

Abstract
Profiling genetic variants—including single nucleotide variants, small insertions and deletions, copy number variations and structural variations (SVs)—from both healthy and diseased individuals is a key component of genetic and biomedical research. SVs are large-scale changes in the genome and involve breakage and rejoining of DNA fragments. They may affect thousands to millions of nucleotides, and can lead to loss, gain and reshuffling of genes as well as regulatory elements. SVs are known to impact gene expression and potentially resulting in altered phenotypes and diseases. Therefore, identifying SVs from human genomes is particularly important. In this review, I describe advantages and disadvantages of the available high throughput assays for the discovery of SVs, which are the most challenging genetic alterations to detect. A practical guide is offered to suggest the most suitable strategies of discovering different types of SVs including common germline variants, rare variants, somatic variants and complex variants. I also discuss factors to be considered, such as cost and performance, for different strategies when designing experiments. Lastly, I present several approaches to identify potential SV artifacts caused by samples, experimental procedures and computational analysis.
Keywords: Genomic rearrangements, chromothripsis, next-generation sequencing, single-molecule sequencing
Introduction
Structural variations (SVs) are an important class of genetic variants in the human genome (Feuk et al., 2006; Stankiewicz and Lupski, 2010). They include deletions, duplications, insertions, inversions, translocations and other more complex forms (Figure 1). Some SVs change the dosage of DNA, such as deletions and duplications, and are also considered as copy number variations (CNVs); while others, such as inversions and balanced translocations, do not change the DNA dosage. The major difference between SVs and CNVs is that SVs always involve breakage and rejoining of DNA fragments. Hence, events like whole chromosomal gains and losses are not considered as SVs. In a given human genome, the germline SVs typically affect an order of magnitude more nucleotides than single nucleotide polymorphisms (SNPs) (Mills et al., 2011). The size of SV can range from dozens to millions of base pairs. Conventionally, SVs with less than 50bp in size are called small insertion/deletions (indels). The strategies of indel discovery are very different from large SVs (Mullaney et al., 2010; Xu, 2018). In this guide, we will only discuss SVs that are larger than 50bp.
Figure 1.
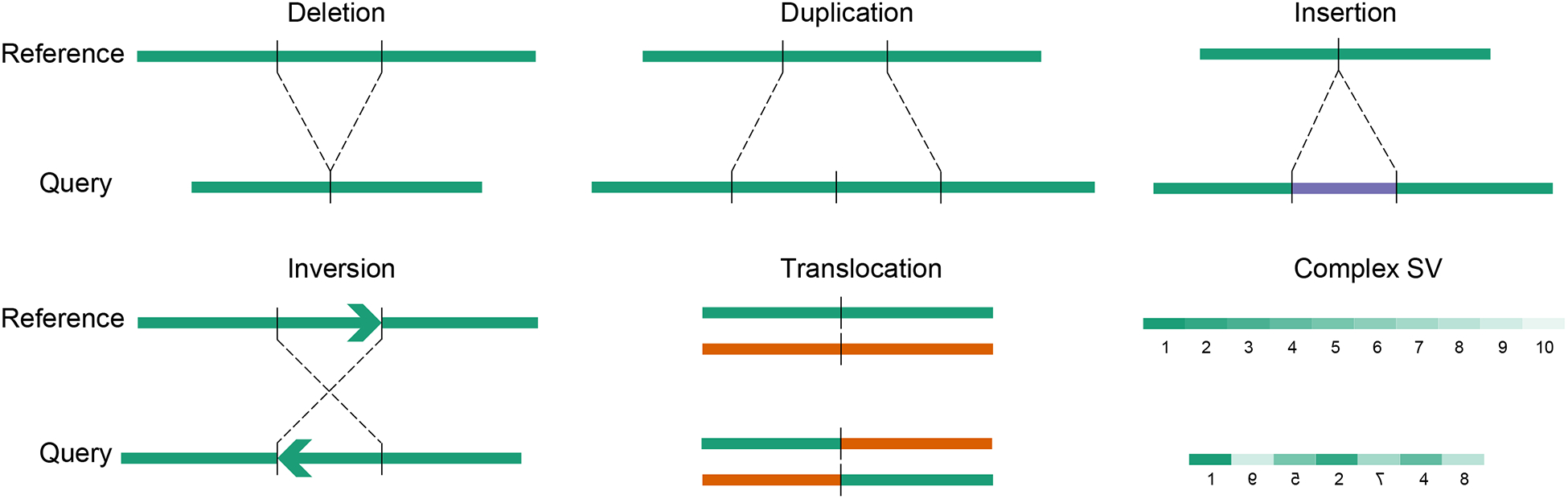
Scheme of different types of SVs.
Other than the simple forms of SVs, more complex SVs are also frequent in normal individuals as well as in patients with genetic disorders, such as duplication-inverted triplication-duplication (Carvalho et al., 2011), insertion or inversion inside deletion (Kidd et al., 2010; Yang et al., 2013), templated insertion (Li et al., 2020), etc. Recently, an extremely complex form of SVs was described in cancer called chromothripsis, in which dozens to hundreds of breakpoints on one or a few chromosomes are involved (Stephens et al., 2011). Such event is considered to occur at one time rather than many simple SVs being accumulated over a long period of time. Chromothripsis was originally reported in many different types of cancers (Stephens et al., 2011; Molenaar et al., 2012; Rausch et al., 2012a), and was also found in germline genomes causing developmental disorders and neuronal disorders (Liu et al., 2011; Chiang et al., 2012). Similar complex events have been named chromoanasythesis (Liu et al., 2011), chromoanagenesis (Holland and Cleveland, 2012) and chromoplexy (Baca et al., 2013) in various contexts.
SVs are usually caused by erroneous DNA replication and DNA damage repair as well as activities of repetitive elements (Cordaux and Batzer, 2009; Lee et al., 2012). A number of molecular mechanisms are known to form SVs in the germline and somatic cells (Hastings et al., 2009; Bunting and Nussenzweig, 2013). These include nonhomologous end joining (NHEJ), alternative end joining (alt-EJ), non-allelic homologous recombination (NAHR), single-strand annealing (SSA), break-induced replication (BIR), fork-stalling and template switching (FoSTeS), etc. Chromothripsis is known to be induced by micronuclei formed via chromosomal segregation error (Zhang et al., 2015a), chromatin bridge due to telomere attrition (Maciejowski et al., 2015), template switching at stalled replication fork (Liu et al., 2011; Yang et al., 2013) and breakage-fusion-bridge (BFB) cycles (Li et al., 2014).
There are two main impacts of SVs: change of DNA dosage and change of DNA order. Firstly, the gains and losses of important genes and regulatory elements due to SVs will impact phenotype and cause diseases (Stankiewicz and Lupski, 2010; Weischenfeldt et al., 2013). The dosage changes of genes and their contributions to diseases have been extensively studied (Zhang et al., 2009; Stankiewicz and Lupski, 2010; Tang and Amon, 2013). Recently, it has been reported that duplications or deletions of enhancers and super-enhancers lead to misregulation of their target genes (MYC, AR, SOX9 and KLF5) and cause diseases such as cancer and sex development disorders (Zhang et al., 2015b; Takeda et al., 2018; Croft et al., 2018; Zhang et al., 2018a). Secondly, the SVs can reorganize the DNA contents and connect two distal fragments together. This will lead to gene fusions and chimeric proteins when two distinct genes are joined into one. Gene fusions are often major cancer-driving events, especially in pediatric cancers and liquid tumors (Mertens et al., 2015). Moreover, the interactions between genes and regulatory elements can also be altered by SVs. A number of oncogenes, such as MYC, BCL2, EVI1, TERT, GFI1, etc., are activated by distal enhancers through somatic SVs (Boxer and Dang, 2001; Gröschel et al., 2014; Davis et al., 2014; Northcott et al., 2014; Valentijn et al., 2015). When enhancer-gene interactions are rewired by various types of SVs such as deletion, duplication or inversion around the WNT6/IHH/EPHA4/PAX3 locus, the misregulated genes can lead to different forms of limb malformation (Lupiáñez et al., 2015). Duplications of different regions near SOX9 can cause sex reversal or limb malformation depending on the types of newly formed gene-enhancer interactions (Franke et al., 2016). Furthermore, inherited rare SVs in cis-regulatory elements are found to be associated with autism (Brandler et al., 2018).
Methods overview
Here, I briefly describe the methods used for SV discovery. The main goal for these methods is to detect unknown SVs. The methods to confirm if a particular SV exist in a genome or not are described in the last section “Validation and genotyping”.
1. Cytogenetics
Cytogenetic testing used to be routinely performed on diagnosis and screening for genetic diseases. Recurrent translocations were found in cancer by cytogenetic analysis decades ago. Karyotyping (Figure 2A) is the most common cytogenetic testing technique (Wan, 2014). Dividing cells are required to view condensed chromosomes at metaphase. Chromosomes are stained or colored probes are hybridized to chromosomes. The chromosomal banding patterns are visualized with microscope. Only large SVs visible under the microscope can be detected, such as deletions, duplications and inversions that are at least 5 Mb in size as well as translocations. Mosaic events (SVs exist in a subset of cells but not all cells) are less likely to be detected. The SV breakpoints are not precise. Complex SVs are typically not detectable. Nowadays, karyotyping is rarely used for SV discovery due to its low sensitivity and precision.
Figure 2.
