

Abstract
Bio3D is a family of R packages for the analysis of biomolecular sequence, structure, and dynamics. Major functionality includes biomolecular database searching and retrieval, sequence and structure conservation analysis, ensemble normal mode analysis, protein structure and correlation network analysis, principal component, and related multivariate analysis methods. Here, we review recent package developments, including a new underlying segregation into separate packages for distinct analysis, and introduce a new method for structure analysis named ensemble difference distance matrix analysis (eDDM). The eDDM approach calculates and compares atomic distance matrices across large sets of homologous atomic structures to help identify the residue wise determinants underlying specific functional processes. An eDDM workflow is detailed along with an example application to a large protein family. As a new member of the Bio3D family, the Bio3D‐eddm package supports both experimental and theoretical simulation‐generated structures, is integrated with other methods for dissecting sequence‐structure–function relationships, and can be used in a highly automated and reproducible manner. Bio3D is distributed as an integrated set of platform independent open source R packages available from: http://thegrantlab.org/bio3d/.
Keywords: allosteric regulation, distance matrix analysis, functional dynamics, molecular dynamics, normal mode analysis, principal component analysis, protein sequence, protein structure, protein structure network, structural bioinformatics
1. INTRODUCTION
Bio3D 1 , 2 is a group of related R packages with a focus on processing, organization, and analysis of biomolecular structures. Major features include search and retrieval interfaces for major bioinformatics databases, sequence, and structure conservation analysis, along with popular computational methods for characterizing and predicting protein structural dynamics. These include principal component analysis (PCA), 3 , 4 , 5 , 6 structure and correlation network analysis (CNA), 7 , 8 , 9 , 10 a wide range of normal mode analysis (NMA) methods, 11 , 12 and new ensemble difference distance matrix (eDDM) analysis. Bio3D also provides utilities to convert and process common file formats in structural bioinformatics and couple these data to the broader R ecosystem for advanced statistical analysis, machine learning, and data visualization. 13
A particular strength of Bio3D is its ability to connect heterogeneous sequence and structural data to advanced methods for predicting internal motions and analyzing functional dynamics across protein families. This enables Bio3D to be used as a powerful tool for the analysis of experimental structures in the PDB. 14 The recent rapid growth of such structures provides an unprecedent opportunity to understand sequence‐structure–function relationships from comparative analysis across large and diverse protein families for which structures are now available. A challenge is to deal with related but “non‐identical” structures that may have different lengths due to variable missing residues, insertions, and deletions. Bio3D has a robust solution for performing analysis across such heterogenous datasets. In Bio3D, all structures to be analyzed are aligned based on a multiple sequence or structural alignment. Then, automatically detected equivalent (aligned) residues across structures are used for various comparative analysis methods such as PCA, network analysis, NMA, and distance matrix analysis. Each of these major methods will be discussed in the context of application to experimental structure sets in subsequent sections (examples of applying these methods to structures derived from molecular simulations, which is typically more straightforward due to their homogenous composition, can be found online). We then present a detailed example application of the new eDDM method and conclude with a discussion of broader perspectives and future directions.
2. BIO3D CURRENT STATUS AND RECENT DEVELOPMENTS
Bio3D was originally developed as a single R package 1 and has recently grown to encompass multiple packages with new and improved functionality (Figure 1). The Bio3D‐core package provides functionality for data processing and basic analysis, including alignment, sequence and structure comparisons, and inter‐conformer analysis with PCA. Additional packages serve as extensions containing related functions that collectively solve a specific data analysis task. These include Bio3D‐nma for ensemble normal mode analysis aimed at predicting and contrasting functional dynamics across protein families, Bio3D‐cna for protein structure and correlation network analysis to characterize correlated protein motions underlying allosteric regulation, Bio3D‐web enabling user‐friendly online interactive analysis of protein structures and their dynamics, Bio3D‐view for interactive 3D visualization, and Bio3D‐eddm for the new ensemble difference distance matrix analysis approach to characterizing functionally significant conformational changes. Collectively these packages represent a comprehensive environment for analysis of sequence‐structure‐dynamics relationships in user‐defined protein structure sets.
FIGURE 1.
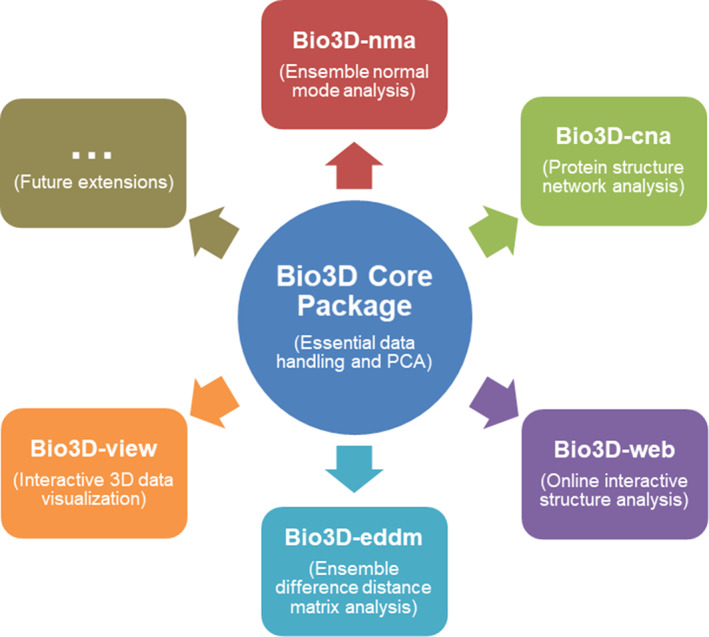
The Bio3D family of R packages
2.1. The Bio3D core package
The Bio3D core package provides functions for data input and output (I/O), format conversion and data manipulation, and basic sequence and structure analysis including database searching, sequence alignment, sequence and structural conservation analysis, as well as multivariate analysis of structural data including PCA and related methods. A fully documented list of Bio3D functionality can be found online: http://thegrantlab.org/bio3d/. Here, we restrict discussion to a minimal set of functions for performing a typical comparative analysis of available experimental structures for a given protein family of interest. This analysis is comprised of four main steps including: (a) structure search and selection, (b) multiple alignment, (c) structure fitting and analysis, and (d) principal component analysis.
