

Abstract
Cellular therapies have shown increasing promise as a cancer treatment. Encouraging results against hematologic malignancies are paving the way to move into solid tumors. In this review, we will focus on T-cell therapies starting from tumor infiltrating lymphocytes (TILs) to optimized T-cell receptor-modified (TCR) cells and chimeric antigen receptor-modified T cells (CAR-Ts). We will discuss the positive preclinical and clinical findings of these approaches, along with some of the persisting barriers that need to be overcome to improve outcomes.
Keywords: CAR-T cells, T-cell receptor-modified (TCR) cells, Cytolytic T cells (CTLs), Tumor infiltrating T cells (TILs), Adoptive cell therapy, hematologic malignancies
1. Introduction
In recent years, the use of cellular therapies has energized the field of oncologic immunotherapy. While immune cells like Natural Killer (NK) and NKT cells are also increasingly utilized for cancer therapy, we will here focus on the use of T lymphocytes. T cells are uniquely positioned for fighting cancer because of their direct effector activity and helper function through the recruitment of other components of the immune response. Furthermore, T lymphocytes can expand ex-vivo and establish a memory compartment, a major property for antitumor surveillance.
Starting with tumor infiltrating lymphocytes (TILs) and ex-vivo expanded cytotoxic T cells (CTLs), and moving to optimized T-cell receptor-modified T cells (TCR-Ts) and chimeric antigen receptor-modified T cells (CAR-Ts), each of these technologies displayed a gradual improvement in our understanding of harnessing the power of immune cellular elements to eradicate cancer. There have been promising results with clinical trials, especially with CAR-Ts, demonstrating long-term efficacy and remission potentials with hematologic malignancies. However, some hurdles hamper T-cell therapies from achieving higher levels of success and require further research. In this review, we will discuss the positive preclinical and clinical findings, along with some of the current barriers of T-cell-based therapies.
2. TILs and ex-vivo expanded CTLs: Dawn of Adoptive Cell Therapy (ACT)
Seminal studies in humans have shown that the presence of cytotoxic (CD8+) and helper (CD4+) T lymphocytes infiltrating tumors and the surrounding stroma is not only indicative of an ongoing antitumor response by the host, but also correlates with clinical outcomes in several tumor types, especially melanoma [1–2]. Tumor-specific CTLs could also be found in the circulation, although at very low frequencies [2]. These initial observations propelled improvements in culture conditions for the ex-vivo expansion of these cells, with the goal of returning them in large numbers to patients to eliminate tumors (Figure 1).
Figure 1.

Overview of the adoptive cell therapy (ACT) process for cancer treatment using tumor-infiltrating lymphocytes (TILs), cytotoxic T lymphocytes (CTL), or genetically modified TCR-T or CAR-T cells. A. For TIL ACT, tumors are resected, and tumor-reactive T cells are isolated and expanded ex vivo. B. CTL therapy relies on enriching and expanding peripheral blood T cells with known TAA specificity. C & D. ACT with TCR-T or CAR-T cells relies on the genetic modification of peripheral blood T cells by viral vectors to express a specific TCR or CAR. Patients are pretreated with a lymphodepletion regime before adoptive transfer of TILs, TCR-T, or CAR-T cells to create space and availability of homeostatic and activating cytokines. Figure created with Biorender.
2.1. Rules of engagement
TILs and CTLs target tumors via their native TCR, that recognize tumor-associated antigens (TAAs), presented as epitopes via the major histocompatibility complex (MHC) on the surface of antigen-presenting cells, an interaction that is highly specific and results in T-cell activation [1,4]. MHC proteins are encoded by the human leukocyte antigen (HLA-A, B, and C) family of genes, which are highly polymorphic, resulting in these T cells being MHC-restricted [3]. TAAs susceptible for TIL- and CTL-targeting comprise of epitopes derived from viral open reading frames (for virus-induced tumors), non-mutated proteins overexpressed in cancer cells, to which T-cell tolerance is incomplete (like the cancer-testis antigens), or peptides that are absent in the normal human genome, so-called neo-antigens. These epitopes can be derived from any antigen, whether it is an intracellular or a cell surface protein.
2.2. Clinical relevance
Because of the high mutational load and accompanying high neo-antigen rates of melanoma [4–7], the adoptive transfer of autologous TILs represents one of the more effective approaches to try and induce complete regression in patients with metastatic melanoma [8–13]. For similar reasons, durable tumor regression have been observed for metastatic melanoma, and partial responses in patients with pulmonary or hepatic metastasis from melanoma, colon cancer, and renal cancer [14]. TILs have also proven effective when targeting immunogenic viral proteins, like in Human Papilloma Virus (HPV)-associated cancers, which overexpress the HPV-E6 and -E7 oncoproteins [17,18]. Equally, CTLs have been found effective against HPV-associated cancers and in eradicating other virus-associated tumors like Hodgkin and Non-Hodgkin lymphomas, and nasopharyngeal carcinoma when they express Epstein Barr Virus-associated antigens [17–19]. They have also proven successful for treatment of hematological cancers, because of their expression of tumor restricted antigens, like Wilms tumor gene product 1 (WT1), preferentially expressed antigen in melanoma (PRAME), and melanoma-associated antigen (MAGE) [20,21].
2.3. Generation of TAA-specific T cells
Traditionally, TILs are isolated from resected tumor specimens, ex-vivo expanded with OKT3, irradiated feeder cells, and IL-2 [1,22,23] and thus retain the targeted antigen specificity to the tumor from which they were isolated. Approximately 6–8 weeks after tumor resection, up to 1011 lymphocytes can be prepared for infusion into patients (Figure 1A) [1,22,23]. Although the expansion can be reduced to 5 weeks, the procedure remains highly specialized and labor intensive, often producing “aged” cells. Additional concerns include the possibility that TILs may be rendered metabolically unfit by the chronic exposure to antigen before their isolation [24,25], and the potential expansion of less effective clones.
CTLs are generated ex-vivo over the course of 2–3 weeks, by repeated stimulations with cytokines and professional antigen presenting cells presenting the specific antigen (Figure 1B) [26]. T-cell clones have also been used, but like TILs they require extensive cultures and may generate exhausted cells. Unless targeting viral-associated antigen, the production of CTLs and clones for clinical purposes remain suboptimal, due to the low frequency of CTLs with adequate TCR affinity in the circulation as well as central tolerance.
To overcome the shortcomings of TIL and CTL therapy, including access to tumors, and expansion to sufficient cell numbers for therapy, research has concentrated on redirecting the antigen specificity to T cells through genetic engineering.
3. TCR-T cells: Genetic modifications make a debut in ACT
The ability to confer antigen specific recognition through the direct expression of ectopic TCRs allows to produce large quantities of TAA-specific T cells from the peripheral blood. In addition, TCR transgenic expression allows the recruitment of the auxiliary molecules of the signal transduction pathway, resulting in T cells being fully activated by a small amount of antigen (Figure 1C) [27].
3.1. Generating effective TCRs
Similarly to TILs and CTLs, candidates for TCR-based T-cell therapy are antigens either over-expressed or associated with tumor differentiation, like MAGE-A3, MAGE-A4 [28], melanoma antigen recognized by T cells (MART)-1 [29], gp100 [30], New York esophageal squamous cell carcinoma-1 (NY-ESO-1) [29], and carcinoembryonic antigens (CEA) [31], or viral antigens like the HPV-E6 and E7 proteins [32,33]. TCR sequences were traditionally identified by isolating single cells and expanding each clone, but major advances in engineering technologies have facilitated the identification and optimization of TCR specificity and affinity. Such approaches rely on using structure-guided modulation of the TCR-peptide-MHC interface, on selecting targeted mutations using computational design to characterize binding parameters and structure, or on rationally designing sequence substitutions in contact areas using known TCR-pMHC structures. In some instances, deep mutational scanning of TCR-pMHC interactions are used to test the effects of amino acid alterations at multiple positions. These techniques are coupled with in-silico binding analyses, to provide a more informed high-throughput screening of engineered TCRs than traditional yeast and phage display technologies [34–41].
3.2. Limitations of TCR-based therapies
TCR-based T cells have produced successful responses in melanoma, myeloma, and HPV-associated cancers [16,28,32,42–47] While these successes have highlighted the feasibility and clinical potency of this strategy, limitations have also progressively emerged.
First, despite having the advantage of targeting any peptide, including those derived from intracellular protein degradation, this strategy remains MHC-restricted, making the TCR sequence only useful for individuals that share the same identical HLA. The majority of TCR-based trials are HLA-A2-restricted because of the prevalence of this haplotype in most human populations [48]. Expanding the targeting of TCR-gene therapies beyond HLA-A2 to a more diverse panel of targetable MHC alleles would make these therapies accessible to diverse populations.
Second, because antigen recognition by TCRs is regulated by MHC-presentation, there is the possibility of tumor escape due to MHC downregulation, or dysfunction of the antigen processing machinery [49]. Identifying TCR restricted for various HLA alleles may not only extend the strategy to a greater subset of patients, but also broaden the repertoire of TCRs to recognize other epitopes from the same antigen, ultimately overcoming the issue of heterogenous MHC expression [50].
Finally, on-target/off-tumor toxicities can occur if TCR-Ts attack antigens expressed on healthy tissues. Two separate clinical trials reported fatal neurotoxicity and cardiotoxicity events [51–53], caused by a MAGE-A3-TCR which was affinity enhanced in the α-chain of the complementarity-determining region (CDR)2 region, generating unforeseen cross-reactions with healthy tissues. An additional safety factor is the potential occurrence of TCR mispairing. Hybrid TCRs, formed from the combination of endogenous and exogenous chains, have the potential to generate unknown reactivity. Additionally, hybrid TCRs, even if not toxic per se, may cause T-cell dysfunction by competing with the intended α/β-pairs for surface and, thus, functional expression [54]. Mitigation strategies are being explored, like downregulating the endogenous TCR by small-interfering RNA (siRNA) and Dicer-substrate small interfering RNA (DsiRNA), which result in high transgene TCR expression with a reduced degree of TCR mispairing [55,56].
