

Abstract
We present two rare cases of mixed large cell neuroendocrine carcinoma and squamous cell carcinoma of the colon. A literature search revealed only three published cases with similar histology but none of these reports provided profound molecular and mutational analyses. Our two cases exhibited a distinct, colon‐like immunophenotype with strong nuclear CDX2 and β‐catenin expression in more than 90% of the tumour cells of both components. We analysed the two carcinomas regarding microsatellite stability, RAS, BRAF and PD‐L1 status. In addition, next‐generation panel sequencing with Ion AmpliSeq™ Cancer Hotspot Panel v2 was performed. This approach revealed mutations in FBXW7, CTNNB1 and PIK3CA in the first case and FBXW7 and RB1 mutations in the second case. We looked for similar mutational patterns in three publicly available colorectal adenocarcinoma data sets, as well as in collections of colorectal mixed neuroendocrine‐non‐neuroendocrine neoplasms (MiNENs) and colorectal neuroendocrine carcinomas. This approach indicated that the FBXW7 point mutation, without being accompanied by classical adenoma–carcinoma sequence mutations, such as APC, KRAS and TP53, likely occurs at a relatively high frequency in mixed neuroendocrine and squamous cell carcinoma and therefore may be characteristic for this rare tumour type. FBXW7 codifies the substrate recognition element of an ubiquitin ligase, and inactivating FBXW7 mutations lead to an exceptional accumulation of its target β‐catenin which results in overactivation of the Wnt‐signalling pathway. In line with previously described hypotheses of de‐differentiation of colon cells by enhanced Wnt‐signalling, our data indicate a crucial role for mutant FBXW7 in the unusual morphological switch that determines these rare neoplasms. Therefore, mixed large cell neuroendocrine and a squamous cell carcinoma can be considered as a distinct carcinoma entity in the colon, defined by morphology, immunophenotype and distinct molecular genetic alteration(s).
Keywords: neuroendocrine carcinoma, squamous cell carcinoma, mutations, distinct entity, FBXW7, colorectal cancer
Introduction
Neuroendocrine carcinomas of the colorectum are rare and highly aggressive tumours with poor clinical outcome. Their incidence is 0.1–0.6% [1, 2]. The percentage of pure squamous cell carcinoma among all colorectal carcinomas is even lower [3, 4]. Here we present two cases of mixed large cell neuroendocrine carcinoma and squamous cell carcinoma in the colon. Previously, only three cases with an identical histology were described in the caecum, rectum and the descending colon [5, 6, 7], but extensive immunohistochemical and molecular profiling was not performed. This is the first report of this rare type of carcinoma that also defines its typical molecular genetic features. Combined neuroendocrine and squamous cell carcinomas also occur in organs with original squamous epithelium, such as the maxillary sinus or the oesophagus [8, 9]. Such neoplasms biologically present tumour development via stages of increasing atypia. On the contrary, mixed neuroendocrine and squamous cell carcinomas in the colon represent a different kind of tumour emergence. In our opinion, these rare carcinomas might be the outcome of progressive malignant transformation of mixed neuroendocrine‐non‐neuroendocrine neoplasms (MiNENs), formerly termed mixed adenoneuroendocrine carcinomas (MANECs) [10]. In accordance with this hypothesis, single cases with an additional squamous carcinoma component are known among high‐grade MiNENs in the colorectum [11]. Alongside accurate morphological evaluation, molecular classification of colorectal cancers with high grade morphology, via immunohistochemistry of mismatch repair proteins and mutational analyses of BRAF and other genes, has proven essential to provide best guidance for patient treatment and therapeutic outcome. Hence, we carefully analysed the present lesions morphologically and immunohistochemically. In order to better understand the pathophysiological mechanisms underlying these rare neoplasms, we additionally applied next‐generation sequencing and compared the mutational results to data sets of classical colorectal adenocarcinoma as well as MiNEN and neuroendocrine carcinomas of the colorectum. Based on next‐generation panel sequencing data and immunohistochemical analyses, our data indicate that mixed neuroendocrine and squamous cell carcinoma may be a distinct new colon cancer entity.
Materials and methods
Tumour specimens, histology and immunohistochemistry
This study was conducted according to the recommendations of the ethics committee of the Medical Faculty of the Ludwig‐Maximilians‐University Munich, Germany and the standards set in the declaration of Helsinki 1975. Archival tissue from two formalin‐fixed and paraffin‐embedded (FFPE) cases of colorectal combined large cell neuroendocrine carcinoma and squamous cell carcinoma were accessed from the Institute of Pathology in Bayreuth as well as from a practice of pathology in Munich. The neoplasms were resected in 2014 (first case) and 2017 (second case). Sections of 5 μm were cut, deparaffinised and stained with H&E for histological preparation. For immunohistochemistry, sections were incubated with prediluted mouse anti‐β‐catenin (14, ready to use, Ventana), rabbit mouse anti‐CK5/6 (D5/16B4, ready to use, Ventana), mouse anti‐MSH‐2 (G219‐1129, ready to use, Ventana), rabbit anti‐MSH‐6 (SP93, ready to use, Ventana), mouse anti‐PMS‐2 (A16‐4, ready to use, Ventana), rabbit anti‐PDL‐1 (SP263, ready to use, Ventana), mouse anti‐CD56 (123C3, ready to use, Ventana), rabbit anti‐synaptophysin (MRQ‐40, ready to use, Ventana), mouse anti‐chromogranin A (LK2H10, ready to use, Ventana), mouse anti‐neuron‐specific enolase (NSE; BBS/NC/VI‐H14, 1:200, Dako, Santa Clara, CA, USA), rabbit anti‐CDX2 (EPR2764y, 1:50, Medac; Bio‐Genex), mouse anti‐MLH‐1 (ES05, 1:100, Leica, Wetzlar, Germany), rabbit anti‐NUT (C52B1, 1:75, Cell Signaling), mouse anti‐p63 (BC4A4, 1:100, Zytomed; Biocare Medical, Pacheco, CA, USA), mouse anti‐p40 (BC28, 1:100, Zytomed, Berlin, Germany), mouse anti‐TTF‐1 (8G7G3/1, 1:200, Agilent, Santa Clara, CA, USA), or mouse anti‐Ki67 antibody (MIB‐1, 1:150, Dako). For staining, a Ventana Benchmark XT autostainer was used. Detection was performed with either ultraView Universal DAB detection kits or optiView DAB IHC detection kits (Ventana Medical Systems, Tuscon, AZ, USA).
