

Abstract
Biology is advanced by producing structural models of biological systems, such as protein complexes. Some systems are recalcitrant to traditional structure determination methods. In such cases, it may still be possible to produce useful models by integrative structure determination that depends on simultaneous use of multiple types of data. An ensemble of models that are sufficiently consistent with the data is produced by a structural sampling method guided by a data‐dependent scoring function. The variation in the ensemble of models quantified the uncertainty of the structure, generally resulting from the uncertainty in the input information and actual structural heterogeneity in the samples used to produce the data. Here, we describe how to generate, assess, and interpret ensembles of integrative structural models using our open source Integrative Modeling Platform program (https://integrativemodeling.org).
Keywords: biophysics, chemical cross‐linking, electron microscopy, integrative structure modeling, model validation, protein complexes, structural biology
Abbreviations
- BS3
chemical crosslinking reagent bissulfosuccinimidyl suberate
- DSS
chemical crosslinking reagent disuccinimidyl suberate
- EM
electron microscopy
- GMM
Gaussian mixture model
- IMP
Integrative Modeling Platform
- mmCIF
(.mmcif) macromolecular Crystallographic Information File—a format for archiving macromolecular experiments and their results
- MRC
(.mrc)—Medical Research Council format—a file format for storing molecular densities
- PDB
Protein Data Bank
- PMI
Python Modeling Interface, a module in IMP
- RMF
(.rmf, .rmf3)—Rich Molecular Format, a file format for storing hierarchical molecular data
- RMSD
root mean squared deviation
- RMSF
root mean squared fluctuation
1. INTRODUCTION
To understand the function of a macromolecular assembly, we must know the structure of its components and their spatial arrangement. Direct experimental determination of such a complex structure is generally difficult, as individual experimental techniques cannot always sufficiently characterize the entire system. For example, crystals of complexes suitable for X‐ray crystallography cannot always be produced. Cryo‐electron microscopy (cryo‐EM) can be used to study large assemblies, but it is often limited to worse than atomic resolution. Finally, molecular biology, biochemistry, and proteomics techniques, such as yeast two‐hybrid and affinity purification mass spectrometry, yield information about the interactions between proteins, but not the positions of these proteins within the assembly or the structures of the proteins themselves.
One approach to solving structures of systems recalcitrant to traditional methods is by combining multiple types of input information, including from varied experiments, physical theories, statistical inferences, and prior structural models. By considering all information simultaneously, modeling in principle maximizes the accuracy, precision, completeness, and efficiency of structure determination. The better the accuracy, precision, and completeness of a model, the larger the variety of questions that can be addressed. For example, a model at a precision of 10 Å may be useful to identify protein interfaces in a multi‐protein complex, however, is not useful for characterizing a small molecule binding pocket. Numerous structures have already been solved using an integrative approach, including the 26S proteasome, 1 the yeast spindle pole body core, 2 and the yeast nuclear pore complex. 3 The approach can also be applied to systems that exist simultaneously in multiple structural states, as demonstrated by the two‐state model of the PhoQ sensor histidine kinase. 4
The integrative modeling workflow proceeds through four stages (Figure 1), described in detail elsewhere. 5 , 6 , 7 Briefly, in the first stage, input information about the structure of the system is gathered. In the second stage, the representation of the model (e.g., atomic or coarse‐grained) and the scoring function that ranks alternative models based on input information are defined; the scoring function is generally a sum of individual spatial restraints, each one dependent on a single data point. In the third stage, models are sampled to find those that best satisfy the input information; sampling is usually performed using stochastic methods such as Monte Carlo sampling. Finally, in the fourth stage, the set of models produced by sampling is assessed. Once a model is assessed, it can be used to address biological questions of interest.
FIGURE 1.
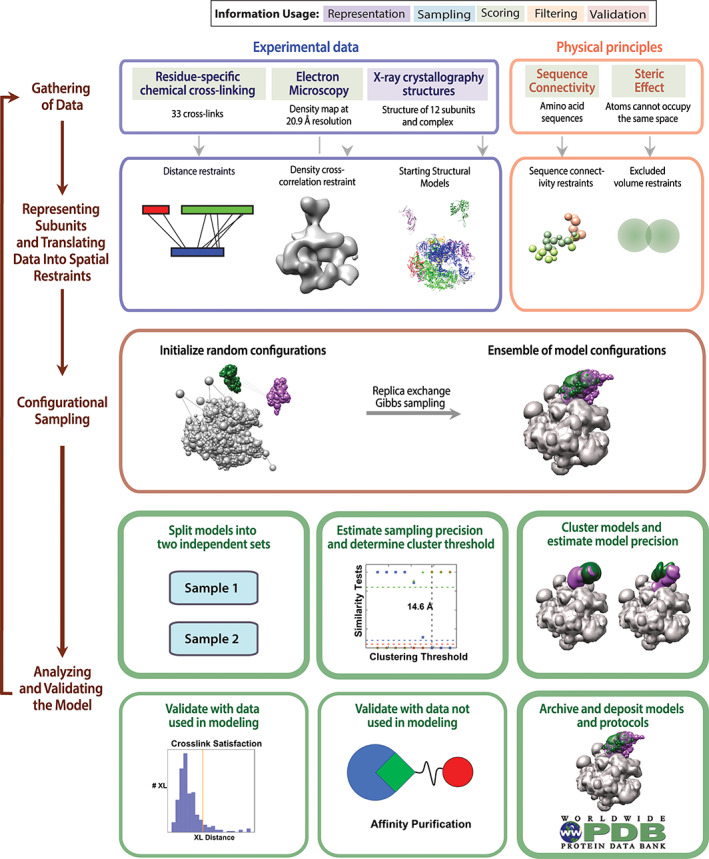
The four stages of integrative modeling. The integrative structure determination procedure proceeds through four stages. First, we collect all information describing the system, including experimental data and physical laws. Second, the representation of the system components is chosen, and each piece of input information is translated into a set of spatial restraints. Third, alternative configurations of the system components are sampled. Fourth, the ensemble of models resulting from sampling are filtered by their fit to the input data, the sampling and model precision computed, and the resulting model ensembles validated against information used and not used in modeling. Should the model not be deemed satisfactory, by either being too imprecise or poorly fit, the process can be iterated, adding more information or increasing sampling, until a satisfactory model is obtained. Steps in the fourth stage covered in this tutorial are outlined with thick lines
Answering different biological questions requires models of different precision. 8 Stochastic sampling methods often used in integrative structure modeling do not output a single structure but, rather, an ensemble of varied models that can be similarly consistent with the input data. This model ensemble defines the precision of the output model, but only if the resolution of sampling (sampling precision) is better than the model precision. In an analogy, sampling of a scoring function landscape is akin to taking an optical microscope image, in which only features larger than the resolution of the microscope are meaningful. Thus, accurately estimating the model and sampling precision are crucial for correctly interpreting a model.