4. CAR-T cells: An evolution of ACT
Since their initial development, CAR-Ts have conquered some inherent weaknesses of the former two methodologies. First, CAR-Ts can be rapidly generated with known tumor specificities within weeks. Second, their tumor recognition is MHC-unrestricted, making them broadly applicable to multiple individuals. Third, unlike TCRs, CARs can bind not only to extracellular proteins but also to surface carbohydrate and glycolipid structures, resulting in an expanded range of targets. However, CAR signaling cannot fully recapitulate the physiologic events that arise from the native TCR engagement and further engineering had to be developed. Regardless, their use has significantly accelerated the development of personalized medicine.
4.1. CAR Structure
The structure of the CAR construct consists of primarily three sections: an extracellular ectodomain, responsible for binding to the targeted antigen, a transmembrane domain, and an endodomain responsible for signal transduction that carries out the effector component of the CAR-T-cell apparatus (Figure 1D).
4.1.1. Extracellular domain.
This portion comprises of a single-chain variable fragment (scFv), traditionally derived from a monoclonal antibody, which is the tumor antigen-binding moiety of the CAR construct. The scFv is usually composed of a variable light (VL) and a variable heavy (VH) region, joined by a linker segment. Work is on-going for deriving these moieties from alternative antigen-binding proteins that are smaller in size, more stable, and less likely to aggregate, which should facilitate the construction of multi-specific recognition motifs. Examples of single domain antibody mimetic proteins are: monobodies, based on the type III domain of fibronectin [57]; affibodies, based on a three-helix bundle Z domain [58]; and DARPins, based on the designed ankyrin repeat protein [59]. A hinge segment flexibly anchors the scFv to the transmembrane domain. Some of the hinges used contributed to activation-induced death, while others led to off-target activation, or played a role in tonic signaling [60–62], suggesting that this region can have significant impact on the overall CAR structure and functionality.
4.1.2. Transmembrane Domain.
The transmembrane domain connects, via the hinge region, the scFv to the signaling endodomain. Initially, transmembrane domains were derived from CD3ζ, CD4, CD8, or CD28 molecules, but more recent second-generation CAR transmembrane domains are derived solely from CD8 and CD28 molecules. Though difficult to compare across each of these subtypes, the transmembrane domains acts as a dynamic part of the CAR construct [63]. Together with the hinge, the transmembrane domain participates in modulating CAR function, which may vary depending on the type or the location of the tumor antigen epitope [61,64].
4.1.3. Endodomains.
This segment is tasked with coordinating and recruiting signaling elements through its immunoreceptor tyrosine-based activation motifs (ITAMs) [65,66]. Initially, CARs only contained a signaling domain, commonly derived from the CD3-ζ chain (1st generation CAR). This iteration of CAR, while showing preclinical cytotoxicity, exhibited only limited persistence, expansion, and antitumor efficacy [67]. Incorporation of co-stimulatory domains from either the CD28 or 4–1BB molecules in tandem to the ζ-chain promoted superior expansion, longevity, and tumor cytotoxicity preclinically and clinically (2nd generation CARs) [68,69]. While there is no clear consensus on which co-stimulatory domain is superior, key intrinsic differences are emerging. CD28-based CAR-Ts appear to proliferate and expand more rapidly and secrete larger amounts of Th1 cytokines like IFN-γ and IL-2; 4–1BB-based CAR-Ts display a more gradual response, but with less exhausted phenotypes and superior persistence in-vivo [70]. These distinctions also reflect specific metabolic pathways. CD28-CAR-Ts express a genetic signature pattern, which coopts an enhanced aerobic glycolytic pathway, resulting in effector memory differentiation and phenotype with less potential for extended survival and persistence [71]. In contrast, 4–1BB-CAR-Ts show increased oxidative phosphorylation capabilities, with enhanced fatty acid oxidation and mitochondrial biogenesis, which leads to more central memory phenotype. Intrinsic cellular kinases like lymphocyte-specific protein tyrosine kinase (LCK) in CD28-CARs and phosphatases such as SHP1 in 4–1BB-CARs also work differentially in each respective construct to further generate distinct modulation of cellular kinetics, metabolism, and persistence of each of these CAR-T types [71,72]. Additional co-stimulatory domains, such as OX40, CD27, and ICOS, have shown promise in preclinical CAR-T cell studies. Combination of multiple co-stimulatory molecules (3rd generation CARs) are also being studied [73,74], but more clinical experience is necessary to prove superiority [75].
4.2. Lessons learned from CAR-Ts in Hematologic Malignancies
The largest clinical experience with CAR-Ts is in hematologic malignancies, especially in those expressing the CD19 molecule, solely found on B-cell lineage cells. Over the course of the past 10 years multiple trials have shown unprecedented high and durable response rates in adults and pediatric patients with acute lymphoblastic leukemia (ALL) and aggressive B-cell non-Hodgkin lymphomas (B-NHL). Overall, these trials have resulted in the approval of a few CD19.CAR-T cell products for ALL, aggressive B-NHL, and mantle cell lymphoma [84–86]. CARs are also in clinical trials for non-B-cell hematological malignancies. For multiple myeloma, CAR-Ts targeting the B-cell maturation antigen (BCMA) are the most advanced in the clinic [76–78]. For Hodgkin Lymphoma, phase I-II trials with CAR-Ts targeting the CD30 antigen have shown safety and encouraging efficacy [79–81].
Despite the successes, these trials are previewing upcoming issues that CAR-based therapy is likely to have when applied to solid tumors. For example, the on-target/off-tumor toxicity of CD19.CAR-Ts produce B-cell aplasia and hypogammaglobulinemia with increased susceptibility to infections that can be long-lasting [82–84]. Supportive care with intravenous immunoglobulin infusions and prophylactic antibiotics help lessen the impact, but approaches are being actively explored to mitigate these on-target/off-tumor toxicities while retaining high clinical efficacy. Examples for B-cell malignancies are CARs targeting the surface immunoglobulin light-chains [85] (kappa or lambda), which are clonally restricted in expression and would spare the reciprocal light chain-expressing, non-malignant B cells. On-target/off-tumor toxicities have also delayed the application of CAR-Ts for the treatment of T-cell leukemia and lymphoma and for acute myeloid leukemia (AML), due to failures in identifying targets not shared by the leukemic and healthy T and myeloid precursor cells [86,87].
Another example is the loss or downregulation of the targeted antigen. Despite CD19 being highly expressed by blasts, antigen escape can still occur because of either selection pressure due to therapy, presence of clone containing a preexisting alternatively spliced CD19 isoform, a mutated CD19 protein, or occurrence of a lineage switch from lymphoid to myeloid [89–91].
Finally, epitope masking has been observed after the accidental transduction of B blasts by the CD19.CAR [90]. Although this is a rare phenomenon, the risk that this may occur in malignancies with circulating tumors, especially T-cell leukemia, should not be underestimated.
4.3. CAR-Ts Take the Stage in Solid Tumors
The application of CAR-Ts in solid tumors remains a work in progress. Solid malignancies exhibit unique impediments to CAR-T-cell therapies and clinical trials have so far displayed only modest activity. The reasons for these tempered results are multifactorial. Some are intrinsic to the CAR strategy and will be described in this section; others are common to the TIL/TCR-Ts approach and will be described in the next section.
4.3.1. Shared expression of TAA
One problem with the physiology of solid tumors is that cell surface targetable antigens on cancer cells are almost invariably also expressed on healthy cells. Unlike blood cancers where tumors usually express a cell-lineage specific marker, solid tumors more commonly express TAA, which while enriched in expression levels on tumor surfaces, tend to exist at low levels on normal tissues. For example, a trial with carboxyanhydrase-IX (CAIX).CAR-Ts in patients with renal cell carcinoma had to be stopped due to dose-limiting hepatotoxicity stemming from low-level expression of the CAIX antigen in normal bile duct epithelial cells [91]. In another patient with metastatic colon cancer, Her2.CAR-Ts caused severe respiratory failure, leading to the patient’s death. Low levels of Her2 on the lung epithelial cells were likely the culprit [92]. To circumvent this problem, researchers are exploring ways to modulate the affinity of the scFv towards its target. The critical question is how one can tune CARs to sense antigen density without impairing efficacy.
One way to modulate CAR signaling is by modifying the scFv affinity for the tumor antigen. However, challenges lie in balancing the efficiency of the CAR construct-mediated activation: if affinities are too weak, the recognition of TAAs can be ineffective; if too augmented, on-target/off-tumor toxicity could prevail (Figure 2A). An alternative approach uses the tyrosine kinase inhibitor dasatinib, which reduces LCK phosphorylation of CARs. This reversible inhibition curtails CAR-T-cell proliferation and cytokine production. Although potentially useful for abrogating toxicities like cytokine release syndrome (CRS) and neurotoxicity [93,94], side effects are expected to reoccur when the drug is withdrawn (Figure 2A). Different strategies looked at tightly regulating the expression of a cross-reactive CAR. One of these methods incorporates combinatorial antigen recognition with split signaling, as well as with balanced T-cell activation and costimulation. In this model, T cells co-express a CAR construct targeting the TAA of interest lacking costimulatory endodomains, and a chimeric costimulatory receptor (CCR) motif, which recognizes a second TAA and provides the costimulatory signal [95] (Figure 2B). Alternatively, adapter-mediated CARs separate the tumor antigen recognition motif and the CAR signaling, which allows for titratable tumor killing and reversible control of the CAR-Ts. Affinity between the adaptor and the CAR (zipper pair), and the affinity between tumor antigen and scFv can also be modified to provide additional tuning of CAR activation [96,97] (Figure 2B). To minimize off-tumor activation, CAR expression can be restricted to the tumor microenvironment (TME). This can be accomplished by incorporating a hypoxia inducible factor (HIF) within the construct. Such CAR molecules are continuously degraded when transduced cells are present in a normoxic environment, but expressed when cells are in hypoxic conditions like upon entering the TME [98] (Figure 2B). Finally, combinatorial logic for the precise recognition of tumors have been explored. For example, T cells have been engineered to express combinatorial circuits, in which a synthetic receptor (Notch) for one antigen induces the expression of a CAR for a second antigen. This increases the threshold of activation of CAR-Ts, so that they are armed and activated in the presence of dual antigen tumor cells (AND-gate strategy) [99] (Figure 2C).
