

Abstract
BACKGROUND
Intrahepatic cholestasis in pregnancy (ICP) is the most common liver disease during pregnancy, and its exact etiology and course of progression are still poorly understood.
AIM
To investigate the link between the gut microbiota and serum metabolome in ICP patients.
METHODS
In this study, a total of 30 patients were recruited, including 15 patients with ICP (disease group) and 15 healthy pregnant patients (healthy group). The serum nontarget metabolomes from both groups were determined. Amplification of the 16S rRNA V3-V4 region was performed using fecal samples from the disease and healthy groups. By comparing the differences in the microbiota and metabolite compositions between the two groups, the relationship between the gut microbiota and serum metabolites was also investigated.
RESULTS
The Kyoto Encyclopedia of Genes and Genomes analysis results showed that the primary bile acid biosynthesis, bile secretion and taurine and hypotaurine metabolism pathways were enriched in the ICP patients compared with the healthy controls. In addition, some pathways related to protein metabolism were also enriched in the ICP patients. The principal coordination analysis results showed that there was a distinct difference in the gut microbiota composition (beta diversity) between the ICP patients and healthy controls. At the phylum level, we observed that the relative abundance of Firmicutes was higher in the healthy group, while Bacteroidetes were enriched in the disease group. At the genus level, most of the bacteria depleted in ICP are able to produce short-chain fatty acids (e.g., Faecalibacterium, Blautia and Eubacterium hallii), while the bacteria enriched in ICP are associated with bile acid metabolism (e.g., Parabacteroides and Bilophila). Our results also showed that specific genera were associated with the serum metabolome.
CONCLUSION
Our study showed that the serum metabolome was altered in ICP patients compared to healthy controls, with significant differences in the bile, taurine and hypotaurine metabolite pathways. Alterations in the metabolization of these pathways may lead to disturbances in the gut microbiota, which may further affect the course of progression of ICP.
Keywords: Intrahepatic cholestasis in pregnancy, Metabolome, Gut microbiota, Bile acids
Core Tip: This study is the first to investigate the consequences of intrahepatic cholestasis in pregnancy (ICP) with respect to the serum metabolome and gut microbiota. Our data suggest that ICP can cause significant changes in the serum metabolome. Changes in the relative abundance of bile acid-related compounds in serum due to ICP may have an impact on gut microbiota, which in turn may threaten the safety of pregnant women and fetuses. Our findings also suggest a mechanism of ICP that is associated with the microbiota via changes in serum metabolites.
INTRODUCTION
Intrahepatic cholestasis in pregnancy (ICP) is the most common liver disease associated with pregnancy. ICP usually develops in the third trimester of pregnancy and is characterized by itching and increased levels of bile acid and/or alanine aminotransferase. The disease spontaneously resolves after delivery; however, it tends to recur in more severe forms in 45%-90% of patients during second pregnancies. The incidence of intrahepatic cholestasis varies among ethnic groups according to geographic region[1,2]. ICP increases the risk of premature delivery, intrauterine asphyxia, fecal staining of amniotic fluid and fetal bradycardia. Women with ICP have a significantly increased risk of preterm birth, stillbirth and admission to the neonatal unit for treatment[3]. Other symptoms that can impact the fetus include neonatal depression and respiratory distress syndrome[4,5]. However, the etiology of cholestasis is poorly understood, and its management is difficult due to the lack of data on its diagnosis, treatment and associated adverse consequences.
At present, the most sensitive biochemical marker for the diagnosis of ICP is the level of total bile acid (TBA), which may be the first or only index measured by laboratory tests. The cut-off point has been defined as a concentration of TBAs higher than 10 μmol/L[3]. Some prospective studies have assessed the risk of complications to fetal development at TBA concentrations higher than 40 μmol/L[3]. In the human body, cholesterol forms primary bile acids through two different pathways. The classical pathway generates cholic acid and chenodeoxycholic acid (CDCA), and at least 75% of bile acids are produced by this pathway; on the other hand, the alternative pathway generates CDCA[6,7]. Primary bile acids are generated and conjugated with taurine and glycine to form conjugated bile acids. Unconjugated bile acids can cross cell membranes by diffusion, while conjugated bile acids need to be actively transported into the bile by a bile salt export pump and stored in the gallbladder. The gallbladder contracts to release bile acids into the intestine, and microorganisms from the gut microbiota continue to metabolize bile acids, glycine and taurine-conjugated cholic acid and CDCA to form secondary bile acids via bile acid hydrolase deconjugation and 7α-dehydroxylation[6,8]. In the terminal ileum, most unconjugated bile acids are absorbed into intestinal epithelial cells by apical sodium-dependent bile acid transporters and then are secreted into the vena portae, eventually reaching the liver through the circulatory system and completing the liver-intestinal circulation.
