

Abstract
Background:
Beta-blockers (BB) are mainstay therapy for heart failure with reduced ejection fraction (HFrEF). However, individual patient responses to BB vary, which may be partially due to genetic variation. The goal of this study was to derive and validate the first polygenic response predictor (PRP) for BB survival benefit in HFrEF patients.
Methods:
Derivation and validation analyses were performed in n=1,436 total HF patients of European descent and with EF <50%. The PRP was derived in a random subset of the Henry Ford Pharmacogenomic Registry (HFPGR; n=248), and then validated in a meta-analysis of the remaining patients from HFPGR (n=247), the TIME-CHF (n=431), and HF-ACTION trial (n=510). The PRP was constructed from a genome-wide analysis of BB*genotype interaction predicting time to all-cause mortality, adjusted for MAGGIC score, genotype, level of BB exposure, and BB propensity score.
Results:
Five-fold cross-validation summaries out to 1000 SNPs identified optimal prediction with a 44 SNP score and cutoff at the 30th percentile. In validation testing (n=1188) greater BB exposure was associated with reduced all-cause mortality in patients with low-PRP score (n=251; HR=0.19 [95% CI=0.04–0.51], p=0.0075), but not high-PRP score (n=937; HR=0.84 [95% CI=0.53–1.3], p=0.448), a difference that was statistically significant (p interaction =0.0235). Results were consistent regardless of atrial fibrillation, EF (≤40% vs. 41–50%), or when examining cardiovascular death.
Conclusions:
Among patients of European ancestry with HFrEF, a PRP distinguished patients who derived substantial survival benefit from BB exposure from a larger group that did not. Additional work is needed prospectively test clinical utility and to develop PRPs for other population groups and other medications.
Keywords: polygenic score, pharmacogenomics, beta blockers, precision medicine
The landmark beta-blocker (BB) clinical trials for heart failure with reduced ejection fraction (HFrEF) showed a significant reduction in the risk of death overall,1–3 but it is important to point out that individual patient responses to BB widely vary. For example, in one randomized clinical trial, the 95% confidence interval for change in left ventricular ejection fraction (LVEF) with carvedilol was −11.1 to +32.9.4 Despite this well-recognized variability BB response,5 current guidelines for HFrEF correctly adopt a “one size fits all” approach, whereby all patients are recommended to be treated with the same target doses of BB,6 because clinical characteristics largely did not impact HFrEF patients’ response to BB therapy in terms of survival.1–3 A relatively recent possible exception to this is atrial fibrillation, which some studies have reported negates BB effectiveness in HFrEF.7, 8 Since clinical factors are largely unable to identify BB responders vs. non-responders, understandably much research has focused on other factors, such as genetic variation, to support precision medicine approaches for BB treatment decisions.9–11 These pharmacogenetic studies, mostly employing the candidate gene approach,12–16 generally support the concept that genetic variation impacts BB response in HFrEF patients but results are inconsistent and no clinically actionable markers are proven to date.17 Although there are some examples where one or two genetic variants (usually in the setting of altered drug metabolism) have sufficient impact on drug response to generate a clinical action (e.g. CYP2C19 and clopidogrel)18, it now appears that common complex phenotypes are likely under the influence of many genetic loci, each variant acting with relatively small impact,19–21 and drug response may be similarly complex and polygenic in nature.
Polygenic risk scores have emerged as a method to aggregate the small effects of numerous genetic variants into a score that reflects the overall genetic risk of a phenotype of interest. For common complex traits, this appears to often capture enough variation for clinical utility, where a smaller number of genome-wide significant “hits” have failed to do so.22, 23 Polygenic scores have now been developed for several common diseases, such as coronary disease and others,23, 24 and these will soon be explored for implementation of targeted population management. Despite this exciting new development, similar analytic methods have largely not yet been successfully adapted to drug response in the setting of prevalent disease. There are emerging examples published,10, 25–34 but to our knowledge, none applied to treatment of HFrEF. Limited adaptation of polygenic scores to drug-response may be due in part to methodologic challenges, such as lack of sufficiently detailed drug exposure data and complexities of analysis. Although cohorts with detailed drug exposure information tend to be smaller in size relative to recent GWAS, a polygenic score approach may offer enhanced power and an efficient approach for constructing polygenic drug-response scores could have broad impact on precision medicine and drug development. The objective of this study was to derive and validate the first PRP for BB-associated survival benefit in patients with HFrEF.
Methods
Datasets and Overall Approach
The current study was approved by the Henry Ford Hospital Institutional Review Board, and all participants provided written informed consent. Three datasets were used: the Henry Ford Heart Failure Pharmacogenomic registry (HFPGR);35 the Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure (TIME-CHF);36 and Heart Failure: A Controlled Trial Investigating Outcomes of Exercise Training (HF-ACTION).37 The data from HFPGR is publicly available via dbGaP (https://www.ncbi.nlm.nih.gov/gap). The remaining data that support the findings of this study may be made available upon reasonable request from qualified researchers trained in human subject confidentiality protocols by contacting Dr. David E. Lanfear (dlanfea1@hfhs.org) at Henry Ford Hospital.
The patient flow from parent study to final analytic cohorts is shown in the consort diagram (Supplemental Figure 1.) Only self-reported white patients were included in this analysis. The parent studies are described briefly below and in detail elsewhere, and each had institutional review board/ethics approval.35–37 A diagram illustrating overall study design is presented in Figure 1. We first created a derivation group upon which the PRP would be constructed, followed by testing in multiple independent datasets to test its performance. Since the HFPGR had the most granular drug-exposure data (derived from pharmacy claims and time-updating), it was randomly divided into two halves: one for derivation (n = 248) and the other to be included in the validation (n = 247). The PRP was derived solely in the derivation subset of HFPGR. Thus, the validation data included the remaining half of the HFPGR (n = 247), TIME-CHF (n = 431), and HF-ACTION (n = 510) for a total of 1188 validation patients.
Figure 1.
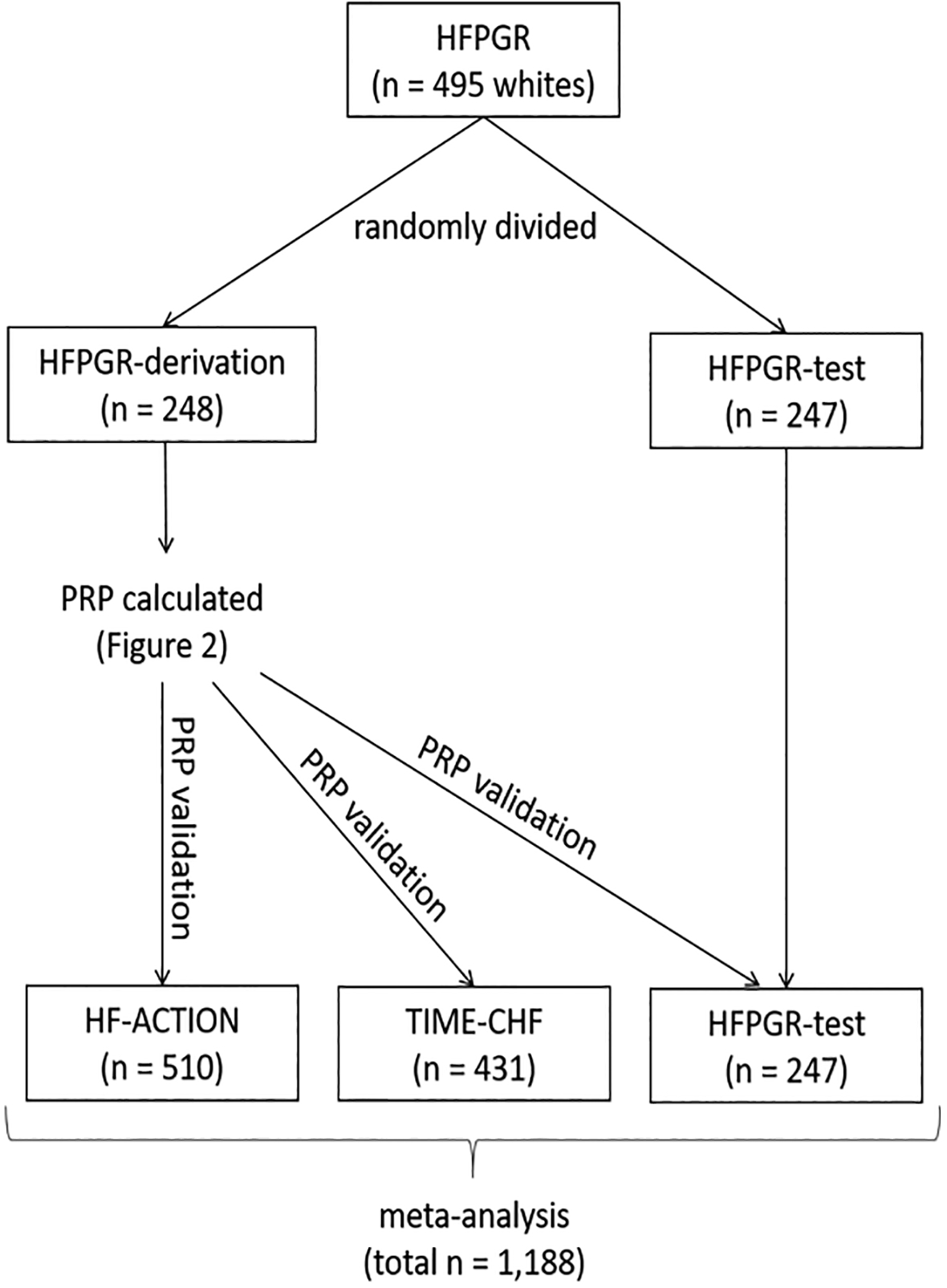
Schematic illustrating the use of the three datasets for polygenic response predictor (PRP) derivation and validation. HF-ACTION = the Heart Failure: A Controlled Trial Investigating Outcomes of Exercise Training;37 HFPGR = Henry Ford Pharmacogenomic Registry35; TIME-CHF = the Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure36; PRP = polygenic response predictor.
