

Abstract
Oral squamous cell carcinoma (OSCC) is the most common malignant tumor of the oral cavity and is usually preceded by a range of premalignant tissue abnormalities termed oral potentially malignant disorders. Identifying malignant transformation is critical for early treatment and consequently improved survival and decreased morbidity. Invadopodia (INV) are specialized subcellular structures required for cancer cell invasion. We developed a new method to visualize INV in keratinocytes using fluorescent immunohistochemistry (FIHC) and semi‐automated images analysis. The presence of INV was used to determine the risk of malignant transformation. We analyzed 145 formalin‐fixed, paraffin‐embedded (FFPE) oral biopsy samples from 95 patients diagnosed as nondysplastic, dysplastic, and OSCC including 49 patients whose lesions transformed to OSCC (progressing) and 46 cases that did not transform to OSCC (control). All samples were stained for Cortactin, tyrosine kinase substrate with five SH3 domains (Tks5) and matrix metallopeptidase 14 (MMP14) using FIHC, imaged using confocal microscopy and analyzed using a multichannel colocalization analysis. The areas of colocalization were used to generate an INV score. Using the INV score, we were able to identify progressing lesions with a sensitivity of 75–100% and specificity of 72–76%. A positive INV score was associated with increased risk of progression to OSCC. Our results suggest that INV markers can be used in conjunction with the current diagnostic standard for early detection of OSCC.
Keywords: epithelial dysplasia, oral cancer, prediction of cancer, invadopodia
Introduction
Oral squamous cell carcinoma (OSCC) is a devastating disease that can affect all mucosal surfaces of the oral cavity and is associated with high morbidity and mortality, particularly in advanced disease [1]. For localized disease without metastasis, the 5‐year survival rate is 80%, but it drops to 59 and 36% in regional and disseminated disease, respectively [2]. Dentists have been fundamental in the identification of early premalignant lesions [3]. However, despite being readily accessible for examination and biopsy, 64% of oral cancer cases are still detected in advanced stages with regional or distant metastasis, leading to high mortality rates that have been relatively stable in the last few decades [4]. The World Health Organization (WHO) has promoted the importance of prevention and early detection as the primary targets to control the oral cancer burden worldwide [5, 6]. Detecting the earliest signs of invasion and identifying progression to OSCC in premalignant lesions would facilitate earlier treatment and significantly decrease morbidity and mortality [7, 8]. Currently, monitoring of oral potentially malignant disorders (OPMD) is based primarily on a comprehensive clinical examination of the oral cavity [3, 9]. If areas of concern are noted during the examination, a biopsy is performed and submitted to standard H&E histopathological examination by light microscopy.
Examination of oral biopsy specimens and histopathological grading of epithelial dysplasia (OED) by light microscopy is the gold standard method to detect and predict malignant transformation and to define clinical management in OPMD [10, 11, 12]. There are many significant limitations to the current gold standard of dysplasia grading. Strong evidence indicates that a model of dysplasia progression to OSCC cannot accurately predict malignant transformation and may lead to inappropriate treatments [11, 13, 14, 15]. Dysplastic lesions are graded from mild to severe depending on the extent of abnormalities in the tissue [10] and a diagnosis of dysplasia carries an overall risk of malignant transformation of 6–36% [16, 17]. Dysplasia grading is affected by low inter‐ and intraobserver agreement [18, 19]. Importantly, OSCC can arise in nondysplastic sites as well as up to 5% of mild (low‐grade) dysplasias [10, 11], while only ~30% of severe (high‐grade) dysplasias progress to carcinoma [17]. A recent report shows that dysplasia has low sensitivity (59.6%) and low specificity (62.1%) to identify prevalent or incident oral cancer and up to 39.6% of cancers arose in patients that had leukoplakia without dysplasia [20].
Standard histopathology relies primarily on gross morphological evidence of invasion. However, at the cellular level, invasive cells in transforming dysplasias have similar genetic abnormalities to invasive OSCC before evidence of invasion can be detected by histopathology [21]. There is an urgent need to improve our ability to identify cases that will transform to OSCC and enable earlier, appropriate treatment of progressing OPMD while reducing treatments for nonprogressing OPMD.
In this study, we use markers of invasive structures present in cancer cells as potential markers of invasion. Early invasive processes involve cancer cell‐mediated degradation of the basement membrane and migration through the extracellular matrix (ECM) [22, 23, 24]. These complex and tightly regulated processes require degradation of the ECM by specialized actin‐rich membrane structures that are termed invadopodia (INV) [25, 26, 27]. INV are specialized actin‐rich subcellular structures that are seen in several invasive cancer cell lines, such as breast, head and neck, prostate, fibrosarcoma, and melanoma [28] and that enable cancer cells to escape the primary tumor. Recent evidence demonstrates direct molecular links between INV assembly and cancer progression in mouse models [29, 30] and humans [31, 32, 33, 34, 35]. The invasive functions of INV are dependent on the activation of several actin‐regulatory proteins (e.g. cortactin, Tks5, and cofilin) and matrix metalloproteases (e.g. MMP14) [26, 27, 36]. Each of these genes is upregulated in cancer cells [37] and the encoded proteins distinctively accumulate at the core of the invadopodium, rationalizing their use as markers for invasion in histological specimens [36, 38, 39].
We developed a high‐resolution, semi‐automated fluorescent immunohistochemistry (FIHC) protocol to detect INV markers in formalin‐fixed, paraffin‐embedded (FFPE) samples and used this approach to identify malignant transformation in OPMD. We used the INV score in a retrospective case–control, operator‐blinded study of oral lesions to determine specificity and sensitivity in the detection of lesions that transformed into carcinoma compared to dysplasia grading.
Materials and methods
Research ethics
All methods and experiments follow the University of Toronto research guidelines. Ethical approval was obtained from the University of Toronto Research Ethics Board #32724.
Patient selection
All cases were selected from the archives of the Toronto Oral Pathology Service (TOPS), Faculty of Dentistry, University of Toronto. All cases were received between January 2008 and December 2017. A total of 110 patients had OSCC with previous biopsies at the same site where they developed OSCC. Out of these 110 selected patients, 50 had available material to be analyzed in this study and were included in the ‘progressing’ group. One case was later removed due to the poor quality of the sections and to preserve the remaining block (49 patients total in the final analysis). We selected samples without evidence of transformation (nonprogressing) based on the review of our biopsy service database to be included as a control group. Cases were matched for histopathological diagnoses and location of the lesions. Four cases were later removed due to lack of material in the block (46 patients total in the final analysis). All cases were reviewed by three pathologists (MM, AA, ABS) before inclusion in the study and the original diagnoses were changed based on consensus between the observers. A total of 12 cases had the original diagnoses revised (see supplementary material, Table S1). All selected cases were stratified using a two‐tier system following the most recent WHO Classification of Head and Neck Tumors [10] to improve reproducibility and statistical analysis. The final diagnoses were: low‐grade dysplasia (LGD; mild epithelial dysplasia), high‐grade dysplasia (HGD; moderate or severe epithelial dysplasia), and nondysplastic lesions [18, 40].
