

Abstract
Genetic Code Expansion (GCE) can use TAG stop codons to guide site-specific incorporation of phosphoserine (pSer) into proteins. To eliminate prematurely truncated peptides, improve yields and enhance the production of multi-phosphorylated proteins, Release Factor 1 (RF1) deficient expression hosts were developed, yet these grew slowly and their use was associated with extensive mis-incorporation of natural amino acids instead of pSer. Here, we merge a healthy RF1-deficient E. coli cell line with a high-efficiency pSer GCE translation system to produce a versatile pSer GCE platform in which only trace mis-incorporation of natural amino acids are detected even when five phosphoserines were introduced into one protein. Approximately 400 and 200 milligrams of singly- and doubly-phosphorylated GFP per liter culture were obtained. Importantly, the lack of truncated protein permits expression of oligomeric proteins and use of N-terminal solubility-enhancing proteins to aid phospho-protein expression and purification. To illustrate the enhanced utility of this system, we produce doubly-phosphorylated STING (Stimulator of Interferon Genes), as well as triply-phosphorylated BAD (Bcl2-associated agonist of cell death) complexed with 14-3-3, in quantity, purity and homogeneity sufficient for structural biology applications. We anticipate the facile access to phosphoproteins enabled by this system, which we call pSer-3.1G, will expand studies of the phospho-proteome.
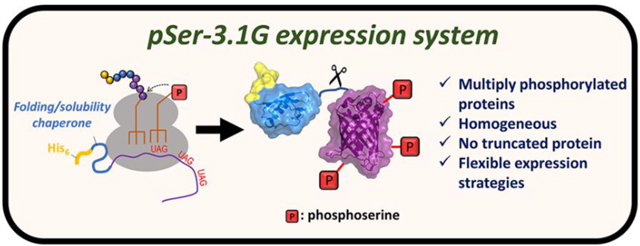
Graphical Abstract
INTRODUCTION
Protein function is tightly regulated by phosphorylation.1 For many proteins, this regulation involves multiple sites of phosphorylation, and distinct functions result from different patterns of phosphorylation. Yet learning how these various patterns impart different functional and structural outputs is challenging due to a lack of effective methods for efficiently making pure forms of multi-phosphorylated proteins. Approaches to circumvent these challenges include mutating sites of phosphorylation to Asp or Glu residues to mimic the negative charge of phosphate, however these do not recapitulate the shape and charge of phosphoserine and so they are not reliable functional mimics.2 Kinases can be used to install authentic phosphates, but their utility is limited because the required kinases are not always known and they often lack the required specificity.3–5
Genetic code expansion (GCE) has emerged as a leading technology for studying protein post-translational modifications because it permits programmable and site-specific incorporation of modified amino acids into recombinantly expressed proteins. With GCE, non-canonical amino acids (ncAAs) are translationally incorporated typically at TAG (amber) stop codons by a suppressing orthogonal tRNACUA, which is amino-acylated by an orthogonal aminoacyl-tRNA synthetase engineered for a specific ncAA.6 Balancing these added translational components to obtain high levels of pure ncAA-protein is not trivial. Of particular importance is ensuring the GCE system does not permit incorporation of natural amino acids at the intended site of ncAA incorporation (a property commonly referred to as fidelity). Also, since the ncAA-tRNACUA must compete with the cell’s Release Factor 1 (RF1, the E. coli protein responsible for terminating translation at TAG codons), early termination instead of ncAA incorporation is common and so truncated peptides are made along with ncAA containing proteins. C-terminal affinity purification tags are typically appended to GCE-produced proteins to ensure that only full-length protein is purified.
Substantial effort has been put into the development of GCE systems for programmable installation of phosphoserine (pSer). Currently published pSer GCE systems are summarized in Supporting Tables 1 and 2, and in Supporting Figs 1–3. The 1st generation proof-of-concept pSer GCE system (called here pSer-1G) proved pSer proteins could be expressed in E. coli,7 but it suffered from low efficiency. This prompted evolution of a second-generation pSer machinery system (pSer-2G) with improved translational components.8 These components were later tested in an RF1-deficient expression host (C321.ΔA) in order to increase efficiency of pSer protein production by eliminating competition between the amber suppressing pSer-tRNAUAG and RF1 (called here pSer-2.1G and referred to elsewhere as “SepOTS”).9 However, in this RF1-deficient pSer-2.1G system a large fraction of the expressed GFP (~30%) contained natural amino acids at the intended and generally permissive site of pSer incorporation, indicating the pSer-2G machinery was not efficient enough to outcompete single site amber codon suppression by endogenous tRNAs. This fidelity worsened when the site of incorporation in GFP was altered, when incorporating pSer into biologically relevant proteins, and when multi-site amber suppression was attempted.9,10 These challenges are accentuated by the slow growth rates and metabolic deficiencies of the RF1-deficient C321.ΔA strain,11,12 leading some users of the pSer-2G machinery to revert back to healthy, RF1-containing strains for high fidelity production of phospho-proteins.10,13
As no pSer-2G system has produced phospho-proteins with the efficiency and fidelity needed for structural biology applications, Chin and colleagues in 2015 developed a third generation pSer GCE system (pSer-3G) that displayed 17-fold greater efficiency than the pSer-2G machinery.14 No mis-incorporation of natural amino acids at pSer sites was detected with this system when expressed in RF1-containing strains of E. coli. The authors noted that use of the RF1-deficient C321.ΔA strain was not necessary for efficient pSer incorporation, and so the pSer-3G system has primarily been employed with RF1-containing expression hosts.15–18 Thus, truncated protein still builds up in this system, constraining users to express phospho-proteins with C-terminal affinity purification tags for most applications.
One commonly overlooked advantage of using RF1-deficient strains for GCE applications is that prematurely truncated protein should not be made during protein expression since these expression hosts do not have a mechanism to terminate translation at TAG stop codons. This lack of truncated peptide during expression would permit placement of affinity tags at either the N- or the C-terminus of the protein-of-interest since full-length protein need not be separated from truncated peptide. This is a crucial advantage of RF1 expression hosts for pSer GCE applications not only because some proteins require unmodified C-termini, but also because recombinant protein expression is routinely enhanced by N-terminal fusions with folding or solubility promoting domains, such as Small Ubiquitin-Like Modifier Protein (SUMO), Glutathione-S-Transferase (GST) or Maltose Binding Protein (MBP)19,20; C-terminal solubility tags are lackluster in comparison to N-terminal fusions because the latter provide an efficient translational initiation context and they can be removed easily by endo-proteases while leaving few or no additional residues on the desired protein.21,22 A second advantage of using RF1-deficient strains for pSer GCE is for the production of phosphorylated oligomers. For example, expression of a dimeric protein with sites of phosphorylation C-terminal to its oligomerizing domain would produce a mixture of complexes consisting of full-length and truncated peptides, which could be difficult or impossible to separate irrespective of the placement of the affinity purification tag. However, high fidelity pSer incorporation (which we define here as >90% of the protein being stoichiometrically phosphorylated) has not been demonstrated in an RF1-deficient expression host.