BB exposure was computed using two different methods depending on the cohort. For both HFPGR cohorts BB exposure was calculated from pharmacy claims (i.e. drug actually dispensed to patient) and was updated over time, as previously described,35, 38 and briefly summarized in the Supplemental Methods section. For TIME-CHF and HF-ACTION BB exposure was calculated from the specific drug and dose at baseline, using the same dose-equivalence scheme as above but implemented without any updates over time and without information on medication dispensing (i.e. assumes patients were receiving the dose prescribed).
The HFPGR enrolled patients from October 2007 through March 2015 at Henry Ford Health System in Detroit, MI, USA. The overall goal of the HFPGR is to discover novel ways to better predict prognosis and response to HFrEF treatments. Patients aged 18 years or older were included if they had health insurance coverage, met the definition for heart failure as defined by the Framingham Heart Study,39 and had at least one documented left ventricular ejection fraction (LVEF); a total of 1760 patients were enrolled. For the current analysis those with LVEF < 50% and self-identified European ancestry were included (n=495). BB exposure was calculated from pharmacy claims and was updated over time, as previously described35, 38
TIME-CHF was a randomized controlled trial that took place at 15 centers in Switzerland and Germany from January 2003 to June 2008 that enrolled a total of 622 patients. The primary objective of the trial was to compare N-terminal pro-B-type natriuretic peptide (NTproBNP)–guided versus symptom-guided treatment for heart failure. To be included in the trial, individuals had to meet the following criteria: age > 60 years, diagnosed with systolic heart failure, New York Heart Association (NYHA) class of II or greater, hospitalization for heart failure within the year prior to enrollment, and an NTproBNP level 2 times the upper limit of normal. For the current analysis, only patients consenting to participate in the genetic substudy with analyzable data and baseline LVEF < 50% were included (n=431). BB exposure was calculated from the specific drug and dose at baseline, with dose standardization performed as previously described.35, 38 Heart failure therapy guided by NTproBNP did not significantly affect the primary outcome.
HF-ACTION was a National Institute of Health funded randomized clinical trial that took place at 82 centers within the United States, Canada, and France from April 2003 through February 2007, enrolling a total of 2331 patients. The primary objective of the trial was to compare usual HFrEF care to usual HFrEF care plus exercise training, but it included a genetic sub-study. The trial included adult HFrEF patients with LVEF ≤ 35% and NYHA class II to IV symptoms despite optimal heart failure therapy for at least 6 weeks. BB exposure was calculated from the specific drug and dose at baseline, with dose standardization performed as previously described.35 In primary analysis, exercise training resulted in non-significant reductions in the primary end point of all-cause mortality or hospitalizations although in the pre-specified adjusted analysis, the exercise intervention significantly reduced the composite primary outcome. We felt this did not justify the need to adjust for trial arm assignment when modeling all-cause mortality (the primary outcome for the current study). Only participants of the genetic sub-study with quality genetic array data, complete clinical data and self-identified as European ancestry were included in the current analysis (n=510).
Genotyping
Blood samples were collected, processed and stored at −70°C. DNA was extracted using standard methods.40 Patients from all three studies were genotyped with the Axiom® Biobank array (Affymetrix, Santa Clara, CA). This array was designed to optimize genome-wide imputation, and it includes ~600,000 genetic variants with the following characteristics: ~300,000 genome-wide variants with minor allele frequencies >1%, ~250,000 low frequency (<1%) coding variants from global exome sequencing projects, and an additional ~50,000 variants for improved African ancestry coverage. All genotyping was performed at the University of Michigan genotyping core lab with standard quality checks performed. In brief, single nucleotide polymorphisms (SNPs) with a minor allele frequency < 0.05, those not in Hardy–Weinberg equilibrium (HWE p < 10−8), multi-allelic sites, and ambiguous SNPs were deleted from the analysis. Samples with sex inconsistencies (i.e., between patient report and genetically determined) or duplicate genotyping were removed. All datasets underwent imputation using the University of Michigan imputation server with Minimac341 and using the cosmopolitan reference panel from the 1000 Genomes Project.42 Imputed SNPs passing a quality threshold of r2 > 0.5 were included for analysis.
Polygenic Response Predictor (PRP) Construction
A combined SNP selection and multivariable data analysis strategy was formulated to derive the PRP based on a GWAS of the HFPGR-derivation cohort, summarized in Figure 2. The primary outcome was time to all-cause mortality. This was modeled using Cox regression focusing in on the SNP-by-BB exposure multiplicative interaction term (SNP*BB) and including the main effects of BB exposure and SNP as covariates. An additional two covariates were also included in the model; to adjust for established clinical risk factors for death we include the MAGGIC score (calculated without BB as a contributing variable)43 and to adjust for potential confounding by indication or treatment bias we included a BB propensity score44 Thus the overall model was Death = MAGGIC + BBpropensity + BBexposure + SNP + SNP*BBexposure. The Q-Q plot of the GWAS result is shown in supplemental Figure 2. BB propensity score was calculated using logistic regression of baseline characteristic variables with the resulting output separated into quartiles and used as an ordinal covariate.45 BB exposure was implemented as a time-dependent covariate (this is specific to HFPGR). The SNPs were then ranked according to the p-value of SNP*BBexposure and the selected SNPs were pruned down to the most significant one within each linkage disequilibrium (LD) block, which was kept for subsequent analysis.
Figure 2.
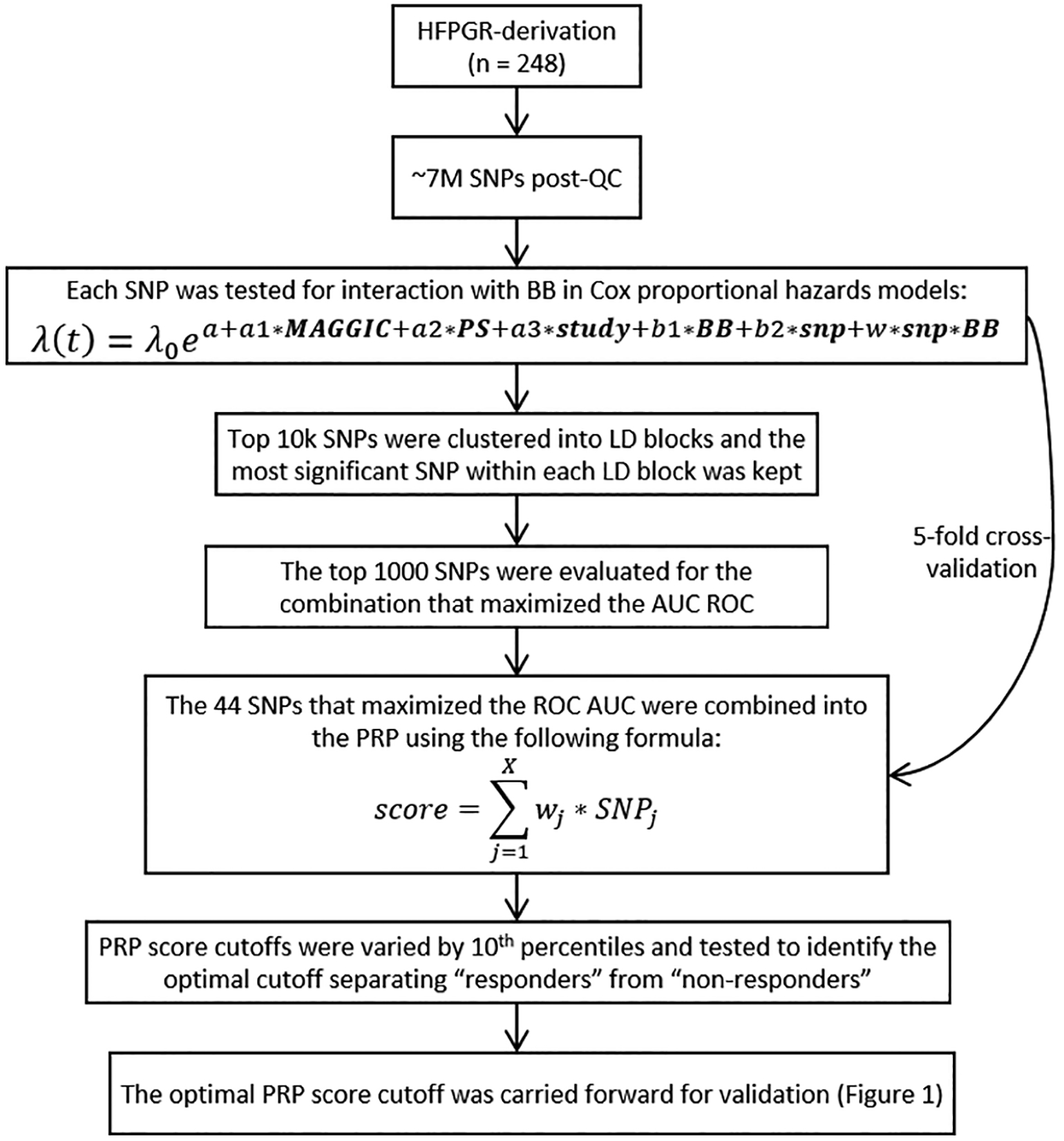
Schematic and formulae for calculating the PRP. AUC = area under the curve; BB = Beta Blocker; LD = linkage disequilibrium; MAGGIC = Meta-Analysis Global Group in Chronic Heart Failure risk score;43 PS = propensity score; QC = quality control; ROC = receiver operating characteristic curve; SNP = single nucleotide polymorphism.