To facilitate all computational aspects of integrative structure determination, we developed the open source Integrative Modeling Platform (IMP) package (https://integrativemodeling.org/) 9 , 10 for constructing and distributing integrative modeling protocols. The modularity and flexibility of IMP allow us to mix‐and‐match alternative representations, scoring function terms, and sampling algorithms, which in turn facilitates addressing difficult modeling problems. IMP allows representing molecules at multiple resolutions, using spatial restraints from almost any type of data, and searching for solutions by a variety of sampling algorithms.
In a previous Tools in Protein Science article, 7 we demonstrated the Python Modeling Interface (PMI) of IMP for defining model representation, adding data‐based restraints, sampling structural models, and clustering and analyzing output ensembles. In this iteration of the tutorial, we discuss the rationale and implementation of previously described techniques for estimating sampling precision and model uncertainty (Figure 1, bottom) 5 , 11 and illustrate them by a sample integrative structure modeling application.
2. INTEGRATIVE MODELING OF THE RNA POLYMERASE II STALK
In this tutorial, we model the positions and orientations of the Rpb4 and Rpb7 subunits in the RNA polymerase II stalk relative to the rest of the complex. The entire complex was previously solved via X‐ray crystallography at 3.8 Å. 12 Here, we utilize biophysical data including chemical cross‐links, a negative‐stain EM map, and physical principles to localize the two subunits of the stalk in relation to the rest of the complex. Users may opt to perform only the analysis portion of the tutorial by skipping Section 2.2.
2.1. Installing the required software
IMP is available for Linux, Mac, and Windows at https://integrativemodeling.org/download.html. Source code, nightly builds, and detailed installation instructions can be found at https://github.com/salilab/imp. Running the tutorial requires a number of Python packages, including numpy, scikit‐learn, hdbscan, and matplotlib. 13
IMP can also be used through Anaconda Python (https://www.anaconda.com/products/individual). To install IMP through Anaconda, use the following command:
conda install -c salilab imp
The python dependencies noted above can be installed using the following commands:
conda install numpy scikit-learn matplotlib pandas conda install -c conda-forge hdbscan conda install -c salilab pyrmsd
If using Anaconda, all python commands shown below should use the Anaconda instance of Python ($ANACONDA_HOME/bin/python)
The code containing the data and scripts for the integrative modeling of the RNA Polymerase II stalk can be found at https://github.com/salilab/imp_analysis_tutorial/. This repository should be downloaded to the same computer where IMP is installed.
After downloading, navigate to the tutorial base directory and download required submodules:
git submodule init git submodule update
2.2. Generating models of RNA polymerase II
The first three stages of integrative modeling of the RNA polymerase II stalk are performed by moving to the ./ranpolii/modeling/ folder, modifying the ./run_rnapolii_modeling.sh file to point to the users' python instance and running the following command:
./run_rnapolii_modeling.sh out_dir N n_steps
where out_dir is the prefix of the output folder, N is the number of independent samples to run, and n_steps is the number of Monte Carlo frames to produce during sampling. On modern machines, n_steps = 10,000 should take less than an hour to run. N must be greater than 1 to perform sampling exhaustiveness testing and N should not exceed the number of computational cores on the computer system used. The system configuration in this tutorial has been found to be sufficiently sampled with N ≥ 32 and n_steps ≥ 50,000.
The script runs N independent instances of the script modeling.py, creating N output directories out_dir0, out_dir1, …, out_dir{N − 1}. Below, we detail the workflow of this modeling script in the context of the four stages of integrative modeling.
2.2.1. Stage 1: Gathering data
The set of information used to create our model is contained in the ./rnapolii/data folder (Figure 2). The components of the system, protein chains and their component residues, are defined in the FASTA file 1WCM.fasta.txt. Two sets of chemical crosslinks 14 , 15 are defined in files polii_xlinks.csv and polii_jura.csv (using BSS and DS3 crosslinkers, respectively) (Figure 2b). A negative‐stain EM density 16 (EMDB 1883) is stored in emd_1883.map.mrc and a corresponding Gaussian mixture model (GMM) to approximate the density written in emd_1883.map.txt (Figure 2c). The atomic‐resolution coordinates of each subunit, and the relative positioning of the components other than Rpb4 and Rpb7 are found in the PDB file 1WCM.pdb 12 (Figure 2d). In addition to this experimental information, we also utilize the physical principles of sequence connectivity and excluded volume.
FIGURE 2.