Noting that a reproductively healthy RF1-deficient strain was recently developed,23 we hypothesized that integrating the features of this fast growing, “truncation free” strain with those of the enhanced pSer-3G translational machinery would lead to an improved ability to produce and purify milligram quantities of protein containing multiple phosphorylation sites. We report here the development and characterization of such an adaptation of the third generation pSer system (which we call here pSer-3.1G) that is compatible with N-terminal affinity purification and solubility-promoting tags, making multiply-phosphorylated proteins accessible at high levels and with high fidelity. To illustrate the enhanced utility of the pSer-3.1G system, we produce two multiply phosphorylated proteins that are of high biomedical interest in quantities and homogeneity sufficient for structural analyses which could not be made with previous pSer GCE systems.
RESULTS
Creating the pSer-3.1G system
Making a healthy, RF1-deficient strain of E. coli compatible with pSer GCE.
We chose to work with the RF1-deficient B-95(DE3) ΔAΔfabR strain because, unlike other RF1-deficient strains, (i) it is derived from BL21(DE3), a traditional work-horse T7-compatible strain for recombinant protein over-expression, and (ii) it does not suffer from growth or metabolism defects associated with other RF1 knockout strains 11,23,24. The reason for its greater vitality is that rather than simply removing all or none of the chromosomal TAG stop codons, in this strain only a select set of 95 which do not overlap with other genes are mutated to TAA.23
To modify this cell line for improved phosphoserine incorporation, we deleted the serB gene in B-95(DE3) ΔAΔfabR using traditional λ-red recombineering protocols25–27 to create the markerless B-95(DE3) ΔAΔfabRΔserB (Supporting Figure 4). This is what we refer to as the pSer-3.1G cell line. The serB gene encodes the phosphoserine phosphatase SerB,28 and by knocking it out, intracellular levels of phosphoserine build up providing a suitable pool for incorporation into proteins without additional supplementation to the growth media.29 The pSer-3.1G strain grows well, with a doubling time of 34±2 min, compared to 31±1 min for its parent strain B-95(DE3) ΔAΔfabR and 29±1 min for BL21(DE3) (Supporting Figure 5).
Adding pSer GCE components to the pSer-3.1G expression strain.
For site-specific pSer incorporation, we started with the original pSer-3G expression system, which utilizes the pKW2-EFSep and pNHD plasmids (Supporting Figures 6B and 6C, Supporting Table 2). pKW2-EFSep encodes the phosphoserine GCE machinery components, while the pNHD plasmid expresses the TAG codon-interrupted gene-of interest under the control of a T7 promoter. Unexpectedly, we found that the pSer-3.1G strain to be particularly sensitive to tetracycline even when harboring the pNHD plasmid (doubling time of ~90 min, Supporting Figure 5). We therefore replaced its tetracycline resistance cassette with an ampicillin resistance cassette to form the pRBC plasmid (Supporting Figure 6A). This lowered the doubling time to 36±1 min in the presence of both antibiotics (ampicillin and chloramphenicol), only marginally slower than the same cells lacking both pSer GCE plasmids (Supporting Figure 5). The mechanistic basis for the improved growth in ampicillin compared to tetracycline is not understood. In addition, these pSer-3.1G cells displayed a 2-fold faster doubling time and grew to twice the final cell density as C321.ΔA cells harboring the same pRBC/pKW2 plasmid combination (Supporting Figure 5).
Characterization of the pSer-3.1G expression system
Efficiency and fidelity of phosphoserine incorporation into sfGFP in the pSer-3.1G system.
Given that the employment of RF1-deficient expression hosts was previously associated with mis-incorporation of natural amino acids at the intended site of phosphorylation,9,10 we first scrutinized pSer incorporation efficiency and fidelity in the pSer-3.1G cell line. To do this, we used the pKW2-EFSep/pRBC plasmid combination to express wild-type super-folder green fluorescent protein30 (sfGFP), sfGFP phosphorylated at position 150 (sfGFP-150TAG), and sfGFP phosphorylated at positions 134 and 150 (sfGFP-134/150TAG) (Figure 1A). For comparison, we also expressed these proteins from the same plasmid combination using the pSer-3G cell line (the RF1-containing BL21(DE3) ΔserB). Using auto-induction media, the pSer-3.1G system produced ~650, 400 and 200 mg of wild-type, singly and doubly phosphorylated sfGFP per liter culture, respectively. Compared to the pSer-3G output, the wild-type sfGFP expression levels were equivalent, but for singly and doubly phosphorylated sfGFP the pSer-3.1G outputs were approximately 2-fold and 3-fold greater, respectively (Figure 1B). A direct comparison of protein production with the pSer-2.1G system (which utilizes the C321.ΔA strain) was not possible because the cell line is not compatible the T7-based expression systems used here.
Figure 1.

Efficiency and fidelity of site-specific pSer incorporation into sfGFP using the pSer-3G and pSer-3.1G systems. (A) Constructs for sfGFP wild-type, 150TAG and 134/150TAG were used to incorporate zero, one or two phosphoserines, respectively. (B) Yields of sfGFP constructs expressed in the pSer-3G (black) and pSer-3.1G (gray) systems. Error bars represent standard deviations of three independent expressions. (C) Coomassie-stained SDS-PAGE (bottom) and Phos-Tag gel (top) for wildtype, singly and doubly phosphorylated-sfGFP produced in the pSer-3G and pSer-3.1G systems. Equal quantities of protein were loaded in each lane. Therefore, band intensity does not reflect protein expression efficiency.
