

Abstract
To improve the description of interactions among the localized d, f electrons in transition metals, we have introduced a ligand-field motivated contribution into the Density Functional Tight Binding (DFTB) model. Referred to as DFTB3+U, the approach treats the d, f electron repulsions with rotationally invariant orbital-orbital interactions and a Hartree-Fock model; this represents a major conceptual improvement over the original DFTB3 approach, which treats the d, f -shell interactions in a highly averaged fashion without orbital level of description. The DFTB3+U approach is tested using a series of nickel compounds that feature Ni(II) and Ni(III) oxidation states. By using parameters developed with the original DFTB3 Hamiltonian and empirical +U parameters (F0/2/4 Slater integrals), we observe that the DFTB3+U model indeed provides substantial improvements over the original DFTB3 model for a number of properties of the nickel compounds, including the population and spin polarization of the d-shell, nature of the frontier orbitals, ligand field splitting and the energy different between low and high spin states at OPBE optimized structures. This proof-of-concept study suggests that with self-consistent parameterization of the electronic and +U parameters, the DFTB3+U model can develop into a promising model that can be used to efficiently study reactive events involving transition metals ion condensed phase systems. The methodology can be integrated with other approximate QM methods as well, such as the extended tight binding (xTB) approach.
Graphical Abstract
DFTB3+U for Transition Metal Systems
I. Introduction
Transition metal ions play important roles in chemistry and biology,1–8 thus it is important to describe their electronic structure and coordination geometry with a high degree of accuracy in computational studies.9–12 Due to the localized nature of the valence shell in transition metals (e.g., 3d), electron correlation effects are significant and need to be well treated for an accurate description.9,11,12 In fact, both static and dynamic correlation effects are significant for transition metal (TM) ions, making them challenging to study using quantum mechanical (QM) methods.9,11–13 Progress in quantum chemistry has seen the development of various Density Functional Approximations (DFAs),13–16 or different methodologies such as DMRG,17–22 and Multi-Configuration Pair-Density Functional Theory23,24, followed very recently by including DMRG (DMRG-PDFT).25 These developments have generated promising results14 for transition metal systems, including multi-metal clusters.20,26–32 Nevertheless, such calculations remain computationally expensive and therefore not readily applicable to problems where extensive sampling of the configurational space is crucial.33 An exciting and emerging direction is to develop machine learning (ML) potentials based on high-level QM methods,34–40 yet application of ML potentials to condensed phase systems is not as straightforward and remains an active area for research.41 Semi-empirical QM methods42–45 can potentially fill the gap between highly accurate quantum chemistry methods and empirical or numerical (e.g., machine learning) potentials. As described below in more detail, while these techniques generally provide reasonable geometries, most current approaches lack accuracy with respect to energetics and electron distribution particularly when comparing different electronic (magnetic) states in transition metal compounds. Here we show a possible solution to this problem by enhancing Density Functional Tight Binding (DFTB) with an explicit description of electron-electron interactions inside the d,f-shell of a transition metal with orbital resolution.
DFTB has attracted much attention in recent years due to its computational efficiency and reasonable accuracy for many applications.42–44 The DFTB245 approach has been parameterized for the first row of transition metal ions by Morokuma and co-workers;46 the method was found to give reasonable structures, while energetics, especially spin state splittings, often have large errors on the order of at least tens of kcal/mol.46 The third-order variant of DFTB (DFTB3)47 has been parameterized for several transition metal ions such as zinc,48 copper49 and nickel50 within the framework of the 3OB parameterization. Obtained structures are general fairly robust, but the metal-ligand binding interactions may exhibit large errors, especially for charged ligands; the robustness of the structures was demonstrated by the much improved metal-ligand interaction energies in single point calculations at higher level theories (e.g., G3B3 or B3LYP). Investigation of the bonding structure in the context of Natural Bonding Orbital analysis51 suggested that the DFTB3/3OB model generally provides physically sound descriptions of metal-ligand interactions, including (pseudo) Jahn-Teller effects,49,50 although the degree of ligand-to-metal charge transfer and certain orbital interactions can be grossly overestimated. Specifically, for nickel, the first parameterization had to resort to distinct Hubbard charge derivatives for different oxidation states to capture reliable structural and energetic properties for the respective oxidation state, and larger errors were observed for the spin state splittings.50
More recently, the xTB model52,53 of Grimme and co-workers has been developed for the entire periodic table, including all transition metal ions. The model has been shown to give impressive structural properties for many transition metal compounds, including transition state structures for organometallic catalysis.54 Similar to DFTB3/3OB, however, the energetics are generally less satisfactory, due in part to the fact that the xTB model was parameterized based on structure and vibrational frequencies.52
Therefore, it is evident that additional development is worthwhile to improve the energetic properties of transition metal compounds at the semi-empirical QM level. In this work, we address the problem of strong d-electron correlation by augmenting the DFTB3 approach with a ligand field (LF) 9,55–58 motivated model to specifically treat the d-d interactions with resolved magnetic quantum numbers (ml). The idea of using LF theory is not new; LF model has been incorporated into force fields for transition metal ions, 59 and it has been integrated with Density Functional Theory60,61 and ab initio wavefunction methods for the interpretation of electronic structure and spectroscopies of metal compounds.62 The philosophy of treating the d and f electrons in transition metal ions separately is also the basis for the DFT+U method in the materials science community. 63–66 Therefore, we expect that introducing the LF motivated model into DFTB3 can improve the latter’s description of transition metal compounds. Indeed, using a set of nickel compounds as exploratory examples, we find that including the additional term into DFTB3 Hamiltonian with 3OB improves many properties related to the electronic structure of both Ni(II) and Ni(III) compounds, such as the occupancy of metal d orbitals and spin-state splitting. While further adjustment in the 3OB parameterization is clearly required since the DFTB3 Hamiltonian is modified, the current findings suggest that the augment DFTB3 with a LF type of model for the d electrons, referred below as DFTB3+U, has the potential to become an effective approach for describing transition metal systems.
