

Abstract
Porcine deltacoronavirus (PDCoV) has been detected sporadically in China since its first description in 2012. In our study, 62 faecal and intestinal samples from pigs with diarrhoea were collected in Guangxi Province, China, during 2017 and 2018. Twelve samples (19.4%, 12/62) were positive for PDCoV. Five complete genomes of PDCoV were then determined, and sequence alignment revealed that the five strains had discontinuous deletions at 400–401 aa in non‐structural protein 2 (NSP2) and 758–760 aa in non‐structural protein 3 (NSP3) compared with the respective proteins in the HKU15‐44 strain. Notably, the CHN‐GX81‐2018 strain contained two insertions in the S gene and 3′‐UTR. Multiple sequence alignment and phylogenetic analysis showed that four strains shared 98.2%–98.4% nucleotide identity with CHN‐AH‐2004 and were classified into a new cluster of China lineage strains, whereas the CHN‐GX81‐2018 strain shared 98.7% nucleotide identity with Vietnam/Binh21/2015 and belonged to the Vietnam/Laos/Thailand lineage. Recombination analyses revealed that four strains were the result of recombination between CHN‐HB‐2014 and Vietnam/Binh21/2015 strains. This study demonstrated the co‐existence of multiple lineages of PDCoV in China, and our findings will aid the reorganization and evolution of the virus.
Keywords: evolution, genetic characterization, non‐structural proteins, PDCoV
Five complete genomes of PDCoV were sequenced and had discontinuous deletions at 400–401 aa in non‐structural protein 2 (NSP2) and 758–760 aa in non‐structural protein 3 (NSP3) compared with the respective proteins in the HKU15‐44 strain. Four strains were classified into a new cluster of China lineage strains, whereas the CHN‐GX81‐2018 strain belonged to the Vietnam/Laos/Thailand lineage.

1. INTRODUCTION
Coronaviruses (CoVs), including four genera of these viruses (alpha‐, beta‐, gamma‐ and delta‐CoVs), are present in a wide variety of animals (Wang, Su, Bi, Wong, & Gao, 2018; Wertheim, Chu, Peiris, Kosakovsky Pond, & Poon, 2013). Five porcine CoVs that cause diarrhoea in pigs have been identified: porcine epidemic diarrhoea virus (PEDV) (Wang, Lan, & Yang, 2016), transmissible gastroenteritis virus (TGEV) (Sirinarumitr, Paul, Kluge, & Halbur, 1996), porcine respiratory coronavirus (PRCV) (Sirinarumitr et al., 1996), porcine enteric alphacoronavirus (PEAV) (Fu et al., 2018; Mandelik et al., 2018; Xu et al., 2019; Zhou et al., 2018) and porcine deltacoronavirus (PDCoV) (Wang, Byrum, & Zhang, 2014). PDCoV, an emerging enveloped, positive‐sense RNA virus, belongs to the genus Deltacoronavirus in the family Coronaviridae and has attracted considerable attention due to its pathogenicity to newborn piglets (Ajayi et al., 2018; Chen et al., 2015). The genome of PDCoV is 25.4 kb in length, containing eight open reading frames (ORFs): four nonstructural proteins (ORF1a, ORF1b, NS6 and NS7) and four structural proteins (the spike (S), envelope (E), membrane (M) and nucleoprotein (N)), arranged in the order 5′‐UTR‐ORF1a‐ORF1b‐S‐E‐M‐NS6‐N‐NS7‐3′‐UTR (Li et al., 2014). The polyproteins pp1a and pp1b, encoded by the ORF1a and ORF1b genes, are cleaved into a total of 15 functional proteins, these proteins, known as NSP2 to NSP16, are involved in viral replication and transcription. (Chen et al., 2015). Among these structural proteins, the S protein is the pivotal surface glycoprotein involved in viral attachment and contains immunologically important B cell epitopes associated with viral neutralization (Dong et al., 2015). Significantly, the S protein is a useful marker for determining the genetic diversity of PDCoVs (Zhai et al., 2016).
