

Abstract
Bioactive lipid mediators resulting from the metabolism of polyunsaturated fatty acids (PUFA) are controlled by many pathways that regulate the levels of these mediators and maintain homeostasis to prevent disease. PUFA metabolism is driven primarily through three pathways. Two pathways, the cyclooxygenase (COX) and lipoxygenase (LO) enzymatic pathways, form metabolites that are mostly inflammatory, while the third route of metabolism results from the oxidation by the cytochrome P450 enzymes to form hydroxylated PUFA and epoxide metabolites. These epoxygenated fatty acids (EpFA) demonstrate largely anti-inflammatory and beneficial properties, in contrast to the other metabolites formed from the degradation of PUFA. Dysregulation of these systems often leads to chronic disease. Pharmaceutical targets of disease focus on preventing the formation of inflammatory metabolites from the COX and LO pathways, while maintaining the EpFA and increasing their concentration in the body is seen as beneficial to treating and preventing disease. The soluble epoxide hydrolase (sEH) is the major route of metabolism of EpFA. Inhibiting its activity increases concentrations of beneficial EpFA, and often disease states correlate to mutations in the sEH enzyme that increase its activity and decrease the concentrations of EpFA in the body. Recent approaches to increasing EpFA include synthetic mimics that replicate biological activity of EpFA while preventing their metabolism, while other approaches focus on developing small molecule inhibitors to the sEH. Increasing EpFA concentrations in the body has demonstrated multiple beneficial effects in treating many diseases, including inflammatory and painful conditions, cardiovascular disease, neurological and disease of the central nervous system. Demonstration of efficacy in so many disease states can be explained by the fundamental mechanism that EpFA have of maintaining healthy microvasculature and preventing mitochondrial and endoplasmic reticulum stress. While there are no FDA approved methods that target the sEH or other enzymes responsible for metabolizing EpFA, current clinical efforts to test for efficacy by increasing EpFA that include inhibiting the sEH or administration of EpFA mimics that block metabolism are in progress.
Keywords: PUFA, sEH, Oxylipin, Epoxide, Lipid metabolism
5.1. Introduction
5.1.1. Production of Hormones and Other Chemical Mediators
Lipids are an important component of human health, providing a source of energy, maintaining cellular integrity and acting as regulators of cell signaling. These bioactive lipids include steroids, diacyl glycerol (DAG), sphingolipids, phosphatidylinositol phosphate (PIP), phosphatidylcholine (PC), and polyunsaturated fatty acids (PUFA). They range in function from energy storage and generation through beta-oxidation (PUFA), cellular proliferation (PIP), insulin regulation (DAG), cellular protection (sphingolipids), pulmonary function (PC), and maintaining cell structure (cholesterol). However, the metabolism of these lipids often produces more potent biological mediators than the parent molecule. For example, PUFAs, which circulate through blood as triglycerides or as free fatty acids, can also be incorporated into adipose tissue or the membranes of cells that are further released upon insult or response to cell signaling, and their metabolism produces potent inflammatory and anti-inflammatory compounds that have profound effects on the body. Due to their potent effects, inhibitors that alter their metabolism represent one of the earliest drug targets of the pharmaceutical industry. For example, aspirin was discovered in the late 1800s although its mechanism of action as both a reversible and irreversible cyclooxygenase (COX) inhibitor that blocked the formation of prostaglandins (PG) was not identified until the early 1970s, and is still debated [1]. Blocking COX activity and the formation of inflammatory PG compounds resulted in one of the largest selling classes of drugs, NSAIDs, on the market today. In addition to COX metabolism, PUFA are also metabolized by the lipoxygenase (LO) and cytochrome P450 (CYP450) enzymes that have more recently attracted attention for their potential in modulating disease. LO inhibitors are targeted for their ability to inhibit the formation of inflammatory leukotrienes, and Zileuton, a 5-LO inhibitor, is used for the treatment of asthma [2] by blocking the formation of inflammatory leukotrienes. In contrast to COX and LO metabolism, the CYP branch of PUFA metabolism results in the formation of both inflammatory hydroxylated metabolites as well as ant-inflammatory fatty-acid epoxides (Fig. 5.1). Disease altering strategies targeting the CYP450 branch of the pathway focuses on increasing these beneficial epoxy fatty acids (EpFA). However, so far, few drugs specifically targeting this pathway have reached the market although several currently approved drugs alter EpFA concentrations through their action on enzymes and their metabolites in the CYP450 branch of the arachidonic acid cascade. This chapter will focus on the biological activity of these largely beneficial lipid epoxides, as well as strategies for developing pharmaceutical interventions to increase their concentrations in the body.
Fig. 5.1.

Metabolic fate of polyunsaturated fatty acids Free fatty acids are primarily metabolized through β-oxidation, Cyclooxygenase (COX), Lipoxygenase (LOX or LO), and CytochromeP450 (CYP450). A simplistic overview of the biological action of the resulting metabolites is shown in parenthesis.
COX 1 and COX 2 metabolize PUFA to PGH2 which is the precursor to other inflammatory prostanoids and thromboxanes that regulate the immune system (PGD2), increase pain and inflammation (PGE2) and control platelet aggregation (PHI2 and TXA2). [108]
5-LO metabolizes PUFA to 5-HpETE or 15-LO to 15-HpETE, both are precursors to inflammatory leukotrienes and cytotoxic leukotoxins. These metabolites are important in exacerbating asthma by acting as powerful bronchoconstrictors, and they can also sustain inflammatory reactions through chemotaxis of inflammatory mediators. LO metabolism of omega-3 fatty acids result in pro-resolving lipid mediators called Resolvins, Protectins and Maresins. [139]
CYP450 metabolizes PUFA to anti-i nflammatory epoxy fatty acids (EpFAs), n-terminal hydroxylated n-HETE, ω−1 oxidation, or allylic hydroxylations. 20-HETE regulates blood pressure by acting as a potent vasoconstrictor in kidneys and preventing sodium reabsorption in nephrons [140, 141]. The biological significance of mid-chain hydoxylations is less understood; however, biological significance has been observed with 12-HETE in corneal inflammation and neovascularization [142, 143]. Formation of EpFA, particularly if induced, is primarily accomplished by CYP2C and CYP2J subfamily; however, other CYP enzymes can generate EETs [144]. The EpFA act as homeostatic regulators to other metabolites in this pathway by stabilizing mitochondria, reducing ROS, decreasing ER-stress [20] and inflammation [15], regulating the vascular endothelium [88] and increasing bronchodilation [145]
5.1.2. Polyunsaturated Fatty Acids: The Essential Fatty Acids
PUFA are named for the presence of two or more double bonds in the mid-long chain carbon backbone that ranges in length from 16 to 24 carbons or longer. They are considered essential because the body cannot synthesize them naturally and must consume them in order to maintain health; however, with the proper precursors, PUFAs can be altered and interconverted. Depletion of either omega-3 or −6 PUFA result in serious side effects such as neuronal and vision impairment, skin anomalies, thrombocytopenia and intellectual disability [3]. PUFA in the body are either circulating as free fatty acids or incorporated into glycerides and cellular membranes. Upon injury or stress, PUFA are released from cell membranes by phospholipase acetyltransferases (PLA) and other enzymes. PLA2 liberates PUFA from the triglycerides to release free fatty acid (FFA), but also PLC and PLD further act to increase FFA in circulation (Fig. 5.2). Once freed from the plasma membrane, FFA are rapidly metabolized by β-oxidation or other enzymatic metabolism [4] (Fig. 5.1). As mentioned above, the three main enzymes responsible for non-catabolic metabolism of PUFA are COX, LO and CYP450. The COX and LO enzymes form primarily inflammatory mediators, and therapeutic interventions focus on blocking the formation of these compounds. In contrast, the CYP450 metabolism results in the formation of both hypertensive and inflammatory hydroxylated compounds, as well as and anti-hypertensive and anti-inflammatory epoxide compounds. Therapeutic interventions discussed here focus on increasing the concentration of the anti-inflammatory epoxide metabolites.
Fig. 5.2.

Formation of EpFA
Polyunsaturated fatty acids (PUFAs) differ in both structure and function based on number of carbons and location of double bonds. They are incorporated as glycerides in fat cells, cellular membranes or circulating micelles and are liberated to free fatty acids by different phospholipases (PL) that act upon different areas of the glyceride or phospholipid (A). EpFA are formed through the oxygenation of FFA by CYP450. The metabolism of Arachidonic Acid (AA, 20:4 n-6) by cytochrome P450 yields EpFAs, epoxyeicosatrienoic acids (EET), which are further degraded by the soluble epoxide hydrolase into dihydroxyeicosatrienoic acids (DHET). The epoxide and diol on the 11,12 position is shown, but similar regioisomers are possible on all the double bonds in the PUFA. The n-6 fatty acid linoleic acid, LA, has been attributed to largely inflammatory epoxides, EPOMES; however, recent studies show that they are only toxic in the presence of sEH, suggesting that the diols of LA, DIHOMES, are responsible for this inflammatory action [137]. The 18:3 omega-3 fatty acid, linolenic acid (ALA), does not seem to have this same inflammatory action. The omega-6 fatty Adrenic Acid, AdA, is named for its abundance in the adrenal gland. Less is known about this PUFA and its metabolites, although the AdA EpFA, dihomoEETs are thought to regulate blood flow to the adrenal gland [138]. n-3 fatty acids alpha eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) form EpFA epoxyeicosatetraenoic acids (EEQs) and epoxydocosapentaenoic acids (EDPs) respectively. Omega-3 EpFA are largely beneficial with anti-inflammatory and pain resolving properties in vitro and in vivo. Little biological activity has been associated with the diols of Ada (dihomoDHET) or the diols of EPA and DHA, dihydroxyeicosatetraenoic acids (DHEQs) and dihydroxydocosapentaenoic acids (DiHDPAs) respectively. (see [86] for review)
5.2. Epoxy Fatty Acids Are Therapeutic Targets for Disease
5.2.1. Epoxy Fatty Acids (EpFA) Are Important Signaling Molecules That Are Regulated by Their Metabolism
EpFA are formed from the activity of CYP450, a large class of metabolizing enzymes that oxidize fatty acids as well as xenobiotics using heme as a co-factor. The addition of molecular oxygen results in the formation of compounds from the epoxidation of double bounds, end terminal hydroxylation or allylic oxidation [5] each with unique biologies and roles in disease. EpFA have positive beneficial effects on maintaining endothelium function, inflammation and cellular oxidative stress. As early as 1986 and continuing through present day, CYP450 metabolites were identified as influencing blood pressure and renal function by regulating vascular smooth muscle proliferation through MAP kinase signaling pathways [6], anti-aggregation of platelets by decreasing leukocyte adhesion to endothelial cells [7], and regulating vascular tone. Vascular tone, in part, is regulated by the balance of hydroxylated and epoxygenated CYP450 PUFA metabolites. The 20-hydroxy-fatty acids (20-HETE) act as vasoconstrictors by hyperpolarizing vascular smooth muscle cells through activation of the protein kinase C pathway, while the epoxy fatty acids hyperpolarize vascular endothelium through activation of voltage-gated potassium (BK) channels. In this way, the two metabolites act as homeostatic regulators to prevent pathological changes in vascular tone [8, 9]. Although increased 20-HETE results in endothelium dysfunction and increased hypertension, increased EpFA decrease blood pressure, but has not been shown to cause hypotension [10, 11]. This provides strong evidence that EpFA are homeostatic regulators of endothelium function [12, 13]. EpFA also show other beneficial biologies, including potent anti-inflammatory effects and decreasing the endoplasmic-reticulum (ER) stress response pathway [14]. EpFA act as anti-inflammatory agents by reducing the nuclear translocation of NF-kappaβ, thereby preventing the transcription of a number of inflammatory cytokines and synthesis of inflammatory eicosanoids [15, 16]. Furthermore, the diol metabolite of EpFA metabolism drives monocyte chemotaxis in response to monocyte chemoattractant protein, MCP-1. Thus preventing the metabolism of EpFA by the sEH further regulates inflammation by decreasing monocyte infiltration [17]. EpFA have demonstrated a wide range of beneficial effects in animal models of disease [18] which is not fully explained by anti-inflammatory or endothelial homeostasis, but possibly explained by the regulation of ER-stress by EpFA. ER-stress is a protective mechanism deployed by the cell to overcome cellular stress and prevent deleterious mutations. The ER-stress pathway is a homeostatic mechanism regulating cellular responses to the presence of increasing misfolded protein resulting from oxidative, physiological or pathological stressors. Although a fundamental mechanism in maintaining homeostasis, this response is often upregulated in disease, and if not controlled, will result in increased inflammation or activation of cell death pathways. Preventing or reducing the inflammatory and apoptotic branches of ER-stress is associated with many beneficial disease treatments [19], and EpFA have demonstrated the ability to reduce ER-stress [20]. These ubiquitous mechanisms (inflammation, endothelial function, and ER-stress) underlie many diseases and are regulated by EpFA, which may explain why preclinical models investigating the role of EpFA indicate beneficial results in many different diseases when these lipid mediators are maintained.
In normal cell systems, and increasingly in disease, EpFA are rapidly removed from circulation either because they are re-incorporated back in cellular membranes, metabolized by beta-oxidation, or further degraded by epoxide hydrolases [21]. Metabolism enzymes generally increase polarity of compounds to aid in their elimination from the body, and the metabolism of PUFA are no different. After metabolism by the CYP450, the EpFA are further degraded by epoxide hydrolases into the corresponding polar vicinal diols which diffuse from the cells or are rapidly conjugated and removed from the body. In many diseases, the sEH activity is upregulated compared to healthy controls, thus inhibiting its activity in disease settings indicate a potential target for increasing EpFA as a potential therapy.
The epoxide hydrolases are catalytically active dimers that convert xenobiotic or PUFA epoxides to corresponding diols through an exothermic, 2-step hydrolysis reaction (Fig. 5.3) [22]. There are four structurally related isozymes in the epoxide hydrolase family of enzymes (EPHX1–4) in mammals, in addition to other enzymes that have hydrolase activity such as the leukotriene A4 hydrolase (LTA4H), cholesterol hydrolase, and Peg1/MEST. LTA4H metabolizes leukotriene A4 into the leukocyte recruiter, leukotriene B4, to help in the resolution of inflammation. While the functions of PEG/MEST and EPHX4 are not known, isozymes EPHX 1–3 are capable of metabolizing EpFA and differ in cellular location and substrate preference. EPHX1 (mEH), is named for its cellular location: it is found in microsomal membrane fractions in tissue and is active on polyaromatic hydrocarbons found in a variety of xenobiotic epoxides and is also capable of metabolizing EpFA. Conversely, EPHX2 is similarly named as the soluble epoxide hydrolase (sEH) after its predominant location in the cytosolic and peroxisomal fractions of the cell. The EH activity is located at the C-terminus portion of the protein, and the N-terminal portion of sEH has phosphatase activity while the N-terminus of mEH is anchored to the membrane. While mEH is capable of metabolizing both aromatic and aliphatic epoxides, catalytic turnover by the sEH is greater for mono and disubstituted epoxides, making EpFA a good substrate for this enzyme. However, trans, di, tri and even tetra-substituted epoxides can be turned over sometimes with a low Km [23]. Cholesterol epoxides and the squalene epoxide precursor to lanosterol are also substrates of mEH, although the cholesterol epoxide hydrolase and lanosterol synthase are the primary routes of metabolism for these compounds [24]. EPHX3 is also capable of hydrolyzing EpFA with similar efficiency (Kcat/Km) as that of sEH, but a > 10x higher Km indicates that EpFA of arachidonic acid (AA) and presumably other PUFA are a weak substrate for EPHX3 compared to their affinity for sEH or mEH [25].
