

Abstract
A novel gas-phase charge and mass manipulation approach is demonstrated to facilitate the mass measurement of high mass complexes within the context of native mass spectrometry. Electrospray ionization applied to solutions generated under native or near-native conditions has been demonstrated to be capable of preserving biologically relevant complexes into the gas phase as multiply-charged ions suitable for mass spectrometric analysis. However, charge state distributions tend to be narrow and extensive salt adduction, heterogeneity, etc. tend to lead to significantly broadened peaks. These issues can compromise mass measurement of high mass bio-complexes, particularly when charge states are not clearly resolved. In this work, we show that the attachment of high mass ions of known mass and charge to populations of ions of interest can lead to well-separated signals that can yield confident charge state and mass assignments from otherwise poorly resolved signals.
Keywords: Native mass spectrometry, ion/ion chemistry, E. coli ribosome, multiply-charged ion attachment
Graphical Abstract
The development of native MS (nMS) has enabled the mass measurement of large biological complexes. Inherent challenges in applying nMS to study large systems are extensive salt adduction and heterogeneity (as seen in the E. coli ribosome particles we studied), which can preclude mass determination of the complex. We present an alternative method to extract mass information from these complicated systems using an ion/ion reaction workflow that involves the attachment of multiply charged ions of known mass and charge.
INTRODUCTION
The observation of gaseous ions comprised of specific non-covalently bound components known to exist together in solution (1,2,3,4) was a remarkable development in electrospray ionization (ESI) mass spectrometry (MS). It indicates the possibility of generating gaseous ions from specific non-covalently bound protein-ligand, protein-protein, and protein-nucleic acid complexes for subsequent characterization by mass spectrometry and ancillary techniques. Since then, a branch of biological mass spectrometry, often referred to as ‘Native MS’, has emerged for the study of large non-covalent complexes (2,3,5,6,7,8). Native MS encompasses mass spectrometry, tandem mass spectrometry (9,10), and ion mobility measurements (11) applied to ions derived from complexes subjected to ESI under conditions designed to preserve the complexes of interest as gas-phase ions. Challenges associated with native MS include the fact that, despite the multiple charging associated with ESI, ions are of relatively high mass-to-charge (m/z) ratios and are generated with relatively narrow charge state distributions, presumably due to their formation via the so-called ‘charged residue mechanism’ (12,13,14). Therefore, technologies amenable to high m/z measurements, such as mass filters operated at low frequency for ion isolation and TOF or electrostatic traps for m/z measurement, are often employed (15,16). Ultimately, the complexity of the sample, which can arise from mixtures of counter-ions, salts and other adducts, and the native heterogeneity of the complex (e.g., post-translational modifications (PTMs), varying identities of components in the complex, etc.) can render the native MS uninterpretable due to an inability to resolve and/or identify charge states. One way to address this problem is to measure m/z and z simultaneously on an ion-by-ion basis, which is the approach taken by charge detection MS (17,18). We present an alternative approach to dealing with the heterogeneity problem that does not rely on single ion analysis/detection and that retains the potential for tandem mass spectrometry and ion mobility using platforms currently in use for native MS with relatively minor modification.
The underlying concept relies on simplifying the mixture of ions generated by ESI via mass-selection of a fraction of the broad distribution of m/z ratios present in the original ESI mass spectrum and subjecting this mixture to an ion attachment reaction that gives rise to a known Δm and Δz. This idea has been described for proton transfer ion/molecule (19) and ion/ion (20) reactions in which Δm = 1 Da and Δz = 1 unit charge. The idea has also been demonstrated with electron transfer ion/ion reaction reagents (21,22). Such small changes in mass and charge, however, can be difficult to resolve when they occur with highly charged high mass heterogeneous mixtures. We describe here ion attachment ion/ion reactions in which Δm and Δz are both much greater than 1 Da and 1 unit charge, respectively. This form of charge and mass manipulation leads to large m/z separations that are much more readily resolved than those associated with single proton or electron transfer. The idea is demonstrated here using a modified commercial quadrupole/time-of-flight (QTOF) tandem mass spectrometer. However, these experiments can, in principle, be executed with any native MS platform capable of executing ion/ion reaction experiments.
Experimental
Sample preparation for native mass spectrometry of bio-complexes.