Figure 2.

Schematic representation of CAR T cell control mechanisms. A. Tuning the affinity of the scFv to tumor antigen and drug-induced inhibition of CAR T cell signaling by dasatinib. B. Split-CAR signaling by adaptors or drug-induced dimerization. HIF-driven CAR expression is regulated by hypoxic environment i.e. tumor microenvironment. The suicide gene iCasp9 can be activated by a dimerizable drug to kill CAR T cells. C. CAR T cells can have autonomous regulation in the killing of targets based on cell surface protein expression by cancerous and healthy cells. Figure created with Biorender.
Suicide genes represent alternative methods to remove CAR-Ts in the event of a life-threatening on-target/off-tumor toxicity, although with the downside of also halting efficacy. Titratable safety switches, such as the inducible caspase 9 (iCasp9), may be more useful to not jeopardize clinical responses, by potentially allowing a small remnant population of functional CAR-Ts behind (Figure 2B) [100,101].
4.3.2. Antigen Expression Levels.
Surface antigen expression presents inter- and intra-heterogeneity, with proportions of cancer cells expressing no or low levels of antigen. This variation affects the binding capacities of CARs, as, in contrast to TCR, they require a higher threshold of antigen expression for activation [102,103]. While an advantage when targeting shared-TAA (see above), a low-activation threshold can be a hinderance in case of tumor heterogeneity, with cells expressing antigens at low levels escaping killing. Enhancing the strength of CAR-signaling should increase activity against low-antigen density tumors [104], but it an undesirable trade-off if resulting in off-tumor toxicities, over-activation, and premature exhaustion. This may imply fine tuning and balancing, and the requirement for tailoring CARs to each tumor model.
An alternative compromise may rely on the use of CAR-Ts that recognize multiple targets to prevent the upfront tumor antigen remodeling driven by the treatment pressure. For tumors with intra-tumor variability, a hierarchy of two or three non-overlapping antigens could be sufficient [105]. While a combination of “monovalent” CAR-Ts, each targeting a highly expressed antigen, to be infused simultaneously would allow flexibility to mix and match, the approach comes with regulatory hurdles and costs, and complex logistics [106]. Alternatively, multiple CAR constructs could be engineered in the same T-cell, although their expression seems overall reduced, and there are concerns about their functionality. Recently bi- or trivalent CAR constructs that recognize 2 or 3 antigens have been developed and tested preclinically with impressive results. To overcome some of the challenges of assembling several scFv’s in one single CAR, such as maintaining optimal protein folding, researchers are sourcing to antibody mimics [57,59].
5. Roadblocks to Effective ACT
Whether ACT occurs in the form of TILs, CTLs, TCR-Ts or CAR-Ts, there are several common barriers when targeting solid tumors. Because CAR-Ts are broadly applicable (as MHC-unrestricted) and readily available (quick turn-around of product through genetic engineering), the obstacles for targeting solid tumors have become better appreciated in the context of this modality.
5.1. Trafficking and infiltration.
For productive homing after infusion, there must be a match between the chemokine released by the tumor and/or its environment (TME) and the receptors that a T-cell expresses. T cells are expected to traffic to lymphoid tissues and bone marrow, but this homing can be suboptimal for certain locations (i.e. brain, pleural or peritoneum spaces). To overcome this obstacle, cells can be delivered locally, with successful results [107,108]. However, not every tumor is accessible for local delivery, and metastases to multiple sites render this approach unpractical. Leveraging on specific chemokine gradients/chemokine receptor pairs represents a possible solution. If the chemokine gradient is unique to the tumor, then the strategy is straightforward, and the match can be artificially created by forcing the expression of homing receptors and/or chemokines. Examples can be found both for TILs [109] and CAR-Ts [110–114], and some of these strategies are reaching clinical application. Making the tumor and/or its TME release chemokines that attract T cells may produce the same results [110,115].
In addition to homing, physical barriers also impede T-cell infiltration into cancer sites, especially in solid malignancies, where cancer-associated stromal cells and extracellular matrix (ECM) remodeling enzymes create highly desmoplastic TMEs [116]. Reducing or degrading the ECM components could assist in tumor eradication. For example, CAR-Ts targeting fibroblast activating protein on stromal cells augmented endogenous antitumor responses in mice [117]. Coexpression by CAR-Ts of enzymes capable of degrading the ECM has also demonstrated enhanced tumoricidal efficacy in preclinical models of stromal rich tumors [118].
5.2. Nutrient Starvation and Hypoxia
The lack of nutrients constitutes a major barrier for T cells in the TME. Indoleamine 2,3 dioxygenase (IDO) is an intracellular enzyme involved in the oxidative catalysis of tryptophan into kynurenine, with a vital regulatory role in mediating effects on innate and adaptive immunity in response to inflammation[119]. In the context of tumor anti-immunity, the IDO pathway is a potential tumor escape mechanism, through induction of cell-cycle arrest and CD8+ T-cell anergy, and by blocking the differentiation of CD4+ T cells to T helper (TH)17, while promoting differentiation, activation, and tolerogenicity of regulatory T cells (Treg) [120,121]. Additionally, kyneurine and its catabolites can inhibit T-cell proliferation, cytokine secretion, and cytotoxicity [122,123]. Tumor-IDO expression can finally orchestrate the recruitment of myeloid derived suppressor cells (MDSCs), which leads to further amino acid depletion through the secretion of arginase-1 [124,125]. By encouraging the differentiation and activation of immunosuppressive Tregs, depleting the local tryptophan supply, and recruiting MDSCs, IDO expression in tumors creates an oppressive and toxic environment for T cells. Engineering of CAR-Ts to become “IDO-insensitive” have been developed [123].
Hypoxia, along with nutrient depletion, is a characteristic trait of the TME. Deficient perfusion due to aberrant angiogenesis and ineffective delivery of oxygen are causative factors for this phenomenon. Hypoxia can negatively affect T-cell metabolism and effector function, by inducing inefficient cellular metabolism and enhancing glycolysis, instead of the tricarboxylic acid cycle [126]. Upregulation of HIF1-alpha further promotes MDSC recruitment, enhancing the immune suppressive environment [127]. Lessening the deleterious effects of hypoxia could help boost T-cell function. Recently, a CAR engineered to intrinsically activate the IL-23 pathway, via expression of p40, resulted in enriched hypoxia-related genes, ultimately leading to enhanced antitumor activity in solid tumor models [128].
5.3. Inhibitory Cytokines and Soluble Factors
Inhibitory cytokines and soluble factors are other prominent hallmarks of the TME. They can directly encumber T-cell cytotoxicity, proliferation and survival, and indirectly hamper them by recruiting immunosuppressive cells such as MDSCs and Tregs.
Chemotherapy regimens or irradiation prior to ATC infusions have proven helpful in the removal of suppressive cells and competing endogenous lymphoid sinks for the homeostatic cytokines, thus creating a favorable milieu supportive for T-cell persistence and function [129]. However, the effects of lymphodepleting conditioning are transient, and neither is poised to reshape inhibitory TMEs.
For example, adenosine and prostaglandin E2 (PGE2) are two of the molecules over-expressed by hypoxic TMEs. Both can subvert executive function of T cells through intracellular activation of protein kinase A (PKA) in a cyclic AMP (cAMP)-dependent fashion [130]. Specifically, the regulatory subunit I-anchoring disruptor (RIAD) is a protein that interrupts the PKA anchorage close to adenylyl cyclase and its expression in mesothelin-directed CAR-Ts has resulted in superior tumor infiltration and cytotoxicity [131].
Transforming growth factor beta (TGFβ), IL-4, and IL-10 are among the more predominant immunosuppressive cytokines secreted into the TME milieu by tumors, Tregs, and tumor-associated macrophages [132–134]. In advanced phases of tumor invasion and metastasis, TGFβ helps to stimulate the epithelial-to-mesenchymal transition of malignant cells, and assists in immune evasion by skewing T cells towards a Th2 phenotype [135]. IL-10 stifles antigen presentation and T-cell differentiation through suppression of antigen presenting cells [136]. Blocking the effects of these cytokines poses a chance to enhance the efficacy of T cells. Augmented tumoricidal effects, both in-vitro and in-vivo, were demonstrated by incorporating a dominant negative TGFβ receptor into antigen-specific CTLs and CAR-Ts [137,138]. The strategy is now in the clinic for tumor-specific CTLs, with very encouraging results [139]. To combat IL-4-mediated immunosuppression in the TME, a chimeric cytokine receptor comprising of an extracellular IL-4 receptor domain fused to an activating IL-7 cytoplasmic domain has been used to enhance persistence and effector function of CAR-Ts [140].
6. Immune Checkpoints to synergize with ACT
CTLA-4 and PD-1 are inhibitory protein receptors expressed by activated T cells, including CAR-Ts. They are categorized as negative regulators, or checkpoints, of T-cell activation, as they blunt T-cell-mediated anti-tumor activity. CTLA-4, a homolog of the T-cell co-stimulatory receptor CD28, dampens T-cell responses by out-competing CD28 for CD80 and CD86 ligands, and disrupting intracellular stimulatory signaling processes [141]. Most of the success with ipilimumab, a CTLA-4 antibody, has been with tumors carrying a high mutational and neo-antigen burden, such as melanoma [142]. PD-1 is also a homolog of both CTLA-4 and CD28; its binding to the B7 ligands PD-L1 and PD-L2, which many tumors constitutively express, causes reduction in T-cell metabolism and proliferation [143]. Analyses with melanoma in a murine model showed that production of proinflammatory cytokines from activated CD8+ T cells correlated with induced upregulation of PD-L1 [144]. There have been encouraging results with blockade of PD-L1 in patients with advanced stage cancers including in combination with TILs [145].