DNA extraction and pyrosequencing
To identify tumour areas, we used sections stained with H&E, which were subsequently used as templates to isolate areas of the combined large cell neuroendocrine and squamous cell carcinoma under microscopic control from deparaffinised serial sections using sterile scalpel blades. Neuroendocrine and squamous components were not micro‐dissected separately. Tumour DNA was extracted with QIAamp DNA Micro Kits and GeneRead DNA FFPE Kits (Qiagen, Hilden, Germany) for consecutive analyses of KRAS, NRAS and BRAF V600E gene mutations as well as panel sequencing, respectively. The mutational status of KRAS exon 2–4, NRAS exon 2–4 and BRAF V600E was analysed by pyrosequencing on a PyroMark Q24 Advanced instrument (Qiagen), as previously described [12].
Panel sequencing
The Ion AmpliSeq Cancer Hotspot Panel v2, covering the mutation hotspots of 50 oncogenes and tumour suppressor genes (Life Technologies, Calsbad, CA, USA), was used for next‐generation panel sequencing following the manufacturer's protocol. 10 ng of Qubit quantified DNA was used for library generation with Ion AmpliSeq Library Kits and Ion Xpress Barcode Adapters (Thermo Fisher, Calsbad, CA, USA). After emulsion PCR and bead purification, multiplexed libraries were then loaded onto 318 chips, and sequenced on an Ion Personal Genome Machine (all Thermo Fisher). For data analysis, sequence reads were mapped to human reference genome hg19 and filtered for non‐synonymous variants using Ion reporter software v5.0 (Thermo Fisher). Annotations, information on pathogenesis and population allele frequencies were retrieved from Ensembl VEP (www.ensembl.org/Homo_sapiens/Tools/VEP).
Results
Case presentations
Case 1
Clinical data and pathological findings
A 51 year old male patient with known ulcerative colitis presented with rectal bleeding and diarrhoea, leading to the diagnosis of a tumour in the sigmoid colon followed by complete surgical resection. The 8 cm large, ulcerated tumour caused luminal stenosis and infiltration of the entire wall into the surrounding adipose tissue. Histology revealed lymphangiosis carcinomatosa, venous invasion and three lymph node metastases. Resection margins were free of tumour cells. Samples showed no signs of ulcerative colitis.
The carcinoma showed a solid growth pattern without gland formation or mucin production. In central areas, the tumour cells exhibited distinct squamous differentiation, whereas large tumour cells in the marginal zone exhibited no specific differentiation. Profound atypia, high rates of apoptosis, and numerous atypical mitoses, with Ki‐67 labelling index up to 90%, were present. Immunohistochemistry revealed strong nuclear expression of CDX2 and β‐catenin in over 90% of tumour cells. Cells with squamous differentiation were positive for cytokeratin 5/6 and p63, whereas the large tumour cells without specific differentiation showed strong positivity for synaptophysin and neuron specific enolase (NSE). Morphological and immunhistochemical findings are shown in Figure 1 and supplementary material, Figure S1. All tumour cells were negative for CD56, chromogranin A, p40 and TTF‐1. To distinguish the lesion from NUT (nuclear protein in testis) midline carcinoma (NMC), we performed NUT immunohistochemistry, which was negative. Immunohistochemistry for hMLH1, hMSH2, hMSH6 and hPMS2 showed nuclear expression in all tumour cells, characterising the neoplasm as a microsatellite stable tumour. In summary, a mixed large cell neuroendocrine and squamous cell carcinoma of the sigmoid colon, pT3, pN1a (3/17), V1, L1, Pn0 was diagnosed.
Figure 1.
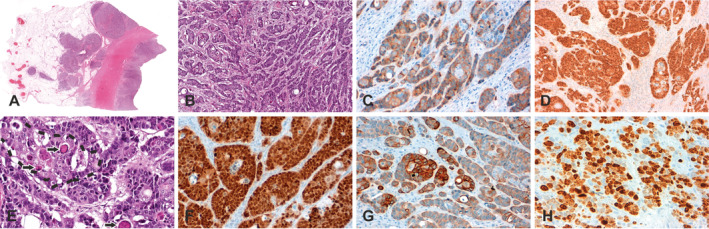
Morphological and immunohistochemical characteristics of the first case of colorectal combined large cell neuroendocrine carcinoma and squamous cell carcinoma pictured in overview (A) and close‐up view (B–H). Examples of neuroendocrine differentiation are shown by immunostaining for synaptophysin (accentuated in marginal areas; C). Tumour cells exhibit strong expression of β‐catenin (D). The squamous component is marked with a dotted line and foci of keratinisation are highlighted by arrows (E). The neoplasm shows intense staining of CDX2 (F). Examples of squamous differentiation as well as proliferation are shown by immunostaining for CK5/6 (accentuated in central areas; G) and Ki67 (H), respectively.
Within the following months of disease, distant metastasis to the liver and the abdominal wall occurred (pM1c [HEP, OTH]) resulting in a final UICC‐stage IVC. Therapy with three courses of panitumumab plus FOLFOX 6, two courses of cisplatin and etoposide and later four courses of bevacizumab and FOLFOXIRI was performed.
Molecular pathology
Because of insufficient therapeutic response, immunohistochemistry for PDL1 and molecular genetic analysis were carried out. PDL1 expression was not detectable in carcinoma cells or in the surrounding stroma. No mutations were present in exons 2, 3 and 4 of the KRAS and NRAS genes and in exon 15 of the BRAF gene. Next‐generation sequencing analysis surveying hotspot regions of 50 oncogenes and tumour suppressor genes detected CTNNB1 (c.110C>G, p.Ser37Cys), PIK3CA (c.1173A>G, p.Ile391Met) and FBXW7 (c.1393C>T, p.Arg465Cys) mutations.