Rabbit pyruvate kinase was purchased from Sigma Aldrich. The lyophilized solid was reconstituted in water to create a stock solution at a concentration of 3 μM (calculated by using the mass of the tetramer). The sample underwent buffer exchange, via centrifugation, once with an ammonium acetate (Sigma Aldrich) buffer adjusted to pH 7 with ammonium hydroxide (Sigma Aldrich), using a 100 kilodalton (kDa) molecular weight cutoff (MWCO) Amicon Ultra 0.5 mL filter (Millipore Sigma). The recovered sample (15 μL) was diluted with the same buffer to achieve the same original concentration from the stock solution. Ferritin from equine spleen (Sigma Aldrich) lyophilized powder was prepared (3 μM) in a similar manner to pyruvate kinase. GroEL (Sigma Aldrich) lyophilized powder preparation was described before in detail, and made to a 1 μM (23). Briefly, the sample was buffer exchanged and underwent acetone precipitation.
E. coli 70S ribosome solution was purchased from New England Biolabs. The original sample, with an initial concentration of 13 μM, was constituted in a buffer containing 10 mM magnesium acetate, which is necessary for the 70S ribosome to be intact in the condensed phase. The sample preparation for the working solutions was described in detail previously (24) and modified accordingly. Briefly, the sample was buffer exchanged 8 times with 150 mM ammonium acetate and 10 mM magnesium acetate (Sigma Aldrich) with the same filter used in the aforementioned section. It is important to note a difference in the preparations. We utilized our reagent anion to perform the charge reduction of the protein in the gas phase, therefore we did not employ the use of the reducing agent, triethylammonium acetate, to the solution.
Sample preparation for the reagent anions.
Oxidized insulin chain A (IcA) from bovine pancreas (average mass, free acid = 2531.66 Da) was purchased from Sigma Aldrich. The lyophilized solid was reconstituted in water. The working solution was a 50:50 mixture of aqueous ammonium hydroxide (pH 11) and HPLC grade methanol (Sigma Aldrich) to give a final concentration of 25 μM. The most abundant charge states were [IcA-5H]5− and [IcA-6H]6−. In experiments where a larger charge reduction was required, we utilized myoglobin from equine skeletal muscle (Sigma Aldrich). Holo-myoglobin (hMb) (average mass=17,567 Da), which retains the heme group, was prepared with piperidine (Sigma Aldrich). We isolated and used the two different charge states in the experiments shown in this manuscript. The final solution was 20 μM holo-myoglobin and 50 mM piperidine in water.
Ion-ion reactions in the mass spectrometer.
All experiments were performed using a QTOF 5600 (SCIEX), which was previously modified to allow for ion/ion reactions (25). The positive and negative ions were generated by utilizing an alternately pulsed nano-electrospray ionization source (nESI) (26). Mutual ion polarity trapping was enabled by applying a supplemental AC to the end plates of the linear ion trap reaction cell, q2. Multiply protonated protein cations and reagent anions were sequentially isolated before being stored in q2 to react over times of 50–100 ms. The reagent anions were isolated during Q1 transmission by conventional RF-DC apex isolation. The protein cations were too high in m/z to be isolated via conventional RF-DC apex isolation. Therefore, sequential resonance ejection ramps in q2 were used to eject ions of lower and higher m/z ratios that bordered the population of interest (27).
Native MS.
Both analyte and reagent ions were generated via nESI from separately pulsed borosilicate glass emitters (28). The large bio-complexes were sprayed in positive mode with approximately +1500 V applied to a platinum wire, which was in contact with the solution. The reagent anions were sprayed in negative mode with an applied voltage of approximately −1400 V. The large complex ions were initially injected into the instrument and isolated (see above) and stored in q2. Subsequently, the reagent anions were injected and isolated during Q1 transmission before being accumulated and stored in q2. The mutual storage time can be adjusted to control the number of sequential adductions. Nitrogen gas was used in q2 at pressures ranging from 6–8 mtorr. Due to the size of the ribosome particles, the difference in DC offsets of Q0 and q2 were increased to as high as 50–70 V to collisionally activate the ions upon injection into q2 to drive off weakly-bound adduct species. A Q0-q2 voltage difference of 5–15 V was used for the reagent anions as minimal salt adduction was observed for these species. Mass calibration over a wide range was done in two stages, using primary and secondary calibrants. Cesium iodide, the primary calibrant, was used to calibrate the instrument over the range of m/z 1,000–10,000. This allowed for an accurate mass determination of the pyruvate kinase tetramer, the secondary calibrant, from the mass spectrum derived under native conditions (see insert to Figure 2a). Finally, the population of pyruvate kinase charge states was reacted with the oxidized insulin chain A, [IcA-6H]6− anion ([IcA-6H]−; Δm = 2526 Da) resulting in a series of peaks extending beyond m/z 100,000. This experiment allowed for the mass scale to be calibrated over the range of m/z 6,500–100,000.