-
Structure search and selection: This step is to prepare the structure set related to a given protein sequence for subsequent analyses. Given a query protein sequence or database identifier, all related structures can be collected from the PDB database via the Bio3D functions blast.pdb() or hmmer(). Identified structures are ordered by decreasing sequence similarity to the query. Users can optionally set a threshold (in terms of E‐value) to select and download structures to be used in subsequent steps. A default threshold is automatically generated by calling the plot. blast() or plot.hmmer() functions. The structure set can be further annotated and filtered by resolution, R‐free, date of deposition etc. using the pdb.annotate()function. Selected structures are then downloaded with get.pdb() and optionally split into individual chains for further analysis (by setting the split = TRUE option). In the following example, structures related to protein kinase A (with PDB ID: 1L3R, line 2) are identified (line 3), annotated (line 6) and downloaded (line 9) for further analysis:
# Extract the query sequence and perform database search. aa <‐ get.seq(“1L3R_E”) blast <‐ blast.pdb(aa) hits <‐ plot(blast) # Annotate and filter BLAST results. annotation <‐ pdb.annotate(hits) pdb.id <‐ with(annotation, subset(hits$pdb.id, resolution<=3)) # Download structures and split into individual chains. files <‐ get.pdb(pdb.id, path="pdbs", split=TRUE)
-
Multiple alignment: In this step, all selected structures are subject to multiple alignment. Bio3D provides numerous options for performing such an alignment. These include the calling of external programs such as MUSCLE, 15 using the MUSCLE algorithm internally as implemented in the Bioconductor “msa” package, 16 and accessing to online alignment servers. All of these methods are implemented in the pdbaln() and seqaln() functions.
# Align structures. pdbs <‐ pdbaln(files) # Optionally trim the alignment to focus on e.g. the ‘kinase domain’. pdbs <‐ trim(pdbs, col.inds=c(88:318)) # Produce an alignment overview figure. plot(pdbs)
-
Structure fitting and analysis: In this step, all aligned structures are fitted (i.e., superposed) using the pdbfit() function based on their invariant structural core as identified by the core. find() function. Routine structure analysis, such as individual residue fluctuations (RMSF) and overall structural deviations (RMSD and TM scores) can be performed at this stage.
# Identify structural invariant core and use it to fit structures. cores <‐ core.find(pdbs) xyz <‐ pdbfit(pdbs, inds=cores)
-
Principal component analysis: In this step fitted structures are compared using principal component analysis (PCA). PCA is a well‐established multivariate statistical technique used to reduce the dimensionality of a complex data set to a more manageable number of principal components (termed PCs). This method is particularly useful for highlighting strong patterns and relationships in large datasets (i.e., revealing major similarities and differences) that are otherwise hard to visualize. In terms of structure analysis, PCA transforms atomic coordinates into a few PCs (typically 2 or 3) that represent directions where the structure set displays the largest collective variances. The projection of structures using the resulting PCs is called a conformer plot. These plots represent an efficient way to interpret inter‐conformer relationships. The “scree plot” that displays the spectrum of eigenvalues (or the structural variance captured by the each PC) sorted in the descending order can be used to identify significant PCs that capture dominant collective variances in the structure set. Another useful output of PCA is a representation of the collective structural motions captured by the top PCs as these are often related to protein function. Alternatively, the relative displacement of a residue described by a PC (termed residue loadings) can be displayed and analyzed. In Bio3D, all these outputs can be easily generated with the pca()function, which is specifically designed for the analysis of structural data. Example PCA results on PKA structures are shown in Figure 2.
# Perform PCA for aligned or non‐gapped positions. pc <‐ pca(xyz, rm.gaps=TRUE) # Perform and view structural clustering in the PC1‐PC2 plane. d <‐ dist(pc$z[, 1:2]) hc <‐ hclust(d) hclustplot(hc, k=2, labels=pdb.id) grps <‐ cutree(hc, k=2) # Generate conformer plot, scree plot, and residue loadings plot. plot(pc, col=grps) plot.bio3d(pc$au[, 1], sse=pdbs2sse(pdbs), ylab="Loadings") # Generate a trajectory showing the collective motion defined by PC1. # The output pc_1.pdb can be opened with PyMol or VMD. mktrj(pc, pc=1, file="pc_1.pdb")
FIGURE 2.
