

Abstract
Histiocytic disorders are a spectrum of rare diseases characterised by the accumulation of macrophage‐, dendritic cell‐, or monocyte‐differentiated cells in various tissues and organs. The discovery of recurrent genetic alterations in many of these histiocytoses has led to their recognition as clonal neoplastic diseases. Moreover, the identification of the same somatic mutation in histiocytic lesions and peripheral blood and/or bone marrow cells from histiocytosis patients has provided evidence for systemic histiocytic neoplasms to originate from haematopoietic stem/progenitor cells (HSPCs). Here, we investigated associations between histiocytic disorders and additional haematological malignancies bearing the same genetic alteration(s) using the nationwide Dutch Pathology Registry. By searching on pathologist‐assigned diagnostic terms for the various histiocytic disorders, we identified 4602 patients with a putative histopathological diagnosis of a histiocytic disorder between 1971 and 2019. Histiocytosis‐affected tissue samples of 187 patients had been analysed for genetic alterations as part of routine molecular diagnostics, including from nine patients with an additional haematological malignancy. Among these patients, we discovered three cases with different histiocytic neoplasms and additional haematological malignancies bearing identical oncogenic mutations, including one patient with concomitant KRAS p.A59E mutated histiocytic sarcoma and chronic myelomonocytic leukaemia (CMML), one patient with synchronous NRAS p.G12V mutated indeterminate cell histiocytosis and CMML, and one patient with subsequent NRAS p.Q61R mutated Erdheim–Chester disease and acute myeloid leukaemia. These cases support the existence of a common haematopoietic cell‐of‐origin in at least a proportion of patients with a histiocytic neoplasm and additional haematological malignancy. In addition, they suggest that driver mutations in particular genes (e.g. N/KRAS) may specifically predispose to the development of an additional clonally related haematological malignancy or secondary histiocytic neoplasm. Finally, the putative existence of derailed multipotent HSPCs in these patients emphasises the importance of adequate (bone marrow) staging, molecular analysis and long‐term follow‐up of all histiocytosis patients.
Keywords: histiocytosis, malignant histiocytic disorders, histiocytic sarcoma, Langerhans cell sarcoma, indeterminate cell histiocytosis, Erdheim–Chester disease, non‐Langerhans‐cell histiocytosis, Langerhans‐cell histiocytosis, leukaemia, lymphoma
Introduction
Histiocytic disorders are a spectrum of rare diseases characterised by the accumulation of macrophage‐, dendritic cell‐, or monocyte‐differentiated cells in various tissues and organs [1]. Based on their clinical, radiographic and histopathological features, a wide variety of different subtypes of histiocytic diseases has been described [1]. Recurrent genetic alterations have been identified in many of these histiocytoses [2], including Langerhans cell histiocytosis (LCH) [3, 4, 5], Erdheim–Chester disease (ECD) [6], indeterminate cell histiocytosis (ICH) [7] and histiocytic sarcoma (HS) [8, 9]. These genetic alterations primarily comprise somatic missense mutations, indels and fusions involving genes that encode proteins of the mitogen‐activated protein kinase (MAPK) signalling pathway [10]. Consequently, many of the histiocytic disorders are now considered clonal neoplastic diseases [11, 12], characterised by constitutive MAPK pathway activation [10].
The recent identification of the same somatic mutation in histiocytic lesions and peripheral blood and/or (CD34+) bone marrow mononuclear cells from patients with disseminated histiocytoses has provided compelling evidence for systemic histiocytic neoplasms to originate from somatically mutated haematopoietic stem/progenitor cells (HSPCs) [13, 14, 15, 16]. These cells are multipotent and have intrinsic proliferative and self‐renewal potential. Therefore, patients with histiocytic neoplasms derived from (long‐lived) somatically mutated HSPCs could be at increased risk of developing additional clonally related haematological neoplasms derived from these cells. Conversely, transformation of antecedent haematological malignancies to secondary histiocytic neoplasms may also occur [17, 18]. In the 1990s, the LCH‐Malignancy Study Group of the Histiocyte Society already reported recurrent associations between LCH and leukaemia or lymphoma [19, 20], which were confirmed in later studies [21, 22]. Likewise, a high prevalence of haematological malignancies was also observed in adults with ECD [23] or HS [24]. Some isolated case reports and small case series described associations between single histiocytic disorders and haematological malignancies harbouring the same genetic alteration(s) [15, 23, 25, 26, 27, 28, 29, 30, 31, 32], supporting a common clonal origin of both diseases. Yet, the rarity of the histiocytic disorders, as well as the reality that adult patients are treated by a diverse range of medical specialists, and the fact that not all histiocytic neoplasms (e.g. ICH) are registered by national cancer registries, have thus far limited a comprehensive study of the occurrence of this phenomenon among a large cohort of patients with different types of histiocytic neoplasm. To address this issue, we requested data from the nationwide Dutch Pathology Registry (PALGA) [33], and retrieved the pathology reports of all patients diagnosed with a histologically confirmed and professionally PALGA‐coded histiocytic disorder in the Netherlands between 1971 and 2019. In this population‐based dataset, we searched for histiocytosis patients with an additional (histologically confirmed) haematological malignancy harbouring the same genetic alteration(s). Using this approach, we identified three cases with different histiocytic neoplasms and acute or chronic myeloid leukaemia bearing identical oncogenic mutations.
Materials and methods
In the Netherlands, all histo‐ and cytopathology reports generated at each of the (current) 43 pathology laboratories are digitally archived in the central network and registry of histo‐ and cytopathology – called PALGA [33]. Personal data are pseudonymised. This internationally unique archive was founded in 1971, achieved nationwide coverage in 1991, and contained more than 76 million pathology reports from over 12 million patients at the end of 2019 (Figure 1A). At sign‐out, the reporting pathologist adds one or more diagnosis coding lines – consisting of a combination of diagnostic terms (referring to the localisation, acquisition technique and abnormality) – to each pathology report. These diagnostic terms are automatically linked with one or more classification codes. These codes were originally related to the Systematised Nomenclature of Medicine, 1982 version, published by the College of American Pathologists. Researchers may request pseudonymised data from the national PALGA database.
Figure 1.
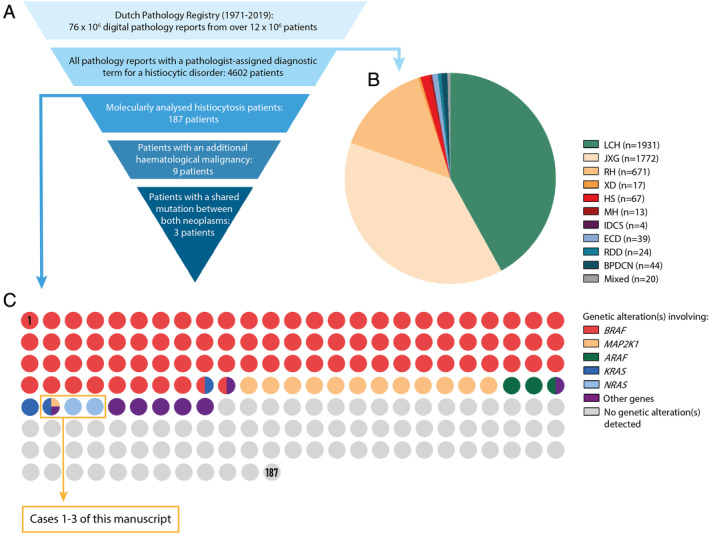
Study methodology. (A) Schematic representation of the funnel method used for patient identification. (B) Pie chart showing the number of identified patients with a pathology report registered in the Dutch Pathology Registry between 1971 and 2019 that contains a pathologist‐assigned diagnostic term for a specific histiocytic disorder. (C) Dot matrix chart displaying the mutational status of the 187 identified histiocytosis patients whose tissue samples were successfully molecularly analysed. Every dot represents one patient. The colour of the dot depicts whether a genetic alteration involving a specific gene has been detected in the histiocytosis‐affected tissue specimen(s) of the represented patient. Dots with multiple colours represent patients with multiple detected genetic alterations involving multiple genes. The three dots inside the orange box represent Cases 1–3, who are described in detail in the results section of this manuscript.
For this study, we retrieved all pathology reports with a pathologist‐assigned diagnostic term for one of the various histiocytic disorders (see supplementary material, Table S1) from the entire PALGA database from its start in 1971 until (and including) 2019. Because HSs and malignant histiocytoses reported before the development and widespread use of immunohistochemistry and clonality testing have later been demonstrated to often comprise lymphomas [34, 35, 36], we decided to exclude pathology reports with a diagnostic term for HS, interdigitating dendritic cell sarcoma (IDCS) or malignant histiocytosis (MH) from 2001 or before. In 2001, the third edition of the World Health Organisation classification of tumours of haematopoietic and lymphoid tissues was published [37], which underscored the importance of excluding a lymphoma before making a diagnosis of a malignant histiocytic disorder. Furthermore, we manually reviewed the digital pathology reports of all patients with one or more pathology reports that (collectively) contained diagnostic terms of two or more histiocytic disorders, to evaluate whether these patients had mixed histiocytosis or whether one diagnosis (e.g. xanthogranuloma) had been changed into another (e.g. ECD) after pathology review and/or histopathological analysis of ensuing tissue samples. Using this approach, we identified n = 4602 patients with a putative diagnosis of a histiocytic disorder (Figure 1B), including n = 1931 with LCH, n = 1772 with juvenile xanthogranuloma (JXG), n = 671 with reticulohistiocytoma (RH), n = 17 with xanthoma disseminatum (XD), n = 67 with HS, n = 13 with MH not otherwise specified, n = 4 with IDCS, n = 39 with ECD, n = 24 with Rosai–Dorfman disease (RDD), n = 44 with blastic plasmacytoid dendritic cell neoplasm (BPDCN) and n = 20 with mixed histiocytosis. Using the pseudonymised patient identification number provided with each pathology report, we also retrieved all additional pathology reports of these 4602 patients with a pathologist‐assigned diagnostic term for any given malignancy.