Figure 2.

Inset (upper right) Positive nESI mass spectrum of pyruvate kinase a) Post-ion/ion mass spectrum after reaction of the 32+ ions of pyruvate kinase with [IcA-6H]6− anions derived from nESI of IcA. b) Post-ion/ion mass spectrum after reaction of the 32+ ions of pyruvate kinase with the [hMb-13H]13− anions derived from nESI of holo-myoglobin.
Results and Discussion
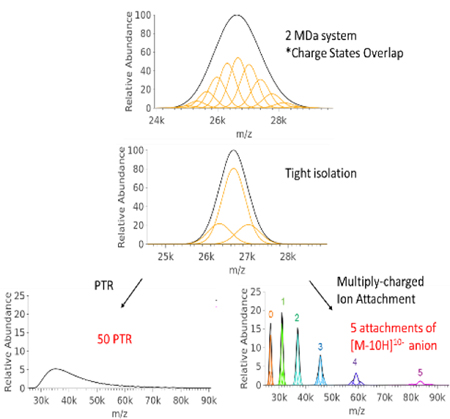
The concept is modeled in Figure 1 for a cationic mixture of ions generated from a particle of average mass 2 MDa, width of 50 kDa (FWHM), charge state range of 70+ to 80+, and analyzer resolution of 10,000 FWHM (see Supplemental Information for a description of the model). Under these conditions, the ESI mass spectrum shows a large unresolved envelope (Figure 1a), which precludes a mass measurement due to a lack of information regarding charge. Figure 1b shows the simulated spectrum after an isolation step centered at the maximum of the distribution with a FWHM width of 900 m/z units. Figure 1c shows the simulated spectrum following up to fifty single proton transfer steps, as might result from ion/ion reactions of the multiply-charged ions with singly charged anions of opposite polarity (20). Figure 1d shows the result after the attachment of up to 5 ions of mass 17,557 Da and unit charge of 10-. This Δm and Δz apply to the 10- charge of hMb, which is one of the reagents used for the data reported here (see below). After the isolation of ions in a selected region of the mass spectrum (Figure 1b), multiple single proton transfer reactions tend to spread the signal among many products (Figure 1c) without baseline resolution of adjacent peaks until close to 50 proton transfers. However, after the attachment of up to five high mass reagent anions of z = 10-, baseline-resolved peaks are apparent that can be used to determine the charge states of the ions that make up the centroids of the peaks without spreading the signal across much of the m/z range. After the fourth attachment, even the individual charge states within the initially selected ion population are beginning to separate sufficiently to be resolved.
Figure 1.

Simulated mass spectra of (a) a hypothetical 2 MDa (50 kDa FWHM) analyte particle with 70 to 80 charges, (b) selection (900 FHWM) of most abundant charge states, (c) up to 50 proton transfer reactions of the selected analyte charge states, and (d) up to 5 attachments (indicated by colored numbers) of the 10− charge states holomyoglobin (17,567 Da) to the selected analyte charge states. Abundance scales in (c) and (d) are relative to (b).
The determination of charge states associated with the peak centroids in Figure 1d, which involves the attachment of reagent ions of known mass and charge to the high mass complex ions, requires a generalization of the relationships originally provided by Mann et al. (29) for the determination of charges from the m/z values of adjacent charge states in an electrospray mass spectrum. For positively charged complexes, the m/z value for a given charge state is given by:
| (1) |
where M is the mass of the neutral molecule, n is the charge state, and x is the average mass of the cationizing agent (e.g., an excess proton). The m/z value of the product generated by the attachment of a single reagent of mass = Δm and charge = Δz is given by:
| (2) |
The magnitude of the charge on the initial analyte ion, n, is given by:
| (3) |
The same relationship applies for any two adjacent attachment products such that M, the mass of the original complex represented by peak apex, can be determined from any of the attachment products via:
| (4) |
where N is the number of attached reagent anions.