Fig. 5.3.

Mechanism of epoxide to diol metabolism by the soluble epoxide hydrolase (sEH)
Two acidic tyrosines are oriented by pi-stacking to bind to and polarize the epoxide moiety. sEH inhibitors functionally mimic transient intermediates and the transition state of the enzyme (middle panel). The NH groups of the urea, amide, carbamate or other electronegative groups possibly encourage a salt bridge formation, for example between a polarized urea and the catalytic aspartic acid, likely stabilized by hydrogen bonds with the tyrosine. [146]
5.2.2. Human Polymorphisms Altering Epoxide Hydrolase Activity Affect EpFA Concentrations and Correlate Epoxides with Disease States
Most EH related mutations in humans that are associated with disease states are associated with EPHX2 (sEH). There are four main mutations that affect enzyme activity, two that do not alter function, and as many as 20 less characterized non-coding mutations that can occur in up to 20% in human populations [26, 27]. The four most common coding mutations result in two gain of function mutations (K55R and C154Y) and two reduced function mutations (R287Q and R103C mutations results in decreased dimerization). These mutations affect the velocity of epoxide hydrolysis while leaving the selectivity for substrate binding and phosphatase activity apparently unchanged [28, 29] Considering that the concentration of the sEH enzyme (ranging from 3 nM in lung to 400 nM in liver) is often higher than that of the EpFA substrate (low nM in tissue [30–32]), and the concentration of EH enzymes, such as EPHX1 and 3, can convert epoxides to diols, it should be taken into consideration that mutations that only slightly affect enzyme efficiency may not significantly affect EpFA concentration. However, mutations in the sEH and prevalence of SNPs associated with different diseases suggest that this enzyme plays an important role in disease, as described below.
Mutations in sEH Are Biomarkers of Anorexia Nervosa
In a genome wide association study of patients with anorexia nervosa (AN), investigators identified rare mutations in non-coding regions of the EPHX2 that correlate with disease susceptibility. The activity of the mutations was not determined, but investigations in the levels of epoxide:diol ratios found they were decreased in ill AN patients compared to recovered AN or healthy populations, suggesting an upregulation in the conversion of epoxides to diols. Furthermore, the loss of function mutation, R287Q, occurred less frequently in AN patients compared to healthy populations, further correlating increased sEH activity to higher risk of severe AN [33–36]. Although the exact mechanism for the involvement of EpFA in AN is unknown, considering the impacts of diet and aversion to food in this disease, understanding the role of altered lipid signaling in this disease is of increasing interest.
Reduced sEH Activity Protects Against Familial Hypercholesterolemia
Familial hypercholesterolemia (FH) is a hereditary disease that causes increased plasma cholesterol concentrations resulting from a defective hepatic low-density lipoprotein receptor (LDLR). In a targeted analysis approach that studied the prevalence of the reduced function sEH mutation, R287Q, in 8 generations of families with FH, Sato et al. discovered that family members with the R287Q mutation had normal cholesterol levels compared to members with the normal 287R allele. Cholesterol was unchanged in healthy humans with the R287Q mutation [37]. Preclinical investigations further support the role that sEH has in reverse cholesterol transport, and possibly explain the protective effects on inhibiting its activity in LDLR related disease. For example, in Ldlr−/− mice that mimic the FH disease, treatment with the sEH inhibitor (sEHI) t-AUCB decreased atherosclerosis plaques through increasing HDL synthesis and efflux of cholesterol from adipose lesions compared to vehicle treated controls. Further investigation demonstrated that sEHI treated mice had increased ATP binding cassette transporter A1, which is responsible for HDL synthesis through efflux of cellular cholesterol to extracellular apoA1 [38].
sEH Mutations Are Associated with Outcome Measures in Vascular Disease
Impaired vascular function contributes to heart disease, age-related vascular decline, blood pressure and stroke. EpFA act as vasodilators by opening calcium-activated potassium channels that relax the vascular smooth muscle and are thought to mitigate vascular disease [39]; therefore, it is hypothesized that decreased EpFA would contribute to heart disease, and in fact gain-of-function sEH polymorphisms are associated with increased cardiovascular diseases. For example, pregnant women with preeclampsia had higher frequency of the K55R gain of function mutation and lower methylation of the EPHX2 promotor region, causing higher expression, than healthy pregnant women [40]. In another study that analyzed sEH polymorphisms with frequency of stroke in African American and Caucasian populations participating in the Atherosclerosis Risk in Communities study, investigators identified a rare EPHX2 mutation in African Americans that was not found in Caucasian populations. The mutation resulted in increased sEH activity and a twofold to threefold increased risk of stroke. Although the infrequency of the mutation complicated statistical analysis, and larger sample sizes were needed to determine significance. Additional haplotypes in the sEH gene correlated with both increased and decreased risk of stroke in both races [26]. Although the effect of these gene sequences on enzyme activity is not known, further studies analyzed site-directed mutagenesis on survival rates of ischemic cells in vivo and found that decreased sEH activity with the R287Q mutant conferred to increased survival after inducing a simulated stroke environment of depleted glucose and oxygen [41]. This mutation was also associated with lower risk of stroke in Europeans [42]. Although a large Danish study failed to identify correlations in sEH SNPs and stroke, myocardial infarction or ischemic heart disease [43]. A large study in Swedish men found correlations between the gain-of-function mutation, K55R, with increased risk of stroke [44]. Additionally, Hawaiian Asians with dementia but without prior ischemic injury compared to healthy age-matched controls had increased 14,15 DHET in cortical brain tissue from patients with dementia; however, heterozygous carriers of the reduced function R287Q SNP had increased markers for plaques compared to healthy patients [45]. These data are at odds for understanding if sEH activity protects or contributes to the progress of AD especially considering that dementia correlates with a decreased EET:DHET ratio, suggesting a negative role of sEH in this disease. However, because the R287Q mutation affects dimerization, it’s possible that these heterozygous carriers still have functional sEH protein.
In addition to the sEH, the mEH also hydrolyses EpFA into corresponding diols, although with much less efficiency. As discussed later, the association of the mEH in the lipophilic endoplasmic reticulum membrane and with the cytochrome P450 that oxidize epoxy fatty acids may result in greater mEH contribution to diol formation than anticipated from the concentration and kinetic constants of the mEH. Mutations in mEH are also associated with several diseases, including cancer, preeclampsia, seizure, neurological disease, drug-dependence and COPD. However, because this enzyme is primarily responsible for metabolizing aromatic xenobiotic epoxides, it is uncertain if these associations are a result of decreased epoxides in the body, or from accumulation of toxic xenobiotics (reviewed in [46]). Interestingly, sEH expression in the brain is mostly localized in glial cells of the brain, while mEH is found more throughout the brain, suggesting either a specific role of sEH or more significant contribution of mEH to EpFA metabolism in the brain [47]. Recent studies demonstrate that sEH activity outside the brain can influence depression. In this study, overexpression of sEH in the liver resulted in increased depression in mice, and genetic deletion of sEH in the liver had a positive effect in treating stress-induced depression. This study suggests that peripheral-acting EpFA can influence CNS diseases [48].
5.2.3. Laboratory Knockout Models Identify Beneficial Effects of Increasing EpFA in Treating Preclinical Models of Disease
A homozygous mouse model deleting EPHX2 stabilize EpFA and further demonstrate biological activity of increasing EpFA in disease. The first incidence using sEH−/− mice observed decreased blood pressure on a high-salt diet compared to wild-type mice [49], and sEH−/− mice are used to further demonstrate that increasing EpFA and decreasing diol formation resolves multiple disease states. For example, sEH null mice demonstrate beneficial effects in decreasing inflammation, maintaining the vascular endothelium, and resolving neuroinflammatory diseases. Specifically, genetic sEH −/− mice demonstrated accelerated wound healing, reduced inflammation in inflammatory bowel disease and LPS-induced inflammatory models, improved insulin signaling in a Type II model of diabetes, reduced cisplatin-induced kidney damage, reduced niacin flushing, reduced arteriosclerosis, smaller infarct size in cerebral artery occlusion/reperfusion injury, decreased hepatic and arterial fibrotic disease, decreased autism, depression, and Parkinson’s disease (see [50, 51] for review). Recently, tissue specific sEH knockout models demonstrate the local effect that EpFA have on tissues. Mice with podocyte specific sEH knockout were protected from hyperglycemia-induced renal injury resulting from high fat diet or STZ-induced diabetic hyperglycemia [52]. In further support of the importance of this pathway in disease, the analgesic effects of morphine were attenuated in CYP-null mice that lack the ability to generate EpFA, and in mice administered compounds that inhibit fatty acid oxidizing CYPs [53–55]. Thus genetic models that increase EpFA concentrations through deletion of their metabolism support the beneficial effects of EpFA in treating disease, and models that decrease the ability of animals to form EpFA reduce these beneficial effects, suggesting an essential role of EpFA in disease.
5.3. Clinical Approaches for Increasing EpFA Concentrations
The EpFA to diol ratio are reduced in several disease sates suggesting that increasing the EpFA concentrations in the body could provide beneficial effects in preventing or treating disease (Table 5.1). There are many different approaches for increasing concentrations of fatty acid epoxides for the treatment of disease. Strategies include direct administration of PUFA or EpFA, inhibition of EpFA metabolism through chemical inhibitors of sEH (sEHI), induction of EpFA formation through CYP modulators, or directly mimicking the fatty acid epoxide.
Table 5.1.
Altered ratios of EpFA: diol correlate to disease outcomes
| Alzheimers disease (AD) [130] |
| AD patients with and without type 2 diabetes (T2D) had increased DHET compared to healthy controls. However, there were no effects following adjustments for multiple comparisons. |
| Arthritis [131] |
| In synovial fluid of arthritic vs. normal joints, 11,12-DHET and 14,15-DHET were higher in affected joints of people with unilateral osteoarthritis. In addition, these and 8,9-DHET were associated with worse progression over 3.3 years. |
| Anorexia Nervosa [34, 35] |
| Ill anorexia nervosa patients have higher DHA diol metabolites 19,20 DiHDPE:EpDPE compared to either recovered AN patients or healthy human subjects, while both ill and recovered AN patients have higher ALA diol metabolites 15,16 DiHODE:EpODE ratios compared to healthy subjects. |
| Peripheral arterial disease [132] |
| Increased 8,9 DHET correlated with increased risk of coronary and cerebrovascular events in patients with peripheral arterial disease. |
| Coronary artery disease [133] |
| Decreased EETs in patients with obstructive coronary artery disease compared to healthy controls |
| Depression [134] |
| In patients with major seasonal depression syndrome, sEH-derived oxylipins (12,13 DiHOME, 7,8- and 19,20 DiHDPE), in addition to other eicosanoids, increased in winter compared to summer-fall, while 14,15 EET and corresponding diol both decreased in the winter. |
| Preeclampsia [135] |
| In preeclamptic women 14,15-DHET was higher in urine samples compared to healthy pregnant women. |
| Vascular dementia [136] |
| In patients with cognitive impairment, an increase in 9,10- and 12,13 DiHOME: EpOME was associated with poor performance in function but not memory. |
5.3.1. PUFA Supplementation
Omega-3 supplementation is a widely investigated approach for improving health or treating disease, and the resulting fatty acid epoxides are thought to account for some of the beneficial effects observed with their supplementation [31]. However, randomized clinical trials often fail to support the efficacy observed in meta-analysis of diet behaviors and disease risk [56]. Increased sEH activity in disease would increase EpFA metabolism, thus limiting efficacy and may account for the inconsistent therapeutic benefits reported with omega-3 supplementation. This is possibly due to lipid peroxidation products in some omega-3 lipids. Omega-3 lipids are inherently unstable and subject to oxidation, often as much as 200% increase in PUFA peroxides after only 22 days of storage [57], and further oxidation results in aldehydes that are not tested for in commercial settings [58]. The oxidized products are associated with cellular stress and increased cellular toxicities and are hard to detect or control outside of sophisticated analytical labs and thus not a well-controlled approach for improving health or treating disease [59]. Similarly, supplementation with EpFA is also subject to oxidation and provides further challenges because EpFA are also rapidly degraded by acid hydrolysis in the stomach or are eliminated through first-pass metabolism in the liver. An alternative administration route, for example direct application to affected tissue, would avoid first-pass metabolism; however, other metabolizing enzymes would still contribute to rapid elimination. For example, enteric coating to bypass the acid in the gut may provide options for treatment of intestinal diseases; however, metabolizing enzymes in the gut or microbiota containing epoxide hydrolases would further limit their effectiveness. Ocular therapy provides another application to avoid first-pass metabolism; however, recent studies have implicated increased sEH expression as contributing to disease progression in a mouse model of diabetic retinopathy as well as in samples from humans with diabetic retinopathy [60]. Thus, ocular application to treat retinopathy would be susceptible to sEH metabolism possibly demonstrating that disease treatment by direct EpFA supplementation will be difficult due to sample stability and metabolic instability.
5.3.2. Increase Formation of EpFA
Clinical and preclinical strategies for investigating biological activity of EpFA have focused on preventing their metabolism either through inhibition of the sEH enzyme or through synthesizing stable EpFA mimics. A third approach involves increasing their formation through CYP450 activity. Preclinical data indirectly suggests that increased CYP activity could reduce certain pathologies. In a mouse model of inflammatory pain, co-administration of a CYP modulator, omeprazole, and the sEHI, TPPU, resulted in increased efficacy compared to either compound administered alone. Omeprazole induces CYA1A1, 1A2, 2B1 and 3A1 and inhibits 2D2 and 2C. CYP2C is a more efficient at forming epoxides of AA (12/8 ratio of epoxidation/hydroxylation) compared to the other CYP isoforms which form hydroxylated metabolites in greater amounts than EpFA; however, because the other isoforms also form EpFA, increasing their activity would also increase EpFA as well as hydroxylated PUFA metabolites [61]. This is consistent with the results published in this study showing that the EpFA and hydroxylated metabolites were increased in the plasma of mice treated with both compounds [62]. Hydroxylated metabolites, especially hydroxylated metabolites of 18:2 linoleic acid (LA), are often associated with increased inflammation and oxidative stress [63–66], which brings up another complication with targeting CYP induction to increase EpFA in that it could also increase the production of inflammatory PUFA metabolites that could negate the beneficial effects of EpFA. Preclinical studies demonstrate that administration of ketoconazole, an inhibitor of CYP2A6, 2C19 and 3A4, reduces pain in an inflammatory mouse model by reducing the formation of hydroxy octadecadienoic acid (HODE), the mono-hydroxy metabolite of LA that causes hyperalgesia through activation of TRPV1 channels [61, 67]. These studies demonstrate that increasing CYP activity is a complicated approach given the promiscuity of the CYP enzymes for forming both anti-inflammatory EpFA and pre-inflammatory hydroxylated PUFA metabolites, as well as influencing the metabolism of other xenobiotics. Thus, increasing CYP activity does not present a viable strategy for clinical utility, and clinical approaches designed to increase EpFA should take into consideration drug-drug interaction potentially affecting EpFA formation if patients are currently taking CYP modulators.