Follow up
The tumour progressed rapidly under bevacizumab plus FOLFOXIRI therapy. Chemotherapy was changed to paclitaxel, carboplatin and palliative care. The patient died 1 year after initial diagnosis of the tumour.
Case 2
Clinical data and pathological findings
A 46 year old female patient without relevant pre‐existing conditions underwent colonoscopy due to diarrhoea with admixed blood. A tumour in the sigmoid colon was found and complete surgical resection performed. The resection specimen showed a 2.5 cm ulcerated tumour. Histology revealed a high‐grade carcinoma with solid growth devoid of glandular differentiation. The transmural infiltration involved the serosa. Five regional lymph node metastases were detected. Lymphangiosis carcinomatosa and venous invasion were present. Resection margins were free of tumour cells. PET‐CT scanning showed diffuse liver metastases.
The histology of the carcinoma exhibited clusters of squamous tumour cells showing immunohistochemical expression of cytokeratin 5/6, but not p63 or p40. A second tumour component showed solid and trabecular growth of large carcinoma cells with strong immunohistochemical expression of synaptophysin and CD56, but negativity for chromogranin A and NSE. All tumour cells exhibited strong cytoplasmic expression of nuclear β‐catenin and CDX2. The mitotic rate was high and the Ki‐67 proliferation index was 80% of tumour cells (Figure 2). No TTF‐1 and NUT expression was detectable by immunohistochemistry. Analysis of hMLH1, hMSH2, hMSH6 and hPMS2 showed nuclear expression in tumour cells. In summary, a mixed large cell neuroendocrine and squamous cell carcinoma of the sigmoid colon devoid of microsatellite instability was diagnosed. The following staging was reported: pT4a, pN2a (5/19), cM1a (HEP), L1, V1, Pn0, R0, UICC‐stage IVA.
Figure 2.
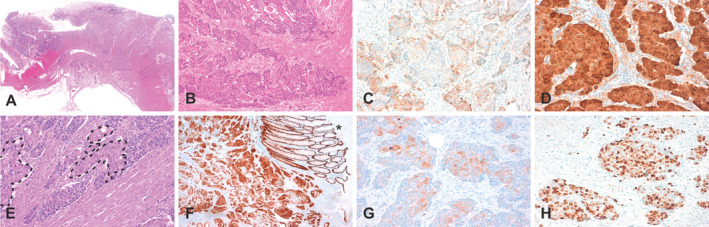
Morphological and immunohistochemical characteristics of the second case of colorectal combined large cell neuroendocrine carcinoma and squamous cell carcinoma pictured in overview (A) and close‐up view (B–H). Examples of neuroendocrine differentiation are shown by immunostaining for synaptophysin (accentuated in marginal areas; C). Tumour cells exhibit strong expression of β‐catenin (D). The squamous component is again marked with dotted lines (E). The overview shows intense staining of CDX2 in tumor and remaining normal colon mucosa (F; asterisk). Examples of squamous differentiation as well as proliferation are shown by immunostaining for CK5/6 (accentuated in central areas; G) and Ki67 (H), respectively.
Molecular pathology
Next‐generation sequencing analysis revealed a FBXW7 (c.1393C>T, p.Arg465Cys) point mutation, as was also true for the first analysed case. In addition, a RB1 (c.2284C>T, p.Gln762Ter) mutation was found. In contrast to the first case, no CTNNB1 and PIK3CA mutations were detected.
Follow up
In accordance with standard guidelines and results from the NORDIC NEC study [13], therapy with five cycles of cisplatin and etoposide followed. Follow‐up PET‐CT scanning showed complete remission of liver metastasis. Three years later one new liver metastasis with strong immunohistochemical expression of NSE was successfully ablated by local brachytherapy.
Data set analyses
Genomic data analysis on three publicly available colorectal adenocarcinoma cohort data sets was performed, employing the cBioPortal as a cancer genomics tool. The TCGA Nature 2012 Study, the updated TCGA Pan Cancer Atlas Study on CRC, and the MSKCC 2018 Cancer Cell Study for metastatic colorectal cancer [14, 15, 16, 17, 18] were screened for other cases with FBXW7, CTNNB, PIK3CA and RB1 mutations. Our search revealed 5–8% CTNNB1 mutations, 13–17% FBXW7 mutations, 20–28% PIK3CA mutations and 3–5% RB1 mutations, respectively. As expected, the classical adenoma–carcinoma sequence mutations, such as APC, KRAS and TP53, outnumber those findings by far (Table 1). In addition, we screened for significant co‐occurrences or mutual exclusivities between FBXW7, CTNNB1, PIK3CA and RB1 mutations in all three data sets, which mostly consist of classic adenocarcinoma cases, in order to explore possible mutational correlations that could potentially also occur in the scarce mixed neoplasms described here. Here again we included most common classical adenoma–carcinoma sequence mutations, such as APC, KRAS and TP53, for comparison. Referring to these, we detected significant co‐occurrence of APC and KRAS and APC and TP53 in two of three data sets. In addition, mutations in the genes coding for APC and CTNNB1 as well as TP53 and PIK3CA related to the classical adenoma–carcinoma sequence were found to be mutually exclusive. Importantly, significant co‐occurrence of FBXW7 and PIK3CA as well as FBXW7 and RB1 mutations, as was found in the scarce neoplasm type described here, was identified in two of the three data sets (Table 2). This points to functional importance of these two mutational interactions also in classical adenocarcinomas. To define similarities and differences between classical colorectal adenocarcinomas, mixed large cell neuroendocrine and squamous cell carcinomas of the colorectum, colorectal MANECs and pure colorectal neuroendocrine carcinomas, we compared frequencies of genetic alterations between those entities (Table 3). In the two cases of mixed large cell neuroendocrine and squamous cell carcinoma described here, and in contrast to MiNENs and classic adenocarcinomas, we noted the absence of APC, KRAS and TP53 mutations, as well as the occurrence of mutations in the FBXW7 gene in both tumours. The frequency of mutations in FBXW7 in particular was markedly lower (16–25%) in classic adenocarcinomas and MiNENs (Table 3), although we cannot exclude the existence of FBXW7 wild‐type, mixed neuroendocrine and squamous cell carcinoma cases from our case report on only two individuals affected by this very rare tumour type. Given that tissue images of colorectal carcinoma cases with FBWX7 mutation were available via cBioPortal within the TCGA Nature 2012 study, these were screened for unusual morphology, such as squamous or neuroendocrine differentiation. However, only two of the reviewed 35 cases showed a tendency toward neuroendocrine differentiation, and none of those had relevant morphological features which would have pointed towards squamous differentiation. Hence, other factors, such as the cell of tumour origin or epigenetic peculiarities might also be needed which, presumably in collaboration with mutant FBXW7, contribute to the occurrence of this very rare, mixed colorectal cancer entity.