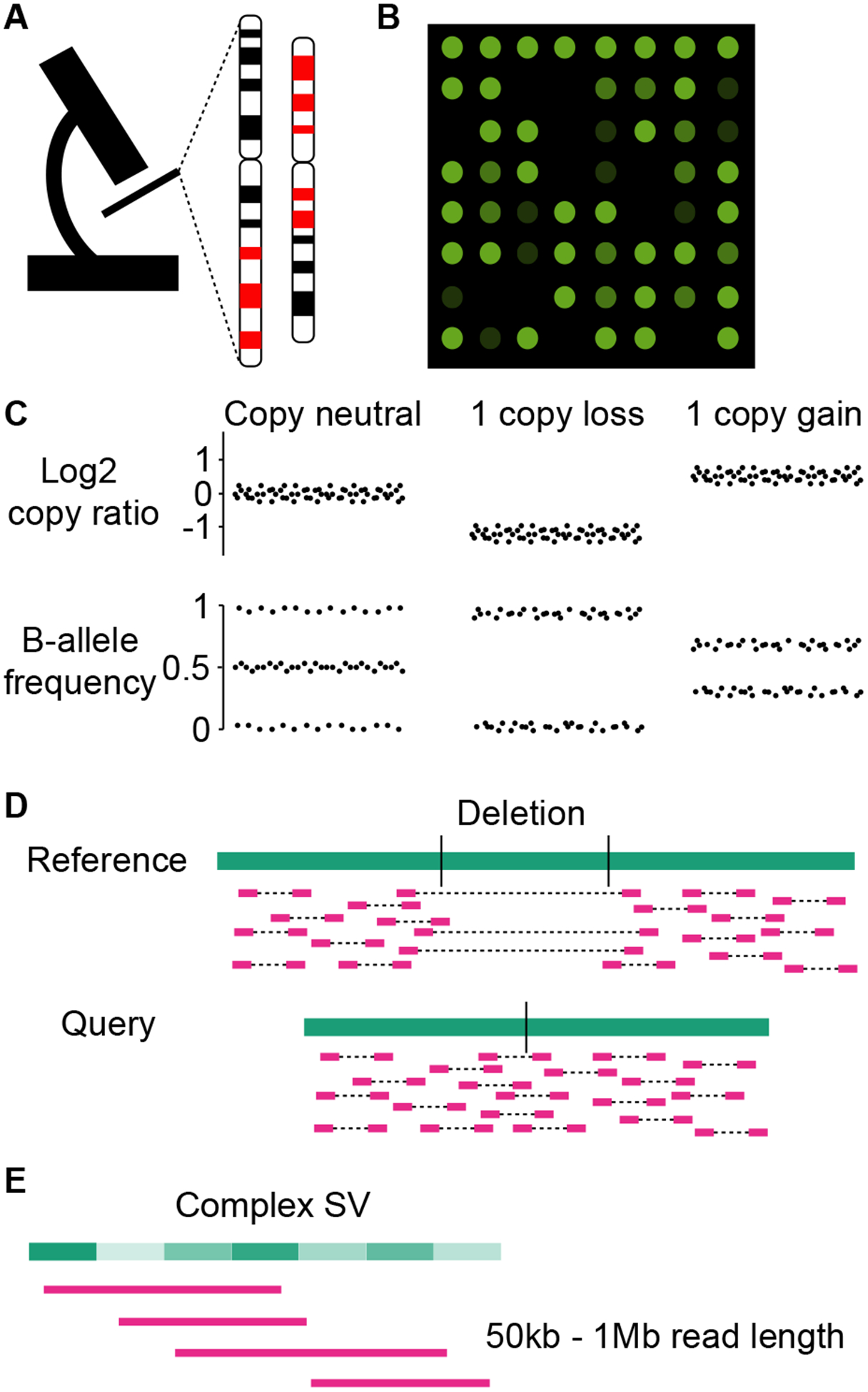
SV detection platforms. A, Karyotyping. B, DNA microarray. C, Scheme of copy ratio and B-allele frequency profiles for copy neutral, one-copy loss and one-copy gain regions. D, Short-read sequencing. E, Long-read sequencing.
2. Microarray
DNA microarray (Figure 2B) is an advanced version of florescent in situ hybridization (FISH) described in Section 5 where probes are designed to hybridize with DNA and the readout is florescent signal (Heller, 2002; Bumgarner, 2013). On a microarray, thousands to millions of probes are printed on a very dense surface. The intensity of florescent signal represents the amount of DNA that can hybridize to the probes. This feature can be used to quantify the copy number of DNA (Figure 2C top panel). Microarray-based Comparative Genomic Hybridization (array CGH) is particularly designed to detect CNVs (Pinkel and Albertson, 2005; Lockwood et al., 2006). SNP genotyping array can also be used to measure DNA copy number as well (Schaaf et al., 2011; Lin et al., 2013). The current cost for human DNA microarray ranges from $100 to $500 per sample depending on the probe density. For SNP array, in addition to probe hybridization intensity, the minor-allele (B-allele) frequencies can also be used to infer CNVs. For example, in copy neutral regions, the germline heterozygous SNPs should have B-allele frequencies of 0.5. In one-copy loss regions, the B-allele frequencies will become 0 or 1. Similarly, they will be 0.33 and 0.67 in one-copy gain regions. Since there is always noise in the florescent signal, CNVs are typically called when there are several consecutive probes supporting the events. CNVs less than 50kb in size are usually undetectable from microarray. The breakpoints cannot be precisely determined. In addition, balanced SVs are not detectable because there is no DNA dosage change. Due to its high throughput nature, microarray was widely used to study CNVs in normal population (Conrad et al., 2005; McCarroll et al., 2008) as well as many diseased individuals (Zack et al., 2013; Sebat et al., 2007; Bochukova et al., 2010). Although microarray has been mostly replaced by second-generation sequencing in scientific research, it is still commonly used in clinical diagnosis for genetic disorders.
3. Second-generation sequencing
More than 10 years ago, a number of second-generation sequencing technologies became available, including pyrosequencing (Roche 454), sequencing by synthesis (Illumina/Solexa), sequencing by ligation (Life Technologies SOLiD), nanoball sequencing (Complete Genomics/BGI), Ion Torrent sequencing (Life Technologies), etc. Over the years, the sequencing quality has improved significantly while the cost continues to drop. Whole-genome sequencing (WGS) has become quite affordable and enabled SV detection to the base pair resolution (Figure 2D). The current cost for library preparation and Illumina sequencing (150bp paired-end) of a 30x human genome is between $800 to $1,300. In second-generation sequencing experiments, genomic DNA is first shredded into small fragments and then sequenced from both ends of the DNA molecules (paired-end sequencing). Sequencing reads are typically short (<500bp). With high coverage WGS, since all genomic regions are sequenced, theoretically, all SVs may be detected including balanced SVs and complex events. However, due to the limitation of short read-length and the repetitive nature of the human genome, SVs in repetitive regions and segmental duplicated regions remain difficult to identify. Furthermore, sampling bias and gaps in reference genome will also affect SV detection. Fresh and fresh-frozen samples are recommended to study SVs for both second- and third-generation platforms. In formalin fixed paraffin embedded (FFPE) samples, DNA is highly degraded and chimeric molecules are abundant. There will be many artifact SVs detected. Therefore, FFPE samples are generally not recommended for SV discovery.
4. Third-generation sequencing/imaging
Third-generation sequencing technologies feature long reads. PacBio Single-molecule real-time (SMRT) sequencing passes single-strand DNA through an immobilized DNA polymerase to detect florescent light. Oxford Nanopore detects bases via ion current when DNA or RNA passes through the protein nanopores. Both technologies can produce tens of kb to Mb sequences at the single molecule level without amplifying the templates. The cost for a 30x human genome sequenced by both PacBio continuous long reads (CLR) and Nanopore PromethION including library preparation ranges between $3,000 to $5,000. A major drawback of long-read sequencing is the high error rates (15% for PacBio and 30% for Nanopore). Hence, long-read sequencing technologies by themselves are great for SV discovery but not optimal for detection of single nucleotide variants (SNVs) or small indels. To overcome the high error rate, the latest HiFi reads from PacBio substantially improved the base quality by sequencing the same molecules multiple times. The cost of a 30x human genome with PacBio HiFi sequencing is about $15,000. An alternative approach is linked-read sequencing offered by 10X Genomics. Long DNA molecules are embedded in individual microfluidic droplets called Gel-bead in EMulsion (GEMs) containing unique barcodes. DNA fragments are sequenced by Illumina short-read sequencing platform and the barcodes can be used to link short reads into longer contigs. The cost of linked-read sequencing is about $2,000 for a 30x human genome. In addition, microscopic imaging can provide genomic information through very long range as well. The genome mapping technology offered by BioNano Genomics labels specific sequence motifs in the genome and scan these labels by imaging. The maps of sequence motifs can then be used for SV detection and genome assembly. However, the resolution of SV breakpoints in optical mapping is much lower than sequencing based approaches. The cost of Bionano Optical Mapping is about $1,500 for a human genome. All of the third-generation sequencing and imaging technologies can overcome the major limit of second-generation short-read sequencing. The long-range information is ideal to resolve complex SVs as well as SVs in repetitive regions (Figure 2E). Note that the third-generation sequencing and imaging platforms depends on high molecular weight (HMW) DNA so that the long DNA molecules can be sequenced or imaged. The HMW DNA extraction costs another $500 to $1,000 per sample on top of the library preparation and sequencing costs. Fresh or fresh-frozen samples are recommended for third-generation sequencing and imaging.