5.3.3. Stabilizing EpFA Through Small Molecule Inhibitors of SEH
To overcome metabolic instability of EpFA, sEH inhibitors (sEHI) were identified that mimic the transition state of the epoxide ring opening. The pharmacophore of sEHI consist of a urea, carbamate, or amine heterocycle that mimic the interaction of the substrate and enzyme by forming one or more hydrogen bonds between the NH and aspartate (Fig. 5.3.) The inhibitors contain hydrophobic regions that allow interaction with the enzyme and maintain high specificity over the mEH, while specifically placed polar side groups increase druggable properties such as water solubility and metabolic stability and increased oral bioavailability. The first generation of sEH inhibitor, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (AUDA) [68], was a small molecule designed to mimic to 14,15 EET with a dodecanoic acid to mimic the aliphatic chain and carboxylic acid present at the α end of the fatty acid, an adamantine to mimic the ω hydrophobic end of the fatty acid, and urea to act as a mimic to the epoxide and also inhibitor to the sEH enzyme [69]. AUDA is a potent sEHI, with an IC50 of 3–60 nM, but is short lived in the body (T1/2 = 2.9 h in canine PK) due to oxidation of the adamantine and beta oxidation of the alkyl chain, thus limiting its use in vivo to conditions where long half-life is not required. Since AUDA also mimics epoxy fatty acids, it shows biological activity in tissues lacking sEH. Such dual action may improve efficacy, but it complicates interpretation when AUDA is used to investigate physiology [70]. Second generation sEHI replaced the adamantine with a more stable benzyl-trifluoromethoxy and alkyl chain with a benzenesulfonamide to maintain potency with the sEH enzyme while improving solubility and PK stability. ( [71] for review). The most potent inhibitors of the sEH are reversible and tight binding with a slow off-rate from the enzyme and sub-nanomolar potency. They also show improvements in PK for increased chances of testing clinical relevance for treating disease. Kinetic studies demonstrate optimization of new inhibitors by targeting drug occupancy time on the enzyme and demonstrate that inhibitors remain bound to the enzyme long after they are detectable by classical PK techniques. These data correlate with in vitro half-life measurements and provide novel techniques for selecting potent lead compounds for clinical development [72].
5.3.4. Mimics of EpFA
As an alternative to blocking the metabolism of EpFA in order to increase their concentrations, another strategy focuses on creating more stable mimics of EpFA while retaining the active moiety of the original compound. Epoxy fatty acid mimics investigated in clinical trials often replace the epoxide with functional groups that retain beneficial effects in vivo while also blocking β-oxidation by adding functional groups to the α-hydroxy portion of the fatty acid. Mimics of epoxy fatty acids remove the complexity of enzyme potency and off-rate kinetic parameters of small molecule sEHI that can complicate translation from the lab bench to clinical efficacy, but due to the unknown target for EpFA activity and complex structure diversity of EpFA, selecting one isomer for development could be overly simplistic. Many of the mimics were based on earlier studies in agricultural chemistry where various groups resistant to epoxide hydration were used to replace the epoxide of natural juvenile hormone while presumably retaining efficacy at the putative receptor [73].
5.4. Clinical Development of Compounds That Alter EpFA Concentrations
One of the first identified biological targets of EpFA action was modulation of the microvasculature and inflammation which underlies many pathological disease states. Clinical targets testing efficacy of increasing EpFA include hypertension, cardiovascular pulmonary disease, stroke, diabetes and pain. Preclinical studies provide evidence that altering concentrations of regulatory lipid mediators formed from metabolism of PUFA could provide beneficial effects in many other diseases, including neurological disease and bone degeneration. Despite the unknown target, multiple clinical trials investigating the therapeutic potential of both small molecule inhibitors of the sEH as well as EpFA mimics have been initiated. These trials are highlighted in Table 5.2 and the mechanisms are explained in detail below along with the benefits and liabilities of these approaches.
Table 5.2.
Clinical development candidates of small molecule sEHI and EpFA mimics
| Small molecule sEH inhibitors | Clinical Trials | |
|---|---|---|
| AUDA | 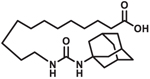 |
Clinical trial
NCT00654966: Microvessel Tone in patients with heart failure. Status: Complete. Healthy humans and patients with heart failure challenged with topical urotensin II, a potent vasoconstrictor, and treated with sEHI [74]. |
| TPPU | This compound has not been investigated in clinical trials but is the most frequently used compound in preclinical research. | |
| AR9281 | 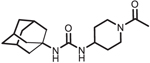 |
Clinical trial
NCT00847899: Evaluation of sEHI in Patients with Hypertension and Impaired Glucose Tolerance. Status: Complete. Single and multiple day testing up to 8 days at doses up to 1.2 g/day (400 mg every 8 h) were well tolerated [129]. Data from Phase II clinical trials not published. |
| GSK2256294 | 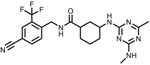 |
Clinical trial
NCT01762774: A Study to Assess the Safety,
Tolerability, PK and PD of Single and Repeat Doses of GSK2256294 in Healthy Volunteers and Adulate Male Moderately Obese Smokers. Status: Complete. Doses were well tolerated and attenuated smoking related endothelial dysfunction. |
| EC1728 (t-TUCB) | Clinical trials are in development for companion animals. | |
| EC5026 | 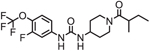 |
Clinical trial
NCT04228302: Safety, Tolerability, and Pharmacokinetics of Oral EC5026 in Healthy Subjects. Status: Enrolling |
| EpFA Mimics | Summary of results | |
| CMX020 | 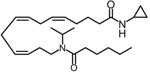 |
Clinical trial ACTRN12615000885594: A Study to Evaluate the Safety and Analgesic Efficacy of Oral CMX-020 in Subjects with Symptoms of Sciatica Resulting from Lumbosacral Radiculopathy. ACTRN12616001435471: A Phase 2 Study to Assess the Efficacy and Safety of CMX-020 in Treating Osteoarthritis. Status: Phase 1 studies complete. Enrolling for Phase 2. |
| OMT-28 | Structure not disclosed |
Clinical trial
NCT03906799: Study on OMT-28 in Maintenance of Sinus Rhythm in Patients with Persistent Atrial Fibrillation. Status: Enrolling |
| Icosabutate |
Clinical trial
NCT04052516: A Phase 2b Study of Icosabutate in Fatty Liver Disease. Status: Phase 1 studies complete. Enrolling for Phase 2b. |
|
| Vascepa (ethyl-EPA) | Status: approved for the treatment of hypertriglyceridemia. | |
5.4.1. Small Molecule sEHI in Clinical Development
The clinical trials registry lists six clinical trials completed with small molecule soluble epoxide hydrolase inhibitors tested in diseases mostly affected by endothelial dysfunction. The following sections will highlight the small molecule sEHI investigated in these clinical strategies to increase EpFA as a mechanism for treating disease.
AUDA
Based on the observation that EpFA open potassium channels and hyperpolarize vascular endothelium resulting in vasodilation [39], investigators in Australia conducted clinical trial NCT00654966 as an exploratory study in humans to determine if AUDA increases EpFA to protect against heart failure by increasing blood flow in the microvasculature. After topical challenge with a potent vasoactive peptide, urotensin II (UII), blood flow was measured in healthy humans or patients with heart failure (HF) treated with and without topical AUDA. UII causes vasodilation and increased blood flow in healthy humans but vasoconstriction and reduced blood flow in patients with HF. AUDA alone caused increase blood flow when administered at the intermediate dose of 0.1 μM in both healthy and HF patient populations. When administered with UII, AUDA was able to reverse the reduced blood flow observed in HF patients, although not back to levels observed in healthy subjects, and significantly increased blood flow in healthy subjects more than when UII was administered alone [74]. The increased vasodilation observed from this study indicate many potential benefits in addition to heart failure that increasing EpFA would have in patients; for example, diabetics and hypertensive patients would benefit from increased vasodilation; however, poor PK probably prevented this molecule from being a viable drug candidate.
AR9281 (UC1153)
AR9281 offered some advantages as a clinical candidate including low IC50 on the rodent sEH, easy synthesis, a surprisingly high water solubility, high selectivity for the sEH, and low mammalian toxicity; however, its poor target occupancy and the speed with which it was metabolized to synthetically complex metabolites were clear liabilities. In preclinical models, AR9281 reduced hypertension and renal injury in an angiotensin induced model of hypertension in rats [75]. Multiple clinical trials were launched by Arete Therapeutics to test the v and efficacy of AR9281 for treating hypertension. Published results from the Phase 1 study found that AR9281 was well tolerated at doses up to 1000 g/day but was rapidly cleared from the body, with a half-life of only 3–5 h in humans. However, high plasma concentrations well above the IC50, and ex vivo assays monitoring sEH activity showing 90% inhibition of the enzyme up to 24 h at the top dose, justified advancing the compound to Phase 2 human efficacy studies. The results of the Phase 2 efficacy studies in hypertensive patients with glucose intolerance were not published, and Arete Therapeutics closed shortly after completing the study, so one assumes that the compound failed to show efficacy. One possible speculation for lack of efficacy was the high metabolic clearance of the compound. For example, the ex vivo determination of the IC90 was based on a 60 min assay; however, AR9281 is a reversible inhibitor that has a kinetic T1/2 on the enzyme of 6 min [76]. Thus, the ex vivo data may not be representative of the in vivo environment where the inhibitor is rapidly metabolized and cleared from the body. Additional measurements of diols in the urine of subjects were combined from all treatments and compared to pretreatment levels and placebo treated subjects as a marker of target engagement. The diols of human subjects treated with AR9281 modestly decreased compared to pre-treated controls, while placebo treated subjects remained unchanged. Epoxide concentrations were not detectable in the urine; thus, the ratio of epoxide:diol change was not monitored and thus impossible to determine if the decrease in diol levels was due to treatment effect or other independent variables. Overall, the data from the Phase 1 trial did not demonstrate convincing data that AR9281 effectively inhibited sEH activity, possibly explaining the lack of efficacy in phase 2 trials. There are situations where rapid clearance such as that demonstrated with adamantane containing compounds like AR9281 and AUDA offer clinical advantages; however, these have not been explored.
GSK225629A4
GSK225629A4 is a potent sEHI that attenuated leukocyte infiltration in mice after exposure to cigarette smoke [77]. Based on this information, clinical trials were initiated to test safety and endothelial dysfunction in both healthy humans and obese smokers at doses of 2–20 mg administered as a single or repeat oral dose for 14-days. Endpoints included safety and endothelial dysfunction measured by forearm venous occlusion plethysmography after intra-articular challenge with vasodilator, bradykinin. Overall, GSK225629A4 was well tolerated with favorable PK and T1/2 equal to 19–30 h. in healthy humans and 41–49 h in overweight smokers. One significant adverse event was reported, but ultimately considered non-drug related as the subject had a history of this event prior to the clinical trial. Otherwise, only mild-moderate adverse events of headache and contact dermatitis were reported. Headache occurred with similar occurrence between the placebo and treated groups, and contact dermatitis around the site of ECG electrode placement was reported in nine healthy patients receiving active compound and none in the placebo or obese smokers receiving active or placebo compound. Ex-vivo sEH activity was measured in the plasma of treated patients as a function of EET hydrolysis after 30-min incubations with 14,15 EET, and investigators found that >80% inhibition of EET to diol hydrolysis was observed after repeat dosing in all dose cohorts. In obese smokers, vasodilation after bradykinin injection was reduced compared to healthy subjects, as expected. After administration of GSK225629A4 vasodilation improved in a dose and time dependent manner in obese smokers after 1 and 14-d administration compared to placebo control. Although some limitations need to be considered, such as small sample size and non-smoking obese control subjects. Furthermore, as an added marker of safety, the sponsor measured VEGF and plasma fibrinogen after single and repeat dosing in response to potential safety concerns from increased angiogenesis [78, 79] and tumor metastasis [80] in preclinical settings. Both plasma VEGF and fibrinogen were similar across all treatment groups potentially indicating that preclinical angiogenesis observations may not be clinically relevant. Overall, these data suggest that GSK225629A4 is a safe compound with potential efficacy in patients with COPD, a disease exacerbated by cigarette smoke, or other disease affected by dysfunction of the microvasculature.
Given the successful safety profile of GSK225629A4, independent investigators have secured rights to initiate clinical trials with the GSK compound to investigate insulin resistance and stroke as described below.
Insulin Resistance
Preclinical data suggests that anti-inflammatory properties of EETs reduce inflammation in adipose tissue resulting in decreased insulin resistance in several, but not all, rodent models [81–83]. Based on this information, a group at Vanderbilt University in Nashville, Tennessee initiated a Phase 2 clinical trial to investigate the therapeutic potential of GSK225629A4 ability to improve insulin sensitivity in response to glucose infusion. The study is currently recruiting patients, and there are no reported data from the study.
Subarachnoid Hemorrhage (SAH)
Numerous preclinical studies have investigated the role of soluble epoxide hydrolase in exacerbating stroke symptoms and the utility of inhibiting the sEH as a potential treatment option. (Reviewed in [84]) As a result of these data, investigators at the Oregon Health and Science University initiated a Phase 1 and 2 clinical trial with GSK225629A4 in patients with aneurysmal SAH to evaluate effects on length of hospital stay, incidence of new stroke, disposition upon discharge and outcome measures. The study recently finished recruiting patients, but no results have been published.
Neuropathic Pain
As new mechanisms for explaining EpFA beneficial effects has been discovered, clinical activities are targeting diseases with complicated etiologies. For example, the newest clinical development programs that are scheduled to start at the end of 2019 focus on treating neuropathic pain, a disease attributed to pathological ER-stress. EC5026 is the newest sEHI being developed for clinical utility. Recent press releases announced FDA approval of an IND application from EicOsis to test their small molecule inhibitor, EC5026, for the treatment of neuropathic pain in humans, and EC1728 for treating inflammatory and neuropathic pain in companion animals. Many review papers have described the therapeutic potential of increasing epoxy fatty acids for the treatment of both neuropathic and inflammatory pain (reviewed in [18, 85, 86]). Considering the devastating impacts of the opioid epidemic and lack of effective and safe pain medications, there is considerable interest in the outcomes of these clinical trials.
5.4.2. EpFA Mimics in Clinical Development
CMX-020 is a mimic similar to AUDA that is also designed around 14,15 EET. This compound maintains most of the original structure except the olefin on the 14,15 carbon is replaced with a dimethylcarbamoyl and the α hydroxy group is replaced with a cyclopropyl amine to prevent beta-oxidation. In preclinical studies, CMX-020 undergoes rapid elimination, with most of the drug eliminated 1-h after administration; however, the compound exhibits potent pain-relieving properties preclinically in the acetic assay writhing assay and tail flick assay, comparable to that of morphine [87]. It is unknown if CMX-020 metabolism results in epoxy-fatty acid mimics as a result of CYP450 epoxidation, or if CMX-020 acts to inhibit the sEH, similar to carbamates such as GSK225629A4. Based on the potent analgesic effects of CMX-020 and despite the rapid elimination profile, multiple clinical studies were initiated to test the safety and efficacy of CMX-020 in treating painful conditions such as osteoarthritis and sciatic nerve pain in Australia. Results have not been published.
Focused mimics based on the structure of one regioisomer of one PUFA describe the challenges of developing a mimic to a fatty acid epoxide. While CMX-20 chose to mimic the omega-6 AA epoxides, other companies, described below, selected omega-3 fatty acids as the background for their mimics. The epoxides of omega-3 fatty acids are thought to account for some of the beneficial effects of omega-3 supplementation in the diet, and animal models show that they have more potent activity in in vitro and animal models than the omega-6 epoxides [31]. In addition to choosing which omega position of the fatty acids to model the mimic, further complications arise when identifying which regioisomer to target. For example, 11,12 and 14,15 EET are commonly associated with having vasodilator activity compared to the other regioisomers of arachidonic acid [88].