Preventing CAR-T-cell tolerance from such negative regulatory mechanisms has become a major driving point to boost effector function. There is substantial data demonstrating how these surface inhibitory markers play a critical role in modulating CAR-T-cell efficacy. In a mesothelioma mouse model, mesothelin.CAR-Ts showed an augmented expression of PD-1 at the tumor site, with resultant hypofunction. Their cytotoxicity was restored with the addition of a PD-L1 antibody in the presence of tumor cells ex vivo [117]. In mice bearing Her2+ tumors, there was a distinct advantage in tumor regression when combining Her2-CAR-Ts with PD-1 antibody [146]. However, clinical studies of combination therapy with CAR-Ts and immune checkpoint inhibitors have failed to recapitulate these promising preclinical results [147]. While combinations may prove effective only in certain tumor models, strategies to overcome the inhibitory pathway directly in T cells are being explored. An example is engineering CAR-Ts to secrete a PD-1-blocking scFv [148]. The secreted scFv improved anti-tumor activity of both CAR-Ts and bystander TILs. A more structural way of safeguarding CAR-Ts against the negative impact of immune checkpoint inhibition is the incorporation of a chimeric switch-receptor, containing the extracellular domain of PD-1 fused to the transmembrane and cytoplasmic domain of the costimulatory molecule CD28. This approach was incorporated into mesothelin.CAR-Ts and resulted in amplified tumor cytotoxicity, producing tumor regression in preclinical models of mesothelioma-bearing mice [149].
7. Conclusions and perspective
The first observation that the immune system plays a role in antitumor activity is attributed to Dr. Coley, who in 1890 linked immune responses to bacterial infections and subsequent remission of cancer in the same patients. It took almost a century to make ACT important tools in the therapy for cancer. TILs and CTLs first demonstrated the curative potential of ACT. The introduction of genetic engineering offered the opportunity to overcome the shortcomings of TIL and CTL therapy, by conferring direct antigen specificity to T cells. The creation of CAR molecules marked a new beginning for innovative advances within ACTs, with CAR-Ts addressing some inherent weaknesses in the former two methodologies.
Although cellular therapy remains in relatively nascent stages of development as a therapeutic modality, we should be encouraged by the exponential advances the field of ACT has reached in the past two decades (Figure 3). Despite the prodigious amount of emerging and encouraging data from clinical studies with TILs, CTLs, TCR-T cells, and CAR-T-cells, the field of cellular therapy still has ample room for growth and improvement. As our understanding of cellular physiology, mechanisms, and processes advances, our ability to tackle roadblocks to ACT and augment their clinical efficacy with innovative approaches will also improve.
Figure 3.
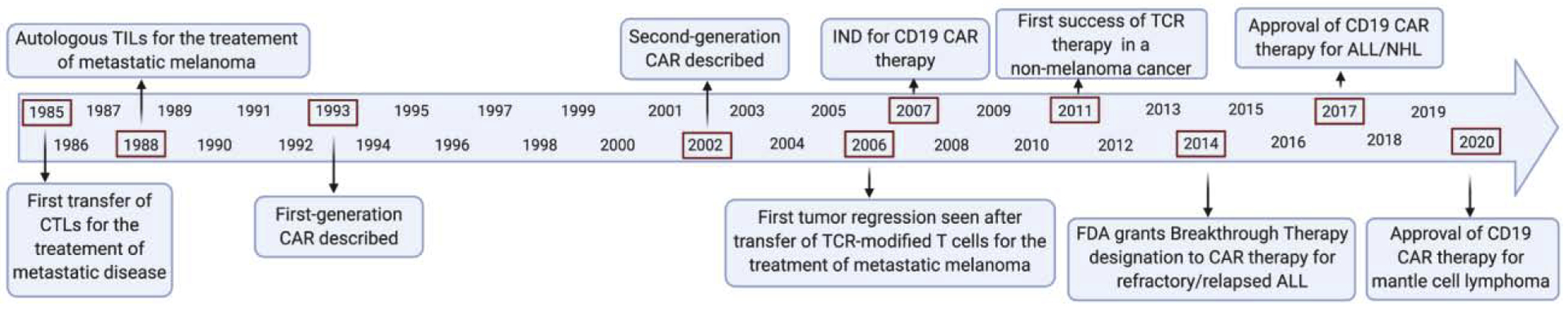
History of adoptive cell therapy (ACT) in cancer. This timeline highlights important developments in the field of adoptive T-cell therapy. Figure created with Biorender.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Rosenberg SA, Spiess P, Lafreniere R, A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes, Science (80-.). 233 (1986) 1318–1321. 10.1126/science.3489291. [DOI] [PubMed] [Google Scholar]
- [2].Romieu R, Baratin M, Kayibanda M, Lacabanne V, Ziol M, Guillet JG, Viguier M, Passive but not active CD8+ T cell-based immunotherapy interferes with liver tumor progression in a transgenic mouse model, J. Immunol 161 (1998) 5133–517. [PubMed] [Google Scholar]
- [3].Krogsgaard M, Davis MM, How T cells “see” antigen, Nat. Immunol 6 (2005) 239–245. 10.1038/ni1173. [DOI] [PubMed] [Google Scholar]
- [4].Lauss M, Donia M, Harbst K, Andersen R, Mitra S, Rosengren F, Salim M, Vallon-Christersson J, Törngren T, Kvist A, Ringnér M, Svane IM, Jönsson G, Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma, Nat. Commun 8 (2017) 1–11. 10.1038/s41467-017-01460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jha S, Rollins MG, Fuchs G, Procter DJ, Hall EA, Cozzolino K, Sarnow P, Savas JN, Walsh D, Mining Exomic Sequencing Data to Identify Mutated Antigens Recognized by Adoptively Transferred Tumor-reactive T cells, Nat. Med 546 (2017) 651–655. 10.1038/nature22814.Trans-kingdom. [DOI] [Google Scholar]
- [6].Sakaguchit K, Appellat E, Yannelli JR, Adema GJ, Miki T, Rosenberg SA, Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection, Proc. Natl. Acad. Sci 91 (1994) 6458–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, Holt RA, Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival, Genome Res. 24 (2014) 743–750. 10.1101/gr.165985.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, Rosenberg SA, Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes, Science (80-.). 298 (2002) 850–854. 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, Morton KE, Laurencot CM, Steinberg SM, White DE, Dudley ME, Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy, Clin. Cancer Res 17 (2011) 4550–4557. 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, Wu R, Lizee G, Mahoney S, Alvarado G, Glass M, Johnson VE, McMannis JD, Shpall E, Prieto V, Papadopoulos N, Kim K, Homsi J, Bedikian A, Hwu WJ, Patel S, Ross MI, Lee JE, Gershenwald JE, Lucci A, Royal R, Cormier JN, Davies MA, Mansaray R, Fulbright OJ, Toth C, Ramachandran R, Wardell S, Gonzalez A, Hwu P, Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients, Clin. Cancer Res 18 (2012) 6758–6770. 10.1158/1078-0432.CCR-12-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, Kubi A, Shoshani N, Zikich D, Ohayon Y, Ohayon D, Shalmon B, Markel G, Yerushalmi R, Apter S, Ben-Nun A, Ben-Ami E, Shimoni A, Nagler A, Schachter J, Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: Intent-to-treat analysis and efficacy after failure to prior immunotherapies, Clin. Cancer Res 19 (2013) 4792–4800. 10.1158/1078-0432.CCR-13-0380. [DOI] [PubMed] [Google Scholar]
- [12].Mehta GU, Malekzadeh P, Shelton T, White DE, John A, Yang JC, Kammula US, Goff SL, Rosenberg SA, Sherry RM, Outcomes of adoptive cell transfer with tumor-infiltrating lymphocytes for metastatic melanoma patients with and without brain metastases, J. Immunother 41 (2018) 241–247. 10.1097/CJI.0000000000000223.Outcomes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, Zlott DA, Yang JC, Sherry RM, Kammula US, Klebanoff CA, Hughes MS, Restifo NP, Langhan MM, Shelton TE, Lu L, Kwong MLM, Ilyas S, Klemen ND, Payabyab EC, Morton KE, Toomey MA, Steinberg SM, White DE, Rosenberg SA, Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma, J. Clin. Oncol 34 (2016) 2389–2397. 10.1200/JCO.2016.66.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, Matory YL, Skibber JM, Shiloni E, Vetto JT, Seipp CA, Simpson C, Reichert CM, Observations on the Systemic Administration of Autologous Lymphokine-Activated Killer Cells and Recombinant Interleukin-2 to Patients with Metastatic Cancer, N. Engl. J. Med 313 (1985) 1485–1492. 10.1056/NEJM198512053132327. [DOI] [PubMed] [Google Scholar]
- [15].Stevanović S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, Robins HS, Robbins PF, Klebanoff CA, Rosenberg SA, Hinrichs CS, Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer, Science (80-.). 356 (2017) 200–205. 10.1126/science.aak9510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Stevanovic S, Helman SR, Wunderlich JR, Langhan MM, Doran SL, Kwong MLM, Somerville RPT, Klebanoff CA, Kammula US, Sherry RM, Yang JC, Rosenberg SA, Hinrichs CS, A Phase II Study of Tumor-infiltrating Lymphocyte Therapy for Human Papillomavirus–associated Epithelial Cancers, Clin. Cancer Res 25 (2018) 1486–1493. 10.1158/1078-0432.CCR-18-2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gottschalk S, Edwards OL, Sili U, Huls MH, Goltsova T, Davis AR, Heslop HE, Rooney CM, Generating CTLS against the subdominant Epstein-Barr virus LMP1 antigen for the adoptive immunotherapy of EBV-associated malignancies, Blood. 101 (2003) 1905–1912. 10.1182/blood-2002-05-1514. [DOI] [PubMed] [Google Scholar]
- [18].Bollard CM, Gottschalk S, Leen AM, Weiss H, Straathof KC, Carrum G, Khalil M, Wu MF, Huls MH, Chang CC, Gresik MV, Gee AP, Brenner MK, Rooney CM, Heslop HE, Complete responses of relapsed lymphoma following genetic modification of tumor-antigen presenting cells and T-lymphocyte transfer, Blood. 110 (2007) 2838–2845. 10.1182/blood-2007-05-091280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ramos CA, Narala N, Vyas GM, Leen AM, Gerdemann U, Sturgis EM, Anderson ML, Savoldo B, Heslop HE, Brenner MK, Rooney CM, Human papillomavirus type 16 E6/E7-specific cytotoxic T lymphocytes for adoptive immunotherapy of HPV-associated malignancies, J. Immunother 36 (2013) 66–76. 10.1097/CJI.0b013e318279652e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Craddock J, Heslop HE, Adoptive cellular therapy with T cells specific for EBV-derived tumor antigens, Update Cancer Ther. 3 (2008) 33–41. 10.1016/j.uct.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zeng G, Li Y, El-Gamil M, Sidney J, Sette A, Wang RF, Rosenberg SA, Robbins PF, Generation of NY-ESO-1-specific CD4+ and CD8+ T cells by a single peptide with dual MHC class I and class II specificities: A new strategy for vaccine design, Cancer Res. 62 (2002) 3630–3635. [PMC free article] [PubMed] [Google Scholar]