Specimen preparation
5 μm sections were prepared from each paraffin block. Slides were heated to 60°C for 30 min and then immersed in antigen retrieval buffer at 98°C for 1 h. Slides were washed with warm water and covered with Tris buffered saline pH 7.6 with 0.05% Tween 20 (TBS‐T) then 0.5% Triton (X100‐Bioshop) for 5 min for permeabilization. The samples were washed with TBS‐T three times and then incubated in sea blocking buffer (Sea Block Serum free, PBS, Abcam, Cambridge, UK) for 2 h. All primary antibodies were purchased from Abcam (Cambridge, MA, USA) and were as follow: anti‐cortactin (ab33333), anti‐FISH ‘Tks5’ (ab118575), and anti‐MMP14 (ab3644). We performed extensive optimization of all antibodies and used a final dilution of 1:100, 1:100, and 1:200 overnight at 4 °C, respectively. The secondary antibodies included Alexa Fluor® labeled donkey anti‐mouse 555 (ab150110, Abcam, USA), donkey anti‐rabbit 488 (ab150073, Abcam, USA) and donkey anti‐goat 647 (ab150139, Abcam, USA). After 1‐h incubation with secondary antibodies at room temperature, slides were washed and stained with DAPI (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) for 30 min. The samples were mounted using ProLong Diamond Antifade Mounting media (Thermo Fisher Scientific) and imaged the same day using a Quorum Spinning Disk Confocal microscope (Quorum Technologies Inc., Puslinch, Canada). Ten images of each slide were taken using ×10 objective and ×1.6 magnification lenses. The image fields were randomly selected and X, Y coordinates were used to prevent overlap.
Image analysis
Image analysis was performed using Volocity 3D Image Analysis Software (PerkinElmer, Waltham, MA, USA). The presence of INV was calculated using colocalization analysis based on a custom algorithm. In brief, channel staining intensity correlation was performed using automatically generated thresholds based on Costes et al [41]. The threshold for colocalization was determined by the linear least‐square fit of the Cortactin/MMP14/Tks5 intensities over all pixels in the image (IG = α × IR + b) and a channel showing the product of the difference of the means was generated (PDM channel). The INV score was defined as = area of PDM in μm2 normalized by the area of the epithelium in the slide. A correction factor was applied to normalize for cell density and size variations using the mean nuclear size of epithelial cells and the number of nuclei/μm2. As shown in Figure 1A, the INV positive areas are usually located at the epithelium–matrix interface.
Figure 1.
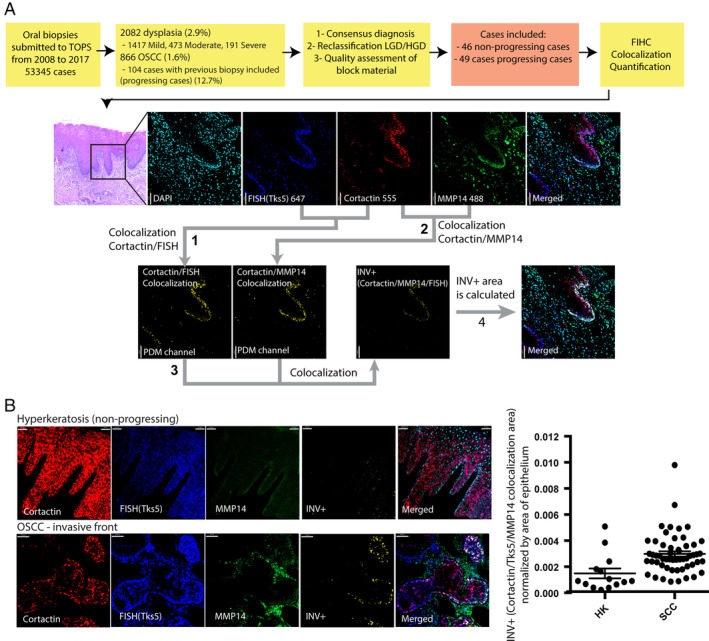
(A) Study design and quantification of INV score in oral lesions. Colocalization analysis between Cortactin and Tks5 and the resulting area of colocalization (1) is shown as the product of the differences of the PDM1 channel means. The same analysis was performed between Cortactin and MMP14 and the resulting area of colocalization (2) is shown as the PDM2 channel. The colocalization of PDM1 and PDM2 (3) was used to calculate the INV score. (B) Representative images showing INV+ areas in the training set – OSCC and nonprogressing lesions (hyperkeratosis) (p < 0.0008, t‐test). All analyzed samples are shown in the right panel. HK, hyperkeratosis; SCC, squamous cell carcinoma.
Determining the inflammatory response
Slides were stained with H&E and evaluated by light microscopy at ×200 magnification. Ten independent fields were quantified for each sample. Due to the limited size of some biopsy specimens and limited material on the block, only 44 nonprogressing and 36 progressing cases had the inflammatory infiltrate analyzed. The inflammatory cells were evaluated by MM and AA according to the following criteria: Severity of inflammation: 0 – mild (minimal), 1 – moderate (focal/dense), 2 – severe (dense/diffuse). Predominant inflammatory cell: 0 – no inflammation, 1 – lymphocytes, 2 – plasma cells, 3 – neutrophils, 4 – no predominant cells. Presence of neutrophils: 0 – no neutrophils, 1 – focal, 2 – diffuse neutrophilic infiltrate. Intraepithelial migration: 0 – no evidence of intraepithelial migration, 1 – focal, 2 – basal/para‐basal layers, 3 – inflammatory cells reaching superficial layers. Distribution of infiltration: 0 – no inflammation, 1 – inflammation adjacent to epithelium, 2 – inflammation contained at the lamina propria, 3 – deep inflammatory infiltrate.
Statistical analysis
Statistical significance was determined as p < 0.05. The results were analyzed using receiver operator characteristic (ROC) curves, Chi square tests and Youden index to determine sensitivity, specificity, and odds/risk. Confidence intervals (CIs) for relative risk were calculated using Koopman asymptotic score; attributable risk CI was calculated using Newcombe/Wilson, odds ratio CI was calculated using Baptista–Pike and sensitivity and specificity using Wilson–Brown. Agreement between observers was analyzed using Pearson's r correlation. Differences between 2 groups were calculated using t‐tests and multiple group comparisons were completed using independent‐samples Kruskal–Wallis test. OSCC progression was analyzed using Kaplan–Meier curves and Cox regression analysis. The results were analyzed using SPSS 26.0 (IBM Software, New York, USA) and PRISM 7.0 (GraphPad software, San Diego, CA, USA.
