

Abstract
The AutoDock suite provides a comprehensive toolset for computational ligand docking and drug design and development. The suite builds on 30 years of methods development, including empirical free energy force fields, docking engines, methods for site prediction, and interactive tools for visualization and analysis. Specialized tools are available for challenging systems, including covalent inhibitors, peptides, compounds with macrocycles, systems where ordered hydration plays a key role, and systems with substantial receptor flexibility. All methods in the AutoDock suite are freely available for use and reuse, which has engendered the continued growth of a diverse community of primary users and third‐party developers.
Keywords: active site prediction, AutoDock, computational docking, computer‐aided drug design, force field, peptide‐docking
1. INTRODUCTION
AutoDock was presented 30 years ago as the first method for docking flexible ligands to proteins. At the time, methods for biomolecular structure prediction were often strictly limited by the nascent state of computational infrastructure, which were limited both in storage space and computational speed. Powerful molecular dynamics methods such as AMBER 1 provided a physics‐based representation, but were limited to short time scales and typically explored local conformational spaces around a starting model. The ground‐breaking DOCK method employed a simplified representation of ligand‐receptor interactions, with rigid ligands and a sphere‐based method for scoring interactions, and was thus able to search the larger conformational spaces required for computational docking studies and drug discovery. 2 AutoDock took an intermediate approach, using a physics‐based force field akin to AMBER, but using it with a rapid volumetric energy evaluation approach to allow docking that explores the large conformational spaces of a flexible ligand. 3
Thirty years later, the AutoDock suite has been used in numerous research and translational medicine efforts around the world. Searching for “AutoDock” at PubMedCentral yields over 7,000 publications and a combined total of 30,000 citations are reported by Google Scholar for the three most‐cited AutoDock suite publications. These include reports focusing on docking, drug design methodology, and primary applications. To get a feeling for the diversity of these applications, we did a survey of applications published in JACS from the PubMedCentral list, finding studies of, for example, analysis of non‐natural substrates of strictosidine synthase, 4 ligand binding in an artificial streptavidin Rh(III) metalloenzyme, 5 binding of covalent DNA intercalators, 6 characterization of the binding of dyes to soluble oligomers of Aβ amyloids, 7 evaluation of targeted covalent inhibitors from a fragment‐based ligand discovery effort to target a colon cancer cell proteome, 8 and virtual screening of glycans against sialoadhesin. 9
Today, users have multiple academic and commercial options for docking (see, for example, several recent reviews 10 , 11 ), and increasingly large docking screens are routinely performed with successful results. 12 , 13 However, the effectiveness of docking and virtual screening is still limited by challenges in both user interaction and the necessary simplifications of the underlying physics. To address these challenges, AutoDock development is proceeding with two goals in mind. First, a strong user‐focused development track has produced validated tools for general use, including GUIs, docking packages, and analysis tools for use by a wide range of expert and non‐expert users. Second, we have continued multiple parallel tracks of development of new methods, using the AutoDock approach as a lab rat to address the ongoing limitations in docking methods, including deficiencies in scoring methods and extension of conformational searches into larger and larger chemical spaces (Figure 1).
FIGURE 1.
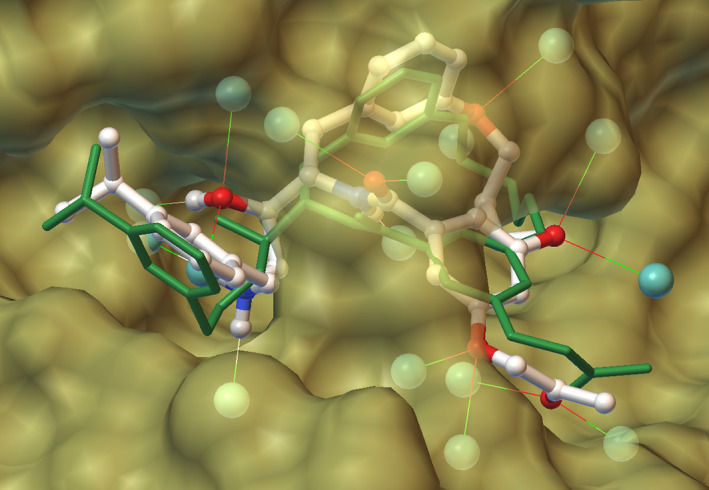
Docking result from the D3R Grand Challenge 4, with a macrocyclic ligand bound to BACE1, using a new approach to gpu‐accelerated docking and a novel hydrated‐ligand model. All possible sites on the ligand are hydrated with ideal geometry (cyan spheres), and after docking all but one overlap with the protein (yellow surface) and are used to evaluate a desolvation contribution to the free energy. The crystallographic pose is shown in green. Image generated in Python Molecule Viewer including AutoDockTools
2. THE AUTODOCK SUITE
Computational docking methods seek to predict the interaction of ligands and macromolecular targets. Docking is typically used as part of a larger exploratory or design pipeline (Figure 2). Two underlying problems must be solved in any effective docking method. First, a force field is required to score trial poses of the complex, hopefully in a way that reflects the underlying energetics of biomolecular interaction. Second, a search method is needed to explore enough of the available conformational space of interaction to ensure that a reproducible and relevant answer is obtained. Early in the development of the AutoDock suite, we made a decision to build our force fields on a strong foundation of physics‐based methods, which have shown much success in prediction of biomolecular structure, interactions, and properties. The challenge has been to simplify these force fields in ways that allow their use in the various search methods needed to explore large docking conformational spaces.
FIGURE 2.