The PRP is a linear combination of SNPs, in which the SNP weights are determined by the estimated Cox regression coefficients in the original model (from the derivation cohort). To select the optimal number of SNPs to include, a series of scores were created using Cox models similar to the above (including MAGGIC, Propensity, BBexposure, SNP and SNP*BBexposure) but sequentially adding more and more SNP and SNP*BBexposure terms, starting with the most highly associated locus and then incrementally adding one SNP at a time, sorted according to the p-value. The SNPs were coded with an additive genetic model (0, 1, or 2 for minor allele count). These scores were tested using 5-fold cross-validation with the merge method46 to select the optimal number of SNPs that would maximize the time-dependent AUC for 1-year survival (estimated from the Cox model). The optimal set of SNPs (and the associated coefficients) were then used to build the final PRP.
To prepare for validation, the PRP was also dichotomized into “responder” and “non-responder” groups. The purpose of converting the continuous PRP into dichotomous categories was to facilitate clinical testing and future implementation of the PRP (i.e., to be able to identify sub-groups of patients where a specific therapeutic action is clearly defined). To accomplish this, we examined the BB survival benefit across a variety of score cutoff thresholds separating “responder” vs. “non-responders”, the performance of each tested in the HFPGR derivation cohort. Performance was judged by examining the BB HRs for high and low PRP patients (separately) with the cutoff at increasing deciles of the PRP within the derivation cohort (i.e. trying the 10th percentile as the cutoff for low vs high, then 20th percentile, and so on). The HR in each PRP group and their separation was examined. Separation was best at 30th percentile in the derivation cohort and the raw score at this level was then carried forward throughout validation testing. For HF-ACTION, 3 SNPS of the PRP did not pass quality metrics, so these were excluded and the PRP was tabulated using the remaining 41 SNPs. The dichotomization point was recalculated using a matching (without those 3 SNPs) PRP in derivation set to identify the corresponding raw score, and this was used as the dichotomization point for HF-ACTION validation testing.
Statistical Analysis and PRP Validation
In validation, the association of BB exposure was tested in PRP stratified groups in each of the 3 validation datasets using Cox models of survival (time to all-cause mortality). Our primary analysis consisted of models adjusted for MAGGIC (again without BB variable)43 and BB propensity score,44, 45 with BB exposure treated as a continuous variable. Given the relatively small sample sizes of the individual datasets, the results from each of the individual validation datasets were combined by meta-analysis using a fixed effect model to provide an overall validation of the PRP (n=1188). We also tested for heterogeneity within the studies of the meta-analysis using Cohran’s Q test and report this along with the Î2 statistic. We then tested models inclusive of both PRP categories with an interaction term (PRP*BB) to calculate an interaction p value. We also present Kaplan-Maier curves in PRP stratified groups comparing baseline BB dose dichotomized into high vs. low exposure at a dose equivalent of <0.5 vs. ≥0.5, corresponding to half the target dose of BB in pivotal clinical trials (e.g. total daily dosage of 100 mg for metoprolol or 25 mg for carvedilol).
Since BB was not randomly assigned, we included propensity score adjustment for baseline BB use in all analyses. The propensity score was constructed using logistic regression predicting baseline BB using the following variables: Age, sex, creatinine, ischemic etiology, stroke, COPD, peripheral vascular disease, atrial fibrillation, and hypertension. The propensity scores were grouped into quartiles and this was used as a covariate (0, 1, 2, 3) in all analytic models. To verify the effectiveness of the propensity score for mitigating bias we performed a balance check by tabulating the weighted standardized difference for each covariate for each study. These ranged from 0.01 to 0.31 (Supplemental Table 1), implying reasonably balanced covariates.47
Secondary analyses included similar models stratified by history of atrial fibrillation, and (separately) by EF category (<40% vs 40–49%) and examining cardiovascular (CV) death. Atrial fibrillation and EF categories were examined in stratified Cox models with the same covariates as above. The time to CV death was analyzed using Fine-Gray models (to account competing risks) and including the same covariates as main survival analyses above. All BB exposures were constructed across agents using dose-equivalency as a proportion of the target dose of agent as previously described.35 For HFPGR-test the BB exposure was time-updating measurements while for TIME-CHF and HF-ACTION the baseline BB dose was used.
Baseline characteristics are shown in total and across cohorts. Continuous variables were summarized by the mean and standard deviation and categorical variables were summarized by counts and percentages. They were compared among the datasets using analysis of variance (ANOVA) for continuous variables and Chi-square test for categorical variables. All statistical analyses were performed using R 3.6.1 (R Foundation, Vienna, Austria). P < 0.05 was considered statistically significant.
Results
Baseline characteristics of the 4 datasets (the 1 derivation and 3 validation, total n = 1,436) are summarized in Table 1. The cohorts significantly differed in relation to all variables assessed, except for the frequency of diabetes and stroke. Notably the median BB dose/exposure varied, with HF-ACTION having a greater median (0.56 dose equivalents, representing half of target dose) compared to the other validation sets (0.28, 0.25 respectively for HFPGR and TIME-CHF). NTproBNP levels were greater in TIME-CHF and least in HF-ACTION. Average follow-up across datasets ranged from 785 to 985 days and there was a total of 332 deaths (23%).
Table 1.
Baseline characteristics of the total, derivation and validation datasets.