In the final output file, we searched all pathology reports with a diagnostic term for a histiocytic disorder for free‐text terms referring to molecular analysis. We manually reviewed the discovered pathology reports and identified n = 187 histiocytosis patients (n = 100 adults; n = 87 children) from whom tissue samples affected by their histiocytic disorder had been analysed for genetic alterations (Figure 1C), including n = 109 patients (n = 51 adults; n = 58 children) with a successfully detected genetic alteration. By manually reviewing all pathology reports of the 187 molecularly analysed patients, we discovered n = 9 patients with an additional (histologically confirmed) haematological malignancy (Table 1). Notably, these patients were all adults. Among them, four patients had presentation of the histiocytic neoplasm and additional haematological malignancy (all lymphomas) in the same tissue specimen (Cases 6–9); mutations were detected in the tissue specimen of Case 9. The resected subcutaneous tumour of this patient, who was known to have chronic lymphocytic leukaemia (CLL), consisted of a proliferation of spindle cells intermingled with many small lymphocytes and was classified as mixed IDCS and small lymphocytic lymphoma (SLL)/CLL. As microdissection of (relatively) pure spindle or small lymphocytic cell populations could not be performed, the existence of shared or unique genetic alterations in the IDCS or SLL/CLL of this patient remains elusive. In addition, no evidence for shared or unique genetic alterations was obtained in a patient with LCH and myelodysplastic syndrome (Case 5), since BRAF mutation analysis of the LCH specimen was negative. In the remaining four cases, a unique BCL2 translocation was identified in the follicular lymphoma (FL) and high‐grade B‐cell lymphoma of a patient who also developed HS without a BCL2 translocation (Case 4). These molecular findings contradict the transformation of FL into HS in this case, which has been reported previously in other patients [17, 18, 38, 39]. Finally, shared genetic alterations were identified in the remaining three patients (Cases 1–3), who presented with different histiocytic neoplasms and acute or chronic myeloid leukaemia harbouring identical oncogenic RAS mutations. The molecular analyses had been performed at three University Medical Centres using targeted next‐generation sequencing (NGS), and were executed as part of routine molecular diagnostics and according to conventional molecular diagnostic laboratory standards for variant allele detection and reporting. DNA samples were isolated from haematoxylin stained tissue slides, except for the acute myeloid leukaemia (AML) and germline DNA samples of Case 3, which were isolated from a 6 ml EDTA bone marrow aspirate and saliva, respectively. Detailed information regarding the applied NGS panels is provided in the supplementary material (Figure S1; Cases 1–3).
Table 1.
All identified histiocytosis patients with an additional haematological malignancy who were analysed for genetic alterations.
| Case | Age * (years) | Histiocytic disorder | Additional haematological malignancy † | Histiocytic disorder specimen(s) | Additional haematological malignancy specimen(s) | Interval | Genetic alteration(s) detected | Shared or unique genetic alteration(s) detected |
|---|---|---|---|---|---|---|---|---|
| 1 | 47 | HS | CMML | Bone marrow; skin | Bone marrow | CMML diagnosed 1 month after HS | Using NGS, mutations were detected in KRAS (p.A59E, VAF 34%) MAP2K1 (p.F53L, VAF 3.8%) and RAF1 (p.S257L, VAF 7.9%) in the HS bone marrow specimen, and in KRAS (p.A59E, VAF 42%), MAP2K1 (p.F53L, VAF 2%) and RAF1 (p.S257L, VAF 0.7%) in the mixed CMML/HS bone marrow specimen | Yes: a shared KRAS mutation, and (presumably) unique MAP2K1 and RAF1 mutations (HS) |
| 2 | 68 | ICH | CMML | Skin | Bone marrow | Diagnosed in the same month | Using NGS, a mutation was detected in NRAS (p.G12V) in both the ICH (VAF 20%) and CMML (VAF 42%) specimen | Yes: a shared NRAS mutation |
| 3 | 61 | ECD | MM and AML | Bone marrow; left femur; left tibia; skin | Bone marrow (MM); bone marrow (AML) | ECD and MM synchronous; AML diagnosed 2 years after ECD/MM | Using NGS, a mutation was detected in NRAS (p.Q61R) in the mixed MM/ECD bone marrow specimen (VAF not reliable), the ECD left tibia and skin specimens (VAF 37% in both samples), and the mixed AML/ECD bone marrow specimen (VAF 44%) | Yes: a shared NRAS mutation (ECD and AML) ‡ |
| 4 | 59 | HS | FL and HGBL | Soft tissue right shoulder | Lymph nodes and bone marrow (FL); mediastinal tumour and bone marrow (HGBL) | HS diagnosed 4 months after FL; HGBL diagnosed 8 months after FL |
No BCL2 translocation (HS); BCL2 translocation (FL); BCL2, BCL6 and MYC translocations (HGBL) |
Yes: a unique BCL2 translocation (FL and HGBL) |
| 5 | 81 | LCH | MDS | Skin; inguinal lymph node | Bone marrow | LCH diagnosed 4.5 years after MDS | No mutations detected in BRAF exon 15 using HRM (LCH) | No |
| 6 | 44 | LCH | FL | Right inguinal lymph node (same specimen) | Right inguinal lymph node | Synchronous | No BCL2 translocation | No |
| 7 | 40 | LCH | HL | Right cervical lymph node (same specimen) | Right cervical lymph node | Synchronous |
No mutations detected using NGS (incl. BRAF exon 15, MAP2K1 exon 2–3, NRAS exon 2–4 and KRAS exon 2–4) |
No |
| 8 | 45 | LCH | HL | Right inguinal lymph node (same specimen); left cervical lymph node (same specimen) | Right inguinal lymph node; left cervical lymph node (relapse) | Synchronous |
No mutations detected in both specimens using NGS (incl. BRAF exon 15, NRAS exon 2–4 and KRAS exon 2–4) |
No |
| 9 | 83 | IDCS | SLL/CLL | Soft tissue right upper arm (same specimen) | Soft tissue right upper arm | Synchronous | No BCL2, BCL6 or MYC translocation(s); using NGS, mutations were detected in MET (p.E1017K, VUS, VAF 8%), TP53 (p.R248W, VAF 35%) and SF3B1 (p.K700E, VAF 29%) | No |
Abbreviations: M, male; F, female; CMML, chronic myelomonocytic leukaemia; MM, multiple myeloma; HGBL, high‐grade B‐cell lymphoma; MDS, myelodysplastic syndrome; HL, Hodgkin lymphoma; SLL/CLL, small lymphocytic lymphoma/chronic lymphocytic leukaemia; NGS, next‐generation sequencing; HRM, high resolution melt analysis; VUS, variant of unknown significance.
Age at diagnosis of the first presenting disorder.
Excluding histiocytosis patients with an additional histiocytic malignancy, such as Langerhans cell sarcoma (LCS), as the histiocytic neoplasms are a spectrum of diseases, with regularly mixed histiocytosis (e.g. LCH/ECD or LCH/LCS).
As the VAF of the NRAS p.Q61R mutation in the mixed MM/ECD bone marrow specimen was unreliable due to poor DNA quality resulting in low number of reads, it could not be established whether the multiple myeloma also harboured the NRAS p.Q61R mutation.
Via the ‘PALGA intermediary procedure’, the pathology laboratories where the histopathological analyses of the tissue samples of Cases 1–3 were originally performed were identified, while complying with EU privacy laws and regulations. This enabled us to retrieve the pseudonymised tissue slides and blocks for central pathology review and additional immunohistochemical investigations. In addition, the pathology laboratories could refer us to the treating physicians of the patients, who subsequently provided us with pseudonymised clinical data and images. This study was approved by the PALGA Scientific Council and Privacy Committee (LZV‐2016‐183) and the Institutional Review Board of the Leiden University Medical Center (B19.074).
Results
Case 1 is a 47‐year‐old male who presented with fatigue, weight loss (18 kg), night sweats and left upper abdominal pain. Complete blood count showed anaemia, thrombocytopenia and monocytosis (2.1 × 109/l monocytes). A PET‐CT scan revealed fluorodeoxyglucose (FDG)‐avid bone lesions and enlarged cervical lymph nodes, as well as extreme splenomegaly (29.5 cm) with diffuse moderately increased metabolism (Figure 2A,B). In addition, FDG‐avid pre‐auricular and scalp skin lesions were noted (Figure 2C,D). A bone marrow biopsy revealed diffuse fibrosis and extensive infiltration by atypical CD163+ CD68+ CD56+ CD1a− histiocytes with a high (80%) Ki67 proliferation index (Figure 3A). A diagnosis of HS was made, and the patient was referred to a University Medical Centre for treatment of this rare disease. Using NGS, KRAS p.A59E (variant allele frequency [VAF] 34%), MAP2K1 p.F53L (VAF 3.8%) and RAF1 p.S257L (VAF 7.9%) mutations were detected in the bone marrow biopsy (Table 2). A pre‐auricular skin biopsy (Figure 2D) and second bone marrow biopsy were taken before start of chemotherapy. While the skin biopsy showed a diffuse dermal proliferation of CD56+ histiocytic cells, confirming HS involvement, the second bone marrow biopsy revealed myelodysplastic/myeloproliferative neoplasia characterised by left‐shifted myelopoiesis, dysplastic features of the erythroid, neutrophilic and megakaryocytic lineages, and MF2‐MF3 fibrosis. CD34 staining showed no increased blast count, whereas CD117 staining revealed 5–10% clustered myeloid precursors and myeloperoxidase (MPO) stained the majority of cells in the bone marrow (Figure 3A). A diagnosis of concomitant chronic myelomonocytic leukaemia (CMML)‐0 was made. A few scattered clusters of atypical (CD163+ CD14+) CD56+ MPO− histiocytes were also observed in the second bone marrow biopsy (Figure 3A), indicative of a small focus of HS. Using NGS, the same KRAS p.A59E mutation (VAF 42%) was detected in this CMML/HS‐affected second bone marrow biopsy (Table 2). The high VAF provides evidence for the presence of the KRAS mutation in the CMML. Interestingly, the RAF1 p.S257L and MAP2K1 p.F53L mutations were only detected at very low frequencies (VAF 0.7 and 2%, respectively), indicating that these mutations were probably HS‐specific. The patient was treated with high‐dose Cytarabine and Mitoxantrone chemotherapy (Cytarabine twice daily 1000 mg/m2, day 1–6; Mitoxantrone 10 mg/m2, day 5–7; one cycle completed), but developed a neutropenic enterocolitis and died due to septic/hyperinflammatory shock 62 days after HS diagnosis.