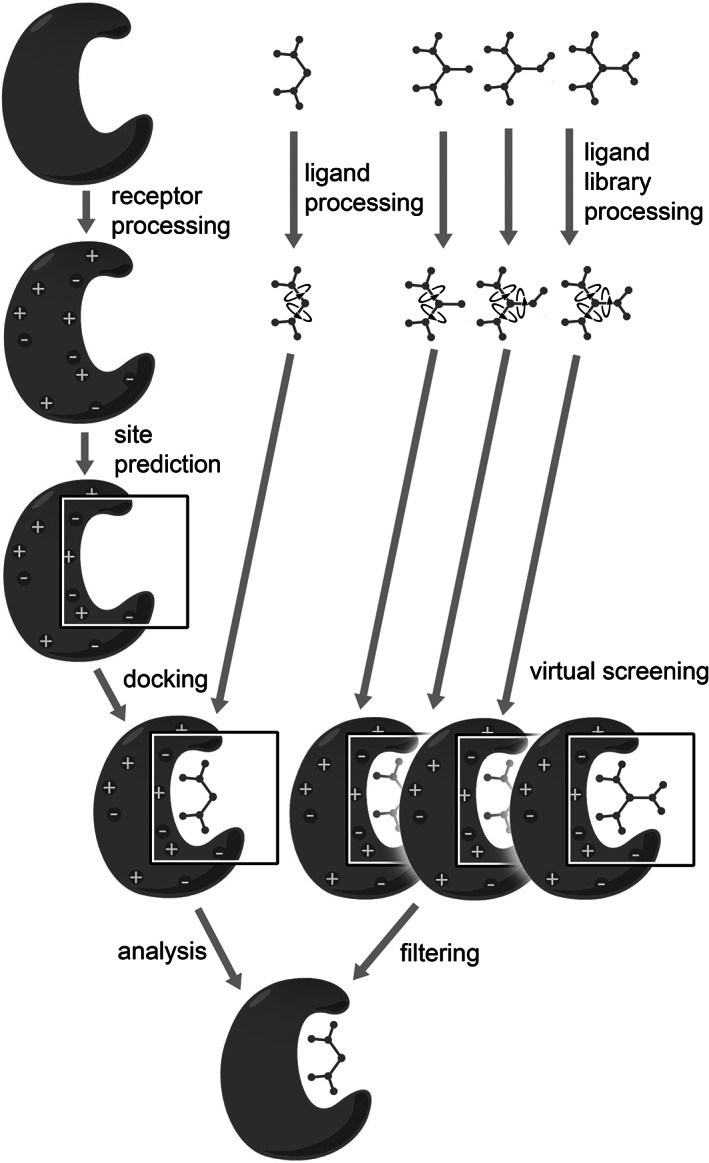
A typical docking pipeline starts with coordinates of a receptor and a ligand, from experimental structure determinations, homology modeling, idealized generation from SMILES, and so forth, shown here schematically at the top. Both receptors and ligands are processed to conform to the representation used in the docking method (assigning atom types and charges, defining modes of flexibility, etc.), and often a preferred binding site on the receptor is identified. The docking engine then predicts energetically favorable poses of the ligand within the receptor binding site. In virtual screening, this whole process happens at a larger scale, preparing and docking an entire library of ligands and then filtering the results to identify the best candidates for further study
The first version of AutoDock combined a volumetric approach to energy evaluation with a simulated annealing search method. Several approximations were required to allow docking in reasonable times on the VAX department‐level computers of the era (Figure 3). Volumetric maps are precalculated for each ligand atom type by scanning probe atoms throughout the space occupied by the target, with the consequence that they impose a limitation of a rigid receptor. 14 In addition, conformational degrees of freedom in the ligand were also limited to torsional rotations, with bond lengths and angles constrained to the geometry of the starting pose. This is based on the assumption that the bound conformation is a torsional variation of the input conformation. Limitations in storage space also required reducing the number of atom types, and thus the number and size of maps to be calculated and stored. The AutoDock suite has grown from this foundation, currently providing multiple docking methods, graphical user interfaces, and analysis tools (Table 1).
FIGURE 3.
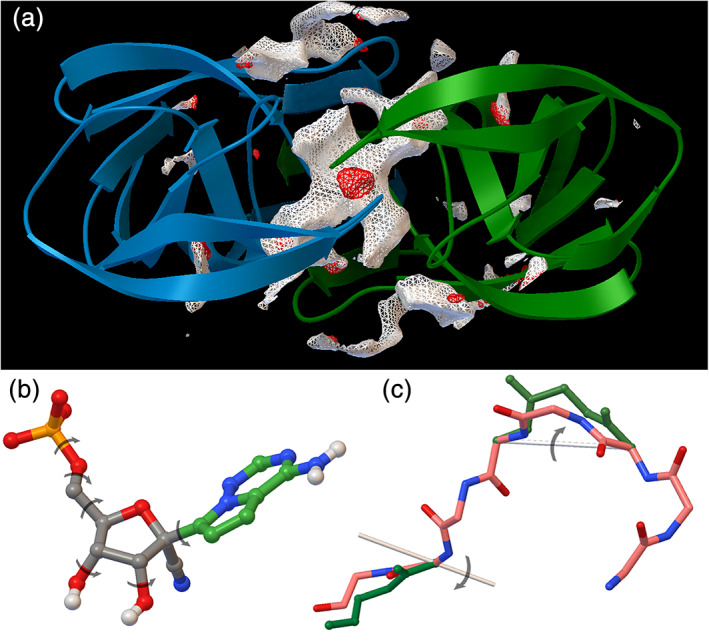
Many simplifications are imposed to improve speed of docking. (a) Interaction energies for probe atoms are calculated volumetrically. Here, favorable locations for carbon (white contours) and oxygen (red contours) are calculated in the active site of a drug‐resistant HIV protease (PDB entry 2hc0). Notice the two lobes off the carbon density corresponding to the P1 and P1' sites, and the prominent oxygen location between the flaps, corresponding to the location of an ordered water in most structures. (b) Only torsional degrees of ligands (here, a monophosphate metabolite of remdesivir) are searched, and limited atom types are used (here, aliphatic carbons in gray and aromatic carbons in green, and only polar hydrogens). (c) Coarser motions are used to simplify conformational search for larger ligands, such as crankshaft motions for peptides. Images generated in Python Molecule Viewer including AutoDockTools
TABLE 1.
Methods in the AutoDock Suite
| Docking engines | ||
| AutoDock4.2/AutoGrid | Ligand docking with empirical free energy force field | Reference 15 |
| AutoDockVina | Rapid, turnkey ligand docking | Reference 19 |
| AutoDockFR/AutoGridFR | Ligand docking with protein flexibility | References 20 and 21 |
| AutoDockCrankPep | Peptide docking | References 25 and 27 |
| AutoDockGPU | GPU‐accelerated version of AutoDock4 | Reference 18 |
| Graphical user interfaces | ||
| AutoDockTools | General GUI for AutoDock programs | Reference 15 |
| Raccoon2 | GUI for virtual screening | Reference 37 |
| AGFRgui | GUI for AutoGridFR and AutoSite | Reference 21 |
| Binding site prediction | ||
| AutoLigand | Prediction of optimal ligands from AutoGrid maps | Reference 30 |
| AutoSite | Rapid prediction of binding sites | Reference 31 |
The current version of AutoDock, AutoDock4 (AD4), retains much of the original concepts of energy evaluation, and has markedly improved the search capabilities, 15 increasing the complexity of ligands that can be docked. Enhancements of the force field have focused on improved geometry of hydrogen bonding and empirical weighting of force field parameters to predict binding free energies, 16 and ongoing developments described in more detail below. Collaboration with computer scientists leads to implementation of a hybrid genetic algorithm/local search method that vastly extends the reach of the conformational search. 17 Recent enhancements include addition of gradients to the force field description and porting of AD4 to graphics processing units (GPUs) to further improve performance. 18
AutoDockVina (ADVina) is a turnkey docking method that captured the 2010 state of the art in docking. 19 Many optimizations are employed to improve the speed of docking, including a piecewise scoring function that is amenable to rapid evaluation and was calibrated using ~1,300 complexes from PDB‐Bind, and a highly optimized search method based on a Monte‐Carlo algorithm and gradient‐based local optimization. Much of the mechanism of docking that is exposed in the AutoDock programs, such as the use of maps for energy evaluation, is hidden in ADVina, making it a good option for use by non‐experts.