Omeicos is focused on developing another epoxy mimic by creating a transition state mimics of the 17,18 EPA omega-3 epoxide. Their lead compound, OMT-28, is actively recruiting patents for a Phase 2 clinical trial to treat atrial fibrillation. Although the exact structure is not disclosed, detailed structure activity relationship studies identified important characteristics needed to exert antiarrhythmic effects. Atrial fibrillation is a type of arrhythmia initiating from the top chambers of the heart and can lead to increased risk of blood clot, stroke, and heart failure. In vitro models using neonatal rat cardiomyocytes have been used to investigate the anti- or arrhythmic effects of certain drugs and demonstrate that omega-3 fatty acids and the R,S isomer of the 17,18 epoxide of EPA decreased the contraction rate in these cells [89], indicating a potential ability to alleviate atrial fibrillation. These studies further demonstrate the complexity of EpFA biology that could potentially complicate the identification of an active mimic.
Other approaches that mimic the PUFA of omega-3 or −6 fatty acids have been developed. For example, Icosabutate, developed by Northsea Therapeutics, and Vascepa (icosapent ethyl, or ethyl-EPA), an approved product sold by Amarin, are structurally engineered fatty acids both being developed to lower triglycerides in the body. Icosabutate mimics EPA with the addition of 2-bromo butyric acid on the α end, while Vascepa adds a methyl group to the α end to prevent β-oxidation; otherwise, both structures are unaltered compared to EPA. Northsea data presented at the International Liver Congress in 2018 show the compound remains in the non-esterified form longer than EPA as expected, but was metabolized primarily by CYP2C enzymes, suggesting that epoxides are likely formed in the metabolism of this compound [90]. Multiple clinical trials were completed with Icosabutate testing the safety and potential drug-drug interactions with CYP inducers and inhibitors, as well as efficacy in hypertriglyceridemia (NCT01893515) and NASH (NCT04052516). Patients with hypertriglyceridemia receiving 600 mg once daily for 12-weeks had significantly lower triglyceride, very low-density lipoprotein cholesterol, and Apo C-III levels [91]. The NASH study recently started dosing and results are not yet available. There is more published information in icosapent ethyl considering that it is an approved drug for reducing the risk of cardiovascular disease in patients with hypertriglyceridemia. In a placebo controlled clinical trial enrolling 8179 patients, administration of 2 g twice daily of icosapent ethyl significantly reduced ischemic events, including reduced incidence of death [92, 93].
Similar to risks in choosing a mimic to omega-3 or 6 fatty acid, identifying the regioisomer and stereoisomer to mimic further complicates the process. Omeicos is in a unique position by having structure-activity relationship information for atrial fibrillation, but without knowing the target of EH activity, translating this information to other disease areas should not be assumed.
5.4.3. New Therapeutic Approaches Targeting EpFA Through Polypharmaceutical Approaches
Pharmaceutical targets to the COX and LO enzymes focus on preventing the formation of inflammatory prostaglandins and leukotrienes, respectively. NSAID drugs inhibit COX-1 and-2 enzymes and are potent anti-inflammatory compounds by preventing the formation of inflammatory prostaglandins; however, these inhibitors are often accompanied by toxic side effects associated with enzyme distribution. COX-1 enzymes are in most cells and protect the GI mucosa by regulating acid secretion through the EP3 receptor. Inhibition of COX-1 increases gastric release, thus causing GI-toxicity and increased ulcer formation. COX-2 enzymes are found in lymphocytes, red blood cells and synovial cells and are thought to regulate pain and inflammation. Selectively inhibiting COX-2 was thought of as a desirable approach to reduce pain and inflammation while avoiding GI complications. However, while selective inhibition of COX-2 avoided gastro-intestinal ulcer formation, chronic use increased the risk of stroke and cardiovascular disease. Further investigation into the mechanism of this toxicity found that COX-1 inhibition decreases the vasoconstrictor thromboxane A2 synthesis; while COX-2 inhibition decreases the vasodilator, prostacyclin. Non-selective inhibitors would decrease both vasoregulators and maintain homeostasis, while selective COX-2 inhibitors would decrease only prostacyclin, resulting in platelet aggregation and vasoconstriction [94, 95]. Vasodilatory effects of EpFA suggest further protection against endothelial dysfunction associated with COX-2 inhibition, but recent studies suggest an additional mechanism for decreasing COX toxicities results from the reduction of ER-stress [96]. NSAIDs increase ER-stress which has been attributed to the toxicities associated with NSAID use, including cardiovascular toxicity [97–99]. ER-stress also increases COX transcription, which creates a feedback inflammatory mechanism perpetuated by COX-2 activation of IRE1a, a key protein involved in activating the ER-stress pathway. Blocking ER-stress, as has been demonstrated with EpFA, prevent the upregulation of COX, and dual inhibition of sEH and COX are an attractive therapeutic strategy for increasing the benefit of NSAIDs while reducing their toxicity. Many preclinical studies demonstrate the advantages of dual COX-sEH inhibition. For example, the sEH inhibitor, t-AUCB, or NSAID inhibitor, Celecoxib, administered alone do not affect tumor volume or metastasis in a Lewis lung carcinoma mouse model of cancer while treatment with both significantly reduced both tumor volume and metastasis. Previous studies demonstrated that sEH null mice have increased metastasis and tumor volume [100], presumably through increased angiogenesis and VEGF expression; however, clinical trials with GSK’s sEHI failed to show translation of VEGF increases in mice to human patients [101]. Furthermore, preclinical studies identify that EETs are angiogenic and induce endothelial cell proliferation [102–104] through metabolism by COX that produces a potent angiogenic metabolite [105]. Thus, dual inhibition would prevent the formation of this metabolite and could explain the added benefit in tumor models. Additional benefits have been observed in improving survival after tumor-induced cytokine surge in mice. Inflammation is a protective mechanism to eliminate foreign toxicants from the body; however, an intense and rapid inflammatory response, as occurs in sepsis, can be deadly. CAR-T therapy is a promising treatment of cancer that activates the body’s immune system to recognize tumor cells as foreign material. The body mounts an aggressive immune attack to eliminate these tumor cells; however, if too effective, the outcomes also result in sepsis and death. Preclinical studies demonstrate that the dual COX/sEHI, p-TUPB, improves survival rate in a cancer cell debris model of cytokine-surge in mice [106].
Dual inhibition with COX and sEH is an attractive target for treating disease by m inimizing the toxicities associated with NSAIDs while also increasing the efficacy of both compounds [107]. Many NSAIDs and coxibs lead to mitochondrial dysfunction, which in turn increases the production of reactive oxygen species. They have also been shown to increase ER-stress [99], which can induce the transcription of COX2 and prostaglandin synthase [108] leading to an increase in inflammatory eicosanoids and cytokines. Thus, the most widely used drugs in the world are themselves inflammatory. Anti-inflammatory sEHI block this inflammation axis both at the level of the mitochondria and the endoplasmic reticulum. Thus, when used with NSAIDS and coxibs should make these drugs not only safer but also improve their anti-inflammatory activity. Multiple rodent studies have shown that sEHI synergize with NSAIDs and coxibs to reduce inflammatory pain and extend the utility of NSAIDs and coxibs to include neuropathic pain. The sEHI also reduce gastrointestinal and cardiovascular side effects of cyclooxygenase inhibitors. Since prescription and over the counter COX inhibitors are so commonly used in pain management, this interaction is likely to be seen in the clinic if patients in trials are continued on standard of care treatment.
Exploiting this interaction commercially will be more complex. Regulatory pathways require that novel therapeutics demonstrate stand-alone efficacy before being combined with other targets, thus the pathway toward human treatment with NSAID-sEHI combinations is complicated. A polypharmaceutical approach: where one compound inhibits both COX and sEH, such as PTUBP, would lessen this challenge; however, the FDA closely monitors NSAID cardiovascular toxicity and requires extensive cardiovascular safety studies and lengthy clinical trials prior to testing in humans, thus developing this dual target under the current regulatory environment would likely be more expensive and risky than other targets. For example, this regulatory scrutiny resulted in the FDA not approving a potent NSAID, etoricoxib, after the FDA advisory committee recommended further cardiovascular risk assessment, despite etoricoxib approval in other countries [109].
Other targets in the fatty acid metabolism pathway provide attractive opportunities for treatments. For example, inhibition of LO is currently approved for the treatment of asthma. Leukotrienes, the main target of LO metabolism, are potent inflammatory agents that potentiate bronchoconstriction and exacerbate an asthma attack. Inhibiting LO activity through direct inhibition (e.g. Zileuton) or through the inhibition of 5-lipoxygnase activating protein (FLAP) is an approved treatment for asthma [110]. sEHI has also shown efficacy for treating asthma [111]. Interestingly, Zileuton and the FLAP inhibitors, Zafirlukast and Montelukast, also have weak activity for inhibiting the sEH (0.9–1.95 μM), but at concentrations likely achieved at therapeutic doses [112]. Identifying improved dual LO-sEHI with greater potency for the sEH is an attractive target for improving asthma treatments.
The fatty acid amide hydrolase enzyme (FAAH) is a complicated target for drug therapy. The endocannabinoids, a bioactive class of lipids formed through the addition of an amide to the carboxylic acid of PUFA. For example, N-arachidonoyl phosphatidylethanolamine (NAPE), and related amides are associated with beneficial effects in a variety of disease models including inflammation, pain, asthma, epilepsy, neurological disease, among others (see [113] for review). Because FAAH metabolizes endocannabinoids, therapeutic approaches of inhibiting this enzyme were considered for treatments of some diseases. Over ten clinical trials have been initiated to test efficacy of FAAH inhibitors, but after significant toxicities and one death were reported in a Phase 1 study of a claimed FAAH inhibitor in France, all clinical trials were put on hold. Although the exact cause of the toxicity is unknown, recent reports suggest that off-target effects on lipases caused the toxicity [114]. Renewed interests in FAAH as a target for pain came after a case study identified a woman with a knock-out mutation in FAAH were thought to result in her inability to feel pain [115]. Administration of both sEH and FAAH inhibitors show synergistic effects in animal models of pain [116] and continued efforts to develop a dual inhibitor suitable for clinical development are ongoing [117].
sEH inhibitors are more effective in reducing pain in the presence of cAMP. Dual phosphodiesterase 4 (PDE4) and sEH inhibition also show synergistic effects in decreasing pain in animal models [118]. PDE inhibitors (PDEi) have been developed for the treatment of inflammation by preventing the metabolism of cyclic adenosine monophosphate (cAMP); however, increasing cAMP has also been associated with increased pain states. Interestingly, PDE4 and 5 inhibitors increase EpFA through lipase activity, releasing arachidonate and possibly EETs from phospholipids, and could possibly explain the mechanism for analgesic effects of PDEi despite increases in cAMP [118]. In laboratory settings, investigators demonstrated that decreasing EpFA concentrations through CYP450 inhibition while treating with PDEi resulted in increased pain states. Conversely, increasing EpFA concentrations through sEHI resulted in synergistic increases in pain relief. Efforts are ongoing to identify dual inhibitors of PDE4 and sEH, and recent advances identified MPPA, a compound with potent inhibition on both enzymes that is also efficacious in an LPS model of inflammatory pain [119].
While some compounds are synthesized specifically to inhibit an enzyme, others are identified after approval as having off-target effects against desirable or undesirable enzymes. For example, sorafenib and regorafenib are receptor tyrosine kinases that have anti-angiogenic properties by inhibiting VEGFR-2 and other kinases. They are approved for the treatment of hepatocellular carcinoma and renal cell carcinoma, and both are potent inhibitors of the sEH (12 and 0.5 nM IC50, respectively). The other approved small molecule inhibitor of VEGFR, sunitinib, is not an sEH inhibitor. Although the biological relevance of this dual activity is difficult to compare with other kinases that do not affect sEH because kinases are in general promiscuous inhibitors of many kinases. Both sorafenib and regorafenib are known for being difficult to formulate and for often serious side effects of their use. Clinical doses of these Raf-1 and pan-kinase inhibitors are likely to inhibit most sEH activity and increase EpFA in vivo. It is likely that without the sEH inhibition, these kinase inhibitors would have even worse side effects. It follows that the side effects of these two kinase inhibitors could be dramatically reduced by increasing dietary ω−3 and limiting dietary ω−6 lipids. The kinase inhibition is not ubiquitous to the urea pharmacophore that inhibits sEH; however, Sorafenib and regorafenib are unique compared to other published sEH inhibitors used in preclinical or clinical studies in their ability to inhibit kinases, as can be predicted from structure activity relationships [120]. Although recent publications indicate that TPPU is a weak inhibitor of p38 kinase (IC50 = 0.98 μM). The p38 kinase is a mitogen-activated protein kinase and activates inflammatory cytokines, and inhibition of this kinase would explain many of the beneficial effects seen in the preclinical studies using this compound; however, p38 is also regulated by oxidative stress, and the study failed to show if the activity was an indirect result of reducing oxidative stress vs. direct inhibition of the kinase given that the in vitro assays were conducted below the IC50 of 100 nM [121].
5.5. Summary and Future Directions for Targeting EpFA as Treatments for Disease
The newest innovations in inhibiting the sEH focus on identifying natural compounds with inhibitory activity, or polypharmaceutical approaches targeting multiple enzyme pathways. The inhibitors being tested for sEH inhibition focus on a central pharmacophore of either a urea, amide, or carbamate, and few new patents have been filed with different pharmacophores. Instead, new technologies focus on identifying new targets of disease, poly-pharmaceutical approaches developing a single compound to inhibit multiple targets [122], or inhibitors isolated from natural products. Inhibiting the sEH affects a regulatory pathway, ER stress, and is a relatively safe target that could offer benefits when combining with other targets. In some cases, efforts for developing dual targets to provide added benefits or safety have been intentional, while others have been discovered as off-target effects after developments. Both approaches are described below.
Current clinical approaches are focused on developing small molecule inhibitors or EpFA mimics; however, future approaches are investigating polypharmaceutical approaches, thus capitalizing on the low toxicity of alerting this pathway. Yet despite widespread interest in targeting this pathway, few new pharmacophores are identified that lack a carbamate or urea as the active moiety. Recently, natural products have been identified as a new source of sEH inhibitors that hold promise for future nutraceutical development; however, these products have relatively poor potency, with IC50s in the μM range, which would require g/day amounts of dosing. Techniques are being considered to improve production and isolation of natural sEHI or identify more potent natural sEHI [123, 124].
Many early sEHI, such as AUDA, were designed as mimics of specific EpFA. This dual action was clearly demonstrated in earlier publications [125]. Subsequent synthesis led to compounds that inhibited the EH without mimicking EpFA or compounds that mimicked specific EpFA without inhibiting sEH. Shen and Hammock discussed the advantages and limitations of each of these approaches [126]. The resulting molecules made biological data much easier to interpret when they were used as physiological probes, and likely simplified patent positions as well. Possibly it is worth reconsidering dual acting compounds. For example, one could have an sEHI pharmacophore which would increase EpFA systemically, while also targeting mimicry of 17,18 EEQ or 19,20 EDP to control atrial fibrillation [127]. Such compounds would offer the advantages of stabilizing all EpFA while allowing direct action of the EpFA mimic in tissue with a reduced ability to epoxidize PUFA.