Results
Characteristics of patients
All biopsy cases from the TOPS that had a confirmed diagnosis of OED or OSCC were reviewed electronically. Patient characteristics are described in Table 1. From 2008 to 2017 there were 53 345 cases in our database. There were 2082 cases with a diagnosis of epithelial dysplasia (3.9% of total): 1417 mild dysplasia, 473 moderate dysplasia, 191 severe dysplasia, and 866 cases of OSCC (1.6% of total). From the 2082 cases of dysplasia analyzed, 64 (3.0%) cases showed a subsequent biopsy confirming malignant transformation. From the 866 OSSC cases, 104 (12.7%) had previous biopsies from the same area before progression. 64 (61.5%) cases had a previous diagnosis of dysplasia (any grade) and 40 (38.5%) had a previous biopsy without evidence of dysplasia. The rate of malignant transformation of mild dysplasia (1.3%) is lower than moderate (6.9%) and severe (5.7%) dysplasia as there were substantially more cases of mild dysplasia (1417) diagnosed compared to moderate (473) and severe dysplasia (191). The results also show a binary distribution (low versus high grade) as moderate and severe dysplasia showed similar rates of transformation. After review of the blocks and available material, 49 patients showing malignant transformation (17 nondysplastic, 32 dysplastic) were included in the ‘progressing’ group. The average time of transformation was 3.5 years (1–15.4 years) from the first biopsy. 46 patients with biopsies matching the diagnoses of the progressing group and without evidence of malignant transformation were selected as controls. The average follow‐up time for the nonprogressing group was 5.9 years (2.9–7.9 years).
Table 1.
Demographics of the groups and characteristics of patient cases (145 cases from 95 patients).
| Count | Age (range) ± SD | Male | Female | ||
|---|---|---|---|---|---|
| Nonprogressing | 46 | 56 (27–84) ±13 | |||
| Low grade dysplasia* £ | 12 | 60 (43–84) ±14 | 7 | 4 | |
| High grade dysplasia** £ | 18 | 60 (40–84) ±13 | 13 | 4 | |
| Nondysplastic | 16 | 50 (27–69) ±10 | 12 | 4 | |
| Progressing | 49 | 65 (29–93) ±13 | |||
| Low grade dysplasia | 13 | 65 (48–93) ±13 | 6 | 7 | |
| High grade dysplasia | 19 | 63 (29–87) ±14 | 13 | 6 | |
| Nondysplastic *** | 17 | 68 (46–79) ±11 | 10 | 7 | |
| OSCC | OSCC* | 50 | 66 (29–93) ±13 | 27 | 23 |
If one patient had multiple biopsies, age was considered at the time of taking each biopsy. Asterisks represent the number of patients missing age (* = 1, ** = 2, *** = 3). (£) represent the number of patients missing gender. Nondysplastic lesions include: hyperkeratosis = 18, nonspecific chronic inflammation = 8, nonspecific ulcer = 3, lichenoid inflammation = 2 and chronic inflammation and candidiasis = 2.
The included samples had the original diagnosis modified to nondysplastic (ND), LGD or HGD. A total of 12 cases had the original diagnoses revised (see supplementary material, Table S1) after consensus. There were no significant differences between the ages of patients with the different diagnoses (p = 0.97) but a significant difference was observed between the mean age of progressing (65 years, range = 29–93) and nonprogressing patients (56 years, range = 27–84) (p < 0.001). These differences were restricted to the cases with nondysplastic (ND) lesions. There were significantly more males than females in all groups reflecting the reported distribution of OSCC in the general population [2, 4].
Developing an approach to quantify INV markers in FFPE samples
INV are specialized subcellular structures that facilitate cellular invasion in carcinomas. In cellular models, the colocalization of INV markers (cortactin, Tks5/FISH, and MMP14) are used to identify these structures and an increased number of INV correlates with increased invasion [27, 36, 42]. We performed a three‐channel colocalization analysis using the product of the differences of the means (PDM) of each pixel between cortactin‐Tks5, cortactin‐MMP14, and between the results of both PDM channels (Figure 1A). To determine if INV markers could differentiate between benign and malignant oral samples, we performed the analysis on a training group that included only OSCC cases and benign, nonneoplastic oral lesions. Our results showed a significant increase in the expression and colocalization of INV markers in malignant lesions (Figure 1B), particularly in areas near the matrix interface, which is the expected localization of these structures.
Quantification of INV markers – the INV score
Next, we used the area of colocalization of the INV markers in the epithelium to create an INV score (see section ‘Materials and methods’). To determine the reliability of the INV values, authors AA and ABS independently evaluated five test samples (50 images) using the INV protocol, with an excellent correlation (Pearson r = 0.93, p < 0.02) (see supplementary material, Figure S1). The remaining sample images were then analyzed following the protocol; the observers did not have access to the diagnosis or the progression status during the analysis (blinded). There was an increase in INV+ areas in lesions that progressed to carcinoma compared to nonprogressing lesions within the different diagnostic groups (ND, LGD, HGD) (Figure 2A–H). Figure 2G summarizes the distribution of INV scores between progressing and nonprogressing cases (n = 95, p < 0.001). INV score was significantly higher in specimens that progressed to OSCC within non dysplastic cases (ND, p < 0.001) and HGD (p < 0.04) (Figure 2H). There were no significant differences in INV between ages (Figure 3A) (p < 0.08, Mann–Whitney U test; p < 0.11 linear regression analysis). Pearson's analysis (0.13, p < 0.1; see supplementary material, Table S2) showed no correlation between age and INV scores. Figure 3B shows the distribution of INV according to the location of the lesion and progression status. There was a significant increase in INV score in progressing lesions in all locations except labial mucosa/lip and the distribution of INV was similar in different locations (for lip only: p < 0.53, for all other areas: p < 0.01 independent‐samples Kruskal–Wallis test). There was a trend towards increased severity of inflammation (p < 0.26, independent‐samples Kruskal–Wallis test) and increased INV (p < 0.07, independent‐samples Kruskal–Wallis test) in tongue samples but did not achieve statistical significance (Figure 3C).
Figure 2.

Representative images of H&E stains, DAPI, Cortactin, Tks5, MMP14, and INV+ areas in lesions that progressed to carcinoma compared to nonprogressing lesions within the different diagnostic groups. (A) Progressing hyperkeratosis, (B) nonprogressing hyperkeratosis, (C) progressing low‐grade dysplasia, (D) nonprogressing low‐grade dysplasia, (E) progressing high‐grade dysplasia, (F) nonprogressing high‐grade dysplasia. (G) Distribution of INV according to progression status (n = 95, p < 0.0001, independent‐samples Mann–Whitney U test). (H) Distribution of INV by diagnosis and progression status (n = 95, p < 0.0002, two‐way ANOVA).
Figure 3.