In the following, we first present the integration of the LF inspired model with DFTB3, leading to the DFTB3+U. This is followed by the analysis of computational results for nickel ions and a set of Ni(II)/Ni(III) compounds to illustrate the impact of including the new term into DFTB3 Hamiltonian. We end by a few concluding remarks.
II. Theory and Computational Methods
In this section, we will give a short overview of DFTB3 methods, and the interested reader is pointed to the literature.33,43–45,47,67,68
II.1. Density Functional Tight Binding (DFTB) Model
The DFTB energy is obtained as an approximation of DFT by replacing the electron density, ρ, by the sum of the reference density ρ0, which is obtained as a sum of precalculated atomic densities and a density fluctuation Δρ,
| (1) |
Expanding the total energy in a Taylor series in Δρ, and following a set of additional approximations,69 the following energy expression is obtained at the third order,
| (2) |
As the first order term depends only on the reference density, the generalized eigenvalue problem needs to be diagonalized only once, while for DFTB2 and DFTB3 it needs to be solved self-consistently.67 DFTB1 is a good approximation when there is no large density fluctuation relative to atoms during bond formation, i.e., for non-polar molecules. For more complex molecules, DFTB2/3 are needed for computing the changes in the electron-electron interactions relative to the reference, as represented in the third and fourth terms of the energy expression. Third order terms involve, Гab, which is the charge derivative of the second order term, γab. 67 Spin-polarization49,68,70,71 and l-dependence of the Hubbard parameters70 have also been incorporated.
II.2. Augment DFTB with a Ligand Field Type of Model for the d,f Electrons
DFTB2 and DFTB3 approaches make strongest approximations when treating electron-electron interactions. In particular these methods only deal with the average interaction among the electrons localized on the atoms. However, for the d or f shells in transition or rare-earth metals, electrons are highly localized and more detailed orbital-orbital interactions are necessary. 9,58 Despite these difficulties, Morokuma and co-workers produced DFTB2 parameters for several first-row transition metals,46 including Fe and Ni while Grimme and co-workers developed a tight-binding model (xTB) for the entire periodic table.52 Both models provided satisfactory results for geometries but not for energetics. Even though their work was certainly a step forward, it is now apparent that further developments are necessary to fully capture the rich chemistry of these systems involving a full register of magnetism, spin-states and other orbital-dependent phenomena like the Jahn-Teller effect.
At the simplest level, the interaction of electrons within d or f shells can be written using the Hartree-Fock method, 72
| (3) |
where α, β, α’, and β’ are indices that run over all the spatial orbitals of the highly localised atomic shell, represented by a basis function χα; σ represents the spin (up or down) and is the corresponding density matrix element. Many classical texts, like that of Griffith,58 show that, when we confine the interactions within a particular shell, and under spherical symmetry, the four-center integrals can be written in term of the Slater integrals (F0, F2, F4 for d-shells and F0, F2, F4 and F6 for f-shells)
| (4) |
where αk can be expressed as a product of Clebsch-Gordan coefficients. Slater integrals are simply related to Racah parameters (A, B and C) commonly used in the ligand field theory, and to parameters U and J in DFT+U:
In these transformations, we follow the usual approximation that fixes the the ratio F4/F2 to a value of 0.625. It is important to note that, in this work where the spin constants Wlal’a have been fixed to values from a previous work,50 the effect of using different F4/F2 values, as well as their ratio, is found to be negligible.
In this work, we propose to add this rotationally invariant ligand-field term to the DFTB3 energy to describe detailed orbital-orbital interactions when active d or f electrons are present in the system. This is a similar prescription to the DFT+U model commonly used in the materials science community. A convenient energy expression can be reached making use of the total density matrix, and spin-polarization density matrix
| (5) |
In this way the first term can be used in simulations in absence of spin polarization while the second term can be added to include magnetic effects.
Comparing the approach taken here to the usual DFT+U73 it is clear that we do not include the correction to avoid double counting d-d interactions. This is partially justified as DFTB is a semi-empirical method where we aim to examine the inclusion of the extra ligand-field or +U term on the description of the spin state splitting with existing basis. Clearly much room for improvement remains with a full reparameterization of the basis, excluding the average d-d/f-f interactions existing in DFTB3 and relying solely on the more detailed description given by the ligand field terms.
III. Results and Discussion
III.1. Atomic Systems: Ni(II) and Ni(III) ions
To examine the impact of the +U contribution on the electronic structure of metal ions, the natural place to start is the atomic systems since they lack the complications due to mixing with the ligand orbitals. Here we focus on both Ni(II) and Ni(III).
The first question we aim to answer is how the increase of interelectronic repulsion influences the energies of the spin states individually as well as the spin state splitting (ΔELS-HS). To this end, we vary the three Slater integral parameters, F0 F2 and F4 individually. As electron-electron repulsion favours the high spin state, it is natural to expect that the general bias of DFTB3 (and many DFT functionals) toward the low spin state will be somewhat counteracted.
As shown in Figure 1 this is, indeed, what is observed in the actual calculations. As expected from LF theory58, it is found that the energies of the spin states, as well as their difference (ΔELS-HS), show perfect linear dependence on the F2/F4 parameters and the high spin state becomes more favoured when the electron-electron repulsion is increased, Figure 1. Somewhat surprisingly, ΔELS-HS also exhibits a linear dependence on F0, which is not the result expected from pure ligand field theory where the difference in energy between the low and high spin states is not dependent on F0. This somewhat surprising result comes from the fact that the ligand-field theory found in classical books like the one by Griffith58 expresses the atomic wavefunction as a combination of multiple Slater determinants while in our case we use a mean-field approach distilled into Eq. 3 where the atomic orbital occupations are fractional. Using Eq. 3, and substituting the four-center integrals using Eq. 4 and setting F2=F4=0 we can show (see SI.docx) that the energy difference between high and low spin states in Ni(III) is twice as large as that for Ni(II), as found numerically in our calculations shown in Figure 1.