Table 1.
Gene alteration frequencies in colorectal adenocarcinoma data sets.
| Genes | TCGA Nature 2012 Study | TCGA Pan Cancer Atlas Study | MSKCC 2018 Cancer Cell Study |
|---|---|---|---|
| APC | 76 | 75 | 76 |
| CTNNB1 | 5 | 7 | 8 |
| FBXW7 | 17 | 17 | 13 |
| KRAS | 42 | 42 | 45 |
| PIK3CA | 20 | 28 | 20 |
| TP53 | 53 | 60 | 73 |
| RB1 | 3 | 5 | 3 |
Values indicate the frequency of gene alterations (in percent) in three different data sets according to The Cancer Genome Atlas Program 2012 (TCGA, [16]), TCGA Pan Cancer Atlas Study [17] and Memorial Sloan Kettering Cancer Center Study (MSKCC, [18]). Classical adenoma–carcinoma sequence mutations, such as APC, KRAS and TP53, are highlighted in orange.
Table 2.
Co‐occurrences and mutual exclusivities of mutated genes in colorectal adenocarcinoma data sets.
| Significant co‐occurrence | Significant mutual exclusivity | |||||
|---|---|---|---|---|---|---|
| Mutated genes | TCGA Nature 2012 Study | TCGA Pan Cancer Atlas Study | MSKCC 2018 Cancer Cell Study | TCGA Nature 2012 Study | TCGA Pan Cancer Atlas Study | MSKCC 2018 Cancer Cell Study |
| APC and CTNNB1 | 0 | 0 | 0 | 0 | 1 (0.014) | 1 (<0.001) |
| APC and KRAS | 0 | 1 (<0.001) | 1 (0.014) | 0 | 0 | 0 |
| APC and PIK3CA | 0 | 0 | 1 (0.019) | 0 | 0 | 0 |
| APC and TP53 | 0 | 1 (<0.001) | 1 (0.022) | 0 | 0 | 0 |
| CTNNB1 and FBXW7 | 0 | 1 (<0.001) | 0 | 0 | 0 | 0 |
| CTNNB1 and PIK3CA | 0 | 1 (<0.001) | 0 | 0 | 0 | 0 |
| CTNNB1 and RB1 | 0 | 1 (<0.001) | 0 | 0 | 0 | 0 |
| FBXW7 and KRAS | 0 | 0 | 1 (0.001) | 0 | 0 | 0 |
| FBXW7 and PIK3CA | 0 | 1 (0.012) | 1 (<0.001) | 0 | 0 | 0 |
| FBXW7 and TP53 | 0 | 0 | 0 | 0 | 0 | 1 (0.013) |
| FBXW7 and RB1 | 0 | 1 (0.014) | 1 (0.001) | 0 | 0 | 0 |
| KRAS and PIK3CA | 1 (<0.001) | 1 (<0.001) | 1 (<0.001) | 0 | 0 | 0 |
| KRAS and TP53 | 0 | 0 | 0 | 0 | 0 | 1 (<0.001) |
| PIK3CA and TP53 | 0 | 0 | 0 | 0 | 1 (<0.001) | 1 (<0.001) |
Values indicate the existence (1) or non‐existence (0) of significant co‐occurrence, or significant mutual exclusivity between the listed mutated genes in three different data sets according to The Cancer Genome Atlas Program 2012 (TCGA, [16]), TCGA Pan Cancer Atlas Study [17] and Memorial Sloan Kettering Cancer Center Study (MSKCC, [18]). No significant finding is shown in red, significant correlation in one data set is marked in orange and significant findings in two or more data sets are highlighted in green. P values are indicated in parenthesis.
Table 3.
Mutations in colorectal neoplasms.
| Entity | AC | MiNEN | MiNEN | NEC | NEC | Combined large cell neuroendocrine carcinoma and squamous cell carcinoma |
|---|---|---|---|---|---|---|
| Source | TCGA, 2012 | Woischke et al, 2017 | Jesinghaus et al, 2017 | Woischke et al, 2017 | Jesinghaus et al, 2017 | Present study |
| Number of cases | 269 | 6 | 19 | 4 | 8 | 2 |
| Mutations | ||||||
| AKT1 | 0 | 0 | 25 | 0 | ||
| APC | 61 | 83 | 16 | 75 | 63 | 0 |
| ATM | 4 | 0 | 14 | 50 | 0 | |
| BRAF | 8 | 16 | 37 | 25 | 25 | 0 |
| CTNNB1 | 1 | (1 out of 2 cases) | ||||
| EGFR | 2 | 16 | 25 | 0 | ||
| ERBB4 | 0 | 0 | 25 | 0 | ||
| FBXW7 | 12 | 16 | 16 | 25 | (2 out of 2 cases) | |
| FGFR2 | 0 | 0 | 25 | 0 | ||
| FLT3 | 5 | 0 | 25 | 0 | ||
| GNAS | 0 | 0 | 25 | 0 | ||
| HRAS | 0 | 0 | 25 | 0 | ||
| IDH1 | 0 | 16 | 0 | 0 | ||
| IDH2 | 1 | 0 | 25 | 0 | ||
| JAK2 | 1 | 0 | 25 | 0 | ||
| KDR | 0 | 16 | 25 | 0 | ||
| KRAS | 35 | 83 | 21 | 100 | 25 | 0 |
| MET | 0 | 33 | 50 | 0 | ||
| NOTCH1 | 0 | 33 | 25 | 0 | ||
| PIK3CA | 16 | 50 | 5 | 25 | (1 out of 2 cases) | |
| PTEN | 5 | 0 | 11 | 0 | 0 | |
| PTPN11 | 1 | 0 | 25 | 0 | ||
| RB1 | 1 | 16 | 50 | (1 out of 2 cases) | ||
| RET | 0 | 33 | 0 | 0 | ||
| SMAD4 | 10 | 0 | 5 | 25 | 0 | |
| SMO | 0 | 0 | 25 | 0 | ||
| TP53 | 45 | 100 | 47 | 75 | 63 | 0 |
| VHL | 0 | 16 | 25 | 0 |
Frequencies of genetic alterations (in percent) of colorectal adenocarcinomas (AC), MiNENs, neuroendocrine carcinomas (NEC) in three studies (The Cancer Genome Atlas Program 2012 (TCGA, [16]), Jesinghaus et al [48] and Woischke et al [47]) in comparison with the genetic alterations of the two cases of mixed large cell neuroendocrine carcinoma and squamous cell carcinoma. Regarding TCGA cases, only putative driver mutations are included. Frequencies are highlighted by a coloured scale ranging from 0% (yellow) to 100%, or out of two for the category of mixed large cell neuroendocrine carcinoma and squamous cell carcinoma (green).