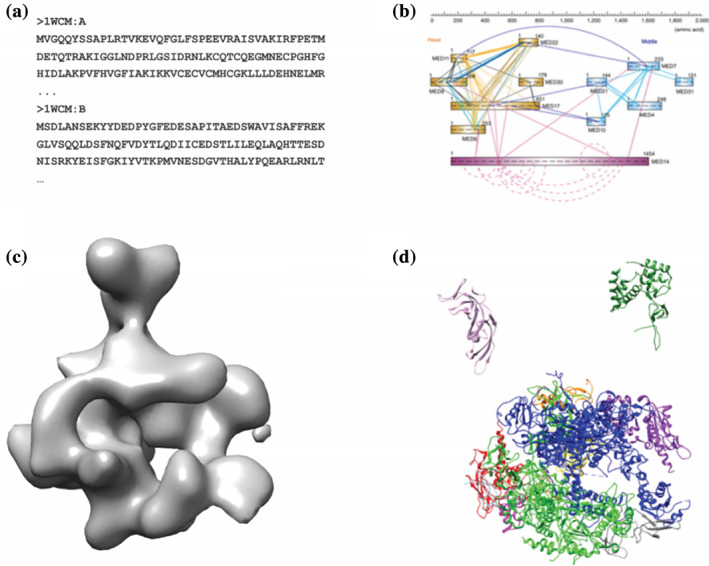
Data used in constructing RNA‐Polymerase II stalk model. (a) Primary sequences of all subunits in the FASTA format. (b) Chemical cross‐linking data, which yields a list of proximate residue pairs. (c) A 3D negative‐stain EM density map of the entire complex. (d) X‐ray crystal structures of each of the subunits. Figure published in a previous Protein Science tutorial 7
2.2.2. Stage 2: Defining representation and constructing scoring function
We represent the RNA polymerase II complex in a multi‐scale fashion. Component residues are simultaneously modeled by coarse grained beads of one‐residue and up to 10‐consecutive‐residues‐per‐bead. Additionally, the density of each component is represented by a GMM for use in the EM restraint. 17 This multi‐scale representation allows restraints of various types to be applied at the most appropriate resolution scales. A balance must be achieved between choosing a representation that is sufficiently detailed to capture structural features that are informed by the data and important for model interpretation while only representing degrees of freedom for which information is available and at a resolution that allows for tractable structural sampling. 18
The restraints are applied to individual resolution scales as appropriate. Beads and GMMs of one or more individual chains or domains are arranged into either a rigid body or a flexible string, based on the crystallographic structures. In a rigid body, all beads and Gaussians representing a structured domain have their relative distances constrained during sampling, while in a flexible string of beads they are restrained by the sequence connectivity. This representation is defined by a topology file, ./rnapolii/data/topology.txt, the format of which is discussed in a previous tutorial. 7 After defining the model representation, we build the restraints by which the individual structural models will be scored based on the input data.
In this system, we utilize four types of restraints. First, we apply an excluded volume restraint (called ExcludedVolumeSphere in the Python script), which prevents beads from occupying the same space. For speed, this restraint is applied to the low‐resolution representation of the system (10 consecutive residues represented by each bead). Second, we restrain components of the system adjacent in sequence by a ConnectivityRestraint. Third, we apply a restraint on the density overlap of a model configuration to the experimental 3D density map. To reduce overhead, this restraint is evaluated by computing the overlap between the GMM representation of the model to a GMM approximation of the experimental density. 19 Finally, we utilize a chemical crosslink restraint (CrossLinkingMassSpectrometryRestraint) to restrain the distances between pairs of residues observed in the two sets of crosslinking data. These restraints are applied to the highest resolution bead modeled for each residue endpoint and are evaluated at upper bound of 21.0 Å, which corresponds to the sum of the crosslinking reagent spacer length (11.4 Å) and the two crosslinked residue side chains. Restraint scores are scaled by a weight term to balance the contribution of each restraint to the overall model score. As a heuristic, weights of 1.0 were applied to the excluded volume, connectivity, and crosslinking restraints while a weight of 80.0 was applied to the electron density restraint. The scoring function is the sum of all restraints.
2.2.3. Stage 3: Sampling
The system is sampled using Markov Chain Monte Carlo 20 with simulated annealing and Gibbs Sampling replica exchange 21 using the number of steps defined above. Using the ./run_rnapolii_modeling.sh script, we initialize multiple independent sampling runs, each starting with random initial configurations, to more efficiently search the scoring function landscape and allow for convergence testing.
2.3. Analyzing output ensembles of RNA polymerase II
To avoid an overinterpretation of a model ensemble, we need to estimate a model precision (i.e., model uncertainty). 5 , 22 Only model features larger than model uncertainty are likely modeled accurately and can thus be interpreted. The model precision can be defined as the variability of the complete ensemble of all good‐scoring models. Thus, the model precision can be computed accurately only by enumeration of all models. The model precision quantifies the uncertainty in the model due to uncertainty in the input information. It can be improved most obviously by increasing the amount of the input information. In practice, we compute an estimated model precision using the variability of the ensemble of sampled good‐scoring models. The accuracy of this estimate depends on the completeness of sampling, which we estimate by computing a sampling precision.
We define the sampling precision as the difference between two sets of sufficiently good‐scoring models, each one independently obtained by a stochastic sampling method. 11 The sampling precision can be improved by increasing the number of computed models and, possibly, by simplifying model representation. When the sampling precision is comparable to or greater than the model precision, the model uncertainty results only from the relative lack of input data and actual structural heterogeneity in the samples used to produce the data, not the lack of structural sampling. In practice, we compute the sampling precision by finding the smallest clustering threshold that produces a statistically similar proportion of models among all clusters, as computed by three criteria for assessing statistical similarity (below).