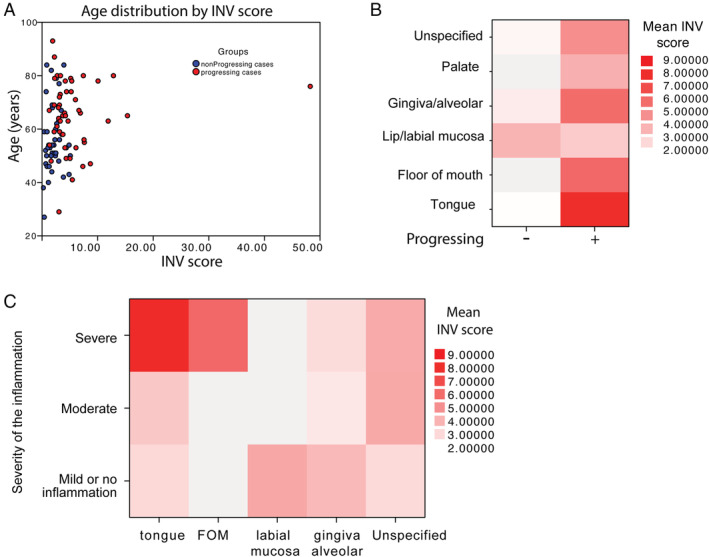
INV score distribution according to age, location and inflammatory infiltrates. (A) INV+ scores according to age (p < 0.081, Mann–Whitney U test) and progression status. (B) Distribution of INV according to location of the lesion (n = 145, p < 0.530, independent‐samples Kruskal–Wallis test), (C) distribution of INV according to location and presence of inflammation. There was trend towards increased severity of inflammation (p < 0.256, independent‐samples Kruskal–Wallis test) and increased INV (p < 0.074, independent‐samples Kruskal–Wallis test) in the tongue samples.
INV, inflammation, and progression
We recently reported a significant increase in inflammatory infiltrates in different grades of dysplasia and showed that neutrophils can directly promote INV‐dependent invasion [43]. We evaluated the characteristics of the inflammatory infiltrate in progressing and nonprogressing cases (Table 2). Our results show an increase in the severity of the inflammatory infiltrate (Figure 4A) in progressing lesions. Progressing cases showed changes in the predominant type of inflammatory cells (Table 2) with an overall increase in neutrophils and intraepithelial migration (Figure 4B,C) in progressing lesions compared to nonprogressing lesions (p < 0.002) but no statistical differences in the location of the infiltrate (superficial, deep) (p < 0.559).
Table 2.
Characteristics of the inflammatory infiltrates according to progression status and diagnosis.
| Nonprogressing | Progressing | Nondysplastic | LGD | HGD | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | % | n | % | n | % | n | % | n | % | ||
| Severity | Mild | 21 | 47.70 | 7 | 19.40 | 13 | 43.30 | 11 | 50.00 | 4 | 14.30 |
| Moderate | 13 | 29.50 | 11 | 30.60 | 6 | 20.00 | 7 | 31.80 | 11 | 39.30 | |
| Severe | 10 | 22.70 | 18 | 50.00 | 11 | 36.70 | 4 | 18.20 | 13 | 46.40 | |
| Predominant cells | No inflammation | 4 | 9.10 | 0 | 0.00 | 3 | 10.00 | 1 | 4.50 | 0 | 0.00 |
| Lymphocytes | 35 | 79.50 | 21 | 58.30 | 18 | 60.00 | 16 | 72.70 | 22 | 78.60 | |
| Plasma cells | 3 | 6.80 | 6 | 16.70 | 3 | 10.00 | 2 | 9.10 | 4 | 14.30 | |
| Neutrophils | 2 | 4.50 | 2 | 5.60 | 2 | 6.70 | 1 | 4.50 | 1 | 3.60 | |
| Mixed | 0 | 0.00 | 7 | 19.40 | 4 | 13.30 | 2 | 9.10 | 1 | 3.60 | |
| Intraepithelial migration | No migration | 5 | 11.40 | 2 | 5.60 | 3 | 10.00 | 4 | 18.20 | 0 | 0.00 |
| Focal | 20 | 45.50 | 4 | 11.10 | 11 | 36.70 | 7 | 31.80 | 6 | 21.40 | |
| Basal layer | 11 | 25.00 | 10 | 27.80 | 4 | 13.30 | 7 | 31.80 | 10 | 35.70 | |
| Superficial layer | 8 | 18.20 | 20 | 55.60 | 12 | 40.00 | 4 | 18.20 | 12 | 42.90 | |
| Neutrophils | No neutrophil | 30 | 68.20 | 8 | 22.20 | 18 | 60.00 | 12 | 54.50 | 8 | 28.60 |
| focal | 11 | 25.00 | 25 | 69.40 | 9 | 30.00 | 9 | 40.90 | 18 | 64.30 | |
| diffuse | 3 | 6.80 | 3 | 8.30 | 3 | 10.00 | 1 | 4.50 | 2 | 7.10 | |
| Distribution | No inflammation | 4 | 9.10 | 0 | 0.00 | 3 | 10.00 | 1 | 4.50 | 0 | 0.00 |
| Close to epithelium | 16 | 36.40 | 23 | 63.90 | 16 | 53.30 | 12 | 54.50 | 11 | 39.30 | |
| Lamina propria | 16 | 36.40 | 5 | 13.90 | 5 | 16.70 | 5 | 22.70 | 11 | 39.30 | |
| Deep | 8 | 18.20 | 8 | 22.20 | 6 | 20.00 | 4 | 18.20 | 6 | 21.40 | |
Figure 4.
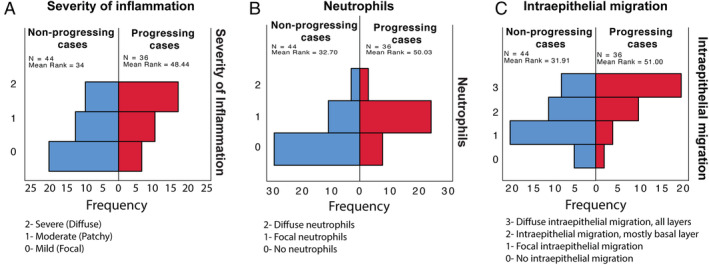
Characteristics of the inflammatory infiltrate in progressing and nonprogressing cases. (A) The severity of inflammation was assessed by two observers and divided in severe (diffuse, 2), moderate (patchy, 1) and mild (focal, 0) (p < 0.001, independent‐samples Mann–Whitney U test), (B) the presence of neutrophils was assessed and categorized as diffuse (2), focal (1) or no neutrophils (0) (p < 0.002, independent‐samples Mann–Mann–Whitney U test), (C) The presence of intraepithelial migration was assessed and categorized as diffuse (all layers, 3), diffuse (basal layer, 2), focal (1) and absent (2). (p < 0.001, independent‐samples Kruskal–Wallis test).