AutoDockFR (ADFR, “Flexible Receptor”) is a parallel development effort that addresses the limitation of a rigid receptor, 20 , 21 building on the earlier FLIPDock program. 22 , 23 AD4 and ADVina include the ability to model user‐defined flexible side chains explicitly, but ADFR creates a more general representation of the receptor, allowing definition of sidechains, loops, and domains undergoing motions during conformational searches. 24 ADFR implements an efficient genetic algorithm that allows the specification of up to 15 flexible side chains in the receptor's binding site. It also supports docking covalent ligands and constraining ligand atoms to predefined positions using harmonic potentials. Building on this work, AutoDock CrankPep (ADCP) is a docking engine developed specifically for docking peptides. 25 ADCP combines Crankite peptide conformation sampling 26 with AutoDock affinity maps for efficient docking of linear or cyclic peptides 27 with up to 20 amino acids starting from their sequence.
The open source availability and modular design of the AutoDock Suite has also promoted its use by numerous third party developers. Examples include Smina, a fork of ADVina that streamlines customization of force fields, 28 and PSO@AutoDock, an implementation of particle swarm optimization within AutoDock3. 29
3. THE PROS AND CONS OF MAPS
Volumetric precalculation of interaction energies was the central innovation that made flexible docking possible in the initial versions of AutoDock, and it continues to be essential for reducing the computational complexity of the docking problem. In the idealized case of a perfect “lock and key” interaction, we might imagine that these maps would contain a perfect image of the ligand in its bound form, defined and held in place through specific and steric interactions with the surrounding receptor. In reality, however, biological macromolecules are composed of atoms with finite size, so the form of the active site is the result of an evolutionary trade‐off between the need to create this perfect binding signature, but limited by the chemical and physical properties of the protein or nucleic acid polymer. Consequently, these maps typically require some chemical intuition for interpretation (Figure 3a).
Nevertheless, interaction maps are powerful tools in the arsenal of methods for drug discovery and design. Building on the idea that the latent image of preferred ligands is contained in these maps, we have developed two methods for predicting binding sites. AutoLigand 30 combines maps for carbon atoms and hydrogen bonding atoms, creating a combined map that determines the best atom type for each location. Then, the user defines a desired size for the ligand and the algorithm finds the best contiguous set of points within the maps with such volume. AutoSite (Figure 4) takes a slightly different approach, locating these ideal binding sites by clustering regions of high affinity. 31 Both methods may be used to identify amenable sites of binding, and to characterize the optimal form of ligands that will bind to the sites.
FIGURE 4.
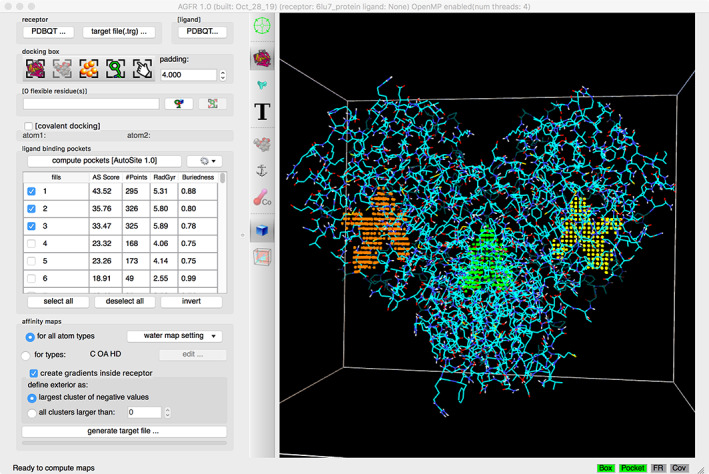
One‐click pocket prediction with AutoSite in the ADFR gui. The three top‐scored pockets of the SARS‐CoV‐II major protease (PDB entry 6lu7) are shown, including a cavity at the dimer interface (green) and the two active sites (yellow and orange)
Over the years, we have also experimented with specialized maps for particular applications. For example, a simple map that holds the distance to the nearest atom may be used to coax ligands into contact with protein, reducing unnecessary exploration of conformations completely surrounded by solvent. Small levels of protein motion may be accommodated during docking through the use of “smoothed” maps that evaluate the minimal energy within a small distance threshold of each point, or by creating maps that combine contributions from multiple conformations of the protein. 32 A more direct approach may also be taken, termed the “Relaxed Complex Method,” where snapshots of a protein are taken from molecular dynamics, and then used in individual docking experiments. 33 The application of AutoDock in a docking ensemble approach played a role in the development of the first clinically approved HIV‐1 integrase inhibitor by Merck. 34
4. GRAPHICAL USER INTERFACES
A responsive front end is absolutely essential to help users chart a path through their applications. AutoDockTools (ADT), built upon the modular graphics methods in MGLTools, has served this purpose for the various versions of AutoDock. 15 It provides graphical tools for adding hydrogens and defining articulation of ligands, preparing flexible and rigid portions of receptors, creating command files, and finally analyzing results of docking simulations. ADT is, however, a tool that is largely designed for users with substantial knowledge of molecular modeling and docking methods, with much of the machinery of AD4 exposed to allow customization for challenging applications. In addition, it is primarily useful for specification of a small number of docking experiments, and is cumbersome when applied to larger problems such as virtual screening. These limitations have been and are being addressed with new tools.
Chimera, 35 PyMOL, 36 and many other third‐party tools have incorporated the ability to specify and write command files for ADVina, providing a turnkey approach to docking for non‐experts. We are currently developing a similar turnkey front end, which provides a point‐and‐click interface that allows users to customize and curate both ligand and receptor coordinates starting from entries from the PDB archive, and specify docking simulations with the resultant choices (see below).
We developed Raccoon as a tool for specifying and managing virtual screens with the AutoDock suite. 37 It uses a flexible database approach to prepare and manage large ligand libraries, has mechanisms for launching and monitoring docking simulations on computing clusters, and, most importantly, has an arsenal of flexible filtering tools to analyze the results and isolate compounds with promise for additional study (Figure 5).
FIGURE 5.