Figure 1.

Dependence of ΔELS-HS on the F0 electron repulsion parameter (a), and F2 electron repulsion parameter (b), for Ni(II) and Ni(III) atomic systems.
Regarding the spin state energetics, the best results in comparison to available experimental data are obtained with F0=0.05–0.055 Hartree with a mean-absolute-error (MAE) of ~13 kcal/mol for both Ni(II) and Ni(III) oxidation states; we need to bear in mind that the double counting of d-d interaction has not been explicitly taken out in this work, thus our value (around 1.3 eV) is very small compared to the usual value of F0 in atoms (tens of eV)74 or the value in solids (around a few (7–9) eV for Ni)75,76. This obtained MAE is competitive with the best DFT results (see Table S5); for example, the MAE is 37 kcal/mol for PBE0 and 8 kcal/mol for OPBE (which showed the best performance out of all used DFAs). However, from a practical point of view using these values of F0 in molecular calculations leads to convergence problems in SCF cycles. While for atomic ions we detect that SCF problems start after F0≥0.05 for Ni(II) and F0>0.1 for Ni(III), we found that robust SCF behaviours for molecules are observed for up to F0=0.035 Hartree. With this F0 value, the average spin-state splitting error becomes 19.3 kcal/mol, in comparison to the range of 8–38.5 kcal/mol for DFT methods (see SI.xlsx, Table S5). Given that standard DFTB3 has a MAE of 32.0 kcal/mol, it is clear that the +U model leads to a significant improvement in the description of the spin state splitting. Finally, it should be noted that the notion of MAE used in this section is not optimal, since we have only two values, the error for Ni(II) and the error for the Ni(III) ion. Since we need the functional with the lowest error for Ni(II) and Ni(III), together, the MAE was used for our general discussion. We will just mention that GGAs have much larger errors for Ni(II), hybrids for Ni(III), while meta-GGAs can show any behaviour; the detailed errors are included in the SI.xlsx.
III.2. Electronic structure of molecular systems: Ni(II)/Ni(III) compounds
Choice of Ni(II) and Ni(III) coordination compounds
To examine the impact of the +U term on the electronic structure of molecular compounds, 85 Ni(II) and 69 Ni(III) compounds have been examined (for details, see SI.xlsx). These include small molecule ligands coordinated using O, C, N, P and S heteroatoms (H2O, OH−, NH3, NH2−, en, AcO−, CN−, NC−, NCS, CO, PH3, PH2−, SH2 and SH−) with varying coordination numbers (CN=1–6). The structures were taken from our previous work50 and were optimized at the PBE/TZP level of theory. We performed single point DFTB3+U energy calculations at the corresponding PBE geometries, which were optimized for each spin state; the same procedure was utilized for various DFT functionals. This choice was made since the centre of the attention in this work revolves around capturing the energetics and the correct electron distribution around the transition metal site rather than the geometry. As discussed in the introduction, DFTB and other approaches (see e.g. Refs. 52, 53) already perform quite adequately for structures and we would like to remove this degree of freedom as a source of error when determining purely electronic properties. A comparison between the geometries of DFTB3+U and DFT is provided in the SI, showing that DFTB3+U has similar performance as DFTB3, despite the fact that most parameters for the DFTB3+U model in this work were optimized previously for the DFTB3 Hamiltonian. Finally, due to the small size of the model ligands, we do not include empirical dispersion when comparing DFTB3(+U) and DFT results. This is supported by the observation that reoptimizing the structures at the PBE+D3 level lead to only small changes in structure and energetics; for example, the impact on the spin-state splitting has a root-mean-square value of 0.48 kcal/mol among all the model compounds studied (see SI). In realistic applications to systems that include large ligands, we expect that including empirical dispersion in DFTB3(+U) is important.
Before discussing the results, we note that the spin states in Ni(II) with most stable and most abundant coordination environments are not always straightforward to compare since they involve different coordination numbers, or, at least, completely different arrangements around the central metal ion. For Ni(III), which has d7 occupation, there is very high metal oxidative power (low energy empty orbitals), resulting in very few known complexes in the CCDC crystal structure database. Despite all these complications, the list of small molecule systems is useful for testing the performance of the DFTB3+U Hamiltonian in comparison to the original DFTB3 approach that does not explicitly treat d-d interactions at the orbital level.
Regarding the choice of F0/F2/F4 parameters, a thorough examination of the influence of F2/F4 parameters (with the fixed F2/F4 ratio) indicates only a small effect on the overall MAE, while F0 has a substantially larger impact. This is in accordance with their relative magnitude9,58 and the portion of e-e interaction that is taken into account by F0 vs. F2/F4 parameters. Therefore, we use a fixed ratio of F2/F4, as often done in the literature. The highest values (in Hartree) that do not lead to SCF problems have been selected: F0=0.035, F2=0.01 and F4/F2 = 0.625. To minimize SCF convergence issues, all calculations have been done with an electronic temperature of 2000K; results with a lower electronic temperature, wherever available, are also included in the SI.
The effect of +U on d-orbital population and spin density
The first set of properties that should be examined includes the d-shell occupation, the total number of d electrons and the number of unpaired d electrons. As seen from Table 1, in an octahedral environment, Ni(II) is expected to have the d8 configuration with 2 unpaired electrons in the high spin (HS) and no unpaired electrons (S=0) in the low spin (LS) state. In the case of Ni(III), in the same octahedral environment, 3 unpaired electrons are expected in the HS state and 1 in the LS state.
