

Abstract
Children from low- and middle-income countries, where there is a high incidence of infectious disease, have the greatest need for the protection afforded by vaccination, but vaccines often show reduced efficacy in these populations. An improved understanding of how age, infection, nutrition, and genetics influence immune ontogeny and function is key to informing vaccine design for this at-risk population. We sought to identify factors that shape immune development in children under five years of age from Tanzania and Mozambique by detailed immunophenotyping of longitudinal blood samples collected during the RTS,S malaria vaccine phase III trial. In these cohorts, the composition of the immune system is dynamically transformed during the first years of life, and this was further influenced by geographical location, with some immune cell types showing an altered rate of development in Tanzanian children compared to Dutch children enrolled in the Generation R population-based cohort study. High titer antibody responses to the RTS,S/AS01E vaccine were associated with an activated immune profile at the time of vaccination, including an increased frequency of antibody-secreting plasmablasts and follicular helper T cells. Anemic children had lower frequencies of recent thymic emigrant T cells, isotype-switched memory B cells and plasmablasts; modulating iron bioavailability in vitro could recapitulate the B cell defects observed in anemic children. Our findings demonstrate that the composition of the immune system in children varies according to age, geographical location and anemia status.
One Sentence Summary:
The composition and function of the immune system in African children is linked to age, location and anemia.
Introduction
Age shapes the composition and function of the human immune system. In particular the first five years of life are a pivotal time in immune development: after birth the immune system changes as thymic output increases, and maternally-derived immune mediators such as antibodies are lost (1, 2). This occurs in parallel with the acquisition and cultivation of the commensal microbiome, encounters with food antigens and exposure to numerous environmental microbes (1). These factors, combined with an individual’s genetics, shape the human immune system (3–6). In this early period of immune development the functional capacity of the immune system appears to be limited, resulting in a reduced ability to generate protective cellular and humoral immunity after vaccination and an increased susceptibility to infectious diseases (2). This results in higher rates of morbidity and mortality from infectious disease in young children world-wide: in 2017, pneumonia, diarrhea and malaria caused ~1.5 million deaths in children under five years of age (7). These infectious diseases disproportionately affect children in low- and middle-income countries (LMICs), particularly those in sub-Saharan Africa (7, 8). A higher pathogen burden, sub-optimal nutrition, impaired maternal health, and poor access to healthcare undoubtedly contribute to this increased burden of infectious disease. However, there is limited understanding of how these factors affect the development and function of the immune system during childhood in LMICs, and how immune system development differs between populations around the world.
In-depth immune phenotyping of peripheral blood samples from large cohorts provides a way to characterize which factors influence the composition of the human immune system. Such studies have estimated that between 20-40% of this inter-individual variation is driven by genetics, demonstrating that non-genetic factors play a dominant role in moulding the composition of the immune system (3, 9–11). Determining the contribution of non-genetic factors to human immunity is highly complex due to the wide range of potential factors and their putative interactions. Nevertheless, an individual’s age, their environment, and chronic viral infections have emerged as key factors that influence human immune variation over time (3–6, 9). While these immunophenotyping studies have increased our understanding of the factors that influence the composition of the human immune profile, it has been difficult to integrate this knowledge in the context of immune function.
This study aimed to characterize how age and other non-genetic factors alter the composition of the immune system of young children living in Tanzania and Mozambique and to link immune status to vaccine responsiveness. Comprehensive immune profiling and transcriptomic analysis was performed on peripheral blood samples collected over a 32-month period from infants and children that were under 5 years of age and participants in a phase III study of the malaria vaccine RTS,S/AS01E (12). This study also aimed to compare immune composition to Dutch children from the Generation R study (5).
Results
Blood leukocyte subsets are stable one month after vaccination
First we compared peripheral blood mononuclear cells (PBMC) samples from the RTS,S trial by flow cytometry. In the RTS,S trial, conducted across seven countries in sub-Saharan Africa, participants were vaccinated three times at monthly intervals with RTS,S/AS01E or a comparator vaccine, followed by a booster dose of vaccine at month 20 (12). PBMC from participants from Tanzania and Mozambique collected at baseline, 3, 21 and 32 months into the trial (Fig. 1A,B, tables S1–2) were analyzed by flow cytometry using both the unsupervised T-Distributed Stochastic Neighbour Embedding (tSNE) method (fig. S1), and by manual gating to identify well-characterized innate and adaptive immune cell populations (Figures S2–5, Tables S3–5). Multidimensional scaling (MDS) analysis of tSNE data from Tanzanian children (5-17 months old at enrollment, n=116) revealed that the immune profile of participants did not differ between vaccine groups at either 3 months (B3) or 21 months (B21) after vaccination (Figure 1C, D). The frequencies of individual tSNE clusters or manual gated populations were not different between participants given the RTS,S or a comparator vaccine at any time point sampled (B0, B3, B21, B32, Figure 1E–H), nor did the type of vaccine administered or the sex of participants significantly contribute to the inter-individual variance observed for immune cell types at any time point (Figure S6A–D). In addition, no cell types or tSNE clusters were changed between baseline and B3 for any group (Fig. 1E, F). Therefore, no vaccine-induced changes to blood immune cells were detected one month after vaccination, indicating that neither the RTS,S or comparator vaccine caused lasting changes to the participant’s immune profile, and that children’s immune profiles are stable over a short timescale of 3 months. Likewise, reanalysis of blood transcriptomes from clinical trial of the RTS,S vaccine in adults from North America (13) showed minimal vaccine-induced gene expression changes remained 21-28 days vaccination (Fig. S6E). These findings are consistent with previous studies showing that changes in the cellular composition of the adult immune system after vaccination occur in the first two weeks after vaccination, followed by a return to baseline (4, 14). However, comparison of the B0 with B21 samples identified 76 tSNE clusters and 25 manual gated cell types that were altered during this timeframe (all vaccine groups combined, Fig. 1G, H), indicating that the immune landscape of children changes over a 21-month period. Consistent with this, fewer strong correlations of cell subset frequencies were observed between baseline and 32 months than between baseline and B3 (Fig. S7–9). This change over a short period is unique to children, as adults have stable immune profiles over a 2-6 year period (3, 4).
Figure 1. No changes to blood leukocyte subsets are detected one month after third vaccination.
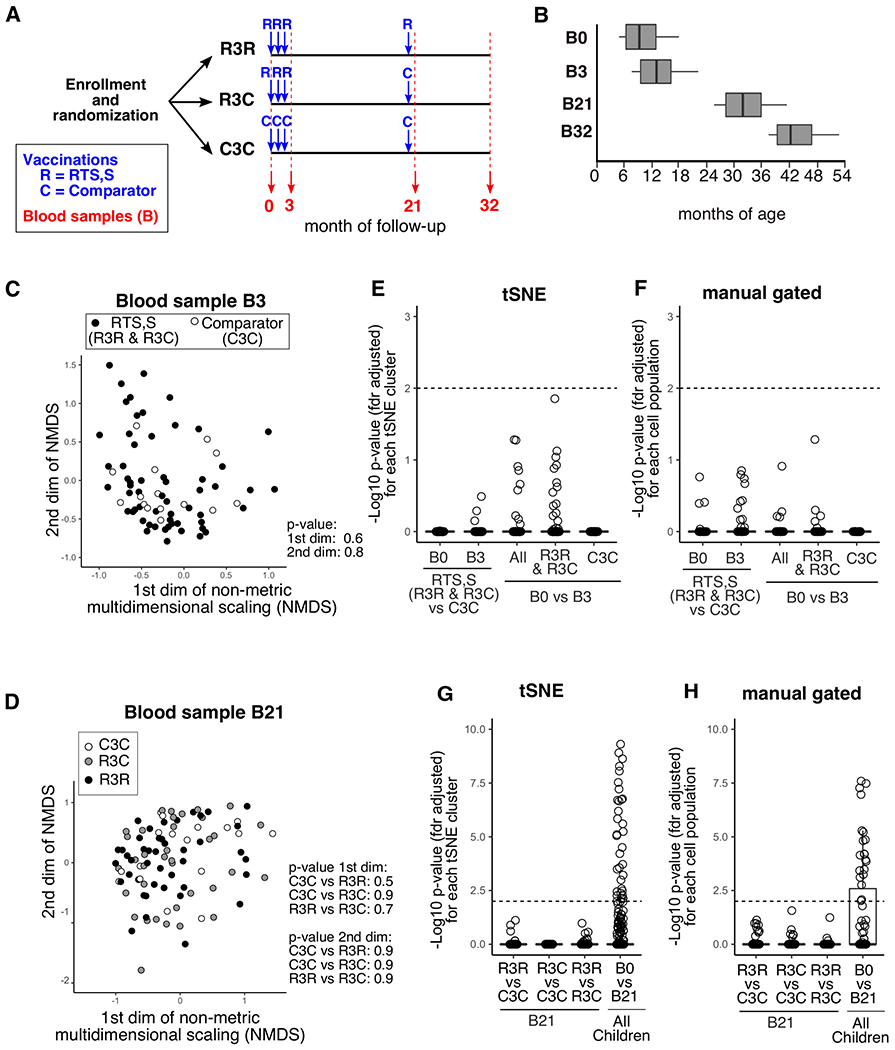
A) The timeline for vaccinations and blood sampling during the vaccine trial follow-up period. The three vaccination groups are shown (R3R, R3C, C3C). Vaccinations are shown in blue (R = RTS,S, C = Comparator). Comparator vaccine differed between age groups (Menjugate for infants, Verorab for children). Red numbers indicate the month of follow-up at which blood samples were collected. B) The distribution of participant age (in months) at each of the blood sampling time-points for Tanzanian children (n=414 total samples). Non-metric multidimensional scaling (NMDS) for Tanzanian children at blood sample 3 (C) or blood sample 21 (D) based on the 336 tSNE clusters and vaccine group (B3; R3R &R3C n = 63, comparator n= 17, B21; C3C n = 18, R3C n= 39, R3R n=44). Samples from R3R and R3C groups were combined at B3 as identical vaccines had been received. The frequencies of 336 tSNE clusters (E, as a proportion of each subpopulation) or 70 manually gated cell subsets for each participant (F, as a % of all leukocytes) were compared between vaccine groups at B0 and B3, and within groups at B3 relative to B0 (All = All Tanzanian children n = 80). Between-group comparisons were analyzed with the Mann-Whitney test, B0 vs B3 used paired data and Wilcoxon signed-rank test. The −log10 p-value is plotted for each cluster or subset (fdr adjusted for multiple comparisons). tSNE clusters (G) or manual gated cell subsets (H) were compared within vaccine group at B21, and in all children (pair-wise) at B21 relative to B0. In E-H any population with an adjusted p-value of less than 0.01 were deemed significantly different, the dotted line indicates padj=0.01.
Dynamic development of the immune system occurs during childhood
To determine how age impacts the blood immune cell composition in the first years of life, we compared the immune profile at B3 of infants (4.8-5.8 months old, n=43) and children (7.5-22 months old, n=55) from Mozambique (Fig. 2A). The two age groups separated from each other by the first MDS dimension of tSNE clusters (Fig. 2B, p < 10−16), and manual gating analysis revealed that 33 immune subsets were different between the age-groups (Fig. 2C). These results, and regression modelling (Fig. 2D), demonstrate that age was the biggest contributor to immune variation between these participants. For example, naïve B cell and CD25+CD127low Treg cell frequencies were elevated in infants compared to children (Fig. 2E). Reciprocally, frequencies of CD4+ T cell memory and T helper cell subsets were increased in children (Fig. 2E). Of note, circulating T follicular helper (cTfh) cells and plasmablasts were more abundant in children (Fig. 2E), cell types that are known to increase transiently after infection or vaccination (4, 14). This could be because children had higher exposure to infections and vaccinations than infants and these cells act as circulating biomarkers of an ongoing immune response in secondary lymphoid tissues (15). Alternatively, these data may indicate that children are more capable than infants of mounting the cellular immune response required for the production of humoral immunity. Consistent with the latter hypothesis, infants had reduced IgG titers to the RTS,S vaccine components hepatitis B surface antigen (HBV.S) and Plasmodium falciparum circumsporozoite surface protein (CSP) compared to children in the trial (Fig. 2F, Fig. S10A–B).