To assess the fidelity of phosphoserine incorporation, purified sfGFP proteins were subjected Phos-tag gel electrophoresis analyses as well as whole-protein mass spectrometry. In Phos-tag gels, phosphorylated proteins migrate incrementally slower with each additional phosphate group.31 Using this approach, approximately ~95% of purified sfGFP-150TAG was phosphorylated, and ~90% of sfGFP-134/150TAG was doubly phosphorylated (Figure 1C). These results from Phos-tag gels were corroborated by whole-protein mass spectrometry, which identified only a trace population of singly phosphorylated protein expressed from the 134/150TAG construct in the pSer-3.1G system (Supporting Fig. 7). In regards to site-specificity, tandem mass-spectrometry analysis of tryptic digested, doubly phosphorylated sfGFP unambiguously identified phosphoserine at residues 134 and 150 (Supporting Figs. 8 and 9).
Dependency of site-of-incorporation on phosphoserine incorporation accuracy.
To assess whether fidelity of the pSer-3.1G system depended on the site of pSer incorporation, we created SUMO-sfGFP fusion constructs with TAG codons at five different sites (Figure 2A). Protein yields differed from ~150 to ~500 mg/L culture among the five different phospho-proteins (Supporting Figure 10), but all proteins showed >95% pSer incorporation (Figure 2B). We conclude that pSer incorporation fidelity by the pSer-3.1G system is largely independent of the targeted site. Interestingly, the extent to which the phosphorylated proteins shift in Phos-tag electrophoresis differed by the site of incorporation (Figure 2B). In addition, an upward shift in SDS-PAGE is observed for the “1x linker” variant, which is not seen in any of the other singly phosphorylated forms of SUMO-sfGFP. The mechanistic basis for this site-dependent electrophoretic shift is not well understood, however it is likely due to altered binding capacity of SDS in the vicinity of the linker region when phosphorylated.32
Figure 2.

Dependency of site-of-incorporation on fidelity of pSer incorporation in the pSer-3.1G system. (A) SUMO-sfGFP constructs containing single TAG sites. (B) Coomassie stained SDS-PAGE (bottom) and Phos-tag gel (top) of the purified SUMO-sfGFP constructs shown in panel A; the wildtype (non-phosphorylated) construct is loaded in between each singly phosphorylated protein for mobility shift reference.
Limits of the pSer-3.1G expression system.
Next, we tested the limits of fidelity in the pSer-3.1G system by introducing, incrementally, up to five TAG codons in the SUMO-sfGFP construct (Figure 3A). An unambiguous mobility decrease in Phos-tag gels was observed for each additional phosphoserine incorporated, and only trace (<10%) sub-stoichiometrically phosphorylated protein was observed in any of these cases, even the quintuply-phosphorylated protein which expressed at ~40 mg per liter culture (Figure 3B, Supporting Figure 11).
Figure 3.

Incorporation of up to five pSer moieties with pSer-3.1G system. (A) SUMO-sfGFP constructs expressed in pSer-3.1G cells containing the spectrum of zero to five TAG sites. (B) Coomassie stained SDS-PAGE (bottom) and Phos-tag (top) gels showing purified unphosphorylated, singly, doubly, triply, quadruply, and quintuply phosphorylated SUMO-sfGFP. Smaller quantities of protein 4x and 5x pSer proteins were loaded to accentuate the pSer-dependent mobility shift. A Phos-tag gel with higher quantities of protein loaded to evaluate presence of trace quantities of substoichiometrically phosphorylated protein populations is shown in Supporting Figure 11.
Evaluation of truncated protein levels using the pSer-3.1G expression system.
Having confirmed faithful, site-specific, context-independent and multi-site pSer incorporation using the RF1-deficient pSer-3.1G system, we next evaluated whether truncated peptide formed during expression. To test this, we created N-terminally His6 - tagged SUMO-sfGFP constructs and introduced one, two and three TAG sites (Figure 4A). By using an N-terminal affinity tag and a SUMO solubilization domain, truncated peptides produced during expression should co-purify with full-length protein which can be visualized by SDS-PAGE. As for above, the pSer-3.1G expressions were compared with pSer-3G expressions, in which truncated peptide would be expected to form and co-purify.
Figure 4.

Evaluation of truncated peptide formation in pSer-3G and pSer-3.1G systems. (A) SUMO-sfGFP constructs containing N-terminal His6 purification tags and zero, one, two or three TAG sites for pSer incorporation. (B) Expression yields per liter culture of His6 -SUMO-sfGFP constructs. Error bars represent standard deviations of three expressions. (C) Coomassie stained SDS-PAGE (bottom) and corresponding Phos-tag gels (top) of purified His6-SUMO-sfGFP constructs expressed in the pSer-3G and pSer-3.1G systems. Equal quantities of fluorescent (full-length) protein were loaded in each lane.
The expression efficiency for these constructs was improved in the pSer-3.1G system compared to the pSer-3G system (Figure 4B) and Phos-tag gel analysis of the purified full-length proteins showed high fidelity (>95%) incorporation for singly, doubly and triply phosphorylated SUMO-sfGFP proteins in both systems (Figure 4C, top panel). Regarding truncation products, none or minimal (<5% total protein) were observed for protein expressed in the pSer-3.1G system, whereas substantial quantities were present in protein purified from the pSer-3G system (Figure 4C, bottom panel). The migration of the truncated peptides from the pSer-3G system were consistent with translational termination occurring at the TAG codons, and the proportion of truncation products to full length protein increased as the number of TAG sites increased.
Taken together, these data confirm faithful multi-site protein phosphorylation with the pSer-3.1G system without the confounding presence of truncation products using a fast growing RF1 deficient expression host. This is, to our knowledge, the first demonstration of faithful genetic incorporation of pSer in an RF1 deficient expression host and is therefore a key step forward compared to the pSer2.1G system. This pSer3.1 system is advantageous compared to the pSer-3G system being fully compatible with N-terminal solubility and affinity purification tags while producing homogenous multi-phosphorylated proteins at higher yields.
Two biologically relevant test cases.
To demonstrate enhanced utility of the pSer-3.1G system, we sought to produce multi-phosphorylated, biologically relevant proteins that (i) could not be made with previous pSer GCE systems and that (ii) researchers have had limited success studying the properties of using either phosphomimetic mutants, kinase-generated phosphorylated forms, or naturally phosphorylated forms isolated from eukaryotic cells. Our first test case is human Stimulator of Interferon Genes (STING), a dimeric receptor that plays an important role in the innate immune response, with phosphoserines in its flexible C-terminal tail (Ser358 and Ser366, Figure 5A).33–37 The mechanism by which STING function is regulated by phosphorylation remains an active subject of study,38,39 with most previous work focusing on the use of phosphomimetics and kinases.34,36,40 Our second test case is phosphorylated BAD (Bcl2-associated agonist of cell death) in complex with the phosphoprotein binder 14-3-3, which inactivates the pro-apoptotic activity of BAD when BAD is phosphorylated by three different kinases at three sites (Ser112, Ser136 and Ser155). BAD and its complex with 14-3-3 have garnered interest as a target for anti-cancer therapeutics41,42 but it has been difficult to study effectively because phosphomimetic mutations are not sufficient for client complexation to 14-3-3,43,44 and the use of kinases or recombinant expression of BAD in eukaryotic systems lead to heterogenous patterns of phosphorylation.45,46
Figure 5.

