

Abstract
To develop a vaccine candidate against coronavirus disease 2019 (COVID-19), we generated a lentiviral vector (LV) eliciting neutralizing antibodies against the Spike glycoprotein of SARS-CoV-2. Systemic vaccination by this vector in mice, in which the expression of the SARS-CoV-2 receptor hACE2 has been induced by transduction of respiratory tract cells by an adenoviral vector, confers only partial protection despite high levels of serum neutralizing activity. However, eliciting an immune response in the respiratory tract through an intranasal boost results in a >3 log10 decrease in the lung viral loads and reduces local inflammation. Moreover, both integrative and non-integrative LV platforms display strong vaccine efficacy and inhibit lung deleterious injury in golden hamsters, which are naturally permissive to SARS-CoV-2 replication and closely mirror human COVID-19 physiopathology. Our results provide evidence of marked prophylactic effects of LV-based vaccination against SARS-CoV-2 and designate intranasal immunization as a powerful approach against COVID-19.
Keywords: lentiviral vectors, intranasal vaccination, mucosal immunity, boost-target, immunoglobulin A, lung inflammation, beta-coronavirus, in vivo Ad5 transduction, respiratory tracts, golden hamsters, neutralizing antibodies
Graphical Abstract
Ku et al. present a lentiviral vaccination vector that encodes a full-length, membrane-anchored form of SARS-CoV-2 Spike glycoprotein and induces neutralizing antibodies and T cell responses. An intranasal boost strategy triggers a localized immune response in the upper respiratory tract that provides disease protection in mouse and hamster COVID-19 models.
Introduction
The new severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that emerged in late 2019 in Wuhan, China, is extraordinarily contagious and fast spreading across the world (Guo et al., 2020). Compared with the previously emerged SARS or Middle East respiratory syndrome (MERS) coronavirus, SARS-CoV-2 poses an unprecedented threat to global health and has tremendous socio-economic consequences. Therefore, developing effective prophylactic vaccines against SARS-CoV-2 is absolutely imperative to contain the spread of the epidemic and to prevent the development of coronavirus disease 2019 (COVID-19)-associated symptoms, such as deleterious inflammation and progressive respiratory failure (Amanat and Krammer, 2020).
Coronaviruses are enveloped, non-segmented positive-stranded RNA viruses characterized by their envelope-anchored Spike (S) glycoprotein (Walls et al., 2020). The SARS-CoV-2 S (SCoV-2) is a (180 kDa)3 homotrimeric class I viral fusion protein that engages the carboxypeptidase angiotensin-converting enzyme 2 (ACE2) expressed on host cells. The monomer of SCoV-2 possesses an ectodomain, a transmembrane anchor domain, and a short internal tail. SCoV-2 is activated by a two-step sequential proteolytic cleavage that initiates fusion with the host cell membrane. After SCoV-2-ACE2 interaction, through a conformational reorganization, the extracellular domain of SCoV-2 is first cleaved at the highly specific furin 682RRAR685 site (Guo et al., 2020; Walls et al., 2020), a key factor determining the pathological features of the virus and linked to furin ubiquitous expression (Wang et al., 2020). The resulting subunits are (1) S1, which harbors the ACE2 receptor binding domain (RBD) with the atomic contacts restricted to the ACE2 protease domain, and (2) S2, which bears the membrane-fusion elements. Similar to that of SCoV-1, the shedding of S1 renders accessible on S2 the second proteolytic cleavage site 797R, namely S2′ (Belouzard et al., 2009). Depending on the cell or tissue type, one or several host proteases (including furin, trypsin, cathepsin, or transmembrane protease serine protease-2 or -4) can be involved in this second cleavage step (Coutard et al., 2020). The consequent “fusogenic” conformational changes of S result in the exposure of a fusion peptide (FP) adjacent to S2′. Insertion of FP into the host cell or vesicle membrane primes the fusion reaction, which in turn leads to viral RNA release into the host cytosol (Lai et al., 2017). The facts that the SCoV-2-ACE2 interaction is the only mechanism thus far identified for the host cell infection by SARS-CoV-2 and that the RBD contains numerous conformational B cell epitopes (Walls et al., 2020) designate this viral envelope glycoprotein as the main target for neutralizing antibodies (NAbs).
Compared with (1) attenuated or inactivated viral vaccine candidates, which require extensive safety testing, (2) nucleic acids with moderate immunogenicity in humans linked to the difficulty of their delivery to targeted immune cells (Hobernik and Bros, 2018), and (3) protein vaccines that require the use of adjuvants and boosting, viral vector vaccines such as adenoviral vectors are interesting vaccine candidates that generate strong immune responses. However, adenoviral vectors are the target of pre-existing immunity in the human population, which largely reduces their immunogenicity (Rosenberg et al., 1998; Schirmbeck et al., 2008). Non-replicative lentiviral vectors (LVs) possess great potential to elicit strong and long-lasting adaptive immunity, including antibody (Ab) responses (Di Nunzio et al., 2012; Hu et al., 2011; Ku et al., 2020; Zennou et al., 2000). These vectors induce very minor inflammation (J.L. et al., unpublished data), and their safety has been demonstrated in humans in a phase 1 HIV-1 vaccine trial (2011-006260-52 EN). In addition, LVs are pseudo-typed with the envelope glycoprotein of vesicular stomatitis virus (VSV-G), to which the human population has barely been exposed. This minimizes the risk of reduced vaccine efficacy linked to a pre-existing cross-reactive immunity ( Hu et al., 2011).
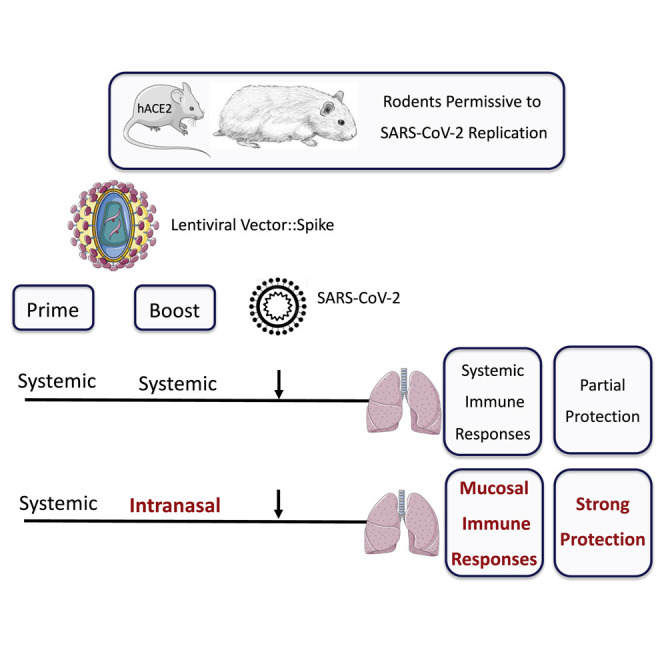
To develop a vaccine candidate able to induce NAbs specific to SCoV-2, we generated LVs coding for (1) S1 alone (LV::S1), (2) the S1-S2 ectodomain without the transmembrane or internal tail domains (LV::S1-S2), or (3) the full-length, membrane-anchored form of S (LV::SFL). We established that LV::SFL gave rise to elevated amounts of NAbs, which inhibit ACE2+ host-cell invasion by SCoV-2-pseudo-typed virions. Anti-SCoV-2 CD8+ T cell effectors were also efficiently induced in LV::SFL-immunized mice. Moreover, in a mouse model in which the expression of human ACE2 (hACE2) was induced in the respiratory tracts by instillation with an adenoviral vector serotype 5 (Ad5), as well as in SARS-CoV-2-susceptible golden hamsters, we demonstrated a strong prophylactic effect of LV::SFL immunization against the replication of a SARS-CoV-2 clinical isolate and a concomitant reduction of infection-related inflammation in the lungs. Importantly, boost-target immunization with LV::SFL via the nasal route was instrumental in the protection efficacy. Our virological, immunological, and histopathological criteria in two preclinical animal models provided the proof-of-principle evidence of marked prophylactic effects of LV-based vaccine strategies against SARS-CoV-2 and the requirement for mucosal immunization to reach vigorous protective lung immunity against COVID-19.
Results
Induction of antibody responses by LVs encoding SARS-CoV-2 Spike protein variants
To develop a vaccine candidate able to induce NAbs against SCoV-2, we generated LVs harboring, under the transcriptional control of the cytomegalovirus (CMV) immediate-early promoter, codon-optimized sequences encoding (1) S1 alone (LV::S1), (2) the S1-S2 ectodomain without the transmembrane or C-terminal short internal tail (LV::S1-S2), and (3) the full-length, membrane-anchored form of S (LV::SFL), all of which harbor the RBD (Figure 1 A; Figure S1), albeit with potential conformational heterogeneities (Yuan et al., 2020). To evaluate the humoral responses induced by these vectors, we immunized C57BL/6 mice (n = 4 per group) by a single intraperitoneal (i.p.) injection of 1 × 107 transduction units (TU) of either an LV or a GFP-encoding LV as a negative control. Anti-SCoV-2 Ab responses were investigated in the sera at weeks 1, 2, 3, 4, and 6 after immunization (Figure 1B). In LV::SFL- or LV::S1-S2-immunized mice, anti-SCoV-2 immunoglobulin G (IgG) was detectable as early as 1 week after immunization and increased progressively until week 6 after immunization to achieve a mean titer (1/dilution) ± SEM of (4.5 ± 2.9) × 106 or (1.5 ± 1) × 106, respectively. In comparison, anti-SCoV-2 IgG titers were 2 orders of magnitude lower, i.e., (7.1 ± 6.1) × 104, in their LV::S1-immunized counterparts. We then evaluated sera for their capacity to neutralize SARS-CoV-2 by using a reliable neutralization assay based on NAb-mediated inhibition of hACE2+ cell invasion by non-replicative LV particle surrogates pseudo-typed with SCoV-2 (Sterlin et al., 2020). Such SCoV-2-pseudo-typed LV particles harbor a luciferase reporter gene, which allows quantitation of the hACE2+ host cell invasion. In this assay, the luciferase activity is inversely proportional to the neutralization efficiency of NAbs present in biological fluids. Fine comparison, by 50% effective concentration (EC50) assays, of the sera from LV::S1-, LV::S1-S2-, or LV::SFL-immunized mice clearly established that LV::SFL was the most potent vector at inducing anti-SCoV-2 NAbs (Figure 1C). Moreover, neutralization activities were correlated with anti-SCoV-2 IgG titers only in the sera of LV::SFL-immunized mice (Figure 1D). These results suggest that the conformation of relevant B cell epitopes is likely to be preserved in SFL but not necessarily in S1 or S1-S2 polypeptides, which could be crucial for giving rise to NAbs. Comparison of the sera from the LV::SFL-immunized mice and a cohort of mildly symptomatic infected individuals living in Crépy-en-Valois, one of the first epidemic zones to appear in France, revealed similar mean neutralizing activities (Figure 1E). These data predict a potentially protective humoral response induced by LV::SFL.
Figure 1.

Induction of anti-SCoV-2 Ab responses by LV
(A) Schematic representation of three forms of SCoV-2 protein (SFL, S1-S2, and S1) encoded by LV injected into mice. RBD, S1/S2 and S2′ cleavage sites, fusion peptide (FP), transmembrane domain (TM), and short internal tail (T) are indicated.