Table 1.
configuration and number of unpaired electrons in d-shell obtained by Mulliken population analysis for DFTB3, DFTB3+U (F0=0.035) and OPBE, together with atomic values. a
| number of electrons in d-shell | ||||||||
|---|---|---|---|---|---|---|---|---|
| Ni(II) | Ni(III) | Ni(II) | Ni(III) | |||||
| HS | LS | HS | LS | HS | LS | HS | LS | |
| atomic values | d8 | d8 | d7 | d7 | 2 | 0 | 3 | 1 |
| DFTB3 | d8.9 | d9.0 | d8.7 | d8.8 | 1.0 | 0 | 1.2 | 0.4 |
| xTB | d8.6 | d8.7 | d8.4 | d8.4 | 1.3 | 0 | 1.6 | 0.6 |
| DFTB3+U | d7.8 | d8.0 | d7.5 | d7.7 | 1.7 | 0 | 2.2 | 0.9 |
| OPBE | d8.3 | d8.5 | d8.2 | d8.2 | 1.3 | 0 | 1.5 | 0.8 |
With the original DFTB3 Hamiltonian and Mulliken population analysis, the population of the d-shell is overestimated while the degree of spin polarization (the number of unpaired d electrons) is underestimated. For both Ni(II) and Ni(III), the d shell population is close to 9.0, and the spin polarization is underestimated by ~1 for Ni(II) and ~1.8/0.6 for HS/LS Ni(III). With the xTB model, which treats the d-shell in a similar fashion as DFTB3, the deviations from the expected values are smaller in magnitude yet still significant; for example, for Ni(III), the d shell population is ~8.4 rather than 7, and the spin polarization is underestimated by ~1.4 and 0.4 for the high spin and low spin states, respectively. In fact, these values are close to those observed for the OPBE functional (for results for other functionals, see SI.xlsx, Table S3 and Table S4).
With the DFTB3+U Hamiltonian and a F0 value of 0.035 Hartree, the results represent a major improvement: both the overall population of the d-shell and the spin polarization are closer to the expected values compared to DFTB3, xTB or OPBE (see Table 1). To better illustrate the impact of the +U contribution, we monitor the populations of the metal d/s shells as functions of the F0 parameter, holding F2/F4 fixed. After examining all the systems, for both oxidation and spin states, we confirm that the total d-population generally decreases as F0 increases, due to a combination of increased s occupancy and larger charge transfers to the ligands. For the example of the HS state of [Ni(NH3)4]2+ in Figure 2 b, for example, the d/s-populations are ~8.85/0.61 for DFTB3, and they change to ~7.96/0.67 for F0=0.035 Hartree, and to ~7.62/0.79 for F0=0.050 Hartree. This general trend is expected, as increased d-d repulsions lead to delocalization of the d electrons to both the metal s orbital and ligand orbitals. Since F0/2/4 parameters also contribute to exchange interactions, d-spin population generally increases with F0, until reaching a maximum and then decreases (see the examples for the HS state of [Ni(NH3)4]2+/3+ in a); the value of F0 that corresponds to the maximum in the d-spin population varies significantly among the different systems.
Figure 2.

Population redistribution between the d and s shells, on Ni centre (with increasing F0 for every subsequent point) for HS state of [Ni(NH3)4]2+/3+and change of spin density on d-shell with F0
MOs contours and ligand field splitting
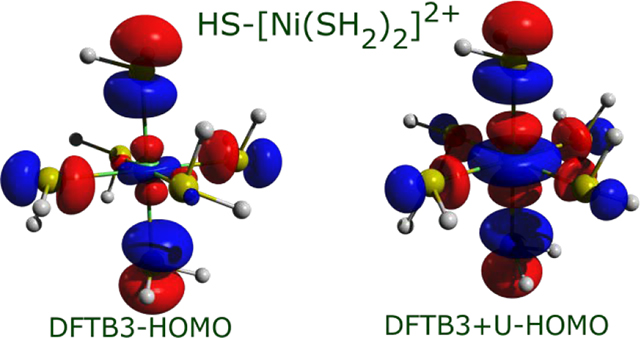
An important aspect of transition metal electronic structure is the localized molecular orbitals (MOs) with dominant d-characters, which are responsible for the peculiar reactivity, optical and magnetic properties of coordination compounds. In the previous paper,50 we have shown that reasonable orbitals and somewhat larger ligand field splittings (LFS) were obtained for Ni(II) octahedral complexes at DFTB3/3OB level of theory. In this work, we have also analysed the +3 oxidation state, and, the low spin state. It turns out that qualitatively expected shape of MOs can be obtained with DFTB3 only for rare cases. The example of HS-[Ni(SH2)6]2+ is given in Figure 3a, which shows that the frontier MOs are largely localized to the ligands. As shown in Figure 3b, the +U Hamiltonian corrects this behaviour and produces the five metal d-orbitals as frontier orbitals.
Figure 3.

An illustration of frontier MOs (isovalue 0.05 a.u.) in HS-[Ni(SH2)6]2+, with a) DFTB3, b) DFTB3+U (F0=0.035 Hartree) and c) DFT-OPBE.
Regarding LFS, it is not straightforward to extract even from (unrestricted) DFT calculations: highest MOs in α-spin usually do not have significant d-contribution and orbitals in β-spin include virtual ones, making the energy predictions difficult, and specialized procedures are often used for estimating LFS.77
Since experimental data for simple octahedral complexes of Ni(III) are not available, we use the [Ni(H2O)6]2+ and [Ni(NH3)6]2+ as examples. As shown in Table 2, DFTB3+U significantly improves the LFS and shows even better performance than OPBE. Experimental results, obtained by fitting the experimental spectra to a LF model, give Δo (10Dq) to be 1.05eV and 1.34 eV for these complexes, respectively.78 Using orbitals in β-spin from the OPBE calculations, values almost twice as the experimental ones are obtained; the same holds for DFTB3. With DFTB3+U, we observe a monotonic decrease in splitting with increasing F0; with F0=0.035, the estimated LFS values are 1.24 eV for [Ni(H2O)6]2+ and 1.38 eV for [Ni(NH3)6]2+, which are in very good agreement with experimental data.