Discussion
In this study, we analysed two mixed large cell neuroendocrine and squamous cell carcinomas of the colorectum by next‐generation sequencing and compared the results with data from three publicly available colorectal adenocarcinoma data sets, as well as from cohorts of colorectal MiNENs and colorectal neuroendocrine carcinomas. This approach revealed a shared FBXW7 mutation and a lack of classical adenoma–carcinoma sequence mutations in both of our cases. This is in contrast to classic adenocarcinomas and MiNENs and therefore represents a molecular signature, which, together with the unique morphological features, may distinguish mixed neuroendocrine carcinoma and squamous carcinoma of the colorectum from other colorectal cancer types. Neuroendocrine carcinomas of colorectal origin represent very rare but highly aggressive tumours with a poor prognosis [1, 2]. Nevertheless, pure squamous cell carcinomas have been reported at an even lower incidence [3, 4, 19]. Since the first pure squamous cell carcinoma in the colorectum was reported by Schmidtmann in 1919 [20], profound literature research provided only 75 more cases to date, stating this neoplasm as extremely rare, with frequencies of 0.1–0.25% of all colorectal carcinomas [3, 4, 19]. Possible causes for this squamous colonic carcinoma are chronic inflammation in the context of ulcerative colitis, schistosomiasis, human papillomavirus infection, abdominal sinus or fistula, or pelvic radiation [4, 21]. Associations between neuroendocrine carcinomas or MiNEN of the colon and ulcerative colitis, as seen in case 1, are sporadically reported [22, 23]. The combination of the two neoplasm types in the colorectal region is highly exceptional and so far very little is known about the underlying mutational landscape of such combined carcinomas. In accordance with the new World Health Organization Classification from 2019, mixed large cell neuroendocrine carcinoma and squamous cell carcinoma in the colorectum is subsumed under the category of MiNENs, formerly named MANECs, in which each component accounts for ≥30% of the neoplasm [24]. Although three case reports of mixed neuroendocrine carcinoma and squamous cell carcinoma of the colorectum in literature do exist [5, 6, 7], only one of those has been assessed for microsatellite stability. In addition, one study examined the mutational status of KRAS and BRAF [5]. However, none of these cases has been analysed regarding its underlying genetic background via next‐generation sequencing. Thus, we performed for the first time next‐generation sequencing‐based multigene panel analysis of mixed large cell neuroendocrine carcinoma and squamous cell carcinoma of the colon.
Our two cases contain several remarkable similarities. One is the striking morphology, showing squamous carcinoma cells in central areas and poorly differentiated large cell neuroendocrine carcinoma in marginal areas, each component accounting for >30% of the tumour. The squamous cell differentiation was demonstrated not only by morphological features, such as intercellular bridges and focal keratinisation, but also by immunohistochemical expression of cytokeratin 5/6 and/or p63, with p63 being positive only in case 1. Cytokeratin 5/6 shows a sensitivity of 84% and a specificity of 79% in the diagnosis of squamous cell carcinoma, and p63 exhibits similar diagnostic performance, with a sensitivity of 81–84% and specificity of 85% [25, 26]. Neuroendocrine differentiation was confirmed by strong immunohistochemical positivity for synaptophysin, which has been approved as the best single marker for neuroendocrine tumours [27]. In accordance with one previous study, we found remarkably strong nuclear expression of CDX2 and β‐catenin in over 90% of tumour cells of both carcinoma cases as well as in both components (neuroendocrine and squamous) of the tumours [7]. The high nuclear abundance of β‐catenin detected here in large cell neuroendocrine carcinomas is very exceptional, but has been reported previously [11]. Besides clinical and morphological aspects, the strong nuclear CDX2 expression detected in the vast majority of carcinoma cells indicates the colon as the primary origin of the lesion, since CDX2 is known as a reliable marker for cancers of intestinal origin [28]. Despite the young age of the patients, both carcinomas were microsatellite stable (MSS), excluding Lynch syndrome.
In one of the cases, we identified a CTNNB1 mutation, which is a key factor in the Wnt signalling pathway and well described in the development of colorectal carcinomas [29, 30]. In one of our cases, there was a mutation in the tumour suppressor gene RB1, which are present in 5.8% of all colorectal cancers (14, 15). To date, no statistically significant impact of RB1 gene mutations on patient prognosis in colorectal cancer has been shown [31]. In addition to CTNNB1 and RB1, a PIK3CA mutation was found in one of the two neoplasms. Mutations in PIK3CA can be detected in various cancer types and have been associated with more aggressive metastatic behaviour in colorectal cancer [32]. However the PIK3CA (c.1173A>G, p.Ile391Met) mutation found here was a variant of uncertain significance (VUS) at the time of diagnosis but is now considered benign [33]. Through analyses of PIK3CA mutations in three colorectal carcinoma data sets we detected a significant co‐occurrence of PIK3CA and KRAS, which supports previous findings on that correlation [34].