(B) Dynamic of anti-SCoV-2 Ab response after LV immunization. C57BL/6 mice (n = 4 per group) received an i.p. injection of 1 × 107 TU of LV::GFP as a negative control, LV::S1, LV::S1-S2, or LV::SFL. Sera were collected 2, 3, 4, and 6 weeks after immunization. Anti-SCoV-2 IgG responses were evaluated by ELISA and expressed as mean endpoint dilution titers.
(C) Neutralization capacity of anti-SCoV-2 Abs induced by LV::SFL immunization. Mouse sera were evaluated in a sero-neutralization assay for determination of EC50 neutralizing titers.
(D) Correlation between the Ab titers and neutralization activity in various experimental groups. Statistical significance was determined by a two-sided Spearman rank-correlation test. NS, not significant.
(E) Head-to-head comparison at a 1:40 dilution between mouse sera taken 3 or 4 weeks after immunization and a cohort of mildly symptomatic individuals living in Crépy-en-Valois, Ile de France. These patients did not seek medical attention and recovered from COVID-19. Results are expressed as mean ± SEM percentages of inhibition of luciferase activity. See also Figure S1.
LV::SFL-immunized C57BL/6 mice (n = 3) also displayed strong anti-SCoV-2 T cell responses, as detected 2 weeks after immunization by IFN-γ ELISPOT-based epitope mapping applied to splenocytes stimulated with distinct pools of 15-mer peptides spanning the full-length SCoV-2 (Figure 2 A). Significant amounts of responding T cells were detected in 6 of 16 peptide pools. Deconvolution of these positive pools allowed identification of S:256–275 (SGWTAGAAAYYVGYLQPRTF), S:536–550 (NKCVNFNFNGLTGTG), and S:576:590 (VRDPQTLEILDITPC) immunodominant epitopes, giving rise to >2,000 spot forming units (SFU) per 1 × 106 splenocytes (Figure 2B). These epitopes elicited CD8+ (but not CD4+) T cells, as assessed by intracellular cytokine staining (Figure 2C). The predominant CD8+ phenotype of these T cells is in accordance with the favored orientation of LV-encoded antigens to the MHC-I presentation pathway (Hu et al., 2011). We also identified S:441–455 (LDSKVGGNYNYLYRL), S:671–685 (CASYQTQTNSPRRAR), and S:991–1005 (VQIDRLITGRLQSLQ) subdominant epitopes, which gave rise to <2,000 SFU per 1 × 106 splenocytes in the ELISPOT assay (Figure 2B).
Figure 2.

Induction of T cell responses by LV::SFL
C57BL/6 mice (n = 3) were immunized with 1 × 107 TU of LV::SFL or a negative control LV via i.p. injection.
(A) Splenocytes collected 2 weeks after immunization were subjected to an IFN-γ ELISPOT with 16 distinct pools of 15-mer peptides spanning the entire SCoV-2 (1–1273 aa) and overlapping each other by 10 aa residues. SFU, spot-forming units.
(B) Deconvolution of the positive peptide pools by ELISPOT applied to splenocytes pooled from three LV::SFL-or control-LV-immunized mice.
(C) Intracellular IFN-γ versus IL-2 staining of CD4+ or CD8+ T splenocytes after stimulation with individual peptides encompassing the immuodominant epitopes.
Establishment of a murine model expressing hACE2 in respiratory tracts
Because SCoV-2 does not interact well with murine ACE2, wild-type laboratory mice are not permissive to replication of SARS-CoV-2 clinical isolates. Therefore, we sought to develop a murine model in which human ACE2 (hACE2) expression was induced in the respiratory tracts and pulmonary mucosa to evaluate the efficacy of the LV::SFL vaccine. This method has been successfully used to establish the expression of human DPP4 for the study of mouse infection with MERS-CoV (Zhao et al., 2014) and also for hACE2 during the preparation of this manuscript (Sun et al., 2020). We generated an Ad5 vector to deliver the gene encoding hACE2 under the transcriptional control of the CMV promoter (Ad5::hACE2). We first checked in vitro the potential of the Ad5::hACE2 vector to transduce HEK293T cells by reverse-transcriptase PCR (RT-PCR) (Figure 3 A). To achieve in vivo transduction of respiratory tract cells, we instilled 2.5 × 109 infectious genome units (IGU) of Ad5::hACE2 into C57BL/6 mice via intranasal (i.n.) administration. Four days later, hACE2 protein expression was detectable in the lung cell homogenate by western blot (Figure 3B). To get more insights into the in vivo expression profile of a transgene administered under these conditions, we instilled the same dose of an Ad5::GFP reporter vector into C57BL/6 mice via i.n. administration. As evaluated by flow cytometry, 4 days after instillation, the GFP reporter was expressed not only in the lung epithelial EpCam+ cells but also in lung immune cells, as tracked by the CD45 pan-hematopoietic marker (Figure 3C), showing that through this approach the transduction was efficiently achieved in epithelial cells, although not restricted to these cells.
Figure 3.

Setup of a murine model expressing hACE2 in the respiratory tracts
(A) Detection of hACE2 expression by RT-PCR in HEK293 T cells transduced with Ad5::hACE2 2 days after transduction. NT, not transduced.
(B) hACE2 protein detection by western blot in lung cell extracts recovered 4 days after i.n. instillation of Ad5::hACE2 or empty Ad5 into C57BL/6 mice (n = 2 per group).
(C) GFP expression in lung cells prepared 4 days after i.n. instillation of Ad5::GFP or PBS into C57BL/6 mice, as assessed by flow cytometry in CD45+ hematopoietic or EpCam+ epithelial cells.
(D) Lung viral loads in mice pretreated with 2.5 × 109 IGU of Ad5::hACE2, control empty Ad5, or PBS and then inoculated with 1 × 105 TCID50 of SARS-CoV-2 via i.n. administration 4 days later. In one group, we inoculated the Ad5::hACE2-pretreated mice with an equivalent amount of heat-killed virus to measure the input viral RNA in the absence of viral replication. Viral load was quantitated by qRT-PCR in the lung homogenates at 2, 4, or 7 dpi. The red line indicates the detection limit.
(E) Percentages of CD45+ cells in the lungs as determined 4 days after pretreatment with various doses of Ad5::hACE2.
(F) Lung viral loads in mice pretreated with various doses of Ad5::hACE2 and then inoculated with 1 × 105 TCID50 of SARS-CoV-2 via i.n. administration 4 days later. Viral loads were determined at 3 dpi. See also Figure S2.
To evaluate the permissibility of such hACE2-transduced mice to SARS-CoV-2 infection, 4 days after i.n. pretreatment with either Ad5::hACE2 or an empty Ad5 control vector, we inoculated C57BL/6 mice with 1 × 105 TCID50 (median tissue-culture infectious dose) of a SARS-CoV-2 clinical isolate (BetaCoV/France/IDF0372/2020) via i.n. administration (Lescure et al., 2020). The lung viral loads, determined 2 days post-inoculation (dpi) by RT-PCR and quantitative real-time PCR (qRT-PCR), were as high as (4.4 ± 1.8) × 109 copies of SARS-CoV-2 RNA in Ad5::hACE2-pretreated mice but only (6.2 ± 0.5) × 105 copies in empty-Ad5-pretreated mice or (4.0 ± 2.9) × 105 copies in non-pretreated mice (Figure 3D). In the latter two control groups, these copy numbers corresponded to the input viral RNA, as determined in Ad5::hACE2-pretreated mice inoculated with equivalent amounts of heat-killed viral particles (Figure 3D). The increased viral load from 106 copies to >109 copies per lung shows the occurrence of active replication of SARS-CoV-2 in vivo. At 4 dpi, the lung viral loads were maintained in Ad5::hACE2-pretreated mice (2.8 ± 1.3 × 109 copies), whereas a drop to (1.7 ± 2.3) × 104 or (3.9 ± 5.1) × 103 copies was observed in empty-Ad5-pretreated or non-pretreated mice, respectively. In Ad5::hACE2-pretreated mice, the viral loads were still detectable at 7 dpi ([1.33 ± 0.9] × 106 copies).
Ad5::hACE-2 i.n. instillation induced CD45+ cell recruitment to the lungs (Figure 3E). However, no pro-inflammatory effect was seen with a lower dose of 4 × 108 IGU/mouse (Figure 3E), which still conferred full permissibility to SARS-CoV-2 replication (Figure 3F), and this dose was therefore chosen for the subsequent experiments described below. In this model of permissive mice, SARS-CoV-2 infection resulted in widespread infiltration of the lung interstitium by mononuclear inflammatory cells, i.e., lymphocytes and macrophages, at 3 dpi (Figure S2). However, in the Ad5::hACE2-pretreated and SARS-CoV-2-inoculated mice, we did not observe any weight loss or clinical signs of disease over the period of 2–7 days after virus inoculation.
These results show that pretreatment of mice with appropriate doses of Ad5::hACE2 can render mice permissive to SARS-CoV-2 replication without inducing Ad5-mediated inflammation, thus providing a valuable model for vaccine or drug studies.
Intranasal boost with LV::SFL protects strongly against SARS-CoV-2 in mice
To investigate the prophylactic potential of LV::SFL against SARS-CoV-2, we gave C57BL/6 mice (n = 4 or 5 per group) an i.p. injection of a single dose of 1 × 107 TU of LV::SFL or a negative control LV (sham). Seven weeks after immunization, mice were pretreated with Ad5::hACE2 and, 4 days later, inoculated with 1 × 105 TCID50 of SARS-CoV-2 via i.n. administration (Figure S3A). At 3 dpi, the lung viral loads in LV::SFL-vaccinated mice were reduced by ∼6.5 fold, i.e., to a mean ± SD of (5.5 ± 3.8) × 108 SARS-CoV-2 RNA copies versus (3.1 ± 1.9) × 109 or (4.3 ± 3.0) × 109 copies in the un- or sham-vaccinated mice, respectively (Figure S3B). Therefore, a single i.p. LV::SFL injection provided only partial protection in the lung despite intense serum NAb activity.
To further improve the prophylactic effect, we evaluated the prime-boost and prime-target approaches. C57BL/6 mice (n = 4 or 5 per group) were primed via the i.p. route with 1 × 107 TU of LV::SFL or a control LV at week 0 and then boosted at week 3 with (1) 1 × 107 TU of the same LV via the i.p. route (LV::SFL i.p.-i.p., prime-boost) or (2) 3 × 107 TU via the i.n. route (LV::SFL i.p.-i.n., prime-target) to attract the mediators of systemic immunity to the lung mucosa (Figure 4 A). Systemic i.p. boosting with LV::SFL resulted in a significant increase in the anti-SCoV-2 IgG titers, which was more obvious when the binding was evaluated against the foldon-trimerized full-length S (Figure 4B, left) than against the S1 or RBD fragment (Figure S4A). In contrast, mucosal targeting with i.n. LV::SFL did not lead to a statistically significant improvement of anti-SCoV-2 IgG titers at the systemic level (Figure 4B, left; Figure S4A). In terms of serum neutralization potential, even though a trend to increase was observed after the i.p. or i.n. boost, the differences did not reach statistical significance (Figure 4B, right).
Figure 4.

Intranasal boost with LV::SFL strongly protects against SARS-CoV-2 in mice
(A) Timeline of the LV-based prime-boost strategy, followed by Ad5::hACE2 pretreatment and SARS-CoV-2 challenge.