Figure 2. Dynamic development of memory B and T cells occurs between 6 months and 2 years of age.

A) The age (in months) of infant and children age-groups from Mozambique during study follow-up. Red box indicates the B3 samples (n = 37 infants, n = 52 children). B) Non-metric multidimensional scaling (NMDS) for children and infants at B3 based on the 336 tSNE clusters, with the coordinates for each dimension compared Mann-Whitney test. C) Heat map showing the manually gated cell subsets (as % of all leukocytes) with an fdr-adjusted p-value of less than 0.001 between children and infants at B3 by Mann-Whitney test. Cell types are shown as rows, and each column represents an individual participant. Rows and columns were clustered using Euclidean distance, and the cell subset frequency converted to z-scores. Individuals without any missing values shown (n = 22 infants, n = 30 children) D) The contribution of sample age group and sex to the cell type variance as determined from linear regression models, shown as a boxplot summary of the 33 cell types shown in C (n= 89 samples). E) Representative cell type frequencies (as % of ‘parent’ population) for infants and children, with p-value from Mann-Whitney test (fdr adjusted for 70 subsets, n = 37 infants, n = 52 children). F) Anti-HBV.S or -CSP IgG titer (B3-B0 AU) from a cohort of RTS,S vaccinated participants from Mozambique (n=198 infants, n= 137 children), with p-value from Mann-Whitney. The 336 tSNE clusters (G) or 70 manually gated cell subsets (H) were compared between infants and children at B3, B21 and B32, and the −log10 fdr-adjusted p-value is shown, with dotted line indicating padj=0.01.
Further longitudinal analyses revealed that the immune profile of infants and children continued to change over the 32-month duration of the vaccine trial (Fig. 2G), indicating ongoing immune development throughout early childhood. To investigate the immune ontogeny over this longer time period, we utilised the larger cohort of samples collected over 32 months from children in Tanzania (n=116, Fig. 3A), which confirmed that age is a major driver of immune variation in this cohort (Fig. 3B). To determine how specific immune cell types change during the first years of life, sample age and sex was modelled against cell type frequency using linear mixed effect regression (LMER)(Fig. 3C, Age range 4.8 months – 4.3 years). This identified 20 cell types that changed in the first years of life in Tanzanian children with age, including increases in Ig isotype-switched memory B cells, T helper cells, and dendritic cell subsets with age (Fig. 3C, D).
Figure 3. Dynamic changes of the immune landscape during early childhood.

A) The age in months of Tanzanian children at blood samples throughout follow-up period (n = 414 in total). B) Non-metric multidimensional scaling (NMDS) for children at B0 (n = 78) and B32 (n = 109) based on the 336 tSNE clusters, with the coordinates for each dimension compared by Mann-Whitney test. Colors correspond to time-point (B0 = white, B32 = black). C) Heatmap of cell subsets which were significantly different between B0 and B32 in Tanzanian children (p<0.0001 and conditional R2 >0.25), with the frequency predicted from LMER models shown normalized to week 20 frequency. p-values for the effect of sex and age determined by ANOVA of LMER models after fdr adjustment shown in grey boxes, and the marginal and conditional R2 of LMER model shown in purple for each cell subset. D) The frequency of four representative cell subsets (as a % of parent population) plotted against age for each sample, with linear-mixed effects regression (LMER) modelling (red line). Marginal R2 and p-value for fit of LMER models shown in boxes above each plot.
The change in the composition of the immune system as children get older was recapitulated using RNA-sequencing of whole PBMC from Tanzanian children from B0 (Age 5.3-17.2 months, n=21) and B32 (Age 37-50 months, n=9) (Fig. 4A–B, data file S1). Blood transcriptional module analysis (16) identified gene signatures of cell division, CD4+ T cell proliferation and B cells as enriched in samples from younger children (B0, Fig. 4C). An erythrocyte differentiation signature was more abundant in younger children indicating the presence of nucleated red blood cells or reticulocytes that are common in infants, and are retained during density centrifugation of whole blood. In older children (B32), signatures of dendritic cell, NK cell and CD4+ T cell activation were enriched, along with hallmarks of NFKB and AP-1 activation and cytokine signalling, suggesting of a higher state of immune activation (Fig. 4D). These data demonstrate that the steady-state blood transcriptome in children is strongly influenced by age, indicative of the profound changes to the immune system that occur during this early period in life.
Figure 4. Blood gene expression signatures reflect innate and adaptive immune cell changes during early childhood.

A) The age (in months) of the Tanzanian children at blood samples B0 and B32 for which RNA sequencing was performed. Dots represent individual samples, B0 (n=21) and B32 (n=9). B) Hierarchical clustering of 2146 differentially expressed (DE) genes between B0 and B32 (1148 upregulated at B0, 1008 upregulated at B32). Significantly different genes had an fdr-adjusted p-value of <0.01 from DESeq2 and an fdr-adjusted p-value of <0.05 from the intensity-difference test, and were clustered using Euclidean distance. Each column represents an individual sample, and genes shown as rows. Enriched blood transcriptional modules (BMTs) at C) B0 and D) B32, represented as colored circles with annotations as text. Colors and connecting lines show gene-sets with related genes, and summary terms shown in text boxes. BTMs enriched from within all expressed genes (17,607) with an enrichment score greater than 1.5 or less and −0.5 and a fdr adjusted p-value of <0.01 (Kolmorogov-Smirnov test) are shown. E) A stylized representation of the Multi-Omics Factor Analysis (MOFA) modelling, which was built using 5000 most variable genes from RNA-Seq and 61 immune cell types measured by flow cytometry for 27 samples (19 B0, 8 B32), and reduces these 5061 parameters to latent factors (LF). F) The percentage of total variance (R2) in the MOFA model explained by flow cytometry or RNA-Seq data sets. G) Scatter plot showing the distribution of samples by the first 2 LF determined by MOFA modelling. Each sample shown as a circle (B0 = white, B32 = black). The percentage of total variance, and the contribution of flow and RNA to each factor are indicated in the axes labels. H) The correlation between the age in weeks for each sample and the sum of values for LF1 and LF2 (Factor sum, B0 = white, B32 = black). Linear regression fit shown by the red line, and R2 and p-value from Pearson correlation are shown. The proportional loading for the 12 cell types with the strongest contribution to I) Factor 1 or K) Factor 2 are shown. The p-values (-log10 fdr-adjusted) for BTMs that were significantly enriched (p-adjusted <0.05) among genes identified by J) Factor 1 and L) Factor 2, and corresponding enrichment scores. I-L) Colour and symbol representing the direction of loading or enrichment in factor space (blue = negative, red = positive).
In addition to age, genetics and other extrinsic factors can contribute inter-individual variation in the immune landscape (9). As many of the age-related changes to immune cell types and gene expression identified in this cohort were highly heterogeneous between individuals, we sought to quantify the contribution of age to immune variation during this dynamic time-frame in young African children. For this we applied multi-omics factor analysis (MOFA) to our flow cytometry and RNA-Seq data (Fig. 4E), which is an unsupervised method to that can deconvolve the sources of heterogeneity in diverse data types (17, 18). The MOFA model attributed the variance in the whole dataset evenly between the RNA-Seq and flow cytometry assays (Fig. 4F). Age clearly separated by the first two MOFA factors, which combined explained 46.2% of the total variance in the dataset, and the first two factors combined strongly correlated the age of each sample (Fig. 4G,H). MOFA showed that a decrease in naïve B cells and an increase in activated dendritic cells, effector memory CD4+ T cells and memory B cells in flow cytometry and transcriptomic data best explained the age-associated immune variance (Fig. 4I–L). This analysis shows that age has a major influence on the immune landscape in the first years of life, with T and B lymphocytes and dendritic cells showing the most dynamic changes.
Comparison of Tanzanian and Dutch children reveals that age and location associate with differences in the immune landscape
To investigate whether these changes over time are characteristic of human immune ontogeny broadly or whether there is an immune signature that is specific to childhood in Tanzania, we reanalyzed published data from a longitudinal immunophenotyping study of Dutch children that were followed from birth to 6 years of age (Generation R cohort, (5)). Children from the same age range in both studies (20-125 weeks of age), and the 19 cell subsets that were measured with the identical marker combinations in both cohorts were included in the analysis (Fig. 5A). LMER modelling of immune cell frequencies against age and sex revealed that some immune cell types were similar between cohorts, but some subsets were influenced by the child’s country of origin (Fig. 5B, C): Vδ2+ cells and CD8+ T effector memory cells showed the same pattern in both cohorts, but the accumulation of memory cells as a proportion of CD4+ and CD8+ T cells differed between Dutch and Tanzanian children. In particular, the frequencies of CD4+ T effector memory cells in Dutch children under 1 year of age were lower, but had converged with Tanzanian children by 2 years of age (Fig. 5B, C). A higher frequency of total memory B cell subsets was also observed in Tanzanian children, however the abundance of immunoglobulin isotypes was variable between the CD27− and CD27+ memory B cell subsets (Fig. 5B, C). Tanzanian children showed a shift towards more IgM, and fewer IgG and IgA expressing cells in the CD27− compartment. In contrast, frequencies of CD27+IgG+ and CD27+IgM+ cells were higher in Tanzanian children (Fig. 5B, C). Together, these data suggest that some immune cells mature as part of a common childhood immune maturation program, but others may be distinct in Tanzanian and Dutch children.
Figure 5. Comparison of Tanzanian and Dutch children reveals that age and location are linked with changes in the immune landscape.