Table 2.
Ligand Field Splitting (LFS) for two octahedral Ni(II) complexes. All energies are given in eV.
| Source | OPBE | DFTB3 | DFTB3+U | Experiment70 |
|---|---|---|---|---|
| Δo(10Dq) for [Ni(H2O)6]2+ | 1.98 | 1.50 | 1.24 | 1.05 |
| Δo(10Dq) for [Ni(NH3)6]2+ | 2.32 | 2.70 | 1.38 | 1.34 |
III.3. Spin State Splitting for Ni(II)/Ni(III) compounds
Before discussing the errors in spin splitting, we note that computed ΔELS-HS with DFTB3+U often exhibits non-monotonic dependence on the F0 value; the ΔELS-HS vs F0 dependence for all the examined systems is summarized in SI.xlsx, Table S6 and Table S7. As illustrations, representative DFTB3+U computed ΔELS-HS results for Ni(II)/Ni(III) compounds with OH− ligands are shown in Figure 4. While [Ni(OH)4]2- and [Ni(OH)3] exhibit a linear dependence on F0, other compounds show clear non-monotonic behaviours between ΔELS-HS and F0. In general, although total energies of different spin states show a linear dependence on F0, small deviations from linearity are sufficient to make a large impact on their difference since ΔELS-HS are several orders of magnitude smaller than the total energies of both spin states.
Figure 4.

Dependence of ΔELS-HS of F0 electron repulsion parameter; monotonic, almost linear dependence for [Ni(OH)4]2- and [Ni(OH)3] and plot with non-monotonic regions for [Ni(OH)3]− and [Ni(OH)4]−.
Table 3 and Table 4 summarize the Mean Absolute Errors (MAE) for all Ni complexes with tight binding-based and DFT methods, respectively (for specific systems see SI.xlsx, Table S6, Table S7, Table S10, Table S11, Table S12 and Table S13). All errors are calculated relative to the OPBE functional, which has been shown to be fairly accurate for spin state energetics.79–81
Table 3.
Variation of MAE in spin-state splitting (ΔELS-HS, in kcal/mol) as the F0 parameter (in Hartree) increases in DFTB3+U; also shown is the MAE for xTB. a
| M | F0 | 0 | 0.005 | 0.01 | 0.015 | 0.02 | 0.025 | 0.03 | 0.035 | 0.04 | 0.045 | 0.05 | xTB |
| Ni(II) | MAE | 21.8 | 19.9 | 18.4 | 15.8 | 14.6 | 14.2 | 13.9 | 13.8 | 14.0 | 14.9 | 16.8 | 18.7 |
| Ni(III) | MAE | 17.3 | 16.6 | 15.5 | 13.9 | 12.7 | 11.5 | 9.9 | 9.0 | 8.6 | 8.9 | 9.4 | 19.6 |
The reference value is OPBE/TZP.
Table 4.
Variation of MAE in spin-state splitting (ΔELS-HS, in kcal/mol) for various DFT functionals.a
| M | GGA | metaGGA | HYBRID | metaHYBRID | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| TPSS | M06-L | B3LYP | PBE0 | X3LYP | B1LYP | B3LYP* | TPSSH | M06 | |||
| MAE | Ni(II) | 1.1–2.0 | 1.6 | 6.9 | 15.2 | 8.5 | 7.0 | 8.9 | 4.0 | 4.6 | 3.8 |
| Ni(III) | 1.4–2.9 | 2.6 | 4.1 | 5.1 | 5.5 | 4.8 | 5.8 | 3.1 | 3.5 | 4.7 | |
The reference value is OPBE/TZP.
For Ni(II) complexes, the MAE of 21.8 kcal/mol for DFTB3 is reduced to 13.8 kcal/mol for DFTB3+U with F0=0.035 Hartree. For Ni(III) systems, the corresponding MAE values are 17.3 kcal/mol and 9.0 kcal/mol, respectively, again supporting the value of the +U contribution. In general, the errors for Ni(III) are smaller than for Ni(II) systems; this is to be expected considering the problems with ligand dissociation and complicated LS state with Ni(II). This provides optimism that results for other transition metal ions, which have simpler spin states (than Ni(II)) and better separated metal d-orbitals (than Ni(III)), will be more satisfactory with DFTB3+U. Compared to the DFTB3 methods, the xTB approach gives spin splittings closer to the original DFTB3; this is expected due to the similar approximations for treating the d-d interactions in these methods.
To put the errors of the tight binding methods in perspective, Table 4 presents MAE for various DFT functionals. All GGA functionals have small errors, which is not surprising since we used a GGA level of theory (OPBE) as the reference. It is interesting to note that B3LYP has a similar level of MAE as DFTB3+U. We note that care needs to be exercised when interpreting these errors. Spin-state splittings in transition metal compounds are difficult to treat properly with any computational method, as post-Hartree-Fock methods strongly depend on the choice of active space, and DFT methods suffer from the unclear choice of the exchange-correlation functionals.14 With electronic spectroscopy of transition metal ions, it is often necessary to calculate the spin forbidden bands (vertical spin state energy difference). It is accepted that transitions with errors up to 2–3000cm−1 (8.5kcal/mol) are considered acceptable, even with the most sophisticated post-HF ab initio methods.82
Attempts to search for simple rationalizations of trends based on, for example, ligand type and polarizability, π-donors and π-acceptors, coordination number, do not lead to any straightforward explanation for the error trends in spin splitting. Furthermore, the non-monotonic behaviour of spin splitting with respect to F0 (Figure 4) is not easily connected with changes in the electronic structure or orbital occupations (as is with atomic systems). Therefore, we illustrate the distinct error vs F0 trends using three examples: [Ni(PH3)3]3+, [Ni(NH3)4]2+ and [Ni(H2O)(SH2)]2+. These complexes cover cases with large, medium and small DFTB3 errors, as well as monotonic and non-monotonic ΔELS-HS vs F0 dependence.