(B) Titers of anti-SCoV-2 IgG as quantitated by ELISA in the sera of C57BL/6 mice primed at week 0 via the i.p. route and boosted at week 3 via the i.p. or i.n. route (left). Titers were determined as a mean endpoint dilution before the boost (week 3) and challenge (week 4). ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; two-way ANOVA followed by Sidak’s multiple-comparison test. NS, not significant. The neutralization capacity of these sera is indicated as EC50 (right). See also Figure S4A.
(C). Lung viral loads at 3 dpi in mice primed (i.p. route) and boosted (i.p. or i.n. route) with LV::SFL. Sham-vaccinated mice received an empty LV. The red line indicates the detection limit. Statistical significance of the differences in the viral loads was evaluated by a two-tailed unpaired t test; ∗p < 0.0139, ∗∗∗p < 0.0088.
(D) Titers of anti-SCoV-2 IgG and IgA Abs determined in the clarified lung homogenates by ELISA with foldon-trimerized SCoV-2 for coating. See also Figure S4B.
(E) Neutralizing activity of the clarified lung homogenates as determined for 1/5 dilution. Statistical significance of the difference was evaluated by a Mann-Whitney U test (∗p < 0.0159). See also Figure S3.
All mice were then pretreated with Ad5::hACE2 and challenged with 0.3 × 105 TCID50 of SARS-CoV-2 via i.n. administration 4 weeks after priming. At 3 dpi, the lung viral loads were significantly lower in LV::SFL-i.p.-i.p.-immunized mice (mean ± SD of [2.3 ± 3.2] × 108 copies of SARS-CoV-2 RNA) than in sham-vaccinated mice ([13.7 ± 7.5] × 108 copies) (Figure 4C) This reduced viral load was similar to that obtained with a single LV::SFL administration (Figure S3B). Most importantly, after i.n. LV::SFL target immunization, a >3 log10 decrease in viral load was observed, and a qRT-PCR assay determined that two of five mice harbored undetectable lung viral loads. We also determined the viral loads in the lungs of these mice by performing a plaque-forming unit (PFU) assay (Figure S4C), which confirmed the same trend in reduction. However, the PFU approach underestimates the amounts of infectious viral particles in the lungs of immunized mice because of the high activity of anti-S NAbs in the lung homogenates of these animals. Anti-SCoV-2 IgG was in fact detected in the clarified lung homogenates of the partially (LV::SFL i.p.-i.p.) or fully (LV::SFL i.p.-i.n.) protected mice. In contrast, anti-SCoV-2 IgA was detectable only in the fully protected LV::SFL i.p.-i.n. mice (Figure 4D). Higher neutralizing activity was detected in the clarified lung homogenates of LV::SFL i.p.-i.n. mice than in their LV::SFL i.p.-i.p. counterparts (Figure 4E). Therefore, increasing the titers of NAbs of the IgG isotype at the systemic level did not improve the protection against SARS-CoV-2. However, a mucosal i.n. target immunization (with the potential to attract immune effectors to the entry point of the virus to the host organism and to induce local IgA Abs) correlated with the inhibition of SARS-CoV-2 replication.
Because of the importance of innate immune hyperactivity in the pathophysiology of COVID-19 (Vabret et al., 2020), we investigated the lung innate immune cell subsets in the non-infected controls and sham-vaccinated or LV::SFL-vaccinated mice inoculated with SARS-CoV-2 (Figure 5 A). At 3 dpi, we detected no difference between the proportions of basophils or natural killer (NK) cells and total lung CD45+ cells among the experimental groups (Figure 5B). In sharp contrast, we detected significantly higher proportions of alveolar macrophages, dendritic cells, neutrophils, and mast cells and a trend toward increased Ly6C+ monocytes and macrophages versus total lung CD45+ cells in sham-vaccinated mice. These observations demonstrate that in this mouse model, lung SARS-CoV-2 loads correlate with the expansion of several inflammation-related innate immune cell subsets, whereas vaccine-mediated protection dampens or prevents the inflammatory reaction.
Figure 5.

LV::SFL vaccination reduces SARS-Co-2-mediated lung inflammation in mice
(A) Cytometric strategy to identify and quantify distinct lung innate immune cell subsets. Lung hematopoietic CD45+ cells were analyzed with the use of Abs specific to surface markers or a combination of surface markers, allowing characterization of innate immune cell populations, via three distinct paths and by sequential gating. Cell populations are highlighted in gray.
(B) Percentages of each cell subset versus total lung CD45+ cells at 3 dpi in mice sham vaccinated or vaccinated with LV::SFL after various prime-boost regimens versus non-infected (NI) controls. All mice were pretreated with Ad5::hACE-2 4 days prior to SARS-CoV-2 inoculation.
(C) Relative log2 fold change in mRNA expression of cytokines and chemokines in mice sham vaccinated or vaccinated with LV::SFL after various prime-boost regimens at 3 dpi. Data were normalized to those of PBS-treated, unchallenged controls. Statistical significance was evaluated by a two-tailed unpaired t test; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
This was corroborated by the reduced inflammatory cytokine and chemokine contents in the RNA extracted from total lung homogenates of mice immunized with LV::SFL i.p.-i.p. (Figure 5C, top) or LV::SFL i.p.-i.n. (Figure 5C, bottom) and protected against SARS-CoV-2. Significant reductions of TNF-α, TGF-β, IL-1β, IL-12p40, IL-17A, IL-33, CCL2, CCL3, CCL5, CXCL9, and CXCL10 contents were observed in the lungs of LV::SFL i.p.-i.p.- versus sham-i.p.-i.p.-vaccinated and challenged mice. Significant reductions were notably detected in the IL-1β, IL-6, CCL2, CCL3, and CXCL5 contents in the lungs of LV::SFL i.p.-i.n.- versus sham-i.p.-i.n.-vaccinated and challenged mice. Therefore, the conferred protection also reduced pulmonary inflammation mediated by SARS-CoV-2 infection, as demonstrated by cytometric and RT-PCR approaches.
Intranasal vaccination with LV::SFL strongly protects against SARS-CoV-2 in golden hamsters
Outbred Mesocricetus auratus, so-called golden hamsters, provide a suitable pre-clinical model for studying COVID-19 pathology because the ACE2 ortholog of this species interacts productively with SCoV-2 by supporting host cell invasion and viral replication (Sia et al., 2020). We thus investigated in this model the protective effect against SARS-CoV-2 infection of vaccination by LV::SFL and by an integrase-deficient, non-integrative version of this vector (NILV) with the prospect of application in future human clinical trials.
For assessment of the prophylactic effect of vaccination after the prime-boost or prime-target regimen, M. auratus hamsters (n = 6 per group) were (1) primed with 1 × 106 TU of LV::SFL via i.p. injection and boosted at week 5 with 4 × 107 TU of LV::SFL via i.n. injection (int LV::SFL i.p.-i.n. low), (2) primed with 1 × 107 TU of LV::SFL via i.p. injection and boosted at week 5 with 4 × 107 TU of LV::SFL via i.n. injection (int LV::SFL i.p.-i.n. high), or (3) primed with 1 × 108 TU of NILV::SFL via intramuscular (i.m.) injection and boosted at week 5 with 1 × 108 TU of NILV::SFL via i.n. injection (NILV::SFL i.m.-i.n.) (Figure 6 A). Sham-vaccinated controls received the same amounts of an empty LV via i.p. and then i.n. routes. Strong and comparable anti-SCoV-2 IgG Abs were detected by ELISA in the sera of hamsters from the three vaccinated groups before and after the i.n. boost (Figure 6B). After boosting and targeting, serology detected neutralization activity in all groups, but the highest EC50 average was observed in the int LV::SFL i.p.-i.n. high individuals. Such levels were comparable to those detected in COVID-19 cases in humans (Figure 6C). All hamsters were challenged with 0.3 × 105 TCID50 of SARS-CoV-2 via i.n. injection at week 5. Up to 16% weight loss was progressively reached at 4 dpi in sham-vaccinated individuals, whereas there was a non-significant loss in the LV::SFL-vaccinated groups (Figure 6D). As assessed by qRT-PCR, at 2 dpi, the lung viral loads of the int LV::SFL i.p.-i.n. low, int LV::SFL i.p.-i.n. high, and NILV::SFL i.m.-i.n. groups were ∼1.2–2 log10 lower than those of sham-vaccinated hamsters (Figure 6E). At 4 dpi, the magnitude of viral-load reductions in the vaccinated groups, compared with the sham group, were statistically significant, even if there was one outlier hamster per group (Figure 6E). Using a PFU assay at 2 and 4 dpi, we detected (6.56 ± 1.88) and (2.94 ± 3.16) × 106 PFU per lung of sham-vaccinated controls. At 2 dpi, the decrease in viral loads was largely statistically significant in all of the LV::SFL-vaccinated hamsters, and at 4 dpi, with the exception of one NILV::SFL i.m.-i.n. hamster, no vaccinated individuals possessed detectable infectious viral particles (Figure S5A).
Figure 6.

Intranasal vaccination with LV::SFL strongly protects against SARS-CoV-2 in golden hamsters
(A) Timeline of prime-boost or prime-target immunization regimen and challenge in hamsters. Sham-vaccinated mice received an empty LV.
(B) Dynamic of anti-SCoV-2 Ab response following immunization. Sera were collected 3, 5 (before the boost), and 6 (after the boost) weeks after priming. Anti-SCoV-2 IgG responses were evaluated by ELISA.
(C) EC50 serum neutralizing titers after the boost or target regimen versus sera from a cohort of asymptomatic (AS), pauci-symptomatic (PS), or symptomatic (S) COVID-19 cases or of hospitalized (H) humans.
(D) Weight follow-up in hamsters either sham- or LV::SFL-vaccinated with diverse regimens. For further clarity, only the individuals reaching 4 dpi are shown. Those sacrificed at 2 dpi had the same mean weight as their counterparts between 0 and 2 dpi.
(E) Lung viral loads at 2 or 4 dpi in LV::SFL-vaccinated hamsters. Statistical significance was evaluated by a two-tailed unpaired t test; ∗p < 0.0402, ∗∗∗∗p < 0.0001. See also Figure S4C.
(F) Relative log2 fold changes in cytokine and chemokine expression in LV::SFL-vaccinated and protected hamsters versus sham-vaccinated individuals, as determined at 4 dpi by qRT-PCR in the total lung homogenates and normalized to untreated controls. Statistical significance was evaluated by one-way ANOVA; ∗p < 0.05, ∗∗p < 0.01. See also Figures S5 and S6.
As evaluated by qRT-PCR in the total lung homogenates of the protected int LV::SFL i.p.-i.n. low, int LV::SFL i.p.-i.n. high, and NILV::SFL i.m.-i.n. groups, the expression of inflammatory IFN-γ and IL-6 cytokines, anti-inflammatory IL-10 cytokine, and CCL2, CCL3, and CXCL10 chemokines was substantially lower at 4 dpi than in their unprotected sham-vaccinated counterparts (Figure 6F). The other inflammatory mediators tested were not significantly modified (Figure S5B).
In sham-vaccinated and challenged hamsters, overall marked multifocal degenerative changes of the bronchiolar epithelium, moderate effacement of the epithelium (associated with mild to moderate mixed inflammation in the airway lumen), and minimal multifocal interstitial mononuclear cell inflammation were detected at both time points (Figure S6A). At 4 dpi, effacement of the respiratory epithelium and mixed inflammation in the alveoli were more severe; moderate fibrin deposits were also noted. In vaccinated hamsters, regardless of the vaccine dose, lung lesions were clearly of lower incidence and severity (Figure S6B). In these animals, only minimal degenerative lesions and minimal effacement of the bronchiolar epithelium and occasionally mixed inflammation in the airway lumen of alveoli and mononuclear infiltrates in the interstitium were observed. These microscopic findings were similar after vaccination with the int LV or NILV.