Of the pool of bile acid, 90%-95% is circulated daily, with approximately six to ten cycles between the intestine and the liver each day producing approximately 0.2-0.6 g of newly synthesized bile acids per day to maintain a stable pool of bile acids[6,7,9]. The intestinal microbiome exerts an enormous function on the process of bile acid circulation, and the process of bile acid deconjugation is mainly performed by bacteria with BSH activity in the intestine. In addition, the intestinal microbiota can indirectly regulate synthesis of bile acid through its effects on receptors like FXR and FGF19[6,7,10]. Under the pathological conditions of dietary obesity[11], cholestatic liver disease[12], gastrointestinal inflammation, cancerization and so on[9], the imbalance of microbe-bile acid mutual effect also occurs. To date, few studies have provided information on whether ICP can affect the gut microbiota and metabolome of pregnant women.
The gastrointestinal tract provides surviving conditions for the gut microbiota, which is f important for the function and metabolic activity of the human body[13,14]. A steady gut microbiota is crucial for maintaining human health, and dysbiosis of gut microbiota plays a critical role in the etiopathogenesis of many diseases, such as ulcerative colitis[13-16]. We speculate that changes in serum metabolism caused by ICP may influence the intestinal tract circumstance and thus the intestinal microbiome. The gut microbiota may represent a mechanism by which ICP affects the health of pregnant women and fetuses. In other areas, there are precedents for treating diseases with fecal microbiota transplantation. In the field of the gut microbiota, we will also find the novel therapeutic strategies in ICP. Identifying microbiota-driven mechanisms that link ICP and serum metabolites holds promise for the use of microbiota intervention tactics in ICP treatment. Here, we researched the serum metabolites and gut microbiota of ICP patients and healthy controls using metabolomics and 16S rRNA sequencing analysis. The relationship of the gut microbiota with ICP-induced differential metabolites was further studied. This study reveals, for the first time, the health consequences of ICP with respect to the gut microbiota and metabolome.
MATERIALS AND METHODS
Study design and samples
This study was carried out at the Shanghai First Maternity and Infant Health Hospital from September 2019 to February 2020. A total of 30 patients (15 ICP patients and 15 healthy pregnant women) were recruited for this study. Signed informed consent was obtained from all study participants. This study was approved by the Ethics Committee of Shanghai First Maternity and Infant Health Hospital (KS2035). Thirty fecal samples were collected with a sterile sampler and were immediately transferred to the laboratory on dry ice. Each sample was dispensed into two 1.5 mL Eppendorf tubes and stored at -80 °C until DNA extraction (carried out in a sterile operating chamber). The diagnosis of ICP was made on the basis of the following criteria: severe itching without eruption; markedly increased concentrations of maternal serum bile acid (> 10 μmol/L); inexistence of obvious pruritus disease; exclusive of other hepatic diseases, such as viral hepatitis, cholelithiasis, fatty liver, hepatotoxic drug consumption and inflammatory bowel disease; and absence of antibiotic therapy prior to fecal sample collection. After the participants’ stools were obtained, their peripheral blood samples were collected within 24 h; the blood was centrifuged 10 min at a speed of 3000 rpm, the serum was carefully collected, and 0.5 mL aliquot was stored at -80 °C.
The clinical data of aspartate aminotransferase, alanine aminotransferase, serum bilirubin (indirect and direct) and TBA were obtained from the paper medical records and electronic medical record system of the Shanghai First Maternity and Infant Health Hospital.
Fecal DNA extraction and high-throughput sequencing
An Omega fecal bacterial genomic DNA extraction kit was used, and the specific procedure was performed according to the instructions. The 16S rRNA V3-V4 region was amplified by PCR using the 341F (5’-CCTACGGGNGGCWGCAG-3’) and 805R (5’-GACTACHVGGGTATCTAATCC-3’) primers. The PCR products were recovered using 2% agarose gels and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, United States). The final 16S rRNA gene amplicon library was sequenced on the MiSeq platform (Illumina) using a 2 × 300 bp paired-end protocol.