It has been previously noted for relatively small proteins that reactions of oppositely charged proteins can result in the formation of a long-lived complex along with proton transfer products (30,31). When the hard-sphere cross-sections are large (i.e., when the reactants are physically large) and when the m/z ratios are large (i.e. the charge densities within the reactant ions are relatively low), the formation of long-lived complexes (i.e., the attachment of an ion to a cation), becomes the exclusive product (32). In the cases of the very large complexes encountered in native MS, ion attachment is essentially the exclusive ion/ion reaction process, provided the charge density of the reagent ion is not too high. This is illustrated in Figure 2 with ions derived from the tetrameric pyruvate kinase complex (approximately 232 kDa) in reactions with two different anionic reagents. The insert in Figure 2a shows the charge state distribution generated via positive ion nESI of pyruvate kinase under native MS conditions. The shaded charge state was selected for subsequent ion/ion reactions with a) the [IcA-6H]6− anion (average Δm = 2525.7 Da) and b) the [hMb-13H]13− anion (Δm = 17,554 Da). The attachment of up to three reagent anions is observed in the case of [IcA-6H]6− (Figure 2a) and up to two reagent anions in the case of [hMb-13H]13− (Figure 2b). (Note that the addition of three anions in the case of [hMb-13H]13− would result in the generation of a negatively-charged complex.) The tabular insert in Figure 2b summarizes the measured m/z values and calculated charges and masses arising from the two experiments using the relationships listed above. The experiment of Figure 2a yielded an average mass of 231,969±2 Da and the experiment of Figure 2b yielded an average mass of 232,134±12 Da. Key observations from this proof-of-principle example are that multiply-charged reagent anions can give rise to exclusive ion attachment (i.e., no proton transfer is observed for either reagent) and that there is a high degree of flexibility in choosing reagents over wide ranges of Δm and Δz, which provides for the ‘tunability’ of the m/z spacings between successive ion attachments. It is also noteworthy that no loss of the non-covalently-bound heme group in hMb is observed in these experiments, which indicates that ion attachment to large complexes results in little or no dissociation of the complex. Despite the large potential energy associated with the attachment of oppositely charged ions, the cooling rates associated with the environment of these ion/ion reactions is apparently high enough to avoid fragmentation of these very large ions. Results of analogous experiments using the [M-6H]6− IcA anion in reactions with cations derived from ferritin (480 kDa) and GroEL (804 kDa) are provided in Supplemental Information Figures S1 and S2, respectively. In all cases, exclusive ion attachment is observed, thereby reinforcing the generality of the observations noted in Figure 2. We note that for analyte complex ions of the sizes illustrated here we have found that every charge state of every polypeptide/protein reagent anion thus far examined has resulted exclusively in attachment. We illustrate the concept here using anions derived from hMb and IcA but point out that we are not currently aware of any polypeptide/protein anion that would not serve as a reagent for this kind of experiment.
The E. coli ribosome constitutes an example of a complex that is inherently heterogeneous (33). The protein complement can vary in stoichiometry, identities of proteins, and PTMs of proteins. There is also heterogeneity in ribosomal RNA that includes both alternative RNA and modifications of RNA. These sources of heterogeneity add to the usual mixture of counter ions, salts, etc. associated with any large bio-complex ion generated under native conditions via ESI (34). The E. coli ribosome is comprised of two components, the smaller 30S and larger 50S subunits, that associate to form the intact 70S ribosome. The presence of magnesium ions is important for the association of the two subunits and the appearance of the mass spectrum can be highly influenced by magnesium ion concentration (3). Figure 3 shows mass spectra collected at concentrations of 0.5 mM Mg2+ (Figure 3a) and 10 mM Mg2+ (Figure 3b). Consistent with previous reports (3,24), at high Mg2+ concentrations (10 mM, in this case) ions associated with the 30S and 50S subunits are present in addition to ions associated with the 70S complex, the latter of which centered at roughly m/z 29,000 (Figure 3b). In our hands, we have not observed exclusive formation of the 70S complex (i.e., no evidence for 30S and 50S cations) under the solution and ion transmission conditions we have attempted. At 0.5 mM Mg2+ (Figure 3a), the 70S signals are largely absent and the ions from the 30S and 50S subunits show somewhat narrower peaks than do the subunits with higher magnesium concentrations, presumably due to extra adduction from counter ions, and non-specific adduction of magnesium (24). In all cases, overlapping charge state distributions are evident, indicating that a mixture of species is present for each of the 30S, 50S, and 70S-related complexes. We note that there is sufficient separation between peaks in the spectra of Figure 3 to estimate the charges of the components via deconvolution. However, the limited numbers of charge states and peak overlap leads to significant uncertainty in charge state assignments (20,35).
Figure 3.

The mass spectra of the E. coli ribosome with (a) 0.5 mM and (b) 10 mM Mg2+ concentrations. The higher concentration preserves the 70S population centered around m/z 29,000.