| Variable | Overall (n = 1,436) |
HFPGR-Derivation (n=248) |
HFPGR-Test (n=247) |
HF-ACTION (n=510) |
TIME-CHF (n=431) |
p-value |
|---|---|---|---|---|---|---|
| Age (years) | 69 (12) | 71 (11) | 71 (10) | 61 (12) | 76 (7.6) | < 0.001 |
| Female sex | 397 (28%) | 72 (29%) | 77 (31%) | 100 (20%) | 148 (34%) | < 0.001 |
| MAGGIC (no BB) | 21.9 (7.30) | 18.8 (6.66) | 17.9 (7.29) | 20.9 (6.57) | 27.2 (5.24) | < 0.001 |
| LVEF (%) | 30.3 (9.7) | 34.9 (10.7) | 37.3 (9.6) | 25.1 (7.1) | 29.7 (7.7) | < 0.001 |
| BMI (kg/m2) | 28.8 (6.3) | 30.6 (7.0) | 30.8 (7.3) | 29.8 (5.8) | 25.4 (4.2) | < 0.001 |
| Heart Rate | 73 (13.7) | 70 (12.3) | 70 (12.9) | 70 (11.3) | 76 (14.3) | <0.001 |
| SBP (mmHg) | 120 (20) | 126 (23) | 126 (20) | 115 (19) | 118 (18) | < 0.001 |
| NTproBNP (ng/L) | 3837 (5099) | 3070 (3085) | 3079 (2916) | 1889 (2549) | 6473 (7216) | < 0.001 |
| Creatinine (mg/dL) | 1.28 (0.62) | 1.19 (0.63) | 1.17 (0.57) | 1.33 (0.75) | 1.34 (0.44) | < 0.001 |
| Ischemic | 849 (59%) | 135 (54%) | 140 (57 %) | 327 (64%) | 247 (57%) | 0.033 |
| COPD | 273 (19%) | 56 (23%) | 59 (24%) | 71 (14%) | 87 (20%) | 0.002 |
| Atrial fibrillation | 450 (31%) | 86 (35%) | 98 (40%) | 127 (25%) | 139 (32%) | < 0.001 |
| History Hypertension | 730 (79%) | 215 (87%) | 211 (85%) | NA | 304 (70%) | <0.001 |
| Stroke | 104 (7.2%) | 15 (6.1%) | 22 (8.9%) | 32 (6.3%) | 35 (8.1%) | 0.432 |
| Peripheral Vascular | 167 (11.6%) | 14 (5.6%) | 25 (10.1%) | 41 (8%) | 87 (20.2%) | <0.001 |
| Diabetes | 486 (34%) | 94 (38%) | 91 (37%) | 154 (30%) | 147 (34%) | 0.119 |
| Follow-up (days) | 887 (499) | 844 (642) | 785 (519) | 988 (380) | 852 (499) | < 0.001 |
| Any Beta Blocker | 1143 (80%) | 161 (65%) | 157 (64%) | 480 (94%) | 345 (80%) | <0.001 |
| Carvedilol | 472 (41%) | 66 (41%) | 45 (29%) | 279 (58%) | 82 (24%) | <0.001 |
| Metoprolol Succinate | 417 (36%) | 57 (35%) | 57 (36%) | 134 (28%) | 169 (49%) | |
| Metoprolol Tartrate | 127 (11%) | 34 (21%) | 44 (28%) | 49 (10%) | 0 (0%) | |
| Bisoprolol | 67 (5.9%) | 0 (0%) | 0 (0%) | 6 (1.3%) | 61 (18%) | |
| Other beta-blockers | 60 (5.2%) | 4 (2.5%) | 11 (7%) | 12 (2.5%) | 33 (9.6%) | |
| Proportion target dose | 0.36 (0.37) | 0.23 (0.29) | 0.22 (0.28) | 0.56 (0.43) | 0.28 (0.27) | <0.001 |
| Death | 332 (23.1%) | 53 (21.4%) | 36 (14.6%) | 78 (15.3%) | 165 (38.3%) | < 0.001 |
BMI = body mass index; COPD = chronic obstructive pulmonary disease; HF-ACTION = the Heart Failure: A Controlled Trial Investigating Outcomes of Exercise Training; HFPGR = Henry Ford Heart Failure Pharmacogenomic Registry; LVEF = left ventricular ejection fraction; NTproBNP = N-terminal pro b-type natriuretic peptide; SBP = systolic blood pressure; TIME-CHF = Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure randomized trial.
The PRP was developed as described in the methods and it optimized after the inclusion of 44 genetic loci. The list of the 44 SNPs comprising the PRP (along with annotation) are provided in Supplemental Table 2. The interaction effect between each of them and BB exposure was associated with mortality (p <10−4). The percentile of PRP that resulted in the best separation between BB responders and non-responders in the HFPGR derivation dataset was the 30th percentile (raw score = 68.14) and this was used as the dichotomization point for validation testing. In HF-ACTION, 3 of the 44 SNPs of the PRP did not have usable genotype calls so the remaining 41 SNPs were used to tabulate the PRP for HF-ACTION participants and the 30th percentile (based on this 41-SNP score) was re-tabulated in the derivation set and used as the dichotomization point for HF-ACTION validation testing. Based on the results from the derivation dataset, patients with a low PRP were hypothesized to be the BB responders in the validation datasets, and patients with a high PRP were hypothesized to be BB non-responders.
The hazard ratios for BB exposure (as a continuous variable incorporating dose) in each of the validation datasets and in the overall validation group (combined via fixed-effect meta-analysis) are displayed in Figure 3. The corresponding survival curves (with BB exposure dichotomized at 50% target dose using the baseline dosing) are shown in Figure 4. The hazard ratio for BB exposure was numerically lower (more protective) in all the low-PRP groups than in the corresponding high-PRP groups, however, the confidence intervals were wide within the individual datasets, reaching significance in TIME-CHF. The entire validation group testing included 1,188 patients overall and a total of 279 deaths (23.5% mortality). This analysis revealed a very strong and statistically significant protective association for BB in the low-PRP group (HR = 0.19 [95% CI = 0.058 – 0.64] p = 0.0075), but no significant association in the high-PRP group (HR = 0.84 [95% CI 0.53 – 1.3] p = 0.45). Formal test of interaction indicated a significant difference between the BB effect between PRP groups (interaction p = 0.0235). Of note, PRP group (high vs. low) was not itself significantly associated with mortality (HR=0.83, p=0.37), and there was no difference in heartrate by PRP category within any of the cohorts (all p>0.05). Tests for heterogeneity in the meta-analysis resulted in a Cochran Q of 2.18 (p=0.34) and Î2 statistic of 0.08, suggesting no significant heterogeneity.
Figure 3.
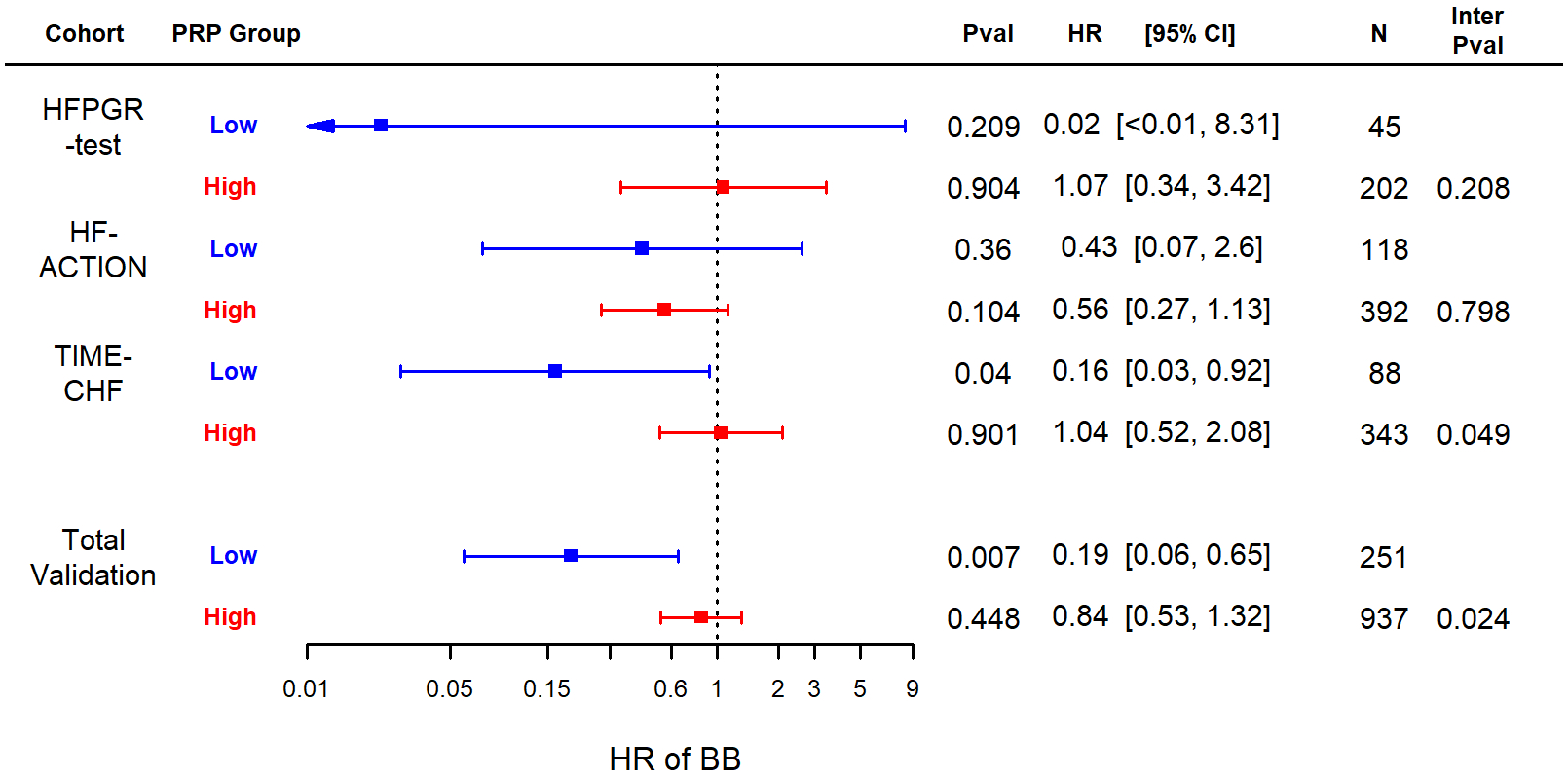
Forest plot of hazard ratios for BB exposure in each of the validation cohorts and for the total validation (meta-analysis). The optimal PRP score cutoff from the derivation dataset was tested in Cox proportional hazards models adjusted for MAGGIC43 and BB propensity score44, 45. Low and high PRP indicate values above or below threshold (30th percentile of the derivation set). BB = Beta Blocker; PRP = polygenic response predictor
Figure 4.