Figure 2.
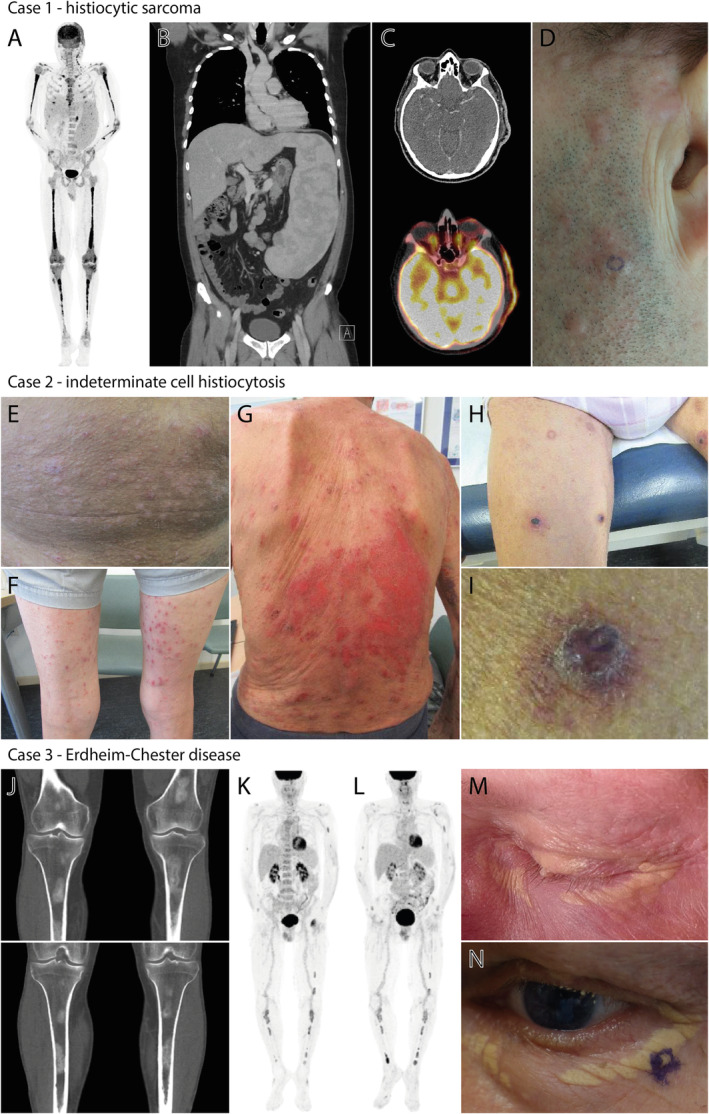
Clinical and radiological features of the patients described in this study. (A,B) Images of the PET‐CT scan made at diagnosis, showing FDG‐avid bone lesions and enlarged cervical lymph nodes, as well as extreme splenomegaly (29.5 cm) with diffuse moderately increased metabolism. (C,D) PET‐CT and clinical images of the pre‐auricular skin lesions. The localisation of the skin biopsy confirming the involvement of HS is encircled in panel (D). (E,F) Skin lesions on the abdomen and lower extremities at diagnosis. (G) Evidently progressed skin lesions at 7 months after initial diagnosis. (H–I) Recurrent skin lesions after treatment with topical corticosteroids and UV‐B phototherapy, with an altered phenotype of typical purple‐red papules. The localisation of the biopsy of one of these papules on the left upper leg is shown in panel (I). (J) Images of the CT‐scan performed at diagnosis, showing bilateral sclerotic femur and tibia lesions. (K) PET‐CT scan showing FDG uptake of ECD‐associated bone lesions after chemotherapy for the patient's multiple myeloma. (L) PET‐CT scan showing slightly increased FDG uptake of existing ECD bone lesions at diagnosis of acute myeloid leukaemia. (M) Peri‐orbital xanthelasma‐like lesions before myeloma‐directed chemotherapy. (N) Peri‐orbital xanthelasma‐like lesions after myeloma‐directed chemotherapy and autologous haematopoietic stem cell transplantation. The localisation of the skin biopsy confirming involvement of ECD is encircled.
Figure 3.
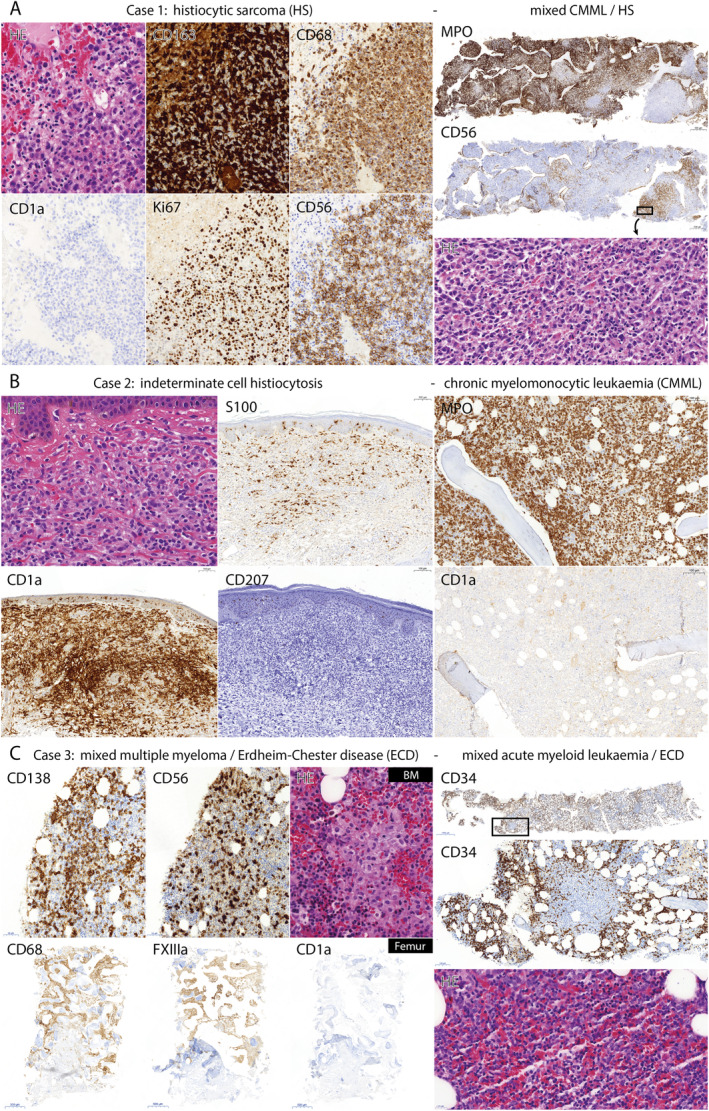
Histopathological features of tissue samples taken from the patients described in this study. (A) Case 1. Photomicrographs of the H&E (×40) and CD163, CD68, CD1a, Ki67 and CD56 (×20) stained first bone marrow biopsy (left), and the MPO, CD56 (×2) and H&E (×40) stained second bone marrow biopsy (right). The first bone marrow biopsy revealed extensive infiltration by atypical CD163+ CD68+ CD56+ CD1a− histiocytes with a high (80%) Ki67 proliferation index, consistent with a diagnosis of HS. The second bone marrow biopsy showed myelodysplastic/myeloproliferative neoplasia, as illustrated by abundant MPO+ cells, supporting a diagnosis of CMML. A few scattered clusters of CD56+ MPO− atypical histiocytic cells were also observed in the second bone marrow biopsy (depicted in the H&E panel), indicative of a small focus of HS. (B) Case 2. Photomicrographs of the H&E (×40), and S100, CD1a and CD207 (×10) stained skin biopsy (left), and the MPO and CD1a (×10) stained bone marrow biopsy (right). The skin biopsy revealed a dermal proliferation of CD1a and (partly) S100 positive histiocytes with overt CD207 negativity, consistent with a diagnosis of ICH. The bone marrow biopsy showed no infiltrating (CD1a+) histiocytes, but displayed a hypercellular bone marrow with substantial expansion of myelopoiesis, as illustrated by abundant MPO+ cells, supporting a diagnosis of synchronous CMML. (C) Case 3. Photomicrographs of the CD138, CD56 (×20) and H&E (×40) stained first bone marrow (BM) biopsy, and the CD68, FXIIIa and CD1a (×2) stained left femur biopsy (both left), as well as the CD34 (×1 and ×10) and H&E (×40) stained bone marrow biopsy taken after 3% of leukocytes in the peripheral blood that appeared as leukemic blasts were detected at a routine follow‐up consultation (right). In the first bone marrow biopsy (left), 10–20% CD138+ CD56+ plasma cells were observed, supporting a diagnosis of MM. In addition, some clusters of histiocytic cells were recognised (depicted in the H&E panel), indicative of a small focus of the soon thereafter diagnosed ECD. The left femur biopsy showed widespread CD68+ FXIIIa+ CD1a− foamy histiocytes, supporting the clinical and radiological diagnosis of ECD. The bone marrow biopsy taken after blasts were detected in the peripheral blood at a routine follow‐up consultation (right) revealed 30% of CD34+ leukaemic blasts, consistent with a diagnosis of acute myeloid leukaemia (AML). Like in the first (MM/ECD‐affected) bone marrow biopsy, a few clusters of histiocytic cells were present in this AML‐affected bone marrow biopsy (depicted in the H&E panel), again indicative of a small focus of ECD.