Expression of phosphorylated STING with the pSer-3.1G system. (A) Cartoon structure of the constitutive STING dimer with flexible C-terminal tail (dotted lines), highlighting the phosphorylation sites Ser358 and Ser366 (PDB code 4ef5). (B) Constructs of His6-SUMO-STING wildtype (wt), 1x (Ser-366TAG), and 2x (Ser-358/366TAG) expressed in pSer-3.1G system. (C) Coomassie stained SDS-PAGE gels of purified STING proteins demonstrating phosphoserine dependent electrophoretic shifts. Un-cropped gels before and after λ-phosphatase treatment are available in Supporting Figure 12.
Test case 1. Production of STING: an oligomeric protein with C-terminal phosphorylation sites.
For oligomeric phospho-proteins such as STING (Figure 5A), expression in the pSer-3G system would result in a mixture of full-length and truncated protein dimers, which may not be easily separated from the desired homomeric full-length complexes. Regardless, our attempts to express wild-type or phosphorylated STING with a C-terminal His6 affinity tag were unsuccessful (data not shown). For efficient expression of STING, it is commonly fused at its N-terminus with SUMO,47 and so we attempted to express in the pSer-3.1G system His6-SUMO-STING constructs with TAG sites at Ser358, and at both Ser358 and Ser366 (Figure 5B). These constructs were expressed, purified, and the SUMO fusion protein was cleaved by ULP1 and removed by subtractive affinity chromatography. Approximately 5–10 mg per liter culture were obtained for wild-type, singly and doubly phosphorylated STING. A shift in electrophoretic mobility corresponding to increasing number of pSer groups was observed in SDS-PAGE (Figure 5C). Interestingly, this shift was not accentuated in Phos-tag gels (Supporting Fig. 12). Incubation with λ-Protein Phosphatase resulted in the putatively phosphorylated proteins all homogenously shifting downward to the apparent mass of wild-type (Supporting Figure 12), confirming that these electrophoretic shifts were phospho-serine dependent. By these electrophoretic analyses, ~90% of the STING-1x and 2x pSer proteins were stoichiometrically phosphorylated, though it appeared the latter was more susceptible to proteolysis (Fig. 5C). Tandem mass spectrometry of trypsin digested STING-2x pSer unambiguously identified sites 358 and 366 as being phosphorylated (Supporting Figure 13).
Test case 2. Production of phosphorylated BAD in complex with 14-3-3.
To further demonstrate the utility of the pSer-3.1G system, we aimed to express BAD in complex with the phospho-protein binder 14-3-3 (Figure 6A). Recombinant expression of native BAD in heterologous hosts is notoriously challenging due to its susceptibility to aggregation and proteolysis.48,49 Methods to produce milligram quantities of wild-type BAD, let alone phosphorylated forms of it, have not been well established. We hypothesized the pSer-3.1G system could be used to improve expression of BAD if it were expressed in its phosphorylated forms simultaneously with 14-3-3. To test this, we created a dual expression plasmid (pRBCduet) in which untagged 14-3-3β is expressed from the first open reading frame and constructs of either wild-type, singly, doubly and triply phosphorylated BAD are expressed from open reading frame 2 (Figure 6B). Even though 14-3-3 is expressed in all cases, it should co-purify only if BAD is authentically phosphorylated at site 136 (Fig. 6A). Additional phosphorylation at sites 112 and 155 lead to the fully mature anti-apoptotic 14-3-3—BAD complex found under cell survival conditions (Fig. 6A).
Figure 6.

Expression of phosphorylated BAD in complex with 14-3-3 using the pSer-3.1G system. (A) Schematic of BAD function showing non-phosphorylated, un-complexed BAD primes the cell for apoptosis, while phosphorylation at site Ser136 is sufficient for complexation with 14-3-3 and inhibition of its pro-apoptotic activity. Additional phosphorylation at site Ser112 stabilizes the complex and the fully mature, anti-apoptotic complex includes a third site of phosphorylation at site Ser155. (B) Dual expression system for expressing 14-3-3β with His6-SUMO-BAD constructs. (C) Coomassie stained SDS-PAGE of purified 14-3-3/BAD complexes (full-gel provided in Supporting Figure 17B). Wild-type BAD, though co-expressed with 14-3-3 (Supporting Figure 17A) does not co-purify with 14-3-3. (D) Coomassie-stained Phos-tag gel of purified His6-SUMO-BAD proteins.
We expressed these constructs in the pSer-3.1G system as described above for other proteins. No detectable quantities of protein were produced when BAD was expressed with a C-terminal affinity purification tag (data not shown), perhaps because the C-terminus of BAD is important for its function.46 However, when BAD was expressed with an N-terminal His6-SUMO fusion in the pSer-3.1G system, significant quantities of phosphorylated BAD in complex with 14-3-3 were purified. These complexes were cleaved with ULP1 and the His6-SUMO tag was removed by subtractive affinity chromatography to yield authentic BAD—14-3-3 complexes (Figure 6C). Approximately 7–15 mg of singly, doubly and triply phosphorylated BAD bound to 14-3-3 were obtained per liter of culture. However only trace quantities of wild-type (un-complexed) BAD could be purified (< 0.25 mg per liter culture), supporting our hypothesis that co-expression of phosphorylated of BAD with 14-3-3 provides a useful strategy to improve BAD expression yields and stability compared to wild-type BAD.