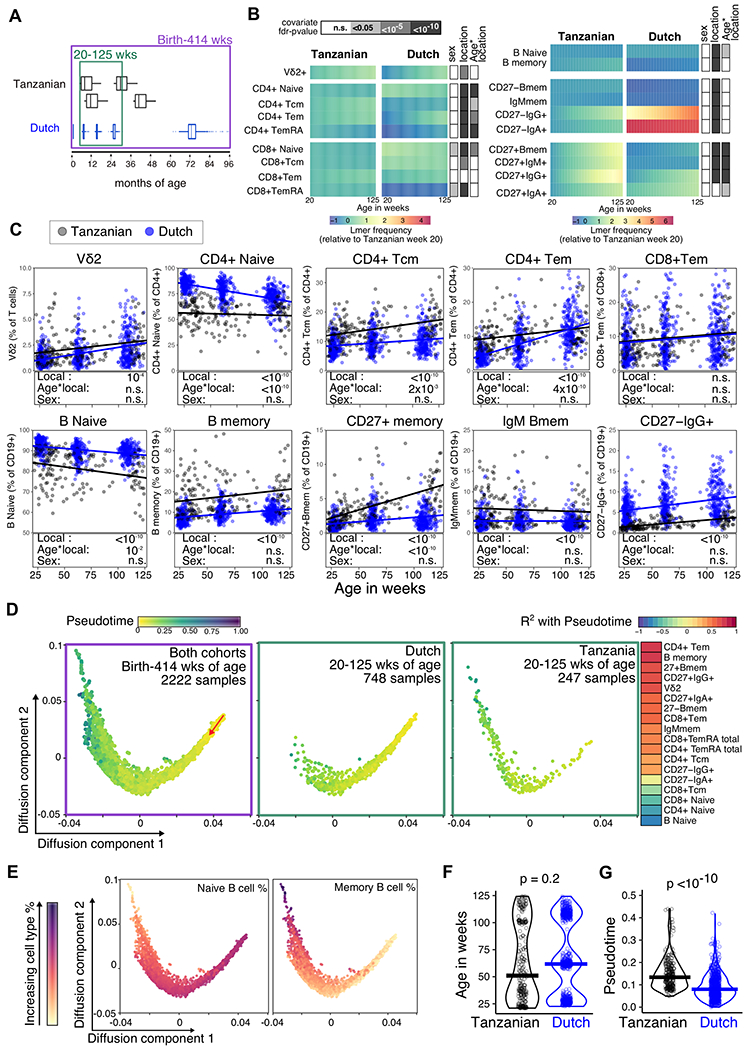
A) The age in months for Tanzanian samples relative to Dutch cohort. The green box indicates the 20-125 week age-range used for age-matched LMER modelling, with 176 total samples from 102 Tanzanian children, and 748 total samples from 504 Dutch children. The purple box represents the birth-414 week age-range used for immune trajectory building. B) Cell subsets that were measured in both cohorts and were significantly altered with age in either cohort are shown as the frequency predicted from LMER models, normalized to the Tanzanian cohort week 20 levels. p-values determined by ANOVA for the effect of sex and location (Tanzanian or Dutch cohorts), and for the interaction between age and location are shown in grey boxes, with fdr adjustment. C) The frequency (% of either B cells, CD4 or CD8 T cells) of representative cell subsets are shown from 20-125 weeks of age for the Tanzanian (Grey) and Dutch samples (blue). Solid lines represent respective LMER models, with p-values from ANOVA for location, age and sex effects shown in boxes below (fdr adjusted for the 19 of cell subsets). D) Diffusion-map dimensionality reduction of Dutch and Tanzanian samples using scaled cellular frequencies and the diffusion-pseudotime algorithm. Each dot represents a sample, color represents the pseudotime output values, and the red arrow indicates the direction of the trajectory that starts with the Dutch newborn samples. First panel shows all samples used for building the trajectory (421 total samples from 157 Tanzania children, 1801 total samples from 1119 Dutch children), and second and third panels show Dutch and Tanzanian samples in the 20-125wk age-range, respectively, used in LMER models in B and C. The correlation coefficients (R2) for the 18 cell types which significantly correlated with the pseudotime are shown as colored boxes. E) The distribution of naïve and memory B cell frequencies along the diffusion-map trajectory are shown for 2222 samples, with color representing the scaled cell type frequency. F) Age (in weeks) and corresponding pseudotime age for samples in the 20-125 wk age–range shown in D, with p-values determined by Mann-Whitney test.
A common trajectory of immune development in neonates has been proposed from studies of newborns in Europe, West Africa and Australasia (6, 19) To determine whether Tanzanian and Dutch children follow a similar path of immune development, we applied a diffusion-pseudotime algorithm(20) to the immunophenotyping data that spanned birth to 8 years of age Children’s immune profiles were distributed along a continuous spectrum that originated with samples from newborn Dutch children and progressed with age (Fig. 5D, Fig. S11). The two cohorts clustered together and followed the same pseudotime path (Fig. 5D), which has been reported to indicate a shared trajectory that is not influenced by technical differences between datasets (21). A pseudotime measure of immunological age was determined for each sample relative to their position along the trajectory, and this position was strongly correlated with a transition from naïve to memory lymphocytes (Fig. 5D, E). 20-125-week-old children from Tanzania had higher pseudotime values than Dutch counterparts of the same age-range (Fig. 5F, G). These data suggest that immune cell types develop along a common pathway throughout childhood, but the rate at which these changes occur can vary between countries.
Immune development during childhood proceeds differently in Tanzania and Mozambique.
The observation that the immune landscape in children is influenced by location at the continental level prompts the hypothesis that there may be differences in immune development between African countries. Children from Mozambique (n=55) clustered distinctly from children of the same age from Tanzania (n=116) by both MDS dimensions of tSNE clusters (B0 samples, Fig. 6A). We identified 31 immune subsets that varied between the sites, and, apart from total and naïve B cells, the altered cell types showed lower frequencies in children from Tanzania. These cells came from diverse immune lineages, including NK cells, monocytes, and memory B and T cells (Fig. 6B). The influence of the different African sites on immune composition was present at baseline (B0) and continued throughout the 32-month study period (Fig. 6C). At baseline children from Mozambique had increased frequencies of activated monocytes, activated CD4+ and CD8+ T cells (HLADR+CD38+) and Th subsets compared to children from Tanzania (Fig. 6D). This was accompanied by elevated cTfh cell and plasmablasts frequencies at baseline (Fig. 6D). Interestingly, the increase in cTfh cells and plasmablasts in children from Mozambique was linked to higher IgG titers to both components of the RTS,S vaccine (Fig. 5E and fig. S12A–B, Tanzania n = 123, Mozambique n=137). This decrease in antibody responses to the RTS,S vaccine in Tanzanians compared to Mozambicans was validated in an independent sub-cohort of infants from the phase 3 trial for which serum antibody titres were measured (fig. S12C–D), indicating that country-specific differences are present from early in life. Despite the close proximity of the two countries within East Africa, our results demonstrate that country of origin alters immune development in children.
Figure 6. Childhood immune development is influenced by location within Africa.

A) Non-metric multidimensional scaling (NMDS) for children at B0 from Tanzania (n= 78) and Mozambique (n=30) based on the 336 tSNE clusters, with the coordinates for each dimension compared by Mann-Whitney test. B) Heat map showing manually gated cell subsets (as % of all leukocytes) with an fdr-adjusted p-value of less than 0.01 between each site at B0 by Mann-Whitney test. Cell types are shown as rows, and each column represents an individual sample. Rows and Columns were clustered using Euclidean distance, and the cell subset frequency converted to z-scores (Mozambique n= 19, Tanzania n=86). C) The 336 tSNE clusters or 70 manually gated cell subsets were compared between samples from Tanzania and Mozambique at B0, B3, B21 and B32 by Mann-Whitney test. The −log10 fdr-adjusted p-value is shown, with the dotted line indicating padj=0.01. D) Representative cell type frequencies for each site (as a % of parent population), with p-value from Mann-Whitney test (fdr adjusted for 70 subsets). E) The increase in Anti-HBV.S or Anti-CSP IgG (B3-B0 AU) at B3 (B3 minus B0) from a larger cohort of RTS,S vaccinated participants from each site (n=137 Mozambique, n= 123 Tanzania), with p-value from Mann-Whitney test. F) The age of children for immunophenotyping samples from each site (Tanzania n = 33, Mozambique n = 94). G) The contribution of sample age, sex, and country (Tanzania or Mozambique) to the cell type variance as determined from linear regression models, shown as a boxplot summary of the 31 cell types shown in B. H) Weight and I) height (z-scores, age-adjusted according to global reference values) for each site (Tanzania n = 359, Mozambique n = 974). J) Blood hemoglobin concentration (g/dL) for each site, with dotted lines representing the various disease categories (Tanzania n = 359, Mozambique n = 974).
Within this study, age was not different between the two sites (Fig. 6F), and despite a trend towards Mozambican children being older, age and sex made a minimal contribution to the country specific variance observed (Fig. 6G). Weight did not differ between these two cohorts (Fig. 6H), and 55% of participants were male at both sites, although Tanzanian children were slightly taller for their age (Fig. 6I). In our transcriptomic dataset from Tanzanian children at B0 we observed enrichment for gene signatures of erythrocyte maturation and oxygen transport, which suggests that anemia may impact the blood transcriptome. This led us to speculate that anemia may influence the geographic differences observed in our study, and we observed lower hemoglobin concentration in children from Tanzania compared to Mozambique (Fig. 6J). This was also reflected in the higher frequency of children characterized as having moderate or severe anemia in Tanzania (60%) compared to Mozambique (44%), based on WHO definitions (22). This prompts the hypothesis that anemia may be one of the factors influencing immune composition in children.
Anemia is linked with an altered composition of the immune system in children and reduced iron bioavailability impairs plasmablast generation
To investigate whether anemia influences the immune landscape we compared gene expression and immune profiles in blood samples between age-matched anemic (<8.5g/dL hemoglobin, n=6) and non-anemic (>10.5g/dL hemoglobin, n=9) children from Tanzania and Mozambique subsampled from within our dataset based on hemoglobin concentration (Fig. 7A). Anemia status influenced the blood transcriptome (Fig. 7B, data file S2), with a strong enrichment of an erythrocyte differentiation and heme biosynthesis gene signature in anemic children (Fig. 7C), indicative of an increase in reticulocytes as a compensatory mechanism during anemia. In non-anemic children, blood transcription modules of mitosis, CD4+ T cell differentiation, B cells and plasma cell were enriched (Fig. 7D), suggesting that anemia is associated with changes to the cellular composition of the immune system. Anemia contributed significantly to the immune variation (Fig. 7E), and the immune cell types altered in anemic children mirrored the transcriptomic analysis; anemic children had fewer recent thymic emigrant CD4+ T cells, IgG memory B cells and plasmablasts than non-anemic children at B0 (Fig. 7F). In this cohort, the anemic status of children changed over time, enabling us to evaluate immune phenotypes in older children who developed anemia later in life. The immune cell types impacted by anemia at B0 were also reduced in anemic children at B21 (Fig. 7G), none of which were anemic at B0.
Figure 7. Anemia is linked to changes in immunophenotype and iron availability directly impacts B cell biology.
