

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a novel etiological agent of coronavirus disease 2019 (COVID-19). Nigella sativa, commonly known as black seed or black cumin, has been a historical and traditional plant since thousands of years. Based on their therapeutic efficacy, the chief components of terpenoids and flavonoids were selected from N. sativa seeds and seed oil. This study was designed to check the antiviral efficacy of N. sativa main phytoconstituents against five potential targets of SARS-CoV-2 using in silico structure-based virtual screening approach. Out of twenty five phytocomponents, ten components showed best binding affinity against two viral proteins viz. N-terminal RNA binding domain (NRBD; PDB ID: 6M3M) of nucleocapsid protein and papain-like protease (PL-PRO; PDB ID: 6W9C) of SARS-CoV-2 using AutoDock 4.2.6, AutoDock Vina and iGEMDOCK. PASS analyses of all ten phytocomponents using Lipinski’s Rule of five showed promising results. Further, druglikeness and toxicity assessment using OSIRIS Data Warrior v5.2.1 software exhibited the feasibility of phytocomponents as drug candidates with no predicted toxicity. Molecular dynamics simulation study of NRBD of SARS-CoV-2 nucleocapsid protein-alpha-spinasterol complex and PL-PRO-cycloeucalenol complex displayed strong stability at 300 K. Both these complexes exhibited constant root mean square deviation (RMSDs) of protein side chains and Cα atoms throughout the simulation run time. Interestingly, PL-PRO and NRBD are key proteins in viral replication, host cell immune evasion and viral assembly. Thus, NRBD and PL-PRO have the potential to serve as therapeutic targets for N. sativa phytoconstituents in drug discovery process against COVID-19.
Keywords: Nigella sativa, phytocomponents, SARS-CoV-2, nucleocapsid protein, papain-like protease, molecular chemoinformatics
Graphical Abstract

Communicated by Ramaswamy H. Sarma
1. Introduction
Coronavirus disease 2019 (COVID-19) is a highly contagious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). It was first identified in December 2019 in Wuhan city of China and has spread globally, thereafter, resulting in the ongoing pandemic (Riou & Althaus, 2020). COVID-19 has spread rapidly in the human population and has caused a high number of deaths globally. According to Center for Disease Control and Prevention, human coronaviruses cause mild to severe infections in humans. But this new virus SARS-CoV2 is a public concern because not much is known about its spread amongst the people and its mechanism of function in the human body.
SARS-CoV-2 is an enveloped positive-sense and single-stranded RNA genome containing virus belonging to Coronaviridae family of β- viruses (Pal et al., 2020). SARS-CoV-2 has shown similarity with severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) viruses. Coronaviruses possess four structural proteins namely spike-S, envelope-E, membrane-M and nucleocapsid-N proteins. The entry of coronavirus into host cells is mediated by the transmembrane S glycoprotein (Li, 2016). S protein contains two functional subunits viz. S1 responsible for binding to the host cell receptor and S2 for fusion of the viral and host membranes which allow the CoV-RNA genome to enter inside the host cells. N-terminal domain (NTD) and C-terminal domain (CTD) are the major structural and functional domains of the nucleocapsid protein, which regulate the replication and transcription of viral RNA. The most important function of the NTD of nucleocapsid protein is RNA binding, while the primary job of the CTD is dimerization (Chang et al., 2016; Walls et al., 2020). Open reading frame (ORF) of CoV RNA genome encodes two large polyproteins PP1a and PP1ab. After processing of PPs by cysteine proteases, 16 non-structural proteins (NSPs) are formed. The C-terminal ends of these PPs are cleaved by chymotrypsin-like cysteine protease (3CL-PRO) and the N-terminal end is processed by the papain-like protease (PL-PRO). The PL-PRO cleaves the N-terminal region of the PPs to generate three NSPs which help in the formation of replicase transcriptase complex for viral propagation (Prajapat et al., 2020). Thus, both NTD of nucleocapsid protein and PL-PRO represent important targets from the perspective of drug discovery.
The incidence of COVID-19 has elevated intense attention not only in India but worldwide. No specific therapeutic is available till date and, therefore, to control the propagation of COVID-19, current management including travel restrictions, social distancing, lockdown, patient isolation and supportive medical care are being used by the governments of pandemic-hit countries. Considering the proportion of the disastrous epidemic, research is being carried out at a break-neck speed, so that future treatment schemes with effective novel therapeutic agents and vaccines can be released immediately. In some countries, anti-HIV and antimalarial drugs are being used as prophylaxis, but these drugs lack the desired biological effects and are not free from adverse effects in clinical trials. Hydroxychloroquine is an antimalarial drug and in case of viral infection, it increases the pH within intracellular vacuoles to inhibit the replication of different viruses by interfering with endosome/lysosome trafficking or viral protein maturation during virion maturation; while, ivermectin is a broad spectrum anti-parasitic drug that paralyzes and kills the parasites (Choudhary & Sharma, 2020). Ivermectin is known to exert its antiviral effect by preventing viral proteins moving in and out of the host cell’s nucleus, which is essential for replication of coronavirus (Caly et al., 2020). Among the repurposed drugs for COVID-19, hydroxychloroquine has been approved by the FDA as an Emergency Use Authorization (EUA) against COVID-19, while ivermectin is an FDA-approved antiparasitic agent with antiviral activity against a broad range of viruses, such as influenza, human immunodeficiency virus (HIV), dengue virus, West Nile virus, and Venezuelan equine encephalitis virus (Choudhary & Sharma, 2020; Heidary & Gharebaghi, 2020). Previous studies have also reported the antiviral effects of hydroxychloroquine and ivermectin against several distinct negative-sense single-strand RNA viruses, including SARS-CoV-2 (Choudhary & Sharma, 2020; Liu et al., 2020). Therefore, hydroxychloroquine and ivermectin are expected to inhibit viral load in patients with COVID-19.
Development of novel drugs is also a time consuming process with an exorbitant cost. Therefore, a planned and systematic approach is needed for rational drug design and discovery to overcome the burden of the pandemic. Bioinformatics has been an integral part of drug development in this age of personalized medicine and cost-effective public health outcomes. In the post genomic era, virtual in silico prediction of promising drug candidates outsourced from the plant kingdom can play a significant role in drug discovery in complementary and alternative medicine, thus proving to be time and cost effective. Medicinal plants and their phytoconstituents offer diverse pharmacological properties and unlimited scope as part of Indian traditional system of medicine i.e. Ayurveda; however, most of them remain to be studied as therapeutic agents against the ongoing pandemic.
Nigella sativa, belonging to family Ranunculaceae , is commonly known as black seed or black cumin. In historical and religious texts, N. sativa is known as a miracle curative herb for all ailments, except death (Yimer et al., 2019). The black seed is used for stimulating the body's energy and helping recovery from fatigue and dispiritedness (Ahmad et al., 2004). N. sativa seeds and oils have wide therapeutic effects against many ailments such as skin diseases, jaundice, gastrointestinal problems, anorexia, conjunctivitis, dyspepsia, rheumatism, diabetes, hypertension, intrinsic hemorrhage, paralysis, amenorrhea, anorexia, asthma, cough, bronchitis, headache, fever, influenza, eczema and cancer (Forouzanfar et al., 2014, Ahmad et al., 2013, Ahmad, Khan, et al., 2020; Yarnell & Abascal, 2011). The seeds and oil of N. sativa contain terpenoids, flavonoids, phenolics, alkaloids, saturated and unsaturated fatty acids (Forouzanfar et al., 2014; Menounos et al., 1986; Yarnell & Abascal, 2011). Because of the rich nutraceuticals in N. sativa, it could be extensively used to prevent and cure COVID-19. Till date, only limited studies have reported the antiviral activities of N. sativa showing protective effect of black seed oil against murine cytomegalovirus infection (Salem & Hossain, 2000) and in silico antiviral activity of some unsaturated/saturated fatty acids against angiotensin-converting enzyme 2 (ACE2) receptor of host cells (Ahmad, Abbasi, et al., 2020). However, none of the studies has reported the antiviral activities of selected terpenoids and flavonoids (Table 1) against SARS-CoV-2 viral proteins viz. NTD of nucleocapsid protein and PLpro of SARS-CoV-2 by employing chemoinformatics tools.
Table 1.
List of ten bioactive components from N. sativa, standard drugs hydroxychloroquine and ivermectin and their respective binding to nucleocapsid protein (PDB ID: 6M3M) and papain-like protease (PL-PRO, PDB ID: 6W9C) of SARS-CoV-2.
| S. No. | Ligands | PDB: ID (SARS-CoV-2) | MF and MW of phyto-components | Molecular Structure | Pub Chem CID |
Chemical Class of Ligands | Source |
|---|---|---|---|---|---|---|---|
| 1. | 24-methylene- cycloartanol | 6M3M 6W9C |
MF: C31H52O MW: 440.7 |
 |
94204 | Pentacyclic triterpenoid | Seed oil |
| 2. | Alpha-spinasterol (spinasterol) |
6M3M 6W9C |
MF: C29H48O MW: 412.7 |
 |
5281331 | Steroid | Seed/seed oil |
| 3. | Beta-amyrin | 6W9C | MF: C30H50O MW: 426.7 |
 |
73145 | Pentacyclic triterpenoid | Seed oil |
| 4. | Beta-sitosterol | 6M3M 6W9C |
MF: C29H50O MW: 414.7 |
 |
222284 | Phytosterol | Seed oil |
| 5. | Campesterol | 6M3M 6W9C |
MF: C28H48O MW: 400.7 |
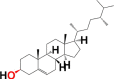 |
173183 | Phytosterols | Seed/seed oil |
| 6. | Citrostadienol (alpha1-sitosterol) |
6M3M 6W9C |
MF: C30H50O MW: 426.7 |
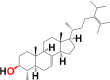 |
9548595 | Sterol | Seed oil |
| 7. | Cycloartenol | 6W9C | MF: C30H50O MW: 426.7 |
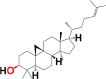 |
92110 | Pentacyclic triterpenoid | Seed oil |
| 8. | Cycloeucalenol | 6M3M 6W9C |
MF: C30H50O MW: 426.7 |
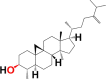 |
101690 | Pentacyclic triterpenoid | Seed oil |
| 9. | Taraxerol | 6M3M 6W9C |
MF: C30H50O MW: 426.7 |
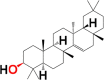 |
92097 | Pentacyclic triterpenoid | Seed oil |
| 10. | Tirucallol | 6W9C | MF: C30H50O MW: 426.7 |
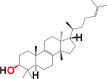 |
101257 | Tetracyclic triterpene | Seed oil |
| 11. | Hydroxy- chloroquine | 6M3M 6W9C |
MF: C18H26ClN3O MW: 335.9 |
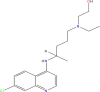 |
3652 | 4-aminoquinoline (Standard drug) |
Chemotherapeutic agent |
| 12. | Ivermectin | 6M3M 6W9C |
MF: C48H74O14 MW: 875.1 | 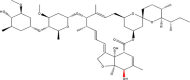 |
6321424 | Macrocyclic lactone (Standard drug) | Derived from Streptomyces avermitilis. |
In the present study, twenty five phytoconstituents from N. sativa were selected for their binding affinity with five target proteins of SARS-CoV-2 using AutoDock, of which ten showed best binding kinetics against viral N-terminal RNA binding domain (NRBD) of nucleocapsid protein and papain-like protease (PL-PRO). MD simulation study of two bound ligand-protein complexes exhibited strong stability while all phytoconstituents displayed druglikeness with no predicted toxicity. Our present findings are further supported by the previously reported antiviral efficacies of a class of terpenoids and flavonoids (Ghildiyal et al., 2020; Naithani et al., 2010; Yang et al., 2020). Thus, NRBD and PL-PRO of SARS-CoV-2 warrant further validation as potential drug targets through wet lab and clinical studies.