5. Validation and genotyping
Validation and genotyping are often performed to confirm the presence of SVs in the query samples or new samples. Usually, validation needs to be performed using an orthogonal method. The widely used methods are PCR/Sanger sequencing, florescent in situ hybridization (FISH), or another sequencing platform that differs from the variant discovery platform. Genotyping can be done by PCR/Sanger sequencing, low coverage WGS or targeted sequencing.
Study design
Both karyotyping and microarray are rarely used for SV discovery any more due to their cost inefficiency, labor intensity and low throughput. Sequencing and imaging have become the go-to choices. According to the brief introduction in the previous section, clearly third-generation sequencing/imaging technologies are superior, but often cost more. Therefore, in a specific study, researchers always need to find the balance between the cost and performance. The performance often depends on the choice of platform and sequencing coverage. Data analysis will certainly play important role as well. Here, I provide some recommendations on different types of variants.
1. CNV detection
If detecting CNV is the major goal, and balanced SVs as well as the precise breakpoints do not matter, low-coverage short-read WGS will be the most cost-efficient method. We have shown that at 2x coverage, Illumina WGS can achieve comparable or even better accuracy and precision than SNP array (TCGA Network, 2012). The sequencing cost will be much less than microarray. However, with such coverage, other types of variants, such as SNVs, cannot be detected. In addition, the breakpoints are not at the base pair resolution.
2. Common germline variants
SVs including CNVs are known to have more drastic effects on gene expression (Stranger et al., 2007; Chiang et al., 2017). Microarray has been used for genome-wide association studies (GWAS) on common germline CNVs (Craddock et al., 2010; Glessner et al., 2009). Common SVs can be used to study their associations with diseases as well. Low-coverage short-read WGS is again a cost-efficient choice for common variant detection. The rationale is that common variants are shared in population. When sequencing many individuals, the signal can be combined for variant discovery. The 1000 Genomes Project has used this strategy to interrogate SVs and the software Genome STRiP was designed for this particular purpose (Handsaker et al., 2011). Once the variants are called, the same sequencing data can be used to genotype the variants in all individuals.
3. De novo variants and rare germline variants
De novo variants are the ones not inherited from parents, and likely occur during germ cell formation of the parents. Rare germline variants are inherited from parents, but the allele frequencies are very low in the general population. Both of these variants may be associated with diseases (Bochukova et al., 2010; Georgieva et al., 2014; Redin et al., 2017). Since rare variants and de novo variants are typically not shared between individuals, their discovery must rely on deep sequencing. High coverage (>20x) short-read WGS is very powerful to detect SVs in non-repetitive regions. However, resolving SVs in repetitive regions is very challenging. A recent study using very high coverage (average depth 65x) PacBio long-read sequencing platform reported that as much as 87% of the germline SVs discovered by long-read sequencing cannot be detected by short-read sequencing (Audano et al., 2019). Most of the SVs missed by short-read sequencing are associated with variable number of tandem repeats (VNTRs). Larger-scale efforts to elucidate the full spectrum of SVs in human genome by integrating multiple sequencing platforms are ongoing. For individual studies, such as to identify disease-associated variants, it is recommended to perform second-generation sequencing initially. If there are reasons to believe the pathogenic SVs are located in the repetitive regions, third-generation sequencing technologies may be considered to extend the search.
4. Mosaic variants and somatic variants
Mosaic variants and somatic variants both refer to variants that occur during cell divisions after the formation of fertilized eggs. Therefore, in an individual, only a subset of his/her cells carry such variants in contrast to germline variants and de novo variants being present in all cells. It is widely accepted that all somatic cells carry somatic mutations to some extent because DNA polymerase cannot replicate DNA 100% accurately. In every cell cycle, a small number of point mutations are accumulated. Sometimes, some cells will acquire growth advantage, expand the cell population (Martincorena et al., 2015; Blokzijl et al., 2016; Lodato et al., 2018), and even become cancerous. Tumor cells from a cancer patient are mostly derived from a common ancestor cell, although in some cases, it is possible for tumors to develop with multiple independent origins. Somatic variants acquired by the common ancestor cell are shared by all tumor cells. These variants are called clonal variants. During the clonal expansion, additional somatic variants occur. The ones occur after the major clonal expansion and not shared by all tumor cells are subclonal variants, which may play important roles in tumor evolution and drug response (Schmitt et al., 2016). Mosaic variants and somatic variants are almost always private and not present in other individuals. Even for the highly frequent pathogenic SVs, such as translocations leading to BCR-ABL1 fusion, the precise locations of translocation junctions are clustered, but still differ between patients (Groffen et al., 1984). So pooling data from multiple individuals will not be helpful for SV discovery. The difficulty in SV detection caused by repetitive regions described previously also applies to mosaic and somatic variants. So, the rationale to choose between short-read and long-read technologies presented in the previous paragraph also applies here. A major issue that is specific to mosaic and somatic variants is sequencing depth. Typically, 30x sequencing coverage is sufficient to confidently detect 99% of the heterozygous variants that are present in all cells. However, for variants present in half of the cells, 60x coverage will be needed to achieve the same sensitivity. As a proof-of-concept, we performed a simple simulation to test how sequencing coverage affects SV detection (Figure 3). Various types of SVs as well as Illumina sequencing reads are simulated, and SVs are called by Meerkat (Yang et al., 2013). As expected, at 30x coverage, almost all SVs are detectable. At 20x coverage, about 80% of the SVs remain detectable. When the coverage is lower than 10x, more than half of the SVs are missed. The undetected SVs are mostly due to the lack of supporting reads. Similar trend was observed in SNV calling by down-sampling a 410x WGS dataset (Kishikawa et al., 2019). Our simulation reflects the best-case scenario. Other factors such as repeats, sequencing bias, sequencing error, chimeric molecules formed during library preparation, etc., are not considered. When deciding sequencing depth, aneuploidy, which is a hallmark of cancer, should be taken into account as well. Many chromosomes in tumor cells have more than two copies. With the same sequencing coverage, the reads spanning the variants in aneuploid chromosomes will be less if the variants are only present on only one copy of the chromosomes. Due to the existence of subclonal variants, in tumor sequencing studies, it is obvious that more somatic variants can always be identified with higher sequencing depth. The choice of sequencing coverage will depend on the importance of the subclonal variants. If the goal is to find cancer-driving events and actionable variants, it will not be necessary to sequence very deep, since the variants present in a very small fraction of cells are probably not the major drivers of the disease. It is recommended to select a coverage that allows 80% of the variants to be detectable. For example, if a tumor has a purity of 50% and is known to be aneuploid, 60x coverage would offer enough depth to detect the majority of clonal SVs and a good portion of subclonal variants. If the main goal is to interrogate the very low frequency variants, it is recommended to test very high coverage, such as 200x, in one or two samples, and down-sample the reads to see if a lower coverage can achieve satisfying results.
Figure 3.
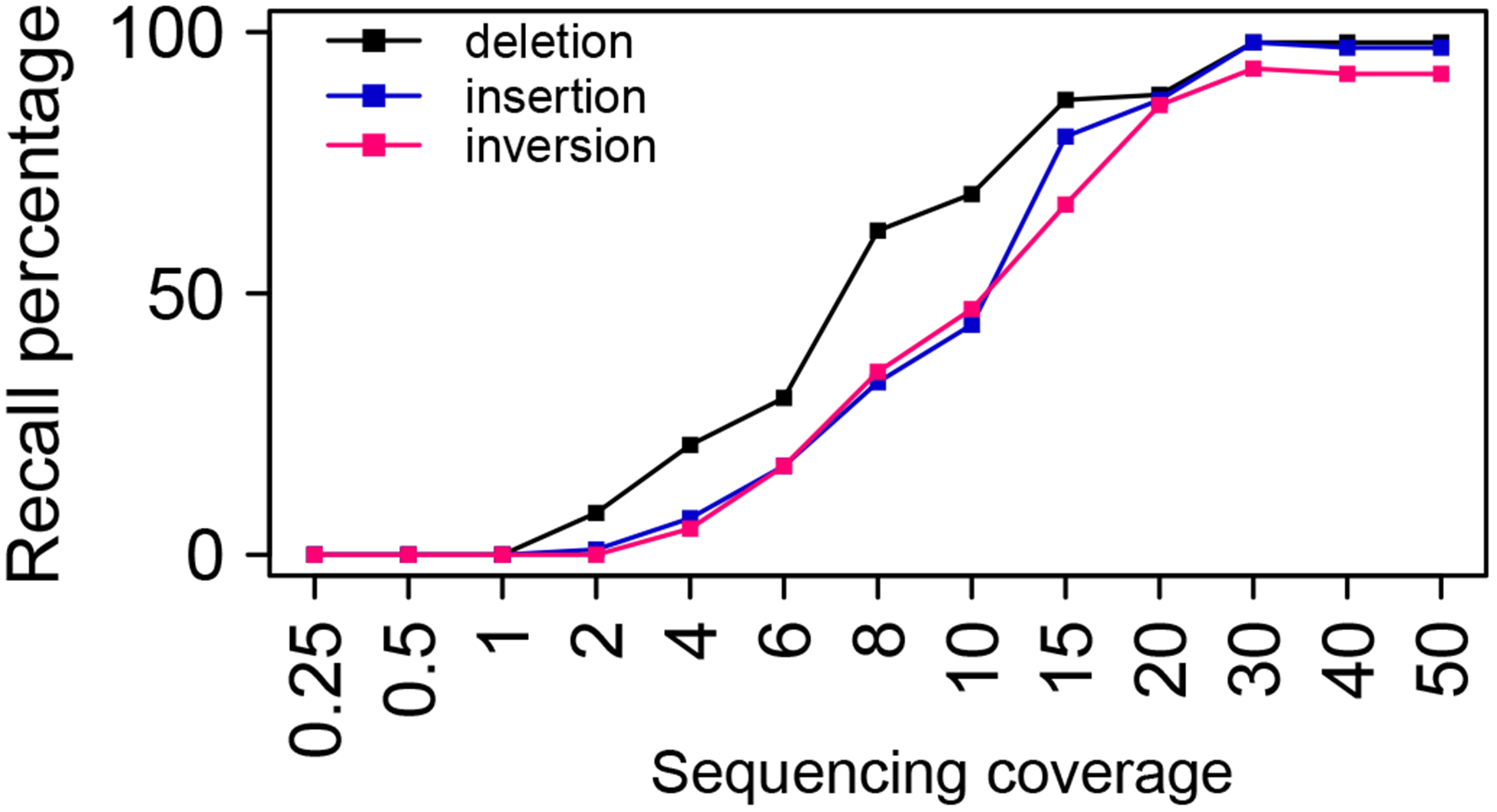
The effect of sequencing coverage on SV detection sensitivity. All simulated SVs are 1kb in size. Synthetic heterozygous SVs are placed at uniquely mappable regions on a pseudo diploid chromosome. Illumina sequencing reads (paired-end 75bp) are simulated.