In an additional experiment (Figure 7 A), we showed that (1) a single i.m. injection of NILV::SFL induced high titers of serum anti-S Abs (Figure 7B) and initiated significant—but partial—levels of protection in the lungs (Figure 7C) and (2) an i.n. boost with NILV::SFL, which did not improve the serum NAb activity (Figure 7D), induced significantly improved protection against SARS-CoV-2, as determined by the lung viral loads detected via qRT-PCR (Figure 7C) and PFU (Figure S7A), as well as weight loss (Figure S7B), at 4 dpi. At 4 dpi, in sham-vaccinated and challenged hamsters, substantial pulmonary lesions, severe parenchyma inflammation, consolidation of pulmonary parenchyma, marked alteration of the bronchiolar epithelium, and moderate effacement of the bronchiolar epithelium were detected (Figure 7E; Figure S7C). In their NILV::SFL-vaccinated counterparts, boosted or not, pulmonary lesions were clearly less severe (Figures 7E–7G).
Figure 7.

Protective efficacy of NILV::SFL in a systemic prime and i.n. boost regimen in hamsters
(A) Timeline of the NILV::SFL prime-boost or prime-target immunization regimen and challenge.
(B) Profile of serum anti-SCoV-2 IgG response following a single (i.m.) injection or a prime (i.m.)-boost (i.n.) immunization with NILV::SFL.
(C) Lung viral loads at 4 dpi with SARS-CoV-2 in controls or NILV::SFL-vaccinated hamsters. Statistical significance was evaluated by a two-tailed unpaired t test; ∗∗p < 0.01.
(D) EC50 serum neutralizing titers after the boost-target regimen.
(E) Lung histological H&E analysis shown in a heatmap recapitulating the histological scores for each parameter defined in Figure S7C and determined for individuals of various groups at 4 dpi.
(F and G) Representative whole-lung section from NILV::SFL i.m.-NILV::SFL i.n. (F) or sham i.m.-sham i.n. (G) hamsters. See also Figure S7.
Altogether, on the basis of a complete set of virological, immunological, and histopathological data, the LV::SFL vector elicited anti-SCoV-2 NAbs and T cell responses and provided robust protection against SARS-CoV-2 infection in two pertinent animal models, particularly when mucosal i.n. administration was included in the vaccinal scheme.
Discussion
Prophylactic strategies are necessary to control SARS-CoV-2 infection, which, 9 months into the pandemic, still continues to spread exponentially without signs of slowing down. It has now been demonstrated that primary infection with SARS-CoV-2 in rhesus macaques leads to protective immunity against re-exposure (Chandrashekar et al., 2020). Numerous vaccine candidates, based on naked DNA (Yu et al., 2020) or mRNA, recombinant protein, replicating or non-replicating viral vectors (including adenoviral Ad5 vector; Zhu et al., 2020), or alum-adjuvanted inactivated virus (Gao et al., 2020), are under active development for COVID-19 prevention. Our immunological rationale for selecting a LV to deliver the gene encoding SCoV-2 antigen was based on its potential to induce in situ expression of heterologous genes and on the ability of this immunization platform to elicit sustained humoral and cell-mediated responses. Unique to LVs is the ability to transduce proliferating and non-dividing cells such as dendritic cells (Esslinger et al., 2002; Firat et al., 2002; He et al., 2005) and thereby serve as a powerful vaccination strategy (Beignon et al., 2009; Buffa et al., 2006; Coutant et al., 2012; Gallinaro et al., 2018; Iglesias et al., 2006) to provoke strong and long-lasting adaptive responses (Cousin et al., 2019; M.-W.K. et al., unpublished data). Notably, in net contrast to many other viral vectors, LVs do not suffer from pre-existing immunity in populations because of their pseudo-typing with VSV-G, to which humans are barely exposed. We recently demonstrated that a single injection of an LV expressing the Zika envelope provides a rapid and durable sterilizing protection against Zika infection (Ku et al., 2020). Our recent comprehensive comparison of LVs to the gold-standard Ad5 immunization vector also documented the superiority of LVs in inducing multifunctional and central memory T cell responses in the mouse model and their strong immunogenicity in outbred rats (M.-W.K. et al., unpublished data), underlining the suitability of LVs for vaccinal applications.
Because laboratory mice are not naturally susceptible to SARS-CoV-2 infection, we set up an in vivo transduction murine model in which hACE2 expression is induced in the respiratory tracts by an i.n. Ad5::hACE2 pretreatment prior to SARS-CoV-2 inoculation. Our system, very close to a recently published protocol (Sun et al., 2020), confirmed that mice become largely permissive to SARS-CoV-2 replication in the lungs and thus represent a model for assessment of vaccine or drug efficacy against this virus. Even though the Ad5::hACE2 model might not fully mimic the physiological ACE2 expression profile and thus might not reflect all the aspects of the pathophysiology of SARS-CoV-2 infection, it provides a pertinent model for evaluating in vivo the effects of anti-viral drugs, vaccine candidates, various mutations, or genetic backgrounds on the SARS-CoV-2 replication. Using a low dose of Ad5::hACE2, we detected no CD45+ cell recruitments 4 days after instillation, indicative of an absence of Ad5-related inflammation before the inoculation of SARS-CoV-2.
In the transduced mouse model that allows high rates of SARS-CoV-2 replication, vaccination by a single i.p. administration of 1 × 107 TU of LV::SFL 6 weeks before virus inoculation was sufficient to inhibit the viral replication by ∼6-fold. Compared with a single administration, further boosting via the systemic route did not afford an improved protection rate. However, systemic priming followed by a mucosal boost efficiently inhibited viral replication and prevented lung inflammation. This protection was correlated with high titers of anti-SCoV-2 IgG and IgA and strong neutralization activity in sera. Additional experiments with appropriate transgenic mice or adoptive immune cell transfer will be necessary for identifying the immunological pathways that contribute to disease severity or protection against SARS-CoV-2. NAbs and cell-mediated immunity, very efficaciously induced with the LV-based vaccine candidate, could synergize to block infection and viral replication.
As this manuscript was being prepared, the laboratory of M.S. Diamond reported that i.n. administration of a Chimpanzee adenoviral-36 vector encoding a stabilized form of SCoV-2 protected upper and lower respiratory tracts against SARS-CoV-2 both in BALB/c mice expressing hACE2 in the lung after i.n. delivery of Ad5::hACE2 and in hACE2-expressing transgenic C57BL/6 mice (Hassan et al., 2020). The authors mentioned that immunization and challenge studies in hamsters and non-human primates (NHPs) are planned and that investigations in humans will also be required for evaluating cross-immunity between this Chimpanzee adenoviral-36 vector and adenoviruses circulating in humans. The latter can in fact be a source of pre-existing immunity, which can interfere with the efficacy of Chimpanzee adenoviral-36 vector. No such limitation exists in the case of VSV-G-pseudo-typed LV vaccinal vector (Hu et al., 2011), as mentioned above.
Ab-dependent enhancement (ADE) of coronavirus entry into host cells has been evoked as a potential obstacle in vaccination against coronaviruses. With DNA (Yu et al., 2020) or inactivated SARS-CoV-2 virus (Gao et al., 2020) vaccination in macaques, no immunopathological exacerbation has been observed. Long-term observation even after a decrease in Ab titer could be necessary for excluding such a hypothesis. In the case of MERS-CoV, it has been reported that one particular RBD-specific neutralizing monoclonal Ab (Mersmab1) could mediate in vitro ADE of MERS-CoV into host cells by mimicking the viral receptor human DPP4 and inducing conformational rearrangements of SMERS (Wan et al., 2020). We believe that it is difficult to compare the polyclonal Ab response and its paratope repertoire complexity with the singular properties of a monoclonal Ab that cannot be representative of the polyclonal response induced by a vaccine. In addition, very contradictory results from the same team reported that a single-dose treatment with a humanized version of Mersmab1 afforded complete protection of a human transgenic mouse model from lethal MERS challenge (Qiu et al., 2016). Therefore, even with an Ab that might facilitate host-cell invasion in vitro in some conditions, not only is there no exacerbation of the infection in vivo, but there is also a notable protection.
It is noteworthy that all vaccine candidates currently under phase 2 and 3 clinical development are planned to be administered via the i.m. route and will thus induce systemic—but not mucosal—immunity. Our results evidenced a strong correlation between the presence of mucosal anti-SCoV-2 IgA and local NAb activity in the respiratory tracts and very robust pulmonary protection. Therefore, vaccine administration via the i.n. route, which is the main entry door of SARS-CoV-2, has to be taken into account when COVID-19 vaccination protocols are established, especially because this route is non-invasive and particularly suitable for mass vaccination of children and elderly people.
On the basis of largely diverse manifestations of COVID-19 in humans, distinct animal models mimicking various settings of human infection are needed. After SARS-CoV-2 inoculation, rhesus macaques develop transient and mild disease (Muñoz-Fontela et al., 2020). It has been shown that an i.m.-i.m. prime-boost strategy with a DNA-based anti-SCoV-2 vaccine candidate in these animals results in lower infectious virus titers in bronchoalveolar lavages and nasal swabs than do sham controls (Yu et al., 2020). In parallel, golden hamsters develop severe lung pathology and rapid weight loss, which sets a high bar for demonstrating prophylactic efficacy (Muñoz-Fontela et al., 2020). In this more stringent model and also in Ad5::hACE2-pretreated mice, although LV::SFL systemic immunization induces strong systemic adaptive immunity and significant protection, only an i.n. boost allows sterilizing protection, which indicates a very high prophylactic efficacy of LV::SFL.
The fact that the current vaccine candidates do not take advantage of i.n. immunization is certainly related to the pro-inflammatory properties of most of the vaccination vectors or the requirement of the encapsulation or adjuvantation of nucleic acids or inactivated vaccines, which raises safety concerns. Unlike most of the other vaccination viral vectors, LVs are non-replicative, non-inflammatory, and effective under their non-integrative variant and do not suffer from pre-existing immunity in human populations. Therefore, LVs are particularly safe and favorable for use in mucosal immunization through the i.n. route, which is essential in immune protection against SARS-CoV-2. Prophylactic vaccination is the most cost-effective and efficient strategy against infectious diseases in general and against emerging coronaviruses in particular. Our results firmly establish that the SCoV-2-encoding LV used in a prime-target protocol is a prominent vaccine strategy against COVID-19 and should be considered for future human clinical trials.