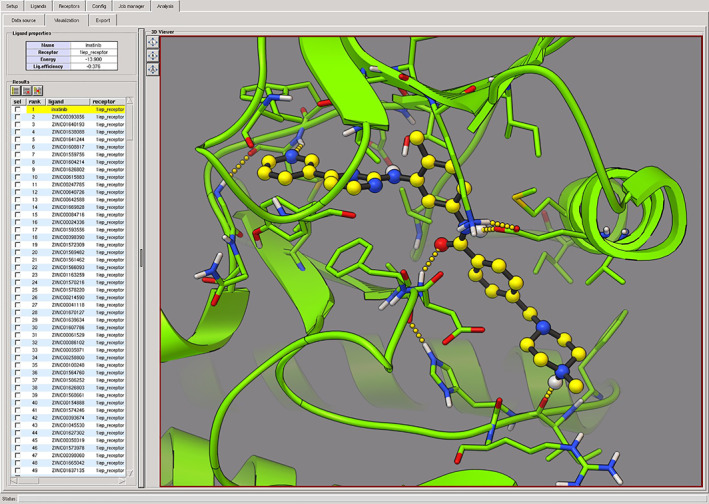
Virtual screening analysis using Raccoon2: docking of a small library from ZINC 62 on the c‐Abl kinase, showing the re‐docked conformation of imatinib (PDB entry 1iep) used as a reference compound
5. ENHANCEMENTS
Biology being biology, there are exceptions to every rule. The core functionality of the AutoDock suite was created to be a general tool, parameterized and validated against a diverse set of drug‐like ligands binding to pocket‐shaped protein binding sites. In our hands, these basic methods will give turnkey docking results for roughly half of new trial systems. In other cases, the biology imposes new aspects that are not effectively addressed in the main suite, so we have spent most of the last three decades creating a series of enhancements to address these challenges.
Solvent effects remain one of the biggest challenges to improving the accuracy and specificity of docking simulations. AD4, ADVina and ADFR all incorporate empirical approaches to estimating the energetic consequences of desolvation, based on measures that approximate the amount of water displaced by a ligand when it binds. These methods use functions with very gentle distance dependence, and do not account for localized effects of bridging water. We have developed a more explicit approach to this problem by attaching waters to all possible positions of interaction on a ligand, then allowing these waters to interact with protein or disappear during the docking simulation 38 (Figure 1).
Specialized potentials may be used to incorporate covalent bonding into docking simulations. These may be approached in several ways. Internal potentials may be added to create bonds within ligands. This has been used successfully to model ligands with flexible macrocycles: 39 the ring is broken at one site, and docking simulation is performed with a custom potential that favors the reconstitution of the original macrocyclic bond. Similarly, building on previous work with covalent docking, 40 we developed our most effective approach to prediction and design of targeted covalent inhibitors. Termed “reactive docking,” the method uses a custom potential between the ligand and the receptor to evaluate ligands for their ability to bind to sites on the protein and subsequently form covalent bonds with adjacent sites of chemical reaction 41 (Figure 6).
FIGURE 6.
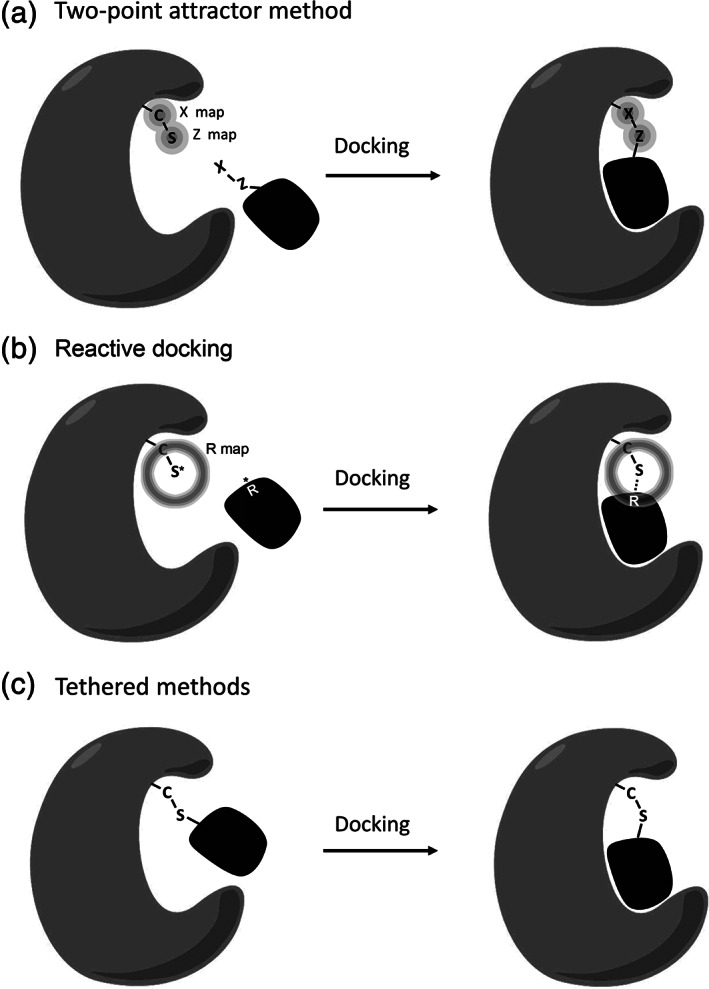
Methods for covalent bonding supported in AD4. (a) The two‐point attractor method uses two specialized maps (X and Z) that have favorable energies at the site of attachment on the protein, driving the ligand docking to the covalent binding location. (b) The reactive docking method uses a custom potential that drives the reactive atom on the ligand (R) to the near‐attack position on the target amino acid. (c) The tethered approach uses the flexible sidechain method to optimize an arbitrary pose of the covalent complex
We designed methods to exploit experimental or otherwise derived information to steer calculations toward establishing precise interactions between the ligand and the protein. For example, cosolvent molecular dynamics analysis 42 has been used to identify binding hot spots on the protein surface and steer ligand poses to establish specific interactions with such regions. 43 Similarly, ADFR supports harmonic constraints that penalize user‐specified ligand atoms from moving away from the predefined position, thus enabling”anchored docking,” where a group of ligand atoms is subjected to such a constraint. It also implements the concept of neighborhood search, where the docking search concentrates on exploring the “neighborhood” of an initial docked pose, the neighborhood being defined by an RMSD cutoff.
The basic force field of the AutoDock suite is parameterized and optimized for standard atom types, when arranged into standard drug‐like molecules. Several laboratories have performed custom parameterizations for specific systems, such as an approach optimized for binding of carbohydrates to proteins. 44 We have focused attention on aspects of the force field that are missing, such as new potentials to specify coordination by metal ions such as zinc. 45 We have also initiated an effort to improve the potentials used to specify hydrogen bonding, starting with a detailed survey of hydrogen bonding strength and directionality using a quantum mechanical analysis of model compounds. 46
6. PERFORMANCE AND CHOOSING THE BEST TOOL
As mentioned above, we have pursued a multi‐track approach to development of the AutoDock suite to allow exploration of diverse new methods. This, along with the fact that many other effective academic and commercial docking methods are currently available, can make it difficult to choose a tool for a particular application. Within the AutoDock suite, we typically suggest ADVina as a first‐line, turnkey approach, since it is fast for typical drug‐like ligands. For systems presenting different non‐conventional challenges, such as chemical reactivity, receptor flexibility, or systems that require ad hoc parametrizations, other tools in the suite may be applied to model them. Table 2 includes suggestions for appropriate tools for a variety of common applications.
TABLE 2.