Phos-tag gel analysis of the His6-SUMO-BAD proteins demonstrated high fidelity pSer incorporation, with only ~10% of BAD in the 2x- and 3x- samples being sub-stoichiometrically phosphorylated (Figure 6D). Tandem mass spectrometry of trypsin digested 3x-BAD unambiguously identified phosphoserine at sites 112, 136 and 155 (Supporting Figs. 14–16). As expected, 14-3-3 did not co-purify with wild-type BAD (Figure 6C) even though 14-3-3 expressed robustly in the culture (Supporting Figure 17), consistent with the minimal functional requirement of Ser136 phosphorylation for complexation.50
DISCUSSION
While significant progress from the original proof-of-concept pSer GCE systems has been made, we still found it challenging to access biologically relevant proteins with multiple sites of phosphorylation, which constitute the majority of phospho-proteins.51 We presumed these limitations were based upon, at least in part, the build-up of prematurely truncated peptide during expression. Premature truncation forces users to express their pSer proteins with a C-terminal affinity tag so that full-length phosphorylated protein can be easily separated from truncated peptide during purification. While this might initially be seen as a minor inconvenience, many proteins cannot be expressed with C-terminal affinity tags. Also, the well-established benefits of N-terminal solubility/folding enhancing fusion proteins (e.g. SUMO, GST, MBP) cannot be leveraged when truncated peptide is present. Their use is particularly important because practical pSer GCE technology requires poly-peptides fold with multiple pSer groups present, in contrast to natural phosphorylation in which these modifications are installed after folding. Lastly, truncated protein prevents pragmatic expression of oligomeric phospho-proteins where mixed complexes of full-length and truncated peptide would co-purify with full-length protein complexes.
Thus, the successful marriage of an efficient pSer GCE machinery system with a healthy RF1-deficient, truncation free expression strain would be of great benefit to the field. Prior attempts to do this used a slow growing and metabolically compromised strain of E. coli with the pSer-2G machinery system, resulting in high rates of mis-incorporation of natural amino acids at the intended sites of phosphorylation.9,10 The pSer 3.1G system described here is advantageous over these prior systems because (i) the expression host grows at fast rates (doubling time ~35–40 min) and (ii) multiple phosphoserine groups can be incorporated into a single peptide with only trace (<10% total protein) mis-incorporation of natural amino acids across all sites of phosphorylation, and (iii) truncation product is prevented or minimized, permitting the use of N-terminal affinity and solubility/folding enhancing fusion proteins. Together these attributes make the pSer-3.1G system highly versatile and provides a key step forward in transitioning pSer GCE from a niche technology with a narrow set of applications to one that can be more easily implemented in routine laboratory workflows for accessing biologically relevant phospho-proteins.
While these are important advancements, other challenges remain to be addressed with regard to pSer GCE technology and application. First, since biologically relevant sites of phosphorylation are often located at structurally flexible or disordered regions of proteins,52 hydrolysis of the phosphate groups and proteolysis of the phospho-protein during in vivo expression may pose a significant challenge for some systems. In an effort to address this concern, we show here that the otherwise unstable and poorly expressing BAD protein could be expressed efficiently if co-translationally phosphorylated and complexed with its stabilizing binding partner 14-3-3. Given that there are over 500 known phospho-protein binding clients of 14-3-3,44 this co-expression strategy of phospho-proteins with 14-3-3 is highly generalizable. Indeed, a recent report showed that the aggregation prone toxin ExoS, which binds 14-3-3 in a phosphorylation-independent manner, could be stabilized by co-expression with 14-3-3.53 With so much interest in understanding the mechanisms of 14-3-3—phospho-protein signaling cascades,54 and a remarkable void of structural information in this area,55 the pSer-3.1G system provides a timely methodological advance for studying these signaling systems regulated by serine phosphorylation. A second challenge still faced by pSer GCE is founded on observations that, even in the absence of RF1 in the pSer-3.1 cell line, the efficiency of phospho-protein production still decreases with increasing numbers of TAG sites (e.g. Figures 1 and 4). In line with previous suggestions,10 we infer from this observation that the competition between the pSer-tRNACUA and RF1 is not inherently the rate limiting step in pSer protein production. Further improvements in the enzymatic properties of the pSer GCE components may therefore be a worthwhile next step, though other unidentified steps in phospho-protein expression could be limiting, such as the translational context of TAG codon suppression,56 or protein folding limitations as eluded to above.
Nevertheless, the improvements provided by the pSer-3.1G GCE system for accessing biologically relevant, multiply phosphorylated proteins should greatly expand our ability to interrogate the complex nature of protein regulation by phosphorylation. Further expansion in this area beyond phosphoserine should be possible by developing strains of E. coli analogous to the pSer-3.1G strain that are compatible with GCE systems for phosphothreonine57 and a non-hydrolyzable analog of phosphoserine.14
MATERIALS AND METHODS
Expanded methods for each of the sections below are provided in the Supporting Information.
Creating the pSer-3.1G cell line
The serB gene in the RF1-deficient B-95(DE3) ΔAΔfabR strain 23 (provided by RIKEN BRC through the National BioResource Project of the MEXT/AMED, Japan) was deleted using traditional λ-red recombineering strategies26,27,58.
Other strains and plasmids for phosphoserine GCE.
The RF1-containing BL21(DE3) ΔserB strain (the pSer-3G cell line, Addgene #34929) and the RF1-deficient C321.ΔA.exp (Addgene #49018) were gifts from Jesse Rinehart and George Church, respectively. The pKW2-EFSep and pNHD plasmids were a gift from Jason Chin. The pRBC plasmid was generated by replacing the tetracycline resistance cassette of pNHD with the ampicillin resistance of the pBAD plasmid (Invitrogen). Genes encoding human 14-3-3β, mouse BAD and human STING were codon optimized for E. coli 59 and synthesized by Integrated DNA Technologies. The dual protein expression plasmid pRBCduet was made by inserting the second T7 promoter sequence of the pACYCduet vector (Invitrogen) in between the NdeI and XhoI restriction sites of pRBC. Assembly of all genes into expression vectors and mutagenesis we done using PPY-based SLiCE techniques 60.
Cell Growth Assessment
Fresh transformations were performed for every expression such that the pKW2-EFSep machinery plasmid and the pRBC plasmids were transformed simultaneously into plasmid free, chemically competent cells.61 Strains containing the indicated plasmid combinations were grown overnight in a buffered, glucose-rich non-inducing medium (ZY-NIM, Supporting Table 4) and diluted to a starting optical density (OD600) of 0.05 into 50 mL of fresh ZY-NIM supplemented with the appropriate antibiotics (chloramphenicol, ampicillin and tetracycline were used at 15, 50 and 12.5 μg/mL, respectively). Cells were grown at 37 °C in baffled flasks shaking at 250 rpm over 8 hours, with OD600 measurements taken every 45 minutes. Cultures were all grown in triplicate.
Phospho-protein expression
Phosphorylated sfGFP and SUMO-sfGFP.