Liquid chromatography-mass spectrometry-based metabolism analysis
Serum samples were separated on an Agilent 1290 Infinity LC Ultra Performance Liquid Chromatography System HILIC column with the following conditions: Column temperature: 25 °C; flow rate: 0.3 mL/min; and mobile phase composition: A: Water 25 mmol/L ammonium acetate 25 mmol/L ammonia; B: Acetonitrile. The gradient elution program was illustrated below: 0-0.5 min, 95% B; 0.5-7 min, B changed linearly from 95% to 65%; 7-8 min, B changed linearly from 65% to 40%; 8-9 min, B maintained at 40%; 9-9.1 min, B changed linearly from 40% to 95%; 9.1-12 min, B maintained at 95%. The samples were placed in an autosampler at 4 °C throughout the analysis. Electrospray ionization(ESI) positive and negative ion modes were used for detection. The samples were analyzed by mass spectrometry with an Agilent 6550 mass spectrometer. The ESI source conditions were illustrated below: Gas temp: 250 °C; drying gas: 16 L/min; nebulizer: 20 psi; sheath gas temp: 400 °C; gas sheath flow: 12 L/min; Vcap: 3000 V; nozzle voltage: 0 V; fragment: 175 V; mass range: 50-1200; acquisition rate: 4 Hz; and cycle time: 250 ms.
Bioinformatics and statistical analysis
Sequencing read pairs were demultiplexed based on the unique molecular barcodes, and the reads were merged using USEARCH Version 8.0. During the merge process, 0 mismatches and a minimum overlap of 50 bases were allowed. Sequences that could not be spliced and chimaeras were removed, and chimaeras were eliminated using UCHIME software. Sequences less than 400 bases in length after splicing were deleted. Operational taxonomy units (OTUs) were clustered using UPARSE53 (version 7.1 http://drive5.com/uparse/) software based on 97% similarity. OTUs were determined by mapping the centroids to the SILVA v128 database. Other analyses were performed using the QIIME 1.9 pipeline[17]. The raw sequencing data have been submitted to the NCBI Sequence Read Archive under accession number PRJNA657645.
Microbiota data principal coordinate analysis ordination plots were based on Bray-Curtis distances, and significance between healthy controls (HC) and ICP was determined by anosim using the vegan package[18]. Significant differences in the relative abundance of the bacterial genera between groups were determined by the Mann-Whitney U test. All P values were adjusted for multiple comparisons with the FDR algorithm, and P values < 0.05 after adjustment for multiple comparisons were considered significant. Microbiome statistical analysis and figure generation were performed in R 4.0 using the ggplot2 package[19]. We used the random Forest package in R for classification[20]. All the differentially expressed metabolites were queried and mapped to pathways based on the online Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.kegg.jp/), and enrichment analysis was performed. A Spearman rank correlation test was performed to analyze the correlation between the significant metabolites and different bacterial genera in the serum and stool samples. The Mann-Whitney U test was used to compare continuous data between two groups, and continuous data were expressed as the median. All the statistical analyses were performed using R 4.0.
RESULTS
Basic characteristics of participants
Thirty subjects were enrolled in the study, including 15 patients with ICP and 15 control subjects. The basic clinical data of the subjects is in Table 1. There was no significant difference in the ages of the ICP patients and the healthy controls. Samples were collected from the ICP patients and healthy controls at approximately 38 wk and approximately 33.4 wk gestation, respectively. In the ICP patients, the median values of alanine aminotransferase, aspartate aminotransferase, TBA and creatinine were markedly higher than those in controls (Table 1).
Table 1.