5. Complex variants
Mildly complex SVs can be reconstructed by integrating copy number profiles and SV junctions (Greenman et al., 2011). The structure of chromothripsis events can still be inferred from short-read sequencing data (Yang et al., 2013), but it is an extremely challenging task. Therefore, long-read sequencing and imaging technologies will be very powerful to uncover the fine structures of very complex SVs. For example, linked-read sequencing was used to resolve haplotypes and somatic SVs in metastatic castration-resistant prostate cancers (Viswanathan et al., 2018); short-read WGS, high-throughput chromosome conformation capture (Hi-C) and optical mapping were integrated to resolve complex SVs and phase multiple SVs to single haplotype (Dixon et al., 2018); and the structures of circular extra-chromosomal DNA in glioblastoma cell lines were determined by the combination of short-read WGS, optical mapping and super-resolution microscopy (Wu et al., 2019). When the main goal is to characterize complex SVs, we recommend combining short- and long-read platforms in order to determine the long-range structure and the precise breakpoints at the same time. A combination of 30x short reads and 10x long reads shall perform well. If clonality and aneuploidy cannot be ignored, higher sequencing depth will be required as described previously.
6. Data recycling
Although WGS is preferred for comprehensive SV detection, other types of sequencing data can still be used to identify SVs. For example, short-read based whole exome sequencing (WES) data are commonly used for CNV detection. In addition, if SV breakpoints are present in the enriched regions being sequenced such as exons, they may be identified using the standard SV calling algorithms. It can be particularly fruitful if the data are readily available on a large number of samples even with limited sensitivity. For examples, rare germline CNVs were studied using nearly 60,000 exomes (Ruderfer et al., 2016), and somatic SVs in cancer were discovered in 5,000 tumor exomes (Yang et al., 2016). Although only 1% of the somatic SV breakpoints are detectable in WES data, a large portion of complex SVs such as chromothripsis can still be detected (Yang et al., 2016). The large sample size in those studies (Ruderfer et al., 2016; Yang et al., 2016) enabled meaningful biological inferences without generating any new data.
Variant calling
By far, there is no gold standard for how to call SVs from sequencing data. Numerous predicting algorithms are available, and the calls made by different algorithms on the same data may differ substantially. No algorithm can out-perform others in all types of SVs and across all size ranges. Numerous published papers and reviews have compared and benchmarked the available tools extensively. However, the benchmarking results also differ in different studies. The main reason is again the lack of gold standard. In this section, I will briefly describe computational tools for variant detection and strategies to determine which tool(s) to use.
Many algorithms have been developed to detect CNVs in short-read WGS data based on read depth, such as BIC-seq (Xi et al., 2011), CNVnator (Abyzov et al., 2011) and Control-FREEC (Boeva et al., 2012). A comprehensive list of CNV detection software can be found at https://bioinformaticshome.com/tools/cnv/cnv.html. Many tools have been reviewed (Zhao et al., 2013; Pirooznia et al., 2015; Hehir-Kwa et al., 2015) and benchmarked (Trost et al., 2018; Zhang et al., 2019). Evaluations of CNV detection tools in exome-sequencing and targeted-sequencing data are also available (Zare et al., 2017; Yao et al., 2019; Kadalayil et al., 2014).
For SV detection using short-read sequencing data, it is recommended to use BWA-MEM (Li, 2013) to align the reads to the reference genome because it can partially align reads. This function is particularly useful to detect reads spanning the SV breakpoints since they are marked as clipped reads. Several strategies can be used to predict SVs from short-read sequencing data (Alkan et al., 2011). Read depth can be used to interrogate SVs with copy number change. The presence of discordant read pairs (reads in a pair mapped to different chromosomes, or in incompatible orientations, or not within the size limit of sequencing library) often suggests the presence of SVs. Reads spanning the SV breakpoint junction (split read) can be used to refine the precise locations of breakpoints. Reads around the breakpoints can be assembled for SV detection. Dozens of computational algorithms have been developed using one or several of the above strategies, such as Meerkat (Yang et al., 2013), DELLY (Rausch et al., 2012b), Manta (Chen et al., 2016), novoBreak (Chong et al., 2016), etc. A comprehensive list of SV detection software can be found at https://omictools.com/structural-variant-detection-category. Several benchmarking studies have tested the performances of many of these tools (Lee et al., 2018; Kosugi et al., 2019; Cameron et al., 2019; Alaei-Mahabadi et al., 2016). If choosing only one SV caller, it is recommended to choose one that implements several SV detection strategies, for examples Meerkat and Manta, because such software often performs better than the ones only use one SV discovery strategy. To achieve the highest accuracy, the best practice is to use several tools and integrate their SV calls to minimize caller-specific bias (Sudmant et al., 2015; Campbell et al., 2020). Apparently, it will significantly increase the computational burden for large datasets. When SVs are called by multiple algorithms, one needs to identify the overlapping calls and merge them into a unified call set. Theoretically, the same SVs should have the same genomic coordinates called by different algorithms. However, read depth and read pair strategies cannot provide the precise locations of breakpoints. In addition, the SV breakpoints are often not blunt ends, but carry homologous sequences or insertions (Yang et al., 2013). Different algorithms handle homology and insertion sequences differently and provide slightly different coordinates. Practically, if two SVs have the same breakpoint orientations at both breakpoint junctions and the distances between the corresponding breakpoints are within 50bp, they can be considered as the same SV. The breakpoint orientation is determined by read mapping orientation. For example, for the deletion shown in Figure 2D, the breakpoint on the left is typically marked with orientation of “1” or “+” because the supporting reads near this breakpoints are mapped to forward strand, while the breakpoint on the right is marked as “−1” or “-”. See ref (Yang et al., 2013) for more examples. One widely used strategy to assemble a unified SV call set is that once the overlapping SVs are determined, caller specific SVs shall be removed. The remaining SVs supported by at least two callers are usually of high quality. Researchers can also consider different ways of combining SVs called by multiple algorithms, such as based on individual SV scores provided by callers. Different SV callers may be weighed differently. For example, if one algorithm is known to produce very few false calls, all SVs detected by this algorithm can be included in the final call set. Some tools run a suite of individual SV callers and provide an ensemble call set, such as Parliament2 (Zarate et al., 2018) and FusorSV (Becker et al., 2018).
For long-read sequencing and imaging platforms, there are a number of vendor provided software as well as open source tools for SV detection, such as SMRT Link (PacBio), EPI2ME (Nanopore), LongRanger (10X Genomics), Bionano Access (Bionano), NanoSV (Cretu Stancu et al., 2017), Picky (Gong et al., 2018), Sniffles (Sedlazeck et al., 2018), SMRT-SV (Chaisson et al., 2015), GROC-SVs (Spies et al., 2017), LinkedSV (Fang et al., 2019), etc. Benchmarking studies have shown significant differences between different platforms and computational tools (Zook et al., 2019; Audano et al., 2019; Chaisson et al., 2019; Luan et al., 2020; De Coster et al., 2019). Therefore, it is recommended to combine multiple tools for SV detection to achieve best sensitivity and accuracy. For SV detection combining multiple platforms, although most pipelines are developed inhouse, a few of them are streamlined and publicly available, such as Multibreak-SV (Ritz et al., 2014) and HySA (Fan et al., 2017).
Quality control
Quality control after SV calling is as important as choosing sequencing platforms and deciding SV calling algorithms in order to achieve satisfying performance in SV detection. Poor quality samples, suboptimal library preparation and sequencing as well as SV calling algorithms may produce artifactual SV calls. Read-level quality control, such as FastQC (Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data), may not pick up issues that affect SV calling. These issues include i) whole-genome amplification can produce artifactual duplications (Yang et al., 2019); ii) an unknown source during library preparation induces small inversion-like SVs with microhomology (Yang et al., 2016; Campbell et al., 2020); iii) random ligation of DNA fragments may produce chimeric molecules; iv) germline SVs may not be properly removed when calling somatic and de novo SVs due to poor-quality control samples or inadequate sequencing depth of control samples; and v) repetitive elements can lead to artifactual SV calls if not handled well. Much of our experiences in identifying artifacts came from comparisons of high coverage (>30x) short-read WGS (Li et al., 2020), lowpass (6–8x) short-read WGS (Zhang et al., 2018b), and short-read based WES (Yang et al., 2016) performed on the same samples of the Cancer Genome Atlas (TCGA) cohort. We compared somatic SVs detected in each approach and identified small duplications and small inversion-like events are often artifacts because they are not observed in the same samples profiled by another sequencing approach. Detailed procedures of identification and filtering of SV artifacts in WES data were described in our previous paper (Yang et al., 2016).