Since the first discovery of PDCoV in 2012 (Woo et al., 2012), outbreaks of this virus have been reported in the United States (Homwong et al., 2016), Canada (Ajayi et al., 2018), China (Dong et al., 2015; Liu et al., 2018; Mai, Feng, et al., 2018; Mai, Li, et al., 2018; Wang, Wang, et al., 2018; Zhang, Liang, et al., 2019), South Korea (Jang, Lee, Kim, & Lee, 2017), Lao People's Democratic Republic (Lorsirigool et al., 2016), Thailand, Vietnam (Janetanakit et al., 2016), Japan (Suzuki, Shibahara, Imai, Yamamoto, & Ohashi, 2018), Taiwan (Hsu et al., 2018) and Mexico (Perez‐Rivera, Ramirez‐Mendoza, Mendoza‐Elvira, Segura‐Velazquez, & Sanchez‐Betancourt, 2019), and these have caused enormous economic losses. Phylogenetic divergence analyses based on the S protein have revealed that the majority of PDCoV strains in the world can be classified into three lineages: the China lineage, the US/Japan/South Korea lineage and the Vietnam/Laos/Thailand lineage. Among these three lineages, strains from the China lineage have been prevalent since 2014 and have led to pandemics in large areas of China between 2014 and 2019 (Zhang, Cheng, et al., 2019). The US/Japan/South Korea lineage was first reported in the United States in 2014 and led to pandemics in large areas of North America, Japan and Korea between 2014 and 2019. In contrast, the Vietnam/Laos/Thailand lineage, which was first reported in Thailand in 2015, has been identified in Southeast Asian countries, including Thailand, Vietnam and Laos (Zhang, Cheng, et al., 2019).
PDCoVs induce major clinical manifestations, including diarrhoea, vomiting, dehydration and lethality, in new‐born piglets (Xu et al., 2018). PDCoVs are geared for rapid variation through mutation or recombination, resulting in the production of new isolates with different levels of pathogenicity and virulence (Perez‐Rivera et al., 2019; Sun et al., 2020; Zhang, Liu, et al., 2019). At present, multiple‐recombination types of PDCoVs exist in Chinese swine herds, and these have caused concern regarding the pathogenicity of the variants. Few studies have reported the presence of multilineage strains in China. However, the pathogenesis of diarrhoea caused by PDCoVs belonging to different lineage strains has not been well studied. The aim of this study was to detect the epidemiological and evolutionary characteristics of PDCoVs between 2017 and 2018 through the analysis of 62 clinical samples from pigs in Guangxi Province, China.
2. MATERIALS AND METHODS
2.1. Sample collection
A total of 62 intestinal samples (jejunum and duodenum tissue sections) and faecal samples were collected from five cities (Nanning, Beihai, Baise, Yulin and Fangcheng gang) between 2017 and 2018. Information on the samples collected in this study is provided in Table 1. All clinical samples were transported to the Guangxi Center for Animal Disease Control and Prevention and stored at −80°C.
TABLE 1.
RT‐PCR detection of PDCoV, PEDV, PoRV, and TGEV in the samples
| Prefecture | No. of farms | No. of samples | No. of positive samples (%) | |||
|---|---|---|---|---|---|---|
| PDCoV | PEDV | TGEV | PoRV | |||
| Yulin | 2 | 10 | 2 (20.0%) | 1 (10.0%) | 0 (0%) | 1 (10.0%) |
| Nanning | 3 | 22 | 6 (27.3%) | 10 (45.5%) | 1 (4.5%) | 2 (9.1%) |
| Beihai | 1 | 12 | 1 (8.3%) | 4 (33.3%) | 0 (0%) | 0 (0%) |
| Baise | 1 | 13 | 3 (23.1%) | 2 (15.4%) | 1 (7.7%) | 0 (0%) |
| Fangcheng gang | 1 | 5 | 0 (0%) | 3 (60.0%) | 1 (20.0%) | 1 (20.0%) |
| Total (%) | 8 | 62 | 12 (19.4%) | 20 (32.3%) | 3 (4.8%) | 4 (6.5%) |
The experimental protocols used for obtaining the clinical pig samples used in this study were obtained from Guangxi Center for Animal Disease Control and Prevention and performed in strict accordance with the Animal Welfare and the Animal Experimental Ethical Committee (Guangxi University, No. Xidakezi2000138). All pig owners agreed to use the samples prior to participation in the study.