For [Ni(PH3)3]3+, the DFTB3 error is ~23 kcal/mol; with F0=0.035 in DFTB3+U, the error is reduced to only 1 kcal/mol. From Figure 5, we observe that ΔELS-HS depends largely linearly on F0, with decreasing errors until the point F0=0.035 is reached (the closest point to the OPBE value, horizontal blue line); the error then enlarges as F0 increases.
Figure 5.

Dependence of ΔELS-HS (left side) and absolute error relative to OPBE (right side) on the F0 electron repulsion parameter in DFTB3+U for [Ni(PH3)3]3+, [Ni(NH3)4]2+ and [Ni(H2O)(SH2)]2+.
For [Ni(NH3)4]2+, the DFTB3 error is 58 kcal/mol, and it remains significant (38 kcal/mol) for DFTB3+U with F0=0.035. Finally, [Ni(H2O)(SH2)]2+ does not show a monotonic ΔELS-HS vs F0 behaviour, resulting in irregular relations of error vs F0; the magnitude of error, however, is modest for all F0 values.
In short, we observe that for systems with medium DFTB3 errors (10–15 kcal/mol), the DFTB3+U model can significantly reduce the error in spin splitting; the ones with too high DFTB3 errors (50–60 kcal/mol), the +U contribution leaves still significant (30–40 kcal/mol) errors even with a reasonable F0 value; and finally, there are ~10% of systems with modest and comparable DFTB3/DFTB3+U errors.
Conclusions
To enable extensive sampling of transition metal containing systems, such as metalloenzymes, it is valuable to develop semi-empirical QM methods. This, however, has remained a major challenge in the area of quantum chemistry for many decades. Motivated by the success of ligand field theory and DFT+U models, we have implemented a DFTB3+U approach to treat the d-d/f-f interactions and tested it using a series of nickel compounds. This should be considered as a proof of concept study as we have used the 3OB set of parameters developed with the original DFTB3 Hamiltonian; moreover, the parameters in the +U contribution (F0/2/4 parameters) are considered empirical since the averaged d-d interactions in the original DFTB3 model have not been excluded. Nevertheless, we see that the DFTB3+U model provides substantial improvements over the original DFTB3 model for a number of properties of the nickel compounds, including the population and spin polarization of the d-shell, nature of the frontier orbitals, ligand field splitting and the energy different between low and high spin states. For some of the properties, the DFTB3+U model can give results that are competitive with some of the best DFT results. On the other hand, there are also cases where the errors at the DFTB3 level are so large (e.g., for the spin splitting) that with a +U model, the magnitude of error remains significant.
The results obtained so far suggest that it is worthwhile further developing the DFTB3+U model by reparameterizing the DFTB3 electronic parameters together with the +U parameters. In particular, the valence basis set and spin-spin coupling parameters should be optimized in the framework of the +U model, with the double-counting d-d/f-f interactions properly excluded. Considering that Ni(II) and Ni(III) represent challenging cases for spin splittings, we anticipate that the DFTB3+U model will lead to more favorable results for other transition metal ions important in biology, such as Mn(II) and Co(II). Finally, we note that while the +U term is entirely an on-site contribution, it can have an impact on the molecular structure indirectly by perturbing the nature of metal-ligand interactions, such as the degree of charge transfer between metal and ligands. Therefore, with further parameterization, the DFTB3+U can potentially provide improved description of structural and energetic properties of transition metal containing complexes over existing semi-empirical QM methods and become a useful tool for efficiently exploring reactive processes involving transition metals in the condensed phase. The methodology can also be integrated with other approximate QM methods such as the extended tight binding approach, which already appears to provide rather reliable structures and frequencies for many transition metal systems.
Supplementary Material
Acknowledgments
This project was supported by the Serbian-German collaboration project (DAAD) number 451-03-01038/2015-09/7 (to MG and ME), the Serbian Ministry of Education, Science and Technological Development (Grants 451-03-68/2020-14/200168 and 451-03-68/2020-14/200026), NIH grant R01-GM106443 (QC) and the Spanish Ministry of Economy and Competitiveness through Grant PGC2018-096955-B-C41 (P.G.-F.).
Footnotes
Supporting Information
Further analysis of spin states for Ni(II) and Ni(III) with the current DFTB3+U model, detailed data for DFTB3, DFTB3+U (d orbital population, spin polarization, spin-state splittings computed with different electronic temperatures, geometries) as well as results from xTB and various DFT functionals are included.