Table 2.
Mutations detected in the histiocytic neoplasms and additional myeloid leukaemias of the three patients presented in this study.
| Case | Material | Diagnosis | Day | Method | Mutation(s) | VAF |
|---|---|---|---|---|---|---|
| 1 | First bone marrow biopsy | HS | 0 | NGS CHPv2.0 | KRAS p.A59E | 30% |
| 1 | First bone marrow biopsy | HS | 0 | NGS PATHv2D |
KRAS p.A59E MAP2K1 p.F53L RAF1 p.S257L |
34% 3.8% 7.9% |
| 1 | Second bone marrow biopsy | CMML/HS | 38 | NGS CHPv2.0 | KRAS p.A59E | 40% |
| 1 | Second bone marrow biopsy | CMML/HS | 38 | NGS PATHv2D |
KRAS p.A59E MAP2K1 p.F53L RAF1 p.S257L |
42% 2% 0.7% |
| 2 | Skin biopsy | ICH | 0 | NGS OPv3.0 | NRAS p.G12V | 20% |
| 2 | Bone marrow biopsy | CMML | 21 | NGS OPv3.0 | NRAS p.G12V | 42% |
| 3 | First bone marrow biopsy | MM/ECD | 0 |
1. NGS DPv5.0 2. NGS DPv5.0 3. Sanger sequencing |
NRAS p.Q61R |
1. 69%* 2. 18%* 3. N/A |
| 3 | Left tibia biopsy | ECD | 175 | NGS DPv5.1 | NRAS p.Q61R | 37% |
| 3 | Skin biopsy | ECD | 479 | NGS DPv5.1 | NRAS p.Q61R | 37% |
| 3 | Bone marrow aspirate | AML/ECD | 836 | NGS Illumina TruSight Myeloid | NRAS p.Q61R | 44% |
Abbreviation: N/A, not available.
VAF is unreliable due to poor DNA quality resulting in low number of reads.
Case 2 is a 68‐year‐old male who presented with pruritic skin lesions on the trunk and extremities that had existed for 3 months (Figure 2E,F). A skin biopsy from the lower right abdomen (Figure 2E) revealed a dermal proliferation of CD1a and (partly) S100 positive histiocytes (Figure 3B) with low (5%) MIB‐1 expression. Notably, eosinophilic granulocytes were not observed. A PET‐CT scan showed no FDG‐avid lesions (images not shown), and a diagnosis of cutaneous LCH was made clinically. However, additional immunohistochemical investigations performed during central pathology review revealed overt CD207 (and MPO) negativity of the dermal histiocytes (Figure 3B), which led to an altered histopathological diagnosis of ICH. Using NGS, a NRAS p.G12V mutation (VAF 20%) was detected in the skin biopsy (Table 2). Complete blood count revealed a mild anaemia, thrombocytopenia and monocytosis, which retrospectively had been present for 7 months (1.9 × 109/l monocytes). A bone marrow biopsy showed no infiltrating (CD1a+) histiocytes, but revealed a hypercellular bone marrow with substantial expansion of myelopoiesis, as illustrated by abundant MPO+ cells (Figure 3B). A diagnosis of concomitant CMML‐0 was made. The same NRAS p.G12V mutation (VAF 42%) was detected in the bone marrow biopsy (Table 2), while additional NGS performed on DNA isolated from unaffected tissue of the patient excluded a germline NRAS mutation. Due to stable, asymptomatic CMML but increasing pruritic ICH skin lesions (Figure 2G), topical corticosteroids and UV‐B phototherapy for the ICH and an active monitoring strategy for the CMML were initiated. Despite initial adequate response of the skin lesions, they recurred as typical purple‐red papules (Figure 2H,I). A skin biopsy of one of these papules (Figure 2I) again showed a dermal infiltrate of CD1a+ CD207− MPO− histiocytes (lacking eosinophilic infiltration), but now with high (50–60%) MIB‐1 expression. Renewed staging by PET‐CT (images not shown) showed FDG‐avid thickened skin at the right lower leg and novel splenomegaly (16 cm). In addition, thickening of the right proximal‐mid ureter wall was observed. Soon thereafter, the patient was diagnosed with an invasive urothelial carcinoma (2 years after ICH diagnosis), and died 9 days after laparoscopic nephro‐ureterectomy due to acute respiratory failure after developing a hospital‐acquired pneumonia.
Case 3 is a 61‐year‐old male who was referred due to fatigue with underlying mild anaemia. A diagnosis of multiple myeloma (MM) International Staging System (ISS) stage 2 was made based on a kappa to lambda free light chain (FLC) ratio of 122.8 in the peripheral blood and 10–20% CD138+ CD56+ kappa‐monoclonal plasma cells in a bone marrow biopsy (Figure 3C). Notably, some clusters of histiocytic cells were also observed in the bone marrow biopsy (Figure 3C). A CT scan showed no osteolytic lesions in the context of MM, but bilateral sclerotic femur and tibia lesions were clearly observed (Figure 2J). A PET‐CT scan revealed FDG‐avid lesions in both femurs, both tibiae, both fibulae, the right ilium and both humeri. Biopsies from sclerotic lesions in the left femur and left tibia showed no signs of MM, but both specimens displayed widespread proliferation of CD68+ FXIIIa+ CD1a− (S100−) histiocytic cells (Figure 3C), supportive of a diagnosis of concomitant ECD. Of note, the patient also had bilateral xanthelasma‐like lesions (Figure 2M), which were now suspected also to be ECD manifestations. NGS performed on DNA isolated from the mixed MM/ECD bone marrow and ECD left tibia biopsies revealed a NRAS p.Q61R mutation in both samples (Table 2), which was confirmed in the MM/ECD bone marrow biopsy by Sanger sequencing (see supplementary material, Case 3). Myeloma treatment consisted of four courses of Bortezomib (1.3 mg/m2; day 1,4,8,11), Thalidomide (100 mg/day; day 1–21 of every 4 weeks) and Dexamethasone (40 mg/day; day 1, 2, 4, 5, 8, 9, 11, 12) and the patient achieved a very good partial response [40] (FLC kappa 27.70 mg/l; FLC ratio 1.45). However, PET‐CT still showed unchanged FDG uptake and sclerosis of the ECD bone lesions (Figure 2K). The patient underwent autologous haematopoietic stem cell transplantation after high dose Melphalan conditioning (200 mg/m2; day −2), and received Lenalidomide as post‐remission myeloma‐directed therapy. The bilateral xanthelasma‐like lesions persisted, and a skin biopsy (Figure 2N) confirmed ECD involvement with again the same NRAS p.Q61R mutation (VAF 37%; Table 2). At a routine follow‐up consultation, 13 months after autologous haematopoietic stem cell transplantation, 3% of leukocytes in the peripheral blood appeared to be leukaemic blasts. The patient underwent bone marrow examination and a diagnosis of AML‐M5 (CD34, CD13, HLA‐DR and MPO positive; CD19, CD10, CD3, CD4, CD5 and CD8 negative) was made based on the presence of 30% of CD34+ leukaemic blasts in a bone marrow biopsy (Figure 3C). As in the earlier collected MM/ECD‐affected bone marrow biopsy, a few clusters of histiocytic cells were present in this AML‐affected bone marrow biopsy (Figure 3C), indicative of a small focus of ECD. A PET‐CT scan showed no extramedullary AML lesions, but revealed slightly increased FDG uptake of existing ECD lesions (Figure 2L). While NGS revealed no recurrent cytogenetic or molecular abnormalities associated with AML in the AML/ECD‐affected bone marrow sample (see supplementary material, Case 3), the same NRAS p.Q61R mutation (VAF 44%) as was previously detected in the ECD lesions was detected in this bone marrow specimen (Table 2). Importantly, the high VAF provides evidence for the presence of the NRAS mutation in AML blasts. Moreover, the NRAS p.Q61R mutation was not detected by NGS in the patient's germline DNA. The patient was treated for his AML with 7 + 3 chemotherapy of Cytarabine (200 mg/m2; day 1–7) and Idarubicine (12 mg/m2; day 1–3), and reached complete remission after one course of induction chemotherapy. Before the second course, he presented however with altered vision of the left eye, which was found to be caused by an invasive Aspergillus fumigatus infection in the left sphenoid sinus that was unresponsive to extensive anti‐fungal treatment and quickly led to cranial nerve II, III, IV, V1 and VI palsies. Eventually, the patient went home with palliative care and died shortly after.
Discussion
In this retrospective population‐based study, we report three patients with different histiocytic neoplasms and additional haematological malignancies bearing identical oncogenic mutations. All patients were diagnosed and treated in different non‐academic as well as tertiary referral hospitals across the Netherlands, emphasising the benefit of our study design based on the nationwide Dutch Pathology Registry. These patients add to the growing list of reported patients with a histiocytic neoplasm and additional haematological cancer bearing the same genetic alteration(s) (Table 3; see supplementary material, Tables S2 and S3). Of note, all three patients had typical histiocytic disease presentations, including bilateral osteosclerotic lesions of the long bones in the patient with ECD (Case 3). This is in contrast to three recently reported ECD‐CMML patients with only xanthelasma‐like lesions [30], which are not specific for ECD [31].
Table 3.