Figure 4 summarizes the results from [hMb-10H]10− ion attachment experiments for the 30S (Figure 4a) and 50S (Figure 4b) species observed from nESI of the 10 mM Mg2+ solution. The mass spectrum is shown as an insert in Figure 4a (see also Figure 3b) and the ions selected for subsequent ion attachment are indicated within a blue box (30S) or red box (50S). The ion attachment experiment for the 30S component (Figure 4a) revealed two major components (863.2 kDa (purple dots) and 802.0 kDa (green dots), respectively) that differ in mass by that of the S1 stalk protein. The stalk protein, RS1 (61.2 kDa), is a labile component that has been observed to dissociate when the 30S ribosome is activated (3,24). The masses indicated above are roughly 1.8% greater than those expected for the intact 30S component (847.5 kDa) and 30S-S1 (786.3 kDa). The difference might be attributed to either non-specific magnesium adduction (24) and/or incomplete desolvation commonly seen on TOF platforms, which typically requires higher amounts of collisional cooling (16, 36) or the presence of an additional roughly 15 kDa component, or the combination of both. We note that at a concentration of 0.5 mM Mg2+, the average masses for the two components noted above were 861.8 kDa and 799.8 kDa, respectively, and a third major component (772.7 kDa) was observed following the ion attachment experiment which was consistent with the largest component minus both the S1 and S2 (26.7 kDa) (24) stalk proteins (see Figure S3 for a summary of the ion attachment experiment associated with the 30S related ions at 0.5 mM Mg2+).
Figure 4.

Post-ion attachment MS spectra of the E. coli ribosome subunits, (a) 30S and (b) 50S, reacting with with hMb. The concentrations of Mg2+ is 10 mM. The inset in panel (a) is the E. coli ribosome precursor spectrum, and the inset in panel (b) is the zoom-in of the 4th attachment population. Note, the colored blue (30S), red (50S) and green (70S) borders are the isolated precursor populations that were reacted with [hMb-10H]10−.
For the 50S subunit (Figure 4b), we detected three major components of masses 1,479.7±0.2 kDa (blue line), 1,413±2 kDa (green line), and 1,399.4±0.6 kDa (red line). The mass difference between the two larger components is consistent with that of the stalk complex [L10(L7/L12)4] (66.5 kDa) (37) and the mass difference between the two smaller components is consistent with that of the L11 ribosomal protein (14.9 kDa) (24). The stalk complex for the 50S subunit is believed to contain multiple dimers (L7/L12)4 (38,39). The dimers are bound to the L10 ribosomal protein, which is also bound to the L11 protein. Consequently, the loss of these components is plausible. The 1,479.7 kDa component is roughly 1.7% higher in mass than the nominal mass of the 50S component (24), which could be due to the presence of additional protein component(s), salts, etc. These components were not well separated after the first two attachments but were resolved in the 3rd and 4th attachments. An expanded region highlighting the 4th [hMb-10H]10− attachment is provided as an insert in Figure 4b. Ion attachment data for the 50S-related ions generated at a concentration of 0.5 mM Mg2+ is provided in Figure S4. No evidence for the two larger intact components noted in Figure 4b was noted at lower Mg2+ concentration. Rather, a fourth component (1,389.5 kDa) was found to be most abundant along with the 1399.7 kDa component (see Figure S4). A prominent signal for a 50S component at relatively low Mg2+ concentration of 1,389.6 kDa has been noted previously and was ascribed to (50S-L10(RL7/12)4) (24). The fact that we see this component as well as larger components might suggest that one or more additional components may be associated with the 50S particle at high Mg2+ concentrations.
Both examples illustrated in Figure 4 involve the presence of mixtures of complexes. A complication in native MS is the fact that multiple combinations of mass and charge can give rise to similar m/z ratios over the narrow charge state ranges typically generated under native MS conditions. This can complicate charge state assignment as well as the identification that mixtures are present. However, ions of significantly different mass will not overlap over wide ranges of charge. The ion attachment approach described here allows for a very wide range of charge to be accessed, thereby resulting the separation of components somewhere along the m/z scale. Regions of significant overlap of the mixture components identified for both the 30S and 50S examples were observed prior to and after one or more ion attachments. However, in both cases, one or more attachments (e.g., attachments 3 and 4) showed separation of the components. A similar situation prevails for the 70S-related ions.