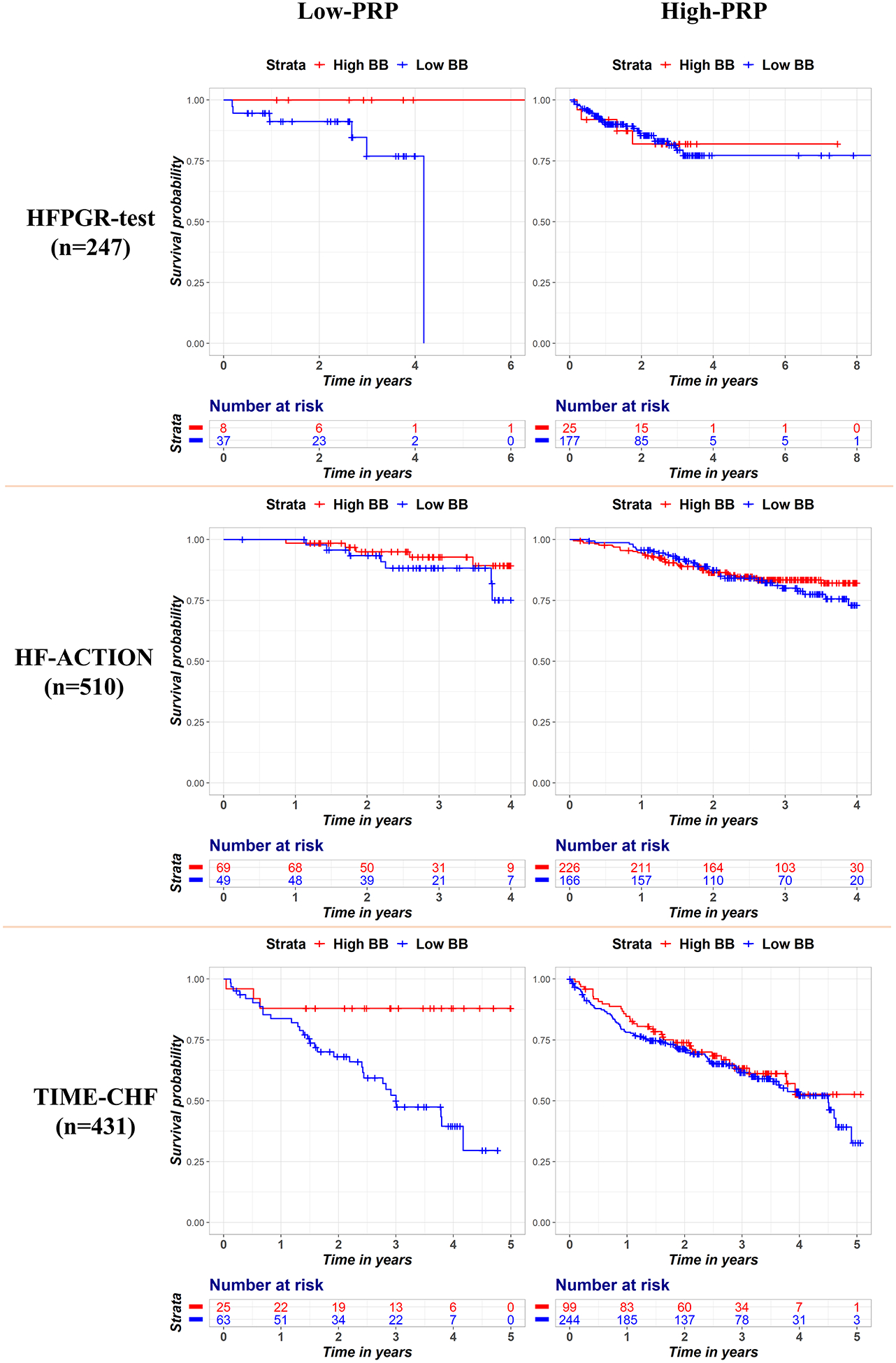
Kaplan Meier survival curves stratified by PRP (high vs. low) and BB exposure for each dataset. The left panels show patients with a low PRP and the right-sided panels show patients with a high PRP. The red curves are patients with high BB exposure (≥50% target dose), and blue curves are patients with low BB exposure. BB = Beta Blocker; PRP = polygenic response predictor
We performed a series of additional sensitivity analyses. Since atrial fibrillation has been associated with impaired BB effectiveness in pooled subgroup analyses of BB pivotal trials, we performed additional modeling stratified by baseline history atrial fibrillation in the validation dataset. Overall, there were 364 patients with history of atrial fibrillation and 824 without. Among those without atrial fibrillation the low-PRP group showed a BB HR of 0.33 (p=0.11) while the corresponding high-PRP group BB HR was 0.73 (p=0.31). In HFrEF patients with comorbid atrial fibrillation, those with a low-PRP had a BB HR of 0.063 (p=0.017) while the comparable high-PRP group showed a BB HR of 0.96 (p=0.92). Similarly, since we included patients with EF<50% in our study but HFrEF has more recently been defined as EF <40%, we also performed secondary analyses stratified by EF category (<40% vs 40–49%). Overall, there were 971 patients with EF<40% and 217 with EF 40–49%, and in both subgroups the PRP trended towards predicting BB response with HR similar to the total validation analysis. In those with EF<40% and low-PRP the BB-HR was 0.23 (p=0.0203) while in the high-PRP patients BB-HR was 0.87 (p=0.57). In patients with baseline EF 40–49% and low-PRP the BB-HR was 0.053 (p=0.23) while in the corresponding high-PRP patients the BB-HR was 0.82 (p=0.76). Finally, we also tested PRP performance in terms of BB exposure association to cardiovascular (CV) death. There were a total of 200 CV death events modeled across the validation cohorts using a Fine-Gray model (to account competing risks) and the same covariates as above. The results are summarized in Table 2. In the total validation meta-analysis the HR of BB in low-PRP was 0.19 (95% CI 0.044 – 0.804) and HR of BB in the high-PRP was 0.89 (95% CI 0.51 – 1.5), and a test of interaction that was statistically significant (p=0.046).
Table 2.
Cox model results for cardiovascular death using competing outcomes approach.
| Cohort | Group | N | HR | P | P (interaction) |
|---|---|---|---|---|---|
| HFPGR-test | Low PRP | 45 | 0.03 | 0.14 | 0.11 |
| High PRP | 202 | 1.62 | 0.57 | ||
| TIME-CHF | Low PRP | 88 | 0.28 | 0.24 | 0.34 |
| High PRP | 343 | 0.84 | 0.68 | ||
| HF-ACTION | Low PRP | 118 | 0.17 | 0.073 | 0.16 |
| High PRP | 392 | 0.74 | 0.47 | ||
| Meta-Analysis | Low PRP | 251 | 0.19 | 0.024 | 0.046 |
| High PRP | 937 | 0.89 | 0.68 |
Discussion
Neither previous candidate gene studies nor clinical predictors have been able to better target BB therapy from the broad indication achieved in HFrEF clinical trials. The PRP, a weighted calculation of genotypes at 44 loci across the genome, was able to do this in a validation group of more than 1100 patients. While polygenic scores specific for drug-response have been described,10, 25–31, 33, 34, 48 ours is the first validated example that we are aware of relevant to HF treatment, and one of the first in cardiovascular disease. Our results are provocative and require additional studies, but the potential implications are wide ranging.
Our approach and findings advance the existing literature on polygenic scores, which have been described for disease risk in variety of settings,49, 50 including some cardiovascular diseases such as coronary artery disease50 and atrial fibrillation.51 Some investigators have taken these scores (for disease-risk) and then layered on drug therapy,25 while we have tried to go a step further by focusing on drug response (i.e. gene*drug interaction). This interaction approach has received less attention and presents unique challenges, such more complex statistical modeling and inherently lower power. The distinction is important however, because disease-risk (like pretest probability) is only one factor impacting treatment effectiveness, and existing evidence suggests distinct genetic architecture for drug response vs. disease-risk.34, 52 Approaches somewhat similar to ours have been described with varying levels of success in other disease areas,10, 25–31, 33, 34, 48 to which our data bring some additional optimism. Our sample size is not dissimilar from many early drug development programs, and the idea that greater definition of the “responder” subset could routinely be had for many medications is obviously very alluring. This approach could amplify effect sizes (shrinking sample size requirements in pivotal trials), and ultimately reduce health care costs and adverse events by avoiding treatment in patients unlikely to be benefitted.