2.3.1. Estimating sampling precision
Here, we estimate the sampling precision for the set of models included in the tutorial repository (8 individual runs of 5,000 steps each) in the ./rnapolii/analysis directory. In addition, we illustrate the results of a more complete sampling protocol (32 individual runs of 50,000 steps each) contained in the ./rnapolii/analysis_extensive directory. The set of models from the latter sampling protocol is not included in the tutorial repository due to the large disk space requirement (33G).
This process takes three steps. In the first step, we split our set of trajectories into two independent sets, identify models that sufficiently satisfy the input restraints (good‐scoring models) and cluster these models based on the scores of individual restraints. In the second step, we extract the good‐scoring models from each cluster identified in the first step. In the third step, we compute the sampling precision for each cluster of good‐scoring models.
For the first step, we navigate to the directory to analyze the set of eight independent modeling runs provided in./rnapolii/modeling/example* using the following command:
python ./run_analysis_trajectories.py ../modeling/ example
where ../modeling/ is the path to the folder containing the output directories and example is the prefix for the output directories to be analyzed. This initial analysis takes up to a few minutes on a single processor for the number of samples in the example directories and scales linearly with the total number of steps over all sampling runs. The run_analysis_trajectories.py script must be modified to analyze the restraints specific to other modeling setups. Documentation for analyzing other standard IMP restraints and custom restraints can be found at https://github.com/salilab/PMI_analysis.
We first assess for sufficient sampling and fit to input data by clustering based on total and individual restraint scores. This assessment is a fast and convenient way to assess millions of models before proceeding to more computationally expensive structure‐based analysis. In this step, the equilibration of score terms in each Markov chain is computed; 23 models produced after equilibration of all scores are pooled and clustered using HDBSCAN, 24 an assumption‐free, density‐based hierarchical clustering algorithm particularly good at defining oddly‐shaped clusters.
The script outputs a large number of data files and plots into the model_analysis folder. We first examine model_analysis/plot_clustering_scores.png to observe the distributions of the various scoring function terms (Figure 3). Distinct, non‐overlapping clusters of solutions, as observed in Figure 3a in the XLs_sum parameter (total crosslink score), can indicate insufficient sampling; there are two distinct sets of solutions with different sets of crosslinks satisfied. In comparison, analysis of the more extensive sampling protocol from analysis_extensive (Figure 3b) shows a continuous distribution, indicating a more complete sampling. 23 , 25 , 26
FIGURE 3.
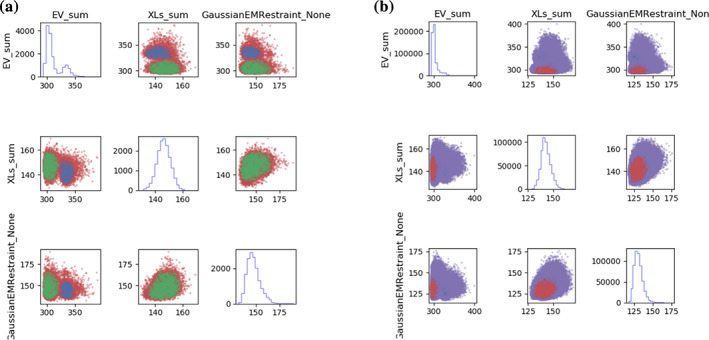
Scores plots for undersampled and extensively sampled systems. Histograms of individual scores are on the diagonal and 2D score plots are on the off‐diagonals. The colors in the 2D plots represent the different clusters. (a) The undersampled modeling scores plots shows two distinct clusters of scores, indicating that the independent simulations did not converge on the same solution. (b) The plot of the extensively sampled modeling shows a continuous distribution of scores that suggests more complete sampling
This protocol also outputs information about the crosslink satisfaction for each cluster. The mean crosslink distance for all crosslinks is plotted in model_analysis/plot_XLs_distance_histogram_cl1.pdf (Figure 4a). Of the 273 total crosslinks, 13 are considered “unsatisfied” in cluster 1 (a crosslink is unsatisfied if no model in a set has a crosslink distance of less than 30 Å). These unsatisfied crosslinks can be identified in the distance distribution for each individual crosslink: model_analysis/plot_XLs_distance_distributions_cl1.pdf (Figure 4b).
FIGURE 4.
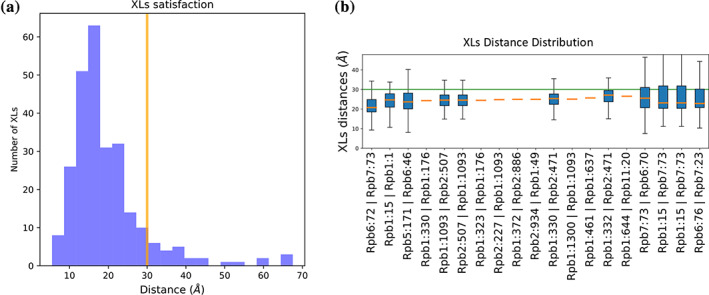
Evaluation of crosslink satisfaction. (a) Histogram of mean crosslink distances for Cluster 0 showing that the majority of observed crosslinks are satisfied at the 30 Å cutoff value (yellow line). (b) Individual crosslink distance distributions for a subset of crosslinks used to model the complex. The identity of each crosslink is noted in the X‐axis label in the format “Protein1:Residue1 | Protein2:Residue2”. The total range of crosslink distances in the cluster are shown by the whiskers while the 25th to 75th percentile is represented by the blue boxed. The median crosslink distance is represented by an orange line. The 30 Å cutoff value is shown in the green line. Crosslinks that show a fixed distance (orange dashes) have both residue endpoints located in the rigid body subcomplex of this model. The figure containing all crosslink distributions is found in ./rnapolii/analysis /model_analysis/ plot_XLs_distance_distributions_cl1.pdf
The model_analysis/summary_hdbscan_clustering.dat cluster file shows numerical results of score‐based clustering (Table 1), including the number of models in each of the two subsets of models (A and B) and the average scores for each restraint. There are two large clusters, one of which satisfies the restraints slightly better than the other (similar to what is observed in Figure 3a). The third cluster is minor and ignored. From this point, we continue analysis with cluster 1; other clusters can be analyzed similarly.