Developing a prognostic model for oral cancer malignant transformation
We analyzed the distribution of INV scores in progressing and nonprogressing groups to determine the sensitivity and specificity of the INV score in detecting lesions that progressed to carcinoma. For patients with multiple biopsies, the samples with the highest INV values were selected for analysis. Receiver operating characteristic curve (ROC) analysis followed by calculation of Youden's index ([Ss + Sp] − 1) was used to determine the cut off value for a positive INV. As seen in Table 3, a positive INV was able to identify progressing cases with a sensitivity of 75% (95% CI 0.62–0.85) and specificity of 76% (0.62–0.86) (Chi square 25.27, p < 0.0001, OR = 9.8, 95% CI 3.83–23.5). Progressing lesions located in the tongue showed the most significant differences in INV scores compared to nonprogressing lesions with a sensitivity of 100% (0.79–1.0 95% CI) and specificity of 72% (0.52–0.85 95% CI) (Chi square 19.64, p < 0.0001).
Table 3.
Sensitivity and specificity.
| (A) INV+ sensitivity and specificity (all samples) | |||
|---|---|---|---|
| Transformed | Nontransformed | Total | |
| INV+ | 37 | 11 | 48 |
| INV− | 12 | 35 | 47 |
| Total | 49 | 46 | 95 |
| Chi square | 25.27, p < 0.0001 | ||
| 95% CI | |||
| Relative risk | 3.019 | 1.881–5.15 | |
| Odds ratio | 9.811 | 3.831–23.5 | |
| Sensitivity | 0.7551 | 0.6191–0.854 | |
| Specificity | 0.7609 | 0.6206–0.8609 | |
| Likelihood ratio | 3.158 | ||
| Area under the curve | 0.8265 | 0.7465–0.9065 | |
| (B) INV+ sensitivity and specificity | |||
|---|---|---|---|
| Transformed | Nontransformed | Total | |
| INV+ | 15 | 7 | 22 |
| INV− | 0 | 18 | 18 |
| Total | 15 | 25 | 40 |
| Chi square | 19.64, p < 0.0001 | ||
| 95% CI | |||
| Relative risk | Infinity | 2–infinity | |
| Odds ratio | Infinity | 7.986–infinity | |
| Sensitivity | 1 | 0.7961–1 | |
| Specificity | 0.72 | 0.5242–0.8572 | |
| Likelihood ratio | 3.571 | ||
| Area under the curve | 0.8833 | 0.7762 to 0.9905 | |
Cut off values for all samples (A) and tongue‐only samples (B) were selected using Youden index ([Ss + Sp] − 1).
To determine the time‐dependent changes in OSCC progression we used Kaplan–Meier estimates and Cox proportional hazards analysis. The follow up time for control (nonprogressing) cases was determined as the date of original biopsy to the end of the data collection period (December 2019). Only two progressing cases had longer follow up of 184 and 185 months to transformation as we included all cases with available material in our cohort. Kaplan–Meier plots show that INV was associated with increased progression to oral cancer (Chi square 17.91, p < 0.0001 Mantel–Cox; Chi square 14.56, p < 0.001 Gehan‐Breslow‐Wilcoxon test) (Figure 5A). The calculated Hazard Ratio (Mantel–Haenszel) was 3.56 (95% CI 1.98–6.42). We used the SPSS automated optimal binning tool to stratify INV according to outcome (progression). The results were used to create a 3‐tier stratification of INV (high, intermediate, low) and we analyzed it using Kaplan–Meier analysis. The results show a marked increase in progression to cancer in the high INV group compared to low INV (Chi square 18.83, p < 0.0001 Mantel–Cox; Chi square 18.79, p < 0.0001 Logrank test for trend; Chi square 13.49, p < 0.0012 Gehan–Breslow–Wilcoxon test) (Figure 5B). High INV detected 20/29 progressing cases while the low risk INV score values excluded 14 out of 46 nonprogressing cases.
Figure 5.
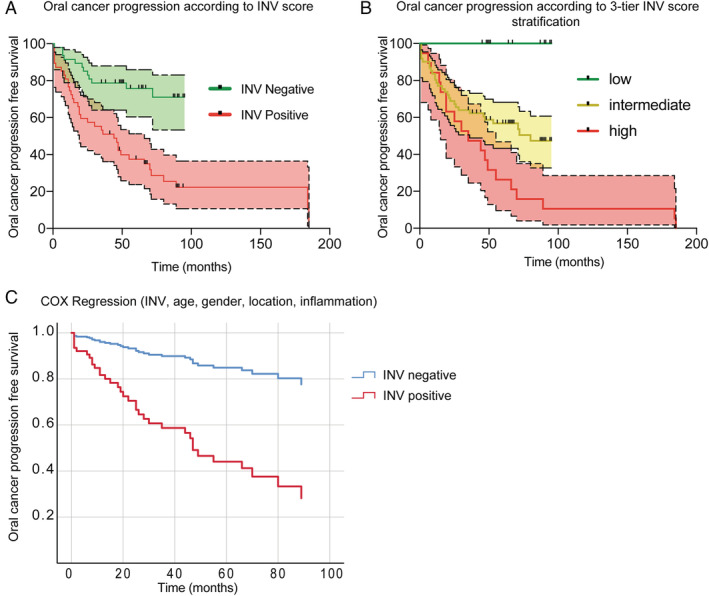
Kaplan–Meier and Cox regression analysis of oral cancer progression‐free survival according to INV. (A) Progression according to positive INV. Cut off values were defined using the Youden index; censored cases are marked with a vertical black line. Chi square 17.91, p < 0.0001 Mantel–Cox; Chi square 14.56, p < 0.001 Gehan–Breslow–Wilcoxon test. (B) Progression according to positive 3‐tier INV score distribution. Stratification of INV was performed using the SPSS optimal binning tool. Chi square 18.83, p < 0.0001 Mantel–Cox; Chi square 18.79, p < 0.0001 Logrank test for trend; Chi square 13.49, p < 0.0012 Gehan–Breslow–Wilcoxon test. (C) Cox regression analysis of oral cancer progression according to INV score status (positive/negative) HR = 8.485; 95% CI 2.855–25.213, p < 0.0001.
Cox regression analysis results are presented in Table 4. We evaluated the contribution of covariates age, gender, location, diagnosis, presence of inflammation and INV in the progression to OSCC. A positive INV score (HR = 8.49; 95% CI 2.86–25.21, p < 0.0001), the presence of dysplasia (p < 0.0016), the presence of neutrophils (HR = 2.72; 95% CI 1.23–6.03 p < 0.013) and age (HR = 1.09; 95% CI 1.044–1.127, p < 0.0001) were independently associated with increased progression to OSCC (Figure 5C).
Table 4.