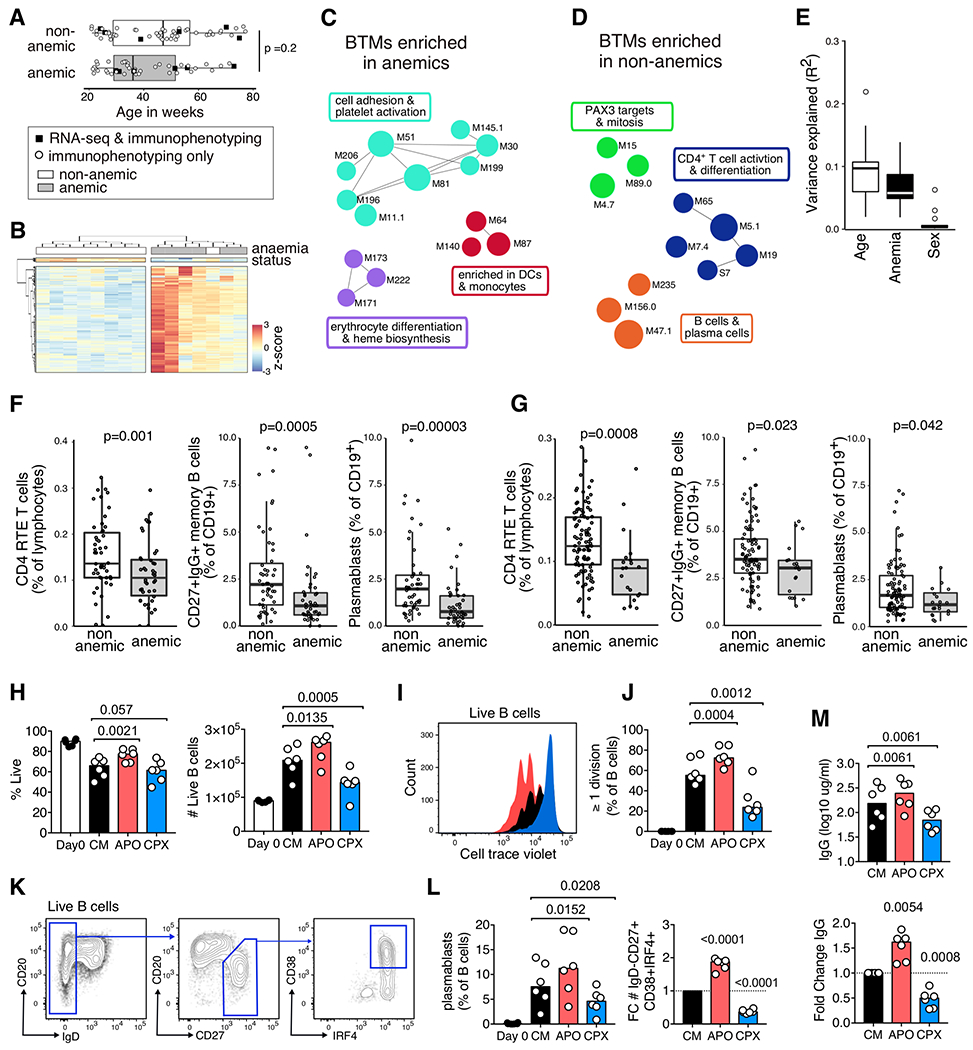
A) The age of anemic (<8.5g/dL hemoglobin, n = 43) and non-anemic (>10.5g/dL hemoglobin, n = 52) from Tanzania and Mozambique at B0. Black squares indicate samples where RNA sequencing was performed on whole PBMC in addition to immunophenotyping. B) Hierarchical clustering of 376 differentially expressed (DE) genes between anemic (n=6) and non-anemic (n = 8) Tanzanian children. Significantly different genes had an fdr-adjusted p-value of <0.01 from DESeq2 and an fdr-adjusted p-value of <0.05 from the intensity-difference test, and were clustered using Euclidean distance. Each column represents an individual sample, and genes shown as rows. Enriched blood transcriptional modules (BMTs) in C) anemic and D) non-anemic children, represented as colored circles with annotations as text. Colors and connecting lines show gene-sets with related genes, and summary terms shown in text boxes. BTMs enriched from within all expressed genes (17,607) with an enrichment score greater than 1.5 or less and −0.5 and a fdr adjusted p-value of <0.01 (Kolmorogov-Smirnov test) are shown. E) The contribution of sample age, anemia status and sex to the cell type variance as determined from linear regression models. Representative cell subset frequencies that were significantly different between anemic and non-anemic children at F) B0 (non-anemic n=52, anemic n = 43) and G) B21 (non-anemic n=99, anemic n = 20), with p-value from Mann-Whitney test. (H-M) Purified B cells from UK adults were stained with CellTraceViolet and incubated in culture media with IL-21 and CD40L (CM) for five days. Some cultures were supplemented with either apo-transferrin (APO) throughout or with ciclopirox olamine (CPX) after 24 hours of culture. H) The percentage and number of live B cells at Day 0 and Day 5 in CM, APO and CPX culture conditions. I) Representative histogram of cell trace violet on day 5 (black = CM, red = APO, blue = CPX), and J) the percentage of cells that have undergone one or more divisions on day 5. K) Plasmablast gating strategy (Live IgD−CD27+CD38+IRF4+ B cells). L) The percentage of plasmablasts, and fold change (FC) relative to CM alone. M) The concentration of IgG in culture supernatants as measured by ELISA, and fold change relative to CM culture conditions. H-M) p-values determined using Holm-Sidak’s multiple comparison testing, except for fold change analyses for which a one-sample t-test was used. Representative of 3 independent experiments with 6-8 UK adult blood samples.
The immunophenotyping data suggested that induction of plasmablasts and memory B cells, important cell types for the protective immunity afforded by vaccination, are negatively impacted in children with anemia. We sought to determine whether the availability of iron could explain the B cell defects observed in anemic children by modulating iron bioavailability during plasmablast differentiation in vitro. Cellular iron was modulated by supplementing media with apo-transferrin (APO) which facilitates iron uptake by lymphocytes, and by limiting intracellular iron using a cytostatic dose of the iron chelator ciclopirox olamine (CPX, Fig. 7H, Fig. S13). Increased cellular iron enhanced B cell proliferation, and conversely proliferation was impaired when iron availability was reduced (Fig. 7 H–J). Likewise, increased bioavailability of iron enhanced plasmablast differentiation (Fig. 7K–L), which was mirrored by an increase of class switched antibody produced in these cultures (Fig. 7M). These data show a direct effect of bioavailable iron on B cell biology in vitro, and suggest that dietary iron deficiency or infection may impair development of adaptive immunity in anemic children.
Discussion
In this study we show that the composition of the immune system in children from sub-Saharan Africa undergoes dynamic changes in the first years of life. This maturation profile differs from that of Dutch children sampled over the same period of early life, suggesting that the rate of immune maturation is influenced by country-specific factors. Consistent with this finding, location within Africa also had a large effect on the composition of the immune system, and was linked to differential responses to the RTS,S/AS01E vaccine. Groups of children who responded with higher antibody titers to the RTS,S vaccine had circulating cells indicative of a more receptive immune system at the time of first vaccination, including increased baseline frequencies of antibody secreting cells and cTfh cells. To determine what underpins this productive immune profile we examined several factors, and found that anemia was linked to poor baseline frequencies of several immune cell types, including memory B cells and plasmablasts. Importantly, we were able to show a direct link between plasmablast formation and antibody production driven by the bioavailability of iron in vitro, demonstrating a direct effect of iron on B cell responsiveness. This indicates that anemia, which is common in children from LMICs, is associated with an altered composition of the immune system in the first years of life. Together, this study shows that the development and function of the immune system in children from sub-Saharan Africa varies according to their age, geographic location and by co-morbidities and nutritional status that cause anemia.
The factors that underpin the difference in immune profiles from children living in Tanzania and Mozambique are likely to be multifactorial; with contributions from genetics, climate, diet, sanitation, maternal health and the differential prevalence of infectious diseases. We observed that children from Mozambique had a more ‘activated’ immune profile than children from Tanzania at baseline. This corresponded to increased antibody production after RTS,S vaccination in children from Mozambique. These findings are consistent with the protective efficacy of Rotavirus vaccination being highly variable between different African sites (23). Disentangling the contributions of environment or genetics at a population level is complex, although twin studies in Gambian infants suggest that environmental factors predominantly influence antibody persistence after vaccination (24). Our findings suggest that an individual’s immunological profile at time of first vaccination may impact the ensuing antibody response, and are not consistent with suggestions that a highly-activated immune system in individuals from LMICs prohibits good responses to vaccination (25–27). Children from sub-Saharan Africa showed altered immune development with age compared to Dutch children, and had accumulated memory/effector lymphocytes at earlier years in life indicative of advanced immunological age relative to biological age. Viral pathogens may underpin the immune development differences between Tanzanian, Mozambican and Dutch children, as infection with Epstein-Barr virus and Cytomegalovirus and HIV exposure have been linked to changes to several of the T and B cell memory populations observed to change in this study (9, 28–30). Differences in the childhood vaccination programs could contribute to country-specific immune landscapes, as the BCG vaccination, given to African but not Dutch newborns, is known to have non-specific effects on innate immunity and lymphocytes(31). It is remains unclear how these and other factors contribute to the reduced responses to rotavirus, cholera, and polio vaccines in infants from LMICs compared to Europe and North America (23, 32–38), however our study indicates that reduced vaccine responses in LMIC children does not result from a delayed or impaired immune development relative to high-income countries.
The prevalence of anemia in preschool aged children in sub-Saharan Africa is over 65%, and represents a major public health problem due to its association with increased risk of death and impaired cognitive development (22). In this study, children from Tanzania were more likely to have moderate to severe anemia than children from Mozambique, and anemia was associated with changes to the PBMC transcriptome and reduced frequencies of CD4+ T cells, IgG+ memory B cells and plasmablasts. The major drivers of childhood anemia are iron and folate deficiency, infectious disease (e.g. malaria, HIV, bacteraemia, schistosomiasis), and inherited hemoglobinopathies (22). It is possible that infectious diseases are responsible for shaping the immune system in anemic children, but it is also possible that anemia directly impacts the composition and responsiveness of the immune system. After infection or immunisation, B and T cells rapidly proliferate and whilst it is known that iron plays a critical role in both cellular metabolism and DNA synthesis (39, 40), the precise role that iron plays in adaptive immune responses remains unclear (41). We show that sufficient bioavailable iron is needed for optimal production of plasmablasts and IgG responses by human B cells in vitro, which is consistent with in vivo work in mice (42–44). In humans, homozygous mutation of transferrin receptor 1 causes a primary immunodeficiency, characterised by hypoglobulinemia and defects in isotype switched memory B cells (45). Together with the data presented here this supports the notion that the low iron state of anemia may impact plasmablast differentiation directly. This impairment in B and T cell responses could explain the poorer vaccine outcomes that have been reported in anemic children and iron deficient adults (44, 46), although further studies are needed to dissect the contributions of iron-deficiency, genetic polymorphisms and co-infections to the reduced plasmablast differentiation we observed in this study.
Childhood vaccination programmes, such as the Expanded Program on Immunization, save millions of lives worldwide by preventing disease caused by pertussis, measles, polio, diphtheria, tuberculosis, and tetanus (47). For many of these early vaccination schedules, several vaccine doses are administered within the first 12 weeks of life. In this study, IgG responses to the RTS,S vaccine were much lower in 6-12 week old infants than children over 5 months of age, consistent with responses to other vaccines given in early life (1, 48). Infants three months into this trial had an ‘inactive’ immune phenotype, consisting of low frequencies of memory B cells, memory CD4+ T cell subsets, and an elevated Treg cell frequency. It has been reported that neonatal CD4+ T cells tend to differentiate into Treg cells (49, 50), which could explain the higher Treg frequency and could promote tolerogenicity of antigen presenting cells and thus limit T cell responses in infants. Tfh cell generation, germinal center formation, and plasma cell survival is impaired in neonatal mice (48, 51–55), and here we show that human infants show reduced cTfh and plasmablast differentiation compared to older children. These immune composition differences likely underpin the reduced responses in infants to the RTS,S vaccine, which based on the phase 3 trial results has been licensed only for use in children at 5-17 months of age (56). Our findings may also be applicable to other vaccines, and suggests a “one size fits all” approaches in vaccinology is suboptimal. It is conceivable that the optimal age range to vaccinate infants and children would need to be determined for each vaccine and location, as American infants with a more mature adaptive immune system elicit stronger responses to diphtheria-tetanus-pertussis (DTP) vaccine (57) but weaker responses to measles-mumps-rubella (MMR) immunization (58).
The data presented here suggests that age, location and anaemia may alter the rate of immune development in children. Like all large-scale human immunology studies however, there are limitations: Collection and immune phenotyping of the Dutch cohort was independent of the RTS,S vaccine trial, because of this it was not possible to determine how differences at the continental level relate to immune system function. . Within the RTS,S trial, country-specific differences in vaccine-specific antibody responses were observed across the seven African (59), however PBMC samples were only available from two countries for use this study. An understanding of what underpins the geographical changes in immune function will require larger scale cohorts, that include multiple countries. Due to the low malaria incidence in Tanzania and Mozambique during the RTS,S vaccine trial, it was not possible to investigate how immune status related to vaccine efficacy and immune function in this cohort. Further, it will be important to understand whether differences in immune profile relates to changes in susceptability to infectious disease. Future studies will clearly be of value to elucidate the influence of genetics, the microbiome, pathogens, and nutrition on immune development, anemia, and vaccine efficacy in different populations around the globe.
Despite the success of childhood immunizations to date, there are still numerous infectious diseases, such as malaria, that require a vaccination solution. It is likely that such solutions will come from a combination of innovative vaccine design and an understanding of the host response to vaccination. RTS,S is the most clinically advanced malaria vaccine, and has now been rolled out in pilot implementation programs in areas of Ghana, Kenya and Malawi (60), but we need to be better informed about what underpins this vaccine’s efficacy at the individual and population level to use it effectively. The data reported here demonstrate that multiple factors influence the immune system of children in sub-Saharan Africa, with age and location having the strongest influence on immune composition and were linked with differential responses to the RTS,S vaccine. Therefore, a better understanding of what underpins productive immune responses at early age has the potential to improve the efficacy of many childhood vaccine approaches in different populations globally.