TABLE 1.
Score‐based clustering results. Individual restraint scores and populations for each of the three clusters determined from the first step of sampling precision estimation (Section 2.3.1)
| Cluster | Total_Score | EV_sum | XLs_sum | GaussianEMRestraint | N_models | N_A | N_B |
|---|---|---|---|---|---|---|---|
| 1 | 601.49 | 303.43 | 146.67 | 147.20 | 11,282 | 4,809 | 6,473 |
| 0 | 625.75 | 333.80 | 142.70 | 144.37 | 2,551 | 2,515 | 36 |
| −1 | 628.66 | 326.14 | 146.15 | 150.48 | 2,167 | 1,476 | 691 |
The second step is initiated by the following command:
python ./run_extract_models.py ../modeling example 1
where ../modeling is the base output directory, example is the output directory prefix, and 0 is the cluster identity to be extracted. Extracting the ~11,000 models from the tutorial example takes up to a few minutes on a single processor.
This script extracts the coordinates and total scores for all of the models from the chosen cluster and separates them into the A and B subsets in preparation for structural clustering. Because structure‐based clustering is CPU and memory intensive, at most 30,000 models are randomly chosen from each cluster. Model coordinates (A_models_clust1.rmf3 and B_models_clust1.rmf3) and model scores (A_models_clust1.txt, and B_models_clust1.txt) for the A and B subsets of models are written to ./model_analysis.
In the final step, we estimate the sampling precision using the following command:
imp_sampcon exhaust -n rnapol \ --rmfA ./model_analysis/A_models_clust1.rmf3 \ --rmfB ./model_analysis/B_models_clust1.rmf3 \ --scoreA ./model_analysis/A_models_clust1.txt \ --scoreB ./model_analysis/B_models_clust1.txt \ -d density_rnapol.txt \ -m cpu_omp -c 4 \ -gp -g 2.0
The script clusters models using a nearest‐neighbor algorithm at increasing cluster thresholds (RMSD values) and computes each of the three criteria for cluster similarity. The options specified are: a name for the system that is used to prefix the output files (option n), the files containing the models and scores generated in step 2 (options rmfA, rmfB, scoreA, scoreB), the file, listing the domains for density map creation (option ‐d density_rnapol.txt, format described below), the parallelization mode for the expensive pairwise RMSD calculation (option m), the number of cores used (option c), and a flag to generate plots using gnuplot (option gp, only if gnuplot is installed). The grid size (option g) defines the step size in Å for increasing the cluster threshold; each cluster threshold from the minimum to the maximum RMSD of the ensemble is tested, so for larger more structurally diverse systems, setting a value of 5 or 10 Å significantly decreases computational time at the expense of precision in the estimate of the sampling precision. Computing the sampling precision for the tutorial modeling example using the above protocol takes a few hours to complete on a typical workstation.
One output of this protocol is localization densities of system components. The localization density is the probability of finding a protein at each position in space, given a set of superposed models of the modeled system; this set is often either the entire ensemble or only a cluster of sufficiently good‐scoring models. Most often, the localization density is provided separately for discrete subunits or their domains. The file density_rnapol.txt is used to define the system components, either by their molecule name (e.g., “Rbp4”) or by a tuple defining the molecule name as well as the first and last residues, (e.g., [“Rbp4”, 5, 100]). The components in each localization density are noted in a Python list and localization named in a dictionary. For this example, we create three localization densities: one for each of the two moving subunits, Rbp4 and Rbp7, and one for the remainder of the complex:
density_custom_ranges={"Rbp4":["Rbp4"],"Rbp7":["Rbp7"], "Rigid_Body":["Rbp1", "Rbp2", "Rbp3", "Rbp5", "Rbp6", "Rbp8", "Rbp9","Rbp10","Rbp11","Rbp12"]}
Additionally, there are options for changing the run folder, model file format (RMF or PDB), file names for the model scores, and localization density voxel size. For models that are not restrained via an EM density map, models should be superposed before clustering, by including the –align option in the above command. The resolution for the localization density is set using the –density_threshold option and should be chosen at a resolution above the known or expected sampling precision (default = 20.0 Å). Advanced options include running the protocol on selected subunits, considering ambiguity in RMSD calculation by listing subunits with multiple copies, and aligning the models before RMSD calculation. The IMP documentation (https://integrativemodeling.org/2.13.0/doc/) contains the complete list of options for this protocol.
The three criteria for cluster similarity are computed at each clustering threshold, tabulated in rnapol.ChiSquare_Grid_Stats.txt and shown visually in rnapol.ChiSquare.pdf (Figure 5a). The three criteria are (a) a p‐value of greater than 0.05 in the χ2 test for homogeneity of proportions, which indicates no statistically significant difference between the sample distributions among all clusters, (b) a value in the χ 2 test of Cramer's V less than 0.1, which indicates an insignificant magnitude of difference between the sample distributions, and (c) the population of models in significant clusters, containing more than 10 models, is greater than 80%. For this example, the smallest clustering threshold that satisfies all three tests is 12.6 Å; this value is reported in the output file rnapol.Sampling_Precision_Stats.txt.
FIGURE 5.