Here, I offer several strategies to identify problematic samples and SVs:
The number of SVs detected in all samples shall be inspected to identify outliers. Samples with extremely high number of SVs require close attention. For example, in one of our study of somatic SVs in 98 tumors, the sample on the left in Figure 4A had much more somatic SVs than other samples in the same cohort. After detailed investigations using other strategies described in this section, this sample turned out to be fine. In our somatic SV study based on WES data, outlier samples were all discarded (Yang et al., 2016) due to artifactual amplification-induced small duplications.
The SV composition shall be inspected. In the same cohort shown in Figure 4A, most samples had a mixture of deletions, duplications, inversions and translocations. However, a few samples carried predominantly deletions which require further investigation. In fact, these deletions were artifacts (see item 3 for more details).
The SV size, microhomology at the breakpoints, number of supporting reads, location of breakpoints, etc. shall be inspected. For deletions in Figure 4A, when we plotted size, microhomology at breakpoints and number of supporting reads, we found a large number of deletions were around 300bp in size with microhomology of more than 10bp (Figure 4B). Compared to other deletions, these small deletions all had very few supporting reads. If these deletions were true somatic SVs, their unique size range and microhomology suggested they were likely being generated via a distinct mutational process. Few supporting reads suggested they were subclonal events that were only present in a subset of tumor cells. It is unlikely that a mutational process only operates in a subset of the cells. We then used a different SV caller and didn’t detect any of these deletions. Therefore, we concluded that the artifactual deletions were caused by the SV caller we used initially. Similarly, the small inversion-like events we identified in WGS and WES in TCGA cohort were also less than 1kb in size with large homology and few supporting reads. For germline SVs, the number of supporting reads is also a very useful measure. The allele fractions of homozygous and heterozygous SVs are 1 and 0.5, respectively. They can be calculated as the numbers of reads supporting SVs divided by read depths at the SV breakpoint junctions, or as the numbers of discordant read pairs divided by the sums of discordant and concordant read pairs. The allele fractions of germline heterozygous SVs should follow a binomial distribution with mean of 0.5. If the allele fractions of SVs detected in a particular sample and/or of a particular type deviate substantially from the binomial distribution, the SVs are very likely to be artifacts.
The number and composition of SVs shall be compared with previous studies of similar cohorts to identify problematic samples and SVs. For germline SVs, a recent large population-based study reported an average of 4,400 germline SVs per individual (Abel et al., 2020) and the Genome Aggregation Database (gnomAD) reported 7,400 germline SVs per individual on average (Collins et al., 2019). Both studies are based on Illumina short reads. The vast majority of germline SVs are deletions and insertions, while inversions and duplications are much less common. Recent comprehensive multi-platform studies reported 13,000 SVs in a trio (Zook et al., 2019) and 27,000 SVs per germline genome (Chaisson et al., 2019). Since many germline SVs in a genome are common in population, studies focusing on germline SVs shall produce SV calls consistent with the above mentioned large-scale comprehensive studies. For somatic SVs in cancer, the Pan-Cancer Analysis of Whole Genome (PCAWG) reported that each tumor carries from 0 to up to 2,000 somatic SVs queried by Illumina high coverage WGS (Campbell et al., 2020). When we studied somatic SVs in cancer using WES data, samples with over 1,000 somatic SVs immediately rang alarm (Yang et al., 2016) because WES only captures about 2% of the genome. Those samples were discarded.
A better approach to identify somatic SV artifacts is to perform mutational signature analysis on SVs. Somatic alterations, including SNVs and SVs, form in tumor cells via different molecular mechanisms, such as external mutagens, internal mutational processes and defects of DNA damage repair. It is mathematically feasible to decompose the mutational signatures if a large number of tumor samples are sequenced. Methods like Non-negative Matrix Factorization (NMF) can be used to extract such signatures (Alexandrov et al., 2013b). NMF has been applied to deconvolute signatures of somatic SNVs (Alexandrov et al., 2013a, 2020) and SVs (Nik-Zainal et al., 2016; Li et al., 2020) in cancer. If systematic artifacts are present in the variant calls, they are likely to be captured by one or more signatures (Alexandrov et al., 2020). In the cohort shown in Figure 4A, we used signeR (Rosales et al., 2016) to decompose somatic SV signatures. All SVs were initially classified into deletions, duplications, inversions and translocations. For non-translocation events, they were then divided into 5 size ranges: <1kb, 1kb-10kb, 10kb-100kb, 100kb-1Mb and >1Mb. For each size range, the SVs were further divided into 7 categories based on homology length and insertion sequence length at the breakpoints (Figure 4C bottom zoomed-in panel). After applying such SV classification, we obtained 3 signatures in the cohort. Signature 1 represented less than 1kb deletions with more than 5bp homology. Signature 2 and 3 were small deletions (predominantly 1kb to 10kb in size) and large duplications (10kb to 1Mb in size) with short homology. If the homology and insertion sequence are not available for the SVs, the SVs can be classified based on event type and size (Nik-Zainal et al., 2016). After identifying signatures, the number of supporting reads and locations of breakpoints shall be inspected to identify possible artifact signatures as described previously. For example, the Signature 1 corresponded to the deletion artifacts shown in Figure 4B. In another study of a different cancer patient cohort, we also identified a somatic small deletion signature with large homology. The deletions were also around 300bp; however, the homology at breakpoints was between 10 to 15bp. The breakpoints of these deletions were all at the boundaries of Alu elements in the reference genome. These deletions were in fact germline Alu polymorphisms. The Alu insertions in the reference genome were not present in the query genome, so they appeared as deletions in the query genomes when compared to the standard reference genome (Figure 4D). The 10–15bp homology reflected the target site duplication (TSD) of Alu insertions. In this particular study, the germline Alu polymorphisms were not properly filtered and remained in the somatic SV call set. In general, if the sequencing quality of matched normal tissue is poor or the read depth is not high enough, germline SVs may be unfiltered. Therefore, in the SV caller we previously developed, Meerkat (Yang et al., 2013), SVs detected in each tumor are filtered against all normal samples merged together so that the common germline variants can be filtered even if one or a few normal samples are of poor quality.
The read alignment shall always be inspected in IGV (Robinson et al., 2011) or other genome browsers for a random subset of samples and SVs, especially the suspicious samples and SVs. For short-read based sequencing, true SVs shall present clear support of discordant read pairs, split reads and sometimes changes in coverage (Figure 4E). Note that read depth won’t change for balanced SVs, such as inversions and balanced translocations. Users may need to change the insert size range setting to properly display the discordant read pairs in IGV. The insert size range can be set as either percentiles (e.g. >99.9% and <0.1%) or actual base pair length (e.g. >800bp and <200bp). For somatic SVs, one shall load the tumor and matched normal genomes at the same time. The somatic SVs should be well supported in the tumor genome, but not in the normal genome (Figure 4E). If similar supporting reads are present in the normal genome, the SV probably is a germline event. If no similar support reads are found in the normal, but the region has many other discordant read pairs, clipped reads and non-unique mapped reads (reads shown as white boxes), this suggests that the read alignment of this region is problematic. The somatic SV called from this region may not be of high confidence.
Figure 4.