Demographic characteristics
|
|
Control, n = 15
|
ICP, n = 15
|
Adjusted P value
|
| Age in yr | 30.0 (28.5-32.0) | 31.0 (30.5-32.5) | 0.3322 |
| Pregnancy in d | 266.0 (248.0-274.5) | 234.0 (227.5-252.0) | 0.0273 |
| Alanine aminotransferase in U/L | 12.0 (8.5-17.0) | 22.0 (12.0-50.0) | 0.0369 |
| Aspartate aminotransferase in U/L | 16.0 (14.0-18.5) | 29.0 (19.5-41.5) | 0.0273 |
| Lactate dehydrogenase in U/L | 177.0 (155.0-217.5) | 208.0 (191.0-217.5) | 0.2196 |
| Total bilirubin in μmol/L | 7.1 (6.1-7.9) | 6.4 (5.9-9.6) | 0.8356 |
| Direct bilirubin in μmol/L | 1.7 (1.2-1.8) | 2.5 (1.6-3.0) | 0.2669 |
| Total bile acids in μmol/L | 4.0 (3.5-5.5) | 26.0 (18.5-38.0) | 0.0000 |
| Urea in mmol/L | 3.8 (3.5-4.0) | 3.9 (3.1-5.0) | 0.6321 |
| Creatinine in μmol/L | 42.0 (39.0-44.5) | 48.0 (44.0-54.0) | 0.0463 |
| Uric acid in μmol/L | 294.0 (253.0-342.0) | 261.0 (236.5-333.0) | 0.4552 |
Continuous data are presented as median and interquartile range in squared parentheses and were tested using the Mann-Whitney U test. P < 0.05 was considered statically significant. The adjusted P value was calculated by the “p.adjust ()” command in R using the “BH” method to control for the false discovery rate. ICP: Intrahepatic cholestasis in pregnancy.
Serum metabolomes of ICP patients and healthy controls are diverse
The serum samples were analyzed by untargeted mass spectrometry, and the abundance profiles were obtained for 64 significantly different serum metabolites (Supplementary Table 1). The serum metabolomes of the ICP patients and healthy controls were clearly separated (Figure 1A). The ICP serum metabolome was characterized mainly by the enrichment of bile acid-related metabolites (e.g., primary and secondary bile acids).
Figure 1.
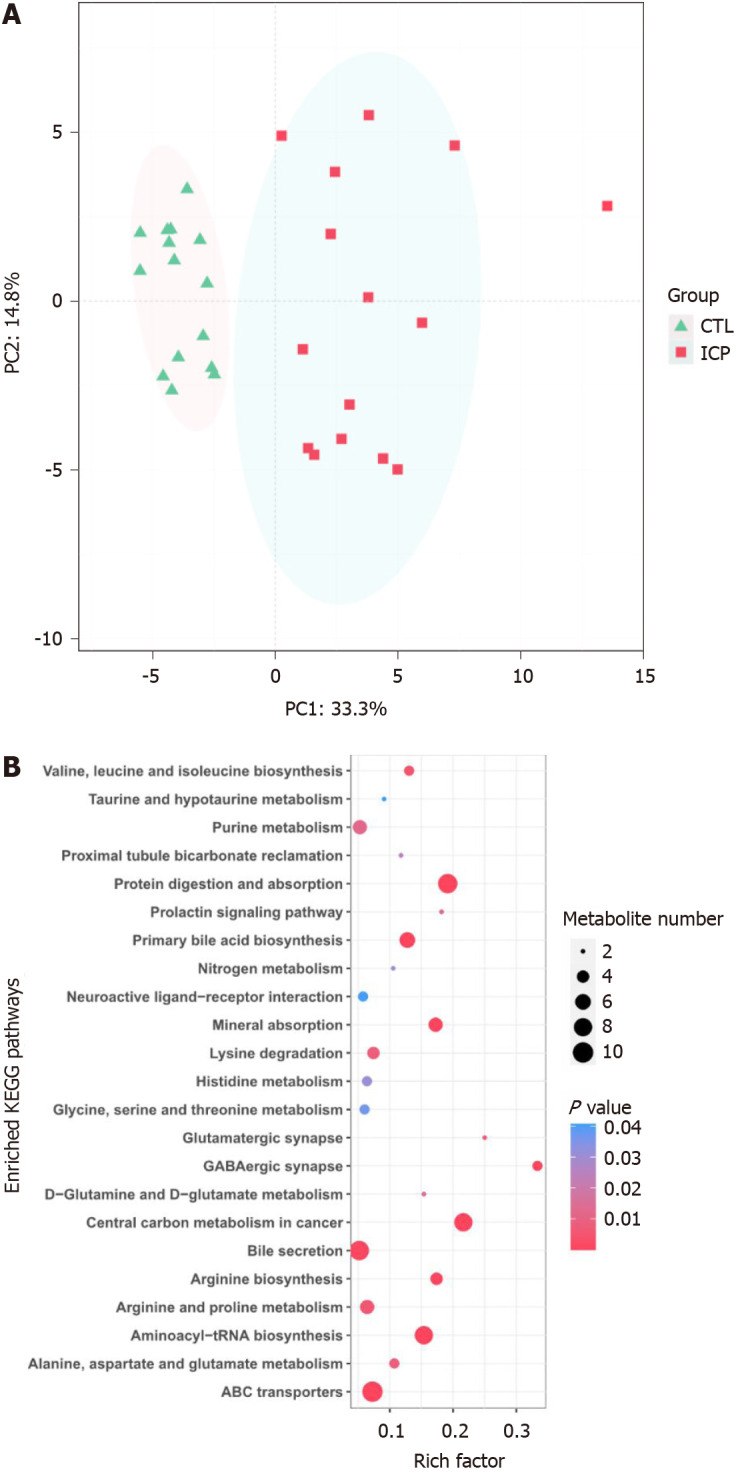
Distinct serum microbiome feature associated with intrahepatic cholestasis in pregnancy patients. A: Bray-Curtis principal analysis of metabolites shows significant differences between intrahepatic cholestasis in pregnancy and healthy controls; B: Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis of metabolites identified in this study. The metabolites identified were subjected to a pathway enrichment analysis using the Kyoto Encyclopedia of Genes and Genomes database. Only significantly enriched Kyoto Encyclopedia of Genes and Genomes functional categories (P < 0.05) are depicted according to their P-values. CTL: Control; ICP: Intrahepatic cholestasis in pregnancy; KEGG: Kyoto Encyclopedia of Genes and Genomes.