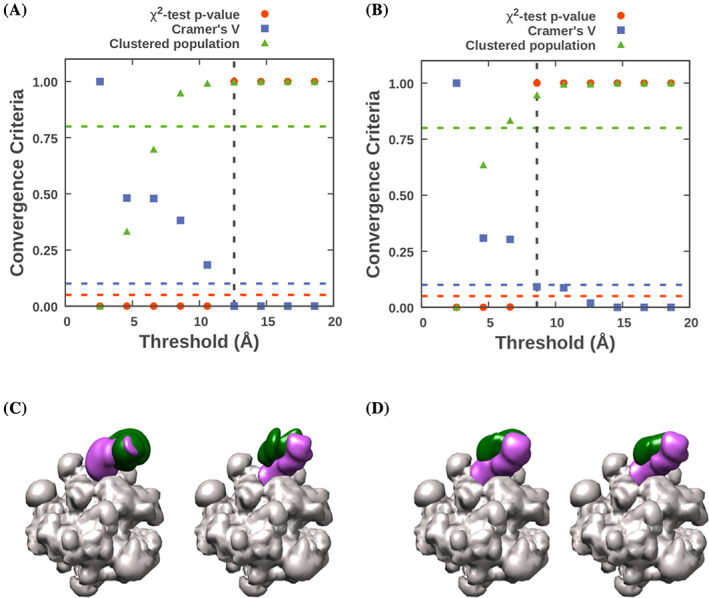
Determination of sampling precision and visual validation for under and rigorously sampled models. The results of the three sampling convergence tests at multiple clustering thresholds are plotted in panel (a) for the undersampled set of models, showing a sampling precision of 12.6 Å and (b) for the rigorously sampled set, showing a sampling precision of 8.6 Å. C) Comparison of the two independent sets of in localization densities of Rpb4 (green volume) and Rpb7 (purple volume) for (c) the undersampled model set shows significant differences between the two, while for D) the pair generated from the extensively sampled set appears identical
In this example, the small number of computed models results in a relatively low sampling precision. The more extensive sampling protocol results in a significantly higher sampling precision of 8.6 Å (Figure 5b).
The set of models are then clustered at the sampling precision and models from each individual cluster placed in folders cluster.0, cluster.1, and so on. The total number of models and the population of the A and B subsamples for each cluster are reported in rnapol.Cluster_Population.txt. For this example, one cluster is found to define our system.
2.3.2. Estimating model precision and its comparison to sampling precision
We estimate the model precision of each output cluster by computing the average RMSD between each configuration in the cluster and the cluster centroid configuration (RMSF). The estimated model precision for the one cluster found above is 8.7 Å, reported in rnapol.Cluster_Precision.txt. To compare this value numerically to our estimated sampling precision, we first multiply it by 1.4, to reflect the general approximate relationship between RMSD and RMSF for globular proteins. 27 Our scaled model precision is 12.2 Å; this is slightly less than our sampling precision of 12.6 Å.
We note that the estimated sampling precision defines the minimum distance at which features in the model can be interpreted. Interpreting a model in terms of its sampling and model precision, similar to interpreting an EM map in terms of its resolution, is meant as a general guideline to inform the length scale at which the position of model components is reliable. Thus, we may conclude that the sampling is sufficiently exhaustive to justify interpreting the features of the output model cluster. Alternatively, if resources allow, we may decide to perform additional sampling to improve the sampling precision.
2.3.3. Interpreting and visualizing individual model clusters
After computing the sampling precision, identifying the cluster models and estimating the model precision, we can use this information to interpret our model at the appropriate resolution scale. Our model now consists of a single ensemble of configurations (in cluster.0), a sampling precision (12.6 Å) and estimated model precision (8.7 Å).
Individual models and the entire model ensembles can be viewed in ChimeraX. 28 The localization density for each individual model component defined in density_rnapol.txt is output in a .mrc file in cluster.0. The centroid structure of the cluster is output in cluster_center_model.rmf3. Localization densities and centroid models for both the A and B sets of independent samples are also computed and deposited in the Sample_A and Sample_B directories in the cluster folder.
As a final validation step, the Sample_A and Sample_B localization densities can be compared visually and via their cross‐correlation coefficient (CCC). The independently computed localization densities from the undersampled set (Figure 5c) have relatively low CCC values, 0.92 for Rbp4 and 0.91 for Rbp7, and are qualitatively different. The set from analysis_extensive (Figure 5d) has higher CCC values, 0.98 for Rpb4 and 0.97 for Rpb7, indicative of the higher sampling precision of that model.
The final, complete, model from analysis_extensive can be visualized by opening model files cluster_center_model.rmf, Rpb4.mrc and Rpb7.mrc from the ./analysis_extensive/cluster.0/ folder in ChimeraX (Figure 6). The localization densities for Rpb4 and Rbp7 are well‐delineated, indicating that we have found their general position within the complex. Identification of the exact binding interfaces is beyond the resolution range of our model and should not be inferred.
FIGURE 6.