Limitations of study
Multiple animal models that mimic various manifestations of human COVID-19 are required. One can hypothesize that the expression of hACE2 in the transduced lungs of Ad5::hACE2-pretreated mice could be inconsistent. However, the routine use of this mouse model in our hands shows that (1) all pretreated animals express hACE2 mRNA (as detected in total lung extracts) and (2) the range of lung SARS-CoV-2 loads is highly reproducible and barely variable in unvaccinated mice. Even though Ad5::hACE2-pretreated and SARS-CoV-2-inoculated mice display no weight loss, lung histopathology, or clinical signs of disease, this model paves the way to quantitating SARS-CoV-2 replication in vivo and to demonstrating its immune control by LV vaccination. The highly susceptible hamster model sets a high bar for demonstrating the efficacy of drugs or vaccine candidates even though it does not mimic the mild or even asymptomatic variants of human COVID-19. Before clinical trials, further development of LV::SFL will include vaccination and challenge experiments in NHPs to confirm protection efficacy against the transient and mild manifestations of the COVID-19 variant developed in this animal model.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-human ACE-2 | R&D systems | Cat# AF933; RRID: AB_35572 |
| HRP-conjugated anti-goat | Abcam | Cat# ab6741 |
| HRP-conjugated anti-β-actin | Abcam | Cat# ab197277 |
| Streptavidin-HRP | Mabtech | Cat# 3310-8-1000 |
| Rat anti-mouse IgA-HRP | Southern Biotech | Cat# 1165-05; RRID: AB_2794657 |
| Rabbit anti-hamster IgG-HRP | Jackson ImmunoResearch Labs | Cat# M37470 |
| Goat anti-mouse IgG-HRP | Jackson ImmunoResearch Labs | Cat# 115-035-075; RRID: AB_2338508 |
| Rat anti-IFN-γ, biotin conjugated, clone R4-6A2 | BD Biosciences | Cat# 551506; RRID: AB_394224 |
| Rat anti-IFN-γ, unconjugated, clone AN-18 | BD Biosciences | Cat# 551309; RRID: AB_394145 |
| Near IR live/dead | Thermo Fisher Scientific | Cat# L34976 |
| Rat anti-CD16/CD32, clone 2.4G2 | BD Biosciences | Cat# 553142; RRID: AB_394657 |
| APC rat anti-IFN-γ, clone XMG1.2 | BD Biosciences | Cat# 554413; RRID: AB 398551 |
| APC-H7 rat anti-mouse CD8a, clone 53-6.7 | BD Biosciences | Cat# 560182; RRID: AB_1645237 |
| PE-Cy7 rat anti-CD4, clone RM4-5 | BD Biosciences | Cat# 552775; RRID: AB_394461 |
| BV605 rat anti-CD45, clone 30-F11 | BD Biosciences | Cat# 563053; RRID: AB_2737976 |
| PE rat anti-CD11b, clone M1/70 eBioscience | Thermo Fisher Scientific | Cat# 12-0112-82; RRID: AB2734869 |
| APC rat anti-CD11b, clone M1/70 | BD Biosciences | Cat# 553312; RRID: AB_398535 |
| PE-Cy7 hamster anti-CD11c, clone N418 | Thermo Fisher Scientific | Cat# 25-0114-82; RRID: AB_469590 |
| eF450 hamster anti-CD11c, clone N418 | Thermo Fisher Scientific | Cat# 48-0114-82; RRID: AB_1548654 |
| BV711 hamster anti-CD103, clone 2E7 | BioLegend | Cat# 121435; RRID: AB_2686970 |
| AF700 rat anti-MHC-II, clone M5/114.15.2 | BioLegend | Cat# 107621; RRID: AB_493726 |
| PerCP-Cy5.5 rat anti-Ly6C, clone HK1.4 | Thermo Fisher Scientific | Cat# 45-5932-80; RRID: AB_2723342 |
| APC Hu anti-Ly-6G, clone REA526 | Miltenyi Biotec | Cat# 130-107-913; RRID: AB_2652821 |
| PE rat anti-Ly6G, clone 1A8 | BioLegend | Cat# 127608; RRID: AB_1186099 |
| FITC rat anti-CD24, clone M1/69 | BD Biosciences | Cat# 561777; RRID: AB_10896486 |
| BV421 rat anti-Siglec-F, clone E50-2440 | BD Biosciences | Cat# 562681; RRID: AB_2722581 |
| BV421 Mo anti-CD64, clone X54-5/7.1 | BioLegend | Cat# 139309; RRID: AB_2562694 |
| PE-Cy7 rat anti-CD117, clone 2B8 | BD Biosciences | Cat# 561681; RRID: AB_10893022 |
| APC hamster anti-FcεR1, clone MAR-1 | BioLegend | Cat# 134316; RRID: AB_10640121 |
| AF700 rat anti-NKp46, clone 29A1.4 | BD Biosciences | Cat# 561169; RRID: AB_10561840 |
| FITC rat anti-CCR3, clone J073E5 | BioLegend | Cat# 144509; RRID: AB_2561608 |
| Bacterial and virus strains | ||
| LV:S1 | this paper | N/A |
| LV::S1-S2 | this paper | N/A |
| LV::SFL | this paper | N/A |
| LV::GFP or empty | this paper | N/A |
| Non-replicative LV::SFL | this paper | N/A |
| LV::GFP | Zennou et al. (2000) | N/A |
| LV::empty | this paper | N/A |
| Ad5::hACE2 | this paper | N/A |
| Non-replicative SCoV-2 pseudo-typed LV particles | Anna et al. (2020) | N/A |
| SARS-CoV-2 (BetaCoV/France/IDF0372/2020) | clinical isolate; Lescure et al. (2020) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Stabilized foldon-trimerized SARS-CoV-2 Spike | Grzelak et al. (2020) | N/A |
| S1 monomer | this paper | N/A |
| RBD subdomain | this paper | N/A |
| SCoV-2-derived 15-mer peptide library | this paper; (Hoffmann et al., 2020); http://www.mimotopes.com |
N/A |
| Critical commercial assays | ||
| ViraPower Adenoviral Promoterless Gateway Expression Kit | Thermo Fisher Scientific | ViraPower Adenoviral Promoterless Gateway Expression Kit |
| Adeno-X rapid Maxi purification kit | Takara Bio | Cat# 631533 |
| QIAamp Viral RNA Mini Kit | QIAGEN | Cat# 52906 |
| Experimental models: cell lines | ||
| HEK293T cells | ATCC | HEK293T cells |
| HEK293T cells transduced to stably express human ACE2 | Grzelak et al. (2020) | N/A |
| Experimental models: organisms/strains | ||
| Mouse: C57BL/6J | Janvier, Le Genest Saint Isle, France | Cat#000664; RRID: IMSR JAX:000664 |
| Hamster: Mesocricetus auratus golden | Janvier, Le Genest Saint Isle, France | Cat# HAMST-M79 |
| Oligonucleotides | ||
| pFLAP plasmid: forward 5′-TGG AGG AGG AGA TAT GAG GG-3′ | this paper | N/A |
| pFLAP plasmid: reverse 5′-CTG CTG CAC TAT ACC AGA CA-3′ | this paper | N/A |
| gadph gene: forward 5′-TCT CCT CTG ACT TCA ACA GC-3′ | this paper | N/A |
| gadph gene: reverse 5′-CCC TGC ACT TTT TAA GAG CC-3′ | this paper | N/A |
| Primers and probes for SARS-CoV-2 load determination | this paper | Table S1 |
| Ad5: forward 5′- TTG TGG TTC TTG CAG ATA TGG C-3′ | Gallaher and Berk (2013) | N/A |
| Ad5: reverse 5′- TCG GAA TCC CGG CAC C-3′ | Gallaher and Berk (2013) | N/A |
| Ad5: probe 5′-[FAM]-CTC ACC TGC CGC CTC CGT TTC C-[BHQ1]-3′ | Gallaher and Berk (2013) | N/A |
| Mouse and hamster cytokine and chemokine primers | this paper | Table S2 |
| Recombinant DNA | ||
| pFlap-ieCMV-WPREm PC | Zennou et al. (2000) | N/A |
| pMK-RQ_S-2019-nCoV | this paper | N/A |
| Software and algorithms | ||
| GraphPad Prism | GraphPad | v9.0.0 |
| FlowJo | FlowJo LLC | v10 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Laleh Majlessi (laleh.majlessi@pasteur.fr).
Materials availability
All plasmids and LV generated in this study will be available under MTA for research use, given a pending patent directed to a vaccine candidate against SARS-CoV2.
Data and code availability
The published article includes all datasets generated and analyzed during this study.
Experimental model and subject details
Mice
Female C57BL/6JRj mice were purchased from Janvier (Le Genest Saint Isle, France) and used between the age of 6 and 10 weeks. They were housed under specific pathogen-free conditions in individually ventilated cages during the immunization period and were then transferred into isolator for SARS-CoV-2 inoculation. Mice were housed in individually filtered cages in isolator until the end of the experiment at the Institut Pasteur animal facilities. Mice were vaccinated with the indicated TU of LV and sera were collected at various time points post-immunization to monitor binding and neutralization activities. Previous to i.m. or i.n. instillations, mice were anesthetized by i.p. injection of a mixture of Ketamine (Imalgene, 50 mg/kg) and Xylazine (Rompun, 50 mg/kg).
Hamsters
Male Mesocricetus auratus golden hamsters (Janvier, Le Genest Saint Isle, France) were purchased mature, i.e., 80-90 gr weight. At the beginning of the immunization regimen they weighed 100 to 120 gr. Hamsters were housed under specific pathogen-free conditions in individually ventilated cages during the immunization period and were transferred into isolators for SARS-CoV-2 inoculation. Hamsters were housed in individually filtered cages inside isolators until the end of the experiment at the Institut Pasteur animal facilities. Hamsters were vaccinated with the indicated TU of LV and their sera were collected at the time points indicated after immunization to monitor Ab activities. Prior to i.m. or i.n. injections, hamsters were anesthetized by i.p. injection of Ketamine (Imalgene, 50 mg/kg) and Xylazine (Rompun, 50 mg/kg).
Ethical approval of animal studies
Experimentation on animals was performed in accordance with the European and French guidelines (Directive 86/609/CEE and Decree 87-848 of 19 October 1987) subsequent to approval by the Institut Pasteur Safety, Animal Care and Use Committee, protocol agreement delivered by local ethical committee (CETEA #DAP20007) and Ministry of High Education and Research APAFIS#24627-2020031117362508 v1.
Patient samples
Serum samples were taken from COVID-19 patients at a period where they were asymptomatic, pauci-symptomatic, symptomatic or hospitalized, as recently described elsewhere (Grzelak et al., 2020). Each participant provided written consent to participate in the study, which was approved by the regional investigational review board (IRB; Comité de Protection des Personnes Ile-de-France VII, Paris, France), according to European guidelines and the Declaration of Helsinki. The study was registered as ClinicalTrials.gov (NCT04325646).
Cell culture
HEK293T and Vero cells were cultured in DMEM media (GIBCO Thermo Fisher Scientific) complemented with 10% heat-inactivated serum (Corning), 100 U/mL penicillin and 100 μg/mL streptomycin (GIBCO Thermo Fisher Scientific) and were incubated at 37°C, 5% CO2.
Method details
Construction of transfer pFLAP plasmids coding for SFL, S1-S2, or S1 proteins
A codon-optimized full-length S (1–1273) sequence was amplified from pMK-RQ_S-2019-nCoV and inserted between BamHI and XhoI sites of pFlap-ieCMV-WPREm. Sequences encoding for S1-S2 (1–1211) or S1 (1–681) were amplified by PCR from the pFlap-ieCMV- SFL-WPREm plasmid and sub-cloned into pFlap-ieCMV-WPREm between the BamHI and XhoI restriction sites (Figure S1). Each of the PCR products were inserted between the native human ieCMV promoter and a mutated Woodchuck Posttranscriptional Regulatory Element (mWPRE) sequence, in which the atg starting codon was mutated to avoid transcription of the downstream truncated “X” protein of Woodchuck Hepatitis Virus, in order to improve the vector safety. Plasmids were amplified in Escherichia coli DH5a in Lysogeny Broth supplemented with 50 μg/mL of kanamycin and purified using the NucleoBond Xtra Maxi EF Kit (Macherey Nagel) and resuspended in Tris-EDTA Endotoxin-Free buffer overnight. Plasmid were quantified with a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific), aliquoted and stored at −20°C. Plasmid DNA were verified by enzymatic digestion and by sequencing the region proximal to the transgene insertion sites.