2.2. RNA extraction and RT‐PCR
Total RNA from intestinal contents and/or faeces was extracted using the UNlQ‐10 Column TRIzol Total RNA Reagent (Sangon Biotech) following the manufacturer's instructions. The PrimeScript RT reagent kit (Takara Co.) was used for RT‐PCR according to the manufacturer's recommended protocol. Previous studies confirmed PDCoV coinfections with PEDV, porcine rotavirus (PoRV) and TGEV in the diarrhoeal specimens from pigs. Specific primers were used for amplifying the PDCoV, PEDV, PoRV and TGEV genes, as previously described (Jang et al., 2017). The experimental design is shown in Figure 1.
FIGURE 1.
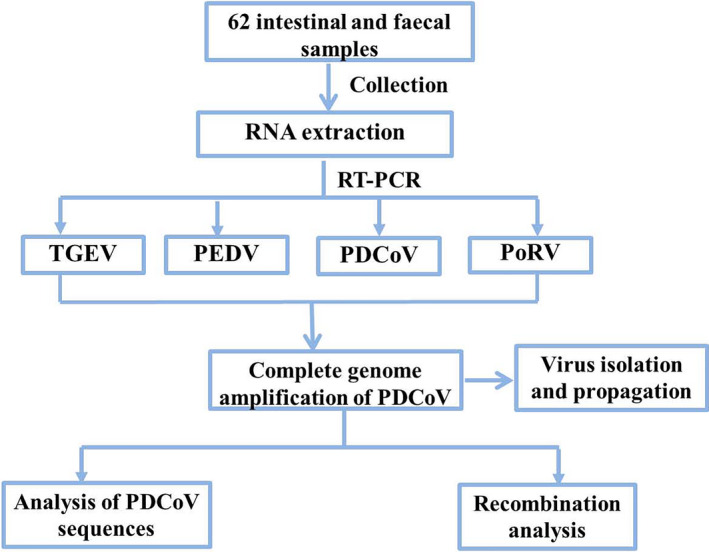
Experimental design
2.3. Complete genome amplification of PDCoV
Twenty‐five pairs of primers were used to amplify the complete PDCoV genomes (Table S1). The amplified products were purified and cloned into pMD‐18T. The cloned products were then sequenced (Takara Co.). For each of the amplified fragments, two clones were sequenced.
2.4. Analysis of PDCoV sequences
Reference PDCoV strains were retrieved from GenBank (Table S2) for genetic analysis. The nucleotide and deduced amino acid sequences were aligned by ClustalW using the Lasergene sequence analysis software package (MegAlign). Phylogenetic trees were generated using the neighbour‐joining (NJ) method with 1000 bootstrap replicates with the MEGA package (version 7.0).
2.5. Recombination analysis
For the investigation of putative recombination events, we identified possible recombination breakpoints using Recombination Detection Program version 4.0 (RDP4). To visualize the recombination events and breakpoints, a similarity analysis was implemented in SimPlot software (v3.5.1) with a 200‐bp window width and a 20‐bp step size.
2.6. Virus isolation and propagation
PDCoV‐positive samples were diluted with serum‐free Dulbecco's Modified Eagle Medium (DMEM) and centrifuged at 4,000g and 4°C for 4 min, and the supernatants were collected and filtered through a syringe filter with a 0.22‐μm pore size. The samples were separately inoculated onto monolayers of LLC‐PK1 cells and ST cells. The virus was determined by RT‐PCR.
3. RESULTS AND DISCUSSION
Previous studies have identified PDCoV in samples from pigs with diarrhoea symptoms co‐infected with PEDV, TGEV and PoRV. In this study, 19.4% (12/62) of the samples were positive for PDCoV, as detected by RT‐PCR, which suggests that PDCoV was prevalent in Guangxi Province. Among the 62 porcine samples examined, 20 (32.3%, 20/62) were positive for PEDV, three (4.8%, 3/62) were positive for TGEV and four (6.5%, 4/62) were positive for PoRV. PDCoV/PEDV coinfections were the most common (12.9%) (Table 1).