Choosing a docking engine
| AutoDock4 | AutoDockGPU | AutoDock Vina | ADFR | ADCP | |
|---|---|---|---|---|---|
| Rapid docking of drug‐like molecules | ✓ | ||||
| Very flexible ligands? (>20 torsions) | ✓ | ✓ | ✓ | ||
| Flexible macrocycle | ✓ | ✓ | |||
| Is the ligand a peptide? | ✓ | ||||
| Are waters important for binding? | ✓ | ✓ | ✓ | ||
| Is the binding site flexible? | ✓ | ✓ | |||
| Is there a metal ion in the site? | ✓ | ✓ | ✓ | ||
| Covalent inhibitor (known site)? | ✓ | ✓ | ✓ | ✓ | |
| Covalent inhibitor (unknown site)? | ✓ | ||||
| Anchored docking | ✓ |
Many third party studies have been presented that quantify the performance of methods within the AutoDock suite and compare them with other available tools. For example, a recent detailed study of five commercial and five academic methods with 2,000 complexes from PDB‐Bind showed a comparable performance of ADVina, Glide, GOLD, and several others, obtaining successful best docked poses in roughly half of systems to within 2.0 A RMSD, and correlation coefficients of roughly 0.5 for energy prediction. 47 These results mirror our own advice given to users, building on the results of our validation studies: 16 , 17 , 19 typically, docking methods will be successful roughly half the time, with better statistics for smaller, less flexible ligands and targets with limited flexibility, and energies are predicted to within about 2–3 kcal/mol, allowing separation of milli‐, micro‐, and nanomolar inhibitors, but not being effective for ranking with finer energetic differences.
7. VIRTUAL SCREENING WITH THE AUTODOCK SUITE
The effectiveness and limitations of current docking methods become most apparent in virtual screening efforts. The current state‐of‐the‐art in the AutoDock suite, and similarly for most current docking methods, provides consistent docked conformations for molecules with roughly a dozen torsional degrees of freedom, with free energies predicted to within about 2–3 kcal/mol, in systems where protein motion does not play a significant role. This level of accuracy has proven sufficient to drive success in virtual screening efforts, with the expectation that between 1 and 10% of predicted virtual hits will turn out to show detectable binding affinity upon experimental testing.
Virtual screening is arguably the major application of the current AutoDock user community, and much of the development effort of the past decade has been focused on improving the infrastructure and results. Raccoon is the primary front end supporting virtual screens that is provided with the AutoDock suite (Figure 5). A variety of web services are also available from third parties, such as MtiOpenScreen 48 and DrugDiscovery@TACC (https://drugdiscovery.tacc.utexas.edu/).
Force field development has also addressed some of the challenges posed by virtual screening. For example, in 2007, we moved to an energetic model based on a thermodynamic cycle that includes explicit evaluation of bound and unbound states. 16 Slightly better predictive results were obtained with a protocol that estimated energies from intramolecular contacts in free ligands as part of this cycle, but this protocol was eventually abandoned for a simpler model that assumes that intramolecular effects are similar in bound and unbound states of the ligand. The fuller model consistently ranked a set of crowded molecules with internal clashes at the top of the list, due to computational instability of the very high energies of the unbound form.
Virtual screening has driven the need for ever larger computing resources, since the larger the haystack, the higher the chance it contains a golden needle. Fortunately, virtual screening can be “embarrassingly parallel” by assigning each compound docking to a different processor running in parallel, giving a practically linear speedup. In 2000 in collaboration with Entropia, a computational startup, we initiated FightAIDS@Home (FAAH) to demonstrate the practical utility of grid‐based computing in drug design. FAAH was the first biomedical project developed for a grid‐based volunteer platform following on from earlier Citizen Science projects like SETI@Home and the GIMPS (Great Internet Mersenne Prime Search) that used volunteer computing to solve astronomical and mathematical problems. With such a resource running AutoDock on thousands of widely distributed processors, we were able to expand our studies of the structural biology of HIV with large virtual screens. In 2005, FAAH joined IBM's World Community Grid, which has amplified the resources available to more than 3 million volunteer CPUs, greatly increasing the sizes of compound libraries searched, as well as increasing the degrees of freedom in models of the protein targets.
Over these past 20 years, application of the AutoDock suite (AD4 and ADVina) on FAAH and local platforms has informed and broadened the approaches to HIV therapeutic development. We have examined the role of protein flexibility in allosteric inhibition in HIV protease 49 and integrase 50 using a panel of molecular dynamics snapshots; evaluated new broadly based (HIV/FIV) protease inhibitors, 51 identified new targets for drug development such as HIV capsid, 52 and explored the mechanisms of HIV drug resistance evolution. 53 With massive numbers of docking results from large FAAH virtual screens, we have utilized machine learning on data generated by FAAH to improve the selection criteria for true positives in the screening. 54 In order to further refine AutoDock screening results with more intensive free energy calculations, FAAH has now started a second phase in collaboration with the Levy laboratory, in which molecular dynamics simulations are used to provide better estimates of the free energy of binding. 55
Recently, in collaboration with IBM World Community Grid we initiated the OpenPandemics‐COVID‐19 Project utilizing AutoDock (https://www.worldcommunitygrid.org/research/opn1/overview.do) to perform large virtual screens against multiple target sites in the SARS‐CoV‐2 proteome to look for new potential COVID‐19 therapeutic candidates, including reactive covalent inhibitors. Beside our own efforts, our docking engines support other World Community Grid projects that run virtual screenings targeting cancer, malaria, and Ebola. The magnitude of these distributed computing efforts poses unique challenges for docking data management and analysis that are being addressed in our current development efforts.
8. PEPTIDE DOCKING
Beginning with the first release of AutoDock, users have pushed the envelope of what can be effectively modeled with computational docking. Peptides, in particular, have been a persistent interest of the user community, but one that requires a creative approach. The motivation is clear: therapeutic peptides have undergone a Renaissance in recent years, as witnessed by the 60 peptide‐based drugs currently approved in major markets, and over 150 in active clinical development. 56 Cyclic peptides are of particular interest, with 40 examples in current clinical use and on average a new one entering the market every year. 57 Docking of peptides, however, presents daunting challenges, both for the search and the scoring.
The size of the solution‐space to be explored during docking grows exponentially with each added variable to optimize, so search methods developed for drug‐like small molecules, which typically have about a dozen rotatable bonds, stand little chance to sample properly the 50 to 100 rotatable bonds in peptides. Peptides also tend to associate with their binding partners in shallow grooves at the surface, while drug‐like molecules more often bind in docking‐friendly deep pockets that minimize their interactions with solvent. Hence, the scoring functions developed and calibrated for small drug‐like molecules often perform poorly for peptides, even short ones. 58
Early workers solved these problems by splitting peptides into fragments, docking them separately, and then choosing poses that could be recombined into a desired peptide sequence. 59 Surprisingly, even protein–protein docking is amenable to this approach. 60 We are currently tackling these challenges by exploring simplified representations of the conformational space based on backbone crankshaft transformations 26 (Figure 3). ADCP, which currently allows consistent docking of peptides with up to 20 amino acids 25 (Figure 7), is at the cutting edge of this field in a recent evaluation of 14 peptide docking programs. 61 ADCP can also cyclize peptides head‐to‐tail (Figure 7a) and/or by forming up to two disulfide bridges when cysteines are present (Figure 7b) thus supporting molecules with multiple cycles. 27 Cyclization is achieved on‐the‐fly during the docking simulation by using potentials to pull the N‐ and C‐termini or cysteine sulfur atoms together while ignoring steric repulsion between these atoms. In the latter case, the pairing of the cysteines does not need to be specified by the user, but instead results from the docking.