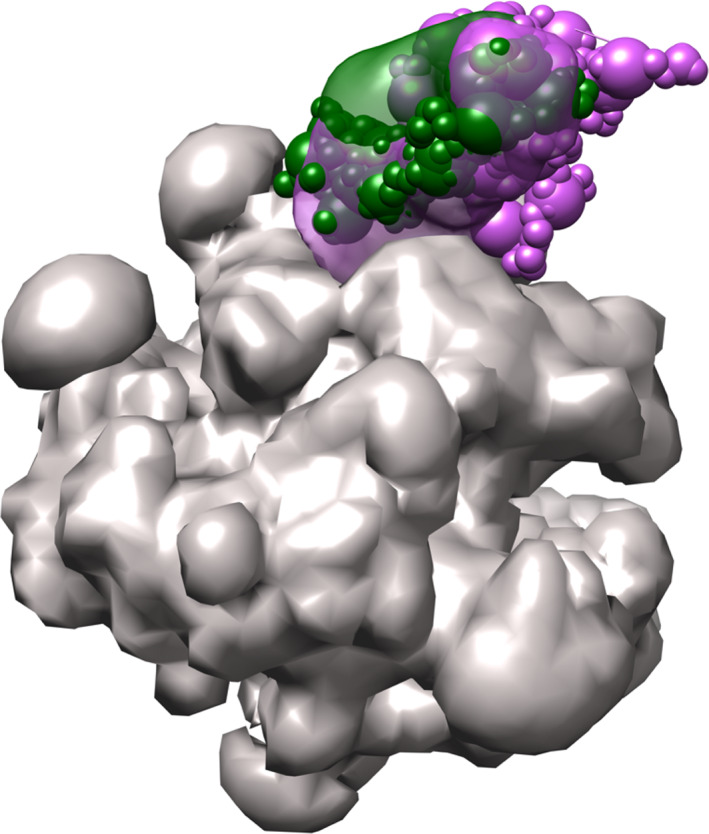
Integrative model of the RNA Polymerase II stalk. The final integrative model for the RNA polymerase II stalk, determined to 8.7 Å precision, is visualized by the localization densities of subunits Rpb4 (green volume) and Rpb7 (purple volume) in relation to the rigid structure of the polymerase base (gray volume). A single representative model of the ensemble, the centroid model, for the two moving components is shown in sphere representation with the same colors. Figure prepared using Chimera
While in this example our analysis resulted in a single cluster, often multiple significant clusters are obtained and should be considered when interpreting the model. As noted above, this model uncertainty is generally a consequence of both a relative lack of input information and actual heterogeneity in the samples used to generate the data for modeling.
Should the model(s) be sufficient to answer the biological question or used in a publication, the model(s) and modeling protocol should be archived so others can reproduce or improve the model with additional information.
2.4. Archiving models and depositing into the wwPDB
With a validated and interpreted model in hand, the final step is archiving the model(s) along with the data and protocols used to create them into the nascent worldwide PDB database for integrative structures (pdb-dev.wwpdb.org). 29 The mmCIF format has been extended to describe all aspects of integrative modeling. To produce an mmCIF file suitable for deposition, the python‐ihm module must be installed (Methods). Navigate to the tutorial repository's ihm‐deposition folder, and run the following command:
python create_ihm_cif_file.py
which will output tutorial_deposition.cif.
A full tutorial describing the deposition of integrative models can be found at https://integrativemodeling.org/tutorials/deposition/. Descriptions of the mmCIF extensions specific for integrative models can be found at http://mmcif.wwpdb.org/dictionaries/mmcif_ihm.dic/Index/ and an overview of the Python library used to create these files from IMP output can be found at https://python-ihm.readthedocs.io/en/latest/.
While the focus of this tutorial is on estimating sampling and model precisions, a complete validation also includes validation of the input data quality, validation of the model by the data used to compute it, validation of the model by the data not used to compute it, in addition to estimating sampling and model precisions. 30 A complete pipeline for these assessments is being constructed under the auspices of the wwPDB and will eventually be used as part of model deposition into the PDB.
3. CONCLUSIONS
Structural models of protein complexes are a powerful tool for advancing our knowledge about their function and evolution. A model needs to be validated before its interpretation, followed by its deposition. The IMP program facilitates generation, validation, and deposition of integrative models. A key aspect of a model is an estimate of its uncertainty (precision); knowledge of model uncertainty enables a user to avoid its overinterpretation.
4. MATERIALS AND METHODS
The scripts and libraries used to complete this tutorial are contained in the IMP analysis tutorial GitHub repository: https://github.com/salilab/imp_analysis_tutorial
Additional software is required to complete the tutorial protocols:
IMP version 2.13.0—https://integrativemodeling.org/
Python‐ihm—https://github.com/ihmwg/python-ihm
UCSF ChimeraX—https://www.cgl.ucsf.edu/chimerax/download.html
RMF reader for ChimeraX—https://cxtoolshed.rbvi.ucsf.edu/apps/chimeraxrmf
ACCESSIBILITY AND ACCESS
All data, scripts and software used for modeling are freely available in the GitHub repositories and links described in Materials and Methods.
AUTHOR CONTRIBUTIONS
Daniel Saltzberg: Conceptualization; software; visualization; writing‐original draft; writing‐review and editing. Shruthi Viswanath: Conceptualization; funding acquisition; software; writing‐original draft; writing‐review and editing. Ignacia Echeverria: Software; writing‐original draft; writing‐review and editing. Ilan Chemmama: Software; writing‐original draft; writing‐review and editing. Benjamin Webb: Software; writing‐original draft; writing‐review and editing. Andrej Sali: Conceptualization; funding acquisition; writing‐original draft; writing‐review and editing.