References
- (1).Atkins P; Overton T Shriver and Atkins’ Inorganic Chemistry; OUP Oxford, 2010. [Google Scholar]
- (2).Miessler GL; Tarr DA Inorganic Chemistry; Pearson Prentice Hall, 2011. [Google Scholar]
- (3).Crans DC; Kostenkova K Communications Chemistry 2020, 3, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Yam VW-W; Chan AK-W; Hong EY-H Nature Reviews Chemistry 2020. [Google Scholar]
- (5).Liang Y; Dong H; Aurbach D; Yao Y Nature Energy 2020. [Google Scholar]
- (6).Seo J; Kim DY; An ES; Kim K; Kim G-Y; Hwang S-Y; Kim DW; Jang BG; Kim H; Eom G; Seo SY; Stania R; Muntwiler M; Lee J; Watanabe K; Taniguchi T; Jo YJ; Lee J; Min BI; Jo MH; Yeom HW; Choi S-Y; Shim JH; Kim JS Science Advances 2020, 6, eaay8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Crichton R Biological Inorganic Chemistry: An Introduction; Elsevier Science, 2007. [Google Scholar]
- (8).Cotton FA; Wilkinson G; Grimes RN; Murillo CA; Bochmann M; Bochmann SLSCSM. Advanced Inorganic Chemistry; Wiley, 1999. [Google Scholar]
- (9).Bersuker IB Electronic Structure and Properties of Transition Metal Compounds: Introduction to the Theory; Wiley, 2010. [Google Scholar]
- (10).Cramer CJ; Truhlar DG Phys Chem Chem Phys 2009, 11, 10757. [DOI] [PubMed] [Google Scholar]
- (11).Neese F; Pantazis DA Spectroscopy and Electronic Structure of Transition Metal Complexes; Royal Society of Chemistry, 2020. [Google Scholar]
- (12).Mingos DMP; Day P; Dahl JP Molecular Electronic Structures of Transition Metal Complexes II; Springer Berlin Heidelberg, 2012. [Google Scholar]
- (13).Verma P; Truhlar DG Trends in Chemistry 2020, 2, 302. [Google Scholar]
- (14).Zhang D; Truhlar DG Journal of Chemical Theory and Computation 2020, 16, 4416. [DOI] [PubMed] [Google Scholar]
- (15).Swart M; Gruden M Accounts of Chemical Research 2016, 49, 2690. [DOI] [PubMed] [Google Scholar]
- (16).Harvey JN Annual Reports Section “C” (Physical Chemistry) 2006, 102, 203. [Google Scholar]
- (17).Chan GK-L; Zgid D In Annual Reports in Computational Chemistry; Wheeler R A., Ed.; Elsevier: 2009; Vol. 5, p 149. [Google Scholar]
- (18).Chan GK-L; Sharma S In Solving the Schrödinger Equation, p 43. [Google Scholar]
- (19).Guo S; Li Z; Chan GK-L Journal of Chemical Theory and Computation 2018, 14, 4063. [DOI] [PubMed] [Google Scholar]
- (20).Marti KH; Ondík IM; Moritz G; Reiher M The Journal of Chemical Physics 2008, 128, 014104. [DOI] [PubMed] [Google Scholar]
- (21).Baiardi A; Reiher M The Journal of Chemical Physics 2020, 152, 040903. [DOI] [PubMed] [Google Scholar]
- (22).Baiardi A; Stein CJ; Barone V; Reiher M Journal of Chemical Theory and Computation 2017, 13, 3764. [DOI] [PubMed] [Google Scholar]
- (23).Li Manni G; Carlson RK; Luo S; Ma D; Olsen J; Truhlar DG; Gagliardi L Journal of Chemical Theory and Computation 2014, 10, 3669. [DOI] [PubMed] [Google Scholar]
- (24).Stoneburner SJ; Truhlar DG; Gagliardi L The Journal of Physical Chemistry A 2020, 124, 1187. [DOI] [PubMed] [Google Scholar]
- (25).Sharma P; Bernales V; Knecht S; Truhlar DG; Gagliardi L Chemical Science 2019, 10, 1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhou C; Gagliardi L; Truhlar DG The Journal of Physical Chemistry A 2019, 123, 3389. [DOI] [PubMed] [Google Scholar]
- (27).Carlson RK; Li Manni G; Sonnenberger AL; Truhlar DG; Gagliardi L Journal of Chemical Theory and Computation 2015, 11, 82. [DOI] [PubMed] [Google Scholar]
- (28).Roemelt M; Pantazis DA Advanced Theory and Simulations 2019, 2, 1800201. [Google Scholar]
- (29).Kurashige Y; Chan GK-L; Yanai T Nature Chemistry 2013, 5, 660. [DOI] [PubMed] [Google Scholar]
- (30).Schurkus H; Chen D-T; Cheng H-P; Chan G; Stanton J The Journal of Chemical Physics 2020, 152, 234115. [DOI] [PubMed] [Google Scholar]
- (31).Li Z; Guo S; Sun Q; Chan GK-L Nature Chemistry 2019, 11, 1026. [DOI] [PubMed] [Google Scholar]
- (32).Li Z; Li J; Dattani NS; Umrigar CJ; Chan GK-L The Journal of Chemical Physics 2019, 150, 024302. [DOI] [PubMed] [Google Scholar]
- (33).Christensen AS; Kubař T; Cui Q; Elstner M Chemical Reviews 2016, 116, 5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Tkatchenko A Nature Communications 2020, 11, 4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Schütt KT; Chmiela S; von Lilienfeld OA; Tkatchenko A; Tsuda K; Müller KR Machine Learning Meets Quantum Physics; Springer International Publishing, 2020. [Google Scholar]
- (36).Schütt KT; Arbabzadah F; Chmiela S; Müller KR; Tkatchenko A Nature Communications 2017, 8, 13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hansen K; Biegler F; Ramakrishnan R; Pronobis W; von Lilienfeld OA; Müller K-R; Tkatchenko A The Journal of Physical Chemistry Letters 2015, 6, 2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).von Lilienfeld OA; Müller K-R; Tkatchenko A Nature Reviews Chemistry 2020, 4, 347. [DOI] [PubMed] [Google Scholar]
- (39).Rupp M; Ramakrishnan R; von Lilienfeld OA The Journal of Physical Chemistry Letters 2015, 6, 3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Montavon G; Rupp M; Gobre V; Vazquez-Mayagoitia A; Hansen K; Tkatchenko A; Müller K-R; Anatole von Lilienfeld O New Journal of Physics 2013, 15, 095003. [Google Scholar]