2. Materials and methods
2.1. Data sources
The present study was carried out at Molecular Chemoinformatics Section, Cell and Tissue Culture Lab, Dept. of Biochemistry, Era’s Lucknow Medical College and Hospital, Era University, Lucknow. A total number of twenty five bioactive components of black cumin (Nigella sativa) were selected from Dr. Duke’s Phytochemical and Ethnobotanical Databases (https://phytochem.nal.usda.gov/phytochem/search/list), comprising thirteen compounds from seed oil and twelve compounds from N. sativa seeds (Table S1). The criteria for selection were based on their respective structure-activity relationships and prospective targeted metabolic pathways. During compound screening, all ubiquitous chemicals were excluded and phytochemicals belonging to the class of terpenoids and flavonoids were selected for the present study. The compounds from seed oil used in the present study were 24-methylene-cycloartanol (CID_94204), Alpha-spinasterol, also known as spinasterol (CID_5281331), arachidonic-acid (CID_444899), beta-amyrin (CID_73145), beta-sitosterol (CID_222284), campesterol (CID_173183), citrostadienol also known as alpha-1-sitosterol (CID_9548595), cycloartenol (CID_92110), cycloeucalenol (CID_101690), taraxerol (CID_92097), thymol(CID_6989), thymoquinone (CID_10281), and tirucallol (CID_101257). The selected phytochemicals from seed source were astragalin (CID_5282102), carvone (CID_7439), D-limonene (CID_440917), nigellicine (CID_11402337), nigellidine (CID_136828302), nigellimine, also known as isosalsolidine (CID_20725), nigellimine-n-oxide (CID_348288664), nigelline, also known as damascenine (CID_21368), nigellone, also known as dithymoquinone (CID_398941), quercetin-3-glucoside, also known as isoquercetin (CID_5280804), rutin (CID_5280805), and thymohydroquinone, also known as thymoquinol (CID_95779). Among the repurposed drugs for COVID-19, hydroxychloroquine and ivermectin are the mainstay for COVID-19 treatment in the present scenario. Though hydroxychloroquine is generally considered safe and side-effects are generally mild with no secondary or associated complications, it has been found to be toxic in SARS-CoV-2 patients with cardiovascular disorders (Touret & de Lamballerie, 2020). As far as ivermectin is concerned, huge uncertainty remains about whether this treatment can be safely and effectively repurposed to tackle the coronavirus. The major concern in using ivermectin as a repurposed drug against COVID-19 is the safety of its use in pregnant females and children below the age of 14. Whereas ivermectin generally does not cause problematic side effects at the currently used doses, there is still limited information about whether much larger doses would also be safe. Therefore, there is a need for better alternatives/substitutes for both these drugs; hence the premise of the current study. Thus, the standard drugs hydroxychloroquine (CID_3652) and ivermectin (CID_6321424) were also included in the present study for comparison of their physicochemical and drug properties with those of the selected N. sativa phytoconstituents.
2.2. Preparation of ligands
All phytochemicals of N. sativa belonging to the class of terpenoids and flavonoids were selected for ligand preparation. PubChem (https://pubchem.ncbi.nlm.nih.gov/) database was used to download 3D structures of all phytochemicals and reference drugs in .sdf format. Before docking, energy minimization of ligands was performed by ChemBio3D Ultra 14.0, with Force Field type MM2 and saved in .pdb format (Ahmad, 2019).
2.3. Preparation of target proteins/macromolecules
The 3D crystal structures of selected SARS-CoV-2 protein targets viz. spike glycoprotein (closed state, PDB ID: 6VXX), spike glycoprotein (open state, PDB ID: 6VYB), N-terminal RNA binding domain of nucleocapsid protein (NRBD, PDB ID: 6M3M), 3 C-like protease (3CL-PRO main protease, PDB ID: 6M03), and papain-like protease (PL-PRO, PDB ID: 6W9C) whose X-ray diffraction structures are available in RCSB database, were downloaded from Protein Data Bank (http://www.rcsb.org/pdb) in .pdb format. Before docking analyses, all 3D protein structures were subjected to refinements and energy minimizations. Whole pdb structures of proteins were used for molecular docking study. The refinement procedure was carried out by addition of missing atoms to the residues, addition of polar hydrogen atoms and Kollman charges, removal of crystallographic water-molecules and external and irrelevant ligands and ions from the protein. During the docking period, the ligands were considered to be flexible and the proteins were considered as rigid. The highest binding energy (most negative) obtained for a ligand was considered as the ligand having maximum binding affinity to a particular target protein.
2.4. Molecular Docking analysis
2.4.1. AutoDock 4.2.6
Molecular docking of selected twenty five phytoconstituents and two standard drugs against five target receptors/proteins of SARS-CoV-2 was performed using AutoDock version 4.2.6 (Morris et al., 1998). Autogrid was used to determine the position of the native ligand on the binding site of protein using grid spacing 0.375 Å and grid coordinates (X, Y and Z) axes at 60 × 60 × 60. Lamarckian genetic algorithm (GA) parameter was employed using 10 runs of the GA criteria and the binding energies of the results were further analyzed (Oprea et al., 2001). After evaluating binding of twenty five phytochemicals with five proteins with AutoDock 4.2.6 software, ten phytochemicals and corresponding two target proteins were selected based on the lowest binding energy i.e. maximum binding affinity for further validation through two additional docking softwares viz. AutoDock Vina and iGEMDOCK version 2.1 (Ahmad, 2019).
2.4.2. AutoDock Vina
Execution of AutoDock Vina is faster than AutoDock 4.2.6, which reduces the size of the conformational space, allowing it to be searched reliably and reduces the computational effort in predictions of binding pockets (Trott & Olson, 2010). AutoDock Vina was used to perform docking simulations, generating 10 conformations of ligand in complex with the receptor, which were finally ranked on the basis of binding energy. The resulting conformations were visualized in Accelrys Biovia Discovery Studio 2017 R2 (Biovia, San Diego, CA, USA).
2.4.3. iGEMDOCK
Target proteins and ligands were further docked with iGEMDOCK version 2.1. The genetic algorithm (GA) parameters, which guided the docking procedure, were set as follows: population size = 200, generations = 70, and number of solutions = 2. After generating a set of poses, the best fit was selected which represented the total binding energy in the form of hydrogen bond (HB), van der Waals forces (VDW), and electrostatic interactions (EI) (Yang & Chen, 2004).
2.5. Analysis and visualization of docked ligand-protein complexes
Based on the obtained lowest binding energy (B.E.) and dissociation constant (Kd), the best orientation (pose) of the ligand-protein interaction was selected for computational analysis and visualization of docking site using Accelrys Biovia Discovery Studio version 2017 R2.
2.6. Prediction of activity spectra for substances (PASS) analysis
PASS analysis program predicts biological activity spectrum of a compound under study based on its structure-activity relationship with a known chemical entity (Ahmad, 2019). In this study, PASS analysis was performed using various online and offline tools as detailed below.
2.6.1. Lipinski’s rule of five
The druglikeness of ten phytochemicals of black cumin and two standard antiviral drugs was evaluated using Lipinski's rule of five (Lipinski, 2004). The parameters of druglikeness such as MW ≤500, logP ≤5, number of hydrogen bond donors (NOHNH) ≤5 and hydrogen bond acceptor sites (NON)≤10, topological polar surface area (TPSA) (≤140 Å2), and number of rotatable bonds (≤10) were determined. In the present study, druglikeness of phytochemicals was calculated using online tool Molinspiration (http://www.molinspiration.com/cgi-bin/properties) and was compared with that of standard reference drugs.
2.6.2. Toxicity potential assessment
Toxicity risk assessment gives an idea about the probable side effects of compounds that may be used for further processing in drug discovery and development. The prediction of different properties of molecules at an early stage is a vital step in drug discovery and development process. Drug-toxicity risk parameters such as druglikeness, mutagenicity, tumorigenicity, reproductive and irritant effects were analyzed by the OSIRIS Data Warrior v5.2.1 (Khan et al., 2018).
2.7. Bioactivity score (BAS) prediction
BAS values suggest a compound’s overall ability to be a potent drug candidate. Molinspiration chemoinformatics, an online tool (https://www.molinspiration.com/cgi-bin/properties), was used to predict the drug scores of the prospective phytoconstituents with respect to several human receptors like GPCRs, ion channels, kinases, nuclear receptors, proteases and enzymes. As a general rule, the higher the bioactivity score, the greater is the probability of the compound being active (Proudfoot, 2002).
2.8. Pharmacokinetic (PK) parameters prediction
The ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity) properties of all components including standard drugs were predicted using online SwissADME software (http://www.swissadme.ch/). This software analyses the important pharmacokinetic properties of a compound like distribution viz. blood-brain barrier (BBB) and skin permeability (LogKp), and its metabolism in terms of it being a P-glycoprotein (P-gp) substrate, Cytochrome P450 viz. CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4 inhibitor as well as its lipophilicity for plasma membrane absorption (Tian et al., 2015, Delaney, 2004).
2.9. Principal component analysis (PCA) of phytoconstituents and standard drugs
PCA is a mathematical method to summarize multidimensional datasets into two or three principal components (inter-correlated quantitative variables) that can be visualized graphically with minimal loss of information. The covariance and correlation matrix are calculated to scale the matrix in such a way that data with high variance are compressed with data having low variance. PCA was performed for defining and visualizing various multidimensional ‘property spaces’ by assigning dimensions to numerical descriptors of molecular structures of phytoconstituents and standard drugs viz. MW, % Absorption and TPSA using OSIRIS Data Warrior v5.2.1. The bar charts and 3D scatter plots of principal components were made to depict druglikeness of N. sativa phytoconstituents to standard drugs using OSIRIS Data Warrior v5.2.1 (Ahmad, 2019).
2.10. Molecular Dynamics (MD) simulation using ligand and receptor molecular dynamics (LARMD) online server
MD simulation is very valuable for understanding the dynamic behaviors of fast internal motions to slow conformational changes at different timescales or even protein folding processes of biological macromolecules. MD simulation is also used to study the effect of explicit solvent molecules on protein structure and stability to obtain time-averaged properties of the biomolecular system, such as density, conductivity and dipolar moment, as well as different thermodynamic parameters, including interaction energies and entropies (Hospital et al., 2015). Ligand and Receptor Molecular Dynamics (LARMD) server provides a user-friendly online protocol (http://chemyang.ccnu.edu.cn/ccb/server/LARMD/index.php/home/index) to investigate and visualize the molecular dynamic property of ligand-driven protein molecule. MD simulation of cycloeucalenol with papain-like protease (PL-PRO) (PDB ID: 6W9C) and alpha-spinasterol with NRBD of nucleocapsid protein (PDB ID: 6M3M) were carried out using online server LARMD. Out of three computational modules namely Nor_mod, Int_mod and Str_mod, Int_mod of LARMD was used to investigate the fluctuation of the protein. The softwares CAVER3.0, AMBER16, MDTraj and Bio3d are integrated into these three modules. The plugins and softwares such as JSmol, Chart.js and MolScript are integrated to visualize and analyze the result on the Web page (de Oliveira & de Oliveira, 2020; Yang et al., 2019). In the LARMD protocol, the AMBER ff14SB force field and general AMBER force field (gaff) were used for amino acid residues and ligands, respectively. The enthalpy and the entropy were calculated by the MM/PB(GB)SA method and empirical method, respectively. The binding free energy (ΔGbind) based on binding energy (ΔEbind), salvation entropy (-TΔSsol) and conformational entropy (-ΔTSconf) was calculated using following equation: ΔGbind = ΔEbind – TΔSsol – TΔSconf. For protein-ligand interactions and the contribution of each residue, the binding energy was decomposed per residue by using the decomposition module of the AMBER16 program. Various parameters such as root-mean-square deviation (RMSD), radius of gyration (Rg), fraction of native contacts Q (x) analysis, root mean square fluctuation (RMSF), B-factor, PCA, MM/PB(GB)SA for energy analysis, hydrogen bonds and decompose analysis were evaluated using LARMD online protocol (Bahar et al., 2010).