FIGURE 7.
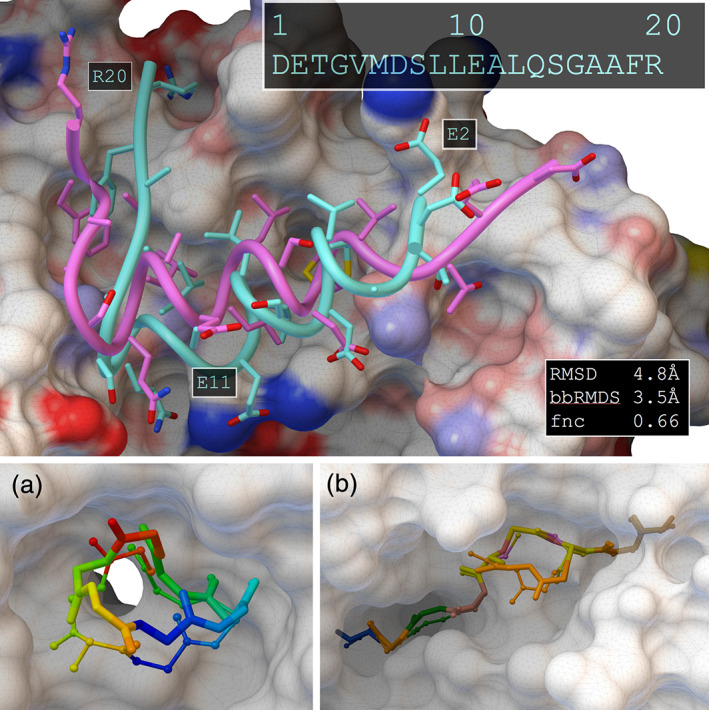
Top panel: best docking solution (cyan) and crystal structure (magenta) of a linear peptide that forms a self‐inhibitory switch in formin mDia1 (PDB entry 2f31). Starting from the sequence, ADCP folds and places the peptide into the receptor. Sidechain‐receptor interactions are predicted well, except for E2, E11, and R20 that find polar patches. Bottom panel: backbone of docked cyclic peptides (ball‐and‐stick) and crystal structure (licorice). (a) Top ranked docked pose for a ubiquitin ligase substrate adaptor protein with an engineered cyclized peptide from one of its targets (PDB entry 3zgc). Residues are colored blue to red to show correct registration. (b) Docking result for an internally disulfide‐linked peptide from Epstein Barr Virus being displayed by MHC (PDB entry 5grd). Residues are color‐coded by residue type and the side chains of the two cysteines show the created disulfide bond
9. CURRENT DEVELOPMENT AND FUTURE DIRECTIONS
Looking to the large body of research and development that cites AutoDock publications, and our ongoing correspondence with users, we find that our primary users tend to be workers in related fields—biochemists, structural biologists, and physicists—who do not have deep expertise in computational chemistry and docking. For this community, the AutoDock suite, and ADVina in particular, is an attractive package given that it is free, easily available, and relatively straight‐forward to get started. This has led to hundreds of reports where docking is used to complement larger structure/function studies. A central focus of our current and planned development is to support this large and growing community of users. We are currently working on a unified graphical front end that will capture the ease of use of ADVina, but will allow turnkey access to the additional levels of functionality available in other tools of the suite. This front end will incorporate nimble tools for managing the many challenges posed by experimental input coordinates: building missing loops and residues, protonation and tautomerization, charge models, handling receptor flexibility, and dozens of other small, but essential, hurdles (Figure 8). The front end will also provide easy tools for managing docking experiments ranging from simple studies of a trial ligand with a receptor to virtual screens, along with analysis tools to filter and interpret results. Finally, the front end will include a comprehensive functionality for capturing provenance for each experiment, ensuring reproducibility of the work that is performed within the suite. We expect that this interface will ultimately provide primary access to the AutoDock suite for all levels of user expertise. We are currently developing pluggable components of this interface that will become part of our envisioned front end, affording an incremental path toward the ultimate goal of a unified environment where users can easily access all the tools developed in our labs.
FIGURE 8.
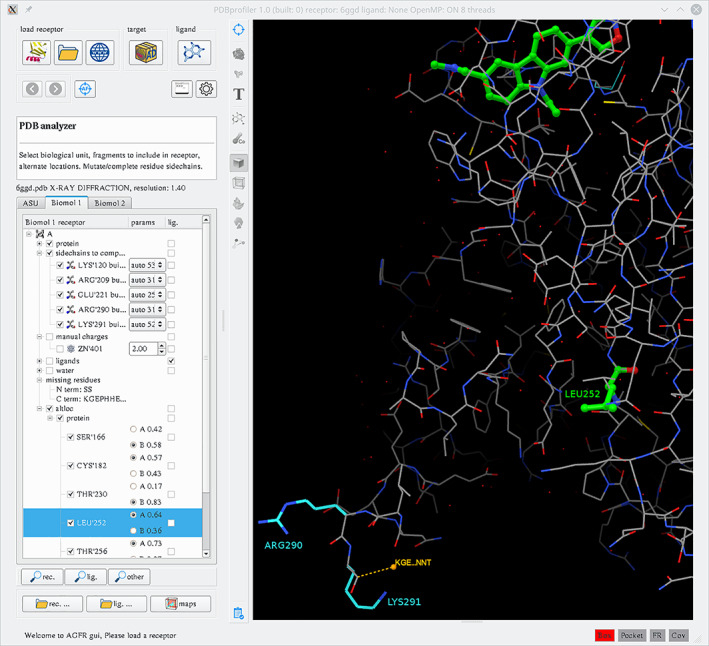
Graphical user interface under development for preparing molecules for docking, shown here with a p53 cancer mutant (PDB entry 6ggd). When the PDB file is loaded, the molecular content is analyzed, classified, and presented in a tree widget allowing the specification of residues that will be included in the receptor or ligand(s). All biomolecules are available along with the asymmetric unit. Standard amino acids with missing side chain atoms are selected for reconstruction and a default rotamer is selected (here, Arg290 and Lys291). Alternate locations are displayed and by default the one with highest occupancy is selected (e.g., Leu252@A). The ligand from the crystal structure is tagged to be prepared as a ligand for docking. Missing segments of amino acids are displayed (orange). The tree widget is linked to the 3D view, allowing the user to focus on potentially problematic areas and inspect the proposed solution
We also enjoy a vibrant and creative community of third‐party developers, and in response have cultivated an open approach to development of the AutoDock suite to support extension and innovation by this important community of users. All components of the AutoDock suite are available under open source licenses and accessible through the AutoDock website (https://autodocksuite.scripps.edu). They are implemented in the C, C++, and Python programming languages. The AutoDock suite is a large software ecosystem composed of numerous software components, many of which can be used independently. Source code and executable for these software components are available in independent repositories. While providing all the software components in a single repository is feasible, keeping them separate supports their independence: i.e., users download the source of a given software component, build and use it without the others being present. If a dependency is introduced inadvertently, this process will fail and alert us of the newly introduced dependency.