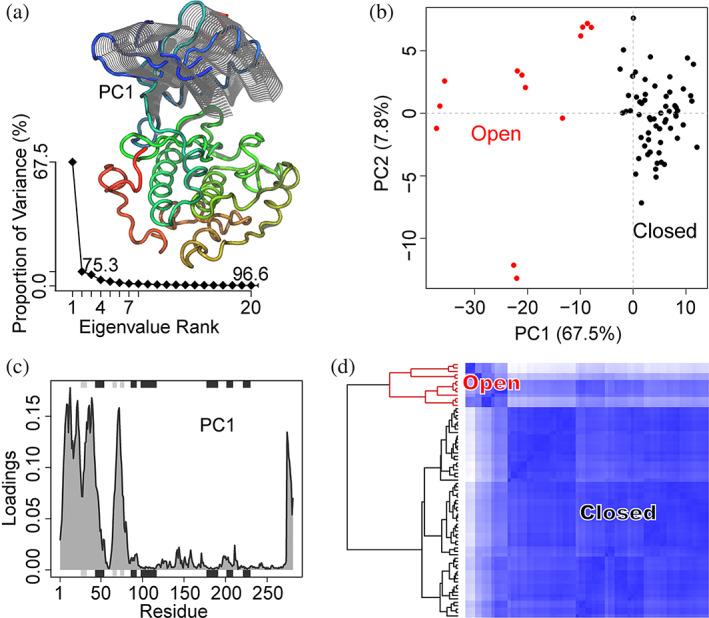
PCA of the PKA kinase domain structures reveals a closing motion of the small lobe along the first principal component (PC1) and two distinct conformational clusters. (a) The collective motion defined by PC1. Grey lines on the small lobe of PKA are generated through a conformational interpolation along PC1. Bottom left is the scree plot of the PCA, which indicates that PC1 is dominant. (b) The conformer plot of all select PKA structures defined by PC1‐PC2. Each point represents a structure and the point color indicates the cluster id from a conformational clustering. (c) Residue contributions or loadings to PC1. (d) Heatmap of inter‐conformer distance matrix and structural clustering calculated in the PC1‐PC2 plane
2.2. Ensemble normal mode analysis with Bio3D‐nma
NMA has emerged as a popular approach for predicting and characterizing the large‐scale internal dynamics of proteins. These low frequency slow motions are often of functional relevance. 17 In addition to conventional single structure NMA, Bio3D can readily perform simultaneous analysis of a large ensemble of structures through our implementation of ensemble normal mode analysis (eNMA). This enables the rapid characterization and comparison of flexibility profiles across homologous structures, without the conventional caveat of potentially overinterpreting the differences between extreme cases using a single artifactual structure. Furthermore, by carefully contrasting the fluctuation profiles, one can provide new information on state‐specific global and local dynamics of potential functional relevance.
In Bio3D‐nma, eNMA is performed using the function nma(), supplying only the aligned pdbs structure ensemble as an input. This function supports versatile popular elastic network models. By default, it implements the efficient C‐alpha based model 11 that enables rapid calculation of modes even for large structural ensembles. We also provide a more accurate all‐atom model obtained through fitting the force constants to a local minimum of multiple high‐resolution structures using the Amber 99SB forcefield. 18 An option to use rotation‐translation block method 19 , 20 or a reduced atomistic model for accelerated calculations is available. The results include aligned eigenvectors and mode fluctuations for all structures in the ensemble. Analysis is facilitated with a range of approaches that aids in the prediction and identifications of distinct patterns of flexibility between different conformational states or even across protein families. Below we outline analysis of the resulting modes through the functions plot() and geostas() for the investigation of fluctuation patterns and dynamic domains, 21 respectively, using PKA as the example (Figure 3):
FIGURE 3.
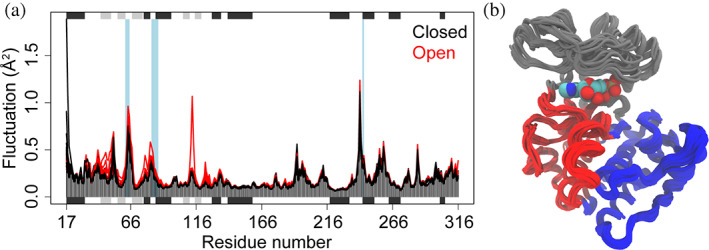
Ensemble normal modes analysis of PKA structures. (a) Residue wise fluctuation profiles across the ensemble of collected PKA structures reveal distinct flexibility patterns for two conformational states. (b) Dissection of dynamic domains obtained through interpolating along the first five modes of the collected PKA structures
# Calculate NMA. modes <‐ nma(pdbs) # Plot fluctuation profiles. plot(modes, pdbs) # Dissect dynamic domains. geostas(modes)
2.3. Ensemble correlation network analysis with Bio3D‐cna
The Bio3D‐cna package performs a range of protein structure and correlation network analysis tasks. Here we introduce the use of Bio3D‐cna for an ensemble correlation network analysis (eCNA). This approach has previously been applied to a wide range of different biological systems. 22 , 23 , 24 The method employs a similar idea to that used in the previous dynamical network method, where a protein structure network is constructed based on a 4.5‐Å contact map and network edges are weighted by values derived from residue dynamic cross‐correlations. 8 However, in eCNA, instead of using a single distance cutoff‐based contact map, network edges are defined by significant correlations across multiple conformational ensembles (e.g., multiple simulation replicas). The objective here is to capture significant and strong correlations that are excluded in the conventional method just because the residue distance is beyond an empirical cutoff. In Bio3D, residue cross‐correlations are calculated with the dccm() function. The dynamical data can be from MD, NMA, multiple experimental structures, or a single multi‐model PDB from, for example, NMR. Significant correlations are then identified with the function filter.dccm(), which inspects both robustness of correlations across simulation replicas and spatial distance between related residues (see ref. 23 for full detail).