Materials and Methods
Study design
The overall objective of this study was to investigate how the composition of the immune system changes throughout childhood in African and Dutch children by using flow cytometry and transcriptomics. This study was performed using cryopreserved PBMC from a subset of participants the RTS,S/AS01E Phase 3 trial (ClinicalTrials.gov NCT00866619), described elsewhere (12). Participants received either three doses of RTS,S/AS01E at months 0, 1, 2 and a booster dose at month 20 (R3R group); three doses of RTS,S/AS01E and a comparator vaccine at month 20 (R3C group); or a comparator vaccine at month 0, 1, 2 and 20 (C3C group). The trial consisted of two age-categories based on participant age at enrolment which were used in this study for consistency; “infants” were 6-12 weeks old and “children” were between 5-17 months of age. The comparator vaccine for infants and children was meningococcal serogroup C conjugate vaccine (Menjugate, Novartis) and rabies (Verorab, Sanofi Pasteur), respectively. Samples analyzed here were collected in two different centers: Manhiça Health Research Center, Fundação Manhiça (FM-CISM, Mozambique), and Ifakara Health Institute and Bagamoyo Research and Training Center (IHI-BRTC, Tanzania). These areas have been described in detail elsewhere (61). Both sites had low-medium malaria transmission intensity at the time of the study (12, 62), and incidence of clinical malaria was low across all vaccine groups. Samples for this study were randomly selected from among participants for which samples were available from all time-points and had no reported malaria episodes throughout the follow-up period (Table S1) to exclude any influence of malaria infection on immune parameters. PBMCs from study month 0, 3, 21 and 32 were available for children (Tanzania and Mozambique), and month 3, 21, and 32 for infants (Mozambique only). A description of the number of participants and samples, sample randomisation, excluded data, vaccine groups, and nested anemia subgroups are outlined in a consort table (Table S2). The analysis of Dutch children involved reanalysing immunophenotyping data from the Generation R study, a prospective population-based cohort study from birth until young adulthood of children born between 2003-2006 (63, 64). For age-matched comparisons to Tanzanian children, a subset of samples from Generation R children between 20-125 weeks of age were analysed (504 out of 1182 children, 748 samples in total). To investigate whether iron influences B cell biology, iron bioavailability was modulated during plasmablast differentiation assays in vitro using healthy UK adult PBMC samples. Investigators conducted all assays, data collection and processing blinded to vaccination group, age-group, study time-point, and study site.
Ethical Approval
Approval for the RTS,S immunological study was obtained from the Ethical Committee of the Hospital Clínic in Barcelona (CEIC, Spain), the National Health and Bioethics Committee (CNBS, Mozambique), the Ethikkommission Beider Basel (EKBB, Switzerland), the National Institutional Review Board (NIMR, Tanzania), the Ifakara Health Institute IRB (IHIIRB, Tanzania), and the PATH’s Research Ethics Committee (REC, USA). Use of RTS,S samples in the UK, and healthy UK adult PBMCs were approved by the UK Health Research Authority (REC reference 17/EE/0063, 15/EE/0071, respectively) and Babraham Institute Human Ethics Committee. The Generation R study received approval from the Medical Ethical Committee of the Erasmus MC, University Medical Center (Rotterdam, the Netherlands). Written informed consent was obtained from parents or guardians of participating children in accordance with the Declaration of Helsinki.
African blood sample preparation and antibody staining
PBMCs were isolated at the African institution laboratories by density gradient centrifugation (65), cryopreserved in liquid nitrogen and shipped to the Babraham Institute where the PBMC were thawed and rested for 2 hours at 37°C. Cell number and viability were measured using a Nucleocounter (NC-3000, Chemometec). Samples with low viability (<50% live cells) were discarded. Two flow cytometry panels (Panels 1 and 2, Table S3) were used to stain between 500,000 and 2 million cells per panel. In brief, cells were incubated with Fc-Receptor blocking antibody for 30 minutes at 4°C, followed by staining with antibody master mixes for 1 hour at 4°C, followed by fixation with 4% paraformaldehyde (Cytofix, BD Biosciences). Fluorescence minus one (FMO) controls were performed for each antibody, in each experiment. Up to 96 PBMC samples were stained per experiment (total of 718 samples over 9 experiments). Participants’ longitudinal samples were tested in the same experiment, and each experiment contained a randomised assortment of samples from each study site, vaccine arm, and age-group to control for batch effects.
African sample flow cytometry and data processing
Samples were acquired on a BD LSR Fortessa 5-laser cytometer, with identical settings for each experiment. 8-peak Rainbow calibration particles (Spherotech) were used to confirm consistent laser power and detector sensitivity in each experiment. Raw .fcs files were pre-processed for quality control to remove events outside the normal range for flow-rate, dynamic range and signal acquisition (Flow AI, (66)). Data files were removed of duplicates and dead cells prior to all analysis, followed by downstream gating for tSNE and manual gating (Fig. S2–5). High inter-assay concordance of control UK blood samples included in each experiment showed limited batch effects in flow cytometry staining (Fig. S14). For tSNE analysis gated subpopulation data (CD3+ T cells, CD19+ B cells, CD4+ T cells, CD8+ T cells, and CD3−CD19− leukocytes) were exported as .csv file, and fluorescence values were arc sin transformed using manually defined fluorescence cut offs and a cofactor value of 50 (Fig S1). For each subpopulation, 1000 cells from each sample were randomly sub-sampled to a maximum of 723,000 cells. Samples with less than 3000 events for subpopulations after FlowAI QC filters were excluded from the analysis (66). The tSNE algorithm was applied to each subpopulation independently (2500 iterations, perplexity=100) using RtSNE (67), followed by clustering using dbscan package. A total of 336 distinct cell clusters characterized by distinct combinations of surface marker expression were identified, and cluster frequencies were calculated for each sample as a proportion the 1000 cells analysed for each subpopulation. Manual gating of 70 well characterized cell subsets was performed using FlowJo software (Table S4–5, and Fig. S2–5). Manually gated data was imported into R using flowWorkspace package (68), and cell subset frequencies were extracted as percentage of total leukocytes, and as a percentage of CD3+, CD19+, CD4+, CD8+, or forward and side-scatter intermediate cells. For the Generation R cohort of Dutch children, existing immunophenotyping data was analyzed for the 19 cell subsets in which the antibodies and gating strategies were in common for both RTS,S and GenR cohorts (5). Cell subsets frequencies as a percentage of CD3+, CD19+, CD4+, or CD8+ cells were used from participants either from birth-414 weeks of age (n= 2010 samples), or between 25 and 125 weeks of age (n=748 samples). For all cohorts, relative frequencies (as a % of total lymphocytes or as a % of ‘parent’ population) were used, as the absolute counts for each cell type were not available.
B cell in vitro cultures and plasmablast differentiation
PBMCs were isolated from healthy UK adult apheresis leukocyte cones by density gradient centrifugation and cryopreserved in liquid nitrogen. PBMC were thawed and rested for two hours at 37°C, stained with CellTrace Violet cell proliferation dye (ThermoFisher Scientific), then B cells were enriched using MagniSort human B cell enrichment kit (ThermoFisher Scientific). Purified B were cultured for 5 days to differentiate plasmablasts. Briefly 100,000 B cells were cultured per well in RPMI culture media (CM) supplemented with 10% heat inactivated foetal calf serum, 10mM HEPES ph 7.4, 1X non-essential amino acids, 1mM sodium pyruvate, 2mM L-glutamine, 100 units/mL Penicillin, 100 units/mL Streptomycin (all from ThermoFisher), 50ng/mL human IL-21 (Peprotech), and 200ng/mL of recombinant human CD40L crosslinked with 50ng/mL anti-Hemagglutinin peptide antibody (both R& D systems). Iron bioavailability was modulated by the addition of 40μg/mL apo-transferrin (APO) for 5 days (ThermoFisher Scientific), or 500nM of Ciclopirox olamine (CPX) added on day 1 of culture, a concentration of CPX that did not impact B cell viability in vitro (Fig. S13). After 5 days of culture at 37°C, culture supernatants were collected and secreted IgG measured by ELISA using a rat anti-human IgG Fc capture antibody (M1310G05, BioLegend), a mouse anti-human IgG-biotin detection antibody (HP6017, BioLegend), Streptavidin-HRP (GE Healthcare), and TMB substrate set (BioLegend). IgG concentration was determined by using a standard curve of human IgG (Sigma). Proliferation and plasmablast differentiation was measured by flow cytometry by staining with Panel 3 (Table S3), with cell number determined using counting beads (123count eBeads, ThermoFisher Scientific). Primary data included in data file S3.
Sequencing and bioinformatics
PBMCs from Tanzanian children at B0 and B32 were stored in RNA-later post-thaw. RNA from samples with high viability (>90%) and sufficient cell number (>3x106 cells) were isolated using RNeasy plus Mini kit (Qiagen). Libraries were generated from samples with high RNA quality (RIIN >7) using TruSeq Stranded mRNA Library prep (Illumina) by Eurofins (n= 30). Libraries were sequenced to an average depth of 24 million 1x50bp single end reads per sample (range 17-34 million) on a HiSeq 2500 (Illumina, v4 chemistry). Raw data was aligned using GRCh38 assembly, reads within CDS quantitated, and differential expressed genes (DE) determined using the DeSeq2 package embedded within the Seqmonk software (https://www.bioinformatics.babraham.ac.uk/projects/seqmonk/). Significantly differentially expressed genes had an fdr-adjusted p-value of <0.01 from DESeq2 and an fdr-adjusted p-value of <0.05 from the intensity-difference test. Heatmaps of DE genes were generated using Pheatmap package in R (69), with hierarchical clustering by Euclidean distance. Blood transcriptional module (BTM) analysis used gene sets previously reported (16) and gene set enrichment tool within the Seqmonk software. BTMs were deemed significant if they had an enrichment z-score of less than −0.5 or greater than 1.5, and an adjusted p-value of <0.05. Enriched BMTs were graphed using the giraph software integrated within Seqmonk. Microarray data from Kazmin et al ((13), GSE89292) was analysed using the GEOquery (70) and limma (71) packages in R for the 14 RTS,S vaccinated north American adults for which there was data available for days 0, 1, 6, 28, 56, 57, 62 and 77. Days 1, 6 and 28 were compared relative to day 0. Days 57, 62 and 77 were compared relative to day 56. Probe sets were deemed significantly differentially expressed if they had a 2-fold change in expression and an fdr-adjusted p-value of less than 0.01.
Multi-Omics Factor Analysis (MOFA)
MOFA is a factor analysis model that enables integration of diverse data sets in a completely unsupervised fashion, and was implemented in R with Python dependencies as previously described (17). Flow cytometry data was pre-processed as follows: cell subset frequencies were converted to z-scores by subtracting the mean and dividing by the standard deviation determined using full African dataset (n = 718 samples) for the 30 samples that also had transcriptomic data. RNA-Seq reads were counted with Rsubread and vsn normalised using DESeq2 package. Genes were ranked according to variance across all 30 samples, and the 5000 most variable genes was used as this produced the most robust MOFA model. Samples with missing data for either flow subsets or gene expression were removed from the analysis resulting in 27 samples. Flow and RNA-Seq data matrices were used to the train model using default settings (number of factors = 25, drop factor threshold 2% of variance), which was then run 10 times independently and the best trained model selected using the EMBO output value. All models converged on seven latent factors, and factor loadings for cell types and genes were determined using MOFA’s TopWeights function, with blood transcriptional modules analysed using the geneset enrichment algorithm implemented in the MOFA package (p<0.05), with gene sets as previously published (16).
Serological data and covariates
Serological data were available for a subset of the total participants from each trial site of the phase III trial. Anti-CSP and anti-HBV.S antibodies were measured by standardised enzyme-linked immunosorbent assays and antigens in a single laboratory (72). Vaccine response was determined by subtracting baseline antibody titre (in arbitrary units) from B3. Additional variables such as participant’s height, weight, and hemoglobin concentration were collected as previously described (12), and available for a subset of participants from each trial site.