- [22].Muul LM, Spiess PJ, Director EP, Rosenberg SA, Identification of specific cytolytic immune responses against autologous tumor in humans bearing malignant melanoma., J. Immunol 138 (1987) 989 LP – 995. http://www.jimmunol.org/content/138/3/989.abstract. [PubMed] [Google Scholar]
- [23].Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA, Simpson C, Carter C, Bock S, Schwartzentruber D, Wei JP, White DE, USE OF TUMOR-INFILTRATING LYMPHOCYTES AND INTERLEUKIN-2 IN THE IMMUNOTHERAPY OF PATIENTS WITH METASTATIC MELANOMA, N. Engl. J. Med 281 (1988) 1676–1680. [DOI] [PubMed] [Google Scholar]
- [24].Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, Delgoffe GM, The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction, Immunity. 45 (2016) 374–388. 10.1016/j.immuni.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Menk AV, Scharping NE, Rivadeneira DB, Calderon MJ, Watson MJ, Dunstane D, Watkins SC, Delgoffe GM, 4–1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses, J. Exp. Med 215 (2018) 1091–1100. 10.1084/jem.20171068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kuznetsova M, Lopatnikova J, Khantakova J, Maksyutov R, Maksyutov A, Sennikov S, Generation of populations of antigen-specific cytotoxic T cells using DCs transfected with DNA construct encoding HER2/neu tumor antigen epitopes, BMC Immunol. 18 (2017) 1–13. 10.1186/s12865-017-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kershaw MH, Westwood JA, Darcy PK, Gene-engineered T cells for cancer therapy, Nat. Rev. Cancer 13 (2013) 525–541. 10.1038/nrc3565. [DOI] [PubMed] [Google Scholar]
- [28].Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalían SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, De Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA, Cancer regression in patients after transfer of genetically engineered lymphocytes, Science (80-.). 314 (2006) 126–129. 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhao Y, Zheng Z, Robbins PF, Khong HT, Rosenberg SA, Morgan RA, Primary Human Lymphocytes Transduced with NY-ESO-1 Antigen-Specific TCR Genes Recognize and Kill Diverse Human Tumor Cell Lines, J. Immunol 174 (2005) 4415–4423. 10.4049/jimmunol.174.7.4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Morgan RA, Dudley ME, Yu YYL, Zheng Z, Robbins PF, Theoret MR, Wunderlich JR, Hughes MS, Restifo NP, Rosenberg SA, High Efficiency TCR Gene Transfer into Primary Human Lymphocytes Affords Avid Recognition of Melanoma Tumor Antigen Glycoprotein 100 and Does Not Alter the Recognition of Autologous Melanoma Antigens, J. Immunol 171 (2003) 3287–3295. 10.4049/jimmunol.171.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DAN, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, Hughes MS, Kammula US, Phan GQ, Lim RM, Wank SA, Restifo NP, Robbins PF, Laurencot CM, Rosenberg SA, T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis, Mol. Ther 19 (2011) 620–626. 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].and Lindsey CSH Drapera M, Kwonga Mei Li, Grosa Alena, Stevanovića Sanja, Trana Eric, Kerkarb Sid, Raffeldb Mark, Rosenberga Steven A., Targeting of HPV-16+ epithelial cancer cells by TCR gene engineered T cells directed against E6, Clin. Cancer Res 21 (2015) 4431–4439. 10.1016/j.physbeh.2017.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jin BY, Campbell TE, Draper LM, Stevanović S, Weissbrich B, Yu Z, Restifo NP, Rosenberg SA, Trimble CL, Hinrichs CS, Engineered T cells targeting E7 mediate regression of human papillomavirus cancers in a murine model, JCI Insight. 3 (2018) 1–12. 10.1172/jci.insight.99488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Robbins PF, Li YF, El-Gamil M, Zhao Y, Wargo JA, Zheng Z, Xu H, Morgan RA, Feldman SA, Johnson LA, Bennett AD, Dunn SM, Mahon TM, Jakobsen BK, Rosenberg SA, Single and Dual Amino Acid Substitutions in TCR CDRs Can Enhance Antigen-Specific T Cell Functions, J. Immunol 180 (2008) 6116–6131. 10.4049/jimmunol.180.9.6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Schmid DA, Irving MB, Posevitz V, Hebeisen M, Posevitz-Fejfar A, Sarria J-CF, Gomez-Eerland R, Thome M, Schumacher TNM, Romero P, Speiser DE, Zoete V, Michielin O, Rufer N, Evidence for a TCR Affinity Threshold Delimiting Maximal CD8 T Cell Function, J. Immunol 184 (2010) 4936–4946. 10.4049/jimmunol.1000173. [DOI] [PubMed] [Google Scholar]
- [36].Howie B, Sherwood AM, Berkebile AD, Berka J, Emerson RO, Williamson DW, Kirsch I, Vignali M, Rieder MJ, Carlson CS, Robins HS, High-throughput pairing of T cell receptor α and β sequences, Sci. Transl. Med 7 (2015) 1–12. 10.1126/scitranslmed.aac5624. [DOI] [PubMed] [Google Scholar]
- [37].Zoete V, Irving M, Ferber M, Cuendet MA, Michielin O, Structure-based, rational design of T cell receptors, Front. Immunol 4 (2013) 1–19. 10.3389/fimmu.2013.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hellman LM, Foley KC, Singh NK, Alonso JA, Riley TP, Devlin JR, Ayres CM, Keller GLJ, Zhang Y, Vander Kooi CW, Nishimura MI, Baker BM, Improving T Cell Receptor On-Target Specificity via Structure-Guided Design, Mol. Ther 27 (2018) 300–313. 10.1016/j.ymthe.2018.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Malecek K, Grigoryan A, Zhong S, Gu WJ, Johnson LA, Rosenberg SA, Cardozo T, Krogsgaard M, Specific Increase in Potency via Structure-Based Design of a TCR, J. Immunol 193 (2014) 2587–2599. 10.4049/jimmunol.1302344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pierce BG, Hellman LM, Hossain M, Singh NK, Vander Kooi CW, Weng Z, Baker BM, Computational Design of the Affinity and Specificity of a Therapeutic T Cell Receptor, PLoS Comput. Biol 10 (2014). 10.1371/journal.pcbi.1003478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Riley TP, Ayres CM, Hellman LM, Singh NK, Cosiano M, Cimons JM, Anderson MJ, Piepenbrink KH, Pierce BG, Weng Z, Baker BM, A generalized framework for computational design and mutational scanning of T-cell receptor binding interfaces, Protein Eng. Des. Sel 29 (2016) 595–606. 10.1093/protein/gzw050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, Lee CCR, Restifo NP, Schwarz SL, Cogdill AP, Bishop RJ, Kim H, Brewer CC, Rudy SF, VanWaes C, Davis JL, Mathur A, Ripley RT, Nathan DA, Laurencot CM, Rosenberg SA, Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen, Blood. 114 (2009) 535–546. 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Goydos JS, Patel M, Shih W, NY-ESO-1 and CTp11 expression may correlate with stage of progression in melanoma, J. Surg. Res 98 (2001) 76–80. 10.1006/jsre.2001.6148. [DOI] [PubMed] [Google Scholar]
- [44].Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, Kammula US, Hughes MS, Restifo NP, Raffeld M, Lee CCR, Levy CL, Li YF, El-Gamil M, Schwarz SL, Laurencot C, Rosenberg SA, Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1, J. Clin. Oncol 29 (2011) 917–924. 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kageyama S, Ikeda H, Miyahara Y, Imai N, Ishihara M, Saito K, Sugino S, Ueda S, Ishikawa T, Kokura S, Naota H, Ohishi K, Shiraishi T, Inoue N, Tanabe M, Kidokoro T, Yoshioka H, Tomura D, Nukaya I, Mineno J, Takesako K, Katayama N, Shiku H, Adoptive transfer of MAGE-A4 T-cell receptor gene-transduced lymphocytes in patients with recurrent esophageal cancer, Clin. Cancer Res 21 (2015) 2268–2277. 10.1158/1078-0432.CCR-14-1559. [DOI] [PubMed] [Google Scholar]
- [46].Stevanović S, Draper LM, Langhan MM, Campbell TE, Kwong ML, Wunderlich JR, Dudley ME, Yang JC, Sherry RM, Kammula US, Restifo NP, Rosenberg SA, Hinrichs CS, Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells, J. Clin. Oncol 33 (2015) 1543–1550. 10.1200/JCO.2014.58.9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Doran SL, Stevanović S, Adhikary S, Gartner JJ, Jia L, Kwong MLM, Faquin WC, Hewitt SM, Sherry RM, Yang JC, Rosenberg SA, Hinrichs CS, T-cell receptor gene therapy for human papillomavirus-associated epithelial cancers: A first-in-human, phase I/II study, J. Clin. Oncol 37 (2019) 2759–2768. 10.1200/JCO.18.02424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].González-Galarza FF, Takeshita LYC, Santos EJM, Kempson F, Maia MHT, Da Silva ALS, Teles E Silva AL, Ghattaoraya GS, Alfirevic A, Jones AR, Middleton D, Allele frequency net 2015 update: New features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations, Nucleic Acids Res. 43 (2015) D784–D788. 10.1093/nar/gku1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Seliger B, Cabrera T, Garrido F, Ferrone S, HLA class I antigen abnormalities and immune escape by malignant cells, Semin. Cancer Biol 12 (2002) 3–13. 10.1006/scbi.2001.0404. [DOI] [PubMed] [Google Scholar]