The most important common feature of the two cases is the FBXW7 point mutation c.1393C>T(p.Arg465Cys). The FBXW7 gene codes for the substrate recognition component of a SCF (SKP1‐CUL1‐F‐box protein) E3 ubiquitin–protein ligase complex, which functions as an ubiquitin ligase marking several dominant oncogenic proteins, including c‐myc, cyclin E, notch and β‐catenin for ubiquitin mediated proteasomal degradation [35, 36]. Loss of function FBXW7 mutations, like the R465C gene variant described here, occur in approximately 11% of colorectal cancers [37]. Mono‐allelic missense alterations, which affect crucial arginine residues, have been reported to be the most common mutant genotypes, even though bi‐allelic inactivation mutations occur [38]. In 2017, Korphaisarn et al showed data suggesting a greater emphasis of FBXW7 missense mutation in comparison to other gene aberrations for patient outcome, linking these mutations, like those found in the above presented two cases, with a strong negative prognostic association [39]. Additional to its role as a key player in maintaining the balance between stem cell resting state and self‐regeneration [40], FBXW7 is a known regulator of Wnt/β‐catenin signalling in pancreatic cancer [41]. Although the latter has not yet been shown in colorectal cancer cells, the concept of FBXW7 controlling Wnt/β‐catenin signalling in colorectal cancer seems plausible, as a correlation between FBXW7 status and Wnt/β‐catenin signalling has been demonstrated in various cancer types [41, 42, 43]. Therefore, we suppose that the detected FBXW7 mutation resulted in malfunctioning of β‐catenin depletion with subsequent β‐catenin accumulation in the nucleus, leading to extreme overactivation of Wnt‐signalling. Due to this excessive activation of the Wnt/β‐catenin pathway, tumour cells in the colon may gain a pronounced plasticity, which may cause the critical switch towards this special combined morphology. Consistent with this hypothesis, de‐differentiation of colon cells by soluble Wnt‐ligand was recently shown by others [44]. Furthermore studies indicated the induction of squamous transdifferentiation through activation of β‐catenin signalling in various tissues [45]. Additionally, this hypothesis is supported by the findings of Davis et al, who showed reinforced Wnt‐signalling through FBXW7 propeller tip mutation and hence a driven tumorigenesis in mouse models [46]. Notably, the R465 gene variant found in our two cases also represents a propeller tip mutation.
Of note, Wnt activating mutations in FBXW7 and CTNNB1 are not restricted to the rare colorectal cancer type identified here, but also occur in classical adenocarcinoma. However, it is widely accepted that the intestinal epithelial cell subtype of cancer origin has a major influence on ultimate tumour characteristics. In neuroendocrine tumours, these cells are most likely represented by neural crest‐derived, precursor (entero)endocrine cells [47]. Different subtypes of these secretory precursor cells localise close to the crypt base, show mixed expression of secretory and bona‐fide intestinal stem cell markers, and possess a high degree of plasticity when confronted by regenerative signals, such as pathway Wnt activation [48, 49]. Importantly, a study by Wang et al revealed that aberrant Wnt activation at an early stage of neurogenin three‐dependent enteroendocrine cell differentiation induces small intestinal adenomas positive for serotonin expression in mice [50]. Given the low frequency of enteroendocrine cells (1–2%), and the short lifespan of their early precursors, this might explain the rare occurrence of neuroendocrine tumours, and the mixed neuroendocrine and squamous cell carcinomas described here, in colorectal cancer patients. Future studies on animal models should clarify if the propeller mutation in FBXW7 alone or in combination with alterations in RB1 or CTNNB1, when occurring in distinct (neuro)endocrine precursor cells of the adult colon, gives rise to the mixed cancer type characterised in our study.
In summary, these data seem to be a first important hint for the tumorigenesis of the mixed neuroendocrine and squamous carcinoma subtype. The underlying FBXW7 mutation might be the connecting element and the trigger for the crucial morphological switch, via overactivation of the canonical Wnt/β‐catenin signalling pathway. Its special relevance is also highlighted by the fact that it appears to reveal co‐occurrence with two mutations, specifically RB1 and PIK3CA, which were also detected in the presented cases. Other genes related to neuroendocrine differentiation, like ASCL1, may also play a role in the development of the neuroendocrine component, especially since ASCL1 is involved in the Notch‐Hes1 axis, which is analogous to the Wnt‐beta catenin signalling pathway, altered by the FBXW7 mutation [51, 52, 53]. Our findings may expedite the understanding of combined tumour development in the colon and in addition help establish awareness for such rare neoplasms, although continuing research, especially with regard to divergent differentiation of neuroendocrine‐ and squamous‐related genes, is necessary to fully decode the development of this combined neoplasm.
In the past, we and others provided evidence that MiNEN do have a monoclonal origin and are not stochastically neighbouring tumours [54, 55]. Furthermore, we found key mutations such as KRAS, TP53 and APC in both tumour components of MiNEN, which indicated a tumour progression similar to the well‐known classical adenoma–carcinoma sequence of colorectal adenocarcinomas [54]. We assume that the large cell neuroendocrine carcinoma, after originating from an adenoma or an adenocarcinoma, developed squamous structures via transdifferentiating processes and hence resulted in a combined large cell neuroendocrine carcinoma and squamous cell carcinoma, in which the original glandular component vanished or was no longer detectable. Interestingly, the initial colon biopsy of the first case showed parts of an ulcerated carcinoma in addition to colon mucosa with distinct serrated morphology, which supports this hypothesis. A different option in the development of the combined morphology, such as chemotherapy‐induced transdifferentiation, as reported in lung cancer, has to be considered as well [56]. However, in our cases chemotherapy took place after the microscopic characterisation of the resected specimen was completed and thus a chemotherapy‐induced switch resulting in the combined morphology seems unlikely.
In conclusion, a mixed large cell neuroendocrine carcinoma and squamous cell carcinoma of the colon can occur, even if it is extremely rare. Furthermore, we provide the histological and genetic evidence for a primary origin of this combined carcinoma in the colon and our data indicate that tumour development might occur via FBXW7 mutation‐triggered tumorigenesis, and very intensive Wnt‐signalling pathway enhancement. In combination with the absence of classical mutations of the adenoma–carcinoma sequence, as well as the notable morphology, this could be a first hint toward a distinct entity and novel subtype of colorectal carcinoma.