Five positive samples were used for further complete sequencing. The complete genomic sequences of CHN‐GX01‐2018, CHN‐GX11‐2018, CHN‐GX12‐2018, CHN‐GX81‐2018 and CHN‐GX09‐2018 were determined and deposited in the GenBank database under the accession numbers MK359104 and MN173779‐MN173782, respectively. The five PDCoV genomes were 25,406 and 25,408 nucleotides (nt) in length, which encompassed the 5′‐untranslated region (UTR) and 3′‐UTR, respectively.
A comparative analysis of the whole genome sequence of those five strains shared 99.1%‐99.9% identity among themselves and 98.1%–98.3%, 97.8%–98.1%, 98.2%–98.4% and 98.1%–98.6% nucleotide identity with HKU15‐44, USA/Iowa136/2015, CHN‐AH‐2004 and Vietnam/Binh21/2015, respectively. The S genes of four PDCoV strains (CHN‐GX01‐2018, CHN‐GX09‐2018, CHN‐GX11‐2018 and CHN‐GX12‐2018) showed 97.4%‐97.7% aa identity with the CHN‐AH‐2004 sequence. The S gene of the CHN‐GX81‐2018 strain shared 96.6%‐96.8% nucleotide identity with the USA/Iowa136/2015 and CHN‐AH‐2004 strains and 98.7% nucleotide identity with the Vietnam/Binh21/2015 strain.
To establish the genetic relationships of the five PDCoV strains investigated in this study with other reference strains, we constructed phylogenetic trees based on their S genes and their complete genomic sequences. The genetic evolution analysis based on the complete genome failed to classify the five strains (Figure 2a), while the genetic evolution analysis based on the S gene showed that these PDCoV strains could be divided into two lineages: CHN‐GX01‐2018, CHN‐GX09‐2018, CHN‐GX11‐2018 and CHN‐GX12‐2018 were classified into the China lineage, and the CHN‐GX81‐2018 strain was classified into the Vietnam/Laos/Thailand lineage (Figure 2b). This study provides the complete sequence of a strain belonging to the Vietnam/Laos/Thailand lineage in China. The China lineage has become predominant in China's pig farms since 2012, but the Vietnam/Laos/Thailand lineage also exists in China. However, the source of the virus has not yet been determined.
FIGURE 2.
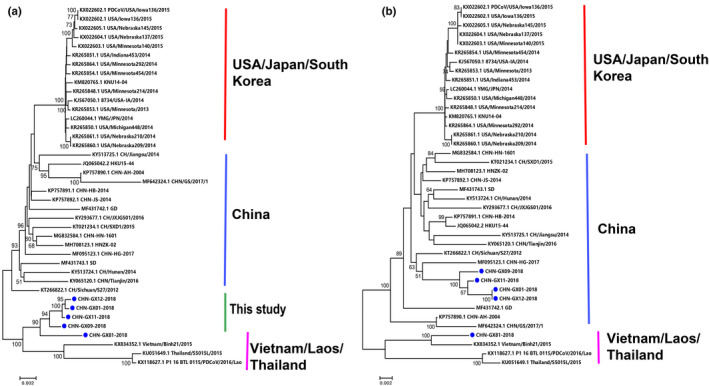
Phylogenetic analyses of PDCoV based on the complete genome (a) and S gene (b) using the neighbour‐joining algorithm and a heuristic search with 1,000 bootstrap replications implemented in MEGA7
In comparison with the strains from the China lineage, the strains from the Vietnam/Laos/Thailand lineage identified the following unique sequence characteristics: 3‐nt (TCT) deletions in the 5′‐UTR, 6‐nt (AGTTTG) and 9‐nt (GAGCCAGTC) deletions in open reading frame 1a/b, 3‐nt (AAT) insertions in the S gene, and 3‐nt (CTC) insertions in the 3′‐UTR. Five strains had a 6‐nt deletion in NSP2, a 9‐nt deletion in the NSP3‐coding region and a 3‐nt (CCC) in the 5′‐UTR. In addition, four PDCoV genomes (CHN‐GX01‐2018, CHN‐GX09‐2018, CHN‐GX11‐2018 and CHN‐GX12‐2018) exhibited a 3‐nt (AAT) deletion in the S gene and a 3‐nt (CTC) insertion in the 3′‐UTR. However, CHN‐GX81‐2018 contained a 3‐nt (AAT) in the S gene and no insertion in the 3′‐UTR (Figure 3). The sequence alignment of the deduced amino acids of the S protein showed that CHN‐GX81‐2018 had the same substitutions as the Vietnam/Laos/Thailand strains at positions 8C/S, 14V/A, 47S/L, 149H/R, 229H/Q, 431D/G, 571V/I and 698 A/S. Four PDCoV genomes (CHN‐GX01‐2018, CHN‐GX09‐2018, CHN‐GX11‐2018 and CHN‐GX12‐2018) had two additional unique substitutions at positions 44S/I and 539 Y/H (Figure S1).