- (41).Noé F; Tkatchenko A; Müller K-R; Clementi C Annual Review of Physical Chemistry 2020, 71, 361. [DOI] [PubMed] [Google Scholar]
- (42).Porezag D; Frauenheim T; Köhler T; Seifert G; Kaschner R Physical Review B 1995, 51, 12947. [DOI] [PubMed] [Google Scholar]
- (43).Cui Q; Elstner M Phys Chem Chem Phys 2014, 16, 14368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Elstner M; Seifert G Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences 2014, 372. [DOI] [PubMed] [Google Scholar]
- (45).Elstner M; Porezag D; Jungnickel G; Elsner J; Haugk M; Frauenheim T; Suhai S; Seifert G Physical Review B 1998, 58, 7260. [Google Scholar]
- (46).Zheng G; Witek HA; Bobadova-Parvanova P; Irle S; Musaev DG; Prabhakar R; Morokuma K; Lundberg M; Elstner M; Köhler C; Frauenheim T Journal of Chemical Theory and Computation 2007, 3, 1349. [DOI] [PubMed] [Google Scholar]
- (47).Gaus M; Cui Q; Elstner M Journal of Chemical Theory and Computation 2011, 7, 931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Lu X; Gaus M; Elstner M; Cui Q The Journal of Physical Chemistry B 2015, 119, 1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Gaus M; Jin H; Demapan D; Christensen AS; Goyal P; Elstner M; Cui Q Journal of Chemical Theory and Computation 2015, 11, 4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Vujović M; Huynh M; Steiner S; Garcia-Fernandez P; Elstner M; Cui Q; Gruden M Journal of Computational Chemistry, 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Lu X; Duchimaza-Heredia J; Cui Q The Journal of Physical Chemistry A 2019, 123, 7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Grimme S; Bannwarth C; Shushkov P Journal of Chemical Theory and Computation 2017, 13, 1989. [DOI] [PubMed] [Google Scholar]
- (53).Bannwarth C; Ehlert S; Grimme S Journal of Chemical Theory and Computation 2019, 15, 1652. [DOI] [PubMed] [Google Scholar]
- (54).Dohm S; Bursch M; Hansen A; Grimme S Journal of Chemical Theory and Computation 2020, 16, 2002. [DOI] [PubMed] [Google Scholar]
- (55).Orgel LE An Introduction to Transition-metal Chemistry: Ligand-field Theory; Methuen, 1966. [Google Scholar]
- (56).Jørgensen CK Modern aspects of ligand field theory; North-Holland. New York: American Elsevier, 1971. [Google Scholar]
- (57).Ballhausen CJ Introduction to Ligand Field Theory; McGraw-Hill, 1962. [Google Scholar]
- (58).Griffith JS The Theory of Transition-Metal Ions; Cambridge University Press, 1964. [Google Scholar]
- (59).Burton VJ; Deeth RJ; Kemp CM; Gilbert PJ Journal of the American Chemical Society 1995, 117, 8407. [Google Scholar]
- (60).Atanasov M; Daul CA Comptes Rendus Chimie 2005, 8, 1421. [Google Scholar]
- (61).Atanasov M; Comba P; Daul CA; Neese F; Springer; Netherlands: Dordrecht, 2008, p 411. [Google Scholar]
- (62).Singh SK; Eng J; Atanasov M; Neese F Coordination Chemistry Reviews 2017, 344, 2. [Google Scholar]
- (63).Anisimov VI; Gunnarsson O Physical Review B 1991, 43, 7570. [DOI] [PubMed] [Google Scholar]
- (64).Anisimov VI; Zaanen J; Andersen OK Physical Review B 1991, 44, 943. [DOI] [PubMed] [Google Scholar]
- (65).Solovyev IV; Dederichs PH; Anisimov VI Physical Review B 1994, 50, 16861. [DOI] [PubMed] [Google Scholar]
- (66).Anisimov VI; Aryasetiawan F; Lichtenstein AI Journal of Physics: Condensed Matter 1997, 9, 767. [Google Scholar]
- (67).Elstner M The Journal of Physical Chemistry A 2007, 111, 5614. [DOI] [PubMed] [Google Scholar]
- (68).Köhler C; Seifert G; Gerstmann U; Elstner M; Overhof H; Frauenheim T Phys Chem Chem Phys 2001, 3, 5109. [Google Scholar]
- (69).Seifert G The Journal of Physical Chemistry A 2007, 111, 5609. [DOI] [PubMed] [Google Scholar]
- (70).Köhler C; Seifert G; Frauenheim T Chemical Physics 2005, 309, 23. [Google Scholar]
- (71).Köhler C; Frauenheim T; Hourahine B; Seifert G; Sternberg M The Journal of Physical Chemistry A 2007, 111, 5622. [DOI] [PubMed] [Google Scholar]
- (72).Helgaker T; Jorgensen P; Olsen J Molecular Electronic-Structure Theory; Wiley, 2014. [Google Scholar]
- (73).Liechtenstein AI; Anisimov VI; Zaanen J Physical Review B 1995, 52, R5467. [DOI] [PubMed] [Google Scholar]
- (74).van der Marel D; Sawatzky GA; Hillebrecht FU Physical Review Letters 1984, 53, 206. [Google Scholar]
- (75).Cococcioni M; de Gironcoli S Physical Review B 2005, 71, 035105. [Google Scholar]
- (76).Mosey NJ; Carter EA Physical Review B 2007, 76, 155123. [Google Scholar]
- (77).Anthon C; Bendix J; Schäffer CE Inorganic Chemistry 2003, 42, 4088. [DOI] [PubMed] [Google Scholar]
- (78).Lever ABP. Inorganic Electronic Spectroscopy; Elsevier, 1984. [Google Scholar]
- (79).Gruden M; Stepanovic S; Swart M Journal of the Serbian Chemical Society 2015, 80, 1399. [Google Scholar]
- (80).Gruden-Pavlović M; Stepanović S; Perić M; Güell M; Swart M Phys Chem Chem Phys 2014, 16, 14514. [DOI] [PubMed] [Google Scholar]
- (81).Stepanović S; Andjelković L; Zlatar M; Andjelković K; Gruden-Pavlović M; Swart M Inorganic Chemistry 2013, 52, 13415. [DOI] [PubMed] [Google Scholar]
- (82).Neese F; Petrenko T; Ganyushin D; Olbrich G Coordination Chemistry Reviews 2007, 251, 288. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.