In summary, lipid epoxides and their mimics or stabilizers are of increasing interest in treating disease due to their many beneficial and homeostatic functions as well as limited observed toxicities. However, their multimodal mechanism of action, including effects on angiogenesis and cancer, raises caution for monitoring safety in clinical trials. Furthermore, few studies report toxicities associated either with direct supplementation of EpFA, or inhibition of sEH; however, one study reported that sEH null mice displayed reduced survival in a cardiac arrest model compared to wild type mice [128] demonstrating one of the few examples of increased toxicities in preclinical models modulating the sEH. While the most recent advances are focused on investigating the polypharmaceutical advantages of increasing EpFA, other approaches that could increase EpFA include altering the release and reincorporation into the cell membrane and could provide attractive benefits to the current clinical approaches being investigated (Fig. 5.4.). Currently small molecule inhibitors of sEH are being tested in clinical settings to investigate the effects of increasing EpFA concentrations in the body as a way of treating pain, diabetes, and reducing the symptoms of subarachnoid hemorrhage. Other strategies focus on mimicking the beneficial EpFA and are targeted in clinical investigations for treating pain, fatty liver disease, hypertriglyceridemia, and atrial fibrillation. Overall, previous clinical trials targeting strategies to increase EpFA lack on-target toxicities, and most studies using sEH knock-out animals lack toxic side effects, which increases confidence in the safety of increasing EpFA concentrations in the body. The combined safety profile and wide-ranging efficacy in treating disease encourage continued investigation in the beneficial effects of this pathway as a treatment option.
Fig. 5.4.

Possible ways of increasing natural epoxy fatty acid chemical mediators to prevent and treat disease Increasing EpFA concentrations in the body have been associated with treating many diseases. The figure above demonstrates potential pathways of increasing their concentrations through increasing their biosynthesis or through preventing their metabolism. EpFA can be increased by increased PUFA consumption, increasing release from lipid membranes, or increased formation through CYP450 activity. Inhibiting the metabolic enzymes that convert EpFA to more polar compounds that are rapidly eliminated from the body, or supplementing the diet with EpFA mimics that prevent degradation by β oxidation are other ways in increasing the concentrations of the beneficial fatty acids
Acknowledgements
This work was supported by the National Institute of Environmental Health Sciences (NIEHS) Grant R35ES030443-01, NIEHS Superfund Research Program P42 ES004699, and T32GM113770 (to CBM). Partial support for clinical development of sEH inhibitors for human medicine comes from the NIEHS SBIR Program R44ES025598, the NIH NINDS Blueprint Neurotherapeutics Network UH2NS094258, and the National Institute On Drug Abuse Award Number UG3DA048767. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors would also like to thank Jogen Atone for input on fig. 5.3.
Footnotes
Conflict of interest statement The University of California holds patents on the sEH inhibitor used in this study as well as their use to treat inflammation, inflammatory pain, and neuropathic pain. KM Wagner, CB McReynolds, and BD Hammock are employees of EicOsis L.L.C., a startup company advancing sEH inhibitors into the clinic.
Contributor Information
Cindy McReynolds, Department of Entomology and Nematology, and U.C. Davis Comprehensive Cancer Center, University of California Davis, Davis, CA, USA EicOsis, Davis, CA, USA.
Christophe Morisseau, Department of Entomology and Nematology, and U.C. Davis Comprehensive Cancer Center, University of California Davis, Davis, CA, USA.
Karen Wagner, Department of Entomology and Nematology, and U.C. Davis Comprehensive Cancer Center, University of California Davis, Davis, CA, USA EicOsis, Davis, CA, USA.
Bruce Hammock, Department of Entomology and Nematology, and U.C. Davis Comprehensive Cancer Center, University of California Davis, Davis, CA, USA.
References
- 1.Vane JR (1971) Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol 231(25):232–235. Epub 1971/06/23. 10.1038/newbio231232a0. PubMed PMID: 5284360 [DOI] [PubMed] [Google Scholar]
- 2.Liu MC, Dube LM, Lancaster J (1996) Acute and chronic effects of a 5-lipoxygenase inhibitor in asthma: a 6-month randomized multicenter trial. Zileuton Study Group. J Allergy Clin Immunol 98(5 Pt 1):859–871. Epub 1996/11/01. 10.1016/s0091-6749(96)80002-9. PubMed PMID: 8939149 [DOI] [PubMed] [Google Scholar]
- 3.J H. Essential fatty acids. Linus Pauling Institute, 2003. [cited 2019]. Available from: https://lpi.oregonstate.edu/mic/other-nutrients/essential-fatty-acids [Google Scholar]
- 4.Zorn K, Oroz-Guinea I, Brundiek H, Bornscheuer UT (2016) Engineering and application of enzymes for lipid modification, an update. Prog Lipid Res 63:153–164. Epub 2016/06/16. 10.1016/j.plipres.2016.06.001. PubMed PMID: 27301784 [DOI] [PubMed] [Google Scholar]
- 5.Konkel A, Schunck WH (2011) Role of cytochrome P450 enzymes in the bioactivation of polyunsaturated fatty acids. Biochim Biophys Acta 1814(1):210–222. Epub 2010/09/28. S1570–9639(10)00258-X [pii], 10.1016/j.bbapap.2010.09.009. PubMed PMID: 20869469 [DOI] [PubMed] [Google Scholar]
- 6.Potente M, Michaelis UR, Fisslthaler B, Busse R, Fleming I (2002) Cytochrome P450 2C9-induced endothelial cell proliferation involves induction of mitogen-activated protein (MAP) kinase phosphatase-1, inhibition of the c-Jun N-terminal kinase, and up-regulation of cyclin D1. J Biol Chem 277(18):15671–15676. Epub 2002/02/28. 10.1074/jbc.M110806200. PubMed PMID: 11867622 [DOI] [PubMed] [Google Scholar]
- 7.Fitzpatrick FA, Ennis MD, Baze ME, Wynalda MA, McGee JE, Liggett WF (1986) Inhibition of cyclooxygenase activity and platelet aggregation by epoxyeicosatrienoic acids. Influence of stereochemistry. J Biol Chem 261(32):15334–15338. Epub 1986/11/15. PubMed PMID: 3095326 [PubMed] [Google Scholar]
- 8.Fan F, Muroya Y, Roman RJ (2015) Cytochrome P450 eicosanoids in hypertension and renal disease. Curr Opin Nephrol Hypertens 24(1):37–46. Epub 2014/11/27. 10.1097/mnh.0000000000000088. PubMed PMID: 25427230; PMCID: PMC4260681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fleming I, Busse R (2006) Endothelium-derived epoxyeicosatrienoic acids and vascular function. Hypertension 47(4):629–633. Epub 2006/02/24. 10.1161/01.hyp.0000208597.87957.89. PubMed PMID: 16490839 [DOI] [PubMed] [Google Scholar]
- 10.Roman RJ, Fan F (2018) 20-HETE: hypertension and beyond. Hypertension 72(1):12–18. Epub 2018/05/16. 10.1161/hypertensionaha.118.10269. PubMed PMID: 29760152; PMCID: PMC6002933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ulu A, Inceoglu B, Yang J, Singh V, Vito S, Wulff H, Hammock BD (2016) Inhibition of soluble epoxide hydrolase as a novel approach to high dose diazepam induced hypotension. J Clin Toxicol 6(3). Epub 2017/03/04. 10.4172/21610495.1000300. PubMed PMID: 28255523; PMCID: PMC5328659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGiff JC, Carroll MA (1991) Cytochrome P450-dependent arachidonate metabolites, renal function and blood pressure regulation. Adv Prostaglandin Thromboxane Leukot Res 21b:675–682. Epub 1991/01/01 [PubMed] [Google Scholar]
- 13.Imig JD, Pham BT, LeBlanc EA, Reddy KM, Falck JR, Inscho EW (2000) Cytochrome P450 and cyclooxygenase metabolites contribute to the endothelin-1 afferent arteriolar vasoconstrictor and calcium responses. Hypertension 35(1 Pt 2):307–312. Epub 2000/01/21. 10.1161/01.hyp.35.1.307 [DOI] [PubMed] [Google Scholar]
- 14.Liu L, Chen C, Gong W, Li Y, Edin ML, Zeldin DC, Wang DW (2011) Epoxyeicosatrienoic acids attenuate reactive oxygen species level, mitochondrial dysfunction, caspase activation, and apoptosis in carcinoma cells treated with arsenic trioxide. J Pharmacol Exp Ther 339(2):451–463. Epub 2011/08/19. 10.1124/jpet.111.180505. PubMed PMID: 21846841; PMCID: PMC3199997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK (1999) Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 285(5431):1276–1279. Epub 1999/08/24. 10.1126/science.285.5431.1276. PubMed PMID: 10455056; PMCID: PMC2720027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai HJ, Kim IH, Tuteja D, Mateo RK, Singapuri A, Davis BB, Low R, Hammock BD, Chiamvimonvat N (2006) Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A 103(49):18733–18738. Epub 2006/11/30. 10.1073/pnas.0609158103. PubMed PMID: 17130447; PMCID: PMC1693731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kundu S, Roome T, Bhattacharjee A, Carnevale KA, Yakubenko VP, Zhang R, Hwang SH, Hammock BD, Cathcart MK (2013) Metabolic products of soluble epoxide hydrolase are essential for monocyte chemotaxis to MCP-1 in vitro and in vivo. J Lipid Res 54(2):436–447. 10.1194/jlr.M031914. PubMed PMID: 23160182; PMCID: 3588870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner KM, McReynolds CB, Schmidt WK, Hammock BD (2017) Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol Ther 180:62–76. Epub 2017/06/24. 10.1016/j.pharmthera.2017.06.006. PubMed PMID: 28642117; PMCID: PMC5677555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin JH, Walter P, Yen TS (2008) Endoplasmic reticulum stress in disease pathogenesis. Ann Rev Pathol 3:399–425. Epub 2007/11/28. 10.1146/annurev.pathmechdis.3.121806.151434. PubMed PMID: 18039139; PMCID: PMC3653419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inceoglu B, Bettaieb A, Trindade da Silva CA, Lee KS, Haj FG, Hammock BD (2015) Endoplasmic reticulum stress in the peripheral nervous system is a significant driver of neuropathic pain. Proc Natl Acad Sci U S A 112(29):9082–9087. Epub 2015/07/08. 10.1073/pnas.1510137112. PubMed PMID: 26150506; PMCID: Pmc4517273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fang X, Kaduce TL, Weintraub NL, Harmon S, Teesch LM, Morisseau C, Thompson DA, Hammock BD, Spector AA (2001) Pathways of epoxyeicosatrienoic acid metabolism in endothelial cells. Implications for the vascular effects of soluble epoxide hydrolase inhibition. J Biol Chem 276(18):14867–14874. Epub 2001/03/30. 10.1074/jbc.M011761200 [DOI] [PubMed] [Google Scholar]
- 22.Nelson JW, Subrahmanyan RM, Summers SA, Xiao X, Alkayed NJ (2013) Soluble epoxide hydrolase dimerization is required for hydrolase activity. J Biol Chem 288(11):7697–7703. Epub 2013/01/31. 10.1074/jbc.M112.429258. PubMed PMID: 23362272; PMCID: PMC3597810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morisseau C (2013) Role of epoxide hydrolases in lipid metabolism. Biochimie 95(1):91–95. Epub 2012/06/23. 10.1016/j.biochi.2012.06.011. PubMed PMID: 22722082; PMCID: PMC3495083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Botham KM, Mayes PA (2016) Cholesterol synthesis, transport, & excretion In: Rodwell VW, Bender DA, Botham KM, Kennelly PJ, Weil PA (eds) Harper’s illustrated biochemistry, 30th edn McGraw-Hill Education, New York [Google Scholar]
- 25.Decker M, Adamska M, Cronin A, Di Giallonardo F, Burgener J, Marowsky A, Falck JR, Morisseau C, Hammock BD, Gruzdev A, Zeldin DC, Arand M (2012) EH3 (ABHD9): the first member of a new epoxide hydrolase family with high activity for fatty acid epoxides. J Lipid Res 53(10):2038–2045. Epub 2012/07/17. 10.1194/jlr.M024448. PubMed PMID: 22798687; PMCID: PMC3435537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fornage M, Lee CR, Doris PA, Bray MS, Heiss G, Zeldin DC, Boerwinkle E (2005) The soluble epoxide hydrolase gene harbors sequence variation associated with susceptibility to and protection from incident ischemic stroke. Hum Mol Genet 14(19):2829–2837. Epub 2005/08/24. 10.1093/hmg/ddi315. PubMed PMID: 16115816; PMCID: PMC1343524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martini RP, Ward J, Siler DA, Eastman JM, Nelson JW, Borkar RN, Alkayed NJ, Dogan A, Cetas JS (2014) Genetic variation in soluble epoxide hydrolase: association with outcome after aneurysmal subarachnoid hemorrhage. J Neurosurg 121(6):1359–1366. Epub 2014/09/13. 10.3171/2014.7.jns131990. PubMed PMID: 25216066; PMCID: PMC4370510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morisseau C, Wecksler AT, Deng C, Dong H, Yang J, Lee KS, Kodani SD, Hammock BD (2014) Effect of soluble epoxide hydrolase polymorphism on substrate and inhibitor selectivity and dimer formation. J Lipid Res. 55(6):1131–1138. Epub 2014/04/29. 10.1194/jlr.M049718. PubMed PMID: 24771868; PMCID: PMC4031944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Przybyla-Zawislak BD, Srivastava PK, Vazquez-Matias J, Mohrenweiser HW, Maxwell JE, Hammock BD, Bradbury JA, Enayetallah AE, Zeldin DC, Grant DF (2003) Polymorphisms in human soluble epoxide hydrolase. Mol Pharmacol 64(2):482–490. Epub 2003/07/19. 10.1124/mol.64.2.482 [DOI] [PubMed] [Google Scholar]