FIGURE 3.
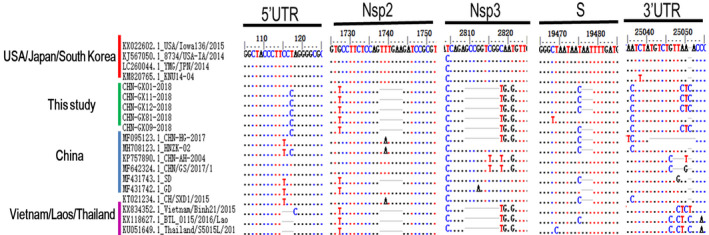
Four main deletions or insertions identified in the complete genome alignment. The multiple sequence alignment was established with ClustalW in BioEdit software
Recombination plays a pivotal role in the evolution of coronaviruses by creating new strains with altered virulence. Previous studies have shown that a large number of new strains were recombined from strains belonging to the China and Vietnam/Laos/Thailand lineages (Dong et al., 2016; Zhang, Liu, et al., 2019; Zhang, Cheng, et al., 2019), e.g., the CHN‐HG‐2017 recombinant strain originated from the CH/SXD1/2015 and Vietnam/HaNoi6/2015 strains. Furthermore, the challenged piglets developed typical symptoms, such as vomiting, anorexia, diarrhoea and lethargy, between 1 and 7 days after inoculation. Our research confirms previous studies describing the discovery of Vietnam/Laos/Thailand lineage strains in China and obtains a Vietnam/Laos/Thailand lineage CHN‐GX81‐2018 strain in China.
Alignments of the complete genes of these PDCoVs with three lineage sequences were analysed using several methods included in RDP4 and confirmed by SimPlot. The genomes of CHN‐GX01‐2018, CHN‐GX09‐2018, CHN‐GX11‐2018 and CHN‐GX12‐2018 shared the same recombination pattern. Four strains were the result of recombination between the CHN‐HB‐2014 (China lineage) and Vietnam/Binh21/2015 (Vietnam/Laos/Thailand lineage) strains. Two recombination breakpoints were identified in CHN‐GX11‐2018: region A located in the NSP2‐NSP3 and region B located in NSP12‐NSP13 (Figure S2 and Table S3).
According to previous reports, efforts to isolate this virus have failed (Hu et al., 2015; Jung, Hu, & Saif, 2016). We thus visually inspected inoculated monolayers of ST and LLC‐PK1 cells, and negative RT‐PCR results were obtained after 10 passages. To date, the Vietnam/Laos/Thailand lineage strain has been successfully isolated in Vietnam (Saeng‐Chuto et al., 2020).
The results of this study demonstrated that multiple lineages of PDCoV, including the China and Vietnam/Laos/Thailand lineages, coexisted in China. An increase in recombination events between different lineages of PDCoV strains could result in greater PDCoV pathogenicity. Further research is needed to elucidate the prevalence and pathogenesis of novel Vietnam/Laos/Thailand‐like strains and to determine the origin and evolution characteristics of the strains.
CONFLICT OF INTEREST
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
AUTHOR CONTRIBUTION
Haixin Huang: Investigation; Methodology. Yanwen Yin: Investigation; Methodology. Wei Wang: Formal analysis; Software. Liang Cao: Formal analysis; Software. Wenchao Sun: Conceptualization; Writing‐original draft. Kaichuang Shi: Project administration; Resources. Huijun Lu: Writing‐review & editing. Ningyi Jin: Writing‐review & editing.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the Guangxi Science and Technology Bureau, China (Grant Number 17204057), the Guangxi Aquatic Animal Husbandry Science and Technology Project (Grant Number 201528017) and the Wenzhou Basic Agricultural Science and Technology Project (Grant Numbers N20180010 and N20190005).