An inspection of our results raises a few observations that are worthy of note. One is that our BB PRP appeared of modest effect in HF-ACTION while the effect estimates in the other two validation cohorts were more dramatic, with the PRP being most statistically significant in TIME-CHF. The individual validation sets each had small sample sizes, so substantial variability across them is expected. Differences in the studies may also play a role. Specifically, HFPGR and TIME-CHF had more patients not treated with BB at baseline, and a lower average BB exposure, compared to HF-ACTION. In HF-ACTION, roughly 95% of patients in the parent study were treated with BB37 and indeed 94% of patients in the ACTION-HF subcohort of the current study. This reflects care more consistent with current guidelines, but the reduced variability in BB exposure (i.e. near uniform use) reduces power to detect gene-drug interactions which is impacted by how many patients are unexposed to treatment. On the other hand, the patients in the TIME-CHF were the highest risk of the 3 studies and it included the highest number and proportion of deaths, providing relatively greater power to detect a differences in BB-associated survival compared to the other two datasets. Despite all this, since each of the individual datasets is likely underpowered the more important consideration is that the overall validation result was quite clear and the direction of the association was consistent across all 3 datasets. Another point of potential interest is that the cut-off for favorable BB-response optimized at roughly 30% of the population. This suggests that most of the benefit of BB occurs in roughly one third of the population while the majority of patients show little, if any, benefit. This may seem counterintuitive for a therapy with population average benefit, but mathematically a smaller ‘super-responder’ group is a plausible explanation for an overall benefit, and proportion is consistent with some data showing that a marked and sustained EF response to BB occurs in only a minority of patients.53 The prospect of identifying a smaller, hyper-responsive subgroup for BB (or eventually other HF treatments) via polygenic scores is tempting to speculate about since this could allow individualized drug therapy and reduce the typical polypharmacy of HF; many patients do not tolerate target doses of all indicated agents54 and these data highlight a potential solution to this real-world difficulty.
Since the PRP was derived by unbiased methodology (not based on pre-existing molecular biological knowledge) it may not be surprising that the loci of interest do not reflect established BB associations; a phenomenon previous seen in pharmacogenomic GWAS.10, 55 Given the number of loci that inform the PRP and the fact that some are of unknown genomic function, a full interrogation of the mechanisms potentially at play remains a future endeavor, however, some pathways that indicate potential biologic plausibility are worth briefly noting. For instance, the top genes from pathway analysis of the PRP include ABCC3, a transporter that is associated with multidrug resistance, has been shown to alter propranolol cellular efflux,56 and could theoretically impact BB absorption or transport. The second gene of interest, ATP2B2a, generated two loci of the PRP and is a plasma membrane Ca+2 transporter. While a specific mechanism is not established, it is well known that Ca homeostasis is critical in heart failure57, 58 and is impacted by BB therapy.59 Finally, variation in PDE5A was also a contributor to PRP and this gene’s product has well known cardiovascular effects though specifics to BB response in HF are unknown.
Our study has some limitations worthy of discussion. First, the BB PRP was only derived and validated in HFrEF patients of European ancestry. Thus, this BB PRP does not apply to patients of other ancestries. While we have previously shown that overall BB response appears similar by race and by genomic ancestry,35 the individual genetic loci that determine this are likely to be very different across these groups due to different linkage patterns or even differing biologic drivers of response. A critical future undertaking is the construction of BB PRP for other ancestral groups (e.g. African or Asian ancestry), or ideally, a score robust to ancestry. While we provide the current functional annotations for the SNPs from bioinformatic databases, future experiments establishing the function and mechanisms of the SNPs and gene products would be needed in order to establish mechanism of the PRP. Another limitation is that the datasets utilized were observational regarding BB use. While BB randomized clinical trials would have been the ideal substrate for our work, this is not possible since it would have been unethical to randomize HFrEF patients to BB vs. placebo, and there is no available genome-wide data from the early pivotal BB trials. We attempted to minimize the treatment bias and other potential confounders by adjusting all models for baseline clinical risk (via MAGGIC risk score) and as well as BB propensity score.44, 45 Lastly, while the relatively smaller size of the derivation set makes possible that our PRP or cut-off point could be further improved, we feel the strategy of keeping the completely distinct validation group relatively larger and including all 3 parent studies therein was preferred in order to maintain adequate validation sample size to test for survival differences and maximize the rigor and generalizability of our findings.
In summary, we developed and validated the first polygenic score for BB survival benefit in HFrEF patients of European ancestry. The score was able to differentiate patients with a greatly enhanced BB-associated survival benefit from a larger group of patients that did not feature a statistically significant survival benefit. These findings challenge the “one size fits all” approach for BB treatment in HFrEF6 and are a step toward precision medicine for HFrEF. Additional replication studies would be reassuring, and work to develop PRPs for other ancestral groups and other medications is warranted. Ultimately, prospective studies would be needed to establish the clinical utility of our BB PRP compared to the current standard of care, ideally via randomized trials.
Supplementary Material
WHAT IS NEW
We generated and validated the first polygenic predictor of beta blocker survival benefit in HFrEF patients derived from unbiased, genome-wide genotyping data. Additional validation and then testing in clinical trials is needed. The current data is limited to European ancestry patients.
WHAT ARE THE CLINICAL IMPLICATIONS
This genetic profile, once proven in trials, could be used to target beta blockers to the subset of HFrEF patients that are likely to benefit and allow a large number of patients to forego unnecessary treatment, fundamentally changing our treatment approach from beta blockers for all to genetically targeted therapy. Parallel work in other ancestral groups is urgently needed.
The general research approach followed here is theoretically applicable to any disease and medication, which could help accelerate broad progress towards clinically useful precision medicine.
Funding Sources:
This research was supported by the National Heart, Lung, and Blood Institute (Lanfear R01HL103871, R01HL132154; Williams R01HL118267, R01HL141845; Sabbah P01HL074237, R01HL132154; Luzum K08HL146990, L30HL110279). Dr. Williams is also supported by the National Institute of Allergy and Infectious Diseases (R01AI079139) and the National Institute of Diabetes and Digestive and Kidney Diseases (R01DK064695, R01DK113003). Dr. Luzum was also supported by a Futures Grant from the American College of Clinical Pharmacy.
Nonstandard Abbreviations and Acronyms
- SNP
Single Nucleotide Polymorphism
- BB
Beta blocker
- MAGGIC
Meta-analysis Global Group in Chronic Heart Failure
- HFPGR
Henry Ford Pharmacogenomic Registry
- HF-ACTION
Heart Failure: A Controlled Trial Investigating Outcomes of Exercise Training
- TIME-CHF
Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure
- GWAS
Genome wide association study
- PRP
Polygenic Response Predictor
- PRS
Polygenic Risk Score
Footnotes
Disclosures: David E. Lanfear is a consultant for Amgen, Janssen, Ortho Diagnostics and DCRI (Novartis) and has participated in clinical trials from Amgen, Bayer, and Janssen, and has submitted a provisional patent request for the beta blocker polygenic score. Jasmine A. Luzum has nothing to disclose. Ruicong She has nothing to disclose. Hongsheng Gui has nothing to disclose. Nicole Zeld has nothing to disclose. Mark P. Donahue has nothing to disclose. Christopher M. O’Connor has nothing to disclose. Kirkwood F. Adams has nothing to disclose. Sandra Sanders-van Wijk has nothing to disclose. Micha T. Maeder has nothing to disclose. Hani N. Sabbah has nothing to disclose. William E. Kraus has nothing to disclose. Hans-Peter Brunner-LaRocca has nothing to disclose. Jia Li has nothing to disclose. L. Keoki Williams has nothing to disclose.