Production and titration of LV
Non-replicative LV were produced in Human Embryonic Kidney (HEK)-293T cells, as previously detailed (Zennou et al., 2000). Briefly, lentiviral particles were produced by transient calcium phosphate co-transfection of HEK293T cells with the vector plasmid pTRIP/sE, a VSV-G Indiana envelope plasmid and an encapsidation plasmid (p8.74 or pD64V for the production of integration-proficient or integration-deficient vectors respectively). Supernatants were harvested at 48 h post-transfection, clarified by 6-min centrifugation at 2500 rpm at 4°C. LV were aliquoted and stored at −80°C. Vector titers were determined by transducing 293T cells treated with aphidicolin. The titer, proportional to the efficacy of nuclear gene transfer, is determined as Transduction Unit (TU)/mL by qPCR on total lysates at day 3 post-transduction, by use of forward 5′-TGG AGG AGG AGA TAT GAG GG-3′ and reverse 5′-CTG CTG CAC TAT ACC AGA CA-3′ primers, specific to pFLAP plasmid and forward 5′-TCT CCT CTG ACT TCA ACA GC-3′ and reverse 5′-CCC TGC ACT TTT TAA GAG CC-3′ primers specific to the host housekeeping gene gadph as previously described (Iglesias et al., 2006).
SARS-CoV-2 inoculation
Hamsters or Ad5::hACE2-pretreated mice were anesthetized by i.p. injection of mixture Ketamine and Xylazine, transferred into a biosafety cabinet 3 where they were inoculated i.n. with BetaCoV/France/IDF0372/2020 SARS-CoV-2 clinical isolate (Lescure et al., 2020), amplified in VeroE6 cells. The strain BetaCoV/France/IDF0372/2020 was supplied by the National Reference Centre for Respiratory Viruses hosted by Institut Pasteur (Paris, France) and headed by Pr. Sylvie van der Werf. The human sample from which strain BetaCoV/France/IDF0372/2020 was isolated has been provided by Dr. X. Lescure and Pr. Y. Yazdanpanah from the Bichat Hospital, Paris, France. During the progression of our experiments, we observed that reducing the input SARS-CoV-2 dose from 1 × 105 to 0.3 × 105 TCID50/individual did not impact the plateau of the lung viral loads reached at 2–4 dpi. Therefore, in many studies we used the lower dose to mimic better the natural infectious dose. The viral inoculum was contained in 20 μL for mice and in 50 μL for hamsters. Animals were then housed in an isolator in BioSafety Level 3 animal facilities of Institut Pasteur. The organs and fluids recovered from the animals infected with live SARS-CoV-2 were manipulated following the approved standard operating procedures of these facilities.
Recombinant SCoV-2 proteins
Codon-optimized nucleotide fragments encoding a stabilized foldon-trimerized version of the SARS-CoV-2 S ectodomain (aa 1–1208), the S1 monomer (aa 16–681) and the RBD subdomain (aa 331–519) both preceded by a murine IgK leader peptide and followed by an 8xHis Tag were synthetized and cloned into pcDNA3.1/Zeo(+) expression vector (Thermo Fisher Scientific). Proteins were produced by transient co-transfection of exponentially growing Freestyle 293-F suspension cells (Thermo Fisher Scientific) using polyethylenimine (PEI)-precipitation method as previously described (Lorin and Mouquet, 2015). Recombinant SCoV-2 proteins were purified by affinity chromatography using the Ni Sepharose Excel Resin according to manufacturer’s instructions (Thermo Fisher Scientific). Protein purity was evaluated by in-gel protein silver-staining using Pierce Silver Stain kit (Thermo Fisher Scientific) following SDS-PAGE in reducing and non-reducing conditions using NuPAGE 3%–8% Tris-Acetate gels (Life Technologies). Purified proteins were dialyzed overnight against PBS using Slide-A-Lyzer dialysis cassettes (10 kDa MW cutoff, Thermo Fisher Scientific). Protein concentration was determined using the NanoDrop One instrument (Thermo Fisher Scientific).
ELISA
Ninety-six-well Nunc Polysorp plates (Nunc, Thermo Fisher Scientific) were coated overnight at 4°C with 100 ng/well of purified SCoV-2 proteins in carbonate-bicarbonate buffer (pH 9.6). The next day, plates were blocked with carbonate buffer containing 1% BSA for 2 h at 37°C. Wells were then washed with PBS containing 0.05% Tween 20 (PBS-T), 1:100-diluted sera or 1:10-diluted lung homogenates in PBS-T containing 1% BSA and four serial ten-to-ten dilutions were added and incubated during 2 h at 37°C. After PBS-T washings, plates were incubated with 1,000-fold diluted peroxydase-conjugated goat anti-mouse IgG (Jackson ImmunoResearch Europe Ltd, Cambridgeshire, United Kingdom) for 1 h. Plates were revealed by adding 100 μL of 3,3′,5,5′-tetramethylbenzidine chromogenic substrate (Eurobio Scientific). Following a 30 min incubation, reaction was stopped by adding 100 μL of 2N H2SO4 and optical densities were measured at 450nm/620nm on a PR3100 reader.
NAb detection
Serial dilutions of heat inactivated sera or clarified lung homogenates were assessed for NAbs via an inhibition assay which uses HEK293T cells transduced to stably express human ACE2 and non-replicative SCoV-2 pseudo-typed LV particles which harbor the reporter luciferase firefly gene, allowing quantitation of the host cell invasion by mimicking fusion step of native SARS-CoV-2 virus (Sterlin et al., 2020). First, 1.5 × 102 TU of SCoV-2 pseudo-typed LV were pre-incubated, during 30 min at room temperature, in U-bottom plates, with serial dilutions of each serum in a final volume of 50 μL in DMEM-glutamax, completed with 10% heat-inactivated FCS and 100 U/mL penicillin and 100 μg/mL streptomycin. The samples were then transferred into clear-flat-bottom 96-well-black-plates (Corning), and each well received 2 × 104 hACE2+ HEK293-T cells, counted in a NucleoCounter NC.200 system (Chemometec, Denmark) contained in 50 μL. After 2 days incubation at 37°C 5% CO2, the transduction efficiency of hACE2+ HEK293-T cells by pseudo-typed LV particles was determined by measuring the luciferase activity, using a Luciferase Assay System (Promega) on an EnSpire plate reader (PerkinElmer), as detailed elsewhere (Sterlin et al., 2020). Results are expressed as percentages of inhibition of luciferase activity compared to the maximum of luciferase activity in the absence of NAbs. Sera were heat-inactivated by 30 min incubation at 56°C and were manipulated in BSL2 conditions.
SFL T cell epitope mapping
In order to map the immuno-dominant epitopes of SCoV-2, peptides spanning the whole SFL (Mimotopes, Australia) were pooled by 16, each containing 15 aa residues overlapping by 10 aa peptides were dissolved in DMSO at a concentration of 2 mg/mL and diluted before use at 1 μg/mL for ELISPOT or 2-5 μg/mL for ICS (Intracellular Cytokine Staining) in RΡΜΙ−1640 medium supplemented with 10% FCS, 100 U/mL penicillin, 100 μg/mL streptomycin, 1 × 10−4 M non-essential amino-acids, 1% vol/vol HEPES, 1 × 10−3 M sodium pyruvate and 5 × 10−5 M of β-mercapto-ethanol. IFN-γ ELISPOT and ICS assays were performed as described previously (Bourgine et al., 2018; Sayes et al., 2016). For ICS, cells were acquired in an Attune NxT Flow cytometer (Thermo Fisher Scientific) and the data were analyzed by FlowJo Software (TreeStar).
Generation of Ad5 gene transfer vectors
The Ad5 gene transfer vectors were produced using the ViraPower Adenoviral Promoterless Gateway Expression Kit (Thermo Fisher Scientific). The sequence containing CMV promoter, BamH1/Xho1 restriction sites and WPRE was PCR amplified from the pTRIPΔU3CMV plasmid, by use of (1) forward primer, encoding the attB1 in the 5′ end, and (2) reverse primer, encoding both the attB2 and SV40 polyA signal sequence in the 5′ end. The attB-PCR product was cloned into the gateway pDONR207 donor vector, via BP Clonase reaction. The hACE2 was amplified from a plasmid derivative of hACE2-expressing pcDNA3.1, while egfp was amplified from pTRIP-ieCMV-eGFP-WPRE. The amplified PCR products were cloned into the pDONR207 plasmid via the BamH1 and Xho1 restriction sites. To obtain the final Ad5 plasmid, the pDONR207 vector, harboring hACE2 or gfp genes, was further inserted into pAd/PL-DEST vector via LR Clonase reaction (Figure S8).
The Ad5 virions were generated by transfecting the E3-transcomplementing HEK293A cell line with pAd CMV-GFP-WPRE-SV40 polyA or pAd CMV-hACE2-WPRE-SV40 polyA plasmid followed by subsequent vector amplification, according to the manufacturer’s protocol (ViraPower Adenoviral Promoterless Gateway Expression Kit, Thermo Fisher Scientific). The Ad5 particles were purified using Adeno-X rapid Maxi purification kit and concentrated with the Amicon Ultra-4 10k centrifugal filter unit. Vectors were resuspended and stored at −80°C in PIPES buffer pH 7.5, supplemented with 2.5% glucose. Ad5 were titrated using a qPCR protocol, as described (Gallaher and Berk, 2013).
Western blot
Expression of hACE2 in the lungs of Ad5::hACE2-transduced mice was assessed by western blotting. One million cells from lung cell suspension were resolved on 4%–12% NuPAGE Bis-Tris protein gels (Thermo Fisher Scientific), then transferred onto a nitrocellulose membrane (Biorad, France). The nitrocellulose membrane was blocked in 5% non-fat milk in PBS-T for 2 h at room temperature and probed overnight with goat anti-hACE2 primary Ab at 1 μg/mL (AF933, R&D systems). Following three wash intervals of 10 min with PBS-T, the membrane was incubated for 1 h at room temperature with HRP-conjugated anti-goat secondary Ab and HRP-conjugated anti-β-actin (ab197277, Abcam). The membrane was washed with PBS-T thrice before visualization with enhanced chemiluminescence via the super signal west femto maximum sensitivity substrate (Thermo Fisher Scientific) on ChemiDoc XRS+ (Biorad, France). PageRuler Plus prestained protein ladder was used as size reference.