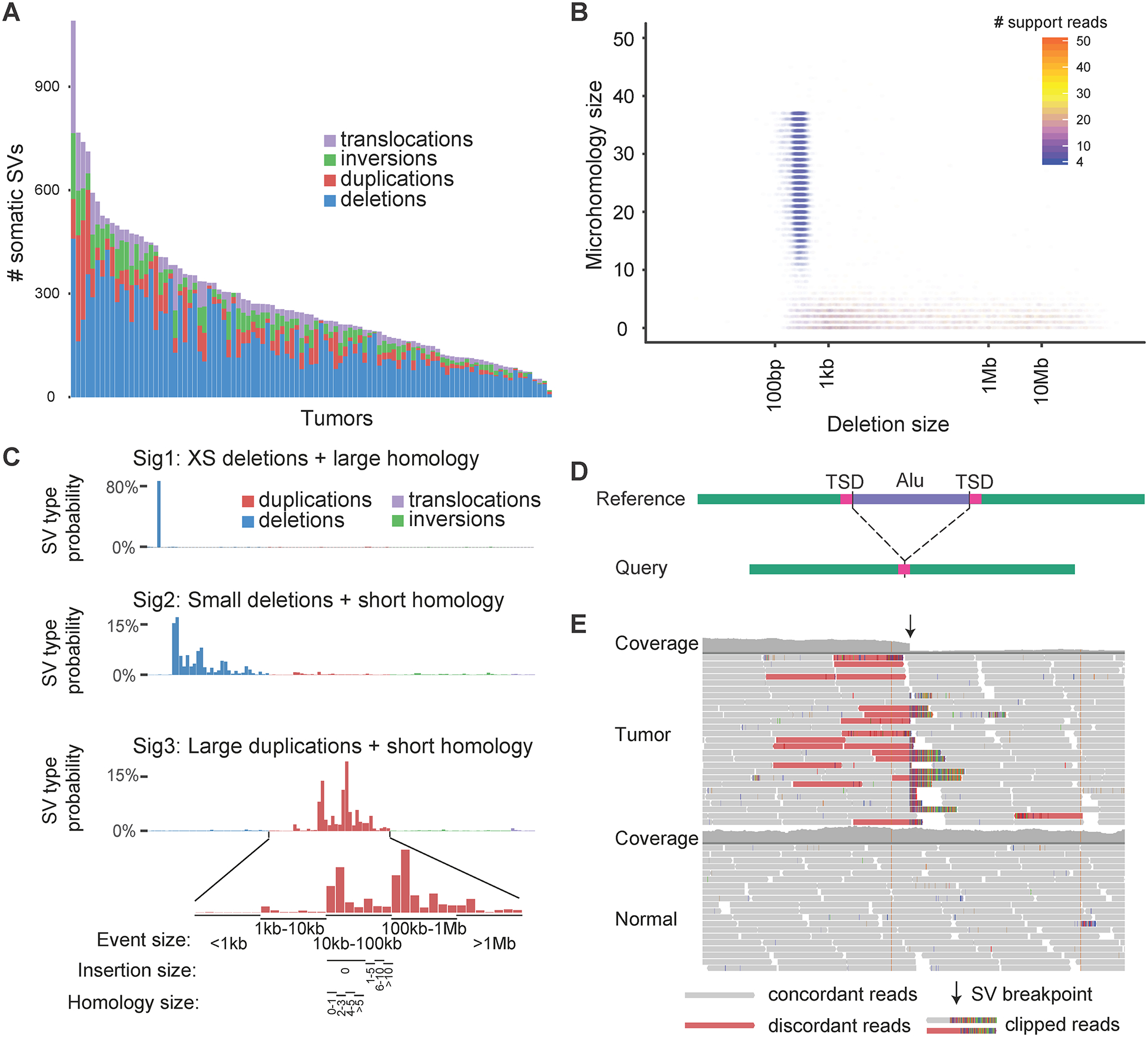
Quality control of SVs detected from sequencing data. A, Numbers and composition of somatic SVs discovered in 98 tumors. B, Distribution of deletion size and microhomology size color-coded by number of supporting reads in 98 tumors. C, Somatic SV signatures in the 98 tumors. D, Scheme of germline Alu polymorphism. The Alu element shown is inserted in the reference genome, but not in the query genome. E, IGV screenshot of a true somatic SV.
Conclusions
SVs are important genetic variants to study population diversity and human diseases. The detection of SVs is particularly challenging compared to other types of variants. Various technologies and platforms are available to interrogate SVs in the human genome. These SV discovery technologies have their own pros and cons. Most of the time, to a specific research project, the major limitation would be budget. Researchers shall decide on their strategies based on their overall goal and cost. After computational detection of SVs, the researchers shall perform rigorous quality control to eliminate possible artifacts caused by sample quality, sequencing/imaging platforms and computational tools.
Acknowledgements
This work was supported by grant R03CA246228-01 from the National Cancer Institute. The author has no conflicts of interest to declare.
References
- Abel HJ, Larson DE, Regier AA, Chiang C, Das I, Kanchi KL, Layer RM, Neale BM, Salerno WJ, Reeves C, et al. 2020. Mapping and characterization of structural variation in 17,795 human genomes. Nature:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abyzov A, Urban AE, Snyder M, and Gerstein M 2011. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Research 21:974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alaei-Mahabadi B, Bhadury J, Karlsson JW, Nilsson JA, and Larsson E 2016. Global analysis of somatic structural genomic alterations and their impact on gene expression in diverse human cancers. Proceedings of the National Academy of Sciences 113:13768–13773. Available at: http://www.pnas.org/lookup/doi/10.1073/pnas.1606220113 [Accessed June 20, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, et al. 2020. The repertoire of mutational signatures in human cancer. Nature 578:94–101. Available at: http://www.ncbi.nlm.nih.gov/pubmed/32025018 [Accessed February 17, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, et al. 2013a. Signatures of mutational processes in human cancer. Nature 500:415–421. Available at: http://www.nature.com/doifinder/10.1038/nature12477 [Accessed June 9, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. 2013b. Deciphering Signatures of Mutational Processes Operative in Human Cancer. Cell Reports 3:246–259. Available at: http://www.ncbi.nlm.nih.gov/pubmed/23318258 [Accessed April 24, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C, Coe BP, and Eichler EE 2011. Genome structural variation discovery and genotyping. Nature Reviews Genetics 12:363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audano PA, Sulovari A, Graves-Lindsay TA, Cantsilieris S, Sorensen M, Welch AME, Dougherty ML, Nelson BJ, Shah A, Dutcher SK, et al. 2019. Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 176:663–675.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data Available at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [Accessed June 27, 2020].
- Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, and Ghandi M 2013. Punctuated evolution of prostate cancer genomes. Cell 153:666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker T, Lee WP, Leone J, Zhu Q, Zhang C, Liu S, Sargent J, Shanker K, Mil-homens A, Cerveira E, et al. 2018. FusorSV: An algorithm for optimally combining data from multiple structural variation detection methods. Genome Biology 19:38 Available at: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-018-1404-6 [Accessed June 27, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, Huch M, Boymans S, Kuijk E, Prins P, et al. 2016. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538:260–264. Available at: http://www.nature.com/doifinder/10.1038/nature19768 [Accessed June 12, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochukova EG, Huang N, Keogh J, Henning E, Purmann C, Blaszczyk K, Saeed S, Hamilton-Shield J, Clayton-Smith J, O’Rahilly S, et al. 2010. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature 463:666–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, Janoueix-Lerosey I, Delattre O, and Barillot E 2012. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics (Oxford, England) 28:423–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer LM, and Dang CV 2001. Translocations involving c-myc and c-myc function. Oncogene 20:5595–5610. [DOI] [PubMed] [Google Scholar]
- Brandler WM, Antaki D, Gujral M, Kleiber ML, Whitney J, Maile MS, Hong O, Chapman TR, Tan S, Tandon P, et al. 2018. Paternally inherited cis-regulatory structural variants are associated with autism. Science 360:327–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumgarner R 2013. Overview of DNA Microarrays: Types, Applications, and Their Future In Current Protocols in Molecular Biology John Wiley & Sons, Inc, Hoboken, NJ, USA: Available at: http://doi.wiley.com/10.1002/0471142727.mb2201s101 [Accessed January 16, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, and Nussenzweig A 2013. End-joining, translocations and cancer. Nature Reviews Cancer 13:443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron DL, Di Stefano L, and Papenfuss AT 2019. Comprehensive evaluation and characterisation of short read general-purpose structural variant calling software. Nature Communications 10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PJ, Getz G, Korbel JO, Stuart JM, Jennings JL, Stein LD, Perry MD, Nahal-Bose HK, Ouellette BFF, Li CH, et al. 2020. Pan-cancer analysis of whole genomes. Nature 578:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CMB, Ramocki MB, Pehlivan D, Franco LM, Gonzaga-Jauregui C, Fang P, McCall A, Pivnick EK, Hines-Dowell S, Seaver LH, et al. 2011. Inverted genomic segments and complex triplication rearrangements are mediated by inverted repeats in the human genome. Nature Genetics 43:1074–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJP, Huddleston J, Dennis MY, Sudmant PH, Malig M, Hormozdiari F, Antonacci F, Surti U, Sandstrom R, Boitano M, et al. 2015. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517:608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJP, Sanders AD, Zhao X, Malhotra A, Porubsky D, Rausch T, Gardner EJ, Rodriguez OL, Guo L, Collins RL, et al. 2019. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nature Communications 10:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, and Saunders CT 2016. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32:1220–1222. Available at: https://academic.oup.com/bioinformatics/article-lookup/doi/10.1093/bioinformatics/btv710 [Accessed February 5, 2020]. [DOI] [PubMed] [Google Scholar]
- Chiang C, Jacobsen JC, Ernst C, Hanscom C, Heilbut A, Blumenthal I, Mills RE, Kirby A, Lindgren AM, Rudiger SR, et al. 2012. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nature Genetics 44:390–397. Available at: 10.1038/ng.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Scott AJ, Davis JR, Tsang EK, Li X, Kim Y, Hadzic T, Damani FN, Ganel L, Montgomery SB, et al. 2017. The impact of structural variation on human gene expression. Nature Genetics 49:692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong Z, Ruan J, Gao M, Zhou W, Chen T, Fan X, Ding L, Lee AY, Boutros P, Chen J, et al. 2016. NovoBreak: Local assembly for breakpoint detection in cancer genomes. Nature Methods 14:65–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RL, Brand H, Karczewski KJ, Zhao X, Alföldi J, Khera AV, Francioli LC, Gauthier LD, Wang H, Watts NA, et al. 2019. An open resource of structural variation for medical and population genetics. bioRxiv:578674. Available at: https://www.biorxiv.org/content/10.1101/578674v1 [Accessed June 28, 2020]. [Google Scholar]
- Conrad DF, Andrews TD, Carter NP, Hurles ME, and Pritchard JK 2005. A high-resolution survey of deletion polymorphism in the human genome. Nature Genetics 38:75–81. [DOI] [PubMed] [Google Scholar]
- Cordaux R, and Batzer MA 2009. The impact of retrotransposons on human genome evolution. Nature Reviews Genetics 10:691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Coster W, De Rijk P, De Roeck A, De Pooter T, D’Hert S, Strazisar M, Sleegers K, and Van Broeckhoven C 2019. Structural variants identified by Oxford Nanopore PromethION sequencing of the human genome. Genome Research 29:1178–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock N, Hurles ME, Cardin N, Pearson RD, Plagnol V, Robson S, Vukcevic D, Barnes C, Conrad DF, Giannoulatou E, et al. 2010. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 464:713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cretu Stancu M, Van Roosmalen MJ, Renkens I, Nieboer MM, Middelkamp S, De Ligt J, Pregno G, Giachino D, Mandrile G, Espejo Valle-Inclan J, et al. 2017. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nature Communications 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft B, Ohnesorg T, Hewitt J, Bowles J, Quinn A, Tan J, Corbin V, Pelosi E, van den Bergen J, Sreenivasan R, et al. 2018. Human sex reversal is caused by duplication or deletion of core enhancers upstream of SOX9. Nature Communications 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, Buhay C, Kang H, Kim SC, and Fahey CC 2014. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 26:319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Xu J, Dileep V, Zhan Y, Song F, Le VT, Yardımcı GG, Chakraborty A, Bann DV, Wang Y, et al. 2018. Integrative detection and analysis of structural variation in cancer genomes. Nature Genetics 50:1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Chaisson M, Nakhleh L, and Chen K 2017. HySA: A hybrid structural variant assembly approach using next-generation and single-molecule sequencing technologies. Genome Research 27:793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Kao C, Gonzalez MV, Mafra FA, Pellegrino da Silva R, Li M, Wenzel SS, Wimmer K, Hakonarson H, and Wang K 2019. LinkedSV for detection of mosaic structural variants from linked-read exome and genome sequencing data. Nature Communications 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuk L, Carson AR, and Scherer SW 2006. Structural variation in the human genome. Nature Reviews Genetics 7:85–97. [DOI] [PubMed] [Google Scholar]