- 30.Morisseau C, Hammock BD (2013) Impact of soluble Epoxide Hydrolase and Epoxyeicosanoids on human health. Annu Rev Pharmacol 53:37–58. 10.1146/annurev-pharmtox-011112-140244. PubMed PMID: WOS:000323040100003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morisseau C, Inceoglu B, Schmelzer K, Tsai HJ, Jinks SL, Hegedus CM, Hammock BD (2010) Naturally occurring monoepoxides of eicosapentaenoic acid and docosahexaenoic acid are bioactive antihyperalgesic lipids. J Lipid Res 51(12):3481–3490. Epub 2010/07/29. 10.1194/jlr.M006007. PubMed PMID: 20664072; PMCID: PMC2975720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ulu A, Harris TR, Morisseau C, Miyabe C, Inoue H, Schuster G, Dong H, Iosif AM, Liu JY, Weiss RH, Chiamvimonvat N, Imig JD, Hammock BD (2013) Anti-inflammatory effects of omega-3 polyunsaturated fatty acids and soluble epoxide hydrolase inhibitors in angiotensin-II-dependent hypertension. J Cardiovasc Pharmacol 62(3):285–297. 10.1097/FJC.0b013e318298e460. PubMed PMID: 23676336; PMCID: 3773051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott-Van Zeeland AA, Bloss CS, Tewhey R, Bansal V, Torkamani A, Libiger O, Duvvuri V, Wineinger N, Galvez L, Darst BF, Smith EN, Carson A, Pham P, Phillips T, Villarasa N, Tisch R, Zhang G, Levy S, Murray S, Chen W, Srinivasan S, Berenson G, Brandt H, Crawford S, Crow S, Fichter MM, Halmi KA, Johnson C, Kaplan AS, La Via M, Mitchell JE, Strober M, Rotondo A, Treasure J, Woodside DB, Bulik CM, Keel P, Klump KL, Lilenfeld L, Plotnicov K, Topol EJ, Shih PB, Magistretti P, Bergen AW, Berrettini W, Kaye W, Schork NJ (2014) Evidence for the role of EPHX2 gene variants in anorexia nervosa. Mol Psychiatry 19(6):724–732. Epub 2013/09/04. 10.1038/mp.2013.91. PubMed PMID: 23999524; PMCID: PMC3852189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shih PB (2017) Integrating multi-omics biomarkers and postprandial metabolism to develop personalized treatment for anorexia nervosa. Prostaglandins Other Lipid Mediat 132:69–76. Epub 2017/02/25. 10.1016/j.prostaglandins.2017.02.002. PubMed PMID: 28232135; PMCID: PMC5565718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shih PB, Yang J, Morisseau C, German JB, Zeeland AA, Armando AM, Quehenberger O, Bergen AW, Magistretti P, Berrettini W, Halmi KA, Schork N, Hammock BD, Kaye W (2016) Dysregulation of soluble epoxide hydrolase and lipidomic profiles in anorexia nervosa. Mol Psychiatry 21(4):537–546. Epub 2015/04/01. 10.1038/mp.2015.26. PubMed PMID: 25824304; PMCID: PMC4591075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J, Shih PB (2018) Fasting and postprandial soluble epoxide hydrolase-associated eicosanoids of remitted patients with eating disorder. Data Brief 17:334–338. Epub 2018/06/08. 10.1016/j.dib.2018.01.028. PubMed PMID: 29876402; PMCID: PMC5988286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato K, Emi M, Ezura Y, Fujita Y, Takada D, Ishigami T, Umemura S, Xin Y, Wu LL, Larrinaga-Shum S, Stephenson SH, Hunt SC, Hopkins PN (2004) Soluble epoxide hydrolase variant (Glu287Arg) modifies plasma total cholesterol and triglyceride phenotype in familial hypercholesterolemia: intrafamilial association study in an eight-generation hyperlipidemic kindred. J Hum Genet 49(1):29–34. Epub 2003/12/16. 10.1007/s10038-003-0103-6 [DOI] [PubMed] [Google Scholar]
- 38.Shen L, Peng H, Peng R, Fan Q, Zhao S, Xu D, Morisseau C, Chiamvimonvat N, Hammock BD (2015) Inhibition of soluble epoxide hydrolase in mice promotes reverse cholesterol transport and regression of atherosclerosis. Atherosclerosis 239(2):557–565. Epub 2015/03/04. 10.1016/j.atherosclerosis.2015.02.014. PubMed PMID: 25733327; PMCID: PMC4527317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Campbell WB, Falck JR, Gauthier K (2001) Role of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factor in bovine coronary arteries. Med Sci Monit Int Med J Exp Clin Res 7(4):578–584. Epub 2001/07/04 [PubMed] [Google Scholar]
- 40.Sari I, Pinarbasi H, Pinarbasi E, Yildiz C (2017) Association between the soluble epoxide hydrolase gene and preeclampsia. Hypertens Pregnancy 36(4):315–325. Epub 2017/10/24. 10.1080/10641955.2017.1388390 [DOI] [PubMed] [Google Scholar]
- 41.Koerner IP, Jacks R, DeBarber AE, Koop D, Mao P, Grant DF, Alkayed NJ (2007) Polymorphisms in the human soluble epoxide hydrolase gene EPHX2 linked to neuronal survival after ischemic injury. J Neurosci 27(17):4642–4649. 10.1523/JNEUROSCI.0056-07.2007. PubMed PMID: 17460077; PMCID: PMC6672984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gschwendtner A, Ripke S, Freilinger T, Lichtner P, Muller-Myhsok B, Wichmann HE, Meitinger T, Dichgans M (2008). Genetic variation in soluble epoxide hydrolase (EPHX2) is associated with an increased risk of ischemic stroke in white Europeans. Stroke 39(5):1593–1596. Epub 2008/03/08. 10.1161/strokeaha.107.502179 [DOI] [PubMed] [Google Scholar]
- 43.Lee J, Dahl M, Grande P, Tybjaerg-Hansen A, Nordestgaard BG (2010) Genetically reduced soluble epoxide hydrolase activity and risk of stroke and other cardiovascular disease. Stroke 41(1):27–33. Epub 2009/11/27. 10.1161/strokeaha.109.567768 [DOI] [PubMed] [Google Scholar]
- 44.Fava C, Montagnana M, Danese E, Almgren P, Hedblad B, Engstrom G, Berglund G, Minuz P, Melander O (2010) Homozygosity for the EPHX2 K55R polymorphism increases the long-term risk of ischemic stroke in men: a study in swedes. Pharmacogenet Genomics 20(2):94–103. Epub 2010/01/13. 10.1097/FPC.0b013e3283349ec9 [DOI] [PubMed] [Google Scholar]
- 45.Nelson JW, Young JM, Borkar RN, Woltjer RL, Quinn JF, Silbert LC, Grafe MR, Alkayed NJ (2014) Role of soluble epoxide hydrolase in age-related vascular cognitive decline. Prostaglandins Other Lipid Mediat 113–115:30–37. Epub 2014/10/04. 10.1016/j.prostaglandins.2014.09.003. PubMed PMID: 25277097; PMCID: PMC4254026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Václavíková R, Hughes DJ, Souček P (2015) Microsomal epoxide hydrolase 1 (EPHX1): gene, structure, function, and role in human disease. Gene 571(1):1–8. Epub 07/26. 10.1016/j.gene.2015.07.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marowsky A, Burgener J, Falck JR, Fritschy JM, Arand M (2009) Distribution of soluble and microsomal epoxide hydrolase in the mouse brain and its contribution to cerebral epoxyeicosatrienoic acid metabolism. Neuroscience 163(2):646–661. Epub 06/18. 10.1016/j.neuroscience.2009.06.033 [DOI] [PubMed] [Google Scholar]
- 48.Qin XH, Wu Z, Dong JH, Zeng YN, Xiong WC, Liu C, Wang MY, Zhu MZ, Chen WJ, Zhang Y, Huang QY, Zhu XH (2019). Liver soluble Epoxide Hydrolase regulates behavioral and cellular effects of chronic stress. Cell Rep. 2019;29(10):3223–3234. e6. Epub 2019/12/05. 10.1016/j.celrep.2019.11.006 [DOI] [PubMed] [Google Scholar]
- 49.Sinal CJ, Miyata M, Tohkin M, Nagata K, Bend JR, Gonzalez FJ (2000) Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J Biol Chem 275(51):40504–40510. Epub 2000/09/26. 10.1074/jbc.M008106200 [DOI] [PubMed] [Google Scholar]
- 50.Harris TR, Hammock BD (2013) Soluble epoxide hydrolase: gene structure, expression and deletion. Gene 526(2):61–74. 10.1016/j.gene.2013.05.008. PubMed PMID: 23701967; PMCID: 3733540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Atone J, Wagner K, Hashimoto K, Hammock B (2019) Prostaglandins and other lipid mediators cytochrome P450 derived epoxidized fatty acids as a therapeutic tool against neuroinflammatory diseases. Prostaglandins Other Lipid Mediat 106385. Epub 2019/11/08. 10.1016/j.prostaglandins.2019.106385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bettaieb A, Koike S, Hsu MF, Ito Y, Chahed S, Bachaalany S, Gruzdev A, Calvo-Rubio M, Lee KSS, Inceoglu B, Imig JD, Villalba JM, Zeldin DC, Hammock BD, Haj FG (2017) Soluble epoxide hydrolase in podocytes is a significant contributor to renal function under hyperglycemia. Biochimica Biophysica Acta Gen Subj 1861(11 Pt A):2758–2765. Epub 2017/08/02. 10.1016/j.bbagen.2017.07.021. PubMed PMID: 28757338; PMCID: PMC5873293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heinricher MM, Maire JJ, Lee D, Nalwalk JW, Hough LB (2010) Physiological basis for inhibition of morphine and improgan antinociception by CC12, a P450 epoxygenase inhibitor. J Neurophysiol 104(6):3222–3230. Epub 2010/10/12. 10.1152/jn.00681.2010. PubMed PMID: 20926616; PMCID: PMC3007650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Conroy JL, Fang C, Gu J, Zeitlin SO, Yang W, Yang J, VanAlstine MA, Nalwalk JW, Albrecht PJ, Mazurkiewicz JE, Snyder-Keller A, Shan Z, Zhang SZ, Wentland MP, Behr M, Knapp BI, Bidlack JM, Zuiderveld OP, Leurs R, Ding X, Hough LB (2010) Opioids activate brain analgesic circuits through cytochrome P450/epoxygenase signaling. Nat Neurosci 13(3):284–286. 10.1038/nn.2497. PubMed PMID: 20139973; PMCID: PMC2828325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hough LB, Nalwalk JW, Yang J, Conroy JL, VanAlstine MA, Yang W, Gargano J, Shan Z, Zhang SZ, Wentland MP, Phillips JG, Knapp BI, Bidlack JM, Zuiderveld OP, Leurs R, Ding X (2011) Brain P450 epoxygenase activity is required for the antinociceptive effects of improgan, a nonopioid analgesic. Pain 152(4):878–887. Epub 2011/02/15. 10.1016/j.pain.2011.01.001. PubMed PMID: 21316152; PMCID: PMC3065546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grey A, Bolland M (2014) Clinical trial evidence and use of fish oil supplements. JAMA Intern Med 174(3):460–462. 10.1001/jamainternmed.2013.12765. PubMed PMID: 24352849 [DOI] [PubMed] [Google Scholar]
- 57.Rymer C, Gibbs RA, Givens DI (2010) Comparison of algal and fish sources on the oxidative stability of poultry meat and its enrichment with omega-3 polyunsaturated fatty acids. Poult Sci 89(1):150–159. 10.3382/ps.2009-00232. PubMed PMID: 20008813 [DOI] [PubMed] [Google Scholar]
- 58.Jackowski SA, Alvi AZ, Mirajkar A, Imani Z, Gamalevych Y, Shaikh NA, Jackowski G (2015) Oxidation levels of North American over-the-counter n-3 (omega-3) supplements and the influence of supplement formulation and delivery form on evaluating oxidative safety. J Nutr Sci 4:e30 10.1017/jns.2015.21. PubMed PMID: 26688721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Albert BB, Cameron-Smith D, Hofman PL, Cutfield WS (2013) Oxidation of marine omega-3 supplements and human health. BioMed Res Int 2013:464921. Epub 04/30. 10.1155/2013/464921. PubMed PMID: 23738326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hu J, Dziumbla S, Lin J, Bibli SI, Zukunft S, de Mos J, Awwad K, Fromel T, Jungmann A, Devraj K, Cheng Z, Wang L, Fauser S, Eberhart CG, Sodhi A, Hammock BD, Liebner S, Muller OJ, Glaubitz C, Hammes HP, Popp R, Fleming I (2017) Inhibition of soluble epoxide hydrolase prevents diabetic retinopathy. Nature 552(7684):248–252. Epub 2017/12/07. 10.1038/nature25013. PubMed PMID: 29211719; PMCID: PMC5828869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ruparel S, Green D, Chen P, Hargreaves KM (2012) The cytochrome P450 inhibitor, ketoconazole, inhibits oxidized linoleic acid metabolite-mediated peripheral inflammatory pain. Mol Pain 8:73 Epub 2012/09/26. 10.1186/17448069-8-73. PubMed PMID: 23006841; PMCID: PMC3488501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goswami SK, Inceoglu B, Yang J, Wan D, Kodani SD, da Silva CA, Morisseau C, Hammock BD (2015). Omeprazole increases the efficacy of a soluble epoxide hydrolase inhibitor in a PGE(2) induced pain model. Toxicol Appl Pharmacol. 289(3):419–427. Epub 2015/11/03. 10.1016/j.taap.2015.10.018. PubMed PMID: 26522832; PMCID: PMC4666679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hennebelle M, Zhang Z, Metherel AH, Kitson AP, Otoki Y, Richardson CE, Yang J, Lee KSS, Hammock BD, Zhang L, Bazinet RP, Taha AY (2017) Linoleic acid participates in the response to ischemic brain injury through oxidized metabolites that regulate neurotransmission. Sci Rep 7(1):4342 Epub 2017/07/01. 10.1038/s41598-017-02914-7. PubMed PMID: 28659576; PMCID: PMC5489485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ramsden CE, Hennebelle M, Schuster S, Keyes GS, Johnson CD, Kirpich IA, Dahlen JE, Horowitz MS, Zamora D, Feldstein AE, McClain CJ, Muhlhausler BS, Makrides M, Gibson RA, Taha AY (2018) Effects of diets enriched in linoleic acid and its peroxidation products on brain fatty acids, oxylipins, and aldehydes in mice. Biochimica Biophysica Acta Mol Cell Biol Lipid 1863(10):1206–1213. Epub 2018/07/28. 10.1016/j.bbalip.2018.07.007. PubMed PMID: 30053599; PMCID: PMC6180905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramsden CE, Ringel A, Feldstein AE, Taha AY, MacIntosh BA, Hibbeln JR, Majchrzak-Hong SF, Faurot KR, Rapoport SI, Cheon Y, Chung YM, Berk M, Mann JD (2012) Lowering dietary linoleic acid reduces bioactive oxidized linoleic acid metabolites in humans. Prostaglandins Leukot Essent Fatty Acids 87(4–5):135–141. Epub 2012/09/11. 10.1016/j.plefa.2012.08.004. PubMed PMID: 22959954; PMCID: PMC3467319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vangaveti VN, Jansen H, Kennedy RL, Malabu UH (2016) Hydroxyoctadecadienoic acids: oxidised derivatives of linoleic acid and their role in inflammation associated with metabolic syndrome and cancer. Eur J Pharmacol 785:70–76. Epub 2015/05/20. 10.1016/j.ejphar.2015.03.096. PubMed PMID: 25987423. [DOI] [PubMed] [Google Scholar]