In our own laboratories and in many others, basic principles of docking and energy evaluation are being discovered and developed. Our own work is currently focused on enhancing the scoring functions to provide more accurate energy evaluation and ranking, including evaluation of machine learning approaches for tuning or underlying these functions, and extending search methods, with peptides being a particular current focus. We have worked under the hood to make the methods of the AutoDock suite modular and extensible to support the work that will continue to expand the functionality and accuracy of docking methods. We expect that the AutoDock Suite will continue to benefit from the improvements in the field, especially the modeling accuracy made possible by the dramatic computing power increase in the recent years. Thanks to the tremendous boost provided by accelerated architectures such as GPUs, it will be possible to perform more expensive and accurate energy evaluations, overcoming some of the limitations imposed by computing in the early days. The tools of the AutoDock suite have been and will continue to be freely available through an open source license at the AutoDock website: https://autodocksuite.scripps.edu, with documentation and tutorials. Detailed protocols for running common applications in AD4 and ADVina have been presented. 37
AUTHOR CONTRIBUTIONS
David Goodsell: Conceptualization; funding acquisition; project administration; writing‐original draft; writing‐review and editing. Michel Sanner: Conceptualization; funding acquisition; project administration; writing‐review and editing. Arthur Olson: Conceptualization; funding acquisition; project administration; writing‐review and editing. Stefano Forli: Conceptualization; funding acquisition; project administration; writing‐review and editing.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
We thank Diogo Santos‐Martins and Giulia Bianco for assistance with the manuscript. Over the past 30 years, AutoDock development has been continuously supported by National Institute of General Medical Sciences‐NIH, most recently by grants GM069832 (Stefano Forli) and GM096888 (Michel Sanner). This is manuscript #30005 from Scripps Research Institute.
Goodsell DS, Sanner MF, Olson AJ, Forli S. The AutoDock suite at 30. Protein Science. 2021;30:31–43. 10.1002/pro.3934
Funding information National Institutes of Health, Grant/Award Numbers: GM069832, GM096888
Contributor Information
David S. Goodsell, Email: goodsell@scripps.edu.
Stefano Forli, Email: forli@scripps.edu.
REFERENCES
- 1. Weiner SJ, Kollman PA, Case DA, et al. A new force field for molecular mechanical simulation of nucleic acids and proteins. J Am Chem Soc. 1984;106:765–784. [Google Scholar]
- 2. Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule‐ligand interactions. J Mol Biol. 1982;161:269–288. [DOI] [PubMed] [Google Scholar]
- 3. Goodsell DS, Olson AJ. Automated docking of substrates to proteins by simulated annealing. Proteins Struct Funct Genet. 1990;8:195–202. [DOI] [PubMed] [Google Scholar]
- 4. Eger E, Simon A, Sharma M, et al. Inverted binding of non‐natural substrates in strictosidine synthase leads to a switch of stereochemical outcome in enzyme‐catalyzed Pictet–Spengler reactions. J Am Chem Soc. 2020;142:792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hassan IS, Ta AN, Danneman MW, et al. Asymmetric δ‐lactam synthesis with a monomeric streptavidin artificial metalloenzyme. J Am Chem Soc. 2019;141:4815–4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Almaqwashi AA, Zhou W, Naufer MN, et al. DNA intercalation facilitates efficient DNA‐targeted covalent binding of phenanthriplatin. J Am Chem Soc. 2019;141:1537–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Salveson PJ, Haerianardakani S, Thuy‐Boun A, et al. Repurposing triphenylmethane dyes to bind to trimers derived from Aβ. J Am Chem Soc. 2018;140:11745–11754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gupta V, Yang J, Liebler DC, Carroll KS. Diverse redoxome reactivity profiles of carbon nucleophiles. J Am Chem Soc. 2017;139:5588–5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nycholat CM, Rademacher C, Kawasaki N, Paulson JC. In silico‐aided design of a glycan ligand of sialoadhesin for in vivo targeting of macrophages. J Am Chem Soc. 2012;134:15696–15699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guedes IA, de Magalhães CS, Dardenne LE. Receptor–ligand molecular docking. Biophys Rev. 2014;6:75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pagadala NS, Syed K, Tuszynski J. Software for molecular docking: A review. Biophys Rev. 2017;9:91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Irwin JJ, Shoichet BK. Docking screens for novel ligands conferring new biology: Miniperspective. J Med Chem. 2016;59:4103–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lyu J, Wang S, Balius TE, et al. Ultra‐large library docking for discovering new chemotypes. Nature. 2019;566:224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goodford PJ. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem. 1985;28:849–857. [DOI] [PubMed] [Google Scholar]
- 15. Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge‐based desolvation. J Comput Chem. 2007;28:1145–1152. [DOI] [PubMed] [Google Scholar]
- 17. Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 18. Santos‐Martins D, Eberhardt J, Bianco G, et al. D3R grand challenge 4: Prospective pose prediction of BACE1 ligands with AutoDock‐GPU. J Comput Aided Mol Des. 2019;33:1071–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2009;31:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ravindranath PA, Forli S, Goodsell DS, Olson AJ, Sanner MF. AutoDockFR: Advances in protein‐ligand docking with explicitly specified binding site flexibility. PLoS Comput Biol. 2015;11:e1004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y, Forli S, Omelchenko A, Sanner MF. AutoGridFR: Improvements on AutoDock affinity maps and associated software tools. J Comput Chem. 2019;40:2882–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhao Y, Sanner MF. FLIPDock: Docking flexible ligands into flexible receptors. Proteins Struct Funct Bioinforma. 2007;68:726–737. [DOI] [PubMed] [Google Scholar]
- 23. Zhao Y, Sanner MF. Protein–ligand docking with multiple flexible side chains. J Comput Aided Mol Des. 2008;22:673–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao Y, Stoffler D, Sanner M. Hierarchical and multi‐resolution representation of protein flexibility. Bioinformatics. 2006;22:2768–2774. [DOI] [PubMed] [Google Scholar]