The eCNA method can be coupled with suboptimal path analysis to predict potentially functional residues. In this analysis, multiple distinct shortest or (sub)optimal paths between pre‐specified sites (termed “source” and “sink,” which are usually functionally relevant such as the active site and an allosteric site) are searched. A path is a set of network edges connecting the two sites, and path length is defined by the sum of edge weights. In eCNA, network edges are weighted by −ln(|cij|), where c ij is the correlation between residues forming the edge. Hence, path length distributions can be used to compare overall coupling strengths between distinct networks (longer paths mean weaker coupling). The importance of a residue is measured by normalized node degeneracy (i.e., the fraction of suboptimal paths going through the node or residue). A typical workflow of eCNA based suboptimal path analysis, using PKA as an example, is give below (see Figure 4 for the result):
FIGURE 4.
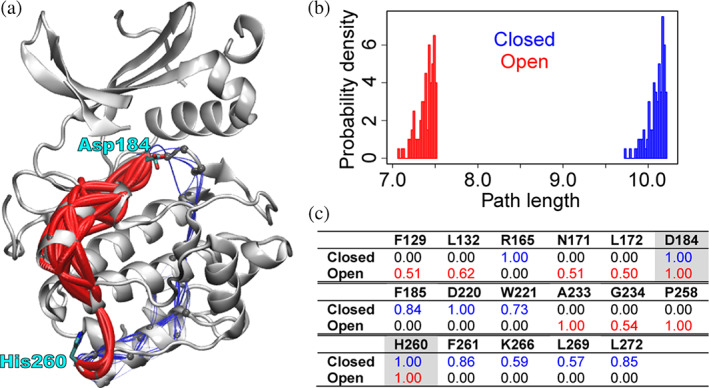
Ensemble correlation network analysis of PKA reveals distinct coupling paths between open and closed structural ensembles. (a) Identified suboptimal paths are viewed as lines mapped on the PKA structure, color coded by the structural ensemble (blue, closed; red, open) used to build the network. Line radii are scaled by path lengths (shorter paths are thicker). (b) Path length distributions. (c) Residues with high (>0.5) normalized node degeneracy (numbers in the table) in either the “open” or “closed” network are shown. The “source/sink” residues are highlighted in grey
# Calculate correlations from an ensemble normal mode analysis. cijs <‐ dccm(modes) # Filter correlation matrices for multiple ensembles defined by ‘grps’. cij <‐ filter.dccm(cijs, xyz=pdbs, fac=grps) # Build networks. ‘cutoff.cij=0’ because ‘cij’ is filtered in the last step. net <‐ cna(cij, cutoff.cij=0) # Find 100 distinct suboptimal paths between the active site # and a potential allosteric site of PKA. pa <‐ cnapath(net, from=142, to=218, k=100) # Plot node degeneracy and path length distributions. plot(pa, pdb=pdb) # View paths in a 3D structure. A ‘pdb’ is generated as the reference. # A script is generated that can be opened with VMD. pdb <‐ pdbs2pdb(pdbs, inds=1, rm.gaps=TRUE, all.atom=TRUE)[[1]] vmd(pa, pdb=pdb, spline=TRUE)
2.4. Bio3D‐web
Bio3D‐web provides a fully online interface to a subset of Bio3D functionality for comparative structure analysis. Methods include a range of conventional sequence and structure conservation assessment methods, as well as inter‐conformer characterization with PCA (Figure 5) and ensemble NMA for comparison of predicted flexibilities and major structural displacements. In contrast to the conventional Bio3D packages, Bio3D‐web does not require any installation or writing of R/Bio3D code. Rather, you explore through an online interactive interface. The design of Bio3D‐web is based on the Shiny web application framework 25 and emphasizes simplicity over exhaustive inclusion of the many additional analysis methods available in the full Bio3D package suite. This effectively reduces the required technical expertise and thus facilitates advanced structural bioinformatics analysis for a broader range of students and researchers. For example, Bio3D‐web is used in undergraduate‐ and graduate‐level bioinformatics and structural biology courses at UC San Diego and elsewhere. In research settings, Bio3D‐web is most often used to quickly explore protein structure datasets; map their structural, conformational, and internal dynamic properties, and thus understand general trends that can inform more specialized analyses.
FIGURE 5.