3. Results
3.1. Docking analysis of phytocomponents of N. sativa against targeted proteins of SARS-CoV-2
AutoDock v4.2.6 was used for docking study of twenty five phytochemicals of N. sativa against five targeted proteins of SARS-CoV-2 (Table S1, S2 and S3). Thus, total one hundred twenty five (25 × 5 = 125) binding combinations were generated for the study of molecular docking analysis (Table S4). Based on the best binding energy and dissociation constant (Kd) of ligand-protein interactions with AutoDock v4.2.6 tool, out of twenty five phytocomponents, ten active components with their corresponding targeted protein receptors were selected for further validation through two additional docking tools viz. AutoDock Vina and iGEMDOCK v2.1 (Table 1). Tables 2 and 3 summarize the various binding energy parameters, dissociation constants (Kd) and interacting amino acid residues participating in the binding pocket of NRBD of nucleocapsid protein and PL-PRO of SARS-CoV-2 with six and ten active components of N. sativa, respectively versus standard drugs hydroxychloroquine and ivermectin through AutoDock v4.2.6, AutoDock Vina and iGEMDOCK v2.1 tools.
Table 2.
Docking interactions of active components of N. sativa with N-terminal RNA binding domain (NRBD) of SARS-CoV-2 nucleocapsid protein (PDB ID: 6M3M) versus standard drugs hydroxychloroquine and ivermectin.
| AutoDock v4.2.6 |
AutoDock Vina |
iGEMDOCK v2.1 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S.No | Ligands | BE (kcal/mol) | Kd | Interacting amino acids | BE (kcal/mol) | Kd | Interacting amino acids | T.E. (kcal/mol) |
VDW | HB | EI | Interacting amino acids |
| 1. | 24-methylene- cycloartanol | −8.29 | 842.64 nM | Ala126,Ala135,Asn49, Asp129, Asp64,Gly125, Gly130, IIe131,IIe132, Lys128,Phe67, Trp109, Trp133,Tyr124, | −9.8 | 76.87 nM | Gln161,Gln84,Glu137 Gly164,Gly70,Ile75,Leu160, Leu162,Leu168, Pro163,Pro81,Ser79,Thr136, Thr161,Thr166,Thr167 |
−96.16 | −91.43 | −4.72 | 0 | Arg69,Gln161,Gln84, Glu137,Glu165,Gly70,Leu160, Leu162,Leu16,Pro163,Pro81,Ser79, Ser80,Thr136,Thr166,Tyr173 |
| 2. | Alpha-spinasterol | −9.54 | 101.42 nM | Ala126,Ala135,Arg69,Asp64,Gln71, Gly130,Gly70,IIe131,IIe132, Leu65,Lys128,Lys66,Phe67,Pro68, Th136,Trp133,Tyr124Val134 | −9.6 | 101.42 nM | Ala107,Asp108,Asp164,Glu161, Glu167,Gly163His89, Leu162,Lys157,Lys92,Pro248,Thr301, Trp108,Trp93,Tyr264,Tyr264,Tyr273 | −91.33 | −84.04 | −7.29 | 0 | Asn109,Gln269,Glu161,Gly160, His89Leu162,Thr158,Val159 |
| 3. | Beta-sitosterol | −8.69 | 426.43 nM | Ala35,Arg69,Asp64,Gln71,Gly130, Gly70,IIe132,Leu65,Lys128, Lys66,Phe67,Pro68,Thr136, Trp109,Trp133,Tyr124,Val134 | −10.4 | 76.87 nM |
Asn76,Gln161,Gln71,Gln84, Gly70Leu160,Leu162, Leu168,Leu57,Pro163,Pro81,Ser79, Thr136,Thr166,THr167,Tyr173 | −88.19 | −84.69 | −3.5 | 0 | Ala126,Arg69,Asn127,Asn155, Gly125,Gly70Ile131,Ile132, Lys128,Phe67,Pro68, Trp133,Tyr124 |
| 4. | Campesterol | −8.57 | 522.64 nM | Ala126,Ala135Arg69, Asn127Asp129, Gln71,Gly70,IIe131 ,Lys128,Lys66,Phe67,Pro68,Trp133, Tyr124,Val134 |
−9.8 | 76.87 nM | Asn76,Gln161,Gln71,Gln84, Glu137,Gly165,Gly70,Leu160, Leu162,Leu168,Pro163Pro81,Ser79, Thr136,Thr166,THr167Thr77,Tyr173 | −93.83 | −90.33 | −3.5 | 0 | Gln161,Gln164,Gln84,Glu137, Gly165,Gly70,Leu162,Pro81,Ser79, Thr136,Thr166,Thr167 |
| 5. | Citrostadienol | −8.43 | 663.45 nM | Ala126,Arg69Asn127,Asn49, Asp129,Gly125,IIe131,IIe132, Lys128,Phe67,Pro152, Thr50,Trp133,Tyr124 | −8.9 | 259.96 nM |
Ala156,Asn127Asn155,Asn76, Asn78,Asp145,Gln161, Ile158,Thr149,Thr50,Trp53,Val159 | −80.13 | −75.88 | −4.24 | 0 | Ala156,Ala157,Asn155Asp145, Glm161,Gly148,His146, Ile158,Thr149,Val159 |
| 6. | Taraxerol | −8.5 | 588.67 nM | Asn49,Asp129,Asp64, Gly130,IIe131IIe132, Leu65,Lys128Lys656Trp133,Tyr124 | −12.1 | 76.87 nM |
Gln161,Gln71Gln84,Glu137, Gly165,Gly70,Ile75,Leu160, Leu162,Leu168,Pro163,Pro74Thr136, Thr166,Thr167,Thr173 | −96.11 | −93.61 | −2.5 | 0 | Gln161,Gln71,Gln84,Glu137, Gly165,Gly70,Ile75Leu160, Leu162,Leu168,Pro163,Pro74, Thr136,Thr166,Tyr173 |
| 7. | Hydroxychloroquine | −3.96 | 1.25 mM | Ala126,Arg69,Asn49,IIe131,IIe132, Lys66Phe67,Thr50,Trp133,Tyr124 | −6.7 | 588.67 nM |
Arg150,Asn151,Asn155, Asn76Gly148,Ile147,Ile158, Ile75,The77,Thr149,Thr50,Trp53 | −78.61 | −75.11 | −3.5 | 0 | Ala156,Asn76,Asp145,Gln161, Gly148,His146,IIe147,IIe158, IIe75,Thr149,Thr77,Val159 |
| 8. | Ivermectin | −9.08 | 220.28 nM | Ala126,Arg89,Arg89,Asn127,Asn154, Asn155,Asn49,Asn76,Asn78,Asp64, Gly117,Gly125,IIe131, IIe132,Leu65Lys128, Lys66,Pro118,Thr50,Trp133,Trp53 | −9.8 | 76.87 nM |
Ala157,Ala174,Ala56,Arg108, Arg150,His60,Pro152, Thr55,Thr58,Tyr110Tyr173,Val159 | −114.23 | −96.98 | −17.28 | 0 | Ala126,Ala51,Arg89,Asn127, Asn154,Asn155,Asn49,Asp129, Asp129,Asp64,Gly130,Ile131, Ile132,Leu65,Lys128,Lys66, Pro118,Thr50,Trp133, Tyr110,Tyr112 |
Table 3.
Docking interactions of active components of N. sativa with papain-like protease (PL-PRO) of SARS-CoV-2 (PDB ID: 6W9C) versus standard antiviral drugs hydroxychloroquine and ivermectin.
| AutoDock v4.2.6 |
AutoDock Vina |
iGEMDOCK v2.1 |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S. No. | Ligands | BE (kcal/mol) | Kd | Interacting amino acids | BE (kcal/mol) | Kd | Interacting amino acids | T.E. (kcal/m ol) | VDW | HB | EI | Interacting amino acids | |
| 1. | 24-methylene- cycloartanol | −8.32 | 800.77 nM | Asn109,Asn109,Cys270,Gln269,Gln269, Gln269,Glu161,Gly160Gly160, Gly161,His89,Leu162,Thr158,Val159 | −9.6 | 101.42 nM | Ala107,Asp108,Asp164,Glu161, Glu167,Gly163His89,Leu162, Lys157,Lys92,Pro248,Thr301, Trp108,Trp93,Tyr264Tyr264,Tyr273 | −84.63 | −78.99 | −5.64 | 0 | Asn109,Gln269,Gly160, Gly161,Leu162,Thr158Val159 | |
| 2. | Alpha-spinasterol | −9.41 | 126.53 nM |
Asn109,Asn109,Asp108,Cys270,Cys270, Gln269,Gln269,Gln269,Glu161,Gly150, Gly160,His89Leu162,Leu162,Val159 | −9.0 | 220.28 nM | Asn109,Asp108,Cys270, Gln269,Glu161, Gly160,Gly160,His89Leu162,Val159 | −91.9152 | −86.94 | −4.97 | 0 | Asn109,Cys270Gln269, Glu161,Gly160, Leu162,Thr158,Val159 | |
| 3. | Beta-amyrin | −8.79 | 357.58 nM | Gln1005,Gln1010,IIe1013,Leu102, Leu763,The1006Thr100,Thr1009,Val1008 | −9.8 | 76.87 nM | Asn109Gln269,Glu161,Gly160, Leu162,Thr158,Val159 | −84.40 | −80.90 | −3.5 | 0 | Ala107,Asn267,Asp108,Asp164, Glu161,Glu167,Gly163,His89Leu162, Lys157,Lys92,Trp106,TYr264,Tyr268 | |
| 4. | Beta-sitosterol | −9.14 | 198.01 nM |
Asn109Asn109,Asn109,Asp108Cys270, Cys270,Gln269,Gln269,Gln269,Glu161 ,Gly160,Gly160,His89,Leu162,Leu162,Val159 | −9.0 | 220.28 nM | Asn109,Gln269,Glu161, Gly160,Gly160, His89,Leu162,Thr158,Val159 | −98.20 | −92.93 | −5.27 | 0 | Asn109,Cys270Gln269,Glu161,Gly160, Gly160,His89,Leu162,Leu162,Thr158 | |
| 5. | Campesterol | −9.71 | 76.87 nM |
Asn109,Asn109,Asn109,Asp108,Cys270Cys270, Gln269,Gln269,Gln269,Glu161,Gly160, Gly160,Leu162,Leu162,Leu162 | −9.2 | 155.9 nM | Asn109,Cys270Gln269,Glu161, Gly160Gly160,His89,Leu162, Thr158,Val159 | −96.65 | −96.65 | 0 | 0 | Asn109,Asp108,Gln269Glu161, Gly160,Gly160,His89,Leu162, Ser85,Thr158,Val159 | |
| 6. | Citrostadienol | −8.98 | 259.96 nM |
Asn109,Asn109,Asn109,Asp108,Cys270, Gln269,Gln269,Glu161Glu161,Gly160, His89,Leu162,Leu162,Leu162, Thr158,Val159,Val159 | −9.5 | 101.42 nM | Ala107,Asn267,Asp108,Asp164, Glu161,Glu167,Gly266,His89,Leu162, Lys157,Lys92,Pro248Trp106, Trp93,Tyr264 | −91.70 | −86.45 | −5.25 | 0 | Ala86,Asn109Asp108,Glu161, Gly160,His89,Ser85,Thr158,Val159 | |
| 7. | Cycloartenol | −8.74 | 389.61 nM | Asn109,Asn109,Cys270,Gln269,Gln269, Gln269,Glu161,Glu161,Gly160,His89, Leu162,Leu162,Thr158,Val159 | −8.8 | 389.61 nM | Asn109,Asp108,Gln269, Glu161,Gly160,His89, Leu162,Thr158Val159,Val159 | −85.32 | −79.14 | −2.17 | 0 | Ala107,Asp108,Asp164,Glu161, Glu167,Gly163,His89,Leu162, Lys157,Lys92,Trp106,Tyr264 | |
| 8. | Cycloeucalenol | −9.65 | 84.23 nM |
Asn109,Asn109,Asp108,Cys270,Gln269, Gln269,Gln269,Glu161,Glu161,Gly160Gly160, Gly160,Leu162,Leu162,Leu162,Val159 | −9.3 | 155.9 nM | Ala107,Asp108,Asp164, Glu161,Glu167,Gly163, His89,Leu162,Lys157, Lys92,Pro248Thr301, Trp106,Trp93,Tyr264,Tyr273 | −102.07 | −93.20 | −8.86 | 0 | Asn109,Asp108,Cys270, Gln269,Gln289Glu161, Gly160,Gly160,His89, Leu162,Val159 | |
| 9. | Taraxerol | −9.29 | 155.9 nM |
Asn109,Asn109,Asn109, Cys270,Gln269Gln269, Gln269,Glu161,Gly160, Gly160,His89,Leu162, Leu162,Thr158,Val159,Val159 | −10.0 | 76.87 nM | Asn109,Cys270, Gln269,Glu161,Gly160, Gly160,Leu162,Thr158,Val159 | −100.31 | −90.34 | −9.96 | 0 | Asn109,Asp108,Gln269,Glu161, Gly160,Leu162,Thr158,Val159 | |
| 10. | Tirucallol | −8.78 | 368.93 nM | Asn109,Asn109,Asn109, Asp108,Cys270,Gln269, Gln269,Gln269,Glu160,Glu161,Glu161, Gly160,Leu162,Leu162,Thr158Val159 | −8.6 | 522.64 nM |
Ala107,Asn267Asn267, Asp108,Asp164, Leu162,Leu289,Pro248, Trp106,Tyr264,Tyr268 | −84.35 | −81.07 | −3.27 | 0 | Asn109,Gln269Glu161,Gly160, Leu162,Thr158,Val159 | |
| 11. | Hydroxychloroquine | −5.93 | 44.86 µM |
Asn109,Asn109,Asn109,Gln269,Gln269, Gln269,Glu161,Glu161,Gly160, Gly160,Leu162,Leu162 | −7.2 | 588.67 nM |
Asn109,Gln269Glu161Gly160, Leu162 | −80.88 | −80.88 | 0 | 0 | Asn109,Cys270Gln269,Glu161, Gly160,Gly160,Leu162 | |
| 12. | Ivermectin | −4.98 | 224.79 µM |
Asp108,Glu161,Glu161,Gly160,Gly160, His89Leu162,Thr158,Thr158, Thr158,Val159,Val159 | −9.3 | 155.9 nM | Ala153,Ala39,Arg82,Asn156, Asn88,Asp76,Cys155,His73,Ile44, Leu36,Lys92Ser78,Thr74, Tyr154,Tyr171 | −103.44 | −82.27 | −21.17 | 0 | Asp108,ASP108,Glu161, Gly160,Gly160,His89,Ser85, Thr158,Val159,Val159 | |
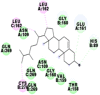
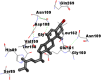
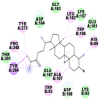
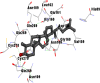
Table 4.