Immunological trajectory and pseudotime analysis
The immune trajectory was assembled as previously described (21). The frequencies for the 19 cell subsets were first normalised for abundance; the mean and s.d. of frequency values for the 10th to 90th percentile of for each cell type for the entire Dutch cohort (1801 samples). These values were used normalize cellular frequencies for both cohorts (frequency-mean10-90 / s.d.10-90). With both cohorts in a combined dataset, principal component analysis was performed. Cell types for which the absolute correlation to PC1 was greater than 0.5 were included (n= 18 cell types). The diffusion maps algorithm was then applied to the scaled frequencies using the destiny R package (20, 73), the resulting diffusion pseudotime values scaled to a range of 0-1,.
Statistical analysis with categorical variables
All statistical tests for cell type frequencies assumed non-parametric data; two group comparisons were made using either two-tailed Mann-Whitney tests, or Wilcoxon tests for paired from the same individual at different time points. False discovery rate (FDR, or Benjamini–Hochberg) adjustment was applied to all multiple testing. tSNE-defined data were used to determine changes to the frequency of individual clusters, with between-group comparisons analyzed with the Mann-Whitney test and paired samples from individuals analysed by Wilcoxon signed-rank test. tSNE-defined cluster frequencies were also used for multi-dimensional scaling (MDS) approaches in which Euclidean distances were calculated from Spearman correlation matrices of all 336 tSNE clusters per sample and multi-dimensional scaling performed using monoMDS from the vegan package (74). Mann-Whitney tests were used to compare MDS co-ordinates between groups. Heatmaps of manual gated cell subsets that were altered by age-group or site were generated using Pheatmap package (69), with hierarchical clustering by Euclidean distances.
Statistical analysis of cell types using linear modelling
Linear regression was used to determine the contribution of vaccine group (Fig. 1), age group (Fig. 2), country (Fig. 6), or anemia (Fig. 7) as categorical variables to cell type frequencies by fitting the following models for each cell subset “i”; MVaccine <- lm(data[,i] ~ vaccine group + patient ID + sex); MAge_group <- lm(data[,i] ~ age group + sex); MCountry <- lm(data[,i] ~ country + age + sex) ; MAnemia <- lm(data[,i] ~ anemia + age + sex). The relative importance for each covariate to the cell subset variance was calculated using the relaimpo R package (75).Linear mixed-effect modelling (LMER) analysis was used to model immune cell subset dynamics over time with the lme4 package using maximum likelihood (76). These models included random intercepts, and enabled the incorporation of longitudinal follow-up data from individual children. Frequencies of each immune parameter were modelled using age (in weeks) as a continuous variable, sex and location (Tanzania/The Netherlands) as categorical variables, and included participant ID (PID) to account for repeated measures over time. In samples from Tanzania samples aged 20-220 weeks (Fig. 4E, n=414), for each cell subset “i” (n=20) the effect of age was tested by ANOVA for following models; MNull <- lmer(data[,i] ~ (1| patient ID)); MSex <- lmer(data[,i] ~ sex +(1| patient ID)); MAge <- lmer(data[,i] ~ age + sex +(1| patient ID). ANOVA p-values were FDR adjusted, and conditional and marginal R2 for MAge were calculated using the MuMIn package (77). For comparison of Dutch and Tanzanian samples, data was combined into a single dataset. The effect of sex and location as a (fixed effect and as an interaction term with age) were tested for each cell subset “i” (n=19) using ANOVA of following models; MNull<-lmer(data[,i] ~ age +(1| patient ID)) ; MSex <-lmer(data[,i] ~ age +sex +(1| patient ID)); MLocation<-lmer(data[,i] ~ age +location +sex +(1| patient ID)) ; MAge*Location<-lmer(data[,i] ~ age*location +sex +(1| patient ID)). Model intercepts and slopes were used to predict cell subset frequencies for 20-220 weeks, data were normalised to week 20 values, and graphed using the Pheatmap package. All statistical analyses were performed with R software version 3.4.1. Normally distributed residuals were confirmed for all linear and mixed-effect models for all cell types.
Supplementary Material
Data file S1: Differentially expressed genes between B0 and B32
Data file S2: Differentially expressed genes between anemic and non-anemic children
Data file S3: Plasmablast differentiation assay raw data
Fig. S1: Pre-gating of cell types for tSNE
Fig. S2: Gating hierarchy Panel 1
Fig. S3: Gating hierarchy Panel 2
Fig. S4: Representative flow plots panel 1
Fig. S5: Representative flow plots panel 2
Fig. S6: No vaccine induced changes are detectable one month after vaccination.
Fig. S7 Strong correlations observed within related cell types
Fig. S8 Cell subset frequency correlations between baseline and B3 time-points
Fig. S9 Cell subset frequency correlations between baseline and B32 time-points
Fig. S10. Anti-CSP and Anti-HBV.S IgG titers before and after vaccination in infants and children from Mozambique
Fig. S11. Distribution of participant age along the diffusion-pseudotime trajectory
Fig. S12. Anti-CSP and Anti-HBV.S IgG titers before and after vaccination in children from Tanzania and Mozambique
Fig. S13. Dose dependent toxicity of ciclopirox olamine (CPX) on B cells in culture
Fig. S14. Highly reproducible flow cytometry data between experiments
Table S1: Participant characteristics
Table S2 CONSORT Table
Table S3. Antibody panels
Table S4: Manual gated cell subset definitions Panel 1
Table S5: Manual gated cell subset definitions Panel 2
Acknowledgements:
We wish to thank all the RTS,S phase 3 vaccine trial participants and their guardians, the MAL055 clinical team and the members of the MAL067 Vaccine Immunology Workgroup. We thank GlaxoSmithKline Biologicals S.A. for their support in the conduct of the MAL067 study. We are grateful to the field and laboratory personnel from the CISM, ISGlobal, and IHI-Bagamoyo Research and Training Center, in particular Laura Puyol, Inocencia Cuamba, Nana Aba Williams, Núria Díez-Padrisa, Hèctor Sanz and Aintzane Ayestaran for their logistics, database and management support. We thank Anne Segonds-Pichon and Elena Vigorito for their advice on statistical methods. We thank the staff of the Babraham Institute Flow Cytometry Facility for their technical assistance. The Generation R Study is conducted by the Erasmus MC, University Medical Center in close collaboration with the Erasmus University Rotterdam and the city of Rotterdam. We gratefully acknowledge the contribution of children and parents.
Funding:
This study was supported by the Biotechnology and Biological Sciences Research Council through Institute Strategic Programme Grant funding (BBS/E/B/000C0427 and BBS/E/B/000C0428), and Core Capability Grant to the Babraham Institute. DLH is supported by a National Health and Medical Research Council Australia Early-Career Fellowship (APP1139911). Funding for the Clinical trial was provided by PATH Malaria Vaccine Initiative (MVI), the National Institute of Allergy and Infectious Diseases (NIAID R01AI095789, HVTN UM1 AI06861, HIPC P30 AI027757), the Instituto de Salud Carlos III (PS11/00423, PI14/01422 and PI17/02044), and the Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR, 2014SGR991). CD was recipient of a Ramon y Cajal Contract from the Ministerio de Economía y Competitividad (RYC-2008-02631). GM was recipient of a Sara Borrell – ISCIII fellowship (CD010/00156) and had the support of the Department of Health, Catalan Government (SLT006/17/00109). CISM is supported by the Government of Mozambique and the Spanish Agency for International Development (AECID). ISGlobal is a member of the CERCA Programme, Generalitat de Catalunya. The general design of Generation R Study is made possible by long-term financial support from the Erasmus MC, University Medical Center, Rotterdam, the Netherlands Organization for Health Research and Development (ZonMw) and the Ministry of Health, Welfare and Sport.
Footnotes
Competing interests:
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Data and materials availability:
The RNAseq data for this study have been deposited in the Gene Expression Omnibus (GEO) database (GSE134985). Flow cytometry data (African and UK samples) and all code are available on GitHub: https://doi.org/10.5281/zenodo.3598467. Flow cytometry data of the Generation R cohort is available upon collaboration request, contact: menno.vanzelm@monash.edu and h.a.moll@erasmusmc.nl. All data associated with this study are present in the paper or the Supplementary Materials.
References and Notes
- 1.Kollmann TR, Kampmann B, Mazmanian SK, Marchant A, Levy O, Protecting the Newborn and Young Infant from Infectious Diseases: Lessons from Immune Ontogeny. Immunity 46, 350–363 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Saso A, Kampmann B, Vaccine responses in newborns. Semin Immunopathol 39, 627–642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brodin P, Jojic V, Gao T, Bhattacharya S, Angel CJ, Furman D, Shen-Orr S, Dekker CL, Swan GE, Butte AJ, Maecker HT, Davis MM, Variation in the human immune system is largely driven by non-heritable influences. Cell 160, 37–47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carr EJ, Dooley J, Garcia-Perez JE, Lagou V, Lee JC, Wouters C, Meyts I, Goris A, Boeckxstaens G, Linterman MA, Liston A, The cellular composition of the human immune system is shaped by age and cohabitation. Nat Immunol 17, 461–468 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van den Heuvel D, Jansen MAE, Nasserinejad K, Dik WA, van Lochem EG, Bakker-Jonges LE, Bouallouch-Charif H, Jaddoe VWV, Hooijkaas H, van Dongen JJM, Moll HA, van Zelm MC, Effects of nongenetic factors on immune cell dynamics in early childhood: The Generation R Study. J Allergy Clin Immunol 139, 1923–1934 e1917 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Olin A, Henckel E, Chen Y, Lakshmikanth T, Pou C, Mikes J, Gustafsson A, Bernhardsson AK, Zhang C, Bohlin K, Brodin P, Stereotypic Immune System Development in Newborn Children. Cell 174, 1277–1292 e1214 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.United Nations Inter-agency Group for Child Mortality Estimation, “Levels & Trends in Child Mortality: Report 2018, Estimates developed by the United Nations Inter-agency Group for Child Mortality Estimation,” (Geneva, 2018). [Google Scholar]