To identify pathways affected by ICP, differential metabolites with known KEGG IDs were used for enrichment analysis of the KEGG pathway. Consistently, the bile secretion, primary bile acid biosynthesis and taurine and hypotaurine metabolism pathways were enriched in the ICP group (Figure 1B). In addition, some pathways related to protein metabolism, such as glycine, serine and threonine metabolism, D-glutamine and D-glutamate metabolism, alanine, aspartate and glutamate metabolism, valine, leucine and isoleucine biosynthesis and arginine and proline metabolism, were enriched in the ICP patients.
Responses of the gut microbiota to intestinal changes in ICP patients
We next investigated whether ICP alters the composition of the gut microbial community. Microbiome analysis of fecal samples from 15 patients with ICP and 15 healthy controls was carried out using 16S rRNA amplification. The two groups did not report any striking differences in the identified operational taxonomy units (Supplementary Table 2). The microbial community richness indicated by the ACE and Chao1 estimators and the community diversity estimated by the Shannon index also showed no significant difference between the ICP and control groups (Supplementary Table 2).
We further performed principal coordination analysis (PCoA) between the groups to investigate potential differences. PCoA (Figure 2) ordination showed that the ICP group was distinctly separated from the HC group (anosim, P = 0.004). At the phylum level, we observed a higher relative abundance of Firmicutes and a markedly lower relative abundance of Bacteroidetes in the healthy group (Figure 3). For Proteobacteria and Actinobacteria, there were no significant differences between the two groups (Figure 3). Overall, the intestinal flora of pregnant women was mainly composed of Firmicutes, Bacteroidetes, Actinobacteria and Proteobacteria, and these phyla are present in at least 95% of these samples. Similarly, at the genus level, the core genera, including Bacteroides, Bifidobacterium, Blautia, Escherichia/Shigella and Faecalibacterium, were present in over 95% of the samples from gestational women (Supplementary Table 3).
Figure 2.