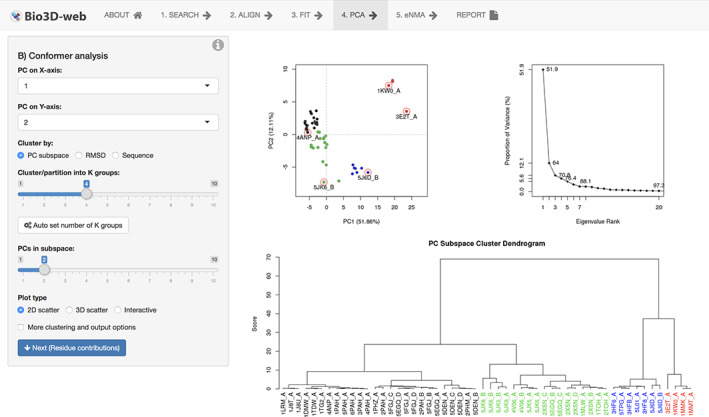
Screenshot of the PCA step in Bio3D‐web. The top navigation menu lists major analysis steps, with the current step “4. PCA” highlighted. Control panels of the particular analysis are on the left, while results are displayed on the right. Here example results display the conformer plot (upper left panel) and cluster dendrogram (lower panel) of the aromatic amino acid hydroxylases including phenylalanine hydroxylase (black), tyrosin hydroxylase (green), tryptophan hydroxalse (blue), and substrate bound “closed” phenylalanine and tryptophan hydroxylase structures (red)
2.5. Ensemble difference distance matrix analysis with Bio3D‐eddm
The Bio3D‐eddm package implements ensemble difference distance matrix analysis (eDDM), a new method for structure comparison. eDDM compares residue‐residue atomic distances across multiple structure sets to identify significant conformational changes that may underlie certain functional processes. A typical analysis comprises the following three major steps:
-
Collecting and preparing the structure set: This step is the same as that described in Steps 1 and 2 of Section 2.1. Note however that no structural fitting (i.e., superposition) is required for an eDDM analysis. Also, in eDDM, aligned structures must include information on all equivalent heavy atoms across structures for subsequent atomic distance calculations. This is setup with an additional call to the read. all() function.
# Read aligned structures with all heavy atoms. pdbs.aa <‐ read.all(pdbs)
-
Calculating difference distance matrices and associated statistics: This step is done with the eddm() function. Distance matrices are calculated first from aligned structures or optionally provided as input. Each entry of a distance matrix represents the minimal atomic distance (based on all heavy atoms) between two residues. Then, distances are compared between structural groups defined by either structural annotations (e.g., the ligand identity associated with each structure obtained from the Bio3D function pdb.annotate()) or a structural clustering analysis. In the latter approach, PCA can be directly applied to the distance matrices using the Bio3D function pca.array() (see example in the next section). Besides difference of mean distances between groups for each residue pair, an assessment of the statistical significance of the difference is performed using a two‐sample Wilcoxon test. In these calculations, residue pairs showing long distances (i.e., non‐interacting) across all structures are omitted. Different methods to “mask” these long distances are available in the eddm() function. The default method calculates “effective” distances, by which changes for long‐range residue pairs are scaled down to zero while changes involving short‐range interacting residues are kept intact.
# Perform eDDM calculations. # Note that ‘grps’ is a pre‐defined variable for structural grouping. tbl <‐ eddm(pdbs.aa, grps=grps)
-
Identifying significant distance changes: This step filters the output of eddm() to focus on statistically significant distance changes. This is done by calling the subset.eddm()function. A significant change is defined by 1) the p‐value of the statistical significance test is below an empirical threshold (e.g., 0.005 or a user defined value, provided through the “alpha” argument of the function) and 2) the absolute distance change is above a threshold (e.g., 1 Å or a user defined values, provided through “beta”). Optionally, only “switching” residues can be reported by turning on the switch. only option. Switching residues are defined as residues that show a group‐level contact (i.e., a contact that persists for a certain fraction of structures in the group, for example, 80%) with one or multiple residues in one group but show a contact with a distinct set of residues in the second group. It is envisaged that such switching residues may serve as potential mediators of allosteric communication, where the residues switch their interacting partners to relay an allosteric “signal.” The result of eDDM analysis can be visualized in both 2D and 3D through utility functions provided in Bio3D‐eddm.
# Identify significant residue distance changes. keys <‐ subset(tbl, alpha=0.005, beta=1.0) # View eDDM results. plot(keys, pdbs=pdbs.aa) pymol(keys, pdbs=pdbs.aa, grps=grps, as="sticks")
3. APPLICATION OF EDDM TO G PROTEIN‐COUPLED RECEPTORS
G protein‐coupled receptors (GPCRs) are essential membrane proteins responsible for regulating many important intracellular signaling pathways. 26 Here, we use the eDDM method to compare available crystallographic structures of the β‐adrenergic receptor (β‐AR) GPCR family. Starting with the sequence of human β2‐adrenergic receptor, related structures are collected from the PDB using BLAST 27 implemented in Bio3D. Low‐resolution structures (>3.5 Å) are excluded, leading to a structure set containing 20 individual β‐adrenergic receptor chains. Structures are clustered into three groups by PCA applied to the distance matrices derived from the structure set (Figure 6a) Inspections reveal that Group 2 contains the known activated GPCR (PDB: 3SN6). 28 Hence, Group‐2 structures are considered as representatives of the “active” state of the receptor and accordingly Group 1 and 3 are considered distinct “inactive” states.
FIGURE 6.