Determination of SARS-CoV-2 viral loads in the lungs
Half of each lung lobes were removed aseptically and frozen at −80°C. Organs were thawed and homogenized for 20 s at 4.0 m/s, using lysing matrix M (MP Biomedical) in 500 μL of ice-cold PBS. The homogenization was performed in an MP Biomedical Fastprep 24 Tissue Homogenizer. The homogenates were centrifuged 10 min at 2000 g for further RNA extraction from the supernatants. Particulate viral RNA was extracted from 70 μL of such supernatants using QIAamp Viral RNA Mini Kit (QIAGEN) according to the manufacturer’s procedure. Viral load was determined following reverse transcription and real-time quantitative TaqMan PCR essentially as described (Corman et al., 2020), using SuperScript III Platinum One-Step Quantitative RT-PCR System (Invitrogen) and primers and probe (Eurofins) targeting SCoV-2 gene as listed in Table S1. In vitro transcribed RNA derived from plasmid “pCI/SARS-CoV envelope” was synthesized using T7 RiboMAX Express Large Scale RNA production system (Promega), then purified by phenol/chloroform extractions and two successive precipitations with isopropanol and ethanol. RNA concentration was determined by optical density measurement, then RNA was diluted to 109 genome equivalents/μL in RNase-free water containing 100 μg/mL tRNA carrier, and stored in single-use aliquots at −80°C. Serial dilutions of this in vitro transcribed RNA were prepared in RNase-free water containing 10 μg/mL tRNA carrier and used to establish a standard curve in each assay. Thermal cycling conditions were (1) reverse transcription at 55°C for 10 min, (2) enzyme inactivation at 95°C for 3 min, and (3) 45 cycles of denaturation/amplification at 95°C for 15 s, 58°C for 30 s. Products were analyzed on an ABI 7500 Fast real-time PCR system (Applied Biosystems). PFU assay was performed as recently described (Case et al., 2020).
Cytometric analysis of lung cells
Lungs from individual mice were treated with 400 U/mL type IV collagenase and DNase I (Roche) for a 30-min incubation at 37°C and homogenized by use of GentleMacs (Miltenyi Biotech) mAbs. Cells were filtered through 100 μm-pore filters and centrifuged at 1200 rpm during 8 min. Cells were then treated with Red Blood Cell Lysing Buffer (Sigma) and washed twice in PBS. Cells were stained as follows. (1) To detect DC, monocytes, alveolar and interstitial macrophages: Near IR Live/Dead (Invitrogen), FcγII/III receptor blocking anti-CD16/CD32 (BD Biosciences), BV605-anti-CD45 (BD Biosciences), PE-anti-CD11b (eBioscience), PE-Cy7-antiCD11c (eBioscience), BV450-anti-CD64 (BD Biosciences), FITC-anti-CD24 (BD Biosciences), BV711-anti-CD103 (BioLegend), AF700-anti-MHC-II (BioLegend), PerCP-Cy5.5-anti-Ly6C (eBioscience) and APC anti-Ly-6G (Miltenyi) mAbs, (2) to detect neutrophils or eosinophils: Near IR DL (Invitrogen), FcγII/III receptor blocking anti-CD16/CD32 (BD Biosciences), PerCP-Vio700-anti-CD45 (Miltenyi), APC-anti-CD11b (BD Biosciences), PE-Cy7-anti-CD11c (eBioscience), FITC-anti-CD24 (BD Biosciences), AF700-anti-MHC-II (BioLegend), PE-anti-Ly6G (BioLegend), BV421-anti-Siglec-F (BD Biosciences), (3) to detect mast cells, basophils, NK: Near IR LD (Invitrogen), BV605-anti-CD45 (BD Biosciences), PE-anti-CD11b (eBioscience), eF450-anti-CD11c (eBioscience), PE-Cy7-anti-CD117 (BD Biosciences), APC-anti-FcεR1 (BioLegend), AF700-anti-NKp46 (BD Biosciences), FITC-anti-CCR3 (BioLegend), without FcγII/III receptor blocking anti-CD16/CD32. Cells were incubated with appropriate mixtures for 25 min at 4°C, washed twice in PBS containing 3% FCS and then fixed with Paraformaldehyde 4% by an overnight incubation at 4°C. The cells were acquired in an Attune NxT cytometer system (Invitrogen) and data were analyzed by FlowJo software (Treestar, OR, USA).
qRT-PCR detection of inflammatory cytokines and chemokines in the lungs of the mice and hamsters
Lung samples from mice or hamsters were added to lysing matrix D (MP Biomedical) containing 1 mL of TRIzol reagent and homogenized at 30 s at 6.0 m/s twice using MP Biomedical Fastprep 24 Tissue Homogenizer. Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s procedure. cDNA was synthesized from 4 μg of RNA in the presence of 2.5 μM of oligo(dT) 18 primers, 0.5 mM of deoxyribonucleotides, 2.0 U of RNase Inhibitor and SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific) in 20 μL reaction. The real-time PCR was performed on QuantStudio 7 Flex Real-Time PCR System (Thermo Fisher Scientific). Reactions were performed in triplicates in a final reaction volume of 10 μL containing 5 μL of iQ SYBR Green Supermix (Biorad, France), 4 μL of cDNA diluted 1:15 in DEPC-water and 0.5 μL of each forward and reverse primers at a final concentration of 0.5 μM (Table S2). The following thermal profile was used: a single cycle of polymerase activation for 3 min at 95°C, followed by 40 amplification cycles of 15 s at 95°C and 30 s 60°C (annealing-extension step). Mice β-globin or hamster ribosomal protein L18 (RLP18) was used as an endogenous reference control to normalize differences in the amount of input nucleic acid. The average C T values were calculated from the technical replicates for relative quantification of target cytokines/chemokines. The differences in the C T cytokines/chemokines amplicons and the C T of the endogenous reference control, termed ΔC T, were calculated to normalize for differences in the quantity of nucleic acid. The ΔC T of the experimental condition compared relatively to the PBS-immunized individuals using the comparative ΔΔC T method. The fold change in gene expression was further calculated using 2−ΔΔC T.
Lung histopathology
Samples from the lung were fixed in formalin for at least 7 days and routinely embedded in paraffin. Five μm thick paraffin sections were stained with hematoxylin eosin & saffron (HE&S) or HE. Microscopic changes were qualitatively described and when applicable scored semiquantitatively, by histopathologists blinded to the conditions, using (1) distribution qualifiers (i.e., focal, multifocal, locally extensive or diffuse), and (2) a five-scale severity grade, i.e., 1: minimal, 2: mild, 3: moderate, 4: marked and 5: severe.
Quantification and statistical analysis
Statistical parameters are presented as means ± SEM or ± SD and the exact number (n) of mice per group are annotated in the corresponding figure legend and in the text. Statistical analysis of virological and immunological data was performed using GraphPad Prism v9.0.0 (GraphPad Software, CA, USA). Differences between two groups were evaluated by using unpaired two-tailed nonparametric Mann-Whitney U test. For statistical analysis of multiple groups, the two-way ANOVA followed by Sidak’s multiple comparisons test was applied (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; ns, not statistically significant).
Acknowledgments
The authors are grateful to Prof. Sylvie van der Werf (National Reference Centre for Respiratory Viruses, Institut Pasteur, France) for providing the BetaCoV/France/IDF0372/2020 SARS-CoV-2 clinical isolate, supplied through the European Virus Archive Global (Evag), funded by the European Union’s Horizon 2020 program under grant agreement no. 653316. The authors thank Dr. Marion Bérard and Pascale Pescher for veterinary advice; Dr. Cyril Planchais for preparing homotrimeric S, S1, and RBD proteins; Marija Backovic for providing the corresponding expression vectors; Damien Batalie for excellent technical assistance in determining viral loads; and Magali Tichit, Sabine Maurin, and Johan Bedel for excellent technical assistance in preparing histological sections. Some histological sections were prepared gratuitously by HISTALIM (Cerba Research - Biomarkers Division, Montpellier, France). This work was also supported by grants from Institut Pasteur, TheraVectys, and Agence Nationale de la Recherche HuMoCID. M.W.K. is part of the Pasteur - Paris University PhD program and received funding from Institut Carnot Pasteur Microbes & Santé and the European Union’s Horizon 2020 program under Marie Sklodowska-Curie grant agreement no. 665807.
Author contributions
Study concept and design, M.-W.K., M.B., F.A., A.M., N.E., L.M., and P.C.; acquisition of data, M.-W.K., M.B., P.A., J.L., K.N., B.V., F.N., P.S., H.T., E.S., F.A., A.M., and L.M.; construction and production of LV and technical support, P.A., F.M., A.N., F.N., C.B., and P.S.; analysis and interpretation of data, M.-W.K., M.B., P.A., J.L., K.N., F.A., A.M., N.E., L.M., and P.C.; histology, D.H., M.L.D., F.G., and L.F.; recombinant S proteins, H.M.; drafting of the manuscript, M.-W.K., M.B., F.G., L.M., and P.C.
Declaration of interests
P.C. is the founder and CSO of TheraVectys. M.-W.K., P.A., J.L., K.N., F.M., A.N., B.V., F.N., and F.A. are employees of TheraVectys. M.-W.K., M.B., P.A., N.E., L.M., and P.C. are inventors of a pending patent directed to a vaccine candidate against SARS-CoV2.
Published: December 16, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.chom.2020.12.010.