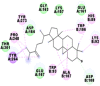
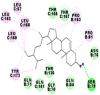
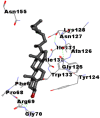
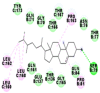
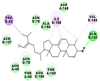
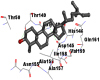
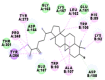
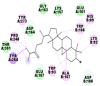
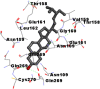
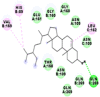
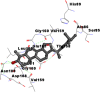
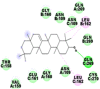
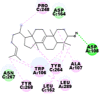
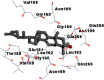
Best docking poses of active components of N. sativa with N-terminal RNA binding domain (NRBD) of SARS-CoV-2 nucleocapsid protein (PDB ID: 6M3M) in comparison to standard drugs hydroxychloroquine and ivermectin. In AutoDock v4.2.6 analyses, ligand is represented as CPK model colored by- H = White, C = Grey, N = Blue, O = Red, S = Yellow, Pink = other elements. In AutoDock Vina, ligand is represented by 2-D line model, whereas in iGEMDOCK v2.1 analyses, ligand is represented by stick model. Green and blue dotted lines represent H- bond.
| S. No. | Ligands | AutoDock v4.2.6 | AutoDock Vina | iGEMDOCK v2.1 |
|---|---|---|---|---|
| 1. | 24-methylene-cycloartanol | 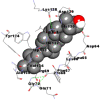 |
 |
 |
| 2. | Alpha-spinasterol | 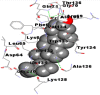 |
 |
 |
| 3. | Beta-sitosterol | 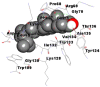 |
 |
 |
| 4. | Campesterol |  |
 |
 |
| 5. | Citrostadienol | 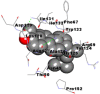 |
 |
 |
| 6. | Taraxerol | 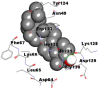 |
 |
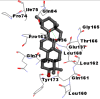 |
| 7. | Hydroxychloroquine | 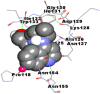 |
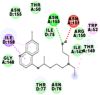 |
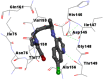 |
| 8. | Ivermectin | 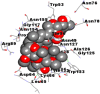 |
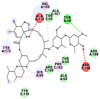 |
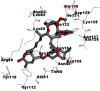 |
As is evident from Table 2, all 6 phytoconstituents and ivermectin exhibited potent binding affinity to NRBD of SARS-CoV-2 nucleocapsid protein. AutoDock v4.2.6 analyses revealed that binding affinities of the phytoconstituents with NRBD of SARS-CoV-2 decreased in the order alpha-spinasterol > beta-sitosterol > campesterol > taraxerol > citrostadienol> 24-methylene-cycloartanol. However, the binding affinity of alpha-spinasterol (BE = −9.54 kcal/mol, Kd = 101.42 nM) was found to be greater than standard drugs ivermectin (BE = −9.08 kcal/mol, Kd = 220.28 nM) and hydroxychloroquine (BE = −3.96 kcal/mol, Kd = 1.25 mM). Thus, both alpha-spinasterol and ivermectin displayed 1000000 x greater affinity than hydroxychloroquine. The results received further confirmation from analysis using AutoDock Vina and iGEMDOCK v2.1 tools (Table 2). On the other hand, the selected phytoconstituents of N. sativa also exhibited potent binding affinity to PL-PRO of SARS-CoV-2. AutoDock v4.2.6 analyses revealed that the binding affinities of the phytoconstituents with PL-PRO of SARS-CoV-2 decreased in the order campesterol > cycloeucalenol > alpha-spinasterol > taraxerol > beta-sitosterol > citrostadienol > beta-amyrin > tirucallol > cycloartenol> 24-methylene-cycloartanol (Table 3). Interestingly, campesterol exhibited a 1000x stronger binding to PL-PRO of SARS-CoV-2 (BE = −9.71 kcal/mol, Kd = 76.87 nM) as compared to standard drugs, hydroxychloroquine (BE = −5.93 kcal/mol, Kd = 44.86 µM) and Ivermectin (BE = −4.98 kcal/mol, Kd = 224.79 µM). However, none of the phytoconstituents was found to interact with the catalytic residues of PL-PRO viz. Cys112, His273, Asp287, Trp107, thereby suggesting the allosteric binding of the Nigella phytoconstituents to viral PL-PRO (Báez-Santos et al., 2015). The docking results obtained from AutoDock v4.2.6, AutoDock Vina and iGEMDOCK v2.1 tools were visualized in Discovery Studio showing prominent interactions between various amino acid residues. Tables 4 and 5 display the best docking poses of six and ten active components of N. sativa, respectively, with NRBD of nucleocapsid protein and PL-PRO of SARS-CoV-2, versus standard drugs hydroxychloroquine and ivermectin.
Table 6.
PASS analysis of major active components of N. sativa versus standard antiviral drugs hydroxychloroquine and ivermectin.
| Lipinski’s rule of 5 (Physicochemical Properties) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| S.No. | Phytoconstituents | % Absorptiona (>50%) |
Topological Polar Surface Area (Å)2 (TPSA)b (<160 Å) |
MW (<500) | c logPc (<5) | Heavy atom count (n atoms) |
Hydrogen Bond Donors (nOHNH) (≤5) |
Hydrogen Bond Acceptors (nON) (≤10) |
Number of Rotatable bonds (≤10) |
Lipinski’s violation |
| 1. | 24-methylene- Cycloartanol |
102.02 | 20.23 | 440.76 | 8.03 | 32 | 1 | 1 | 5 | 1 |
| 2. | Alpha-spinasterol | 102.02 | 20.23 | 412.70 | 7.87 | 30 | 1 | 1 | 5 | 1 |
| 3. | Beta-amyrin | 102.02 | 20.23 | 426.73 | 8.02 | 31 | 1 | 1 | 0 | 1 |
| 4. | Beta-sitosterol | 102.02 | 20.23 | 414.72 | 8.62 | 30 | 1 | 1 | 6 | 1 |
| 5. | Campesterol | 102.02 | 20.23 | 400.69 | 8.30 | 29 | 1 | 1 | 5 | 1 |
| 6. | Citrostadienol | 102.02 | 20.23 | 426.73 | 8.15 | 31 | 1 | 1 | 5 | 1 |
| 7. | Cycloartenol | 102.02 | 20.23 | 426.73 | 8.21 | 31 | 1 | 1 | 4 | 1 |
| 8. | Cycloeucalenol | 102.02 | 20.23 | 426.73 | 7.62 | 31 | 1 | 1 | 5 | 1 |
| 9. | Taraxerol | 102.02 | 20.23 | 426.73 | 8.02 | 31 | 1 | 1 | 0 | 1 |
| 10. | Tirucallol | 102.02 | 20.23 | 426.73 | 8.48 | 31 | 1 | 1 | 4 | 1 |
| 11. | Hydroxychloroquine | 92.16 | 48.38 | 335.88 | 4.00 | 23 | 2 | 4 | 9 | 0 |
| 12. | Ivermectin | 50.31 | 170.09 | 875.11 | 4.58 | 62 | 3 | 14 | 8 | 3 |
Note:.
aPercentage Absorption was calculated as: % Absorption =109- [0.345xTopological Polar Surface Area].
bTopological polar surface area (defined as a sum of surfaces of polar atoms in a molecule).
cLogarithm of compound partition coefficient between n-octanol and water.
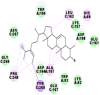
Table 5.