Cox regression analysis of oral cancer progression free survival.
| B | SE | Wald | df | Sig. | Exp(B) | 95% CI | ||
|---|---|---|---|---|---|---|---|---|
| Lower | Upper | |||||||
| Positive INV | 2.138 | 0.556 | 14.808 | 1 | 0.001 | 8.485 | 2.855 | 25.213 |
| Age | 0.081 | 0.019 | 17.465 | 1 | 0.001 | 1.085 | 1.044 | 1.127 |
| Histopathologic diagnosis | 8.243 | 2 | 0.016 | |||||
| LGD | 1.661 | 0.58 | 8.209 | 1 | 0.004 | 5.266 | 1.69 | 16.408 |
| HGD | 1.052 | 0.515 | 4.161 | 1 | 0.041 | 2.862 | 1.042 | 7.861 |
| Severity of inflammation | 0.357 | 0.256 | 1.945 | 1 | 0.163 | 1.429 | 0.865 | 2.36 |
| Neutrophils | 1.002 | 0.405 | 6.104 | 1 | 0.013 | 2.723 | 1.23 | 6.028 |
| Gender | −0.18 | 0.401 | 0.202 | 1 | 0.653 | 0.835 | 0.381 | 1.832 |
| Location | 0.008 | 0.07 | 0.013 | 1 | 0.911 | 1.008 | 0.879 | 1.156 |
SE, standard error; df, degrees of freedom.
Discussion
The importance of monitoring and prognostication of OPMD
Monitoring the progression of OPMD is essential to improve patient survival and decrease the morbidity of OSCC and this is one of the most significant challenges in the context of oral cancer. It is widely known that the implementation of screening programs can effectively reduce mortality in high‐risk individuals [44]. Our data are in keeping with data reported in the literature. Chatuverdi et al recently conducted a retrospective analysis of patients with oral leukoplakia to determine histopathologic predictors of progression [20]. The results revealed that dysplasia showed low sensitivity (59.6%) and low specificity (62.1%) to identify prevalent or incident oral cancer and up to 39.6% of cancers arose in biopsies of leukoplakia without dysplasia. Speight et al showed that, despite the high sensitivity, there was a very low specificity to detect lesions that have a high likelihood of progression to cancer [45, 46]. A review by Warnakulasuriya et al [47] showed that malignant transformation of OPMD can vary significantly from 0.13 to 42.2% while the specificity of the current model of dysplasia grading can be as low as 3%. Specific clinical and histopathologic features of OPMD are associated with increased risk of transformation and may not show classic features of dysplasia [48]. Muller has shown that particular histopathologic architecture is critical in determining clinical behaviour and progression of oral lesions [48].
Our reported sensitivity and specificity values of ~75% and up to 100% in tongue lesions are significantly higher than dysplasia grading and may be very useful in prognostication, as our proposed INV score was able to detect high risk lesions that were originally not identified as OPMD, e.g. hyperkeratosis and nonspecific ulcer. Our proposed INV score can be further optimized and used in conjunction with dysplasia grading to improve risk stratification of OPMD. We appreciate that many more steps are needed to fully determine how to incorporate these markers in practice and we will continue to study this in detail. We have not evaluated the clinical features of the cases of leukoplakia used in this study and further studies are needed to understand the presence of INV with specific clinical features.
Current available prognostic tools
In the context of OSCC, several approaches for estimating progression and prognosis have been used with varying success in oral cancer. Some of these technologies include analysis of DNA abnormalities [49, 50], proteomics [51], serum or salivary markers [52, 53] and histopathology/immunohistochemistry [54, 55]. Each of these methods exhibits significant shortcomings that preclude routine use in clinical settings. Considering tissue markers of invasion/malignant transformation in OSCC, numerous candidates have been examined, including metalloproteases, integrins, VEGF, and members of the S100 family of proteins [55, 56]. Tests based on histopathology alone are almost always nonquantifiable and depend on an operator‐based assessment of staining, which makes them challenging to implement in practice. At the genetic level, chromosomal polysomy, p53 protein expression, loss of heterozygosity at chromosome 3p or 9p 43 and miRNA expression profiles [57, 58] have been used as prognostic markers. However, most tests have failed clinical implementation due to technical challenges, lack of correlation to the disease mechanism and poor performance. In summary, the main problems with current tests/markers are: (1) single marker strategy instead of a multiplex approach, (2) lack of understanding/correlation with mechanisms of progression, (3) lack of reproducibility, and (4) failed implementation due to technical challenges and high cost.
We designed the INV score to be used as a semi‐automated, operator‐blinded method to minimize bias and increase reproducibility. The only input required from the operator is the manual selection of the epithelium in the images and future use of artificial intelligence can overcome this limitation. After the selection of the epithelium, the protocol automatically generates the area of colocalization used to calculate the INV score. Our results show an excellent agreement between scores from two different operators, highlighting the reproducibility of the protocol. All the steps of the protocol have been optimized to be performed using conventional laboratory equipment and FFPE samples, which are part of the patient's current standard of care and no changes in current protocols are needed. During the optimization phase, we analyzed up to 10‐year‐old FFPE samples as well as samples with variable fixation time and no changes were noted in the results. FIHC staining and analysis take 48 h after the receipt of the slide. Our proposed protocol can also be optimized for use in other cancers that are associated with increase in the expression of INV markers such as breast cancer [34, 35].
Our study and can be implemented in a conventional laboratory with access to a confocal microscope. We understand the potential challenges in relying on FIHC and confocal microscopes including the logistics associated with running a multi‐user facility, training and its high procurement cost. Our proposed analysis cannot be accurately performed in IHC stained samples due to the lack of linearity between expression and intensity and the inability to perform multichannel staining on the same slide. To address this issue, we are currently optimizing our analysis to use epifluorescence‐based slide scanners and microscopes that are commonly used in pathology laboratories.
Inflammation, location, and INV
There was a positive correlation between neutrophils, mixed and dense inflammation and increased INV. In all locations examined, there was a significant increase in INV in progressing lesions except for lip/labial mucosa. There are two reasons to explain these differences. First, there are only six lip cases in our dataset. Second, lip carcinomas are commonly associated with direct UV‐damage to the lower lip vermillion, which is a different pathogenesis from intraoral carcinomas and is associated with better outcomes [59, 60]. Exclusion of lip vermillion cases improved the INV sensitivity to 0.76 (95% CI 0.6278–0.864) and specificity to 0.77 (95% CI 0.6301–0.8716). Although the INV score did not show a positive correlation with inflammation in general, our results showed a very high INV score in tongue lesions with moderate to severe inflammation (Figure 3D), supporting our previous findings that the presence of inflammation can increase INV‐dependent invasion (malignant transformation) [43]. More cases are needed to determine if this observation is specific to tongue cancer.
Limitations of the model
This is a proof of principle study and the main limitation is the number of cases included in the analysis. The scores provided are based on best case scenarios and larger prospective studies are needed to confirm the results described here. The follow up data used here are based on our own database analysis and may miss patients from the control (nonprogressing) group that may have progressed to cancer. Our method requires a tissue sample with undisturbed epithelium; therefore, it does not eliminate the need for a proper biopsy. Additionally, further prospective studies are needed to determine if patients with positive INV or high‐risk scores would benefit from more aggressive treatment, including excisional biopsies or rebiopsy, to include margins to decrease malignant transformation. Our results at least suggest that a cohort of ‘low risk’ patients can be adequately monitored clinically without the necessity for further surgery. Further studies are also needed to include other nonneoplastic, nondysplastic, and inflammatory conditions, e.g. lichen planus, that have a risk of malignant transformation.
