI] [PubMed] [Google Scholar]
- (99).Pak CY (2000) Correction of thiazide-induced hypomagnesemia by potassium-magnesium citrate from review of prior trials. Clin Nephrol 54, 271–275. [PubMed] [Google Scholar]
- (100).Franz KB (2004) A functional biological marker is needed for diagnosing magnesium deficiency. J Am Coll Nutr 23, 738S–741S. [DOI] [PubMed] [Google Scholar]
- (101).Liebscher DH, Liebscher DE (2004) About the misdiagnosis of magnesium deficiency. J Am Coll Nutr 23, 730S–731S. [DOI] [PubMed] [Google Scholar]
- (102).Tong GM, Rude RK (2005) Magnesium deficiency in critical illness. J Intensive Care Med 20, 3–17. [DOI] [PubMed] [Google Scholar]
- (103).Gitelman HJ, WELT LG (1969) Magnesium deficiency. Annu Rev Med 20, 233–242. [DOI] [PubMed] [Google Scholar]
- (104).Rex DK, Sullivan AW, Perkins AJ, Vemulapalli KC (2020) Colorectal polyp prevalence and aspirational detection targets determined using high definition colonoscopy and a high level detector in 2017. Dig Liver Dis 52, 72–78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.