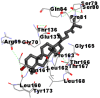
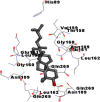
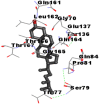
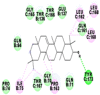
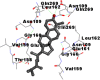
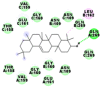
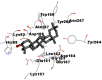
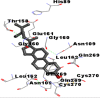
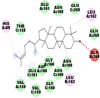
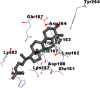
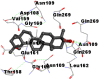
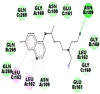
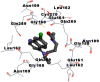
Best docking poses of active components of N. sativa with papain-like protease (PL-PRO) of SARS-CoV-2 (PDB ID: 6W9C) versus standard drugs hydroxychloroquine and ivermectin.
| S. No. | Ligands | AutoDock v4.2.6 | AutoDock Vina | iGEMDOCK v2.1 |
|---|---|---|---|---|
| 1. | 24-methylene-cycloartanol | 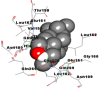 |
 |
 |
| 2. | Alpha-spinasterol | 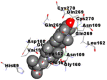 |
 |
 |
| 3. | Beta-amyrin | 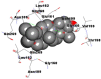 |
 |
 |
| 4. | Beta-sitosterol | 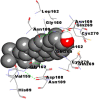 |
 |
 |
| 5. | Campesterol | 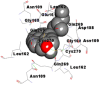 |
 |
 |
| 6. | Citrostadienol | 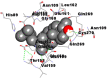 |
 |
 |
| 7. | Cycloartenol | 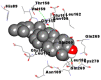 |
 |
 |
| 8. | Cycloeucalenol | 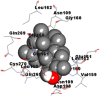 |
 |
 |
| 9. | Taraxerol | 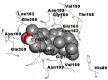 |
 |
 |
| 10. | Tirucallol | 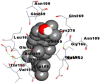 |
 |
 |
| 11. | Hydroxychloroquine | 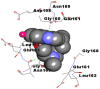 |
 |
 |
| 12. | Ivermectin | 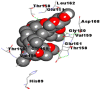 |
 |
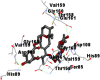 |
As far as interaction of cycloeucalenol with PL-PRO and alpha-spinasterol with NRBD is concerned, it is noteworthy that cycloeucalenol and alpha-spinasterol had almost similar binding sites on the two proteins as the two reference drugs viz. hydroxychloroquine and ivermectin. Differences in interacting amino acid residues in binding pockets are due to variations in the functional groups and basic chains of cycloeucalenol and alpha-spinasterol. This difference in SAR causes the variation in interacting amino acid residues. Binding studies of cycloeucalenol to PL-PRO and alpha-spinasterol to NRBD when compared with those of reference drugs hydroxychloroquine and ivermectin to the same proteins revealed that cycloeucalenol and alpha-spinasterol bind at or near the site where the reference drugs bind, albeit with greater affinity in both the cases (Tables 2 and 3).
3.2. PASS analysis of selected phytocomponents using Lipinski’s rule of five
Lipinski’s rule describes molecular properties of a compound which are important for lead optimization and selectivity of a potential orally active drug candidate in clinical applications. Table 6 shows the PASS analysis of all ten phytocomponents of N. sativa versus standard drugs hydroxychloroquine and ivermectin in terms of their physicochemical properties by applying Lipinski’s rule of five. Generally, an orally active compound should have no more than one Lipinski's violation otherwise its bioavailability is compromised. Interestingly, all 10 phytoconstituents from N. sativa exhibited only 1 Lipinski’s violation in comparison to hydroxychloroquine which did not show any Lipinski's violation. On the other hand, ivermectin displayed 3 violations of Lipinski’s rule of five.
3.3. Druglikeness and toxicity potential assessment
Table 7 depicts druglikeness and toxicity calculations of N. sativa phytoconstituents versus standard drugs hydroxychloroquine and ivermectin by OSIRIS data warrior. The results indicated that phytocomponents 24-methylene-cycloartanol, alpha-spinasterol, beta-amyrin, beta-sitosterol, campesterol, cycloeucalenol and taraxerol are safe to use with no predicted toxicity. However, citrostadienol and cycloartenol displayed irritant effects, while tirucallol exhibited adverse effect on the reproductive system. Hydroxychloroquine showed mutagenic effect but ivermectin did not show any predicted toxicity. A positive value obtained for alpha-spinasterol in druglikeness evaluation indicated that this molecule predominantly contains fragments present in commercial drugs. As expected, both hydroxychloroquine and ivermectin exhibited positive scores for druglikeness (Table 7).
Table 7.
Druglikeness and toxicity calculations of N. sativa phytoconstituents versus standard drugs hydroxychloroquine and ivermectin.
| Druglikeness and Toxicity parameters | ||||||
|---|---|---|---|---|---|---|
| S. No. | Compounds Name | Druglikeness | Mutant | Tumurogenic | Reproductive effective | Irritant |
| 1. | 24-Methylene-Cycloartanol | −9.2281 | N | N | N | N |
| 2. | Alpha-Spinasterol | 1.2217 | N | L | N | N |
| 3. | Beta-Amyrin | −2.4858 | N | N | N | N |
| 4. | Beta-Sitosterol | −4.475 | N | N | N | N |
| 5. | Campesterol | −8.1908 | N | N | N | N |
| 6. | Citrostadienol | −5.602 | N | N | N | H |
| 7. | Cycloartenol | −4.1078 | N | N | N | H |
| 8. | Cycloeucalenol | −7.633 | N | N | N | N |
| 9. | Taraxerol | −2.422 | N | N | N | N |
| 10. | Tirucallol | −4.1331 | N | N | H | H |
| 11. | Hydroxychloroquine | 5.7266 | H | N | N | N |
| 12. | Ivermectin | 5.2314 | N | N | N | N |
N- No toxicity.
L- Low toxicity.
H- High toxicity.
3.4. Bioactivity scores (BAS) of N. sativa phytoconstituents
The predicted BAS of all 10 phytoconstituents of N. sativa and their comparison with those of standard drugs are summarized in Table 8. As a general rule, a molecule having BAS >0.00 is most likely to possess considerable biological activities, while compounds having values between −0.50 and 0.00 are presumed to be moderately active and compounds having BAS < −0.50, are expected to be inactive. The results of the present study demonstrated that all N. sativa phytoconstituents are biologically active molecules because none of the phytoconstituents had bioactivity scores <-0.50. Thus, all N. sativa phytocomponents are capable of producing the physiological actions by multiple mechanisms after interacting with GPCR ligands, nuclear receptor ligands or by acting as inhibitors of proteases and other enzymes. All of the phytoconstituents displayed considerable activity as protease inhibitors as evident from their positive BAS of >0.00, except taraxerol which was found to be moderatively active as a protease inhibitor (BAS 0.00). Interestingly, most phytoconstiteuents showed potent binding to papain like protease of SARS-CoV-2 (PDB ID: 6W9C), thus supporting their role as potential viral protease inhibitors. On the other hand, ivermectin was predicted to be inactive as a protease inhibitor (BAS −1.89) and this was further validated with molecular docking analysis in which ivermectin exhibited 1000x less affinity for PL-PRO (Kd 224.79 µM) as compared to the N. sativa phytoconstituents which had their respective Kd with respect to SARS-CoV-2 PL-PRO in nM (Table 3). A similar trend was seen for the behavior of N. sativa phytoconstituents as nuclear receptor ligands (NRLs). All of them displayed positive BAS scores >0.00, which means they are expected to be considerably active as NRLs. The results were in agreement with the obtained docking scores for N. sativa phytoconstituents which had their respective Kd with respect to NRBD of SARS-CoV-2 nucleocapsid protein in nM (Table 2). Interestingly, ivermectin was predicted to be inactive as a NRL (BAS −2.94) whereas hydroxychloroquine was predicted to be moderately active as a NRL (BAS −0.12) and this was further validated with molecular docking analysis in which hydroxychloroquine exhibited 1000000x less affinity for nuclear receptor i.e. NRBD of SARS-CoV-2 nucleocapsid protein (Kd 1.25 mM) as compared to the N. sativa phytoconstituents which had their respective Kd in nM with respect to NRBD of SARS-CoV-2 nucleocapsid protein (Table 2). Though BAS as enzyme inhibitors were found to be >0.00 for all phytoconstituents, the highest score (0.66) was observed for citrostadienol and cycloartenol followed by tirucallol (0.64) and cycloeucalenol (0.61).
Table 8.
Bioactivity scores of N. sativa phytoconstituents versus standard drugs hydroxychloroquine and ivermectin.
| Parameters of bioactivity score (BAS) |
|||||||
|---|---|---|---|---|---|---|---|
| S. No. | Phytocomponents | GPCR Ligand | Ion Channel Modulator | Kinase Inhibitor | Nuclear Receptor Ligand | Protease Inhibitor | Enzyme Inhibitor |
| 1. | 24-Methylene-Cycloartanol | 0.14 | 0.11 | −0.37 | 0.90 | 0.06 | 0.60 |
| 2. | Alpha-Spinasterol | 0.18 | 0.05 | −0.30 | 0.68 | 0.06 | 0.53 |
| 3. | Beta-Amyrin | 0.22 | −0.05 | −0.31 | 0.67 | 0.11 | 0.56 |
| 4. | Beta-Sitosterol | 0.14 | 0.04 | −0.50 | 0.73 | 0.07 | 0.51 |
| 5. | Campesterol | 0.11 | 0.01 | −0.48 | 0.71 | 0.01 | 0.50 |
| 6. | Citrostadienol | 0.15 | 0.15 | −0.34 | 0.89 | 0.13 | 0.66 |
| 7. | Cycloartenol | 0.21 | 0.10 | −0.40 | 0.86 | 0.14 | 0.66 |
| 8. | Cycloeucalenol | 0.14 | 0.14 | −0.37 | 0.92 | 0.10 | 0.61 |
| 9. | Taraxerol | 0.21 | 0.02 | −0.20 | 0.54 | 0.00 | 0.49 |
| 10. | Tirucallol | 0.18 | −0.05 | −0.39 | 0.82 | 0.06 | 0.64 |
| 11. | Hydroxychloroquine | 0.35 | 0.30 | 0.44 | −0.12 | 0.12 | 0.15 |
| 12. | Ivermectin | −2.49 | −2.86 | −3.23 | −2.94 | −1.89 | −2.53 |
3.5. ADMET properties of phytocomponents
To check the pharmacokinetic feasibility of selected phytoconstituents from N. sativa as prospective drug candidates, their ADMET properties viz. absorption, distribution, metabolism, excretion and toxicity were calculated using online SwissADME software (Table 9). Based on the calculated LogP value, all components were found to be lipid soluble (lipophilic) which indicates good absorption of all components across skin. Interestingly, neither of the phytocomponents displayed blood-brain barrier (BBB) permeability nor were predicted to act as permeability-glycoprotein (P-gp) substrates. P-gp is an ATP-dependent bioavailability protein pump that removes drugs from biological systems. The normal excretion of drugs back into the gut lumen by P-gp decreases the pharmacokinetics and efficacy of pharmaceutical drugs (which are said to be P-gp substrates). Since none of the N. sativa phytoconstituents were found to behave as P-gp substrates, they may be expected to persist in the cells and show their intracellular pharmacological (antiviral) effect, since the virus is also an intracellular pathogen. Interestingly, some cancer and virally infected cells have been found to express large amounts of P-gp. On the other hand, ivermectin was predicted to behave as a P-gp substrate, thereby raising the possibility of development of drug resistance in the near future against ivermectin by SARS-CoV-2 infected cells.
Table 9.
ADMET properties calculated for N. sativa phytoconstituents versus standard drugs hydroxychloroquine and ivermectin.
| S.No. | Phytocomponents | Lipophilicity (Consensus Log Po/w) |
BBB permeant | P-gp substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | Log Kp (skin permeation) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1. | 24-methylene-cycloartanol | 7.79 | No | No | No | No | No | No | No | −1.67 cm/s |
| 2. | Alpha-spinasterol (spinasterol) | 6.88 | No | No | No | No | No | No | No | −2.92 cm/s |
| 3. | Beta-amyrin | 7.18 | No | No | No | No | No | No | No | −2.41 cm/s |
| 4. | Beta-sitosterol | 7.19 | No | No | No | No | No | No | No | −2.20 cm/s |
| 5. | Campesterol | 6.90 | No | No | No | No | No | No | No | −2.50 cm/s |
| 6. | Citrostadienol (alpha1-sitosterol) | 7.26 | No | No | No | No | No | No | No | −2.49 cm/s |
| 7. | Cycloartenol | 7.51 | No | No | No | No | No | No | No | −1.96 cm/s |
| 8. | Cycloeucalenol | 7.45 | No | No | No | No | No | No | No | −1.87 cm/s |
| 9. | Taraxerol | 7.22 | No | No | No | No | No | No | No | −2.30 cm/s |
| 10. | Tirucallol | 7.42 | No | No | No | No | No | No | No | −2.58 cm/s |
| 11. | Hydroxychloroquine | 3.37 | Yes | No | Yes | No | No | Yes | No | −5.81 cm/s |
| 12. | Ivermectin | 4.35 | No | Yes | No | No | No | No | No | −7.14 cm/s |
Cytochromes P450 (CYPs) are a superfamily of major metabolic enzymes involved in the biotransformation of xenobiotics. Drugs and other xenobiotics can act as both substrates and inhibitors of cytochromes P450 and they are involved in the metabolism of most medications. Drugs or compounds that inhibit the five classes of CYPs viz. CYP3A4, CYP1A2, CYP2C9, CYP2C19 and CYP2D6 would cause an increase in their plasma concentrations, thus contributing to improved bioavailability. In the present study, however, none of N. sativa phytocontistuents were found to act as inhibitors of the any of five classes of CYPs versus hydroxychloroquine which was found to act as inhibitor of CYP1A2 and CYP2D6 (Table 9). Skin permeability (Kp) is widely used to quantitatively describe the rate of chemical permeation through the outermost layer (epidermis) of the skin. Interestingly, all 10 phytocomponents showed negative Kp value which indicates less possibility of topical absorption of these phytoconstituents.