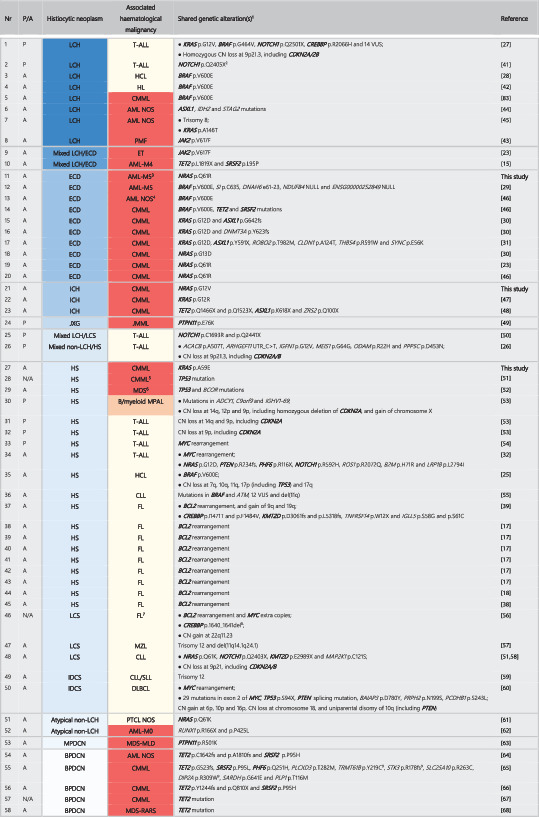
Overview of reported patients with histiocytic neoplasms and additional haematological malignancies bearing the same genetic alteration(s) as evidenced by DNA sequencing and/or DNA methylation profiling.
|
Abbreviations: P, paediatric; A, adult; N/A, not available; LCS, Langerhans cell sarcoma; non‐LCH, non‐Langerhans cell histiocytosis; NOS, not otherwise specified; MPDCN, mature plasmacytoid dendritic cell neoplasm; T‐ALL, T‐cell acute lymphoblastic leukaemia; HCL, hairy cell leukaemia; PMF, primary myelofibrosis; ET, essential thrombocytosis; JMML, juvenile myelomonocytic leukaemia; MPAL, mixed phenotype acute leukaemia; MZL, marginal zone lymphoma; DLBCL, diffuse large B‐cell lymphoma; PTCL, peripheral T‐cell lymphoma; MLD, multilineage dysplasia; RARS, refractory anaemia with ring sideroblasts.
1Genes mutated, deleted and/or translocated in two or more patients are depicted in bold.
2A NRAS p.G12S mutation and homozygous deletion at 9p21, including CDKN2A, was also detected in the T‐ALL sample using WES and SNP array analysis (Kato M, et al. Br J Haematol 2016). LCH specimens were not available for these analyses.
3The patient was initially diagnosed with concurrent ECD and multiple myeloma. Molecular analysis of the multiple myeloma was precluded by the absence of cryopreserved material.
4AML with at least phenotypic monocytic differentiation.
5The patient also had a mediastinal germ cell tumour (MGCT). The existence of a common precursor was suggested by the demonstration of the same TP53 mutation in all three neoplasms and identical chromosomal aberrations in the HS and the MGCT.
6The patient had a history of a metastatic non‐seminomatous germ cell tumour with yolk sac component (NSGCT) with identical mutations in TP53 and BCOR, along with isochromosome 12p (shared with the MDS) and a unique mutation in RRAS2.
7The patient also developed DLBCL seven years before and several months after LCS diagnosis. Molecular analysis of DLBCL specimens was precluded by the absence of material.
8In addition, 11 variants that are presumably germline polymorphisms were detected.
9These mutations are depicted as such in Figure 2A of the manuscript (Patnaik MM, et al. Blood Cancer J 2018), but other mutations in TRMT61B (p.T219C), STK3 (p.N207fs) and DIP2A (p.R373W) are described in the main text of the article.
As routine molecular analysis of (specific) histiocytic disorders has only rather recently found its way into clinical practice, the three patients described in this study may comprise only the tip of the iceberg of all histiocytosis patients in our PALGA cohort (1971–2019) with an additional haematological malignancy bearing the same genetic alteration(s). Accordingly, all three patients were diagnosed with their first presenting disorder from 2017 onwards. In addition, this study is limited by the fact that PALGA diagnostic terms do not exist for all histiocytic disorders, such as ICH, and that pathologists may sometimes assign incorrect or non‐specific coding terms to relevant pathology reports. As a consequence, this study may not include all patients with a histologically confirmed histiocytic disorder. Finally, haematological malignancies are not always histologically confirmed with a trephine biopsy, but may also be diagnosed based on laboratory tests, cytological evaluation and/or flow cytometric analysis alone. Thus, not all haematological malignancies of the patients from our study may have been registered by PALGA. Despite these limitations, we could still identify three patients with different histiocytic neoplasms associated with diverse haematological malignancies bearing identical oncogenic mutations, underscoring the rare but indefinite occurrence of this phenomenon across the spectrum of histiocytic neoplasms.
Importantly, the detected variant allele frequencies point out that the identified RAS mutations were present in both the specified histiocytic neoplasms and myeloid leukaemias, suggesting a common clonal origin. Nevertheless, we acknowledge the lack of flow sorted cells to unequivocally demonstrate the phenotype of the mutated cells [31]. Notably, we could exclude the possibility of a germline RAS variant in Cases 2 and 3. For Case 1, the probability of a germline variant is very low, considering the VAF of 30–42% of the KRAS mutation that was detected in four separate NGS analyses (Table 2). Unfortunately, we could not establish whether the MM of Case 3 harboured the same NRAS mutation as the ECD and AML of this patient due to the absence of cryopreserved cells. Yet, other studies have already demonstrated shared genetic alterations in histiocytic neoplasms associated with various lymphoid neoplasms (Table 3), including MM (see supplementary material, Table S3) [51]. Putative mechanisms for this phenomenon include the existence of a common HSPC, and dedifferentiation or transdifferentation of lineage‐committed haematopoietic cells [69]. In accordance with a previous observation in LCH [19], lymphoid neoplasms often preceded the histiocytic neoplasm in the reported cases (see supplementary material, Tables S2 and S3; n = 45/57), whereas myeloid neoplasms were more frequently diagnosed concurrently (n = 7/34) or after the diagnosis of the histiocytic neoplasm (n = 14/34). In addition, interesting differences in association patterns between the different histiocytic neoplasms were recognisable in our comprehensive overview of published cases (Table 3; see supplementary material, Tables S2 and S3), with ECD only being associated with myeloid leukaemia (n = 10/10) and malignant histiocytoses often associated with lymphoid neoplasms, and particularly FL (n = 20/52).
In addition to the development of clonally related histiocytic neoplasms and lymphoid or myeloid malignancies through trans‐/dedifferentiation or from a common haematopoietic precursor cell or clone, alternative mechanisms may explain the high prevalence of haematological malignancies in (especially adult) histiocytosis patients. A multi‐institutional cohort study of adult patients with ECD or mixed histiocytosis by Papo et al demonstrated that, in 10/12 patients with an additional myeloid malignancy that were molecularly characterised using NGS, the histiocytic neoplasm and myeloid malignancy harboured distinct kinase mutations [23]. These findings suggest that the two neoplasms were not clonally related and arose at different stages of haematopoietic development in these patients. Interestingly, the histiocytosis patients with an additional myeloid neoplasm were significantly older at ECD diagnosis than the histiocytosis patients without an additional myeloid neoplasm. Therefore, the authors hypothesised that the high prevalence of (seemingly unrelated) myeloid malignancies in (particularly older) adult histiocytosis patients could be due to the well‐described association between aging and clonal hematopoiesis (CH) [70], given that CH is known to be associated with increased frequency of myeloid neoplasms because of acquisition of somatic mutations in genes commonly mutated in myeloid neoplasms. Accordingly, prospective analysis of bone marrow samples by NGS revealed that almost half of adult ECD patients harbour mutations in myeloid cancer‐ and CH‐associated genes [71, 72], such as TET2. These mutations may represent early events in the development of ECD and of additional myeloid malignancies in ECD patients. In this hypothetical model, the histiocytic neoplasm and additional myeloid malignancy may independently derive from a different CH subclone, although development from a shared (e.g. TET2 mutated [15, 72]) CH (sub)clone is also possible.
Unfortunately, all three patients described in this manuscript died with active (histiocytic) disease, despite treatment in tertiary referral hospitals according to current standard‐of‐care protocols. Similarly, many of the previously reported patients with a histiocytic neoplasm and additional haematological cancer bearing the same genetic alteration(s) died within several months or a few years after diagnosis of the last presenting disorder (see supplementary material, Tables S2 and S3). Collectively, these observations underscore the poor prognosis for such patients. Novel therapeutic options are thus urgently needed. Recent studies have demonstrated safety and remarkable efficacy of BRAF [73, 74, 75, 76, 77, 78] and MEK [78, 79] inhibition in patients with BRAF‐mutated or BRAF wildtype histiocytic neoplasms, respectively. Moreover, dramatic responses to selective inhibition of RET (Selpercatinib) or ALK (Crizotinib) have been observed in two patients with histiocytic neoplasms driven by rare rearrangements involving one of these genes [10], and high response rates have been described in ECD patients treated with the mTOR inhibitor Sirolimus [80]. Although there is a well‐described risk of paradoxical activation of ERK signalling in (pre)malignant cells bearing MAPK pathway mutations other than BRAF V600E upon BRAF inhibition [81, 82], the use of targeted therapeutics in patients with histiocytoses and additional haematological malignancies bearing the same kinase alterations may result in a beneficial response across both conditions [23]. Accordingly, one patient with multi‐organ LCH and CMML‐1 harbouring the same BRAF p.V600E mutation was successfully treated with the BRAF inhibitor Vemurafenib [83]. Furthermore, one patient with NRAS p.Q61R mutated ECD and CMML‐1 who was treated with Trametinib – a MEK inhibitor – has been reported by Papo et al [23]; they showed a complete metabolic response of the ECD by PET CT after 2 months, in addition to improved monocyte and platelet counts.