Supplemental information
References
- Amanat F., Krammer F. SARS-CoV-2 vaccines: status report. Immunity. 2020;52:583–589. doi: 10.1016/j.immuni.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anna F., Goyard S., Lalanne A.I., Nevo F., Gransagne M., Souque P., Louis D., Gillon V., Turbiez I., Bidard F.-C., et al. High seroprevalence but short-lived immune response to SARS-CoV-2 infection in Paris. Eur. J. Immunol. 2020 doi: 10.1002/eji.202049058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beignon A.S., Mollier K., Liard C., Coutant F., Munier S., Rivière J., Souque P., Charneau P. Lentiviral vector-based prime/boost vaccination against AIDS: pilot study shows protection against Simian immunodeficiency virus SIVmac251 challenge in macaques. J. Virol. 2009;83:10963–10974. doi: 10.1128/JVI.01284-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belouzard S., Chu V.C., Whittaker G.R. Activation of the SARS coronavirus Spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl. Acad. Sci. USA. 2009;106:5871–5876. doi: 10.1073/pnas.0809524106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgine M., Crabe S., Lobaina Y., Guillen G., Aguilar J.C., Michel M.L. Nasal route favors the induction of CD4+ T cell responses in the liver of HBV-carrier mice immunized with a recombinant hepatitis B surface- and core-based therapeutic vaccine. Antiviral Res. 2018;153:23–32. doi: 10.1016/j.antiviral.2018.02.019. [DOI] [PubMed] [Google Scholar]
- Buffa V., Negri D.R., Leone P., Borghi M., Bona R., Michelini Z., Compagnoni D., Sgadari C., Ensoli B., Cara A. Evaluation of a self-inactivating lentiviral vector expressing simian immunodeficiency virus gag for induction of specific immune responses in vitro and in vivo. Viral Immunol. 2006;19:690–701. doi: 10.1089/vim.2006.19.690. [DOI] [PubMed] [Google Scholar]
- Case J.B., Bailey A.L., Kim A.S., Chen R.E., Diamond M.S. Growth, detection, quantification, and inactivation of SARS-CoV-2. Virology. 2020;548:39–48. doi: 10.1016/j.virol.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrashekar A., Liu J., Martinot A.J., McMahan K., Mercado N.B., Peter L., Tostanoski L.H., Yu J., Maliga Z., Nekorchuk M., et al. SARS-CoV-2 infection protects against rechallenge in rhesus macaques. Science. 2020;369:812–817. doi: 10.1126/science.abc4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corman V., Bleicker T., Brünink S., Drosten C. Diagnostic detection of 2019-nCoV by real-time RT-PCR. 2020. https://www.who.int/docs/default-source/coronaviruse/protocol-v2-1.pdf [DOI] [PMC free article] [PubMed]
- Cousin C., Oberkampf M., Felix T., Rosenbaum P., Weil R., Fabrega S., Morante V., Negri D., Cara A., Dadaglio G., Leclerc C. Persistence of integrase-deficient lentiviral vectors correlates with the induction of STING-independent CD8+ T cell responses. Cell Rep. 2019;26:1242–1257.e7. doi: 10.1016/j.celrep.2019.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutant F., Sanchez David R.Y., Félix T., Boulay A., Caleechurn L., Souque P., Thouvenot C., Bourgouin C., Beignon A.S., Charneau P. A nonintegrative lentiviral vector-based vaccine provides long-term sterile protection against malaria. PLoS ONE. 2012;7:e48644. doi: 10.1371/journal.pone.0048644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutard B., Valle C., de Lamballerie X., Canard B., Seidah N.G., Decroly E. The Spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 2020;176:104742. doi: 10.1016/j.antiviral.2020.104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nunzio F., Félix T., Arhel N.J., Nisole S., Charneau P., Beignon A.S. HIV-derived vectors for therapy and vaccination against HIV. Vaccine. 2012;30:2499–2509. doi: 10.1016/j.vaccine.2012.01.089. [DOI] [PubMed] [Google Scholar]
- Esslinger C., Romero P., MacDonald H.R. Efficient transduction of dendritic cells and induction of a T-cell response by third-generation lentivectors. Hum. Gene Ther. 2002;13:1091–1100. doi: 10.1089/104303402753812494. [DOI] [PubMed] [Google Scholar]
- Firat H., Zennou V., Garcia-Pons F., Ginhoux F., Cochet M., Danos O., Lemonnier F.A., Langlade-Demoyen P., Charneau P. Use of a lentiviral flap vector for induction of CTL immunity against melanoma. Perspectives for immunotherapy. J. Gene Med. 2002;4:38–45. doi: 10.1002/jgm.243. [DOI] [PubMed] [Google Scholar]
- Gallaher S.D., Berk A.J. A rapid Q-PCR titration protocol for adenovirus and helper-dependent adenovirus vectors that produces biologically relevant results. J. Virol. Methods. 2013;192:28–38. doi: 10.1016/j.jviromet.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallinaro A., Borghi M., Bona R., Grasso F., Calzoletti L., Palladino L., Cecchetti S., Vescio M.F., Macchia D., Morante V., et al. Integrase Defective Lentiviral Vector as a Vaccine Platform for Delivering Influenza Antigens. Front. Immunol. 2018;9:171. doi: 10.3389/fimmu.2018.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q., Bao L., Mao H., Wang L., Xu K., Yang M., Li Y., Zhu L., Wang N., Lv Z., et al. Development of an inactivated vaccine candidate for SARS-CoV-2. Science. 2020;369:77–81. doi: 10.1126/science.abc1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzelak L., Temmam S., Planchais C., Demeret C., Tondeur L., Huon C., Guivel-Benhassine F., Staropoli I., Chazal M., Dufloo J., et al. A comparison of four serological assays for detecting anti-SARS-CoV-2 antibodies in human serum samples from different populations. Sci. Transl. Med. 2020;12:eabc3103. doi: 10.1126/scitranslmed.abc3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.R., Cao Q.D., Hong Z.S., Tan Y.Y., Chen S.D., Jin H.J., Tan K.S., Wang D.Y., Yan Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak - an update on the status. Mil. Med. Res. 2020;7:11. doi: 10.1186/s40779-020-00240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan A.O., Case J.B., Winkler E.S., Thackray L.B., Kafai N.M., Bailey A.L., McCune B.T., Fox J.M., Chen R.E., Alsoussi W.B., et al. A SARS-CoV-2 infection model in mice demonstrates protection by neutralizing antibodies. Cell. 2020;182:744–753.e4. doi: 10.1016/j.cell.2020.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y., Zhang J., Mi Z., Robbins P., Falo L.D., Jr. Immunization with lentiviral vector-transduced dendritic cells induces strong and long-lasting T cell responses and therapeutic immunity. J. Immunol. 2005;174:3808–3817. doi: 10.4049/jimmunol.174.6.3808. [DOI] [PubMed] [Google Scholar]
- Hobernik D., Bros M. DNA vaccines—how far from clinical use? Int. J. Mol. Sci. 2018;19:3605. doi: 10.3390/ijms19113605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A., et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B., Tai A., Wang P. Immunization delivered by lentiviral vectors for cancer and infectious diseases. Immunol. Rev. 2011;239:45–61. doi: 10.1111/j.1600-065X.2010.00967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias M.C., Frenkiel M.P., Mollier K., Souque P., Despres P., Charneau P. A single immunization with a minute dose of a lentiviral vector-based vaccine is highly effective at eliciting protective humoral immunity against West Nile virus. J. Gene Med. 2006;8:265–274. doi: 10.1002/jgm.837. [DOI] [PubMed] [Google Scholar]
- Ku M.W., Anna F., Souque P., Petres S., Prot M., Simon-Loriere E., Charneau P., Bourgine M. A Single Dose of NILV-Based Vaccine Provides Rapid and Durable Protection against Zika Virus. Mol. Ther. 2020;28:1772–1782. doi: 10.1016/j.ymthe.2020.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A.L., Millet J.K., Daniel S., Freed J.H., Whittaker G.R. The SARS-CoV Fusion Peptide Forms an Extended Bipartite Fusion Platform that Perturbs Membrane Order in a Calcium-Dependent Manner. J. Mol. Biol. 2017;429:3875–3892. doi: 10.1016/j.jmb.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescure F.X., Bouadma L., Nguyen D., Parisey M., Wicky P.H., Behillil S., Gaymard A., Bouscambert-Duchamp M., Donati F., Le Hingrat Q., et al. Clinical and virological data of the first cases of COVID-19 in Europe: a case series. Lancet Infect. Dis. 2020;20:697–706. doi: 10.1016/S1473-3099(20)30200-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorin V., Mouquet H. Efficient generation of human IgA monoclonal antibodies. J. Immunol. Methods. 2015;422:102–110. doi: 10.1016/j.jim.2015.04.010. [DOI] [PubMed] [Google Scholar]
- Muñoz-Fontela C., Dowling W.E., Funnell S.G.P., Gsell P.S., Riveros-Balta A.X., Albrecht R.A., Andersen H., Baric R.S., Carroll M.W., Cavaleri M., et al. Animal models for COVID-19. Nature. 2020;586:509–515. doi: 10.1038/s41586-020-2787-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H., Sun S., Xiao H., Feng J., Guo Y., Tai W., Wang Y., Du L., Zhao G., Zhou Y. Single-dose treatment with a humanized neutralizing antibody affords full protection of a human transgenic mouse model from lethal Middle East respiratory syndrome (MERS)-coronavirus infection. Antiviral Res. 2016;132:141–148. doi: 10.1016/j.antiviral.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg S.A., Zhai Y., Yang J.C., Schwartzentruber D.J., Hwu P., Marincola F.M., Topalian S.L., Restifo N.P., Seipp C.A., Einhorn J.H., et al. Immunizing patients with metastatic melanoma using recombinant adenoviruses encoding MART-1 or gp100 melanoma antigens. J. Natl. Cancer Inst. 1998;90:1894–1900. doi: 10.1093/jnci/90.24.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayes F., Pawlik A., Frigui W., Gröschel M.I., Crommelynck S., Fayolle C., Cia F., Bancroft G.J., Bottai D., Leclerc C., et al. CD4+ T Cells Recognizing PE/PPE Antigens Directly or via Cross Reactivity Are Protective against Pulmonary Mycobacterium tuberculosis Infection. PLoS Pathog. 2016;12:e1005770. doi: 10.1371/journal.ppat.1005770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmbeck R., Reimann J., Kochanek S., Kreppel F. The immunogenicity of adenovirus vectors limits the multispecificity of CD8 T-cell responses to vector-encoded transgenic antigens. Mol. Ther. 2008;16:1609–1616. doi: 10.1038/mt.2008.141. [DOI] [PubMed] [Google Scholar]
- Sia S.F., Yan L.M., Chin A.W.H., Fung K., Choy K.T., Wong A.Y.L., Kaewpreedee P., Perera R.A.P.M., Poon L.L.M., Nicholls J.M., et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature. 2020;583:834–838. doi: 10.1038/s41586-020-2342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterlin D., Mathian A., Miyara M., Mohr A., Anna F., Claër L., Quentric P., Fadlallah J., Devilliers H., Ghillani P., et al. IgA dominates the early neutralizing antibody response to SARS-CoV-2. Sci. Transl. Med. 2020:eabd2223. doi: 10.1101/2020.06.10.20126532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J., Zhuang Z., Zheng J., Li K., Wong R.L., Liu D., Huang J., He J., Zhu A., Zhao J., et al. Generation of a broadly useful model for COVID-19 pathogenesis, vaccination, and treatment. Cell. 2020;182:734–743.e5. doi: 10.1016/j.cell.2020.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabret N., Britton G.J., Gruber C., Hegde S., Kim J., Kuksin M., Levantovsky R., Malle L., Moreira A., Park M.D., et al. Sinai Immunology Review Project Immunology of COVID-19: current state of the science. Immunity. 2020;52:910–941. doi: 10.1016/j.immuni.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 Spike glycoprotein. Cell. 2020;181:281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y., Shang J., Sun S., Tai W., Chen J., Geng Q., He L., Chen Y., Wu J., Shi Z., et al. Molecular mechanism for antibody-dependent enhancement of coronavirus entry. J. Virol. 2020;94:e02015–e02019. doi: 10.1128/JVI.02015-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Qiu Y., Li J.Y., Zhou Z.J., Liao C.H., Ge X.Y. A unique protease cleavage site predicted in the Spike protein of the novel pneumonia coronavirus (2019-nCoV) potentially related to viral transmissibility. Virol. Sin. 2020;35:337–339. doi: 10.1007/s12250-020-00212-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Tostanoski L.H., Peter L., Mercado N.B., McMahan K., Mahrokhian S.H., Nkolola J.P., Liu J., Li Z., Chandrashekar A., et al. DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science. 2020;369:806–811. doi: 10.1126/science.abc6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M., Liu H., Wu N.C., Lee C.D., Zhu X., Zhao F., Huang D., Yu W., Hua Y., Tien H., et al. Structural basis of a shared antibody response to SARS-CoV-2. Science. 2020;369:1119–1123. doi: 10.1126/science.abd2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zennou V., Petit C., Guetard D., Nerhbass U., Montagnier L., Charneau P. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell. 2000;101:173–185. doi: 10.1016/S0092-8674(00)80828-4. [DOI] [PubMed] [Google Scholar]
- Zhao J., Li K., Wohlford-Lenane C., Agnihothram S.S., Fett C., Zhao J., Gale M.J., Jr., Baric R.S., Enjuanes L., Gallagher T., et al. Rapid generation of a mouse model for Middle East respiratory syndrome. Proc. Natl. Acad. Sci. USA. 2014;111:4970–4975. doi: 10.1073/pnas.1323279111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F.C., Li Y.H., Guan X.H., Hou L.H., Wang W.J., Li J.X., Wu S.P., Wang B.S., Wang Z., Wang L., et al. Safety, tolerability, and immunogenicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: a dose-escalation, open-label, non-randomised, first-in-human trial. Lancet. 2020;395:1845–1854. doi: 10.1016/S0140-6736(20)31208-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated and analyzed during this study.