3.6. PCA of various phytoconstituents and standard drugs
PCA is a mathematical method to summarize multidimensional datasets into two or three principal components that can be visualized graphically with minimal loss of information. PCA was performed using OSIRIS Data Warrior v5.2.1 on three most variable properties viz. TPSA, %ABS and MW by applying linear correlation. The bar charts and 3D scatter plots of principal components to depict druglikeness of the phytoconstituents versus standard drugs were made in OSIRIS Data Warrior v5.2.1 and Discovery Studio Visualizer 2017 R2, respectively (Figure 1A and B). As depicted earlier, MWs of all the phytocomponents of N. sativa were <500 and, therefore, it can be expected that all the phytocomponents would be easy to transport, diffusible and readily absorbed. However, the MW of standard drug ivermectin was found to be >500 thereby showing serious limitations in its transportation, diffusion and absorption. As is evident from Figure 1A and B, all the phytoconstituents of N. sativa appear close to each other in scatter and 3D plot, which means that the N. sativa phytoconstituents have more or less similar properties in the context of TPSA, %ABS and MW versus standard reference drugs. Table 10 represents the Bravais-Pearson (linear correlation) coefficient of N. sativa phytoconstituents versus standard drugs. This type of matrix correlation represents ‘druglike’ property of the phytoconstituents.
Figure 1.

PCA of leadlikeness of N. sativa phytoconstituents versus antiviral standard drugs hydroxychloroquine and ivermectin (A) Scatter plot (B) 3 D point plot.
Table 10.
Bravais-Pearson (linear correlation) coefficient of N. sativa phytoconstituents versus standard drugs hydroxychloroquine and ivermectin.
| Properties | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % Ab | 1 | −1 | −0.927 | 0.739 | −0.908 | −0.958 | −1 | −0.496 | −0.597 | 0.992 | −0.126 | 2.74E-04 | |
| TPSA | 2 | −1 | 0.928 | −0.737 | 0.909 | 0.957 | 1 | 0.494 | 0.599 | −0.992 | 0.123 | 2.76E-04 | |
| MW | 3 | −0.927 | 0.928 | −0.449 | 1 | 0.784 | 0.912 | 0.273 | 0.848 | −0.967 | −0.255 | −2.04E-06 | |
| logP | 4 | 0.739 | −0.737 | −0.449 | −0.407 | −0.894 | −0.764 | −0.646 | 0.0812 | 0.654 | −0.717 | 0.175 | |
| natoms | 5 | −0.908 | 0.909 | 1 | −0.407 | 0.752 | 0.891 | 0.241 | 0.873 | −0.953 | −0.302 | 0.00361 | |
| nOHNH | 6 | −0.958 | 0.957 | 0.784 | −0.894 | 0.752 | 0.969 | 0.609 | 0.343 | −0.915 | 0.399 | −0.0561 | |
| nON | 7 | −1 | 1 | 0.912 | −0.764 | 0.891 | 0.969 | 0.514 | 0.565 | −0.986 | 0.164 | −0.00802 | |
| Rb | 8 | −0.496 | 0.494 | 0.273 | −0.646 | 0.241 | 0.609 | 0.514 | 0.0808 | −0.429 | 0.556 | 0.0925 | |
| LV | 9 | −0.597 | 0.599 | 0.848 | 0.0812 | 0.873 | 0.343 | 0.565 | 0.0808 | −0.691 | −0.705 | 0.156 | |
| pc1 | 10 | 0.992 | −0.992 | −0.967 | 0.654 | −0.953 | −0.915 | −0.986 | −0.429 | −0.691 | −1.23E-10 | 1.14E-09 | |
| pc2 | 11 | −0.126 | 0.123 | −0.255 | −0.717 | −0.302 | 0.399 | 0.164 | 0.556 | −0.705 | −1.23E-10 | 3.36E-09 | |
| pc3 | 12 | 2.74E-04 | 2.76E-04 | −2.04E-06 | 0.175 | 0.00361 | 0.0561 | −0.00802 | 0.0925 | 0.156 | 1.14E-09 | 3.36E-09 |
3.7. Structure activity relationship (SAR)
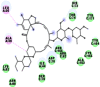
N. sativa contains various phytochemicals belonging to the class of terpenoids and flavonoids; the major ones have been listed in Table S1. Out of twenty five phytocomponents, ten components showed strong binding affinity with targeted proteins of SARS-CoV-2. Based on the structural relationship, all ten components can be divided into three parent configurations as shown in Figure 2. All compounds displayed a similar backbone structure with four rings arranged in a specific molecular configuration. This steroidal backbone is derived from sterol cycloartenol in plants cells. Cycloartenol is an important triterpenoid of the class sterol, which is the starting point for the synthesis of almost all plant steroids. Further, cycloartenol is derived from the cyclization of the triterpene squalene having molecular formula C30H48. In this study, the differential binding kinetics obtained for alpha-spinasterol (MF: C29H48O; MW: 412.7), beta-sitosterol (MF: C29H50O; MW: 414.7) and campesterol (MF: C28H48O; MW: 400.7) with N-terminal RNA binding domain (NRBD) of SARS-CoV-2 nucleocapsid protein (PDB ID: 6M3M) may be attributed to the variation in the number of alkyl groups (Figure 2A) in their backbone structures which might affect hydrogen bonding within the binding site of the targeted viral protein(s). Similarly, the variations in binding kinetics obtained for campesterol, cycloeucalenol (MF: C30H50O; MW: 426.7) and alpha-spinasterol with papain-like protease (PL-PRO; PDB ID: 6W9C) of SARS-CoV-2 may also be attributed to the variation in the number of alkyl side groups in their backbone structures which might affect hydrogen bonding within the binding pockets of the amino acid residues in target protein(s). In addition, another reason for differential SARs among various types of N. sativa phytoconstituents might be attributed to structural differences in alkene and cycloalkane groups along with spatial and stereochemical configurations of alkane groups, which cause the structural rearrangement as shown in Figure 2. These structural variations might be responsible for a better complementary fit of the phytocomponents in the binding pocket of the viral protein(s).
Figure 2.

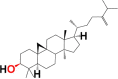
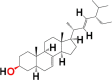
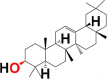
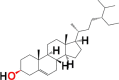
Structural differences in 10 phytocomponents of N. sativa divided into three groups: Group (A) (b) alpha-spinasterol, (d) beta-sitosterol, (e) campesterol, (f) citrostadienol and (j) tirucallol; Group (B) (c) beta-amyrin and (i) taraxerol; Group (C) (a) 24-methylene-cycloartanol, (g) cycloartenol and (h) cycloeucalenol.
3.8. Molecular dynamics (MD) simulation
MD simulation provides information about structural and conformational fluctuations over time and thermodynamics of biological molecules and their complexes. Figures 3 and 4 and Supplementary Figures S1, S2, S3 & S4 describe the MD simulation analysis of cycloeucalenol with PL-PRO (PDB ID: 6W9C) and alpha-spinasterol with NRBD of nucleocapsid protein (PDB ID: 6M3M) of SARS-CoV-2, respectively. The stability of both protein-ligand complexes was assessed through the trajectory analysis obtained through RMSD, RMSF, radius of gyration (Rg), and fraction of native contacts (Qx) analysis over a time frame of 4000ps (4 ns).
Figure 3.

RMSD values of SARS-CoV-2 PL-PRO (PDB ID: 6W9C) complexed with cycloeucalenol were analyzed as a function of time at 300 K. Values were calculated with the use of Cα atoms. (A) Ligand-protein conformation (B) RMDS of receptor and ligand (C) RMSD histogram of receptor (D) RMSD histogram of ligand (E) Radiation of Gyration- Rg value (F) Fraction of native contacts analysis of SARS-CoV-2 PL-PRO (PDB ID: 6W9C) with cycloeucalenol, over a time frame of 4000ps (4 ns) (G) B-factor value (changing from blue to red with increase in value) (H) RMSF value of each residue and (I) B-factor analysis of defined complex.
Figure 4.

RMSD values of NRBD of SARS-CoV-2 nucleocapsid protein (PDB ID: 6M3M) comlexed with alpha-spinasterol were analyzed as a function of time at 300 K. Values were calculated with the use of Cα atoms. (A) Ligand-protein conformation (B) RMDS of receptor and ligand (C) RMSD histogram of receptor (D) RMSD histogram of ligand (E) Radiation of Gyration- Rg value (F) Fraction of Native Contacts Analysis of SARS-CoV-2 NRBD of nucleocapsid protein (PDB ID: 6M3M) with alpha-spinasterol, over a time frame of 4000ps (4 ns) (G) B-factor value (changing from blue to red with increase in value) (H) RMSF value of each residue and (I) B-factor analysis of defined complex.
RMSD is the measurement of the average distance between the atoms of the overlaid structures. Often, equalized RMSD plots indicate that the system is in equilibrium. In the present study, MD simulation analyses showed a satisfactory stability profile at 300 K temperature.Cycloeucalenol-PL-PRO complex displayed very low deviation in RMSD from 1-3.6 Å throughout the 4 ns simulation (Figure 3). Likewise alpha-spinasterol with NRBD of SARSCoV-2 complex also exhibited less deviation in RMSD from 1-2.6 Å throughout the 4 ns time scale (Figure 4). Results from RMSD analysis of both complexes suggested that the deviation in the RMSDs was low; which indicated good stability and compactness of both protein-ligand complexes. The radius of gyration (Rg) of the body on the axis of rotation is considered to be the radial distance of a point from the axis of rotation. It is among the most significant indicators that are commonly used in the prediction of the structural activity and folding behavior of a macromolecule. Once the folding state of the protein is changed, the gyration radius would be affected. Rg of cycloeucalenol-PL-PRO of SARS-CoV-2 was found to be around 30.8, and it was 31.0 for alpha-spinasterol-NRBD of SARS-CoV-2 complex throughout the 4 ns simulations which suggested that there was little change in the compactness of the complex structure during the simulation indicating strong structural stability of both ligand-protein complexes (Figures 3E and 4E). Further, to calculate the average fluctuation of all residues during simulations, RMSFs of both target proteins were plotted using ligand-protein complexes. RMSF values are used to determine the atomic positional fluctuation of each residue via calculation based on the C-alpha (Cα) atom of them. The comparative analysis of RMSF trajectories revealed that all the residues in the complex model of cycloeucalenol-PL-PRO of SARS-CoV-2 fluctuated between 4-8 Å (Figure 3H), while in case of alpha-spinasterol with NRBD of SARS-CoV-2, RMSF value was found to fluctuate between 5-15 Å (Figure 4H). In the folding process, certain cases of non-native interactions are considered to be irrelevant and there are certain simulations as well as folding models which support that only native contacts are energetically favorable. Therefore, fraction of native contacts Q (x) helps in capturing the transition states remarkably well for all proteins along with a folding free energy barrier. In the present study, the Qx value was found to be larger than 95% in both complexes. This result indicates the relative flexibility and increased stability of both complexes throughout the simulation period (Figures 3F and 4F). B-factor, also termed as the temperature factor, which is similar to RMSF and is used to describe the attenuation of x-ray scattering resulting due to thermal motion. The result of B-factors of both complexes revealed that cycloeucalenol-PL-PRO of SARS-CoV-2 fluctuated around 2000 while alpha-spinasterol-NRBD of SARS-CoV-2 fluctuated around 5000 of B-factors, which indicated thermal stability of the complex.