Cells grown overnight in ZY-NIM were used to inoculate ZY-Auto-inducting Media (ZY-AIM, Supporting Table 4). After 24 hrs, expression efficiency was measured by in-cell fluorescence.62 Proteins were purified following standard metal affinity chromatography as outlined in the Supporting Information. Phos-tag gels were poured immediately before use and run according to manufacturer recommendations. Whole-protein mass spectrometry was performed with an FT LTQ mass spectrometer at the Mass Spectrometry Facility at Oregon State University. The site of pSer incorporation was confirmed by in-gel tryptic digestion followed by analysis on an OrbiTrap Elite mass spectrometer with collision induced dissociation of the peptide (Fred Hutchinson Cancer Research Center, Seattle, WA). All data were analyzed with Proteome Discoverer v2.2 using SEQUEST-HT as the protein database search algorithm, Percolator for peptide validation, and ptmRS for modification site localization.
Phospho-STING expression and phosphatase treatment.
Phosphorylated human STING (residues 139–379) was expressed from the pRBC plasmid with pKW2-EFSep in the pSer-3.1G cell line in 2xYT media at 25 °C for 6 hrs using 1 mM IPTG to induce expression. Shorter expression times compared to the sfGFP proteins described above were used to minimize STING proteolysis and phosphoserine hydrolysis (data not shown). Reported protein yields of STING expression are based on purifications from two independent expressions. Site-specific incorporation of pSer was confirmed by tandem mass spectrometry as done for phosphorylated sfGFP. Phosphatase assays were performed with λ phosphatase (NEB) in a solution containing 1x Protein Metallophosphatase (PMP) buffer and 1mM MnCl2.
Co-expression of M. musculus phospho-BAD with 14-3-3β.
pRBCduet vectors co-expressing untagged human 14-3-3β and M. musculus BAD were transformed with pKW2-EFSep in the pSer-3.1G cell line and expressed in ZY-AIM media at 18 °C for 20 hrs. Reported protein yields of purified BAD—14-3-3 complexes and phosphoserine incorporation fidelity are based on purifications from three independent expressions. Site-specific incorporation of pSer at sites 136 and 155 was confirmed by tandem mass spectrometry as described above. An OrbiTrap Fusion mass spectrometer equipped with electron transfer dissociation (ETD) was used to identify site 112 as phosphorylated. ETD data were manually interpreted.
Supplementary Material
Acknowledgements.
We thank P. Andrew Karplus (Oregon State University) for discussions during manuscript preparation, Yongwei Zhang (Albert Einstein College of Medicine) for the PPY strain used for SLiCE cloning, and Lisa Jones (Fred Hutch Proteomics Facility) for assistance with collecting tandem mass spectrometry data. The OrbiTrap Fusion mass spectrometer used in this research was purchased with a generous grant from the MJ Murdock Charitable Trust.
Funding. This work was supported by the National Institute of Health [1R01GM114653–01 to R.A.M]; the Medical Research Foundation at Oregon Health Sciences University [to R.B.C]; and the Collins Medical Trust [to R.B.C.]. The Proteomics Facility at the Fred Hutchinson Cancer Research Center is funded by a National Institutes of Health Cancer Center Support Grant [P30 CA015704].
Footnotes
Conflict of Interest. The authors declare no conflicts of interest.
REFERENCES
- (1).Pawson T; Scott JD Protein Phosphorylation in Signaling−−50 Years and Counting. Trends Biochem. Sci. 2005, 30 (6), 286–290. [DOI] [PubMed] [Google Scholar]
- (2).Dephoure N; Gould KL; Gygi SP; Kellogg DR Mapping and Analysis of Phosphorylation Sites: A Quick Guide for Cell Biologists. Mol. Biol. Cell 2013, 24 (5), 535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).del Peso L; González-García M; Page C; Herrera R; Nuñez G Interleukin-3-Induced Phosphorylation of BAD through the Protein Kinase Akt. Science 1997, 278 (5338), 687–689. [DOI] [PubMed] [Google Scholar]
- (4).Canman CE; Lim DS; Cimprich KA; Taya Y; Tamai K; Sakaguchi K; Appella E; Kastan MB; Siliciano JD Activation of the ATM Kinase by Ionizing Radiation and Phosphorylation of p53. Science 1998, 281 (5383), 1677–1679. [DOI] [PubMed] [Google Scholar]
- (5).Zhu G; Liu Y; Shaw S Protein Kinase Specificity: A Strategic Collaboration between Kinase Peptide Specificity and Substrate Recruitment. Cell Cycle 2005, 4 (1), 52–56. [DOI] [PubMed] [Google Scholar]
- (6).Dumas A; Lercher L; Spicer CD; Davis BG Designing Logical Codon Reassignment - Expanding the Chemistry in Biology. Chem. Sci. 2015, 6 (1), 50–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Park H-S; Hohn MJ; Umehara T; Guo L-T; Osborne EM; Benner J; Noren CJ; Rinehart J; Soll D Expanding the Genetic Code of Escherichia Coli with Phosphoserine. Science (80-. ). 2011, 333 (6046), 1151–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lee S; Oh S; Yang A; Kim J; Söll D; Lee D; Park H-S A Facile Strategy for Selective Incorporation of Phosphoserine into Histones. Angew. Chemie Int. Ed. 2013, 52 (22), 5771–5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pirman NL; Barber KW; Aerni HR; Ma NJ; Haimovich AD; Rogulina S; Isaacs FJ; Rinehart J A Flexible Codon in Genomically Recoded Escherichia Coli Permits Programmable Protein Phosphorylation. Nat. Commun. 2015, 6 (1), 8130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).George S; Aguirre JD; Spratt DE; Bi Y; Jeffery M; Shaw GS; O’Donoghue P Generation of Phospho-Ubiquitin Variants by Orthogonal Translation Reveals Codon Skipping. FEBS Lett. 2016, 590 (10), 1530–1542. [DOI] [PubMed] [Google Scholar]