- [50].Bethune MT, Li XH, Yu J, McLaughlin J, Cheng D, Mathis C, Moreno BH, Woods K, Knights AJ, Garcia-DIaz A, Wong S, Hu-Lieskovan S, Puig-Saus C, Cebon J, Ribas A, Yang L, Witte ON, Baltimore D, Isolation and characterization of NY-ESO-1-specific T cell receptors restricted on various MHC molecules, Proc. Natl. Acad. Sci. U. S. A 115 (2018) E10702–E10711. 10.1073/pnas.1810653115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, Phan GQ, Hughes MS, Kammula US, Miller AD, Hessman CJ, Stewart AA, Restifo NP, Quezado MM, Alimchandani M, Rosenberg AZ, Nath A, Wang T, Bielekova B, Wuest SC, Akula N, McMahon FJ, Wilde S, Mosetter B, Schendel DJ, Laurencot CM, Rosenberg SA, Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy, J. Immunother 36 (2013) 133–151. 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, Binder-Scholl GK, Smethurst DP, Gerry AB, Pumphrey NJ, Bennett AD, Brewer JE, Dukes J, Harper J, Tayton-Martin HK, Jakobsen BK, Hassan NJ, Kalos M, June CH, Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma, Blood. 122 (2013) 863–871. 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Cameron BJ, Gerry AB, Dukes J, V Harper J, Bianchi FC, Grand F, Brewer JE, Gupta M, Bossi G, Vuidepot A, Powlesland AS, Legg A, Adams J, Bennett AD, Pumphrey NJ, Williams DD, Kulikovskaya I, Levine BL, Riley JL, Varela- A, Stadtmauer EA, Rapoport AP, Linette GP, June CH, Hassan NJ, Kalos M, Jakobsen BK, Identification of a Titin-Derived HLA-A1–Presented Peptide as a Cross-Reactive Target for Engineered MAGE A3–Directed T Cells, Sci. Transl. Med 5 (2013). 10.1126/scitranslmed.3006034.Identification. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Landoni E, Smith CC, Fuca G, Chen Y, Sun C, Vincent BG, Metelitsa LS, Dotti G, Savoldo B, A High-Avidity T-cell Receptor Redirects Natural Killer T-cell Specificity and Outcompetes the Endogenous Invariant T-cell Receptor, Cancer Immunol. Res 8 (2020) 57–69. 10.1158/2326-6066.CIR-19-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sun Q, Zhang X, Wang L, Gao X, Xiong Y, Liu L, Wei F, Yang L, Ren X, T-cell receptor gene therapy targeting melanoma-associated antigen-A4 by silencing of endogenous TCR inhibits tumor growth in mice and human, Cell Death Dis. 10 (2019). 10.1038/s41419-019-1717-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Campillo-Davo D, Fujiki F, Van Den Bergh JMJ, De Reu H, Smits ELJM, Goossens H, Sugiyama H, Lion E, Berneman ZN, Van Tendeloo V, Efficient and non-genotoxic RNA-based engineering of human T cells using tumor-specific t cell receptors with minimal TCR mispairing, Front. Immunol 9 (2018) 1–14. 10.3389/fimmu.2018.02503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ahn S, Li J, Sun C, Gao K, Hirabayashi K, Li H, Savoldo B, Liu R, Dotti G, Cancer immunotherapy with T cells carrying bispecific receptors that mimic antibodies, 2019. 10.1158/2326-6066.CIR-18-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Han X, Cinay GE, Zhao Y, Guo Y, Zhang X, Wang P, Adnectin-Based Design of Chimeric Antigen Receptor for T Cell Engineering, Mol. Ther 25 (2017) 2466–2476. 10.1016/j.ymthe.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Balakrishnan A, Rajan A, Salter AI, Kosasih PL, Wu Q, Voutsinas J, Jensen MC, Pluckthun A, Riddell SR, Multispecific targeting with synthetic ankyrin repeat motif chimeric antigen receptors, Clin. Cancer Res 25 (2019) 7506–7516. 10.1158/1078-0432.CCR-19-1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hombach A, Hombach AA, Abken H, Adoptive immunotherapy with genetically engineered T cells: Modification of the IgG1 Fc spacer domain in the extracellular moiety of chimeric antigen receptors avoids off-target activation and unintended initiation of an innate immune response, Gene Ther. 17 (2010) 1206–1213. 10.1038/gt.2010.91. [DOI] [PubMed] [Google Scholar]
- [61].Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, Jensen MC, Riddell SR, The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity, Cancer Immunol. Res 3 (2015) 125–135. 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Watanabe N, Bajgain P, Sukumaran S, Ansari S, Heslop HE, Rooney CM, Brenner MK, Leen AM, Vera JF, Fine-tuning the CAR spacer improves T-cell potency, Oncoimmunology. 5 (2016) 1–14. 10.1080/2162402X.2016.1253656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE, The Optimal Antigen Response of Chimeric Antigen Receptors Harboring the CD3ζ Transmembrane Domain Is Dependent upon Incorporation of the Receptor into the Endogenous TCR/CD3 Complex, J. Immunol 184 (2010) 6938–6949. 10.4049/jimmunol.0901766. [DOI] [PubMed] [Google Scholar]
- [64].Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O’Neill A, Irlam J, Chester KA, Kemshead JT, Shaw DM, Embleton MJ, Stern PL, Gilham DE, The role of extracellular spacer regions in the optimal design of chimeric immune receptors: Evaluation of four different scFvs and antigens, J. Immunother 28 (2005) 203–211. 10.1097/01.cji.0000161397.96582.59. [DOI] [PubMed] [Google Scholar]
- [65].Eshhar Z, Waks T, Gross G, Schindler DG, Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the γ or ζ subunits of the immunoglobulin and T-cell receptors, Proc. Natl. Acad. Sci. U. S. A 90 (1993) 720–724. 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hamerman JA, Lanier LL, Inhibition of immune responses by ITAM-bearing receptors., Sci. STKE 2006 (2006) 1–8. 10.1126/stke.3202006re1. [DOI] [PubMed] [Google Scholar]
- [67].Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S, Rogers-Freezer L, Chen CC, Yang JC, Rosenberg SA, Hwu P, A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer, Clin. Cancer Res 12 (2006) 6106–6115. 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Savoldo B, Ramos CA, Enli Liu MPM, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z, Liu H, Grilley B, Rooney CM, Heslop HE, Brenner MK, Dotti G, CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients, J. Clin. Invest 121 (2011) 1–5. 10.1172/JCI46110DS1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, Campana D, Riley JL, Grupp SA, June CH, Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo, Mol. Ther 17 (2009) 1453–1464. 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Van Der Stegen SJC, Hamieh M, Sadelain M, The pharmacology of second-generation chimeric antigen receptors, Nat. Rev. Drug Discov 14 (2015) 499–509. 10.1038/nrd4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, Patel PR, Guedan S, Scholler J, Keith B, Snyder N, Blair I, Milone MC, June CH, Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells, Immunity. 44 (2016) 380–390. 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- [72].Sun C, Shou P, Du H, Hirabayashi K, Chen Y, Herring LE, Ahn S, Xu Y, Suzuki K, Li G, Tsahouridis O, Su L, Savoldo B, Dotti G, THEMIS-SHP1 Recruitment by 4–1BB Tunes LCK-Mediated Priming of Chimeric Antigen Receptor-Redirected T Cells, Cancer Cell. 37 (2020) 216–225.e6. 10.1016/j.ccell.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M, Chimeric antigen receptors combining 4–1BB and CD28 signaling domains augment PI 3 kinase/AKT/Bcl-X L activation and CD8 T cell-mediated tumor eradication, Mol. Ther 18 (2010) 413–420. 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Yi Z, Prinzing BL, Cao F, Gottschalk S, Krenciute G, Optimizing EphA2-CAR T Cells for the Adoptive Immunotherapy of Glioma, Mol. Ther. - Methods Clin. Dev 9 (2018) 70–80. 10.1016/j.omtm.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ramos CA, Rouce R, Robertson CS, Reyna A, Narala N, Vyas G, Mehta B, Zhang H, Dakhova O, Carrum G, Kamble RT, Gee AP, Mei Z, Wu MF, Liu H, Grilley B, Rooney CM, Heslop HE, Brenner MK, Savoldo B, Dotti G, In Vivo Fate and Activity of Second-versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas, Mol. Ther 26 (2018) 2727–2737. 10.1016/j.ymthe.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, Vogl DT, Weiss BM, Dengel K, Nelson A, Plesa G, Chen F, Davis MM, Hwang WT, Young RM, Brogdon JL, Isaacs R, Pruteanu-Malinici I, Siegel DL, Levine BL, June CH, Milone MC, B cell maturation antigen–specific CAR T cells are clinically active in multiple myeloma, J. Clin. Invest 129 (2019) 2210–2221. 10.1172/JCI126397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Holstein SA, Lunning MA, CAR T-Cell Therapy in Hematologic Malignancies: A Voyage in Progress, Clin. Pharmacol. Ther 107 (2020) 112–122. 10.1002/cpt.1674. [DOI] [PubMed] [Google Scholar]
- [78].Xu J, Chen LJ, Yang SS, Sun Y, Wu W, Liu YF, Xu J, Zhuang Y, Zhang W, Weng XQ, Wu J, Wang Y, Wang J, Yan H, Bin Xu W, Jiang H, Du J, Ding XY, Li B, Li JM, Fu WJ, Zhu J, Zhu L, Chen Z, Fan XH, Hou J, Li JY, Mi JQ, Chen SJ, Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma, Proc. Natl. Acad. Sci. U. S. A 116 (2019) 9543–9551. 10.1073/pnas.1819745116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Wang CM, Wu ZQ, Wang Y, Guo YL, Dai HR, Wang XH, Li X, Zhang YJ, Zhang WY, Chen MX, Zhang Y, Feng KC, Liu Y, Li SX, Yang QM, Han WD, Autologous T cells expressing CD30 chimeric antigen receptors for relapsed or refractory hodgkin lymphoma: An open-label phase i trial, Clin. Cancer Res 23 (2017) 1156–1166. 10.1158/1078-0432.CCR-16-1365. [DOI] [PubMed] [Google Scholar]