Author contributions statement
CW conceived and carried out experiments, drafted the article and contributed substantially to conception and design of the study and interpretation of data. TK and JN contributed substantially to conception of the study and interpretation of data and revised the article critically for important intellectual content. PJ, AJ, JK, SE, CJA and MV carried out experiments, analysed data and revised the article critically. All authors were involved in writing the paper and had final approval of the submitted and published versions.
Supporting information
Figure S1. Morphological characteristics from case 1 in close‐up view
Acknowledgement
We thank G Charell and J Kövi for excellent technical assistance. Open access funding enabled and organized by Projekt DEAL.
No conflicts of interest were declared.
References
- 1. Bernick PE, Klimstra DS, Shia J, et al Neuroendocrine carcinomas of the colon and rectum. Dis Colon Rectum 2004; 47: 163–169. [DOI] [PubMed] [Google Scholar]
- 2. Lai CC, Wang CW, Chang C, et al Neuroendocrine carcinomas of the colon and rectum: result of a 15‐year experience. J Soc Colon Rectal Surgeon (Taiwan) 2008; 19: 87–95. [Google Scholar]
- 3. Frizelle FA, Hobday KS, Batts KP, et al Adenosquamous and squamous carcinoma of the colon and upper rectum: a clinical and histopathologic study. Dis Colon Rectum 2001; 44: 341–346. [DOI] [PubMed] [Google Scholar]
- 4. Audeau A, Han HW, Johnston MJ, et al Does human papilloma virus have a role in squamous cell carcinoma of the colon and upper rectum? Eur J Surg Oncol 2002; 28: 657–660. [DOI] [PubMed] [Google Scholar]
- 5. Hassan U, Mozayani B, Wong NA. Primary combined neuroendocrine carcinoma (small‐cell type) and squamous cell carcinoma of the colon. Histopathology 2016; 68: 755–766. [DOI] [PubMed] [Google Scholar]
- 6. Vardas K, Papadimitriou G, Chantziara M, et al Mixed large cell neuroendocrine carcinoma with squamous cell carcinoma of the rectum: report of a rare case and review of the literature. Int J Surg Case Rep 2013; 4: 1076–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Elkbuli A, Dowd B, McKenney M, et al Mixed neuroendocrine and squamous cell carcinoma of the colon: a case report and literature review. Int J Surg Case Rep 2019; 60: 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Franchi A, Rocchetta D, Palomba A, et al Primary combined neuroendocrine and squamous cell carcinoma of the maxillary sinus: report of a case with Immunohistochemical and molecular characterization. Head Neck Pathol 2015; 9: 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang L, Sun X, Zou Y, et al Small cell type neuroendocrine carcinoma colliding with squamous cell carcinoma at esophagus. Int J Clin Exp Pathol 2014; 7: 1792–1795. [PMC free article] [PubMed] [Google Scholar]
- 10. Reu S, Neumann J, Kirchner T. Gastrointestinal mixed adenoneuroendocrine carcinomas. An attempt at classification of mixed cancers. Pathologe 2012. Feb; 33: 31–38. [DOI] [PubMed] [Google Scholar]
- 11. Bae HI, Lee C, Jo YM, et al Gastric mixed adenoneuroendocrine carcinoma with squamous differentiation: a case report. J Pathol Transl Med 2016; 50: 318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Modest P, Ricard I, Heinemann V, et al Outcome according to KRAS‐, NRAS‐ and BRAF‐mutation as well as KRAS mutation variants: pooled analysis of five randomized trials in metastatic colorectal cancer by the AIO colorectal cancer study group. Ann Oncol 2016; 27: 1746–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sorbye H, Welin S, Langer SW, et al Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): the NORDIC NEC study. Ann Oncol 2013; 24: 152–160. [DOI] [PubMed] [Google Scholar]
- 14. Gao J, Aksoy BA, Dogrusoz U, et al Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pI1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cerami E, Gao J, Dogrusoz U, et al The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cancer Genome Atlas Network . Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hoadley KA, Yau C, Hinoue T, et al Cell‐of‐origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 2018; 173: 291–304.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yaeger R, Chatila WK, Lipsyc MD, et al Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 2018; 33: 125–136.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Scaringi S, Bisogni D, Messerini L, et al Squamous cell carcinoma of the middle rectum: report of a case and literature overview. Int J Surg Case Rep 2015; 7C: 127–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schmidtmann M. Zcor, Kenntnis seltener Krebsformen. Virchows Arch Path Anat 1919; 226: 100–118. [Google Scholar]
- 21. Lundquest DE, Marcus JN, Thorson AG, et al Primary squamous cell carcinoma of the colon arising in a villous adenoma. Hum Pathol 1988; 19: 362–364. [DOI] [PubMed] [Google Scholar]
- 22. Guadagno E, De Rosa F, Borrelli G, et al High‐grade MiNEN in a long‐standing history of ulcerative colitis: an unexpected evolution. Inflamm Bowel Dis 2019; 25: e38–e39. [DOI] [PubMed] [Google Scholar]
- 23. Bolzacchini E, Chini C, Cortelezzi C, et al Poorly differentiated neuroendocrine carcinoma of the sigmoid tract in long‐standing ulcerative colitis: report of a case and review of the literature. Int J Surg Pathol 2018; 26: 479–483. [DOI] [PubMed] [Google Scholar]
- 24. Inzani F, Petrone G, Rindi G. The new World Health Organization classification for pancreatic neuroendocrine neoplasia. Endocrinol Metab Clin North Am 2018; 47: 463–470. [DOI] [PubMed] [Google Scholar]
- 25. Terry J, Leung S, Laskin J, et al Optimal immunohistochemical markers for distinguishing lung adenocarcinomas from squamous cell carcinomas in small tumor samples. Am J Surg Pathol 2010; 34: 1805–1811. [DOI] [PubMed] [Google Scholar]
- 26. Kaufmann O, Fietze E, Mengs J, et al Value of p63 and cytokeratin 5/6 as immunohistochemical markers for the differential diagnosis of poorly differentiated and undifferentiated carcinomas. Am J Clin Pathol 2001; 116: 823–830. [DOI] [PubMed] [Google Scholar]