Author contributions statement
ABS acquired, analyzed and interpreted data, and wrote the manuscript. AA acquired data, provided technical support and wrote the manuscript. DE acquired data and provided technical support. MM conceived, designed and supervised the study, developed the methodology, provided technical support, reviewed the manuscript, and prepared the final figures and tables.
Supporting information
Figure S1. Correlation of INV score between two independent observers
Table S1. List of cases with an updated diagnosis after review
Table S2. Pearson correlation analysis between progression and diagnosis, age, gender, INV score, lesion location and inflammation
Acknowledgements
The authors would like to thank Dr. Grace Bradley for the assistance in selecting the cases. This work is supported by Canadian Cancer Society Innovation grant 704248. MM is supported by the Bertha Rosenstadt Endowment and Connaught New Investigator Award from the University of Toronto.
No conflicts of interest were declared.
References
- 1. Canadian Cancer Statistics Advisory Committee . Canadian Cancer Statistics. Canadian Cancer Society: Toronto, ON, 2019, 2019. Available from cancer.ca/Canadian-Cancer-Statistics-2019-EN. [Google Scholar]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017; 67 7–30. [DOI] [PubMed] [Google Scholar]
- 3. Abadeh A, Ali AA, Bradley G, et al Increase in detection of oral cancer and precursor lesions by dentists: evidence from an oral and maxillofacial pathology service. J Am Dent Assoc 2019; 150 531–539. [DOI] [PubMed] [Google Scholar]
- 4. Howlader N, Noone AM, Krapcho M, et al SEER Cancer Statistics Review, 1975‐2016. In: Bbased on November 2018 SEER data submission, posted to the SEER web site, April 2019. National Cancer Institute: Bethesda, MD, 2019. [Google Scholar]
- 5. Petersen PE. Oral cancer prevention and control – the approach of the World Health Organization. Oral Oncol 2009; 45 454–460. [DOI] [PubMed] [Google Scholar]
- 6. Mignogna MD, Fedele S, Lo Russo L. The world cancer report and the burden of oral cancer. Eur J Cancer Prev 2004; 13 139–142. [DOI] [PubMed] [Google Scholar]
- 7. Ebrahimi A, Murali R, Gao K, et al The prognostic and staging implications of bone invasion in oral squamous cell carcinoma. Cancer 2011; 117 4460–4467. [DOI] [PubMed] [Google Scholar]
- 8. Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol 2009; 45 301–308. [DOI] [PubMed] [Google Scholar]
- 9. Lingen MW, Abt E, Agrawal N, et al Evidence‐based clinical practice guideline for the evaluation of potentially malignant disorders in the oral cavity: a report of the American Dental Association. J Am Dent Assoc 2017; 148 712–27.e10. [DOI] [PubMed] [Google Scholar]
- 10. El‐Naggar AK, Chan JKC, Grandis JR, et al WHO Classification of Head and Neck Tumors (4th edn). International Agency for Research on Cancer (IARC): Lyon, 2017. [Google Scholar]
- 11. Brennan M, Migliorati CA, Lockhart PB, et al Management of oral epithelial dysplasia: a review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2007; 103 S19.e1–S19.e2. [DOI] [PubMed] [Google Scholar]
- 12. Silverman S, Kerr AR, Epstein JB. Oral and pharyngeal cancer control and early detection. J Cancer Educ 2010; 25 279–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cowan CG, Gregg TA, Napier SS, et al Potentially malignant oral lesions in Northern Ireland: a 20‐year population‐based perspective of malignant transformation. Oral Dis 2001; 7 18–24. [PubMed] [Google Scholar]
- 14. Holmstrup P, Vedtofte P, Reibel J, et al Long‐term treatment outcome of oral premalignant lesions. Oral Oncol 2006; 42 461–474. [DOI] [PubMed] [Google Scholar]
- 15. Dost F, Lê Cao K, Ford PJ, et al Malignant transformation of oral epithelial dysplasia: a real‐world evaluation of histopathologic grading. Oral Surg Oral Med Oral Pathol Oral Radiol 2014; 117 343–352. [DOI] [PubMed] [Google Scholar]
- 16. Lumerman H, Freedman P, Kerpel S. Oral epithelial dysplasia and the development of invasive squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1995; 79 321–329. [DOI] [PubMed] [Google Scholar]
- 17. Ho MW, Risk JM, Woolgar JA, et al The clinical determinants of malignant transformation in oral epithelial dysplasia. Oral Oncol 2012; 48 969–976. [DOI] [PubMed] [Google Scholar]
- 18. Kujan O, Khattab A, Oliver RJ, et al Why oral histopathology suffers inter‐observer variability on grading oral epithelial dysplasia: an attempt to understand the sources of variation. Oral Oncol 2007; 43 224–231. [DOI] [PubMed] [Google Scholar]
- 19. Abbey LM, Kaugars GE, Gunsolley JC, et al Intraexaminer and interexaminer reliability in the diagnosis of oral epithelial dysplasia. Oral Surgery, Oral Med Oral Pathol Oral Radiol 1995; 80 188–191. [DOI] [PubMed] [Google Scholar]
- 20. Chaturvedi AK, Udaltsova N, Engels EA, et al Oral leukoplakia and risk of progression to oral cancer: a population‐based cohort study. J Natl Cancer Inst 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cervigne NK, Machado J, Goswami RS, et al Recurrent genomic alterations in sequential progressive leukoplakia and oral cancer: drivers of oral tumorigenesis? Hum Mol Genet 2014; 23 2618–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gimona M, Buccione R, Courtneidge SA, et al Assembly and biological role of podosomes and invadopodia. Curr Opin Cell Biol 2008; 20 235–241. [DOI] [PubMed] [Google Scholar]
- 23. Yamaguchi H, Lorenz M, Kempiak S, et al Molecular mechanisms of invadopodium formation: the role of the N‐WASP‐Arp2/3 complex pathway and cofilin. J Cell Biol 2005; 168 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta 2007; 1773 642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen WT. Proteolytic activity of specialized surface protrusions formed at rosette contact sites of transformed cells. J Exp Zool 1989; 251 167–185. [DOI] [PubMed] [Google Scholar]