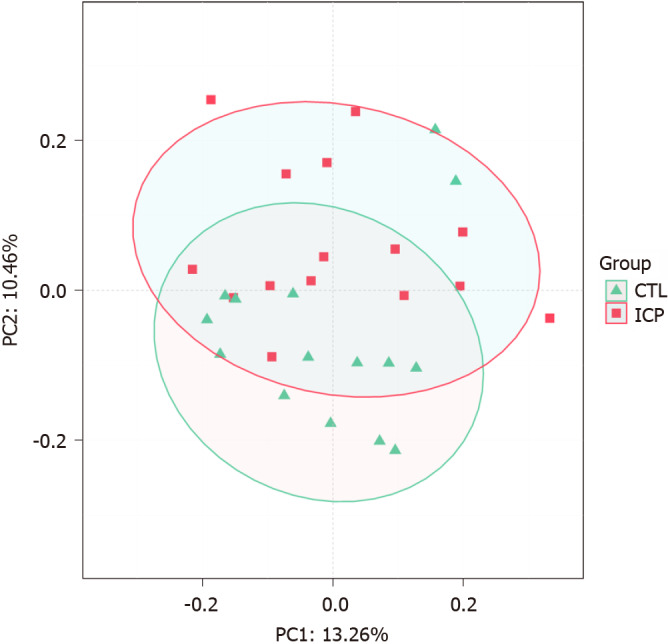
Distinct gut microbiota feature associated with intrahepatic cholestasis in pregnancy patients. Principal coordinate analysis score plot based on Bray-Curtis distance at the phylum level. CTL: Control; ICP: Intrahepatic cholestasis in pregnancy.
Figure 3.
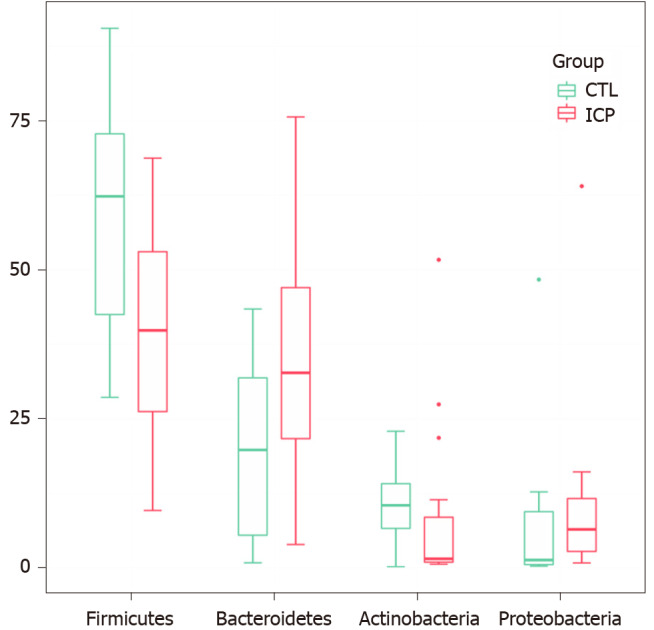
Changes of gut microbiota at the phylum level between intrahepatic cholestasis in pregnancy and healthy groups. Box plots show the relative abundance of the main phylum in microbiota. The center line denotes the median. Points outside the whiskers represent outlier samples. CTL: Control; ICP: Intrahepatic cholestasis in pregnancy.
To identify the main differences in genera between the HC and ICP groups, we used random forests for classification analysis and built classification models. The top 20 genera that had a major role in classifying the groups were identified by the random forest model (Figure 4). At the genus level, the ICP group exhibited a decrease in the relative abundance of Faecalibactium, Bifidobacterium and Blautia and an increase in the relative abundance of Parabacteroides, Bilophila, Bacteroides and Escherichia/Shigella.
Figure 4.

Genera important for differentiating intrahepatic cholestasis in pregnancy and healthy control were identified using random Forest package. A: The ranking of genera according to mean decrease gini were obtained from the random forest algorithm using default parameters. Genera with differences in abundance between the two cohorts are shown in red (intrahepatic cholestasis in pregnancy enriched) and green (control enriched); B: Comparison of the relative abundances of the main different genera of the gut microbiota in the two groups. Box plots show the abundance of the main difference genera in microbiota. The center lines the median. Points outside the whiskers represent outlier samples.
Correlations between the serum metabolome and gut microbiome
To further explore the links between the gut microbiota and serum metabolome, the Spearman correlation coefficient was computed for 64 differential metabolites and 50 bacterial taxa (Supplementary Figure 1). We further reduced the potential targets to major genera and bile acid-related metabolites, and it was found that bacteria such as Bacteroides, which are enriched in ICP patients, were positively correlated with increased bile acids, while bacteria such as Faecalibacterium, which are enriched in HC, were negatively correlated with increased bile acids (Figure 5). This finding suggests that the observed alterations in the gut microbiota of ICP patients are associated with serum metabolites.
Figure 5.