PCA is used to detect nature of conformational differences, while magnitude of pairwise cross-correlation coefficients indicates system's atomic variations associated with each other. As shown in Figure S1 and S3, the correlated residues are blue colored whereas, non-correlated residues are in red. The light pink and light blue lines represent pairwise residues with higher correlated coefficient (>0.8) and with higher non-correlated coefficient (<-0.4). The schematic representation of secondary structures are present on the top and right margins of dynamic residue cross-correlation map appearing in black helices , grey strands and white loops . MM/PB(GB)SA result analysis mainly comprises electrostatic energy (ELE), van der Waals contribution (VDW), total gas phase energy (GAS), non-polar and polar contributions to solvation (PBSOL/GBSOL). The finally recorded binding free energy (deltaPB/deltaGB) is calculated from PBTOT/GBTOT and entropy (TS) as shown in Figures S2 A and S4 A. Further, hydrogen bond analysis includes the hydrogen bond acceptor and donor atoms, average distance (AvgDist), angle (AvgAng), and proportion (Frac) as shown in Figures S2 B and S4 B. Moreover, the results of decompose comprises of electrostatic energy which is calculated by the MM force field (TELE), van der Waals contribution from MM (TVDW), sum of non-polar and polar contributions to solvation (TGBSOL), total gas phase energy (TGAS) and final estimation of binding free energy from TGBTOT. Depending on the TGBTOT energy, the residues with contribution are ranked into top 10 decompose calculations which are arranged from top to bottom in heatmap (Figures S2 C and S4 C).
4. Discussion
Coronavirus disease (COVID-19) is an infectious pandemic that emerged from China and caused deadly outbreaks in USA, UK, Europe and middle east and far east countries including India and Pakistan (Hassan et al., 2020). The ongoing SARS-CoV-2 pandemic has spawned extensive research in identifying therapeutic targets and development of therapeutic drugs without any side effects. Although various antiviral and antimalarial drugs viz. umifenovir, remdesivir, lopinavir, favipiravir, ritonavir, ivermectin, hydroxychloroquine, chloroquine and azithromycin are being used currently to treat COVID-19 (Patil et al., 2020), however, these drugs cause various critical side effects including nervousness, poor concentration, nausea, vomiting, and diarrhoea. In addition, various risk factors such as the elderly and people suffering from pre-existing medical conditions like heart disease, respiratory disease or diabetes have a higher risk of dying from COVID-19 (https://www.drugs.com/condition/covid-19.html). Therefore, finding board-spectrum inhibitors that may reduce the effects of human corona virus infection along with reducing side effects remains a challenging research focus. Complementary and alternative medicine entails a variety of herbal plants, which may have potential for alternative drug development against COVID-19. The plant-derived products also act as immunomodulators without undesirable side effects. Further, virtual screening methods of ligand-protein interactions using computer-assisted drug design tools such as molecular docking and MD simulation analysis are the preliminary steps that lead to further development of potential therapeutic compounds in drug development process.
In the present study, an attempt has been made to explore the antiviral potential of selected phytochemicals of N. sativa belonging to the class of terpenoids and flavonoids against five target proteins of SARS-CoV-2 especially NRBD of nucleocapsid protein and PL-PRO using molecular and chemoinformatic tools and in silico approaches. For this study, twenty five phytocomponents were selected from black cumin seeds and oil based on their structure-activity relationships and prospective targeted metabolic pathways using Dr. Duke’s Phytochemical and Ethnobotanical Database. Based on the best binding energies and Kd of ligand-protein interactions with AutoDock v4.2.6, out of twenty five phytocomponents, ten active components with their targeted proteins viz. NRBD of nucleocapsid protein and PL-PRO of SARS-CoV-2 were selected for further validation using two other molecular docking tools viz. AutoDock Vina and iGEMDOCK v2.1. Interestingly, these two proteins play key roles in viral replication and assembly in host cells and, as such, can be used as therapeutic targets for antiviral drug discovery. Both PL-PRO and 3CL-PRO are involved in the processing of viral polyproteins (PPs) in a coordinated manner which is essential for viral replication. However, PL-PRO has the additional function of stripping ubiquitin and ubiquitin-like protein ISG15 (Interferon-stimulated gene 15) from host-cell proteins to help CoV to escape the host innate immune responses (Báez-Santos et al., 2015). Therefore, targeting PL-PRO with antiviral drugs might have an advantage in not only inhibiting viral replication but also inhibiting the dysregulation of signaling cascades in infected cells that might lead to cell death of surrounding uninfected cells. Nucleocapsid protein consists of three distinct but highly conserved parts: N terminal domain (NTD), Ser/Arg (SR)-rich central linker (CL) and C terminal domain (CTD). The most important function of NTD of nucleocapsid protein is RNA binding, while CTD acts as a dimerization domain, and, thus, also helps in packaging of SARS-CoV viral RNA into a long helical nucleocapsid structure or ribonucleoprotein (RNP) complex, which plays a fundamental role during viral self-assembly (Chang et al., 2014).
Results of molecular docking analysis using AutoDock v4.2.6 have shown that alpha-spinasterol (BE = −9.54 kcal/mol, Kd = 101.42 nM) has the best binding affinities with NRBD of SARS-CoV-2 followed by beta-sitosterol (BE = −8.69 kcal/mol, Kd = 426.43 nM) and campesterol (BE = −8.57 kcal/mol, Kd = 522.64 nM). However, taraxerol (BE = −12.1 kcal/mol, Kd = nM) and 24-methylene-cycloartanol (TE = −96.16 kcal/mol) exhibited best binding affinity as analyzed by AutoDock Vina and iGEMDOCK v2.1, respectively. In case of binding interaction with PL-PRO of SARS-CoV-2, campesterol (BE = −9.7 kcal/mol, Kd = 76.87 nM) has shown the best binding affinities followed by cycloeucalenol (BE = −9.65 kcal/mol, Kd = 84.23 nM) and alpha-spinasterol (BE = −9.41 kcal/mol, Kd = 126.53 nM) as analyzed by AutoDock v4.2.6. On the other hand, taraxerol (BE = −10.0 kcal/mol) and cycloeucalenol (TE = −102.07 kcal/mol) exhibited best binding affinity with PL-PRO as analyzed by AutoDock Vina and iGEMDOCK v2.1, respectively. Interestingly, this study is in agreement with a previously published report where steroidal glycoalkaloids from Solanum nigrum have shown similar kind of variation in binding energy with their selected protein targets (Ahmad, 2019). These results suggest that minor variations in binding affinity of the phytocomponents is because of the differences in the generation of grid boxes and determination of binding pockets on the target proteins by these softwares on account of slight differences in selection criteria. This has led to a difference in interacting amino acid residues in the binding pockets of NRBD of nucleocapsid protein and PL-PRO of SARS-CoV-2 as is evident from Tables 2 and 3. On the basis of their binding energies and Kd values, campesterol, alpha-spinasterol and cycloeucalenol have been found to be the most effective phytocomponents in N. sativa against SARS-CoV-2. In this study, hydroxychloroquine and ivermectin were selected as standard reference drugs since they have shown antiviral effects against several distinct negative-sense single-strand RNA viruses, including SARS-CoV-2 (Heidary & Gharebaghi, 2020; Rebeaud & Zores, 2020).
MD simulations are valuable methods for understanding the dynamic behavior of biological macromolecules at different timescales. RMSD is always non-negative, and a value of 0, although never achieved in practice, indicates a perfect fit to the data. In general, lower the RMSD, better the model is in comparison to the target structure. When a dynamic system fluctuates about a well-defined average position, the RMSD from the average over a time frame can be referred to as the RMSF. Interestingly, in the present study, the high affinity complexes viz. alpha-spinasterol with NRBD of nucleocapsid protein and cycloeucalenol with PL-PRO of SARS-CoV-2 complex displayed a very low deviation of 0.5-2.0 Å and 0.1-2.6 Å, respectively, throughout the 4 ns time scale (Figures 3 and 4), which indicated a good stability of both protein-ligand complexes. Moreover, results from RMSF, Rg, and Qx analyses over a time frame of 4 ns along with B-factors suggested the thermodynamic stability of both complexes.
In the drug discovery context, all ten active phytocomponents were tested for their druglikeness using Lipinski’s rule of five. Lipinski’s rule of five predicts that strong absorption/permeation is more likely when the MW <500, the calculated LogP (cLog P) ≤5.0, there are ≤5 H-bond donors and ≤10 H-bond acceptors. Generally, an orally active compound should have no more than one Lipinski's violation otherwise its bioavailability is compromised (Lipinski, 2004). Interestingly, all ten phytoconstituents from N. sativa exhibited only one Lipinski’s violation. Therefore, it can be postulated that all ten phytocomponents have the potential to be evaluated further from a drug development perspective. Further, toxicity potential assessment is essential for avoiding unsuitable substances for further drug screening in order to initiate in vitro and in vivo evaluation (Parasuraman, 2011). The phytocomponents were screened for their mutagenic, tumorigenic, irritant and reproductive toxicity risk assessment. Most of the phytocomponents such as 24-methylene-cycloartanol, alpha-spinasterol, beta-amyrin, beta-sitosterol, campesterol, cycloeucalenol and taraxerol were found to be safe with no predicted toxicity. Further, all phytocomponents displayed a lipophilic nature which indicated good absorption and transport kinetics through the gut. Principal component analysis revealed that all phytoconstituents of N. sativa fell close to each other and also near to standard drug hydroxychloroquine in scatter and 3D plots representing their drug like properties.
In conclusion, NRBD of nucleocapsid protein and PL-PRO of SARS-CoV-2 have been revealed as important drug targets for N. sativa phytoconstituents . As revealed in section 3.4 and discussion section, the selected phytoconstituents of N. sativa were found to behave as protease inhibitors going by their BAS scores of >0.00, thus, lending credibility to the selection of viral PL-PRO as a drug target in the present study. The most effective phytocomponents viz. campesterol, cycloeucalenol, alpha-spinasterol and beta-sitosterol exhibited high affinities against NRBD and PL-PRO of SARS-CoV-2. Owing to their bioavilability, druglikeness and almost zero toxic and mutagenic effects, these phytoconstituents can be explored further in vitro and in vivo as potential antiviral agents for the treatment of COVID-19 .
Supplementary Material
Acknowledgements
The authors gratefully acknowledge Dr. Meraj Ahmad, Professor, Dept. of Sociology, University of Lucknow, Lucknow, India, for being the inspiration and motivation behind the present work in view of his exceptional work and contribution in the area of social sciences, humanities and public health. Maqusood Ahamed is also grateful to the Researchers Supporting Project number (RSP- 2020/129), King Saud University, Riyadh, Saudi Arabia.
Glossary
Abbreviations
- ADMET
Absorption, Distribution, Metabolism, Excretion and Toxicity
- BE
Binding energy
- COVID-19
Coronavirus disease 2019
- Kd
Dissociation constant
- MD
Molecular Dynamics
- NRBD
N-terminal RNA binding domain
- PCA
Principal component analysis
- PL-PRO
Papain-like protease
- 3CL-PRO
3C-like protease
- SAR
Structure-activity relationship
- RMSD
Root mean square deviation
- RMSF
Root mean square fluctuation
- SARS-CoV-2
Severe acute respiratory syndrome coronavirus 2
Funding Statement
The authors gratefully acknowledge Dr. Meraj Ahmad, Professor, Dept. of Sociology, University of Lucknow, Lucknow, India, for being the inspiration and motivation behind the present work in view of his exceptional work and contribution in the area of social sciences, humanities and public health. Maqusood Ahamed is also grateful to the Researchers Supporting Project number (RSP- 2020/129), King Saud University, Riyadh, Saudi Arabia.