Huang H, Yin Y, Wang W, et al. Emergence of Thailand‐like strains of porcine deltacoronavirus in Guangxi Province, China. Vet Med Sci. 2020;6:854–859. 10.1002/vms3.283
Haixin Huang and Yanwen Yin are contributed equally to this work.
The peer review history for this article is available at https://publons.com/publon/10.1002/vms3.283
Contributor Information
Wenchao Sun, Email: sunwenchao131@163.com.
Kaichuang Shi, Email: shikaichuang@126.com.
DATA AVAILABILITY STATEMENT
No related materials.
REFERENCES
- Ajayi, T. , Dara, R. , Misener, M. , Pasma, T. , Moser, L. , & Poljak, Z. (2018). Herd‐level prevalence and incidence of porcine epidemic diarrhoea virus (PEDV) and porcine deltacoronavirus (PDCoV) in swine herds in Ontario, Canada. Transboundary and Emerging Diseases, 65(5), 1197–1207. 10.1111/tbed.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Q. , Gauger, P. , Stafne, M. , Thomas, J. , Arruda, P. , Burrough, E. , … Welch, M. (2015). Pathogenicity and pathogenesis of a United States porcine deltacoronavirus cell culture isolate in 5‐day‐old neonatal piglets. Virology, 482, 51–59. 10.1016/j.virol.2015.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, N. , Fang, L. , Yang, H. , Liu, H. , Du, T. , Fang, P. , … Xiao, S. (2016). Isolation, genomic characterization, and pathogenicity of a Chinese porcine deltacoronavirus strain CHN‐HN‐2014. Veterinary Microbiology, 196, 98–106. 10.1016/j.vetmic.2016.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, N. , Fang, L. , Zeng, S. , Sun, Q. , Chen, H. , & Xiao, S. (2015). Porcine deltacoronavirus in mainland China. Emerging Infectious Diseases, 21(12), 2254–2255. 10.3201/eid2112.150283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, X. , Fang, B. , Liu, Y. , Cai, M. , Jun, J. , Ma, J. , … Zhang, G. (2018). Newly emerged porcine enteric alphacoronavirus in southern China: Identification, origin and evolutionary history analysis. Infection, Genetics and Evolution, 62, 179–187. 10.1016/j.meegid.2018.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homwong, N. , Jarvis, M. C. , Lam, H. C. , Diaz, A. , Rovira, A. , Nelson, M. , & Marthaler, D. (2016). Characterization and evolution of porcine deltacoronavirus in the United States. Preventive Veterinary Medicine, 123, 168–174. 10.1016/j.prevetmed.2015.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, T. H. , Liu, H. P. , Chin, C. Y. , Wang, C. , Zhu, W. Z. , Wu, B. L. , & Chang, Y. C. (2018). Detection, sequence analysis, and antibody prevalence of porcine deltacoronavirus in Taiwan. Archives of Virology, 163(11), 3113–3117. 10.1007/s00705-018-3964-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, H. , Jung, K. , Vlasova, A. N. , Chepngeno, J. , Lu, Z. , Wang, Q. , & Saif, L. J. (2015). Isolation and characterization of porcine deltacoronavirus from pigs with diarrhea in the United States. Journal of Clinical Microbiology, 53(5), 1537–1548. 10.1128/JCM.00031-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janetanakit, T. , Lumyai, M. , Bunpapong, N. , Boonyapisitsopa, S. , Chaiyawong, S. , Nonthabenjawan, N. , … Amonsin, A. . (2016). Porcine deltacoronavirus, Thailand, 2015. Emerging Infectious Diseases, 22(4), 757–759. 10.3201/eid2204.151852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang, G. , Lee, K. K. , Kim, S. H. , & Lee, C. (2017). Prevalence, complete genome sequencing and phylogenetic analysis of porcine deltacoronavirus in South Korea, 2014–2016. Transboundary and Emerging Diseases, 64(5), 1364–1370. 10.1111/tbed.12690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, K. , Hu, H. , & Saif, L. J. (2016). Porcine deltacoronavirus infection: Etiology, cell culture for virus isolation and propagation, molecular epidemiology and pathogenesis. Virus Research, 226, 50–59. 