- 67.Patwardhan AM, Scotland PE, Akopian AN, Hargreaves KM (2009) Activation of TRPV1 in the spinal cord by oxidized linoleic acid metabolites contributes to inflammatory hyperalgesia. Proc Natl Acad Sci U S A 106(44):18820–18824. Epub 2009/10/22. 10.1073/pnas.0905415106. PubMed PMID: 19843694; PMCID: PMC2764734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.MUAH Henry Krum. Evaluation of the effects of urotensin-II and soluble epoxide hydrolase inhibitors on skin microvessel tone in patients with heart failure, and in healthy volunteers (NCT00654966) 2011. [cited 2019 October 1]. Available from: https://clinicaltrials.gov/ct2/show/NCT00654966?term=soluble+epoxide+hydrolase&rank=1 [Google Scholar]
- 69.Morisseau C, Goodrow MH, Newman JW, Wheelock CE, Dowdy DL, Hammock BD (2002) Structural refinement of inhibitors of urea-based soluble epoxide hydrolases. Biochem Pharmacol 63(9):1599–1608. Epub 2002/05/15. 10.1016/s0006-2952(02)00952-8. PubMed PMID: 12007563. [DOI] [PubMed] [Google Scholar]
- 70.Tsai HJ, Hwang SH, Morisseau C, Yang J, Jones PD, Kasagami T, Kim IH, Hammock BD (2010) Pharmacokinetic screening of soluble epoxide hydrolase inhibitors in dogs. Eur J Pharm Sci. 40(3):222–238. Epub 2010/04/03. 10.1016/j.ejps.2010.03.018. PubMed PMID: 20359531; PMCID: PMC3285443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kodani SD, Hammock BD (2015) The 2014 Bernard B. Brodie Award Lecture Epoxide Hydrolases: drug metabolism to therapeutics for chronic pain. Drug Metab Dispos. 10.1124/dmd.115.063339. PubMed PMID: 25762541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee KS, Liu JY, Wagner KM, Pakhomova S, Dong H, Morisseau C, Fu SH, Yang J, Wang P, Ulu A, Mate CA, Nguyen LV, Hwang SH, Edin ML, Mara AA, Wulff H, Newcomer ME, Zeldin DC, Hammock BD (2014) Optimized inhibitors of soluble epoxide hydrolase improve in vitro target residence time and in vivo efficacy. J Med Chem 57(16):7016–7030. 10.1021/jm500694p. PubMed PMID: 25079952; PMCID: 4148150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Henrick CA, Staal GB, Siddall JB (1973) Alkyl 3,7,11-trimethyl-2,4-dodecadienoates, a new class of potent insect growth regulators with juvenile hormone activity. J Agric Food Chem 21(3):354–359. Epub 1973/05/01. 10.1021/jf60187a043. PubMed PMID: 4708794 [DOI] [PubMed] [Google Scholar]
- 74.Tran L, Kompa AR, Wang BH, Krum H (2012) Evaluation of the effects of urotensin II and soluble epoxide hydrolase inhibitor on skin microvessel tone in healthy controls and heart failure patients. Cardiovasc Ther 30(5):295–300. Epub 2011/09/03. 10.1111/j.1755-5922.2011.00282.x. PubMed PMID: 21884016 [DOI] [PubMed] [Google Scholar]
- 75.Imig JD, Carpenter MA, Shaw S (2009) The soluble Epoxide Hydrolase inhibitor AR9281 decreases blood pressure, ameliorates renal injury and improves vascular function in Hypertension. Pharmaceuticals (Basel, Switzerland) 2(3):217–227. Epub 2009/12/18. 10.3390/ph2030217. PubMed PMID: 27713235; PMCID: PMC3978544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee KS, Morisseau C, Yang J, Wang P, Hwang SH, Hammock BD (2013) Forster resonance energy transfer competitive displacement assay for human soluble epoxide hydrolase. Anal Biochem 434(2):259–268. Epub 2012/12/12. 10.1016/j.ab.2012.11.015. PubMed PMID: 23219719; PMCID: PMC3632402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Podolin PL, Bolognese BJ, Foley JF, Long E 3rd, Peck B, Umbrecht S, Zhang X, Zhu P, Schwartz B, Xie W, Quinn C, Qi H, Sweitzer S, Chen S, Galop M, Ding Y, Belyanskaya SL, Israel DI, Morgan BA, Behm DJ, Marino JP Jr., Kurali E, Barnette MS, Mayer RJ, Booth-Genthe CL, Callahan JF (2013) In vitro and in vivo characterization of a novel soluble epoxide hydrolase inhibitor. Prostaglandins Other Lipid Mediat 104–105:25–31. 10.1016/j.prostaglandins.2013.02.001. PubMed PMID: 23434473 [DOI] [PubMed] [Google Scholar]
- 78.Fleming I, Rueben A, Popp R, Fisslthaler B, Schrodt S, Sander A, Haendeler J, Falck JR, Morisseau C, Hammock BD, Busse R (2007) Epoxyeicosatrienoic acids regulate Trp channel dependent Ca2+ signaling and hyperpolarization in endothelial cells. Arterioscler Thromb Vasc Biol 27(12):2612–2618. 10.1161/ATVBAHA.107.152074. PubMed PMID: 17872452 [DOI] [PubMed] [Google Scholar]
- 79.Pozzi A, Macias-Perez I, Abair T, Wei S, Su Y, Zent R, Falck JR, Capdevila JH (2005) Characterization of 5,6- and 8,9-epoxyeicosatrienoic acids (5,6- and 8,9-EET) as potent in vivo angiogenic lipids. J Biol Chem 280(29):27138–27146. Epub 2005/05/27. doi: 10.1074/jbc.M501730200. PubMed PMID: 15917237 [DOI] [PubMed] [Google Scholar]
- 80.Panigrahy D, Edin ML, Lee CR, Huang S, Bielenberg DR, Butterfield CE, Barnes CM, Mammoto A, Mammoto T, Luria A, Benny O, Chaponis DM, Dudley AC, Greene ER, Vergilio JA, Pietramaggiori G, Scherer-Pietramaggiori SS, Short SM, Seth M, Lih FB, Tomer KB, Yang J, Schwendener RA, Hammock BD, Falck JR, Manthati VL, Ingber DE, Kaipainen A, D’Amore PA, Kieran MW, Zeldin DC. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J Clin Investig. 2012;122(1):178–191. 10.1172/JCI58128. PubMed PMID: 22182838; PMCID: 3248288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dai M, Wu L, Wang P, Wen Z, Xu X, Wang DW (2017) CYP2J2 and its metabolites EETs attenuate insulin resistance via regulating macrophage polarization in adipose tissue. Sci Rep 7:46743 Epub 2017/04/26. 10.1038/srep46743. PubMed PMID: 28440284; PMCID: PMC5404269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li R, Xu X, Chen C, Wang Y, Gruzdev A, Zeldin DC, Wang DW (2015) CYP2J2 attenuates metabolic dysfunction in diabetic mice by reducing hepatic inflammation via the PPARgamma. Am J Physiol Endocrinol Metab 308(4):E270–E282. Epub 2014/11/13. 10.1152/ajpendo.00118.2014. PubMed PMID: 25389363; PMCID: PMC4329496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang Y, Dong R, Chen Z, Hu D, Fu M, Tang Y, Wang DW, Xu X, Tu L (2018) Endothelium-specific CYP2J2 overexpression attenuates age-related insulin resistance. Aging Cell 17(2). Epub 2018/01/11. 10.1111/acel.12718. PubMed PMID: 29318723; PMCID: PMC5847864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zarriello S, Tuazon JP, Corey S, Schimmel S, Rajani M, Gorsky A, Incontri D, Hammock BD, Borlongan CV (2019) Humble beginnings with big goals: small molecule soluble epoxide hydrolase inhibitors for treating CNS disorders. Prog Neurobiol 172:23–39. Epub 2018/11/18. 10.1016/j.pneurobio.2018.11.001. PubMed PMID: 30447256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wagner K, Inceoglu B, Hammock BD (2011) Soluble epoxide hydrolase inhibition, epoxygenated fatty acids and nociception. Prostaglandins Other Lipid Mediat 96(1–4):76–83. Epub 2011/08/23. 10.1016/j.prostaglandins.2011.08.001. PubMed PMID: 21854866; PMCID: PMC3215909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wagner K, Vito S, Inceoglu B, Hammock BD (2014) The role of long chain fatty acids and their epoxide metabolites in nociceptive signaling. Prostaglandins Other Lipid Mediat 113–115:2–12. Epub 2014/09/23. 10.1016/j.prostaglandins.2014.09.001. PubMed PMID: 25240260; PMCID: PMC4254344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brostrom L, Falck JR, inventor Arachidonic Acid Analogs and Methods for Analgesi Treatment Using Same patent 8,658,632 B2. 2012 [Google Scholar]
- 88.Campbell WB, Gebremedhin D, Pratt PF, Harder DR (1996) Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res 78(3):415–423. Epub 1996/03/01. 10.1161/01.res.78.3.415. PubMed PMID: 8593700 [DOI] [PubMed] [Google Scholar]
- 89.Falck JR, Wallukat G, Puli N, Goli M, Arnold C, Konkel A, Rothe M, Fischer R, Muller DN, Schunck WH (2011) 17(R),18(S)-epoxyeicosatetraenoic acid, a potent eicosapentaenoic acid (EPA) derived regulator of cardiomyocyte contraction: structure-activity relationships and stable analogues. J Med Chem 54(12):4109–4118. Epub 2011/05/20. 10.1021/jm200132q. PubMed PMID: 21591683; PMCID: PMC3122156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fraser DA, Wang XY, Skjaeret T, Kastelein JP, Schuppan D (2018) A structurally engineered fatty acid, icosabutate, displays optimised absorption, distribution and metabolism properties for targeting hepatic inflammation and normalises elevated liver enzymes in dyslipidemic patients. Available from: https://www.northseatherapeutics.com/wp-content/uploads/2018/04/EASL-ILC2018-NST.pdf [Google Scholar]
- 91.Bays HE, Hallen J, Vige R, Fraser D, Zhou R, Hustvedt SO, Orloff DG, Kastelein JJ (2016) Icosabutate for the treatment of very high triglycerides: A placebo-controlled, randomized, double-blind, 12-week clinical trial. J Clin Lipidol 10(1):181–91.e1–2. Epub 2016/02/20. 10.1016/j.jacl.2015.10.012. PubMed PMID: 26892135 [DOI] [PubMed] [Google Scholar]
- 92.Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, Doyle RT Jr., Juliano RA, Jiao L, Granowitz C, Tardif JC, Ballantyne CM (2019) Cardiovascular risk reduction with Icosapent ethyl for hypertriglyceridemia. N Engl J Med 380(1):11–22. Epub 2018/11/13. 10.1056/NEJMoa1812792. PubMed PMID: 30415628. [DOI] [PubMed] [Google Scholar]
- 93.Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, Doyle RT Jr., Juliano RA, Jiao L, Granowitz C, Tardif JC, Gregson J, Pocock SJ, Ballantyne CM (2019) Effects of Icosapent ethyl on Total ischemic events: from REDUCE-IT. J Am Coll Cardiol 73(22):2791–2802. Epub 2019/03/23. 10.1016/j.jacc.2019.02.032. PubMed PMID: 30898607 [DOI] [PubMed] [Google Scholar]
- 94.Wallace JL (2000) How do NSAIDs cause ulcer disease? Bailliere Best Pract Res Clin Gastroenterol 14(1):147–159. Epub 2000/04/05. PubMed PMID: 10749095 [DOI] [PubMed] [Google Scholar]
- 95.Becker RC (2005) COX-2 inhibitors. Tex Heart Inst J 32(3):380–383. Epub 2006/01/06. PubMed PMID: 16392224; PMCID: PMC1336714 [PMC free article] [PubMed] [Google Scholar]
- 96.Groenendyk J, Paskevicius T, Urra H, Viricel C, Wang K, Barakat K, Hetz C, Kurgan L, Agellon LB, Michalak M (2018) Cyclosporine A binding to COX-2 reveals a novel signaling pathway that activates the IRE1alpha unfolded protein response sensor. Sci Rep 8(1):16678 Epub 2018/11/14. 10.1038/s41598-018-34891-w. PubMed PMID: 30420769; PMCID: PMC6232179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ghosh R, Alajbegovic A, Gomes AV (2015) NSAIDs and cardiovascular diseases: role of reactive oxygen species. Oxidative Med Cell Longev 2015:536962. Epub 2015/10/13. 10.1155/2015/536962. PubMed PMID: 26457127; PMCID: PMC4592725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ohyama K, Shiokawa A, Ito K, Masuyama R, Ichibangase T, Kishikawa N, Imai K, Kuroda N (2012) Toxicoproteomic analysis of a mouse model of nonsteroidal anti-inflammatory drug-induced gastric ulcers. Biochem Biophys Res Commun 420(1):210–215. Epub 2012/03/20. 10.1016/j.bbrc.2012.03.009. PubMed PMID: 22426477 [DOI] [PubMed] [Google Scholar]
- 99.Tsutsumi S, Gotoh T, Tomisato W, Mima S, Hoshino T, Hwang HJ, Takenaka H, Tsuchiya T, Mori M, Mizushima T (2004) Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death Differ 11(9):1009–1016. Epub 2004/05/08. 10.1038/sj.cdd.4401436. PubMed PMID: 15131590 [DOI] [PubMed] [Google Scholar]
- 100.Panigrahy D, Greene ER, Pozzi A, Wang DW, Zeldin DC (2011) EET signaling in cancer. Cancer Metastasis Rev 30(3–4):525–540. Epub 2011/10/20. 10.1007/s10555-011-9315-y. PubMed PMID: 22009066; PMCID: PMC3804913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lazaar AL, Yang L, Boardley RL, Goyal NS, Robertson J, Baldwin SJ, Newby DE, Wilkinson IB, Tal-Singer R, Mayer RJ, Cheriyan J (2016) Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br J Clin Pharm 81(5):971–979. Epub 2015/12/02. 10.1111/bcp.12855. PubMed PMID: 26620151; PMCID: PMC4834590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Askari A, Thomson SJ, Edin ML, Zeldin DC, Bishop-Bailey D (2013) Roles of the epoxygenase CYP2J2 in the endothelium. Prostaglandins Other Lipid Mediat. 107:56–63. Epub 2013/03/12. 10.1016/j.prostaglandins.2013.02.003. PubMed PMID: 23474289; PMCID: PMC3711961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fleming I (2011) The cytochrome P450 pathway in angiogenesis and endothelial cell biology. Cancer Metastasis Rev 30(3–4):541–555. Epub 2011/10/20. doi: 10.1007/s10555-011-9302-3. PubMed PMID: 22009065 [DOI] [PubMed] [Google Scholar]
- 104.Imig JD (2012) Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev 92(1):101–130. Epub 2012/02/03. 10.1152/physrev.00021.2011. PubMed PMID: 22298653; PMCID: PMC3613253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rand AA, Rajamani A, Kodani SD, Harris TR, Schlatt L, Barnych B, Passerini AG, Hammock BD (2019) Epoxyeicosatrienoic acid (EET)-stimulated angiogenesis is mediated by epoxy hydroxyeicosatrienoic acids (EHETs) formed from COX-2. J Lipid Res. Epub 2019/10/24. 10.1194/jlr.M094219. PubMed PMID: 31641036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gartung A, Yang J, Sukhatme VP, Bielenberg DR, Fernandes D, Chang J, Schmidt BA, Hwang SH, Zurakowski D, Huang S, Kieran MW, Hammock BD, Panigrahy D (2019) Suppression of chemotherapy-induced cytokine/lipid mediator surge and ovarian cancer by a dual COX-2/sEH inhibitor. Proc Natl Acad Sci U S A 116(5):1698–1703. Epub 2019/01/17. 10.1073/pnas.1803999116. PubMed PMID: 30647111; PMCID: PMC6358686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schmelzer KR, Inceoglu B, Kubala L, Kim IH, Jinks SL, Eiserich JP, Hammock BD (2006) Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A 103(37):13646–13651. Epub 2006/09/05. 10.1073/pnas.0605908103. PubMed PMID: 16950874; PMCID: PMC1564210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chopra S, Giovanelli P, Alvarado-Vazquez PA, Alonso S, Song M, Sandoval TA, Chae CS, Tan C, Fonseca MM, Gutierrez S, Jimenez L, Subbaramaiah K, Iwawaki T, Kingsley PJ, Marnett LJ, Kossenkov AV, Crespo MS, Dannenberg AJ, Glimcher LH, Romero-Sandoval EA, Cubillos-Ruiz JR (2019) IRE1alpha-XBP1 signaling in leukocytes controls prostaglandin biosynthesis and pain. Science 365(6450). Epub 2019/07/20. 10.1126/science.aau6499. PubMed PMID: 31320508. [DOI] [PubMed] [Google Scholar]