- [80].Ramos CA, Ballard B, Zhang H, Dakhova O, Gee AP, Mei Z, Bilgi M, Wu M-F, Liu H, Grilley B, Bollard CM, Chang BH, Rooney CM, Brenner MK, Heslop HE, Dotti G, Savoldo B, Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes, J. Clin. Invest 127 (2017) 3462–3471. 10.1172/JCI94306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Ramos CA, Grover NS, Beaven AW, Lulla PD, Wu M-F, Ivanova A, Wang T, Shea TC, Rooney CM, Dittus C, Park SI, Gee AP, Eldridge PW, McKay KL, Mehta B, Cheng CJ, Buchanan FB, Grilley BJ, Morrison K, Brenner MK, Serody JS, Dotti G, Heslop HE, Savoldo B, Anti-CD30 CAR-T Cell Therapy in Relapsed and Refractory Hodgkin Lymphoma, J. Clin. Oncol (2020) JCO.20.01342. 10.1200/jco.20.01342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, Jäger U, Jaglowski S, Andreadis C, Westin JR, Fleury I, Bachanova V, Foley SR, Ho PJ, Mielke S, Magenau JM, Holte H, Pantano S, Pacaud LB, Awasthi R, Chu J, Anak Ö, Salles G, Maziarz RT, Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma, N. Engl. J. Med 380 (2019) 45–56. 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- [83].Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, Timmerman JM, Stiff PJ, Friedberg JW, Flinn IW, Goy A, Hill BT, Smith MR, Deol A, Farooq U, McSweeney P, Munoz J, Avivi I, Castro JE, Westin JR, Chavez JC, Ghobadi A, Komanduri KV, Levy R, Jacobsen ED, Witzig TE, Reagan P, Bot A, Rossi J, Navale L, Jiang Y, Aycock J, Elias M, Chang D, Wiezorek J, Go WY, Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-Cell lymphoma, N. Engl. J. Med 377 (2017) 2531–2544. 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA, Chimeric antigen receptor T cells for sustained remissions in leukemia, N. Engl. J. Med 371 (2014) 1507–1517. 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ramos CA, Heslop HE, Dotti G, Ramos CA, Savoldo B, Torrano V, Ballard B, Zhang H, Dakhova O, Liu E, Carrum G, Kamble RT, Gee AP, Mei Z, Wu M, Liu H, Grilley B, Rooney CM, Clinical responses with T lymphocytes targeting malignancy-associated k light chains Find the latest version : Clinical responses with T lymphocytes targeting malignancy-associated κ light chains, J. Clin. Invest 126 (2016) 2588–2596. 10.1172/JCI86000.chronic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Mardiana S, Gill S, CAR T Cells for Acute Myeloid Leukemia: State of the Art and Future Directions, Front. Oncol 10 (2020) 1–12. 10.3389/fonc.2020.00697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Alcantara M, Tesio M, June CH, Houot R, CAR T-cells for T-cell malignancies: challenges in distinguishing between therapeutic, normal, and neoplastic T-cells, Leukemia. 32 (2018) 2307–2315. 10.1038/s41375-018-0285-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Yu H, Sotillo E, Harrington C, Wertheim G, Paessler M, Maude S, Rheingold S, A Grupp S, Thomas-Tikhonenko A, Pillai V, Repeated loss of target surface antigen after immunotherapy in primary mediastinal large B cell lymphoma, Am. J. Hematol 92 (2017) E10–E11. 10.1002/ajh.24590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, Smithers H, Jensen MC, Riddell SR, Maloney DG, Turtle CJ, Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy, Blood. 127 (2016) 2406–2410. 10.1182/blood-2015-08-665547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, Klichinsky M, Shestova O, Patel PR, Kulikovskaya I, Nazimuddin F, Bhoj VG, Orlando EJ, Fry TJ, Bitter H, Maude SL, Levine BL, Nobles CL, Bushman FD, Young RM, Scholler J, Gill SI, June CH, Grupp SA, Lacey SF, Melenhorst JJ, Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell, Nat. Med 24 (2018) 1499–1503. 10.1038/s41591-018-0201-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Lamers CHJ, Sleijfer S, Vulto AG, Kruit WHJ, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E, Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience., J. Clin. Oncol 24 (2006). 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- [92].Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA, Case report of a serious adverse event following the administration of t cells transduced with a chimeric antigen receptor recognizing ERBB2, Mol. Ther 18 (2010) 843–851. 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, Mades A, Sadelain M, Einsele H, Hudecek M, The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells, Sci. Transl. Med 11 (2019). 10.1126/scitranslmed.aau5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Weber EW, Lynn RC, Sotillo E, Lattin J, Xu P, Mackall CL, Pharmacologic control of CAR-T cell function using dasatinib, Blood Adv. 3 (2019) 711–717. 10.1182/bloodadvances.2018028720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M, Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells, Nat. Biotechnol 31 (2013) 71–75. 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, Yu J, Scholler N, Powell DJ, A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor, Cancer Res. 72 (2012) 1844–1852. 10.1158/0008-5472.CAN-11-3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Cho JH, Collins JJ, Wong WW, Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses, Cell. 173 (2018) 1426–1438.e11. 10.1016/j.cell.2018.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, Duclert A, Duchateau P, Poirot L, An oxygen sensitive self-decision making engineered CAR T-cell, Sci. Rep 7 (2017) 1–8. 10.1038/srep39833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, Lim WA, Precision Tumor Recognition by T Cells with Combinatorial Antigen-Sensing Circuits, Cell. 164 (2016) 770–779. 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, Liu H, Cruz CR, Savoldo B, Gee AP, Schindler J, Krance RA, Heslop HE, Spencer DM, Rooney CM, Brenner MK, Inducible apoptosis as a safety switch for adoptive cell therapy, N. Engl. J. Med 365 (2011) 1673–1683. 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Diaconu I, Ballard B, Zhang M, Chen Y, West J, Dotti G, Savoldo B, Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells, Mol. Ther 25 (2017) 580–592. 10.1016/j.ymthe.2017.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Feuerstein BG, Waldman FM, Feuerstein BG, Benz CC, Szöllösi J, Balázs M, ERBB-2 (HER2/neu) Gene Copy Number, p185HER2 Overexpression, and Intratumor Heterogeneity in Human Breast Cancer, Cancer Res. 55 (1995) 5400–5407. [PubMed] [Google Scholar]
- [103].Harris DT, Hager MV, Smith SN, Cai Q, Stone JD, Kruger P, Lever M, Dushek O, Schmitt TM, Greenberg PD, Kranz DM, Comparison of T Cell Activities Mediated by Human TCRs and CARs That Use the Same Recognition Domains, J. Immunol 200 (2018) 1088–1100. 10.4049/jimmunol.1700236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, Myklebust JH, Kadapakkam M, Weber EW, Tousley AM, Richards RM, Heitzeneder S, Nguyen SM, Wiebking V, Theruvath J, Lynn RC, Xu P, Dunn AR, Vale RD, Mackall CL, Tuning the Antigen Density Requirement for CAR T-cell Activity, Cancer Discov. 10 (2020) 702–723. 10.1158/2159-8290.CD-19-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, Zhang YJ, Baskin DS, Merchant FA, Brawley VS, Byrd TT, Krebs S, Wu MF, Liu H, Heslop HE, Gottachalk S, Yvon E, Ahmed N, Combinational Targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma, Mol. Ther 21 (2013) 2087–2101. 10.1038/mt.2013.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Fernández de Larrea C, Staehr M, Lopez AV, Ng KY, Chen Y, Godfrey WD, Purdon TJ, Ponomarev V, Wendel H-G, Brentjens RJ, Smith EL, Defining an Optimal Dual-Targeted CAR T-cell Therapy Approach Simultaneously Targeting BCMA and GPRC5D to Prevent BCMA Escape–Driven Relapse in Multiple Myeloma, Blood Cancer Discov. 1 (2020) 146–154. 10.1158/2643-3230.bcd-20-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, Kurien A, Priceman SJ, Wang X, Harshbarger TL, D’Apuzzo M, Ressler JA, Jensen MC, Barish ME, Chen M, Portnow J, Forman SJ, Badie B, Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy, N. Engl. J. Med 375 (2016) 2561–2569. 10.1056/nejmoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, Khare PD, Thorn M, Ma Q, Stainken BF, Assanah EO, Davies R, Espat NJ, Junghans RP, Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases, Clin. Cancer Res 21 (2015) 3149–3159. 10.1158/1078-0432.CCR-14-1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Peng W, Ye Y, Rabinovich BA, Liu C, Lou Y, Zhang M, Whittington M, Yang Y, Overwijk WW, Lizée G, Hwu P, Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses, Clin. Cancer Res 16 (2010) 5458–5468. 10.1158/1078-0432.CCR-10-0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, Bouchier-Hayes L, Savoldo B, Dotti G, Armed Oncolytic Virus Enhances Immune Functions of Chimeric Antigen Receptor-Modified T Cells in Solid Tumors, Cancer Res. 74 (2014) 5195–5205. 10.1158/0008-5472.CAN-14-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, Foster AE, Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b, J. Immunother 33 (2010) 780–788. 10.1097/CJI.0b013e3181ee6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Luo H, Su J, Sun R, Sun Y, Wang Y, Dong Y, Shi B, Jiang H, Li Z, Coexpression of IL7 and CCL21 Increases Efficacy of CAR-T Cells in Solid Tumors without Requiring Preconditioned Lymphodepletion, Clin. Cancer Res 26 (2020) 5494–5505. 10.1158/1078-0432.ccr-20-0777. [DOI] [PubMed] [Google Scholar]
- [113].Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K, IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor, Nat. Biotechnol 36 (2018) 346–351. 10.1038/nbt.4086. [DOI] [PubMed] [Google Scholar]