References
- 1.Investigators M-H. Effect of metoprolol cr/xl in chronic heart failure: Metoprolol cr/xl randomised intervention trial in congestive heart failure (merit-hf). Lancet. 1999;353:2001–2007 [PubMed] [Google Scholar]
- 2.The cardiac insufficiency bisoprolol study ii (cibis-ii): A randomised trial. Lancet. 1999;353:9–13 [PubMed] [Google Scholar]
- 3.Packer M, Fowler MB, Roecker EB, Coats AJ, Katus HA, Krum H, Mohacsi P, Rouleau JL, Tendera M, Staiger C, Holcslaw TL, Amann-Zalan I, DeMets DL, Carvedilol Prospective Randomized Cumulative Survival Study G. Effect of carvedilol on the morbidity of patients with severe chronic heart failure: Results of the carvedilol prospective randomized cumulative survival (copernicus) study. Circulation. 2002;106:2194–2199 [DOI] [PubMed] [Google Scholar]
- 4.Metra M, Giubbini R, Nodari S, Boldi E, Modena MG, Dei Cas L. Differential effects of beta-blockers in patients with heart failure: A prospective, randomized, double-blind comparison of the long-term effects of metoprolol versus carvedilol. Circulation. 2000;102:546–551 [DOI] [PubMed] [Google Scholar]
- 5.Roden DM, George AL Jr. The genetic basis of variability in drug responses. Nat Rev Drug Discov. 2002;1:37–44 [DOI] [PubMed] [Google Scholar]
- 6.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Jr., Colvin MM, Drazner MH, Filippatos GS, Fonarow GC, Givertz MM, Hollenberg SM, Lindenfeld J, Masoudi FA, McBride PE, Peterson PN, Stevenson LW, Westlake C. 2017 acc/aha/hfsa focused update of the 2013 accf/aha guideline for the management of heart failure: A report of the american college of cardiology/american heart association task force on clinical practice guidelines and the heart failure society of america. Circulation. 2017;136:e137–e161 [DOI] [PubMed] [Google Scholar]
- 7.Rienstra M, Damman K, Mulder BA, Van Gelder IC, McMurray JJ, Van Veldhuisen DJ. Beta-blockers and outcome in heart failure and atrial fibrillation: A meta-analysis. JACC. Heart failure 2013;1:21–28 [DOI] [PubMed] [Google Scholar]
- 8.Kotecha D, Holmes J, Krum H, Altman DG, Manzano L, Cleland JG, Lip GY, Coats AJ, Andersson B, Kirchhof P, von Lueder TG, Wedel H, Rosano G, Shibata MC, Rigby A, Flather MD, Beta-Blockers in Heart Failure Collaborative G. Efficacy of beta blockers in patients with heart failure plus atrial fibrillation: An individual-patient data meta-analysis. Lancet. 2014;384:2235–2243 [DOI] [PubMed] [Google Scholar]
- 9.Talameh JA, Lanfear DE. Pharmacogenetics in chronic heart failure: New developments and current challenges. Current heart failure reports. 2012;9:23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shahin MH, Conrado DJ, Gonzalez D, Gong Y, Lobmeyer MT, Beitelshees AL, Boerwinkle E, Gums JG, Chapman A, Turner ST, Cooper-DeHoff RM, Johnson JA. Genome-wide association approach identified novel genetic predictors of heart rate response to beta-blockers. J Am Heart Assoc. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parikh KS, Fiuzat M, Davis G, Neely M, Blain-Nelson P, Whellan DJ, Abraham WT, Adams KF Jr., Felker GM, Liggett SB, O’Connor CM, Bristow MR. Dose response of beta-blockers in adrenergic receptor polymorphism genotypes. Circ Genom Precis Med. 2018;11:e002210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson JA, Liggett SB. Cardiovascular pharmacogenomics of adrenergic receptor signaling: Clinical implications and future directions. Clin Pharmacol Ther. 2011;89:366–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shin J, Johnson JA. Pharmacogenetics of beta-blockers. Pharmacotherapy. 2007;27:874–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiuzat M, Neely ML, Starr AZ, Kraus WE, Felker GM, Donahue M, Adams K, Pina IL, Whellan D, O’Connor CM. Association between adrenergic receptor genotypes and beta-blocker dose in heart failure patients: Analysis from the hf-action DNA substudy. Eur J Heart Fail. 2013;15:258–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cresci S, Dorn GW 2nd, Jones PG, Beitelshees AL, Li AY, Lenzini PA, Province MA, Spertus JA, Lanfear DE. Adrenergic-pathway gene variants influence beta-blocker-related outcomes after acute coronary syndrome in a race-specific manner. J Am Coll Cardiol. 2012;60:898–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luzum JA, English JD, Ahmad US, Sun JW, Canan BD, Sadee W, Kitzmiller JP, Binkley PF. Association of genetic polymorphisms in the beta-1 adrenergic receptor with recovery of left ventricular ejection fraction in patients with heart failure. J Cardiovasc Transl Res. 2019;12:280–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr., Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL. 2013 accf/aha guideline for the management of heart failure: A report of the american college of cardiology foundation/american heart association task force on practice guidelines. J Am Coll Cardiol. 2013;62:e147–239 [DOI] [PubMed] [Google Scholar]
- 18.Claassens DMF, Vos GJA, Bergmeijer TO, Hermanides RS, van ‘t Hof AWJ, van der Harst P, Barbato E, Morisco C, Tjon Joe Gin RM, Asselbergs FW, Mosterd A, Herrman JR, Dewilde WJM, Janssen PWA, Kelder JC, Postma MJ, de Boer A, Boersma C, Deneer VHM, Ten Berg JM. A genotype-guided strategy for oral p2y12 inhibitors in primary pci. N Engl J Med. 2019;381:1621–1631 [DOI] [PubMed] [Google Scholar]
- 19.Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nature genetics. 2008;40:695–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibson G On the utilization of polygenic risk scores for therapeutic targeting. PLoS genetics. 2019;15:e1008060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tansey KE, Guipponi M, Hu X, Domenici E, Lewis G, Malafosse A, Wendland JR, Lewis CM, McGuffin P, Uher R. Contribution of common genetic variants to antidepressant response. Biological psychiatry. 2013;73:679–682 [DOI] [PubMed] [Google Scholar]
- 22.Che R, Motsinger-Reif AA. Evaluation of genetic risk score models in the presence of interaction and linkage disequilibrium. Frontiers in genetics. 2013;4:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kathiresan S, Melander O, Anevski D, Guiducci C, Burtt NP, Roos C, Hirschhorn JN, Berglund G, Hedblad B, Groop L, Altshuler DM, Newton-Cheh C, Orho-Melander M. Polymorphisms associated with cholesterol and risk of cardiovascular events. The New England journal of medicine. 2008;358:1240–1249 [DOI] [PubMed] [Google Scholar]
- 24.Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, Kaplan L, Bennett D, Li Y, Tanaka T, Voight BF, Bonnycastle LL, Jackson AU, Crawford G, Surti A, Guiducci C, Burtt NP, Parish S, Clarke R, Zelenika D, Kubalanza KA, Morken MA, Scott LJ, Stringham HM, Galan P, Swift AJ, Kuusisto J, Bergman RN, Sundvall J, Laakso M, Ferrucci L, Scheet P, Sanna S, Uda M, Yang Q, Lunetta KL, Dupuis J, de Bakker PI, O’Donnell CJ, Chambers JC, Kooner JS, Hercberg S, Meneton P, Lakatta EG, Scuteri A, Schlessinger D, Tuomilehto J, Collins FS, Groop L, Altshuler D, Collins R, Lathrop GM, Melander O, Salomaa V, Peltonen L, Orho-Melander M, Ordovas JM, Boehnke M, Abecasis GR, Mohlke KL, Cupples LA. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41:56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Natarajan P, Young R, Stitziel NO, Padmanabhan S, Baber U, Mehran R, Sartori S, Fuster V, Reilly DF, Butterworth A, Rader DJ, Ford I, Sattar N, Kathiresan S. Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation. 2017;135:2091–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shahin MH, Gong Y, McDonough CW, Rotroff DM, Beitelshees AL, Garrett TJ, Gums JG, Motsinger-Reif A, Chapman AB, Turner ST, Boerwinkle E, Frye RF, Fiehn O, Cooper-DeHoff RM, Kaddurah-Daouk R, Johnson JA. A genetic response score for hydrochlorothiazide use: Insights from genomics and metabolomics integration. Hypertension. 2016;68:621–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gong Y, McDonough CW, Wang Z, Hou W, Cooper-DeHoff RM, Langaee TY, Beitelshees AL, Chapman AB, Gums JG, Bailey KR, Boerwinkle E, Turner ST, Johnson JA. Hypertension susceptibility loci and blood pressure response to antihypertensives: Results from the pharmacogenomic evaluation of antihypertensive responses study. Circulation. Cardiovascular genetics 2012;5:686–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hettige NC, Cole CB, Khalid S, De Luca V. Polygenic risk score prediction of antipsychotic dosage in schizophrenia. Schizophrenia research. 2016;170:265–270 [DOI] [PubMed] [Google Scholar]