10.1016/j.virusres.2016.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, B. J. , Zuo, Y. Z. , Gu, W. Y. , Luo, S. X. , Shi, Q. K. , Hou, L. S. , … Fan, J.‐H. . (2018). Isolation and phylogenetic analysis of porcine deltacoronavirus from pigs with diarrhoea in Hebei province, China. Transboundary and Emerging Diseases, 65(3), 874–882. 10.1111/tbed.12821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, G. , Chen, Q. , Harmon, K. M. , Yoon, K. J. , Schwartz, K. J. , Hoogland, M. J. , … Zhang, J. (2014). Full‐length genome sequence of porcine deltacoronavirus strain USA/IA/2014/8734. Genome Announcements, 2(2), e00278‐14 10.1128/genomeA.00278-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorsirigool, A. , Saeng‐chuto, K. , Temeeyasen, G. , Madapong, A. , Tripipat, T. , Wegner, M. , … Nilubol, D. (2016). The first detection and full‐length genome sequence of porcine deltacoronavirus isolated in Lao PDR. Archives of Virology, 161(10), 2909–2911. 10.1007/s00705-016-2983-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai, K. , Feng, J. , Chen, G. , Li, D. , Zhou, L. , Bai, Y. , … Ma, J. (2018). The detection and phylogenetic analysis of porcine deltacoronavirus from Guangdong Province in Southern China. Transboundary and Emerging Diseases, 65(1), 166–173. 10.1111/tbed.12644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai, K. , Li, D. , Wu, J. , Wu, Z. , Cheng, J. , He, L. , … Ma, J. (2018). Complete genome sequences of two porcine deltacoronavirus strains, CHN‐GD16‐03 and CHN‐GD16‐05, isolated in Southern China, 2016. Genome Announcements, 6(4), e01545‐17 10.1128/genomeA.01545-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelik, R. , Sarvas, M. , Jackova, A. , Salamunova, S. , Novotny, J. , & Vilcek, S. (2018). First outbreak with chimeric swine enteric coronavirus (SeCoV) on pig farms in Slovakia – lessons to learn. Acta Veterinaria Hungarica, 66(3), 488–492. 10.1556/004.2018.043 [DOI] [PubMed] [Google Scholar]
- Perez‐Rivera, C. , Ramirez‐Mendoza, H. , Mendoza‐Elvira, S. , Segura‐Velazquez, R. , & Sanchez‐Betancourt, J. I. (2019). First report and phylogenetic analysis of porcine deltacoronavirus in Mexico. Transboundary and Emerging Diseases, 66(4), 1436–1441. 10.1111/tbed.13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeng‐Chuto, K. , Jermsutjarit, P. , Stott, C. J. , Vui, D. T. , Tantituvanont, A. , & Nilubol, D. (2020). Retrospective study, full‐length genome characterization and evaluation of viral infectivity and pathogenicity of chimeric porcine deltacoronavirus detected in Vietnam. Transboundary and Emerging Diseases, 67(1), 183–198. 10.1111/tbed.13339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirinarumitr, T. , Paul, P. S. , Kluge, J. P. , & Halbur, P. G. (1996). In situ hybridization technique for the detection of swine enteric and respiratory coronaviruses, transmissible gastroenteritis virus (TGEV) and porcine respiratory coronavirus (PRCV), in formalin‐fixed paraffin‐embedded tissues. Journal of Virological Methods, 56(2), 149–160. 10.1016/0166-0934(95)01901-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, W. , Wang, L. , Huang, H. , Wang, W. , Cao, L. , Zhang, J. , … Lu, H. (2020). Genetic characterization and phylogenetic analysis of porcine deltacoronavirus (PDCoV) in Shandong Province, China. Virus Research, 278, 197869 10.1016/j.virusres.2020.197869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, T. , Shibahara, T. , Imai, N. , Yamamoto, T. , & Ohashi, S. (2018). Genetic characterization and pathogenicity of Japanese porcine deltacoronavirus. Infection, Genetics and Evolution, 61, 176–182. 10.1016/j.meegid.2018.