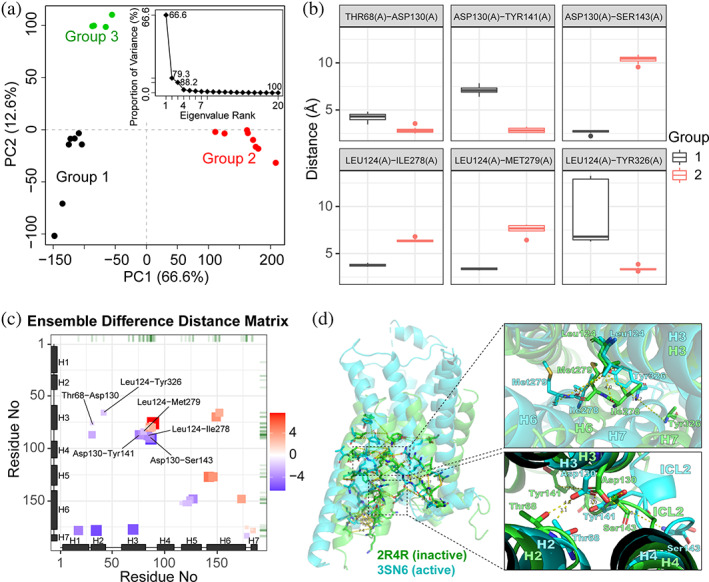
Application of eDDM to GPCRs. (a) PCA of distance matrices derived from β‐AR structures. Each point represents an individual structure projected into the PC1‐PC2 subspace, where PC1 and PC2 represent the directions where atomic distances have the largest collective variance (with the percentage of total variance captured by each PC indicated in the axis label). Structures are clustered into three groups colored differently. Inset, the scree plot showing the spectrum of eigenvalues of the PCA. (b) Box‐whisker plots of select residue‐residue atomic distances from an eDDM analysis of the structures used in A. (c) The 2D “tile” plot of the eDDM analysis result showing the distribution and magnitudes of identified significant residue distance changes (from Group 1 to Group 2; unit: Å). (d) Molecular mapping of key distance changes. On the right are close views of top key changes and associated residues. Representative structures are from PDB 2R4R (for Group 1), 30 which is a structure bound with an inverse agonist and so representing the inactive state of the GPCR, and PDB 3SN6 (for Group 2), 28 an activated GPCR structure. The N‐terminal T4 lysozyme of 3SN6 is omitted for clarity
Totally 36 significant residue‐residue distance changes are identified, involving 34 unique residues. For brevity here we compare Group 1 and Group 2 only, which display the largest separation along the dominant PC (i.e., PC1; Figure 6a). Only switching residues, that is, residues showing distinct contact networks between groups, are reported (see Figure 6b for select residue pairs). A dominant cluster of distance changes is discovered to be around the intracellular loop 2 (ICL2) of the receptor, which connects transmembrane (TM) helices H3 and H4 (Figure 6c). Interestingly, there are significant increases of distance between H3/H5 and the N‐terminus (cytoplasmic end) of H6 upon the transition from Group 1 (inactive) to Group 2 (active) (red tiles vertically aligned H6 in Figure 6c). The result is consistent with existing model of GPCR activation, where an outward movement of the cytoplasmic end of H6 is required to accommodate G‐protein binding. 28
Mapping of identified key residues on GPCR structures reveals dynamic regions that connect the extracellular orthosteric site to the intracellular G‐protein binding surface (Figure 6d). The top two distinct regions (ranked by the maximal absolute distance change associated with each switching residue) are around ICL2 (directly interacting G proteins) and the middle of TM helices closer toward the extracellular ligand binding site. In the first region, Asp130 of H3 switches interactions from Ser143 to Tyr141 in ICL2 upon activation, mainly due to the relatively large displacement of ICL2 (Figure 6d). Asp130 is part of the highly conserved E/DRY motif of GPCRs that is known to be essential for GPCR function. 28 In the second region, Leu124 of H3 is highlighted as a key switching residue, which interacts with Ile278 and Met279 of H6 in the inactive state whereas it switches to interact with Tyr326 of H7 upon activation. Tyr326 is part of the highly conserved NPxxY motif that has been proposed to be important for GPCR activation. 28 Tyr326 is also identified as a key mediator of GPCR activation by a previous analysis of different GPCR family structures. 29 Interestingly, the locations of identified “key residues” in this previous work are similar to those revealed by eDDM. Additional sites identified by eDDM here may be specific to β‐adrenergic receptors and would require further investigation to reveal any potential functional significance. Detailed instruction and code to reproduce this complete analysis is available as Supporting Information.
4. PERSPECTIVES AND FUTURE DIRECTIONS
Bio3D provides a versatile integrated environment with unique capabilities for examining the structural dynamic mechanisms of evolutionary related proteins. In this article we have reviewed some of the major functionalities of Bio3D and introduced the new eDDM method implemented recently in Bio3D. This approach takes internal atomic distances as input and so is free of the requirement for structural superimposition that is usually required in conventional Cartesian coordinate‐based approaches. The eDDM method captures both subtle and large‐scale conformational changes and is potentially a sensitive tool to detect functionally related protein structural dynamic changes. By comparing structural ensembles, eDDM can distinguish significant changes from those changes likely to be due to statistical uncertainties; hence, the method is more robust than simple comparisons between individual structures. Bio3D‐eddm and other Bio3D packages, including the Bio3D core package, Bio3D‐nma, Bio3D‐cna, and Bio3D‐web are open source and freely available.