- Franke M, Ibrahim DM, Andrey G, Schwarzer W, Heinrich V, Schöpflin R, Kraft K, Kempfer R, Jerković I, Chan WL, et al. 2016. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538:265–269. [DOI] [PubMed] [Google Scholar]
- Georgieva L, Rees E, Moran JL, Chambert KD, Milanova V, Craddock N, Purcell S, Sklar P, McCarroll S, Holmans P, et al. 2014. De novo CNVs in bipolar affective disorder and schizophrenia. Human molecular genetics 23:6677–6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, et al. 2009. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459:569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong L, Wong CH, Cheng WC, Tjong H, Menghi F, Ngan CY, Liu ET, and Wei CL 2018. Picky comprehensively detects high-resolution structural variants in nanopore long reads. Nature Methods 15:455–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenman CD, Pleasance ED, Newman S, Yang F, Fu B, Nik-Zainal S, Jones D, Lau KW, Carter N, Edwards PAW, et al. 2011. Estimation of rearrangement phylogeny for cancer genomes. Genome Research 22:346–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, and Grosveld G 1984. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell 36:93–99. [DOI] [PubMed] [Google Scholar]
- Gröschel S, Sanders MA, Hoogenboezem R, de Wit E, Bouwman BAM, Erpelinck C, van der Velden VHJ, Havermans M, Avellino R, and van Lom K 2014. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157:369–381. [DOI] [PubMed] [Google Scholar]
- Handsaker RE, Korn JM, Nemesh J, and McCarroll SA 2011. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nature Genetics 43:269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Lupski JR, Rosenberg SM, and Ira G 2009. Mechanisms of change in gene copy number. Nature Reviews Genetics 10:551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehir-Kwa JY, Pfundt R, and Veltman JA 2015. Exome sequencing and whole genome sequencing for the detection of copy number variation. Expert Review of Molecular Diagnostics 15:1023–1032. [DOI] [PubMed] [Google Scholar]
- Heller MJ 2002. DNA Microarray Technology: Devices, Systems, and Applications. Annual Review of Biomedical Engineering 4:129–153. Available at: http://www.annualreviews.org/doi/10.1146/annurev.bioeng.4.020702.153438 [Accessed January 16, 2020]. [DOI] [PubMed] [Google Scholar]
- Holland AJ, and Cleveland DW 2012. Chromoanagenesis and cancer: mechanisms and consequences of localized, complex chromosomal rearrangements. Nature Medicine 18:1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadalayil L, Rafiq S, Rose-Zerilli MJJ, Pengelly RJ, Parker H, Oscier D, Strefford JC, Tapper WJ, Gibson J, Ennis S, et al. 2014. Exome sequence read depth methods for identifying copy number changes. Briefings in Bioinformatics 16:380–392. Available at: 10.1093/bib/bbu027. [DOI] [PubMed] [Google Scholar]
- Kidd JM, Graves T, Newman TL, Fulton R, Hayden HS, Malig M, Kallicki J, Kaul R, Wilson RK, and Eichler EE 2010. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell 143:837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishikawa T, Momozawa Y, Ozeki T, Mushiroda T, Inohara H, Kamatani Y, Kubo M, and Okada Y 2019. Empirical evaluation of variant calling accuracy using ultra-deep whole-genome sequencing data. Scientific Reports 9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi S, Momozawa Y, Liu X, Terao C, Kubo M, and Kamatani Y 2019. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biology 20:117 Available at: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1720-5 [Accessed February 4, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AY, Ewing AD, Ellrott K, Hu Y, Houlahan KE, Bare JC, Espiritu SMG, Huang V, Dang K, Chong Z, et al. 2018. Combining accurate tumor genome simulation with crowdsourcing to benchmark somatic structural variant detection. Genome Biology 19:188 Available at: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-018-1539-5 [Accessed July 31, 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette LJ, Lohr JG, Harris CC, Ding L, Wilson RK, et al. 2012. Landscape of Somatic Retrotransposition in Human Cancers. Science 337:967–971. Available at: http://www.ncbi.nlm.nih.gov/pubmed/22745252 [Accessed July 29, 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Available at: http://arxiv.org/abs/1303.3997 [Accessed June 20, 2020].
- Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, Khurana E, Waszak S, Korbel JO, Haber JE, et al. 2020. Patterns of somatic structural variation in human cancer genomes. Nature 2020 578:7793 578:112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Schwab C, Ryan SL, Papaemmanuil E, Robinson HM, Jacobs P, Moorman AV, Dyer S, Borrow J, Griffiths M, et al. 2014. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature 508:98–102. Available at: http://www.nature.com/doifinder/10.1038/nature13115 [Accessed April 24, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CF, Naj AC, and Wang LS 2013. Analyzing copy number variation using SNP array data: Protocols for calling CNV and association tests. Current Protocols in Human Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Erez A, Nagamani SCS, Dhar SU, Kołodziejska KE, Dharmadhikari AV, Cooper ML, Wiszniewska J, Zhang F, Withers MA, et al. 2011. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 146:889–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockwood WW, Chari R, Chi B, and Lam WL 2006. Recent advances in array comparative genomic hybridization technologies and their applications in human genetics. European Journal of Human Genetics 14:139–148. [DOI] [PubMed] [Google Scholar]
- Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, Kwon M, Sherman MA, Vitzthum CM, Luquette LJ, Yandava CN, et al. 2018. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359:555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan M-W, Zhang X-M, Zhu Z-B, Chen Y, and Xie S-Q 2020. Evaluating Structural Variation Detection Tools for Long-Read Sequencing Datasets in Saccharomyces cerevisiae. Frontiers in Genetics 11:159 Available at: https://www.frontiersin.org/article/10.3389/fgene.2020.00159/full [Accessed June 20, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. 2015. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161:1012–1025. Available at: http://www.ncbi.nlm.nih.gov/pubmed/25959774 [Accessed August 18, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, Li Y, Bosco N, Campbell PJ, and de Lange T 2015. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 163:1641–54. Available at: http://www.ncbi.nlm.nih.gov/pubmed/26687355 [Accessed July 5, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, et al. 2015. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348:880–886. Available at: http://www.ncbi.nlm.nih.gov/pubmed/25999502 [Accessed February 7, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PIW, Maller JB, Kirby A, et al. 2008. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nature Genetics 40:1166–1174. [DOI] [PubMed] [Google Scholar]
- Mertens F, Johansson B, Fioretos T, and Mitelman F 2015. The emerging complexity of gene fusions in cancer. Nature Reviews Cancer 15:371–381. [DOI] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, et al. 2011. Mapping copy number variation by population-scale genome sequencing. Nature 470:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J, Westerman BA, van Arkel J, et al. 2012. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483:589–593. Available at: http://www.nature.com/articles/nature10910 [Accessed February 14, 2019]. [DOI] [PubMed] [Google Scholar]
- Mullaney JM, Mills RE, Pittard WS, and Devine SE 2010. Small insertions and deletions (INDELs) in human genomes. Human Molecular Genetics 19:R131–R136. Available at: https://academic.oup.com/hmg/article-lookup/doi/10.1093/hmg/ddq400 [Accessed January 2, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network TCGAR 2012. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, et al. 2016. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534:47–54. Available at: http://www.nature.com/doifinder/10.1038/nature17676 [Accessed November 8, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Lee C, Zichner T, Stütz AM, Erkek S, Kawauchi D, Shih DJH, Hovestadt V, Zapatka M, and Sturm D 2014. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511:428–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkel D, and Albertson DG 2005. COMPARATIVE GENOMIC HYBRIDIZATION. Annual Review of Genomics and Human Genetics 6:331–354. Available at: http://www.annualreviews.org/doi/10.1146/annurev.genom.6.080604.162140 [Accessed January 17, 2020]. [DOI] [PubMed] [Google Scholar]