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. , Lan, X. , & Yang, B. (2016). Molecular Epidemiological investigation of porcine kobuvirus and its coinfection rate with PEDV and SaV in northwest China. BioMed Research International, 2016, 7590569 10.1155/2016/7590569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Byrum, B. , & Zhang, Y. (2014). Porcine coronavirus HKU15 detected in 9 US States, 2014. Emerging Infectious Diseases, 20(9), 1594–1595. 10.3201/eid2009.140756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Su, S. , Bi, Y. , Wong, G. , & Gao, G. F. (2018). Bat‐origin coronaviruses expand their host range to pigs. Trends in Microbiology, 26(6), 466–470. 10.1016/j.tim.2018.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Wang, Y. , Baloch, A. R. , Pan, Y. , Tian, L. , Xu, F. , … Zeng, Q. (2018). Detection and genetic characterization of porcine deltacoronavirus in Tibetan pigs surrounding the Qinghai‐Tibet Plateau of China. Transboundary and Emerging Diseases, 65(2), 363–369. 10.1111/tbed.12819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheim, J. O. , Chu, D. K. , Peiris, J. S. , Kosakovsky Pond, S. L. , & Poon, L. L. (2013). A case for the ancient origin of coronaviruses. Journal of Virology, 87(12), 7039–7045. 10.1128/JVI.03273-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. , Lau, S. K. , Lam, C. S. , Lau, C. C. , Tsang, A. K. , Lau, J. H. , … Zheng, B. J. (2012). Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. Journal of Virology, 86(7), 3995–4008. 10.1128/JVI.06540-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Z. , Zhang, Y. , Gong, L. , Huang, L. , Lin, Y. , Qin, J. , … Cao, Y. (2019). Isolation and characterization of a highly pathogenic strain of porcine enteric alphacoronavirus causing watery diarrhoea and high mortality in newborn piglets. Transboundary and Emerging Diseases, 66(1), 119–130. 10.1111/tbed.12992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Z. , Zhong, H. , Zhou, Q. , Du, Y. , Chen, L. I. , Zhang, Y. , … Cao, Y. (2018). A highly pathogenic strain of porcine deltacoronavirus caused watery diarrhea in newborn piglets. Virologica Sinica, 33(2), 131–141. 10.1007/s12250-018-0003-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai, S.‐L. , Wei, W.‐K. , Li, X.‐P. , Wen, X.‐H. , Zhou, X. , Zhang, H. E. , … Wang, D. (2016). Occurrence and sequence analysis of porcine deltacoronaviruses in southern China. Virology Journal, 13, 136 10.1186/s12985-016-0591-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Liang, Q. , Li, B. , Cui, X. , Wei, X. , Ding, Q. , … Hu, H. (2019). Prevalence, phylogenetic and evolutionary analysis of porcine deltacoronavirus in Henan province, China. Preventive Veterinary Medicine, 166, 8–15. 10.1016/j.prevetmed.2019.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M.‐J. , Liu, D.‐J. , Liu, X.‐L. , Ge, X.‐Y. , Jongkaewwattana, A. , He, Q.‐G. , & Luo, R. (2019). Genomic characterization and pathogenicity of porcine deltacoronavirus strain CHN‐HG‐2017 from China. Archives of Virology, 164(2), 413–425. 10.1007/s00705-018-4081-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Cheng, Y. , Xing, G. , Yu, J. , Liao, A. , Du, L. , … Gu, J. (2019). Detection and spike gene characterization in porcine deltacoronavirus in China during 2016–2018. Infection, Genetics and Evolution, 73, 151–158. 10.1016/j.meegid.2019.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, P. , Fan, H. , Lan, T. , Yang, X.‐L. , Shi, W.‐F. , Zhang, W. , … Ma, J.‐Y. (2018). Fatal swine acute diarrhoea syndrome caused by an HKU2‐related coronavirus of bat origin. Nature, 556(7700), 255–258. 10.1038/s41586-018-0010-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
No related materials.