ACKNOWLEDGMENTS
This work was funded by grants NIH R01GM083960, NIH P41GM109824, NSF DBI‐1756250 and NSF DBI‐1832184 of AS and core funds from DAE India through NCBS/TIFR of SV. We would like to thank the current and former contributors to IMP. This tutorial is based, in part, on our previously published reviews. 6 , 7
Saltzberg DJ, Viswanath S, Echeverria I, Chemmama IE, Webb B, Sali A. Using Integrative Modeling Platform to compute, validate, and archive a model of a protein complex structure. Protein Science. 2021;30:250–261. 10.1002/pro.3995
Funding information Division of Biological Infrastructure, Grant/Award Numbers: DBI‐175625, DBI‐1832184; National Centre for Biological Sciences; National Institute of General Medical Sciences, Grant/Award Numbers: P41GM109824, R01GM083960
REFERENCES
- 1. Lasker K, Forster F, Bohn S, et al. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc Natl Acad Sci U S A. 2012;109:1380–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Viswanath S, Bonomi M, Kim SJ, et al. The molecular architecture of the yeast spindle pole body core determined by Bayesian integrative modeling. Mol Biol Cell. 2017;28:3298–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim SJ, Fernandez‐Martinez J, Nudelman I, et al. Integrative structure and functional anatomy of a nuclear pore complex. Nature. 2018;555:475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Molnar K, Bonomi M, Pellarin R, et al. Cys‐scanning disulfide crosslinking and Bayesian modeling probe the transmembrane signaling mechanism of the histidine kinase, PhoQ. Structure. 2014;22:1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rout MP, Sali A. Principles for integrative structural biology studies. Cell. 2019;177:1384–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Saltzberg D, Greenberg CH, Viswanath S, et al. Modeling biological complexes using integrative modeling platform. Methods Mol Biol. 2019;2022:353–377. [DOI] [PubMed] [Google Scholar]
- 7. Webb B, Viswanath S, Bonomi M, et al. Integrative structure modeling with the integrative modeling platform. Protein Sci. 2018;27:245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294:93–96. [DOI] [PubMed] [Google Scholar]
- 9. Webb B, Lasker K, Schneidman‐Duhovny D, Tjioe E, Phillips J, Kim SJ, Velazquez‐Muriel J, Russel D, Sali A Modeling of proteins and their assemblies with the integrative modeling platform In: Methods in molecular biology, New York, NY: Humana Press; 2011. pp. 377–397. Available from: http://salilab.org/pdf/Webb_MethodsMolBiol_2011.pdf. [DOI] [PubMed] [Google Scholar]
- 10. Russel D, Lasker K, Webb B, et al. Putting the pieces together: Integrative structure determination of macromolecular assemblies. PLoS Biol. 2012;10:e1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Viswanath S, Chemmama I, Cimermancic P, Sali A. Assessing exhaustiveness of stochastic sampling for integrative modeling of macromolecular structures. Biophys J. 2017;113:2344–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Armache K‐J, Mitterweger S, Meinhart A, Cramer P. Structures of complete RNA polymerase II and its subcomplex, Rpb4/7. J Biol Chem. 2005;280:7131–7134. [DOI] [PubMed] [Google Scholar]
- 13. Hunter JD. Matplotlib: A 2D graphics environment. Comput Sci Eng. 2007;9:90–95. [Google Scholar]
- 14. Trnka MJ, Baker PR, Robinson PJJ, Burlingame AL, Chalkley RJ. Matching cross‐linked peptide spectra: Only as good as the worse identification. Mol Cell Proteomics. 2014;13:420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen ZA, Jawhari A, Fischer L, et al. Architecture of the RNA polymerase II–TFIIF complex revealed by cross‐linking and mass spectrometry. EMBO J. 2010;29:717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Czeko E, Seizl M, Augsberger C, Mielke T, Cramer P. Iwr1 directs RNA polymerase II nuclear import. Mol Cell. 2011;42:261–266. [DOI] [PubMed] [Google Scholar]
- 17. Kawabata T. Multiple subunit fitting into a low‐resolution density map of a macromolecular complex using a Gaussian mixture model. Biophys J. 2008;95:4643–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Viswanath S, Sali A. Optimizing model representation for integrative structure determination of macromolecular assemblies. Proc Natl Acad Sci U S A. 2019;116:540–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Greenberg CH, Kollman J, Zelter A, et al. Structure of γ‐tubulin small complex based on a cryo‐EM map, chemical cross‐links, and a remotely related structure. J Struct Biol. 2016;194:303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Metropolis N, Ulam S. The Monte Carlo method. J Am Stat Assoc. 1949;44:335–341. [DOI] [PubMed] [Google Scholar]
- 21. Habeck M, Nilges M, Rieping W. Replica‐exchange Monte Carlo scheme for bayesian data analysis. Phys Rev Lett. 2005;94:018105. [DOI] [PubMed] [Google Scholar]
- 22. Schneidman‐Duhovny D, Pellarin R, Sali A. Uncertainty in integrative structural modeling. Curr Opin Struct Biol. 2014;28:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chodera JD. A simple method for automated equilibration detection in molecular simulations. J Chem Theory Comput. 2016;12:1799–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McInnes L, Healy J, Astels S. hdbscan: Hierarchical density based clustering. J Open Source Softw. 2017;2:205. [Google Scholar]
- 25. Gutierrez C, Chemmama IE, Mao H, et al. Structural dynamics of the human COP9 signalosome revealed by cross‐linking mass spectrometry and integrative modeling. Proc Natl Acad Sci U S A. 2020;117:4088–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kwon Y, Kaake RM, Echeverria I, et al. Structural basis of CD4 downregulation by HIV‐1 Nef. Nat Struct Mol Biol. 2020;27:822–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kuzmanic A, Zagrovic B. Determination of ensemble‐average pairwise root mean‐square deviation from experimental B‐factors. Biophys J. 2010;98:861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goddard TD, Huang CC, Meng EC, et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis: UCSF ChimeraX visualization system. Protein Sci. 2018;27:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vallat B, Webb B, Westbrook J, Sali A, Berman HM. Archiving and disseminating integrative structure models. J Biomol NMR. 2019;73:385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berman HM, Burley SK, Chiu W, et al. Outcome of a workshop on archiving structural models of biological macromolecules. Structure. 2006;14:1211–1217. [DOI] [PubMed] [Google Scholar]