Shared RAS mutations were found in some other previously reported cases with a histiocytic neoplasm and haematological malignancy bearing the same genetic alteration(s) as well (Table 3), and were primarily detected in histiocytic neoplasms associated with myeloid leukaemia (n = 11/15). In contrast, shared NOTCH1 mutations or shared CDKN2A deletions were almost exclusively present in patients with histiocytic neoplasms associated with T‐cell acute lymphoblastic leukaemia (n = 4/5 and n = 5/8, respectively; see supplementary material, Tables S2 and S3). As expected, shared BCL2 rearrangements were frequently detected in patients with malignant histiocytoses associated with FL (n = 19/20; see supplementary material, Tables S2 and S3), and shared TET2 mutations were abundant in BPDCN associated with additional myeloid malignancies (n = 5/5), as these are the most prevalent genetic alterations in FL [84] and BPDCN [85], respectively. Although limited by their descriptive nature, these distinct distribution patterns suggest that mutations in particular genes may specifically predispose to the development of an additional clonally related haematological malignancy or secondary histiocytic neoplasm.
In addition to the shared genetic alteration(s), another interesting finding is the additional somatic mutations detected only in the secondary neoplasm. These mutations may have contributed to the divergent lineage differentiation of the common HSPC, or the dedifferentiation or lineage switch of the primary lymphoid [17, 69] or histiocytic [58, 61, 86] neoplasm. For example, in Case 1, the MAP2K1 and RAF1 mutations were (presumably) only present in the HS and not in the CMML, suggesting that these mutations may have contributed to the development of the HS from a common KRAS mutated HSPC or primary CMML clone. Similar situations of one or more shared genetic alterations and additional unique mutations in the histiocytic neoplasm and/or associated haematological malignancy have been reported in other cases [15, 18, 23, 25, 26, 27, 29, 31, 32, 39, 41, 44, 45, 47, 52, 53, 54, 55, 56, 57, 59, 60, 63, 64, 65, 66, 67, 87, 88, 89, 90, 91, 92, 93, 94, 95]. The histiocytic neoplasms often harboured unique mutations in genes encoding proteins of the MAPK signalling pathway, including NRAS [52, 55], KRAS [39, 53, 56], BRAF [15, 23, 44, 45, 54, 92, 94] and RAF1 [32], again demonstrating the importance of constitutive MAPK pathway activation in the pathogenesis of the histiocytic neoplasms [3, 96].
In conclusion, our data further support the existence of a common haematopoietic cell‐of‐origin in at least a proportion of patients with a histiocytic neoplasm and additional haematological malignancy, and emphasise the importance of adequate (bone marrow) staging, prospective molecular analysis and long‐term follow‐up of each histiocytosis patient. Future studies should investigate whether the shared genetic alterations can be traced in HSPCs, more downstream committed precursor cells and/or mature blood cells (e.g. classical monocytes of CMML). In addition, the temporal effect of particular driver mutations on the differentiation of such haematopoietic cells needs to be explored to further unravel the mechanisms underlying the co‐occurrence of histiocytic neoplasms and additional haematological malignancies.
Author contributions statement
HSL, BR, KH, JAML, PM and MDL cared for the patients and provided clinical data. KMH, STP, RMV and KHL performed histopathological analyses and molecular testing of FFPE tissue specimens. PJMV performed molecular testing of peripheral blood, bone marrow and saliva samples. AHB helped in selecting the PALGA search strategy and assisted in obtaining the pathology specimens for histopathological review and additional analyses. PGK, PCWH and AGSH wrote the PALGA study protocol. PGK analysed the PALGA dataset. PCWH performed the central pathology review. AGSH supervised the study. PGK, PCWH and AGSH drafted the manuscript. PGK made the figures and tables. All authors contributed to the final version of the manuscript.
Supporting information
Figure S1. Coverage of various genes by the different NGS panels used to analyse the affected tissue specimens of the three cases presented in this study
Table S1. Available PALGA diagnostic terms for the various histiocytic disorders
Table S2. Reported patients with histiocytic neoplasms associated with additional haematological malignancies bearing the same genetic alteration(s) as demonstrated by DNA sequencing and/or DNA methylation profiling
Table S3. Reported patients with histiocytic neoplasms associated with additional haematological malignancies bearing the same genetic alteration(s) as demonstrated by techniques other than DNA sequencing and/or DNA methylation profiling
Case 1. Detailed sequencing data
Case 2. Detailed sequencing data
Case 3. Detailed sequencing data
Acknowledgements
We thank clinical molecular biologists in pathology L. Kroeze (Radboud UMC) and W.R.R. Geurts‐Giele (Erasmus MC), and research technician in molecular pathology J.A. Reinten (Amsterdam UMC) for NGS data collection and/or analysis. We thank pathologist F.H. van Nederveen (Laboratory for Pathology, Dordrecht, The Netherlands) for providing archived tissue samples. We thank the personnel of the immunolaboratory of the Department of Pathology of the LUMC for performing additional immunohistochemical investigations. This study was financially supported by structural research funding from Histiocytose Nederland and Stichting 1000 kaarsjes voor Juultje (Dr. Astrid G.S. van Halteren).
No conflicts of interest were declared.
Contributor Information
Paul G Kemps, Email: p.g.kemps@lumc.nl.
Astrid GS van Halteren, Email: a.g.s.van_halteren@lumc.nl.
References
- 1. Emile J‐F, Abla O, Fraitag S, et al Revised classification of histiocytoses and neoplasms of the macrophage‐dendritic cell lineages. Blood 2016; 127 : 2672–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Durham BH. Molecular characterization of the histiocytoses: neoplasia of dendritic cells and macrophages. Semin Cell Dev Biol 2019; 86 : 62–76. [DOI] [PubMed] [Google Scholar]
- 3. Badalian‐Very G, Vergilio J, Degar BA, et al Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010; 116 : 6–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nelson DS, Quispel W, Badalian‐Very G, et al Somatic activating ARAF mutations in Langerhans cell histiocytosis. Blood 2014; 123 : 3152–3155. [DOI] [PubMed] [Google Scholar]
- 5. Nelson DS, van Halteren A, Quispel WT, et al MAP2K1 and MAP3K1 mutations in langerhans cell histiocytosis. Genes Chromosomes Cancer 2015; 54 : 361–368. [DOI] [PubMed] [Google Scholar]
- 6. Emile J‐F, Diamond EL, Hélias‐Rodzewicz Z, et al Recurrent RAS and PIK3CA mutations in Erdheim–Chester disease. Blood 2014; 124 : 3016–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brown RA, Kwong BY, McCalmont TH, et al ETV3‐NCOA2 in indeterminate cell histiocytosis: clonal translocation supports sui generis. Blood 2015; 126 : 2344–2345. [DOI] [PubMed] [Google Scholar]
- 8. Shanmugam V, Griffin GK, Jacobsen ED, et al Identification of diverse activating mutations of the RAS‐MAPK pathway in histiocytic sarcoma. Mod Pathol 2019; 32 : 830–843. [DOI] [PubMed] [Google Scholar]
- 9. Egan C, Nicolae A, Lack J, et al Genomic profiling of primary histiocytic sarcoma reveals two molecular subgroups. Haematologica 2020; 105 : 951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Durham BH, Lopez Rodrigo E, Picarsic J, et al Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med 2019; 25 : 1839–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fletcher CDM, Bridge JA, Hogendoorn PCW, et al WHO Classification of Tumours of Soft Tissue and Bone, Volume 5 (4th edn). IARC, 2013. [Google Scholar]
- 12. Swerdlow S, Campo E, Harris N, et al WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Volume 2 (Revised 4th edn). IARC, 2017. [Google Scholar]
- 13. Berres M‐L, Lim KPH, Peters T, et al BRAF‐V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 2014; 211 : 669–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Milne P, Bigley V, Bacon CM, et al Hematopoietic origin of Langerhans cell histiocytosis and Erdheim–Chester disease in adults. Blood 2017; 130 : 167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Durham BH, Roos‐Weil D, Baillou C, et al Functional evidence for derivation of systemic histiocytic neoplasms from hematopoietic stem/progenitor cells. Blood 2017; 130 : 176–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xiao Y, van Halteren AG, Lei X, et al Bone marrow‐derived myeloid progenitors as driver mutation carriers in high‐ and low‐risk Langerhans cell histiocytosis. Blood 2020; 136: 2188–2199. [DOI] [PubMed] [Google Scholar]
- 17. Feldman AL, Arber DA, Pittaluga S, et al Clonally related follicular lymphomas and histiocytic/dendritic cell sarcomas: evidence for transdifferentiation of the follicular lymphoma clone. Blood 2008; 111 : 5433–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brunner P, Rufle A, Dirnhofer S, et al Follicular lymphoma transformation into histiocytic sarcoma: indications for a common neoplastic progenitor. Leukemia 2014; 28 : 1937–1940. [DOI] [PubMed] [Google Scholar]