- 8.Chao F, You D, Pedersen J, Hug L, Alkema L, National and regional under-5 mortality rate by economic status for low-income and middle-income countries: a systematic assessment. Lancet Glob Health 6, e535–e547 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liston A, Carr EJ, Linterman MA, Shaping Variation in the Human Immune System. Trends Immunol 37, 637–646 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Orru V, Steri M, Sole G, Sidore C, Virdis F, Dei M, Lai S, Zoledziewska M, Busonero F, Mulas A, Floris M, Mentzen WI, Urru SA, Olla S, Marongiu M, Piras MG, Lobina M, Maschio A, Pitzalis M, Urru MF, Marcelli M, Cusano R, Deidda F, Serra V, Oppo M, Pilu R, Reinier F, Berutti R, Pireddu L, Zara I, Porcu E, Kwong A, Brennan C, Tarrier B, Lyons R, Kang HM, Uzzau S, Atzeni R, Valentini M, Firinu D, Leoni L, Rotta G, Naitza S, Angius A, Congia M, Whalen MB, Jones CM, Schlessinger D, Abecasis GR, Fiorillo E, Sanna S, Cucca F, Genetic variants regulating immune cell levels in health and disease. Cell 155, 242–256 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roederer M, Quaye L, Mangino M, Beddall MH, Mahnke Y, Chattopadhyay P, Tosi I, Napolitano L, Terranova Barberio M, Menni C, Villanova F, Di Meglio P, Spector TD, Nestle FO, The genetic architecture of the human immune system: a bioresource for autoimmunity and disease pathogenesis. Cell 161, 387–403 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.RTS.S Clinical Trials Partnership, Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. Lancet 386, 31–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kazmin D, Nakaya HI, Lee EK, Johnson MJ, van der Most R, van den Berg RA, Ballou WR, Jongert E, Wille-Reece U, Ockenhouse C, Aderem A, Zak DE, Sadoff J, Hendriks J, Wrammert J, Ahmed R, Pulendran B, Systems analysis of protective immune responses to RTS,S malaria vaccination in humans. Proc Natl Acad Sci U S A 114, 2425–2430 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bentebibel SE, Lopez S, Obermoser G, Schmitt N, Mueller C, Harrod C, Flano E, Mejias A, Albrecht RA, Blankenship D, Xu H, Pascual V, Banchereau J, Garcia-Sastre A, Palucka AK, Ramilo O, Ueno H, Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci Transl Med 5, 176ra132 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hill DL, Pierson W, Bolland DJ, Mkindi C, Carr EJ, Wang J, Houard S, Wingett SW, Audran R, Wallin EF, Jongo SA, Kamaka K, Zand M, Spertini F, Daubenberger C, Corcoran AE, Linterman MA, The adjuvant GLA-SE promotes human Tfh cell expansion and emergence of public TCRbeta clonotypes. J Exp Med, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li S, Rouphael N, Duraisingham S, Romero-Steiner S, Presnell S, Davis C, Schmidt DS, Johnson SE, Milton A, Rajam G, Kasturi S, Carlone GM, Quinn C, Chaussabel D, Palucka AK, Mulligan MJ, Ahmed R, Stephens DS, Nakaya HI, Pulendran B, Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat Immunol 15, 195–204 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Argelaguet R, Velten B, Arnol D, Dietrich S, Zenz T, Marioni JC, Buettner F, Huber W, Stegle O, Multi-Omics Factor Analysis-a framework for unsupervised integration of multi-omics data sets. Mol Syst Biol 14, e8124 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Argelaguet R, Clark SJ, Mohammed H, Stapel LC, Krueger C, Kapourani CA, Imaz-Rosshandler I, Lohoff T, Xiang Y, Hanna CW, Smallwood S, Ibarra-Soria X, Buettner F, Sanguinetti G, Xie W, Krueger F, Gottgens B, Rugg-Gunn PJ, Kelsey G, Dean W, Nichols J, Stegle O, Marioni JC, Reik W, Multi-omics profiling of mouse gastrulation at single-cell resolution. Nature 576, 487–491 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee AH, Shannon CP, Amenyogbe N, Bennike TB, Diray-Arce J, Idoko OT, Gill EE, Ben-Othman R, Pomat WS, van Haren SD, Cao KL, Cox M, Darboe A, Falsafi R, Ferrari D, Harbeson DJ, He D, Bing C, Hinshaw SJ, Ndure J, Njie-Jobe J, Pettengill MA, Richmond PC, Ford R, Saleu G, Masiria G, Matlam JP, Kirarock W, Roberts E, Malek M, Sanchez-Schmitz G, Singh A, Angelidou A, Smolen KK, Consortium E, Brinkman RR, Ozonoff A, Hancock REW, van den Biggelaar AHJ, Steen H, Tebbutt SJ, Kampmann B, Levy O, Kollmann TR, Dynamic molecular changes during the first week of human life follow a robust developmental trajectory. Nat Commun 10, 1092 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haghverdi L, Buttner M, Wolf FA, Buettner F, Theis FJ, Diffusion pseudotime robustly reconstructs lineage branching. Nat Methods 13, 845–848 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Alpert A, Pickman Y, Leipold M, Rosenberg-Hasson Y, Ji X, Gaujoux R, Rabani H, Starosvetsky E, Kveler K, Schaffert S, Furman D, Caspi O, Rosenschein U, Khatri P, Dekker CL, Maecker HT, Davis MM, Shen-Orr SS, A clinically meaningful metric of immune age derived from high-dimensional longitudinal monitoring. Nat Med 25, 487–495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.World Health Organisation, “Iron deficiency anaemia: assessment prevention and control,” (Geneva, 2001). [Google Scholar]
- 23.Madhi SA, Cunliffe NA, Steele D, Witte D, Kirsten M, Louw C, Ngwira B, Victor JC, Gillard PH, Cheuvart BB, Han HH, Neuzil KM, Effect of human rotavirus vaccine on severe diarrhea in African infants. N Engl J Med 362, 289–298 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Marchant A, Pihlgren M, Goetghebuer T, Weiss HA, Ota MO, Schlegel-Hauter SE, Whittle H, Lambert PH, Newport MJ, Siegrist CA, Medical G Research Council Gambia Twin Study, Predominant influence of environmental determinants on the persistence and avidity maturation of antibody responses to vaccines in infants. J Infect Dis 193, 1598–1605 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Fine PE, Variation in protection by BCG: implications of and for heterologous immunity. Lancet 346, 1339–1345 (1995). [DOI] [PubMed] [Google Scholar]
- 26.Kityo C, Makamdop KN, Rothenberger M, Chipman JG, Hoskuldsson T, Beilman GJ, Grzywacz B, Mugyenyi P, Ssali F, Akondy RS, Anderson J, Schmidt TE, Reimann T, Callisto SP, Schoephoerster J, Schuster J, Muloma P, Ssengendo P, Moysi E, Petrovas C, Lanciotti R, Zhang L, Arevalo MT, Rodriguez B, Ross TM, Trautmann L, Sekaly RP, Lederman MM, Koup RA, Ahmed R, Reilly C, Douek DC, Schacker TW, Lymphoid tissue fibrosis is associated with impaired vaccine responses. J Clin Invest 128, 2763–2773 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muyanja E, Ssemaganda A, Ngauv P, Cubas R, Perrin H, Srinivasan D, Canderan G, Lawson B, Kopycinski J, Graham AS, Rowe DK, Smith MJ, Isern S, Michael S, Silvestri G, Vanderford TH, Castro E, Pantaleo G, Singer J, Gillmour J, Kiwanuka N, Nanvubya A, Schmidt C, Birungi J, Cox J, Haddad EK, Kaleebu P, Fast P, Sekaly RP, Trautmann L, Gaucher D, Immune activation alters cellular and humoral responses to yellow fever 17D vaccine. J Clin Invest 124, 3147–3158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abu-Raya B, Kollmann TR, Marchant A, MacGillivray DM, The Immune System of HIV-Exposed Uninfected Infants. Front Immunol 7, 383 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van den Heuvel D, Jansen MA, Bell AI, Rickinson AB, Jaddoe VW, van Dongen JJ, Moll HA, van Zelm MC, Transient reduction in IgA(+) and IgG(+) memory B cell numbers in young EBV-seropositive children: the Generation R Study. J Leukoc Biol 101, 949–956 (2017). [DOI] [PubMed] [Google Scholar]
- 30.van den Heuvel D, Jansen MA, Dik WA, Bouallouch-Charif H, Zhao D, van Kester KA, Smits-te Nijenhuis MA, Kolijn-Couwenberg MJ, Jaddoe VW, Arens R, van Dongen JJ, Moll HA, van Zelm MC, Cytomegalovirus- and Epstein-Barr Virus-Induced T-Cell Expansions in Young Children Do Not Impair Naive T-cell Populations or Vaccination Responses: The Generation R Study. J Infect Dis 213, 233–242 (2016). [DOI] [PubMed] [Google Scholar]
- 31.de Bree LCJ, Koeken V, Joosten LAB, Aaby P, Benn CS, van Crevel R, Netea MG, Non-specific effects of vaccines: Current evidence and potential implications. Semin Immunol 39, 35–43 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Armah GE, Breiman RF, Tapia MD, Dallas MJ, Neuzil KM, Binka FN, Sow SO, Ojwando J, Ciarlet M, Steele AD, Immunogenicity of the pentavalent rotavirus vaccine in African infants. Vaccine 30 Suppl 1, A86–93 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Patriarca PA, Wright PF, John TJ, Factors affecting the immunogenicity of oral poliovirus vaccine in developing countries: review. Rev Infect Dis 13, 926–939 (1991). [DOI] [PubMed] [Google Scholar]
- 34.Hallander HO, Paniagua M, Espinoza F, Askelof P, Corrales E, Ringman M, Storsaeter J, Calibrated serological techniques demonstrate significant different serum response rates to an oral killed cholera vaccine between Swedish and Nicaraguan children. Vaccine 21, 138–145 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Richie EE, Punjabi NH, Sidharta YY, Peetosutan KK, Sukandar MM, Wasserman SS, Lesmana MM, Wangsasaputra FF, Pandam SS, Levine MM, O’Hanley PP, Cryz SJ, Simanjuntak CH, Efficacy trial of single-dose live oral cholera vaccine CVD 103-HgR in North Jakarta, Indonesia, a cholera-endemic area. Vaccine 18, 2399–2410 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Suharyono, Simanjuntak C, Witham N, Punjabi N, Heppner DG, Losonsky G, Totosudirjo H, Rifai AR, Clemens J, Lim YL, et al. , Safety and immunogenicity of single-dose live oral cholera vaccine CVD 103-HgR in 5-9-year-old Indonesian children. Lancet 340, 689–694 (1992). [DOI] [PubMed] [Google Scholar]
- 37.Vesikari T, Karvonen A, Prymula R, Schuster V, Tejedor JC, Cohen R, Meurice F, Han HH, Damaso S, Bouckenooghe A, Efficacy of human rotavirus vaccine against rotavirus gastroenteritis during the first 2 years of life in European infants: randomised, double-blind controlled study. Lancet 370, 1757–1763 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Parker EP, Ramani S, Lopman BA, Church JA, Iturriza-Gomara M, Prendergast AJ, Grassly NC, Causes of impaired oral vaccine efficacy in developing countries. Future Microbiol 13, 97–118 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neckers LM, Cossman J, Transferrin receptor induction in mitogen-stimulated human T lymphocytes is required for DNA synthesis and cell division and is regulated by interleukin 2. Proc Natl Acad Sci U S A 80, 3494–3498 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pearce EL, Poffenberger MC, Chang CH, Jones RG, Fueling immunity: insights into metabolism and lymphocyte function. Science 342, 1242454 (2013).24115444 [Google Scholar]
- 41.Armitage AE, Moretti D, The Importance of Iron Status for Young Children in Low- and Middle-Income Countries: A Narrative Review. Pharmaceuticals (Basel) 12, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsui C, Martinez-Martin N, Gaya M, Maldonado P, Llorian M, Legrave NM, Rossi M, MacRae JI, Cameron AJ, Parker PJ, Leitges M, Bruckbauer A, Batista FD, Protein Kinase C-beta Dictates B Cell Fate by Regulating Mitochondrial Remodeling, Metabolic Reprogramming, and Heme Biosynthesis. Immunity 48, 1144–1159 e1145 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watanabe-Matsui M, Muto A, Matsui T, Itoh-Nakadai A, Nakajima O, Murayama K, Yamamoto M, Ikeda-Saito M, Igarashi K, Heme regulates B-cell differentiation, antibody class switch, and heme oxygenase-1 expression in B cells as a ligand of Bach2. Blood 117, 5438–5448 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Jiang Y, Li C, Wu Q, An P, Huang L, Wang J, Chen C, Chen X, Zhang F, Ma L, Liu S, He H, Xie S, Sun Y, Liu H, Zhan Y, Tao Y, Liu Z, Sun X, Hu Y, Wang Q, Ye D, Zhang J, Zou S, Wang Y, Wei G, Liu Y, Shi Y, Eugene Chin Y, Hao Y, Wang F, Zhang X, Iron-dependent histone 3 lysine 9 demethylation controls B cell proliferation and humoral immune responses. Nat Commun 10, 2935 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jabara HH, Boyden SE, Chou J, Ramesh N, Massaad MJ, Benson H, Bainter W, Fraulino D, Rahimov F, Sieff C, Liu ZJ, Alshemmari SH, Al-Ramadi BK, Al-Dhekri H, Arnaout R, Abu-Shukair M, Vatsayan A, Silver E, Ahuja S, Davies EG, Sola-Visner M, Ohsumi TK, Andrews NC, Notarangelo LD, Fleming MD, Al-Herz W, Kunkel LM, Geha RS, A missense mutation in TFRC, encoding transferrin receptor 1, causes combined immunodeficiency. Nat Genet 48, 74–78 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brussow H, Sidoti J, Dirren H, Freire WB, Effect of malnutrition in Ecuadorian children on titers of serum antibodies to various microbial antigens. Clin Diagn Lab Immunol 2, 62–68 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.World Health Organisation, “Global vaccine action plan 2011-2020.,” (Geneva, 2013). [Google Scholar]