- 109.Sasich LD, Barasain MA, Al Kudsi MA (2008) The cardiovascular risks of etoricoxib (Arcoxia). Ann Saudi Med 28(2):141–142. Epub 2008/04/10. 10.5144/0256-4947.2008.141. PubMed PMID: 18398284; PMCID: PMC6074531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bruno F, Spaziano G, Liparulo A, Roviezzo F, Nabavi SM, Sureda A, Filosa R, D’Agostino B (2018) Recent advances in the search for novel 5-l ipoxygenase inhibitors for the treatment of asthma. Eur J Med Chem 153:65–72. Epub 2017/11/15. 10.1016/j.ejmech.2017.10.020. PubMed PMID: 29133059 [DOI] [PubMed] [Google Scholar]
- 111.Yang J, Bratt J, Franzi L, Liu JY, Zhang G, Zeki AA, Vogel CF, Williams K, Dong H, Lin Y, Hwang SH, Kenyon NJ, Hammock BD (2015) Soluble epoxide hydrolase inhibitor attenuates inflammation and airway hyperresponsiveness in mice. Am J Respir Cell Mol Biol 52(1):46–55. 10.1165/rcmb.2013-0440OC. PubMed PMID: 24922186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gobel T, Diehl O, Heering J, Merk D, Angioni C, Wittmann SK, Buscato E, Kottke R, Weizel L, Schader T, Maier TJ, Geisslinger G, Schubert-Zsilavecz M, Steinhilber D, Proschak E, Kahnt AS (2019) Zafirlukast is a dual modulator of human soluble Epoxide Hydrolase and Peroxisome Proliferator-activated receptor gamma. Front Pharmacol 10:263 Epub 2019/04/06. 10.3389/fphar.2019.00263. PubMed PMID: 30949053; PMCID: PMC6435570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Watkins BA (2019) Diet, endocannabinoids, and health. Nutr Res (New York, NY) 70:32–39. Epub 2019/07/10. 10.1016/j.nutres.2019.06.003. PubMed PMID: 31280882 [DOI] [PubMed] [Google Scholar]
- 114.van Esbroeck ACM, Janssen APA, Cognetta AB, Ogasawara D, Shpak G, van der Kroeg M, Kantae V, Baggelaar MP, de Vrij FMS, Deng H, Allara M, Fezza F, Lin Z, van der Wel T, Soethoudt M, Mock ED, den Dulk H, Baak IL, Florea BI, Hendriks G, DePetrocellis L, Overkleeft HS, Hankemeier T, De Zeeuw CI, Di Marzo V, Maccarrone M, Cravatt BF, Kushner SA, van der Stelt M (2017) Activity-based protein profiling reveals off-target proteins of the FAAH inhibitor BIA 10–2474. Science 356(6342):1084–1087. Epub 2017/06/10. 10.1126/science.aaf7497. PubMed PMID: 28596366; PMCID: PMC5641481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Habib AM, Okorokov AL, Hill MN, Bras JT, Lee MC, Li S, Gossage SJ, van Drimmelen M, Morena M, Houlden H, Ramirez JD, Bennett DLH, Srivastava D, Cox JJ (2019) Microdeletion in a FAAH pseudogene identified in a patient with high anandamide concentrations and pain insensitivity. Br J Anaesth 123(2):e249–e253. Epub 2019/04/02. 10.1016/j.bja.2019.02.019. PubMed PMID: 30929760; PMCID: PMC6676009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sasso O, Wagner K, Morisseau C, Inceoglu B, Hammock BD, Piomelli D (2015) Peripheral FAAH and soluble epoxide hydrolase inhibitors are synergistically antinociceptive. Pharmacol Res 97:7–15. Epub 2015/04/18. 10.1016/j.phrs.2015.04.001. PubMed PMID: 25882247; PMCID: PMC4464910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kodani SD, Bhakta S, Hwang SH, Pakhomova S, Newcomer ME, Morisseau C, Hammock BD (2018) Identification and optimization of soluble epoxide hydrolase inhibitors with dual potency towards fatty acid amide hydrolase. Bioorg Med Chem Lett. Epub 2018/01/26. 10.1016/j.bmcl.2018.01.003. PubMed PMID: 29366648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Inceoglu B, Wagner K, Schebb NH, Morisseau C, Jinks SL, Ulu A, Hegedus C, Rose T, Brosnan R, Hammock BD (2011) Analgesia mediated by soluble epoxide hydrolase inhibitors is dependent on cAMP. Proc Natl Acad Sci U S A 108(12):5093–5097. 10.1073/pnas.1101073108. PubMed PMID: 21383170; PMCID: 3064364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Blocher R, Wagner KM, Gopireddy RR, Harris TR, Wu H, Barnych B, Hwang SH, Xiang YK, Proschak E, Morisseau C, Hammock BD (2018) Orally available soluble Epoxide Hydrolase/Phosphodiesterase 4 dual inhibitor treats inflammatory pain. J Med Chem 61(8):3541–3450. Epub 2018/04/04. 10.1021/acs.jmedchem.7b01804. PubMed PMID: 29614224; PMCID: PMC5933862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hwang SH, Wecksler AT, Zhang G, Morisseau C, Nguyen LV, Fu SH, Hammock BD (2013) Synthesis and biological evaluation of sorafenib- and regorafenib-like sEH inhibitors. Bioorg Med Chem Lett 23(13):3732–3737. Epub 05/15. 10.1016/j.bmcl.2013.05.011. PubMed PMID: 23726028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liang Z, Zhang B, Xu M, Morisseau C, Hwang SH, Hammock BD, Li QX (2019) 1-Trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea, a selective and potent dual inhibitor of soluble epoxide hydrolase and p38 kinase intervenes in Alzheimer’s Signaling in human nerve cells. ACS Chem Neurosci 10(9):4018–4030. Epub 2019/08/06. 10.1021/acschemneuro.9b00271. PubMed PMID: 31378059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Reddy AS, Zhang S (2013) Polypharmacology: drug discovery for the future. Expert Rev Clin Pharmacol 6(1):41–47. Epub 2013/01/01. 10.1586/ecp.12.74. PubMed PMID: 23272792; PMCID: PMC3809828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kim JH, Morgan AM, Tai BH, Van DT, Cuong NM, Kim YH (2016) Inhibition of soluble epoxide hydrolase activity by compounds isolated from the aerial parts of Glycosmis stenocarpa. J Enzyme Inhib Med Chem 31(4):640–644. Epub 2015/10/08. 10.3109/14756366.2015.1057719. PubMed PMID: 26444316 [DOI] [PubMed] [Google Scholar]
- 124.Sun YN, Kim JH, Li W, Jo AR, Yan XT, Yang SY, Kim YH (2015) Soluble epoxide hydrolase inhibitory activity of anthraquinone components from aloe. Bioorg Med Chem 23(20):6659–6665. Epub 2015/09/16. 10.1016/j.bmc.2015.09.003. PubMed PMID: 26372074 [DOI] [PubMed] [Google Scholar]
- 125.Dimitropoulou C, West L, Field MB, White RE, Reddy LM, Falck JR, Imig JD (2007) Protein phosphatase 2A and Ca2+−activated K+ channels contribute to 11,12-epoxyeicosatrienoic acid analog mediated mesenteric arterial relaxation. Prostaglandins Other Lipid Mediat 83(1–2):50–61. Epub 2007/01/30. 10.1016/j.prostaglandins.2006.09.008. PubMed PMID: 17259072 [DOI] [PubMed] [Google Scholar]
- 126.Shen HC, Hammock BD (2012) Discovery of inhibitors of soluble epoxide hydrolase: a target with multiple potential therapeutic indications. J Med Chem 55(5):1789–1808. 10.1021/jm201468j. PubMed PMID: 22168898; PMCID: 3420824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Arnold C, Markovic M, Blossey K, Wallukat G, Fischer R, Dechend R, Konkel A, von Schacky C, Luft FC, Muller DN, Rothe M, Schunck WH (2010) Arachidonic acid-metabolizing cytochrome P450 enzymes are targets of {omega}−3 fatty acids. J Biol Chem 285(43):32720–32733. Epub 2010/08/25. 10.1074/jbc.M110.118406. PubMed PMID: 20732876; PMCID: PMC2963419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hutchens MP, Nakano T, Dunlap J, Traystman RJ, Hurn PD, Alkayed NJ (2008) Soluble epoxide hydrolase gene deletion reduces survival after cardiac arrest and cardiopulmonary resuscitation. Resuscitation 76(1):89–94. Epub 2007/08/31. 10.1016/j.resuscitation.2007.06.031. PubMed PMID: 17728042; PMCID: PMC2585367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chen D, Whitcomb R, MacIntyre E, Tran V, Do ZN, Sabry J, Patel DV, Anandan SK, Gless R, Webb HK (2012) Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects. J Clin Pharmacol 52(3):319–328. Epub 2011/03/23. 10.1177/0091270010397049. PubMed PMID: 21422238 [DOI] [PubMed] [Google Scholar]
- 130.Morris JK, Piccolo BD, John CS, Green ZD, Thyfault JP, Adams SH (2019) Oxylipin profiling of alzheimer’s disease in nondiabetic and type 2 diabetic elderly. Metabolites 9(9). Epub 2019/09/08. 10.3390/metabo9090177. PubMed PMID: 31491971; PMCID: PMC6780570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Valdes AM, Ravipati S, Pousinis P, Menni C, Mangino M, Abhishek A, Chapman V, Barrett DA, Doherty M(2018) Omega-6 oxylipins generated by soluble epoxide hydrolase are associated with knee osteoarthritis. J Lipid Res 59(9):1763–1770. Epub 2018/07/11. 10.1194/jlr.P085118. PubMed PMID: 29986999; PMCID: PMC6121933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Caligiuri SPB, Aukema HM, Ravandi A, Lavallee R, Guzman R, Pierce GN (2017) Specific plasma oxylipins increase the odds of cardiovascular and cerebrovascular events in patients with peripheral artery disease. Can J Physiol Pharmacol 95(8):961–968. Epub 2017/07/18. 10.1139/cjpp-2016-0615. PubMed PMID: 28714336 [DOI] [PubMed] [Google Scholar]
- 133.Theken KN, Schuck RN, Edin ML, Tran B, Ellis K, Bass A, Lih FB, Tomer KB, Poloyac SM, Wu MC, Hinderliter AL, Zeldin DC, Stouffer GA, Lee CR (2012) Evaluation of cytochrome P450-derived eicosanoids in humans with stable atherosclerotic cardiovascular disease. Atherosclerosis 222(2):530–536. Epub 2012/04/17. 10.1016/j.atherosclerosis.2012.03.022. PubMed PMID: 22503544; PMCID: PMC3361525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hennebelle M, Otoki Y, Yang J, Hammock BD, Levitt AJ, Taha AY, Swardfager W (2017) Altered soluble epoxide hydrolase-derived oxylipins in patients with seasonal major depression: an exploratory study. Psychiatry Res 252:94–101. Epub 2017/03/05. 10.1016/j.psychres.2017.02.056. PubMed PMID: 28259037; PMCID: PMC5611448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Santos JM, Park JA, Joiakim A, Putt DA, Taylor RN, Kim H (2017) The role of soluble epoxide hydrolase in preeclampsia. Med Hypotheses 108:81–85. Epub 2017/10/23. 10.1016/j.mehy.2017.07.033. PubMed PMID: 29055406 [DOI] [PubMed] [Google Scholar]
- 136.Yu D, Hennebelle M, Sahlas DJ, Ramirez J, Gao F, Masellis M, Cogo-Moreira H, Swartz RH, Herrmann N, Chan PC, Pettersen JA, Stuss DT, Black SE, Taha AY, Swardfager W (2019) Soluble epoxide hydrolase-derived linoleic acid Oxylipins in serum are associated with periventricular White matter Hyperintensities and vascular cognitive impairment. Transl Stroke Res 0(5):522–533. Epub 2018/11/18. 10.1007/s12975-018-0672-5. PubMed PMID: 30443886 [DOI] [PubMed] [Google Scholar]
- 137.Moghaddam MF, Grant DF, Cheek JM, Greene JF, Williamson KC, Hammock BD (1997) Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat Med 3(5):562–5666. Epub 1997/05/01. PubMed PMID: 9142128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kopf PG, Zhang DX, Gauthier KM, Nithipatikom K, Yi XY, Falck JR, Campbell WB (2010) Adrenic acid metabolites as endogenous endothelium-derived and zona glomerulosa-derived hyperpolarizing factors. Hypertension 55(2):547–554. Epub 2009/12/30. 10.1161/hypertensionaha.109.144147. PubMed PMID: 20038752; PMCID: PMC2819927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Serhan CN (2014) Pro-resolving lipid mediators are leads for resolution physiology. Nature 510(7503):92–101. Epub 2014/06/06. 10.1038/nature13479. PubMed PMID: 24899309; PMCID: PMC4263681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.McGiff JC, Quilley J (1999) 20-HETE and the kidney: resolution of old problems and new beginnings. Am J Phys 277(3 Pt 2):R607–R623. Epub 1999/09/14. PubMed PMID: 10484476 [DOI] [PubMed] [Google Scholar]
- 141.Roman RJ (2002) P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82(1):131–185. Epub 2002/01/05. 10.1152/physrev.00021.2001. PubMed PMID: 11773611 [DOI] [PubMed] [Google Scholar]
- 142.Mezentsev A, Mastyugin V, Seta F, Ashkar S, Kemp R, Reddy DS, Falck JR, Dunn MW, Laniado-Schwartzman M (2005) Transfection of cytochrome P4504B1 into the cornea increases angiogenic activity of the limbal vessels. J Pharmacol Exp Ther 315(1):42–50. Epub 2005/07/13. 10.1124/jpet.105.088211. PubMed PMID: 16009741 [DOI] [PubMed] [Google Scholar]
- 143.Seta F, Patil K, Bellner L, Mezentsev A, Kemp R, Dunn MW, Schwartzman ML (2007) Inhibition of VEGF expression and corneal neovascularization by siRNA targeting cytochrome P450 4B1. Prostaglandins Other Lipid Mediat 84(3–4):116–127. Epub 2007/11/10. 10.1016/j.prostaglandins.2007.05.001. PubMed PMID: 17991614; PMCID: PMC2128778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fromel T, Kohlstedt K, Popp R, Yin X, Awwad K, Barbosa-Sicard E, Thomas AC, Lieberz R, Mayr M, Fleming I (2013) Cytochrome P4502S1: a novel monocyte/macrophage fatty acid epoxygenase in human atherosclerotic plaques. Basic Res Cardiol 108(1):319 Epub 2012/12/12. 10.1007/s00395-012-0319-8. PubMed PMID: 23224081 [DOI] [PubMed] [Google Scholar]
- 145.Weintraub NL, Fang X, Kaduce TL, VanRollins M, Chatterjee P, Spector AA (1999) Epoxide hydrolases regulate epoxyeicosatrienoic acid incorporation into coronary endothelial phospholipids. Am J Phys; 277(5):H2098–H2108. Epub 1999/11/24. 10.1152/ajpheart.1999.277.5.H2098. PubMed PMID: 10564166 [DOI] [PubMed] [Google Scholar]
- 146.Morisseau C, Hammock BD (2005) Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol; 45:311–33. 10.1146/annurev.pharmtox.45.120403.095920. PubMed PMID: 15822179 [DOI] [PubMed] [Google Scholar]