The extent of heterogeneity for both the 30S and 50S subunits can propagate to the 70S-related ions, so there is little surprise that overlapping mixture components appear to be present in the 70S region of the mass spectrum shown in the insert of Figure 4a. A similar result was found in both the E. coli (24) and Thermus thermophilus (40) ribosomes. In our 70S ribosome ion attachment mass spectrum (Figure 5), we used a tighter isolation and thus greater separation was noted for every successive attachment reaction. Nevertheless, a significant extent of overlap is observed even after four attachment reactions. We found at least three distinct populations. There are almost certainly more than three but the three we report here are the most abundant. We did not observe a clearly resolvable component with a molecular weight larger than the expected mass of the intact ribosome (2,302 kDa). The masses of the three assigned components were 2,271±2 kDa, 2,223±1kDa, and 2,209±1 kDa. The difference in mass between the largest and smallest components is consistent with the 30S S1 protein and the difference in mass between the two smaller components is consistent with the 50S L11 protein mentioned above. If the largest component corresponds to the loss of the large subunit stalk complex (70S-[L10(L7/L12)4]), the mass measurement is roughly 1.5% high relative to the predicted value (2,236 kDa), which is consistent with the errors noted above for the 30S and 50S subunits ionized with 10 mM Mg2+. We note that, given the widths of the peaks observed here, that a 70S-S1 (predicted value of 2,241 kDa) product could also be present.
Figure 5.

Post-ion attachment mass spectrum of 70S ribosome-related ions reacting with [hMb−10H]10−. The 70S ribosome precursor population (green border) are shown in the insert to Figure 4a. The insert shows an expansion of the fourth and fifth anion attachment regions.
Conclusion
Electrospray ionization applied to solutions that mimic physiological conditions has enabled the development of native MS. Such conditions, however, lead to relatively narrow charge state distributions and, broadened signals due to extensive salt adduction. These consequences can complicate mass measurement due to uncertainties in charge state assignment and/or extensive signal overlap that precludes the resolution of individual charge states. The situation is exacerbated for inherently heterogeneous complexes that generate mixtures with overlapping charge state distributions. A strategy introduced here to address these problems, and demonstrated with ions generated from the E. coli ribosome, relies on the isolation of a sub-population of ions of interest and the attachment of a large highly-charged ion of opposite polarity of known mass and charge. The large defined change in mass and charge in a single step allows for the separation of ions of initially similar mass-to-charge ratios but different masses and charges. The charges of the selected ions can be determined via measurement of the m/z values of the successive ion attachment peaks. Charge state assignment accuracy is enhanced by the generation of related ions via successive reagent ion attachments over wide ranges of mass-to-charge. The net result is improved ability to determine the masses of high-mass heterogeneous complexes in native mass spectrometry.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by the National Institute of General Medical Sciences under Grant GM R37–45372. The authors acknowledge SCIEX, and particularly Mr. Frank Londry, for modifying the instrument to enable these ion/ion reaction experiments, Dr. James Hager, also of SCIEX for helpful discussions, Prof. Vicki Wysocki and her group for advice in conducting native mass spectrometry experiments, and Mr. Hsi-Chun Chao for developing conditions for the generation of holo-myoglobin reagent anions.
Funding Sources
National Institute of General Medical Sciences under Grant GM R37–45372
Footnotes
ASSOCIATED CONTENT
Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
Simulation description, Ferritin (Figure S1), GroEL (Figure S2, low Mg2+ 30S (Figure S3), and low Mg2+ 50S (Figure S4) data.