- 48.Siegrist CA, Aspinall R, B-cell responses to vaccination at the extremes of age. Nat Rev Immunol 9, 185–194 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Wang G, Miyahara Y, Guo Z, Khattar M, Stepkowski SM, Chen W, “Default” generation of neonatal regulatory T cells. J Immunol 185, 71–78 (2010). [DOI] [PubMed] [Google Scholar]
- 50.de Roock S, Hoeks SB, Meurs L, Steur A, Hoekstra MO, Prakken BJ, Boes M, de Kleer IM, Critical role for programmed death 1 signaling and protein kinase B in augmented regulatory T-cell induction in cord blood. J Allergy Clin Immunol 128, 1369–1371 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Belnoue E, Pihlgren M, McGaha TL, Tougne C, Rochat AF, Bossen C, Schneider P, Huard B, Lambert PH, Siegrist CA, APRIL is critical for plasmablast survival in the bone marrow and poorly expressed by early-life bone marrow stromal cells. Blood 111, 2755–2764 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Debock I, Jaworski K, Chadlaoui H, Delbauve S, Passon N, Twyffels L, Leo O, Flamand V, Neonatal follicular Th cell responses are impaired and modulated by IL-4. J Immunol 191, 1231–1239 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Mastelic B, Kamath AT, Fontannaz P, Tougne C, Rochat AF, Belnoue E, Combescure C, Auderset F, Lambert PH, Tacchini-Cottier F, Siegrist CA, Environmental and T cell-intrinsic factors limit the expansion of neonatal follicular T helper cells but may be circumvented by specific adjuvants. J Immunol 189, 5764–5772 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Pihlgren M, Tougne C, Bozzotti P, Fulurija A, Duchosal MA, Lambert PH, Siegrist CA, Unresponsiveness to lymphoid-mediated signals at the neonatal follicular dendritic cell precursor level contributes to delayed germinal center induction and limitations of neonatal antibody responses to T-dependent antigens. J Immunol 170, 2824–2832 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Gatto D, Pfister T, Jegerlehner A, Martin SW, Kopf M, Bachmann MF, Complement receptors regulate differentiation of bone marrow plasma cell precursors expressing transcription factors Blimp-1 and XBP-1. J Exp Med 201, 993–1005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.World Health Organisation, “Update on RTS,S Malaria Vaccine Implementation Programme and framework for decision-making,” (Geneva, 2018). [Google Scholar]
- 57.Strombeck A, Lundell AC, Nordstrom I, Andersson K, Adlerberth I, Wold AE, Rudin A, Earlier infantile immune maturation is related to higher DTP vaccine responses in children. Clin Transl Immunology 5, e65 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Strombeck A, Lundell AC, Nordstrom I, Andersson K, Adlerberth I, Wold AE, Rudin A, Delayed adaptive immunity is related to higher MMR vaccine-induced antibody titers in children. Clin Transl Immunology 5, e75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.White MT, Verity R, Griffin JT, Asante KP, Owusu-Agyei S, Greenwood B, Drakeley C, Gesase S, Lusingu J, Ansong D, Adjei S, Agbenyega T, Ogutu B, Otieno L, Otieno W, Agnandji ST, Lell B, Kremsner P, Hoffman I, Martinson F, Kamthunzu P, Tinto H, Valea I, Sorgho H, Oneko M, Otieno K, Hamel MJ, Salim N, Mtoro A, Abdulla S, Aide P, Sacarlal J, Aponte JJ, Njuguna P, Marsh K, Bejon P, Riley EM, Ghani AC, Immunogenicity of the RTS,S/AS01 malaria vaccine and implications for duration of vaccine efficacy: secondary analysis of data from a phase 3 randomised controlled trial. Lancet Infect Dis 15, 1450–1458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adepoju P, RTS S malaria vaccine pilots in three African countries. Lancet 393, 1685 (2019). [DOI] [PubMed] [Google Scholar]
- 61.International Network for the continuous Demographic Evaluation of Populations and their Health, Population and Health in Developing Countries. (International Development Research Centre (IDRC), Population and Health in Developing Countries, 2002). [Google Scholar]
- 62.RTS.S. Clinical Trials Partnership, Agnandji ST, Lell B, Soulanoudjingar SS, Fernandes JF, Abossolo BP, Conzelmann C, Methogo BG, Doucka Y, Flamen A, Mordmuller B, Issifou S, Kremsner PG, Sacarlal J, Aide P, Lanaspa M, Aponte JJ, Nhamuave A, Quelhas D, Bassat Q, Mandjate S, Macete E, Alonso P, Abdulla S, Salim N, Juma O, Shomari M, Shubis K, Machera F, Hamad AS, Minja R, Mtoro A, Sykes A, Ahmed S, Urassa AM, Ali AM, Mwangoka G, Tanner M, Tinto H, D’Alessandro U, Sorgho H, Valea I, Tahita MC, Kabore W, Ouedraogo S, Sandrine Y, Guiguemde RT, Ouedraogo JB, Hamel MJ, Kariuki S, Odero C, Oneko M, Otieno K, Awino N, Omoto J, Williamson J, Muturi-Kioi V, Laserson KF, Slutsker L, Otieno W, Otieno L, Nekoye O, Gondi, Otieno A, Ogutu B, Wasuna R, Owira V, Jones D, Onyango AA, Njuguna P, Chilengi R, Akoo P, Kerubo C, Gitaka J, Maingi C, Lang T, Olotu A, Tsofa B, Bejon P, Peshu N, Marsh K, Owusu-Agyei S, Asante KP, Osei-Kwakye K, Boahen O, Ayamba S, Kayan K, Owusu-Ofori R, Dosoo D, Asante I, Adjei G, Adjei G, Chandramohan D, Greenwood B, Lusingu J, Gesase S, Malabeja A, Abdul O, Kilavo H, Mahende C, Liheluka E, Lemnge M, Theander T, Drakeley C, Ansong D, Agbenyega T, Adjei S, Boateng HO, Rettig T, Bawa J, Sylverken J, Sambian D, Agyekum A, Owusu L, Martinson F, Hoffman I, Mvalo T, Kamthunzi P, Nkomo R, Msika A, Jumbe A, Chome N, Nyakuipa D, Chintedza J, Ballou WR, Bruls M, Cohen J, Guerra Y, Jongert E, Lapierre D, Leach A, Lievens M, Ofori-Anyinam O, Vekemans J, Carter T, Leboulleux D, Loucq C, Radford A, Savarese B, Schellenberg D, Sillman M, Vansadia P, First results of phase 3 trial of RTS,S/AS01 malaria vaccine in African children. N Engl J Med 365, 1863–1875 (2011). [DOI] [PubMed] [Google Scholar]
- 63.Jaddoe VW, Mackenbach JP, Moll HA, Steegers EA, Tiemeier H, Verhulst FC, Witteman JC, Hofman A, The Generation R Study: Design and cohort profile. Eur J Epidemiol 21, 475–484 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Kooijman MN, Kruithof CJ, van Duijn CM, Duijts L, Franco OH, van IMH, de Jongste JC, Klaver CC, van der Lugt A, Mackenbach JP, Moll HA, Peeters RP, Raat H, Rings EH, Rivadeneira F, van der Schroeff MP, Steegers EA, Tiemeier H, Uitterlinden AG, Verhulst FC, Wolvius E, Felix JF, Jaddoe VW, The Generation R Study: design and cohort update 2017. Eur J Epidemiol 31, 1243–1264 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moncunill G, De Rosa SC, Ayestaran A, Nhabomba AJ, Mpina M, Cohen KW, Jairoce C, Rutishauser T, Campo JJ, Harezlak J, Sanz H, Diez-Padrisa N, Williams NA, Morris D, Aponte JJ, Valim C, Daubenberger C, Dobano C, McElrath MJ, RTS S /AS01E Malaria Vaccine Induces Memory and Polyfunctional T Cell Responses in a Pediatric African Phase III Trial. Front Immunol 8, 1008 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Monaco G, Chen H, Poidinger M, Chen J, de Magalhaes JP, Larbi A, flowAI: automatic and interactive anomaly discerning tools for flow cytometry data. Bioinformatics 32, 2473–2480 (2016). [DOI] [PubMed] [Google Scholar]
- 67.van der Maaten LJP, Hinton GE, Visualizing High-Dimensional Data Using t-SNE. Journal of Machine Learning Research 9, 2579–2605 (2008). [Google Scholar]
- 68.Finak G, Jiang M, flowWorkspace: Infrastructure for representing and interacting with gated and ungated cytometry data sets. R package version 3.30.0. , (2018). [Google Scholar]
- 69.Kolde R, pheatmap: Pretty Heatmaps. R package version 1.0.10, (2018). [Google Scholar]
- 70.Davis S, Meltzer PS, GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 23, 1846–1847 (2007). [DOI] [PubMed] [Google Scholar]
- 71.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Swysen C, Vekemans J, Bruls M, Oyakhirome S, Drakeley C, Kremsner P, Greenwood B, Ofori-Anyinam O, Okech B, Villafana T, Carter T, Savarese B, Duse A, Reijman A, Ingram C, Frean J, Ogutu B, C. Clinical Trials Partnership, Development of standardized laboratory methods and quality processes for a phase III study of the RTS, S/AS01 candidate malaria vaccine. Malar J 10, 223 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Angerer P, Haghverdi L, Buttner M, Theis FJ, Marr C, Buettner F, destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241–1243 (2016). [DOI] [PubMed] [Google Scholar]
- 74.Oksanen J, Blanchet F, Kindt R, Legendre P, Minchin P, O’Hara R, vegan: Community Ecology Package. R package version 2.4-4, (2015). [Google Scholar]
- 75.Gromping U, Relative importance for linear regression in R: The package relaimpo. Journal of Statistical Software 17, (2006). [Google Scholar]
- 76.Bates D, Maechler M, Bolker B, Walker S, Fitting Linear Mixed-Effects Models Using lme4. Journal of Statistical Software 67, 1–48 (2015). [Google Scholar]
- 77.Bartoń K, MuMIn: Multi-Model Inference. R package v1.42.1., (2018). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data file S1: Differentially expressed genes between B0 and B32
Data file S2: Differentially expressed genes between anemic and non-anemic children
Data file S3: Plasmablast differentiation assay raw data
Fig. S1: Pre-gating of cell types for tSNE
Fig. S2: Gating hierarchy Panel 1
Fig. S3: Gating hierarchy Panel 2
Fig. S4: Representative flow plots panel 1
Fig. S5: Representative flow plots panel 2
Fig. S6: No vaccine induced changes are detectable one month after vaccination.
Fig. S7 Strong correlations observed within related cell types
Fig. S8 Cell subset frequency correlations between baseline and B3 time-points
Fig. S9 Cell subset frequency correlations between baseline and B32 time-points
Fig. S10. Anti-CSP and Anti-HBV.S IgG titers before and after vaccination in infants and children from Mozambique
Fig. S11. Distribution of participant age along the diffusion-pseudotime trajectory
Fig. S12. Anti-CSP and Anti-HBV.S IgG titers before and after vaccination in children from Tanzania and Mozambique
Fig. S13. Dose dependent toxicity of ciclopirox olamine (CPX) on B cells in culture
Fig. S14. Highly reproducible flow cytometry data between experiments
Table S1: Participant characteristics
Table S2 CONSORT Table
Table S3. Antibody panels
Table S4: Manual gated cell subset definitions Panel 1
Table S5: Manual gated cell subset definitions Panel 2