- 26. Oser M, Mader CC, Gil‐Henn H, et al Specific tyrosine phosphorylation sites on cortactin regulate Nck1‐dependent actin polymerization in invadopodia. J Cell Sci 2010; 123 3662–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Magalhaes MA, Larson DR, Mader CC, et al Cortactin phosphorylation regulates cell invasion through a pH‐dependent pathway. J Cell Biol 2011; 195 903–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bergman A, Condeelis JS, Gligorijevic B. Invadopodia in context. Cell Adh Migr 2014; 8 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gligorijevic B, Wyckoff J, Yamaguchi H, et al N‐WASP‐mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J Cell Sci 2012; 125 724–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gil‐Henn H, Patsialou A, Wang Y, et al Arg/Abl2 promotes invasion and attenuates proliferation of breast cancer in vivo. Oncogene 2013; 32 2622–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Eckert MA, Lwin TM, Chang AT, et al Twist1‐induced invadopodia formation promotes tumor metastasis. Cancer Cell 2011; 19 372–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baik M, French B, Chen Y‐C, et al Identification of invadopodia by TKS5 staining in human cancer lines and patient tumor samples. MethodsX 2019; 6 718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Karamanou K, Franchi M, Vynios D, et al Epithelial‐to‐mesenchymal transition and invadopodia markers in breast cancer: Lumican a key regulator. Semin Cancer Biol 2020; 62 125–133. [DOI] [PubMed] [Google Scholar]
- 34. Meirson T, Genna A, Lukic N, et al Targeting invadopodia‐mediated breast cancer metastasis by using ABL kinase inhibitors. Oncotarget 2018; 9 22158–22183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen YC, Baik M, Byers JT, et al TKS5‐positive invadopodia‐like structures in human tumor surgical specimens. Exp Mol Pathol 2019; 106 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ayala I, Baldassarre M, Giacchetti G, et al Multiple regulatory inputs converge on cortactin to control invadopodia biogenesis and extracellular matrix degradation. J Cell Sci 2008; 121 369–378. [DOI] [PubMed] [Google Scholar]
- 37. Wang W, Goswami S, Lapidus K, et al Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res 2004; 64 8585–8594. [DOI] [PubMed] [Google Scholar]
- 38. Artym VV, Zhang Y, Seillier‐Moiseiwitsch F, et al Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res 2006; 66 3034–3043. [DOI] [PubMed] [Google Scholar]
- 39. Sharma VP, Eddy R, Entenberg D, et al Tks5 and SHIP2 regulate invadopodium maturation, but not initiation, in breast carcinoma cells. Curr Biol 2013; 23 2079–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Warnakulasuriya S, Reibel J, Bouquot J, et al Oral epithelial dysplasia classification systems: predictive value, utility, weaknesses and scope for improvement. J Oral Pathol Med 2008; 37 127–133. [DOI] [PubMed] [Google Scholar]
- 41. Costes SV, Daelemans D, Cho EH, et al Automatic and quantitative measurement of protein‐protein colocalization in live cells. Biophys J 2004; 86 3993–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Oser M, Yamaguchi H, Mader CC, et al Cortactin regulates cofilin and N‐WASp activities to control the stages of invadopodium assembly and maturation. J Cell Biol 2009; 186 571–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goertzen C, Mahdi H, Laliberte C, et al Oral inflammation promotes oral squamous cell carcinoma invasion. Oncotarget 2018; 9 29047–29063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sankaranarayanan R, Ramadas K, Thomas G, et al Effect of screening on oral cancer mortality in Kerala, India: a cluster‐randomised controlled trial. Lancet 2005; 365 1927–1933. [DOI] [PubMed] [Google Scholar]
- 45. Speight PM, Khurram SA, Kujan O. Oral potentially malignant disorders: risk of progression to malignancy. Oral Surg Oral Med Oral Pathol Oral Radiol 2018; 125 612–627. [DOI] [PubMed] [Google Scholar]
- 46. Speight PM. Update on oral epithelial dysplasia and progression to cancer. Head Neck Pathol 2007; 1 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Warnakulasuriya S, Ariyawardana A. Malignant transformation of oral leukoplakia: a systematic review of observational studies. J Oral Pathol Med 2016; 45 155–166. [DOI] [PubMed] [Google Scholar]
- 48. Müller S. Oral epithelial dysplasia, atypical verrucous lesions and oral potentially malignant disorders: focus on histopathology. Oral Surg Oral Med Oral Pathol Oral Radiol 2018; 125 591–602. [DOI] [PubMed] [Google Scholar]
- 49. Foy JP, Pickering CR, Papadimitrakopoulou VA, et al New DNA methylation markers and global DNA hypomethylation are associated with oral cancer development. Cancer Prev Res (Phila) 2015; 8 1027–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee JJ, Hong WK, INVtelman WN, et al Predicting cancer development in oral leukoplakia: ten years of translational research. Clin Cancer Res 2000; 6 1702–1710. [PubMed] [Google Scholar]
- 51. Wang Z, Feng X, Liu X, et al Involvement of potential pathways in malignant transformation from oral leukoplakia to oral squamous cell carcinoma revealed by proteomic analysis. BMC Genomics 2009; 10 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Korostoff A, Reder L, Masood R, et al The role of salivary cytokine biomarkers in tongue cancer invasion and mortality. Oral Oncol 2011; 47 282–287. [DOI] [PubMed] [Google Scholar]
- 53. El‐Naggar AK, Mao L, Staerkel G, et al Genetic heterogeneity in saliva from patients with oral squamous carcinomas: implications in molecular diagnosis and screening. J Mol Diagn 2001; 3 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kawaguchi H, El‐Naggar AK, Papadimitrakopoulou V, et al Podoplanin: a novel marker for oral cancer risk in patients with oral premalignancy. J Clin Oncol 2008; 26 354–360. [DOI] [PubMed] [Google Scholar]
- 55. Winter J, Pantelis A, Reich R, et al Risk estimation for a malignant transformation of oral lesions by S100A7 and Doc‐1 gene expression. Cancer Invest 2011; 29 478–484. [DOI] [PubMed] [Google Scholar]
- 56. Kaur J, Matta A, Kak I, et al S100A7 overexpression is a predictive marker for high risk of malignant transformation in oral dysplasia. Int J Cancer 2014; 134 1379–1388. [DOI] [PubMed] [Google Scholar]
- 57. Li Z, Liu Z, Dong S, et al miR‐506 inhibits epithelial‐to‐mesenchymal transition and angiogenesis in gastric cancer. Am J Pathol 2015; 185 2412–2420. [DOI] [PubMed] [Google Scholar]
- 58. Sun G, Liu Y, Wang K, et al miR‐506 regulates breast cancer cell metastasis by targeting IQGAP1. Int J Oncol 2015; 47 1963–1970. [DOI] [PubMed] [Google Scholar]
- 59. Gallagher RP, Lee TK, Bajdik CD, et al Ultraviolet radiation. Chronic Dis Can 2010; 29 51–68. [PubMed] [Google Scholar]
- 60. Biasoli É, Valente VB, Mantovan B, et al Lip cancer: a Clinicopathological study and treatment outcomes in a 25‐year experience. J Oral Maxillofac Surg 2016; 74 1360–1367. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Correlation of INV score between two independent observers
Table S1. List of cases with an updated diagnosis after review
Table S2. Pearson correlation analysis between progression and diagnosis, age, gender, INV score, lesion location and inflammation