Disclosure statement
The authors declare that they have no competing interests.
References
- Ahmad, R. (2019, Jan). Steroidal glycoalkaloids from Solanum nigrum target cytoskeletal proteins: An in silico analysis. PeerJ., 7, e6012. 10.7717/peerj.6012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad, S., Abbasi, H. W., Shahid, S., Gul, S., & Abbasi, S. W. (2020, June). Molecular Docking, Simulation and MM-PBSA Studies of Nigella Sativa Compounds: A Computational Quest to identify Potential Natural Antiviral for COVID-19 Treatment. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1775129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad, A., Husain, A., Mujeeb, M., Khan, S. A., Najmi, A. K., Siddique, N. A., Damanhouri, Z. A., & Anwar, F. (2013, May). A review on therapeutic potential of Nigella sativa: A miracle herb. Asian Pacific Journal of Tropical Biomedicine, 3(5), 337–352. 10.1016/S2221-1691(13)60075-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad, R., Khan, M. A., Srivastava, A. N., Gupta, A., Srivastava, A., Jafri, T. R., Siddiqui, Z., Chaubey, S., Khan, T., & Srivastava, A. K. (2020, Jan). Anticancer Potential of Dietary Natural Products: A Comprehensive Review. Anti-Cancer Agents in Medicinal Chemistry, 20(2), 122–236. 10.2174/1871520619666191015103712 [DOI] [PubMed] [Google Scholar]
- Ahmad, Z, Ghafoor, A., & Aslam, M. (2004, May). Nigella sativa–A potential commodity in crop diversification traditionally used in healthcare. Project on Introduction of Medicinal Herb and Species as Crop. Ministry of Food, Agriculture and Livestock, Pakistan, 2, 6–10. [Google Scholar]
- Báez-Santos, Y. M., John, S. E., & Mesecar, A. D. (2015, Mar). The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antiviral Research, 115, 21–38. 10.1016/j.antiviral.2014.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar, I., Lezon, T. R., Bakan, A., & Shrivastava, I. H. (2010). Normal mode analysis of biomolecular structures: Functional mechanisms of membrane proteins. Chemical Reviews, 110(3), 1463–1497. 10.1021/cr900095e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caly, L., Druce, J. D., Catton, M. G., Jans, D. A., & Wagstaff, K. M. (2020, April). The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antiviral Research, 178, 104787. 10.1016/j.antiviral.2020.104787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C. K., Hou, M. H., Chang, C. F., Hsiao, C. D., & Huang, T. H. (2014, Mar). The SARS coronavirus nucleocapsid protein-forms and functions. Antiviral Research, 103, 39–50. 10.1016/j.antiviral.2013.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C. K., Lo, S. C., Wang, Y. S., & Hou, M. H. (2016). Recent insights into the development of therapeutics against coronavirus diseases by targeting N protein. Drug Discovery Today, 21(4), 562–572. Apr 1 10.1016/j.drudis.2015.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary, R., & Sharma, A. K. (2020, April). Potential use of hydroxychloroquine, ivermectin and azithromycin drugs in fighting COVID-19: Trends, scope and relevance. New Microbes and New Infections, 35, 100684. 10.1016/j.nmni.2020.100684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira, M. D., & de Oliveira, K. M. (2020). Comparative Computational Study of SARS-CoV-2 Receptors Antagonists from Already Approved Drugs. Cep, 11981(8), 7. [Google Scholar]
- Delaney, J. S. (2004, May). ESOL: Estimating aqueous solubility directly from molecular structure. Journal of Chemical Information and Computer Sciences, 44(3), 1000–1005. 10.1021/ci034243x [DOI] [PubMed] [Google Scholar]
- Forouzanfar, F., Bazzaz, B. S., & Hosseinzadeh, H. (2014, Dec). Black cumin (Nigella sativa) and its constituent (thymoquinone): A review on antimicrobial effects. Iranian Journal of Basic Medical Sciences, 17(12), 929–938. [PMC free article] [PubMed] [Google Scholar]
- Ghildiyal, R., Prakash, V., Chaudhary, V. K., Gupta, V., & Gabrani, R. (2020). Phytochemicals as Antiviral Agents: Recent Updates. InPlant-derived Bioactives. (pp. 279–295). Springer. [Google Scholar]
- Hassan, T. A., Hollander, S., van Lent, L., & Tahoun, A. (2020, April). Firm-level exposure to epidemic diseases: Covid-19, SARS, and H1N1. National Bureau of Economic Research. 10.36687/inetwp119 [DOI] [Google Scholar]
- Heidary, F., & Gharebaghi, R. (2020). Ivermectin: A systematic review from antiviral effects to COVID-19 complementary regimen. The Journal of Antibiotics, 73(9), 593–590. Jun 12: 10.1038/s41429-020-0336-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hospital, A., Goñi, J. R., Orozco, M., & Gelpí, J. L. (2015). Molecular dynamics simulations: Advances and applications. Advances and Applications in Bioinformatics and Chemistry : AABC, 8, 37–47. 10.2147/AABC.S70333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, T., Ahmad, R., Azad, I., Raza, S., Joshi, S., & Khan, A. R. (2018, Aug). Computer-aided drug design and virtual screening of targeted combinatorial libraries of mixed-ligand transition metal complexes of 2-butanone thiosemicarbazone. Computational Biology and Chemistry, 75, 178–195. 10.1016/j.compbiolchem.2018.05.008 [DOI] [PubMed] [Google Scholar]
- Li, F. (2016, Sep). Structure, function, and evolution of coronavirus spike proteins. Annual Review of Virology, 3(1), 237–261. 10.1146/annurev-virology-110615-042301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski, C. A. (2004, Dec). Lead- and drug-like compounds: the rule-of-five revolution . Drug Discovery Today. Technologies, 1(4), 337–341. 10.1016/j.ddtec.2004.11.007 [DOI] [PubMed] [Google Scholar]
- Liu, K., Tang, M., Liu, Q., Han, X., Jin, H., Zhu, H., Li, Y., He, L., Ji, H., & Zhou, B. (2020, Mar). Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discovery, 6(1), 1–4. 10.1038/s41421-020-0156-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menounos, P., Staphylakis, K., & Gegiou, D. (1986, Jan). The sterols of Nigella sativa seed oil. Phytochemistry, 25(3), 761–763. 10.1016/0031-9422(86)88046-3 [DOI] [Google Scholar]
- Morris, G. M., Goodsell, D. S., Halliday, R. S., Huey, R., Hart, W. E., Belew, R. K., & Olson, A. J. (1998, Nov). Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry, 19(14), 1639–1662. [DOI] [Google Scholar]
- Naithani, R., Mehta, R. G., Shukla, D., Chandersekera, S. N., & Moriarty, R. M. (2010). Antiviral activity of phytochemicals: A current perspective. In Dietary Components and Immune Function. (pp. 421–468). Humana Press. [Google Scholar]
- Oprea, T. I., Davis, A. M., Teague, S. J., & Leeson, P. D. (2001, Sep). Is there a difference between leads and drugs? A historical perspective. Journal of Chemical Information and Computer Sciences, 41(5), 1308–1315. 10.1021/ci010366a [DOI] [PubMed] [Google Scholar]
- Pal, M., Berhanu, G., Desalegn, C., & Kandi, V. (2020, Mar). Severe acute respiratory syndrome Coronavirus-2 (SARS-CoV-2): An update. Cureus, 12(3),e7423. 10.7759/cureus.7423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parasuraman, S. (2011, Apr). Toxicological screening. Journal of Pharmacology & Pharmacotherapeutics, 2(2), 74–79. 10.4103/0976-500X.81895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil, S., Hofer, J., Ballester, P. J., Fattakhova, E., DiFlumeri, J., Campbell, A., & Oravic, M. (2020). Drug Repurposing for Covid-19: Discovery of Potential Small-Molecule Inhibitors of Spike Protein-ACE2 Receptor Interaction Through Virtual Screening and Consensus Scoring. 10.26434/chemrxiv.12482435.v1 [DOI]
- Prajapat, M., Sarma, P., Shekhar, N., Avti, P., Sinha, S., Kaur, H., Kumar, S., Bhattacharyya, A., Kumar, H., Bansal, S., & Medhi, B. (2020, Jan). Drug targets for corona virus: A systematic review. Indian Journal of Pharmacology, 52(1), 56–65. 10.4103/ijp.IJP_115_20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot, A. E. (2002). Chemokine receptors: Multifaceted therapeutic targets. Nature Reviews. Immunology, 2(2), 106–115. Feb 10.1038/nri722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebeaud, M. E., & Zores, F. (2020, Apr). SARS-CoV-2 and the Use of Chloroquine as an Antiviral Treatment. Frontiers in Medicine, 7, 184. 10.3389/fmed.2020.00184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riou, J., & Althaus, C. L. (2020, Jan). Pattern of early human-to-human transmission of Wuhan 2019 novel coronavirus (2019-nCoV), December 2019 to January 2020. Eurosurveillance, 25(4), 2000058. 10.2807/1560-7917.ES.2020.25.4.2000058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem, M. L., & Hossain, M. S. (2000, Sep). Protective effect of black seed oil from Nigella sativa against murine cytomegalovirus infection. International Journal of Immunopharmacology, 22(9), 729–740. 10.1016/S0192-0561(00)00036-9 [DOI] [PubMed] [Google Scholar]
- Tian, S., Wang, J., Li, Y., Li, D., Xu, L., & Hou, T. (2015, Jun). The application of in silico drug-likeness predictions in pharmaceutical research. Adv Drug Deliv Rev, 86, 2–10. 10.1016/j.addr.2015.01.009 [DOI] [PubMed] [Google Scholar]
- Touret, F., & de Lamballerie, X. (2020, Mar). Of chloroquine and COVID-19. Antiviral Research, 177, 104762. 10.1016/j.antiviral.2020.104762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott, O., & Olson, A. J. (2010, Jan). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls, A. C., Park, Y. J., Tortorici, M. A., Wall, A., McGuire, A. T., & Veesler, D. (2020, Mar). Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell, 181(2), 281–292.e6. 10.1016/j.cell.2020.02.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. M., & Chen, C. C. (2004, May). GEMDOCK: a generic evolutionary method for molecular docking. Proteins, 55(2), 288–304. 10.1002/prot.20035 [DOI] [PubMed] [Google Scholar]
- Yang, W., Chen, X., Li, Y., Guo, S., Wang, Z., & Yu, X. (2020, Mar). Advances in Pharmacological Activities of Terpenoids. Natural Product Communications, 15(3), 1934578X2090355. 10.1177/1934578X20903555 [DOI] [Google Scholar]
- Yang, J. F., Wang, F., Chen, Y. Z., Hao, G. F., & Yang, G. F. (2019, Dec). LARMD: Integration of bioinformatic resources to profile ligand-driven protein dynamics with a case on the activation of estrogen receptor. Briefings in Bioinformatics, 21(6), 2206-2218. 10.1093/bib/bbz141 [DOI] [PubMed] [Google Scholar]
- Yarnell, E., & Abascal, K. (2011, Apr). Nigella sativa: Holy herb of the middle East. Alternative and Complementary Therapies, 17(2), 99–105. 10.1089/act.2011.17203 [DOI] [Google Scholar]
- Yimer, E. M., Tuem, K. B., Karim, A., Ur-Rehman, N., & Anwar, F. (2019, May). Nigella sativa L.(black cumin): a promising natural remedy for wide range of illnesses. Evidence-Based Complementary and Alternative Medicine : eCAM, 2019, 1528635. 10.1155/2019/1528635 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.