- 27. Ramage JK, Ahmed A, Ardill J, et al Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours (NETs). Gut 2012; 61: 6–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Lott LB, Morrison C, Suster S, et al CDX2 is a useful marker of intestinal‐type differentiation: a tissue microarray‐based study of 629 tumors from various sites. Arch Pathol Lab Med 2005; 129: 1100–1105. [DOI] [PubMed] [Google Scholar]
- 29. Clevers H. Wnt/beta‐catenin signaling in development and disease. Cell 2006; 127: 469–480. [DOI] [PubMed] [Google Scholar]
- 30. Alomar SY, Mansour L, Abuderman A, et al β‐Catenin accumulation and S33F mutation of CTNNB1 gene in colorectal cancer in Saudi Arabia. Pol J Pathol 2016; 67: 156–162. [DOI] [PubMed] [Google Scholar]
- 31. Ye J, Lin M, Zhang C, et al Tissue gene mutation profiles in patients with colorectal cancer and their clinical implications. Biomed Rep 2020; 13: 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Müller MF, Ibrahim AE, Arends MJ. Molecular pathological classification of colorectal cancer. Virchows Arch 2016; 469: 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. National Center for Biotechnology Information . ClinVar [VCV000135038.1]. Accessed 27 June 2020. Available from: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000135038.1
- 34. Imamura Y, Lochhead P, Yamauchi M, et al Analyses of clinicopathological, molecular, and prognostic associations of KRAS codon 61 and codon 146 mutations in colorectal cancer: cohort study and literature review. Mol Cancer 2014; 13: 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Welcker M, Orian A, Grim JE, et al A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c‐Myc and cell size. Curr Biol 2004; 14: 1852–1857. [DOI] [PubMed] [Google Scholar]
- 36. Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell 2014; 26: 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perry JM, Li L. Self‐renewal versus transformation: Fbxw7 deletion leads to stem cell activation and leukemogenesis. Genes Dev 2008; 22: 1107–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Davis H, Tomlinson I. CDC4/FBXW7 and the ‘just enough’ model of tumourigenesis. J Pathol 2012; 227: 131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Korphaisarn K, Morris VK, Overman MJ, et al FBXW7 missense mutation: a novel negative prognostic factor in metastatic colorectal adenocarcinoma. Oncotarget 2017; 8: 39268–39279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang Z, Inuzuka H, Fukushima H, et al Emerging roles of the FBW7 tumour suppressor in stem cell differentiation. EMBO Rep 2011; 13: 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jiang JX, Sun CY, Tian S, et al Tumor suppressor Fbxw7 antagonizes WNT signaling by targeting β‐catenin for degradation in pancreatic cancer. Tumour Biol 2016; 37: 13893–13902. [DOI] [PubMed] [Google Scholar]
- 42. Wu WJ, Shi J, Hu G, et al Wnt/β‐catenin signaling inhibits FBXW7 expression by upregulation of microRNA‐770 in hepatocellular carcinoma. Tumour Biol 2016; 37: 6045–6051. [DOI] [PubMed] [Google Scholar]
- 43. Chen Y, Li Y, Xue J, et al Wnt‐induced deubiquitination FoxM1 ensures nucleus β‐catenin transactivation. EMBO J 2016; 35: 668–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Merrell AJ, Stanger BZ. Adult cell plasticity in vivo: de‐differentiation and transdifferentiation are back in style. Nat Rev Mol Cell Biol 2016; 17: 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Miyoshi K, Shillingford JM, Le Provost F, et al Activation of β‐catenin signaling in differentiated mammary secretory cells induces transdifferentiation into epidermis and squamous metaplasias. Proc Natl Acad Sci U S A 2002; 99: 219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Davis H, Lewis A, Behrens A, et al Investigation of the atypical FBXW7 mutation spectrum in human tumours by conditional expression of a heterozygous propellor tip missense allele in the mouse intestines. Gut 2014; 63: 792–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schimmack S, Svejda B, Lawrence B, et al The diversity and commonalities of gastroenteropancreatic neuroendocrine tumors. Langenbecks Arch Surg 2011; 396: 273–298. [DOI] [PubMed] [Google Scholar]
- 48. Sei Y, Lu X, Liou A, et al A stem cell marker‐expressing subset of enteroendocrine cells resides at the crypt base in the small intestine. Am J Physiol Gastrointest Liver Physiol 2011; 300: G345–G356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Van Es J, Sato T, Van de Wetering M, et al Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol 2012; 14: 1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang Y, Giel‐Moloney M, Rindi G, et al Enteroendocrine precursors differentiate independently of Wnt and form serotonin expressing adenomas in response to active beta‐catenin. Proc Natl Acad Sci U S A 2007; 104: 11328–11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Song Y, Lin M, Liu Y, et al Emerging role of F‐box protein in the regulation of epithelial‐mesenchymal transition and stem cells in human cancers. Stem Cell Res Ther 2019; 10: 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kopan R, Ilagan MX. The canonical notch signaling pathway: unfolding the activation mechanism. Cell 2009; 137: 216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Von Arx C, Capozzi M, López‐Jiménez E, et al Updates on the role of molecular alterations and NOTCH signalling in the development of neuroendocrine neoplasms. J Clin Med 2019; 8: 1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Woischke C, Schaaf CW, Yang HM, et al In‐depth mutational analyses of colorectal neuroendocrine carcinomas with adenoma or adenocarcinoma components. Mod Pathol 2017; 30: 95–103. [DOI] [PubMed] [Google Scholar]
- 55. Jesinghaus M, Konukiewitz B, Keller G, et al Colorectal mixed adenoneuroendocrine carcinomas and neuroendocrine carcinomas are genetically closely related to colorectal adenocarcinomas. Mod Pathol 2017; 30: 610–619. [DOI] [PubMed] [Google Scholar]
- 56. Manca P, Russano M, Pantano F, et al Change from lung adenocarcinoma to small cell lung cancer as a mechanism of resistance to afatinib. Oncotarget 2017; 8: 59986–59990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Morphological characteristics from case 1 in close‐up view