- 29.Alemany-Navarro M, Costas J, Real E, Segalas C, Bertolin S, Domenech L, Rabionet R, Carracedo A, Menchon JM, Alonso P. Do polygenic risk and stressful life events predict pharmacological treatment response in obsessive compulsive disorder? A gene-environment interaction approach. Translational psychiatry. 2019;9:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mega JL, Stitziel NO, Smith JG, Chasman DI, Caulfield M, Devlin JJ, Nordio F, Hyde C, Cannon CP, Sacks F, Poulter N, Sever P, Ridker PM, Braunwald E, Melander O, Kathiresan S, Sabatine MS. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: An analysis of primary and secondary prevention trials. Lancet. 2015;385:2264–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strauss DG, Vicente J, Johannesen L, Blinova K, Mason JW, Weeke P, Behr ER, Roden DM, Woosley R, Kosova G, Rosenberg MA, Newton-Cheh C. Common genetic variant risk score is associated with drug-induced qt prolongation and torsade de pointes risk: A pilot study. Circulation. 2017;135:1300–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewis JP, Backman JD, Reny JL, Bergmeijer TO, Mitchell BD, Ritchie MD, Dery JP, Pakyz RE, Gong L, Ryan K, Kim EY, Aradi D, Fernandez-Cadenas I, Lee MTM, Whaley RM, Montaner J, Gensini GF, Cleator JH, Chang K, Holmvang L, Hochholzer W, Roden DM, Winter S, Altman RB, Alexopoulos D, Kim HS, Gawaz M, Bliden KP, Valgimigli M, Marcucci R, Campo G, Schaeffeler E, Dridi NP, Wen MS, Shin JG, Fontana P, Giusti B, Geisler T, Kubo M, Trenk D, Siller-Matula JM, Ten Berg JM, Gurbel PA, Schwab M, Klein TE, Shuldiner AR, Investigators I. Pharmacogenomic polygenic response score predicts ischaemic events and cardiovascular mortality in clopidogrel-treated patients. Eur Heart J Cardiovasc Pharmacother. 2020;6:203–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wimberley T, Gasse C, Meier SM, Agerbo E, MacCabe JH, Horsdal HT. Polygenic risk score for schizophrenia and treatment-resistant schizophrenia. Schizophrenia bulletin. 2017;43:1064–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia-Gonzalez J, Tansey KE, Hauser J, Henigsberg N, Maier W, Mors O, Placentino A, Rietschel M, Souery D, Zagar T, Czerski PM, Jerman B, Buttenschon HN, Schulze TG, Zobel A, Farmer A, Aitchison KJ, Craig I, McGuffin P, Giupponi M, Perroud N, Bondolfi G, Evans D, O’Donovan M, Peters TJ, Wendland JR, Lewis G, Kapur S, Perlis R, Arolt V, Domschke K, Breen G, Curtis C, Sang-Hyuk L, Kan C, Newhouse S, Patel H, Baune BT, Uher R, Lewis CM, Fabbri C. Pharmacogenetics of antidepressant response: A polygenic approach. Progress in neuro-psychopharmacology & biological psychiatry. 2017;75:128–134 [DOI] [PubMed] [Google Scholar]
- 35.Luzum JA, Peterson E, Li J, She R, Gui H, Liu B, Spertus JA, Pinto YM, Williams LK, Sabbah HN, Lanfear DE. Race and beta-blocker survival benefit in patients with heart failure: An investigation of self-reported race and proportion of african genetic ancestry. J Am Heart Assoc. 2018;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pfisterer M, Buser P, Rickli H, Gutmann M, Erne P, Rickenbacher P, Vuillomenet A, Jeker U, Dubach P, Beer H, Yoon SI, Suter T, Osterhues HH, Schieber MM, Hilti P, Schindler R, Brunner-La Rocca HP, Investigators T-C. Bnp-guided vs symptom-guided heart failure therapy: The trial of intensified vs standard medical therapy in elderly patients with congestive heart failure (time-chf) randomized trial. JAMA. 2009;301:383–392 [DOI] [PubMed] [Google Scholar]
- 37.O’Connor CM, Whellan DJ, Lee KL, Keteyian SJ, Cooper LS, Ellis SJ, Leifer ES, Kraus WE, Kitzman DW, Blumenthal JA, Rendall DS, Miller NH, Fleg JL, Schulman KA, McKelvie RS, Zannad F, Pina IL, Investigators H-A. Efficacy and safety of exercise training in patients with chronic heart failure: Hf-action randomized controlled trial. JAMA : the journal of the American Medical Association. 2009;301:1439–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lanfear DE, Hrobowski TN, Peterson EL, Wells KE, Swadia TV, Spertus JA, Williams LK. Association of beta-blocker exposure with outcomes in heart failure differs between african american and white patients. Circ Heart Fail. 2012;5:202–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKee PA, Castelli WP, McNamara PM, Kannel WB. The natural history of congestive heart failure: The framingham study. The New England journal of medicine. 1971;285:1441–1446 [DOI] [PubMed] [Google Scholar]
- 40.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic acids research. 1988;16:1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, Schlessinger D, Stambolian D, Loh PR, Iacono WG, Swaroop A, Scott LJ, Cucca F, Kronenberg F, Boehnke M, Abecasis GR, Fuchsberger C. Next-generation genotype imputation service and methods. Nature genetics. 2016;48:1284–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. A global reference for human genetic variation. Nature. 2015;526:68–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pocock SJ, Ariti CA, McMurray JJ, Maggioni A, Kober L, Squire IB, Swedberg K, Dobson J, Poppe KK, Whalley GA, Doughty RN, Meta-Analysis Global Group in Chronic Heart F. Predicting survival in heart failure: A risk score based on 39 372 patients from 30 studies. European heart journal. 2013;34:1404–1413 [DOI] [PubMed] [Google Scholar]
- 44.Rosenbaum PR, Rubin DB. The central role of the propensity score in observational studies for causal effects. Biometrika. 1983;70:41–55 [Google Scholar]
- 45.D’Agostino RB Jr. Propensity score methods for bias reduction in the comparison of a treatment to a non-randomized control group. Statistics in medicine. 1998;17:2265–2281 [DOI] [PubMed] [Google Scholar]
- 46.Simon RM, Subramanian J, Li MC, Menezes S. Using cross-validation to evaluate predictive accuracy of survival risk classifiers based on high-dimensional data. Briefings in bioinformatics. 2011;12:203–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cohen J Statistical power analysis for the behavioral sciences. New York: Academic Press; 1977. [Google Scholar]
- 48.Lewis JP, Backman JD, Reny JL, Bergmeijer TO, Mitchell BD, Ritchie MD, Dery JP, Pakyz RE, Gong L, Ryan K, Kim EY, Aradi D, Fernandez-Cadenas I, Lee MTM, Whaley RM, Montaner J, Gensini GF, Cleator JH, Chang K, Holmvang L, Hochholzer W, Roden DM, Winter S, Altman R, Alexopoulos D, Kim HS, Gawaz M, Bliden K, Valgimigli M, Marcucci R, Campo G, Schaeffeler E, Dridi NP, Wen MS, Shin JG, Fontana P, Giusti B, Geisler T, Kubo M, Trenk D, Siller-Matula JM, Ten Berg JM, Gurbel PA, Schwab M, Klein TE, Shuldiner AR. Pharmacogenomic polygenic response score predicts ischemic events and cardiovascular mortality in clopidogrel-treated patients. European heart journal. Cardiovascular pharmacotherapy 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Torkamani A, Wineinger NE, Topol EJ. The personal and clinical utility of polygenic risk scores. Nature reviews. Genetics 2018;19:581–590 [DOI] [PubMed] [Google Scholar]
- 50.Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT, Kathiresan S. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weng LC, Preis SR, Hulme OL, Larson MG, Choi SH, Wang B, Trinquart L, McManus DD, Staerk L, Lin H, Lunetta KL, Ellinor PT, Benjamin EJ, Lubitz SA. Genetic predisposition, clinical risk factor burden, and lifetime risk of atrial fibrillation. Circulation. 2018;137:1027–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dorling L, Kar S, Michailidou K, Hiller L, Vallier AL, Ingle S, Hardy R, Bowden SJ, Dunn JA, Twelves C, Poole CJ, Caldas C, Earl HM, Pharoah PD, Abraham JE. The relationship between common genetic markers of breast cancer risk and chemotherapy-induced toxicity: A case-control study. PloS one. 2016;11:e0158984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Metra M, Nodari S, Parrinello G, Giubbini R, Manca C, Dei Cas L. Marked improvement in left ventricular ejection fraction during long-term beta-blockade in patients with chronic heart failure: Clinical correlates and prognostic significance. American Heart Journal. 2003;145:292–299 [DOI] [PubMed] [Google Scholar]
- 54.Ghanem CI, Manautou JE. Modulation of hepatic mrp3/abcc3 by xenobiotics and pathophysiological conditions: Role in drug pharmacokinetics. Curr Med Chem. 2019;26:1185–1223 [DOI] [PubMed] [Google Scholar]
- 55.McDonough CW, Gong Y, Padmanabhan S, Burkley B, Langaee TY, Melander O, Pepine CJ, Dominiczak AF, Cooper-Dehoff RM, Johnson JA. Pharmacogenomic association of nonsynonymous snps in siglec12, a1bg, and the selectin region and cardiovascular outcomes. Hypertension. 2013;62:48–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jarvinen E, Troberg J, Kidron H, Finel M. Selectivity in the efflux of glucuronides by human transporters: Mrp4 is highly active toward 4-methylumbelliferone and 1-naphthol glucuronides, while mrp3 exhibits stereoselective propranolol glucuronide transport. Mol Pharm. 2017;14:3299–3311 [DOI] [PubMed] [Google Scholar]
- 57.Sun YL, Hu SJ, Wang LH, Hu Y, Zhou JY. Effect of beta-blockers on cardiac function and calcium handling protein in postinfarction heart failure rats. Chest. 2005;128:1812–1821 [DOI] [PubMed] [Google Scholar]
- 58.Mackiewicz U, Maczewski M, Konior A, Tellez JO, Nowis D, Dobrzynski H, Boyett MR, Lewartowski B. Sarcolemmal ca2+-atpase ability to transport ca2+ gradually diminishes after myocardial infarction in the rat. Cardiovascular research. 2009;81:546–554 [DOI] [PubMed] [Google Scholar]
- 59.Lowes BD, Gilbert EM, Abraham WT, Minobe WA, Larrabee P, Ferguson D, Wolfel EE, Lindenfeld J, Tsvetkova T, Robertson AD, Quaife RA, Bristow MR. Myocardial gene expression in dilated cardiomyopathy treated with beta-blocking agents. N Engl J Med. 2002;346:1357–1365 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.