A number of previously implemented software solutions (including multiple web servers 31 , 32 , 33 , 34 , 35 and standalone software packages 36 , 37 , 38 , 39 , 40 , 41 , 42 ) offer related solutions including single structure NMA, MD or protein structure‐based network analysis. However, these typically lack extensive coupling to major biomolecular databases and methods for evolutionary and comparative analysis intrinsic to Bio3D. Bio3D also includes many commonly used functions for simulation analysis and specifically tailored plotting and visualization functionality as well as coupling to the well‐developed R environment for statistical analysis, machine learning and graphics. We believe that this combination currently offers unparalleled capabilities for both exploratory interactive and large‐scale batch analysis of structural dynamic mechanisms in biomolecular systems.
Future development of Bio3D aims to further improve the capability and scalability of the package family. A major direction is to fully support multiple chain‐based analyses, including biological assembly‐based sequence and structure searching, alignment, and analysis. Also, internal functions for improved interactive 3D structure visualization will be developed to enhance the experience of integrated data exploration. This is related to the on‐going development of the Bio3D‐view package. Other potential future developments include connections to sequence databases and servers for directly mapping structural dynamics to next‐generation sequencing data, more efficient memory management for analyses of especially large systems and long MD trajectories, continued improvement of I/O including the support of the recent MMTF (MacroMolecular Transmission Format) file format. 43 Many of these features are already under development for the next major version of Bio3D.
AUTHOR CONTRIBUTIONS
Barry J. Grant: Conceptualization; data curation; formal analysis; funding acquisition; investigation; methodology; project administration; resources; software; supervision; validation; visualization; writing‐original draft; writing‐review and editing. Lars Skjærven: Conceptualization; data curation; formal analysis; investigation; methodology; project administration; resources; software; validation; visualization; writing‐original draft; writing‐review and editing. Xin‐Qiu Yao: Conceptualization; data curation; formal analysis; investigation; methodology; project administration; resources; software; supervision; validation; visualization; writing‐original draft; writing‐review and editing.
Supporting information
Supporting Information 1 Detailed instructions and accompanying code for example eDDM analysis.
ACKNOWLEDGMENT
We thank DrsGuido Scarabelli, Shashank Jariwala, and Hongyang Li for beneficial discussion and the broader Bio3D community for instructive feedback.
Grant BJ, Skjærven L, Yao X‐Q. The Bio3D packages for structural bioinformatics. Protein Science. 2021;30:20–30. 10.1002/pro.3923
Contributor Information
Barry J. Grant, Email: bjgrant@ucsd.edu.
Lars Skjærven, Email: larsss@gmail.com.
Xin‐Qiu Yao, Email: xyao4@gsu.edu.
DATA AVAILABILITY STATEMENT
Binaries and platform independent source code along detailed installation instruction of Bio3D core and extension packages is available at: http://thegrantlab.org/bio3d/.
REFERENCES
- 1. Grant BJ, Rodrigues AP, ElSawy KM, McCammon JA, Caves LS. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics. 2006;22:2695–2696. [DOI] [PubMed] [Google Scholar]
- 2. Skjærven L, Yao X‐Q, Scarabelli G, Grant BJ. Integrating protein structural dynamics and evolutionary analysis with Bio3D. BMC Bioinf. 2014;15:399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Aalten DM, Conn DA, de Groot BL, Berendsen HJ, Findlay JB, Amadei A. Protein dynamics derived from clusters of crystal structures. Biophys J. 1997;73:2891–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Caves LS, Evanseck JD, Karplus M. Locally accessible conformations of proteins: Multiple molecular dynamics simulations of crambin. Protein Sci. 1998;7:649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grant BJ, McCammon JA, Caves LSD, Cross RA. Multivariate analysis of conserved sequence‐structure relationships in kinesins: Coupling of the active site and a tubulin‐binding sub‐domain. J Mol Biol. 2007;368:1231–1248. [DOI] [PubMed] [Google Scholar]
- 6. Gorfe AA, Grant BJ, McCammon JA. Mapping the nucleotide and isoform‐dependent structural and dynamical features of Ras proteins. Structure. 2008;16:885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. del Sol A, Fujihashi H, Amoros D, Nussinov R. Residues crucial for maintaining short paths in network communication mediate signaling in proteins. Mol Syst Biol. 2006;2(2006):0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sethi A, Eargle J, Black AA, Luthey‐Schulten Z. Dynamical networks in tRNA:Protein complexes. Proc Natl Acad Sci U S A. 2009;106:6620–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Di Paola L, De Ruvo M, Paci P, Santoni D, Giuliani A. Protein contact networks: An emerging paradigm in chemistry. Chem Rev. 2013;113:1598–1613. [DOI] [PubMed] [Google Scholar]
- 10. Bhattacharyya M, Ghosh S, Vishveshwara S. Protein structure and function: Looking through the network of side‐chain interactions. Curr Protein Pept Sci. 2016;17:4–25. [DOI] [PubMed] [Google Scholar]
- 11. Hinsen K, Petrescu AJ, Dellerue S, Bellissent‐Funel MC, Kneller GR. Harmonicity in slow protein dynamics. Chem Phys. 2000;261:25–37. [Google Scholar]