- 25. Zhang Y, Sanner MF. AutoDock CrankPep: Combining folding and docking to predict protein–peptide complexes. Bioinformatics. 2019;35:5121–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Podtelezhnikov AA, Wild DL. CRANKITE: A fast polypeptide backbone conformation sampler. Source Code Biol Med. 2008;3:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y, Sanner MF. Docking flexible cyclic peptides with AutoDock CrankPep. J Chem Theory Comput. 2019;15:5161–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Koes DR, Baumgartner MP, Camacho CJ. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J Chem Inf Model. 2013;53:1893–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Namasivayam V, Günther R. pso@autodock: A fast flexible molecular docking program based on Swarm Intelligence: pso@autodock for fast flexible docking. Chem Biol Drug Des. 2007;70:475–484. [DOI] [PubMed] [Google Scholar]
- 30. Harris R, Olson AJ, Goodsell DS. Automated prediction of ligand‐binding sites in proteins. Proteins Struct Funct Bioinforma. 2007;70:1506–1517. [DOI] [PubMed] [Google Scholar]
- 31. Ravindranath PA, Sanner MF. AutoSite: An automated approach for pseudo‐ligands prediction—From ligand‐binding sites identification to predicting key ligand atoms. Bioinformatics. 2016;32:3142–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Österberg F, Morris GM, Sanner MF, Olson AJ, Goodsell DS. Automated docking to multiple target structures: Incorporation of protein mobility and structural water heterogeneity in AutoDock: Automated docking to multiple target structures. Proteins Struct Funct Bioinforma. 2002;46:34–40. [DOI] [PubMed] [Google Scholar]
- 33. Lin J‐H, Perryman AL, Schames JR, McCammon JA. The relaxed complex method: Accommodating receptor flexibility for drug design with an improved scoring scheme. Biopolymers. 2003;68:47–62. [DOI] [PubMed] [Google Scholar]
- 34. Schames JR, Henchman RH, Siegel JS, Sotriffer CA, Ni H, McCammon JA. Discovery of a novel binding trench in HIV integrase. J Med Chem. 2004;47:1879–1881. [DOI] [PubMed] [Google Scholar]
- 35. Goddard TD, Huang CC, Meng EC, et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Seeliger D, de Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des. 2010;24:417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, Olson AJ. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc. 2016;11:905–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Forli S, Olson AJ. A force field with discrete displaceable waters and desolvation entropy for hydrated ligand docking. J Med Chem. 2012;55:623–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Forli S, Botta M. Lennard‐Jones potential and dummy atom settings to overcome the AUTODOCK limitation in treating flexible ring systems. J Chem Inf Model. 2007;47:1481–1492. [DOI] [PubMed] [Google Scholar]
- 40. Bianco G, Forli S, Goodsell DS, Olson AJ. Covalent docking using AutoDock: Two‐point attractor and flexible side chain methods: Covalent docking with AutoDock. Protein Sci. 2016;25:295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Backus KM, Correia BE, Lum KM, et al. Proteome‐wide covalent ligand discovery in native biological systems. Nature. 2016;534:570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ghanakota P, Carlson HA. Driving structure‐based drug discovery through cosolvent molecular dynamics: Miniperspective. J Med Chem. 2016;59:10383–10399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arcon JP, Modenutti CP, Avendaño D, et al. AutoDock bias: Improving binding mode prediction and virtual screening using known protein–ligand interactions. Bioinformatics. 2019;35:3836–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hill AD, Reilly PJ. A Gibbs free energy correlation for automated docking of carbohydrates. J Comput Chem. 2008;29:1131–1141. [DOI] [PubMed] [Google Scholar]
- 45. Santos‐Martins D, Forli S, Ramos MJ, Olson AJ. AutoDock4 Zn: An improved AutoDock force field for small‐molecule docking to zinc metalloproteins. J Chem Inf Model. 2014;54:2371–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Santos‐Martins D, Forli S. Charting hydrogen bond anisotropy. J Chem Theory Comput. 2020;16:2846–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang Z, Sun H, Yao X, et al. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: The prediction accuracy of sampling power and scoring power. Phys Chem Chem Phys. 2016;18:12964–12975. [DOI] [PubMed] [Google Scholar]
- 48. Labbé CM, Rey J, Lagorce D, et al. MTiOpenScreen: A web server for structure‐based virtual screening. Nucleic Acids Res. 2015;43:W448–W454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tiefenbrunn T, Forli S, Baksh MM, et al. Small molecule regulation of protein conformation by binding in the flap of HIV protease. ACS Chem Biol. 2013;8:1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Perryman AL, Forli S, Morris GM, et al. A dynamic model of HIV integrase inhibition and drug resistance. J Mol Biol. 2010;397:600–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lin Y‐C, Perryman AL, Olson AJ, Torbett BE, Elder JH, Stout CD. Structural basis for drug and substrate specificity exhibited by FIV encoding a chimeric FIV/HIV protease. Acta Cryst. 2011;D67:540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Craveur P, Gres AT, Kirby KA, et al. Novel intersubunit interaction critical for HIV‐1 core assembly defines a potentially targetable inhibitor binding pocket. mBio. 2019;10:e02858–e02818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bühler B, Lin Y‐C, Morris G, et al. Viral evolution in response to the broad‐based retroviral protease inhibitor TL‐3. J Virol. 2001;75:9502–9508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Belew RK, Forli S, Goodsell DS, O'Donnell TJ, Olson AJ. Fragment‐based analysis of ligand dockings improves classification of actives. J Chem Inf Model. 2016;56:1597–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Deng N, Forli S, He P, et al. Distinguishing binders from false positives by free energy calculations: Fragment screening against the flap site of HIV protease. J Phys Chem B. 2015;119:976–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lau JL, Dunn MK. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg Med Chem. 2018;26:2700–2707. [DOI] [PubMed] [Google Scholar]
- 57. Zorzi A, Deyle K, Heinis C. Cyclic peptide therapeutics: Past, present and future. Curr Opin Chem Biol. 2017;38:24–29. [DOI] [PubMed] [Google Scholar]
- 58. Rentzsch R, Renard BY. Docking small peptides remains a great challenge: An assessment using AutoDock Vina. Brief Bioinform. 2015;16:1045–1056. [DOI] [PubMed] [Google Scholar]
- 59. Friedman AR, Roberts VA, Tainer JA. Predicting molecular interactions and inducible complementarity: Fragment docking of fab‐peptide complexes. Proteins Struct Funct Genet. 1994;20:15–24. [DOI] [PubMed] [Google Scholar]
- 60. Stoddard BL, Koshland DE. Prediction of the structure of a receptor–protein complex using a binary docking method. Nature. 1992;358:774–776. [DOI] [PubMed] [Google Scholar]
- 61. Weng G, Gao J, Wang Z, et al. Comprehensive evaluation of fourteen docking programs on protein–peptide complexes. J Chem Theory Comput. 2020;16:3959–3969. [DOI] [PubMed] [Google Scholar]
- 62. Sterling T, Irwin JJ. ZINC 15 – Ligand discovery for everyone. J Chem Inf Model. 2015;55:2324–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]