- 19. Egeler RM, Neglia JP, Aricò M, et al Acute leukemia in association with Langerhans cell histiocytosis. Med Pediatr Oncol 1994; 23 : 81–85. [DOI] [PubMed] [Google Scholar]
- 20. Egeler RM, Neglia JP, Aricò M, et al The relation of Langerhans cell histiocytosis to acute leukemia, lymphomas, and other solid tumors. The LCH‐Malignancy Study Group of the Histiocyte Society. Hematol Oncol Clin North Am 1998; 12 : 369–378. [DOI] [PubMed] [Google Scholar]
- 21. Goyal G, Shah MV, Hook CC, et al Adult disseminated Langerhans cell histiocytosis: incidence, racial disparities and long‐term outcomes. Br J Haematol 2018; 182 : 579–581. [DOI] [PubMed] [Google Scholar]
- 22. Ma J, Laird JH, Chau KW, et al Langerhans cell histiocytosis in adults is associated with a high prevalence of hematologic and solid malignancies. Cancer Med 2019; 8 : 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Papo M, Diamond EL, Cohen‐Aubart F, et al High prevalence of myeloid neoplasms in adults with non‐Langerhans cell histiocytosis. Blood 2017; 130 : 1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kommalapati A, Tella SH, Durkin M, et al Histiocytic sarcoma: a population‐based analysis of incidence, demographic disparities, and long‐term outcomes. Blood 2018; 131 : 265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Michonneau D, Kaltenbach S, Derrieux C, et al BRAF V600E mutation in a histiocytic sarcoma arising from hairy cell leukemia. J Clin Oncol 2014; 32 : e117–e121. [DOI] [PubMed] [Google Scholar]
- 26. Waanders E, Hebeda KM, Kamping EJ, et al Independent development of lymphoid and histiocytic malignancies from a shared early precursor. Leukemia 2016; 30 : 955–958. [DOI] [PubMed] [Google Scholar]
- 27. Kato M, Seki M, Yoshida K, et al Genomic analysis of clonal origin of Langerhans cell histiocytosis following acute lymphoblastic leukaemia. Br J Haematol 2016; 175 : 169–172. [DOI] [PubMed] [Google Scholar]
- 28. Loghavi S, Khoury JD. Langerhans cell histiocytosis in a patient with hairy cell leukemia: a tale of divergence. Blood 2017; 129 : 1563. [DOI] [PubMed] [Google Scholar]
- 29. Ghobadi A, Miller CA, Li T, et al Shared cell of origin in a patient with Erdheim–Chester disease and acute myeloid leukemia. Haematologica 2019; 104 : e373–e375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bonnet P, Chasset F, Moguelet P, et al Erdheim–Chester disease associated with chronic myelomonocytic leukemia harboring the same clonal mutation. Haematologica 2019; 104 : e530–e533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Goyal G, Liu Y, Ravindran A, et al Concomitant Erdheim–Chester disease and chronic myelomonocytic leukaemia: genomic insights into a common clonal origin. Br J Haematol 2019; 187 : bjh.16177. [DOI] [PubMed] [Google Scholar]
- 32. Roloff GW, Baron JI, Neppalli VT, et al Next‐generation sequencing delineates clonal origins and informs therapeutic strategies in acute lymphoblastic leukemia and histiocytic sarcoma. JCO Precis Oncol 2019: 1–8. [DOI] [PubMed] [Google Scholar]
- 33. Casparie M, Tiebosch ATMG, Burger G, et al Pathology databanking and biobanking in The Netherlands, a central role for PALGA, the nationwide histopathology and cytopathology data network and archive. Cell Oncol 2007; 29 : 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Isaacson PG, O'Connor NT, Spencer J, et al Malignant histiocytosis of the intestine: a T‐cell lymphoma. Lancet 1985; 2 : 688–691. [DOI] [PubMed] [Google Scholar]
- 35. Van Der Valk P, Van Oostveen JW, Stel HV, et al Phenotypic and genotypic analysis of large‐cell lymphomas, formerly classified as true histiocytic lymphoma: identification of an unusual group of tumors. Leuk Res 1990; 14 : 337–346. [DOI] [PubMed] [Google Scholar]
- 36. Wilson MS, Weiss LM, Gatter KC, et al Malignant histiocytosis a reassessment of cases previously reported in 1975 based on paraffin section immunophenotyping studies. Cancer 1990; 66 : 530–536. [DOI] [PubMed] [Google Scholar]
- 37. Jaffe ES, Harris N, Stein H, et al WHO Classification of Tumours: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues, Volume 3 (3rd edn). IARC, 2001. [Google Scholar]
- 38. Zeng W, Meck J, Cheson BD, et al Histiocytic sarcoma transdifferentiated from follicular lymphoma presenting as a cutaneous tumor. J Cutan Pathol 2011; 38 : 999–1003. [DOI] [PubMed] [Google Scholar]
- 39. Péricart S, Waysse C, Siegfried A, et al Subsequent development of histiocytic sarcoma and follicular lymphoma: cytogenetics and next‐generation sequencing analyses provide evidence for transdifferentiation of early common lymphoid precursor – a case report and review of literature. Virchows Arch 2019; 476 : 609–614. [DOI] [PubMed] [Google Scholar]
- 40. Kumar S, Paiva B, Anderson KC, et al International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol 2016; 17 : e328–e346. [DOI] [PubMed] [Google Scholar]
- 41. Yokokawa Y, Taki T, Chinen Y, et al Unique clonal relationship between T‐cell acute lymphoblastic leukemia and subsequent Langerhans cell histiocytosis with TCR rearrangement and NOTCH1 mutation. Genes Chromosomes Cancer 2015; 54 : 409–417. [DOI] [PubMed] [Google Scholar]
- 42. Haefliger S, Bihl M, Krasniqi F, et al PET‐positive bone lesion due to Langerhans cell histiocytosis after BEACOPP therapy for Hodgkin lymphoma: how anamnesis, histopathological accuracy, and molecular analysis could resolve a clinical dilemma. Ann Hematol 2018; 97 : 355–357. [DOI] [PubMed] [Google Scholar]
- 43. Bonometti A, Bagnoli F, Fanoni D, et al JAK2‐mutated Langerhans cell histiocytosis associated with primary myelofibrosis treated with ruxolitinib. Hum Pathol 2018; 73 : 171–175. [DOI] [PubMed] [Google Scholar]
- 44. Khurana S, Sluzevich JC, He R, et al Association between high‐grade myelodysplastic syndrome and cutaneous Langerhans cell histiocytosis suggested by next‐generation sequencing. JAMA Dermatol 2020; 156: 817–819. [DOI] [PubMed] [Google Scholar]
- 45. Wang X, Wang Z. Revealing homologous clonality by synchronous trisomy 8 in Langerhans cell histiocytosis and acute myeloid leukemia. Blood 2019; 134 : 5040–5040. [Google Scholar]
- 46. Tzankov A, Kremer M, Leguit R, et al Histiocytic cell neoplasms involving the bone marrow: summary of the workshop cases submitted to the 18th Meeting of the European Association for Haematopathology (EAHP) organized by the European Bone Marrow Working Group, Basel 2016. Ann Hematol 2018; 97 : 2117–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Loghavi S, Curry JL, Garcia‐Manero G, et al Chronic myelomonocytic leukemia masquerading as cutaneous indeterminate dendritic cell tumor: expanding the spectrum of skin lesions in chronic myelomonocytic leukemia. J Cutan Pathol 2017; 44 : 1075–1079. [DOI] [PubMed] [Google Scholar]
- 48. Santos‐Briz A, Medina‐Miguelañez M, Moyano‐Bueno D, et al Indeterminate dendritic cell tumor as cutaneous involvement of chronic myelomonocytic leukemia successfully treated with phototherapy. Am J Dermatopathol 2020; 42: 876–880. [DOI] [PubMed] [Google Scholar]
- 49. Bátai B, Krizsán S, Gángó A, et al Juvenile myelomonocytic leukaemia presentation after preceding juvenile xanthogranuloma harbouring an identical somatic PTPN11 mutation, Pediatr Blood Cancer 2020: e28368. [DOI] [PubMed] [Google Scholar]
- 50. Rodig SJ, Payne EG, Degar BA, et al Aggressive Langerhans cell histiocytosis following T‐ALL: clonally related neoplasms with persistent expression of constitutively active NOTCH1. Am J Hematol 2008; 83 : 116–121. [DOI] [PubMed] [Google Scholar]
- 51. Facchetti F, Pileri SA, Lorenzi L, et al Histiocytic and dendritic cell neoplasms: what have we learnt by studying 67 cases. Virchows Arch 2017; 471 : 467–489. [DOI] [PubMed] [Google Scholar]
- 52. Tashkandi H, Dogan A. Histiocytic sarcoma arising in patient with history of clonally‐related germ cell tumour and myelodysplastic syndrome. Br J Haematol 2020; 188 : 482–482. [DOI] [PubMed] [Google Scholar]
- 53. Bleeke M, Johann P, Gröbner S, et al Genome‐wide analysis of acute leukemia and clonally related histiocytic sarcoma in a series of three pediatric patients. Pediatr Blood Cancer 2020; 67 : e28074. [DOI] [PubMed] [Google Scholar]
- 54. Venkataraman V, Massoth LR, Sullivan RJ, et al Secondary histiocytic sarcoma with BRAF V600E mutation after T‐cell acute lymphoblastic leukemia in a very young child with dramatic response to dabrafenib and trametinib. Pediatr Blood Cancer 2020; 67 : e28200. [DOI] [PubMed] [Google Scholar]
- 55. Burger JA, Landau DA, Taylor‐Weiner A, et al Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun 2016; 7 : 11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Choi SM, Andea AA, Wang M, et al KRAS mutation in secondary malignant histiocytosis arising from low grade follicular lymphoma. Diagn Pathol 2018; 13 : 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ambrosio MR, De Falco G, Rocca BJ, et al Langerhans cell sarcoma following marginal zone lymphoma: expanding the knowledge on mature B cell plasticity. Virchows Arch 2015; 467 : 471–480. [DOI] [PubMed] [Google Scholar]
- 58. Xerri L, Adélaïde J, Popovici C, et al CDKN2A/B deletion and double‐hit mutations of the MAPK pathway underlie the aggressive behavior of Langerhans cell tumors. Am J Surg Pathol 2018; 42 : 150–159. [DOI] [PubMed] [Google Scholar]