- (11).Lajoie MJ; Rovner AJ; Goodman DB; Aerni H-R; Haimovich AD; Kuznetsov G; Mercer JA; Wang HH; Carr PA; Mosberg JA; et al. Genomically Recoded Organisms Expand Biological Functions. Science 2013, 342 (6156), 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wannier TM; Kunjapur AM; Rice DP; McDonald MJ; Desai MM; Church GM Adaptive Evolution of Genomically Recoded Escherichia Coli. Proc. Natl. Acad. Sci. 2018, 115 (12), 3090–3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Venkat S; Sturges J; Stahman A; Gregory C; Gan Q; Fan C Genetically Incorporating Two Distinct Post-Translational Modifications into One Protein Simultaneously. ACS Synth. Biol. 2018, 7 (2), 689–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Rogerson DT; Sachdeva A; Wang K; Haq T; Kazlauskaite A; Hancock SM; Huguenin-Dezot N; Muqit MMK; Fry AM; Bayliss R; et al. Efficient Genetic Encoding of Phosphoserine and Its Nonhydrolyzable Analog. Nat. Chem. Biol. 2015, 11 (7), 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Mukherjee M; Sabir S; O’Regan L; Sampson J; Richards MW; Huguenin-Dezot N; Ault JR; Chin JW; Zhuravleva A; Fry AM; et al. Mitotic Phosphorylation Regulates Hsp72 Spindle Localization by Uncoupling ATP Binding from Substrate Release. Sci. Signal. 2018, 11 (543), eaao2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Burgess SG; Mukherjee M; Sabir S; Joseph N; Gutiérrez-Caballero C; Richards MW; Huguenin-Dezot N; Chin JW; Kennedy EJ; Pfuhl M; et al. Mitotic Spindle Association of TACC3 Requires Aurora-A-dependent Stabilization of a Cryptic Α-helix. EMBO J. 2018, 37 (8), e97902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Huguenin-Dezot N; De Cesare V; Peltier J; Knebel A; Kristaryianto YA; Rogerson DT; Kulathu Y; Trost M; Chin JW Synthesis of Isomeric Phosphoubiquitin Chains Reveals That Phosphorylation Controls Deubiquitinase Activity and Specificity. Cell Rep. 2016, 16 (4), 1180–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dickson C; Fletcher AJ; Vaysburd M; Yang J-C; Mallery DL; Zeng J; Johnson CM; McLaughlin SH; Skehel M; Maslen S; et al. Intracellular Antibody Signalling Is Regulated by Phosphorylation of the Fc Receptor TRIM21. Elife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Butt TR; Edavettal SC; Hall JP; Mattern MR SUMO Fusion Technology for Difficult-to-Express Proteins. Protein Expr. Purif. 2005, 43 (1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Pandey A; Shin K; Patterson RE; Liu X-Q; Rainey JK Current Strategies for Protein Production and Purification Enabling Membrane Protein Structural Biology. Biochem. Cell Biol. 2016, 94 (6), 507–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Waugh DS Making the Most of Affinity Tags. Trends Biotechnol. 2005, 23 (6), 316–320. [DOI] [PubMed] [Google Scholar]
- (22).Malhotra A Tagging for Protein Expression. In Methods in enzymology; 2009; Vol. 463, pp 239–258. [DOI] [PubMed] [Google Scholar]
- (23).Mukai T; Hoshi H; Ohtake K; Takahashi M; Yamaguchi A; Hayashi A; Yokoyama S; Sakamoto K Highly Reproductive Escherichia Coli Cells with No Specific Assignment to the UAG Codon. Sci. Rep. 2015, 5 (1), 9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Johnson DBF; Wang C; Xu J; Schultz MD; Schmitz RJ; Ecker JR; Wang L Release Factor One Is Nonessential in Escherichia Coli. ACS Chem. Biol. 2012, 7 (8), 1337–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Murphy KC; Campellone KG Lambda Red-Mediated Recombinogenic Engineering of Enterohemorrhagic and Enteropathogenic E. Coli. BMC Mol. Biol. 2003, 4 (1), 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Datsenko KA; Wanner BL One-Step Inactivation of Chromosomal Genes in Escherichia Coli K-12 Using PCR Products. Proc. Natl. Acad. Sci. 2000, 97 (12), 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).St-Pierre F; Cui L; Priest DG; Endy D; Dodd IB; Shearwin KE One-Step Cloning and Chromosomal Integration of DNA. ACS Synth. Biol. 2013, 2 (9), 537–541. [DOI] [PubMed] [Google Scholar]
- (28).Ravnikar PD; Somerville RL Genetic Characterization of a Highly Efficient Alternate Pathway of Serine Biosynthesis in Escherichia Coli. J. Bacteriol. 1987, 169 (6), 2611–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Steinfeld JB; Aerni HR; Rogulina S; Liu Y; Rinehart J Expanded Cellular Amino Acid Pools Containing Phosphoserine, Phosphothreonine, and Phosphotyrosine. ACS Chem. Biol. 2014, 9 (5), 1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Pédelacq J-D; Cabantous S; Tran T; Terwilliger TC; Waldo GS Engineering and Characterization of a Superfolder Green Fluorescent Protein. Nat. Biotechnol. 2006, 24 (1), 79–88. [DOI] [PubMed] [Google Scholar]
- (31).Kinoshita E; Kinoshita-Kikuta E; Takiyama K; Koike T Phosphate-Binding Tag, a New Tool to Visualize Phosphorylated Proteins. Mol. Cell. Proteomics 2006, 5 (4), 749–757. [DOI] [PubMed] [Google Scholar]
- (32).Lee C-R; Park Y-H; Kim Y-R; Peterkofsky A; Seok Y-J The Molecular Mechanism of the Phosphorylation-Dependent Mobility Shift. Bull. Korean Chem. Soc 2013, 34 (7), 2063. [Google Scholar]
- (33).Liu S; Cai X; Wu J; Cong Q; Chen X; Li T; Du F; Ren J; Wu Y-T; Grishin N V; et al. Phosphorylation of Innate Immune Adaptor Proteins MAVS, STING, and TRIF Induces IRF3 Activation. Science 2015, 347 (6227), aaa2630. [DOI] [PubMed] [Google Scholar]
- (34).Tanaka Y; Chen ZJ STING Specifies IRF3 Phosphorylation by TBK1 in theCytosolic DNA Signaling Pathway. Sci. Signal. 2012, 5 (214), ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Li Z; Liu G; Sun L; Teng Y; Guo X; Jia J; Sha J; Yang X; Chen D; Sun Q PPM1A Regulates Antiviral Signaling by Antagonizing TBK1-Mediated STING Phosphorylation and Aggregation. PLOS Pathog. 2015, 11 (3), e1004783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Tsuchiya Y; Jounai N; Takeshita F; Ishii KJ; Mizuguchi K Ligand-Induced Ordering of the C-Terminal Tail Primes STING for Phosphorylation by TBK1. EBioMedicine 2016, 9, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zhao B; Shu C; Gao X; Sankaran B; Du F; Shelton CL; Herr AB; Ji J-Y; Li P Structural Basis for Concerted Recruitment and Activation of IRF-3 by Innate Immune Adaptor Proteins. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (24), E3403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Shang G; Zhang C; Chen ZJ; Bai X; Zhang X Cryo-EM Structures of STING Reveal Its Mechanism of Activation by Cyclic GMP–AMP. Nature 2019, 567 (7748), 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zhang C; Shang G; Gui X; Zhang X; Bai X; Chen ZJ Structural Basis of STING Binding with and Phosphorylation by TBK1. Nature 2019, 567 (7748), 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Konno H; Konno K; Barber GN Cyclic Dinucleotides Trigger ULK1 (ATG1) Phosphorylation of STING to Prevent Sustained Innate Immune Signaling. Cell 2013, 155 (3), 688–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Danial NN BAD: Undertaker by Night, Candyman by Day. Oncogene 2008, 27 (S1), S53–S70. [DOI] [PubMed] [Google Scholar]