- 12. Atilgan AR, Durell SR, Jernigan RL, Demirel MC, Keskin O, Bahar I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys J. 2001;80:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. R Core Team . R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing, 2019. https://www.r-project.org [Google Scholar]
- 14. Berman HM, Westbrook J, Feng Z, et al. The protein data Bank. Nucleic Acids Res. 2000;28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bodenhofer U, Bonatesta E, Horejs‐Kainrath C, Hochreiter S. Msa: An R package for multiple sequence alignment. Bioinformatics. 2015;31:3997–3999. [DOI] [PubMed] [Google Scholar]
- 17. Wako H, Endo S. Normal mode analysis as a method to derive protein dynamics information from the protein data Bank. Biophys Rev. 2017;9:877–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Durand P, Trinquier G, Sanejouand Y‐H. A new approach for determining low‐frequency normal modes in macromolecules. Biopolymers. 1994;34:759–771. [Google Scholar]
- 20. Tama F, Gadea FX, Marques O, Sanejouand YH. Building‐block approach for determining low‐frequency normal modes of macromolecules. Proteins. 2000;41:1–7. [DOI] [PubMed] [Google Scholar]
- 21. Romanowska J, Nowinski KS, Trylska J. Determining geometrically stable domains in molecular conformation sets. J Chem Theory Comput. 2012;8:2588–2599. [DOI] [PubMed] [Google Scholar]
- 22. Scarabelli G, Grant BJ. Kinesin‐5 allosteric inhibitors uncouple the dynamics of nucleotide, microtubule, and neck‐linker binding sites. Biophys J. 2014;107:2204–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yao XQ, Malik RU, Griggs NW, et al. Dynamic coupling and allosteric networks in the alpha subunit of heterotrimeric G proteins. J Biol Chem. 2016;291:4742–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H, Yao XQ, Grant BJ. Comparative structural dynamic analysis of GTPases. PLoS Comput Biol. 2018;14:e1006364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang W, Cheng J, Allaire J, Xie Y, McPherson J (2020) shiny: Web Application Framework for R. https://shiny.rstudio.com/reference/shiny/1.4.0/shiny-package.html
- 26. Pierce KL, Premont RT, Lefkowitz RJ. Seven‐transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. [DOI] [PubMed] [Google Scholar]
- 27. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. [DOI] [PubMed] [Google Scholar]
- 28. Rasmussen SG, DeVree BT, Zou Y, et al. Crystal structure of the beta2 adrenergic receptor‐Gs protein complex. Nature. 2011;477:549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Venkatakrishnan AJ, Deupi X, Lebon G, et al. Diverse activation pathways in class a GPCRs converge near the G‐protein‐coupling region. Nature. 2016;536:484–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rasmussen SGF, Choi H‐J, Rosenbaum DM, et al. Crystal structure of the human β2 adrenergic G‐protein‐coupled receptor. Nature. 2007;450:383–387. [DOI] [PubMed] [Google Scholar]
- 31. Eyal E, Yang L‐W, Bahar I. Anisotropic network model: Systematic evaluation and a new web interface. Bioinformatics. 2006;22:2619–2627. [DOI] [PubMed] [Google Scholar]
- 32. Hollup SM, Salensminde G, Reuter N. WEBnm@: A web application for normal mode analyses of proteins. BMC Bioinf. 2005;6:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Felline A, Seeber M, Fanelli F. webPSN v2.0: A webserver to infer fingerprints of structural communication in biomacromolecules. Nucleic Acids Res. 2020;48:W94–W103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aydinkal RM, Sercinoglu O, Ozbek P. ProSNEx: A web‐based application for exploration and analysis of protein structures using network formalism. Nucleic Acids Res. 2019;47:W471–W476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chakrabarty B, Parekh N. NAPS: Network analysis of protein structures. Nucleic Acids Res. 2016;44:W375–W382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hinsen K. The molecular modeling toolkit: A new approach to molecular simulations. J Comput Chem. 2000;21:79–85. [Google Scholar]
- 37. Bakan A, Meireles LM, Bahar I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics. 2011;27:1575–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zimmermann MT, Kloczkowski A, Jernigan RL. MAVENs: Motion analysis and visualization of elastic networks and structural ensembles. BMC Bioinf. 2011;12:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roe DR, Cheatham TE 3rd. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9:3084–3095. [DOI] [PubMed] [Google Scholar]
- 40. Van Wart AT, Durrant J, Votapka L, Amaro RE. Weighted implementation of suboptimal paths (WISP): An optimized algorithm and tool for dynamical network analysis. J Chem Theory Comput. 2014;10:511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tiberti M, Invernizzi G, Lambrughi M, Inbar Y, Schreiber G, Papaleo E. PyInteraph: A framework for the analysis of interaction networks in structural ensembles of proteins. J Chem Inf Model. 2014;54:1537–1551. [DOI] [PubMed] [Google Scholar]
- 42. Bhattacharyya M, Bhat CR, Vishveshwara S. An automated approach to network features of protein structure ensembles. Protein Sci. 2013;22:1399–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bradley AR, Rose AS, Pavelka A, et al. MMTF—an efficient file format for the transmission, visualization, and analysis of macromolecular structures. PLoS Comput Biol. 2017;13:e1005575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information 1 Detailed instructions and accompanying code for example eDDM analysis.
Data Availability Statement
Binaries and platform independent source code along detailed installation instruction of Bio3D core and extension packages is available at: http://thegrantlab.org/bio3d/.