- [114].Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, Heslop HE, Brenner MK, Dotti G, Savoldo B, T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model, Blood. 113 (2009) 6392–6402. 10.1182/blood-2009-03-209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Moon EK, Wang LCS, Bekdache K, Lynn RC, Lo A, Thorne SH, Albelda SM, Intratumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines, Oncoimmunology. 7 (2018). 10.1080/2162402X.2017.1395997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Pickup MW, Mouw JK, Weaver VM, The extracellular matrix modulates the hallmarks of cancer, EMBO Rep. 15 (2014) 1243–1253. 10.15252/embr.201439246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, Kapoor V, Scholler J, Puré E, Milone MC, June CH, Riley JL, Wherry EJ, Albelda SM, Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors, Clin. Cancer Res 20 (2014) 4262–4273. 10.1158/1078-0432.CCR-13-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, Ittmann MM, Marchetti D, Dotti G, Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes, Nat. Med 21 (2015) 524–529. 10.1038/nm.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Mbongue JC, Nicholas DA, Torrez TW, Kim NS, Firek AF, Langridge WHR, The role of indoleamine 2, 3-dioxygenase in immune suppression and autoimmunity, Vaccines. 3 (2015) 703–729. 10.3390/vaccines3030703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Munn DH, Mellor AL, Indoleamine 2,3 dioxygenase and metabolic control of immune responses, Trends Immunol. 34 (2013) 137–143. 10.1016/j.it.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, Orabona C, Bianchi R, Belladonna ML, Volpi C, Santamaria P, Fioretti MC, Puccetti P, The Combined Effects of Tryptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor ζ-Chain and Induce a Regulatory Phenotype in Naive T Cells, J. Immunol 176 (2006) 6752–6761. 10.4049/jimmunol.176.11.6752. [DOI] [PubMed] [Google Scholar]
- [122].Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB, Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase, J. Exp. Med 196 (2002) 459–468. 10.1084/jem.20020121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, Heslop HE, Brenner MK, Rooney CM, Ramos CA, Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs, Blood. 125 (2015) 3905–3916. 10.1182/blood-2015-01-621474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, Merghoub T, Wolchok JD, Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner, Cell Rep. 13 (2015) 412–424. 10.1016/j.celrep.2015.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Serafini P, Borrello I, Bronte V, Myeloid suppressor cells in cancer: Recruitment, phenotype, properties, and mechanisms of immune suppression, Semin. Cancer Biol 16 (2006) 53–65. 10.1016/j.semcancer.2005.07.005. [DOI] [PubMed] [Google Scholar]
- [126].Zhang Y, Ertl HCJ, Starved and asphyxiated: How can CD8+ T cells within a tumor microenvironment prevent tumor progression, Front. Immunol 7 (2016) 1–7. 10.3389/fimmu.2016.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Wang R, Green DR, Metabolic checkpoints in activated T cells, Nat. Immunol 13 (2012) 907–915. 10.1038/ni.2386. [DOI] [PubMed] [Google Scholar]
- [128].Ma X, Shou P, Smith C, Chen Y, Du H, Sun C, Porterfield Kren N, Michaud D, Ahn S, Vincent B, Savoldo B, Pylayeva-Gupta Y, Zhang S, Dotti G, Xu Y, Interleukin-23 engineering improves CAR T cell function in solid tumors, Nat. Biotechnol (2020) 10.1038/s41587-019-0398-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Garcia-Manero G, Yang H, Kuang S-Q, O’Brien S, Thomas D, Kantarjian H, Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy, Trends Immunol. 26 (2005) 111–117. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3624763/pdf/nihms412728.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Su Y, Huang X, Raskovalova T, Zacharia L, Lokshin A, Jackson E, Gorelik E, Cooperation of adenosine and prostaglandin E2 (PGE2) in amplification of cAMP-PKA signaling and immunosuppression, Cancer Immunol. Immunother 57 (2008) 1611–1623. 10.1007/s00262-008-0494-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Newick K, O’brien S, Sun J, Kapoor V, Maceyko S, Lo A, Puré E, Moon E, Albelda SM, Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase a localization, Cancer Immunol. Res 4 (2016) 541–551. 10.1158/2326-6066.CIR-15-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Levy L, Hill CS, Alterations in components of the TGF-β superfamily signaling pathways in human cancer, Cytokine Growth Factor Rev. 17 (2006) 41–58. 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- [133].Ogden CA, Pound JD, Batth BK, Owens S, Johannessen I, Wood K, Gregory CD, Enhanced Apoptotic Cell Clearance Capacity and B Cell Survival Factor Production by IL-10-Activated Macrophages: Implications for Burkitt’s Lymphoma, J. Immunol 174 (2005) 3015–3023. 10.4049/jimmunol.174.5.3015. [DOI] [PubMed] [Google Scholar]
- [134].ei Ito S, Shirota H, Kasahara Y, Saijo K, Ishioka C, IL-4 blockade alters the tumor microenvironment and augments the response to cancer immunotherapy in a mouse model, Cancer Immunol. Immunother 66 (2017) 1485–1496. 10.1007/s00262-017-2043-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Chevaun WPS Morrison D, Parvani Jenny G, The relevance of the TGF-β Paradox to EMT-MET program, Cancer Lett. 23 (2013) 1–7. 10.1038/jid.2014.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Hamidullah B Changkija, R. Konwar, Role of interleukin-10 in breast cancer, Breast Cancer Res. Treat 133 (2012) 11–21. 10.1007/s10549-011-1855-x. [DOI] [PubMed] [Google Scholar]
- [137].Zhang L, Yu Z, Muranski P, Palmer DC, Restifo NP, Rosenberg SA, Morgan RA, Inhibition of TGF-β signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy, Gene Ther. 20 (2013) 575–580. 10.1038/gt.2012.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Lacuesta K, Buza E, Hauser H, Granville L, Pule M, Corboy G, Finegold M, Weiss H, Chen SY, Brenner MK, Heslop HE, Rooney CM, Bollard CM, Assessing the safety of cytotoxic T lymphocytes transduced with a dominant negative transforming growth factor-β receptor, J. Immunother 29 (2006) 250–260. 10.1097/01.cji.0000192104.24583.ca. [DOI] [PubMed] [Google Scholar]
- [139].Bollard CM, Tripic T, Cruz CR, Dotti G, Gottschalk S, Torrano V, Dakhova O, Carrum G, Ramos CA, Liu H, Wu MF, Marcogliese AN, Barese C, Zu Y, Lee DY, O’Connor O, Gee AP, Brenner MK, Heslop HE, Rooney CM, Tumor-specific t-cells engineered to overcome tumor immune evasion induce clinical responses in patients with relapsed hodgkin lymphoma, J. Clin. Oncol 36 (2018) 1128–1139. 10.1200/JCO.2017.74.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Leen AM, Sukumaran S, Watanabe N, Mohammed S, Keirnan J, Yanagisawa R, Anurathapan U, Rendon D, Heslop HE, Rooney CM, Brenner MK, Vera JF, Reversal of Tumor Immune Inhibition Using a Chimeric Cytokine Receptor, Mol. Ther 22 (2014) 1211–1220. 10.1038/mt.2014.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Egen JG, Kuhns MS, Allison JP, CTLA-4: New insights into its biological function and use in tumor immunotherapy, Nat. Immunol 3 (2002) 611–618. 10.1038/ni0702-611. [DOI] [PubMed] [Google Scholar]
- [142].Hodi, M.D. FS, O’Day, M.D. SJ, McDermott, M.D. DF, Weber, M.D. RW, Sosman, M.D. JA, Haanen, M.D. JB, Gonzalez, M.D. R, Robert, M.D. C, Schadendorf, M.D. D, Hassel, M.D. JC, Akerley, M.D. W, Van den Eertwegh, M.D., Ph.D. AJM, Lutzky, M.D. J, Lorigan, M.D. P, Vaubel, M.D. JM, Linette, M.D., Ph.D. GP, Hogg, M.D. D, Ottensmeier, M., P.D. CH Yellin, M.D. MJ, Nichol, M.B., Ch.B. GM, Hoos, M.D., Ph.D. A, Urba, M.D. WJ, Improved Survival with Ipilimumab in Patients with Metastatic Melanoma, N. Engl. J. Med 363 (2010) 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA, Selective effects of PD-1 on Akt and ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation, Sci. Signal 5 (2012). 10.1126/scisignal.2002796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF, Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells, Sci. Transl. Med 5 (2013). 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Brahmer JR, Tykodi SS, Chow LQM, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM, Safety and activity of anti-PD-L1 antibody in patients with advanced cancer, N. Engl. J. Med 366 (2012) 2455–2465. 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].John LB, Devaud C, Duong CPM, Yong CS, Beavis PA, Haynes NM, Chow MT, Smyth MJ, Kershaw MH, Darcy PK, Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells, Clin. Cancer Res 19 (2013) 5636–5646. 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- [147].Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, Liu H, Wu MF, Mei Z, Gee A, Mehta B, Zhang H, Mahmood N, Tashiro H, Heslop HE, Dotti G, Rooney CM, Brenner MK, CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma, Mol. Ther 25 (2017) 2214–2224. 10.1016/j.ymthe.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, Song M, Miele MM, Li Z, Wang P, Yan S, Xiang J, Ma X, Seshan VE, Hendrickson RC, Liu C, Brentjens RJ, Targeted delivery of a PD-1-blocking scFV by CAR-T cells enhances anti-tumor efficacy in vivo, Nat. Biotechnol 36 (2018) 847–858. 10.1038/nbt.4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, Newick K, Lo A, June CH, Zhao Y, Moon EK, A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors, Cancer Res. 76 (2016) 1578–1590. 10.1158/0008-5472.CAN-15-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]