- 59. Fraser CR, Wang W, Gomez M, et al Transformation of chronic lymphocytic leukemia/small lymphocytic lymphoma to interdigitating dendritic cell sarcoma. Am J Clin Pathol 2009; 132 : 928–939. [DOI] [PubMed] [Google Scholar]
- 60. Ochi Y, Hiramoto N, Yoshizato T, et al Clonally related diffuse large B‐cell lymphoma and interdigitating dendritic cell sarcoma sharing MYC translocation. Haematologica 2018; 103 : e553–e556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Machan S, Córdoba R, Carvajal N, et al Atypical histiocytic lesion preceding a peripheral T‐cell lymphoma involving the skin exhibiting the same molecular alterations. Am J Dermatopathol 2019; 41 : 148–154. [DOI] [PubMed] [Google Scholar]
- 62. Al Mugairi A, Al Turki S, Salama H, et al Isolated bone marrow non‐Langerhans cell histiocytosis preceding RUNX1‐mutated acute myeloid leukemia: case report and literature review. Am J Clin Pathol 2019; 151 : 638–646. [DOI] [PubMed] [Google Scholar]
- 63. Bodmer A, Menter T, Juskevicius D, et al Sharing of a PTPN11 mutation by myelodysplastic bone marrow and a mature plasmacytoid dendritic cell proliferation provides evidence for their common myelomonocytic origin. Virchows Arch 2017; 470 : 469–473. [DOI] [PubMed] [Google Scholar]
- 64. Luskin MR, Kim AS, Patel SS, et al Evidence for separate transformation to acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasm from a shared ancestral hematopoietic clone. Leuk Lymphoma 2020; 61: 2258–2261. [DOI] [PubMed] [Google Scholar]
- 65. Patnaik MM, Lasho T, Howard M, et al Biallelic inactivation of the retinoblastoma gene results in transformation of chronic myelomonocytic leukemia to a blastic plasmacytoid dendritic cell neoplasm: shared clonal origins of two aggressive neoplasms. Blood Cancer J 2018; 8 : 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brunetti L, Di Battista V, Venanzi A, et al Blastic plasmacytoid dendritic cell neoplasm and chronic myelomonocytic leukemia: a shared clonal origin. Leukemia 2017; 31 : 1238–1240. [DOI] [PubMed] [Google Scholar]
- 67. Sukegawa S, Sakata‐Yanagimoto M, Matsuoka R, et al Blastic plasmacytoid dendritic cell neoplasm accompanied by chronic myelomonocytic leukemia successfully treated with azacitidine. Rinsho Ketsueki 2018; 59 : 2567–2573. [DOI] [PubMed] [Google Scholar]
- 68. Krause JR, Baugh L, Swink A, et al Blastic plasmacytoid dendritic cell neoplasm following acquired erythropoietic protoporphyria. Baylor Univ Med Cent Proc 2017; 30 : 450–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stoecker MM, Wang E. Histiocytic/dendritic cell transformation of B‐cell neoplasms: pathologic evidence of lineage conversion in differentiated Hematolymphoid malignancies. Arch Pathol Lab Med 2013; 137 : 865–870. [DOI] [PubMed] [Google Scholar]
- 70. Shlush LI. Age‐related clonal hematopoiesis. Blood 2018; 131 : 496–504. [DOI] [PubMed] [Google Scholar]
- 71. Haroche J, Poulain S, Marceau‐Renaut A, et al Clonal hematopoiesis in Erdheim–Chester disease. Blood 2017; 130 : 3788–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Papo M, Emile J‐F, Maciel TT, et al Erdheim–Chester disease: a concise review. Curr Rheumatol Rep 2019; 21 : 66. [DOI] [PubMed] [Google Scholar]
- 73. Haroche J, Cohen‐Aubart F, Emile J‐F, et al Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim–Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 2013; 121 : 1495–1500. [DOI] [PubMed] [Google Scholar]
- 74. Haroche J, Cohen‐Aubart F, Emile J‐F, et al Reproducible and sustained efficacy of targeted therapy with Vemurafenib in patients with BRAF V600E‐mutated Erdheim–Chester disease. J Clin Oncol 2015; 33 : 411–418. [DOI] [PubMed] [Google Scholar]
- 75. Diamond EL, Subbiah V, Lockhart AC, et al Vemurafenib for BRAF V600‐mutant Erdheim–Chester disease and Langerhans cell histiocytosis. JAMA Oncol 2018; 4 : 384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bhatia A, Ulaner G, Rampal R, et al Single‐agent dabrafenib for BRAFV600E‐mutated histiocytosis. Haematologica 2018; 103 : e177–e180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kieran MW, Geoerger B, Dunkel IJ, et al A phase I and pharmacokinetic study of oral dabrafenib in children and adolescent patients with recurrent or refractory BRAF V600 mutation‐positive solid tumors. Clin Cancer Res 2019; 25 : 7294–7302. [DOI] [PubMed] [Google Scholar]
- 78. Bhatia A, Hatzoglou V, Ulaner G, et al Neurologic and oncologic features of Erdheim–Chester disease: a 30‐patient series. Neuro Oncol 2020; 22: 979–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Diamond EL, Durham BH, Ulaner GA, et al Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature 2019; 567 : 521–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pegoraro F, Maniscalco V, Peyronel F, et al Long term follow up of mTOR inhibition for Erdheim–Chester disease. Blood 2020; 135 : 1994–1997. [DOI] [PubMed] [Google Scholar]
- 81. Poulikakos PI, Zhang C, Bollag G, et al RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild‐type BRAF. Nature 2010; 464 : 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Callahan MK, Rampal R, Harding JJ, et al Progression of RAS‐mutant leukemia during RAF inhibitor treatment. N Engl J Med 2012; 367 : 2316–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Konstantinou MP, Lucas P, Uthurriague C, et al Langerhans cell histiocytosis associated with chronic myelomonocytic leukemia both harboring the same BRAF V600E mutation: efficacy of vemurafenib. Eur Acad Dermatol Venereol 2020; 10.1111/jdv.16850 [DOI] [PubMed] [Google Scholar]
- 84. Carbone A, Roulland S, Gloghini A, et al Follicular lymphoma. Nat Rev Dis Prim 2019; 5 : 83. [DOI] [PubMed] [Google Scholar]
- 85. Menezes J, Acquadro F, Wiseman M, et al Exome sequencing reveals novel and recurrent mutations with clinical impact in blastic plasmacytoid dendritic cell neoplasm. Leukemia 2014; 28 : 823–829. [DOI] [PubMed] [Google Scholar]
- 86. Fernandez‐Pol S, Bangs CD, Cherry A, et al Two cases of histiocytic sarcoma with BCL2 translocations and occult or subsequent follicular lymphoma. Hum Pathol 2016; 55 : 39–43. [DOI] [PubMed] [Google Scholar]
- 87. Wetzler M, Kurzrock R, Goodacre AM, et al Transformation of chronic lymphocytic leukemia to lymphoma of true histiocytic type. Cancer 1995; 76 : 609–617. [DOI] [PubMed] [Google Scholar]
- 88. Bassarova A, Trøen G, Fosså A, et al Transformation of B cell lymphoma to histiocytic sarcoma: somatic mutations of PAX‐5 gene with loss of expression cannot explain transdifferentiation. J Hematop 2009; 2 : 135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mori M, Matsushita A, Takiuchi Y, et al Histiocytic sarcoma and underlying chronic myelomonocytic leukemia: a proposal for the developmental classification of histiocytic sarcoma. Int J Hematol 2010; 92 : 168–173. [DOI] [PubMed] [Google Scholar]
- 90. Shao H, Xi L, Raffeld M, et al Clonally related histiocytic/dendritic cell sarcoma and chronic lymphocytic leukemia/small lymphocytic lymphoma: a study of seven cases. Mod Pathol 2011; 24 : 1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Muslimani A, Chisti MM, Blenc AM, et al Langerhans/dendritic cell sarcoma arising from hairy cell leukemia: a rare phenomenon. Ann Hematol 2012; 91 : 1485–1487. [DOI] [PubMed] [Google Scholar]
- 92. Chen W, Jaffe R, Zhang L, et al Langerhans cell sarcoma arising from chronic lymphocytic lymphoma/small lymphocytic leukemia: lineage analysis and braf v600e mutation study. N Am J Med Sci 2013; 5 : 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Buser L, Bihl M, Rufle A, et al Unique composite Hematolymphoid tumor consisting of a pro‐T lymphoblastic lymphoma and an indeterminate dendritic cell tumor: evidence for divergent common progenitor cell differentiation. Pathobiology 2014; 81 : 199–205. [DOI] [PubMed] [Google Scholar]
- 94. Frauenfeld L, Bonzheim I, Wirths S, et al Clonal evolution of chronic lymphocytic leukemia to Langerhans cell histiocytosis: a case report. Virchows Arch 2019; 475 : 795–798. [DOI] [PubMed] [Google Scholar]
- 95. Skala SL, Ye JC, Stumph J, et al Combined tumors in hematolymphoid neoplasms: case series of histiocytic and Langerhans cell sarcomas arising from low‐grade B‐cell lymphoma. Clin Pathol 2019; 12 : 2632010X1987841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Chakraborty R, Hampton OA, Shen X, et al Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood 2014; 124 : 3007–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Coverage of various genes by the different NGS panels used to analyse the affected tissue specimens of the three cases presented in this study
Table S1. Available PALGA diagnostic terms for the various histiocytic disorders
Table S2. Reported patients with histiocytic neoplasms associated with additional haematological malignancies bearing the same genetic alteration(s) as demonstrated by DNA sequencing and/or DNA methylation profiling
Table S3. Reported patients with histiocytic neoplasms associated with additional haematological malignancies bearing the same genetic alteration(s) as demonstrated by techniques other than DNA sequencing and/or DNA methylation profiling
Case 1. Detailed sequencing data
Case 2. Detailed sequencing data
Case 3. Detailed sequencing data