REFERENCES
- [1].Katta V, Chait BT, Observation of the heme-globin complex in native myoglobin by electrospray-ionization mass spectrometry. J. Am. Chem. Soc. 1991, 113, 8534–8535. [Google Scholar]
- [2].Rostom AA, Robinson CV, Detection of the Intact GroEL Chaperonin Assembly by Mass Spectrometry. J. Am. Chem. Soc. 1999, 121, 4718–4719. [Google Scholar]
- [3].Rostom AA, Fucini P, Benjamin DR, Juenemann R, Nierhaus KH, Hartl FU, Dobson CM, Robinson CV, Detection and selective dissociation of intact ribosomes in a mass spectrometer. Proc. Nat. Acad. Sci. 2000, 97, 5185–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Forsberg E, Fang M, Siuzdak G, Measuring Intact Viable Microbes with Electrospray Ionization Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 14–20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Leney AC, Heck AJR, Native Mass Spectrometry: What is in the Name? J. Am. Soc. Mass Spectrom. 2017, 28, 5–13. [DOI] [PubMed] [Google Scholar]
- [6].Heck AJR, Native mass spectrometry: a bridge between interactomics and structural biology. Nat. Methods 2008, 5, 927. [DOI] [PubMed] [Google Scholar]
- [7].Hernández H, Robinson CV, Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nat. Protoc. 2007, 2, 715. [DOI] [PubMed] [Google Scholar]
- [8].Schachner LF, Ives AN, McGee JP, Melani RD, Kafader JO, Compton PD, Patrie SM, Kelleher NL, Standard Proteoforms and Their Complexes for Native Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2019. 30, 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].E Belov M, Damoc E, Denisov E, Compton PD, Horning S, Makarov AA, Kelleher NL, From Protein Complexes to Subunit Backbone Fragments: A Multi-stage Approach to Native Mass Spectrometry. Anal. Chem. 2013, 85, 11163–11173. [DOI] [PubMed] [Google Scholar]
- [10].Li H, Nguyen HH, Ogorzalek Loo RR, Campuzano IDG, Loo JA, An integrated native mass spectrometry and top-down proteomics method that connects sequence to structure and function of macromolecular complexes. Nat. Chem. 2018, 10, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Konijnenberg A, Butterer A, Sobott F, Native ion mobility-mass spectrometry and related methods in structural biology. BBA - Proteins Proteom. 2013, 1834, 1239–1256. [DOI] [PubMed] [Google Scholar]
- [12].Konermann L, Ahadi E, Rodriguez AD, Vahidi S, Unraveling the Mechanism of Electrospray Ionization. Anal. Chem. 2013, 85, 2–9. [DOI] [PubMed] [Google Scholar]
- [13].Kebarle P, Verkerk UH, Electrospray: From ions in solution to ions in the gas phase, what we know now. Mass Spectrom. Rev. 2009, 28, 898–917. [DOI] [PubMed] [Google Scholar]
- [14].Fernandez J de la Mora, Electrospray ionization of large multiply charged species proceeds via Dole’s charged residue mechanism. Anal. Chim. Acta 2000, 406, 93–104. [Google Scholar]
- [15].Fort KL, van de Waterbeemd M, Boll D, Reinhardt-Szyba M, Belov ME, Sasaki E, Zschoche R, Hilvert D, Makarov AA, Heck AJR, Expanding the structural analysis capabilities on an Orbitrap-based mass spectrometer for large macromolecular complexes. Analyst 2018, 143, 100–105. [DOI] [PubMed] [Google Scholar]
- [16].Snijder J, Rose RJ, Veesler D, Johnson JE, Heck AJR, Studying 18 MDa Virus Assemblies with Native Mass Spectrometry. Angew. Chem. Int. Edit. 2013, 52, 4020–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Keifer DZ, Shinholt DL, Jarrold MF, Charge Detection Mass Spectrometry with Almost Perfect Charge Accuracy. Anal. Chem. 2015, 87, 10330–10337. [DOI] [PubMed] [Google Scholar]
- [18].Schultz JC, Hack CA, Benner WH, Mass Determination of Megadalton-DNA Electrospray Ions Using Charge Detection Mass Spectrometry. J. Am. Soc. Mass Spectrom. 1998, 9, 305–313. [DOI] [PubMed] [Google Scholar]
- [19].McLuckey SA, Goeringer DE, Ion/Molecule Reactions for Improved Effective Mass Resolution in Electrospray Mass Spectrometry. Anal. Chem. 1995, 67, 2493–2497. [DOI] [PubMed] [Google Scholar]
- [20].Laszlo KJ, Bush MF, Analysis of Native-Like Proteins and Protein Complexes Using Cation to Anion Proton Transfer Reactions (CAPTR). J. Am. Soc. Mass Spectrom. 2015, 26, 2152–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Abzalimov RR, Kaltashov IA, Electrospray Ionization Mass Spectrometry of Highly Heterogeneous Protein Systems: Protein Ion Charge State Assignment via Incomplete Charge Reduction. Anal. Chem. 2010, 82, 7523–7526. [DOI] [PubMed] [Google Scholar]