- Pirooznia M, Goes F, and Zandi PP 2015. Whole-genome CNV analysis: Advances in computational approaches. Frontiers in Genetics 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Jones DTW, Zapatka M, Stütz AM, Zichner T, Weischenfeldt J, Jäger N, Remke M, Shih D, Northcott PA, et al. 2012a. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 148:59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, and Korbel JO 2012b. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28:i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redin C, Brand H, Collins RL, Kammin T, Mitchell E, Hodge JC, Hanscom C, Pillalamarri V, Seabra CM, Abbott MA, et al. 2017. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nature Genetics 49:36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz A, Bashir A, Sindi S, Hsu D, Hajirasouliha I, and Raphael BJ 2014. Characterization of structural variants with single molecule and hybrid sequencing approaches. Bioinformatics 30:3458–3466. Available at: 10.1093/bioinformatics/btu714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP 2011. Integrative genomics viewer. Nature Biotechnology 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales RA, Drummond RD, Valieris R, Dias-Neto E, and da Silva IT 2016. signeR: an empirical Bayesian approach to mutational signature discovery. Bioinformatics 33:8–16. Available at: 10.1093/bioinformatics/btw572. [DOI] [PubMed] [Google Scholar]
- Ruderfer DM, Hamamsy T, Lek M, Karczewski KJ, Kavanagh D, Samocha KE, Daly MJ, Macarthur DG, Fromer M, and Purcell SM 2016. Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nature Genetics 48:1107–1111. Available at: http://www.ncbi.nlm.nih.gov/pubmed/27533299 [Accessed March 3, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CP, Wiszniewska J, and Beaudet AL 2011. Copy Number and SNP Arrays in Clinical Diagnostics. Annual Review of Genomics and Human Genetics 12:25–51. Available at: http://www.annualreviews.org/doi/10.1146/annurev-genom-092010-110715 [Accessed January 17, 2020]. [DOI] [PubMed] [Google Scholar]
- Schmitt MW, Loeb LA, and Salk JJ 2016. The influence of subclonal resistance mutations on targeted cancer therapy. Nature Reviews Clinical Oncology 13:335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. 2007. Strong association of de novo copy number mutations with autism. Science 316:445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, Von Haeseler A, and Schatz MC 2018. Accurate detection of complex structural variations using single-molecule sequencing. Nature Methods 15:461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies N, Weng Z, Bishara A, McDaniel J, Catoe D, Zook JM, Salit M, West RB, Batzoglou S, and Sidow A 2017. Genome-wide reconstruction of complex structural variants using read clouds. Nature Methods 14:915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, and Lupski JR 2010. Structural Variation in the Human Genome and its Role in Disease. Annual Review of Medicine 61:437–455. [DOI] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. 2011. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144:27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazlsy C, Thorne N, Redon R, Bird CP, De Grassi A, Lee C, et al. 2007. Relative impact of nucleotide and copy number variation on gene phenotypes. Science 315:848–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Fritz MHY, et al. 2015. An integrated map of structural variation in 2,504 human genomes. Nature 526:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda DY, Spisák S, Seo JH, Bell C, O’Connor E, Korthauer K, Ribli D, Csabai I, Solymosi N, Szállási Z, et al. 2018. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 174:422–432.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YC, and Amon A 2013. Gene copy-number alterations: A cost-benefit analysis. Cell 152:394–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B, Walker S, Wang Z, Thiruvahindrapuram B, MacDonald JR, Sung WWL, Pereira SL, Whitney J, Chan AJS, Pellecchia G, et al. 2018. A Comprehensive Workflow for Read Depth-Based Identification of Copy-Number Variation from Whole-Genome Sequence Data. American Journal of Human Genetics 102:142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentijn LJ, Koster J, Zwijnenburg DA, Hasselt NE, van Sluis P, Volckmann R, van Noesel MM, George RE, Tytgat GAM, and Molenaar JJ 2015. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nature Genetics 47:1411–1414. [DOI] [PubMed] [Google Scholar]
- Viswanathan SR, Ha G, Hoff AM, Wala JA, Carrot-Zhang J, Whelan CW, Haradhvala NJ, Freeman SS, Reed SC, Rhoades J, et al. 2018. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 174:433–447.e19. Available at: http://www.ncbi.nlm.nih.gov/pubmed/29909985 [Accessed February 7, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan TSK 2014. Cancer cytogenetics: Methodology revisited. Annals of Laboratory Medicine 34:413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt J, Symmons O, Spitz F, and Korbel JO 2013. Phenotypic impact of genomic structural variation: insights from and for human disease. Nature Reviews Genetics 14:125–138. [DOI] [PubMed] [Google Scholar]
- Wu S, Turner KM, Nguyen N, Raviram R, Erb M, Santini J, Luebeck J, Rajkumar U, Diao Y, Li B, et al. 2019. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 575:699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi R, Hadjipanayis AG, Luquette LJ, Kim T-M, Lee E, Zhang J, Johnson MD, Muzny DM, Wheeler D. a, Gibbs R. a, et al. 2011. Copy number variation detection in whole-genome sequencing data using the Bayesian information criterion. Proceedings of the National Academy of Sciences of the United States of America 108:E1128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C 2018. A review of somatic single nucleotide variant calling algorithms for next-generation sequencing data. Computational and Structural Biotechnology Journal 16:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Lee M-S, Lu H, Oh D-Y, Kim YJ, Park D, Park G, Ren X, Bristow CA, and Haseley PS 2016. Analyzing Somatic Genome Rearrangements in Human Cancers by Using Whole-Exome Sequencing. The American Journal of Human Genetics 98:843–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Luquette LJ, Gehlenborg N, Xi R, Haseley PS, Hsieh C-H, Zhang C, Ren X, Protopopov A, Chin L, et al. 2013. Diverse mechanisms of somatic structural variations in human cancer genomes. Cell 153:919–929. Available at: http://linkinghub.elsevier.com/retrieve/pii/S0092867413004510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Wang S, Lee JJ-K, Lee S, Lee E, Shinbrot E, Wheeler DA, Kucherlapati R, and Park PJ 2019. An enhanced genetic model of colorectal cancer progression history. Genome Biology 20:168 Available at: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1782-4 [Accessed October 23, 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao R, Yu T, Qing Y, Wang J, and Shen Y 2019. Evaluation of copy number variant detection from panel-based next-generation sequencing data. Molecular Genetics and Genomic Medicine 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang C-Z, Wala J, and Mermel CH 2013. Pan-cancer patterns of somatic copy number alteration. Nature Genetics 45:1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate S, Carroll A, Krashenina O, Sedlazeck FJ, Jun G, Salerno W, Boerwinkle E, and Gibbs R 2018. Parliament2: Fast Structural Variant Calling Using Optimized Combinations of Callers. bioRxiv:424267. Available at: https://www.biorxiv.org/content/10.1101/424267v1 [Accessed June 27, 2020]. [Google Scholar]
- Zare F, Dow M, Monteleone N, Hosny A, and Nabavi S 2017. An evaluation of copy number variation detection tools for cancer using whole exome sequencing data. BMC Bioinformatics 18:286 Available at: https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-017-1705-x [Accessed June 20, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C-Z, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, and Pellman D 2015a. Chromothripsis from DNA damage in micronuclei. Nature 522:179–184. Available at: http://www.nature.com/doifinder/10.1038/nature14493 [Accessed July 5, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Gu W, Hurles ME, and Lupski JR 2009. Copy Number Variation in Human Health, Disease, and Evolution. Annual Review of Genomics and Human Genetics 10:451–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Bai W, Yuan N, and Du Z 2019. Comprehensively benchmarking applications for detecting copy number variation. PLOS Computational Biology 15:e1007069 Available at: https://dx.plos.org/10.1371/journal.pcbi.1007069 [Accessed June 20, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Choi PS, Francis JM, Gao GF, Campbell JD, Ramachandran A, Mitsuishi Y, Ha G, Shih J, Vazquez F, et al. 2018a. Somatic superenhancer duplications and hotspot mutations lead to oncogenic activation of the KLF5 transcription factor. Cancer Discovery 8:108–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Choi PS, Francis JM, Imielinski M, Watanabe H, Cherniack AD, and Meyerson M 2015b. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nature Genetics 48:176–182. Available at: http://www.nature.com/doifinder/10.1038/ng.3470 [Accessed August 3, 2017]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yang L, Kucherlapati M, Chen F, Hadjipanayis A, Pantazi A, Bristow CA, Lee EA, Mahadeshwar HS, Tang J, et al. 2018b. A Pan-Cancer Compendium of Genes Deregulated by Somatic Genomic Rearrangement across More Than 1,400 Cases. Cell Reports 24:515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Wang Q, Wang Q, Jia P, and Zhao Z 2013. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: Features and perspectives. BMC Bioinformatics 14:S1 Available at: https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-14-S11-S1 [Accessed June 20, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zook JM, Hansen NF, Olson ND, Chapman LM, Mullikin JC, Xiao C, Sherry S, Koren S, Phillippy AM, Boutros PC, et al. 2019. A robust benchmark for germline structural variant detection. bioRxiv:664623. Available at: https://www.biorxiv.org/content/10.1101/664623v1 [Accessed June 20, 2020]. [Google Scholar]