- (42).Pandey V; Wang B; Mohan CD; Raquib AR; Rangappa S; Srinivasa V; Fuchs JE; Girish KS; Zhu T; Bender A; et al. Discovery of a Small-Molecule Inhibitor of Specific Serine Residue BAD Phosphorylation. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (44), E10505–E10514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Macdonald A; Campbell DG; Toth R; McLauchlan H; Hastie CJ; Arthur JSC Pim Kinases Phosphorylate Multiple Sites on Bad and Promote 14-3-3 Binding and Dissociation from Bcl-XL. BMC Cell Biol. 2006, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Johnson C; Crowther S; Stafford MJ; Campbell DG; Toth R; MacKintosh C Bioinformatic and Experimental Survey of 14-3-3-Binding Sites. Biochem. J. 2010, 427 (1), 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Lizcano JM; Morrice N; Cohen P Regulation of BAD by cAMP-Dependent Protein Kinase Is Mediated via Phosphorylation of a Novel Site, Ser155. Biochem. J. 2000, 349 (Pt 2), 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (46).Hekman M; Albert S; Galmiche A; Rennefahrt UEE; Fueller J; Fischer A; Puehringer D; Wiese S; Rapp UR Reversible Membrane Interaction of BAD Requires Two C-Terminal Lipid Binding Domains in Conjunction with 14-3-3 Protein Binding. J. Biol. Chem. 2006, 281 (25), 17321–17336. [DOI] [PubMed] [Google Scholar]
- (47).Zhang X; Shi H; Wu J; Zhang X; Sun L; Chen C; Chen ZJ Cyclic GMP-AMP Containing Mixed Phosphodiester Linkages Is an Endogenous High-Affinity Ligand for STING. Mol. Cell 2013, 51 (2), 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Hinds MG; Smits C; Fredericks-Short R; Risk JM; Bailey M; Huang DCS; Day CL Bim, Bad and Bmf: Intrinsically Unstructured BH3-Only Proteins That Undergo a Localized Conformational Change upon Binding to Prosurvival Bcl-2 Targets. Cell Death Differ. 2007, 14 (1), 128–136. [DOI] [PubMed] [Google Scholar]
- (49).Rexford A; Zorio DAR; Miller BG Biochemical and Biophysical Investigations of the Interaction between Human Glucokinase and pro-Apoptotic BAD. PLoS One 2017, 12 (2), e0171587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Masters SC; Yang H; Datta SR; Greenberg ME; Fu H 14-3-3 Inhibits Bad-Induced Cell Death through Interaction with Serine-136. Mol. Pharmacol. 2001, 60 (6), 1325–1331. [DOI] [PubMed] [Google Scholar]
- (51).Sadowski I; Breitkreutz B-J; Stark C; Su T-C; Dahabieh M; Raithatha S; Bernhard W; Oughtred R; Dolinski K; Barreto K; et al. The PhosphoGRID Saccharomyces Cerevisiae Protein Phosphorylation Site Database: Version 2.0 Update. Database (Oxford). 2013, 2013, bat026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Iakoucheva LM; Radivojac P; Brown CJ; O’Connor TR; Sikes JG; Obradovic Z; Dunker AK The Importance of Intrinsic Disorder for Protein Phosphorylation. Nucleic Acids Res. 2004, 32 (3), 1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Karlberg T; Hornyak P; Pinto AF; Milanova S; Ebrahimi M; Lindberg M; Püllen N; Nordström A; Löverli E; Caraballo R; et al. 14-3-3 Proteins Activate Pseudomonas Exotoxins-S and -T by Chaperoning a Hydrophobic Surface. Nat. Commun. 2018, 9 (1), 3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Pennington K; Chan T; Torres M; Andersen J The Dynamic and Stress-Adaptive Signaling Hub of 14-3-3: Emerging Mechanisms of Regulation and Context-Dependent Protein–protein Interactions. Oncogene 2018, 37 (42), 5587–5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Sluchanko NN Association of Multiple Phosphorylated Proteins with the 14-3-3 Regulatory Hubs: Problems and Perspectives. J. Mol. Biol. 2018, 430 (1), 20–26. [DOI] [PubMed] [Google Scholar]
- (56).Chemla Y; Ozer E; Algov I; Alfonta L Context Effects of Genetic Code Expansion by Stop Codon Suppression. Curr. Opin. Chem. Biol. 2018, 46, 146–155. [DOI] [PubMed] [Google Scholar]
- (57).Zhang MS; Brunner SF; Huguenin-Dezot N; Liang AD; Schmied WH; Rogerson DT; Chin JW Biosynthesis and Genetic Encoding of Phosphothreonine through Parallel Selection and Deep Sequencing. Nat. Methods 2017, 14 (7), 729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Murphy KC; Campellone KG Lambda Red-Mediated Recombinogenic Engineering of Enterohemorrhagic and Enteropathogenic E. Coli. BMC Mol. Biol. 2003, 4 (1), 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Chung B; Lee D-Y Computational Codon Optimization of Synthetic Gene for Protein Expression. BMC Syst. Biol. 2012, 6 (1), 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Zhang Y; Werling U; Edelmann W SLiCE: A Novel Bacterial Cell Extract-Based DNA Cloning Method. Nucleic Acids Res. 2012, 40 (8), e55–e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Inoue H; Nojima H; Okayama H High Efficiency Transformation of Escherichia Coli with Plasmids. Gene 1990, 96 (1), 23–28. [DOI] [PubMed] [Google Scholar]
- (62).Peeler JC; Mehl RA Site-Specific Incorporation of Unnatural Amino Acids as Probes for Protein Conformational Changes. In Methods in molecular biology (Clifton, N.J.); 2012; Vol. 794, pp 125–134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.