- [22].Zhao Y, Abzalimov RR, Kaltashov IA, Interactions of Intact Unfractionated Heparin with Its Client Proteins Can Be Probed Directlu Using Native Electrospray Ionization Mass Spectrometry. Anal. Chem. 2016, 88, 1711–1718. [DOI] [PubMed] [Google Scholar]
- [23].Zhou M, Jones CM, Wysocki VH, Dissecting the Large Noncovalent Protein Complex GroEL with Surface-Induced Dissociation and Ion Mobility-Mass Spectrometry. Anal. Chem. 2013, 85, 8262–8267. [DOI] [PubMed] [Google Scholar]
- [24].van de Waterbeemd M, Fort KL, Boll D, Reinhardt-Szyba M, Routh A, Makarov A, Heck AJR , High-fidelity mass analysis unveils heterogeneity in intact ribosomal particles. Nat. Methods 2017, 14, 283–286. [DOI] [PubMed] [Google Scholar]
- [25].Xia Y, Wu J, McLuckey SA, Londry FA, Hager JW, Mutual storage mode ion/ion reactions in a hybrid linear ion trap. J. Am. Soc. Mass Spectrom. 2005, 16, 71–81. [DOI] [PubMed] [Google Scholar]
- [26].Liang X, Xia Y, McLuckey SA, Alternately Pulsed Nanoelectrospray Ionization/Atmospheric Pressure Chemical Ionization for Ion/Ion Reactions in an Electrodynamic Ion Trap. Anal. Chem. 2006, 78, 3208–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].McLuckey SA, Goeringer DE, Glish GL, Selective Ion Isolation/Rejection Over a Broad Mass Range in the Quadrupole Ion Trap. J. Am. Soc. Mass Spectrom. 1991, 2, 11–21. [DOI] [PubMed] [Google Scholar]
- [28].Xia Y, Liang X, McLuckey SA, Pulsed dual electrospray ionization for ion/ion reactions. J. Am. Soc. Mass Spectrom. 2005, 16, 1750–1756. [DOI] [PubMed] [Google Scholar]
- [29].Mann M, Meng CK, Fenn JB, Interpreting Mass Spectra of Multiply Charge Ions. Anal. Chem. 1989, 61, 1702–1708. [Google Scholar]
- [30].McLuckey SA, Huang TY, Ion/ion reactions: new chemistry for analytical MS. Anal. Chem. 2009, 81, 8669–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Prentice BM, McLuckey SA, Gas-phase ion/ion reactions of peptides and proteins: acid/base, redox, and covalent chemistries. Chem. Commun. 2013, 49, 947–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].M Wells J, Chrisman PA, McLuckey SA, Formation and Characterization of Protein−Protein Complexes in Vacuo. J. Am. Chem. Soc. 2003, 125, 7238–7249. [DOI] [PubMed] [Google Scholar]
- [33].McKay AR, Ruotolo BT, Ilag LL, Robinson CV, Mass Measurements of Increased Accuracy Resolve Heterogeneous Populations of Intact Ribosomes. J. Am. Chem. Soc. 2006, 128, 11433–11442. [DOI] [PubMed] [Google Scholar]
- [34].Freeke J, Robinson CV, Ruotolo BT, Residual counter ions can stabilise a large protein complex in the gas phase. Int. J. Mass Spectrom. 2010, 298, 91–98. [Google Scholar]
- [35].Liepold L, Oltrogge LM, Suci PA, Young MJ, Douglas T, Correct Charge State Assignment of Native Electrospray Spectra of Protein Complexes. J. Am. Soc. Mass Spectrom. 2009, 20, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chernushevich IV, Thomson BA, Collisional Cooling of Large Ions in Electrospray Mass Spectrometry. Anal. Chem. 2004, 76, 1754–1760. [DOI] [PubMed] [Google Scholar]
- [37].Gordiyenko Y, Deroo S, Zhou M, Videler H, Robinson CV, Acetylation of L12 Increases Interactions in the Escherichia Coli Ribosomal Stalk Complex. J. Mol. Biol. 2008, 380, 404–414. [DOI] [PubMed] [Google Scholar]
- [38].Markus CW, Wim M, Structure and Function of the Acidic Ribosomal Stalk Proteins. Curr. Protein Pept. Sci. 2002, 3, 93–106. [DOI] [PubMed] [Google Scholar]
- [39].Diaconu M, Kothe U, Schlunzen F, Fischer N, Harms JM, Tonevitsky AG, Stark H, Rodnina MV, Wahl MC, Structural basis for the function of the ribosomal L7/12 stalk in factor binding and GTPase activation. Cell 2005,121, 991–1004. [DOI] [PubMed] [Google Scholar]
- [40].Ilag LL, Videler H, McKay AR, Sobott F, Ficini P, Nierhaus KH, Robinson CV, Heptameric (L12)6/L10 rather than canonical pentameric complexes are found by tandem MS of intact ribosomes from thermophilic bacteria. Proc. Natl. Acad. Sci. USA 2005, 102, 8192–8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.