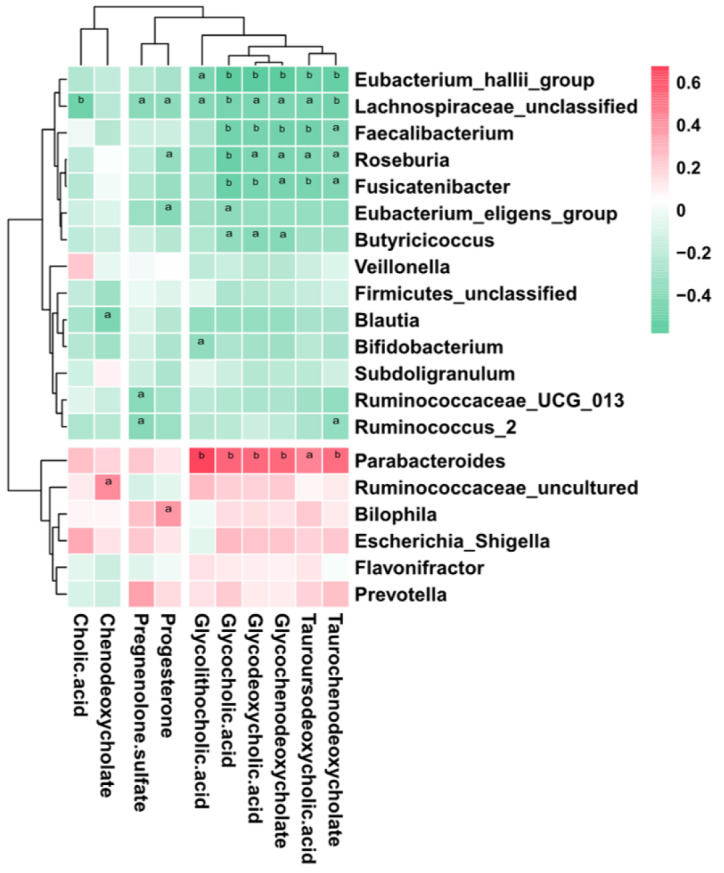
Association of key genera associated with disease with bile acid-related metabolites. Genera important for differentiating healthy controls/intrahepatic cholestasis in pregnancy status were identified using random Forest package. The significance level in the correlation test is denoted as: aP < 0.05; bP < 0.01.
DISCUSSION
In this study, the changes of serum metabolome and gut microbiota in ICP pregnant women and healthy pregnant women were analyzed. Clinically, serum TBA, alanine aminotransferase and aspartate aminotransferase were observably increased in the ICP patients (Table 1). The accumulation of bile acids in hepatocytes leads to hepatotoxicity and the release of aminotransferases, bilirubin and alkaline phosphatase into the serum[21]. Serum aminotransferase levels were elevated in 60%-85% of patients. By KEGG enrichment analysis of differential metabolites, we found that bile acid synthesis and secretion pathways were significantly upregulated in ICP patients; in addition, glucose metabolism and amino acid energy metabolism pathways also showed differences, indicating that patients experienced metabolic disorders. In terms of the gut microbiota, we observed that Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria were the dominant phyla, and Bacteroides, Bifidobacterium, Blautia, Escherichia/Shigella, and Faecalibacterium were the preponderant genera in both groups. The composition of these taxa is in accordance with previous research from Koren et al[22].
We observed that Faecalibacterium, Blautia and Eubacterium hallii, which are genera known to contain butyrate-producing bacteria, were depleted in ICP patients. Faecalibacterium prausnitzii is one of the most abundant bacterial species in the colons of healthy adults, and this species accounts for more than 5% of the total bacterial population[23,24]. F. prausnitzii is known for its immunomodulatory properties and is effective in improving intestinal inflammation and intestinal barrier function[25-28]. In addition, this species produces large amounts of butyrate[23], a short-chain fatty acid (SCFA) that is significant in gut physiology[26,29]. Butyrate has significant health benefits because it is a source of energy for colonocytes and promotes cell differentiation, inhibits colonic inflammatory responses, lowers luminal pH and improves tight junction assembly[30-33]. F. prausnitzii also produces several bioactive molecules that affect inflammation and intestinal barrier function, such as shikimic acid, salicylic acid and a microbial anti-inflammatory molecule[25,27].
Blautia species are also well-known as some of the butyrate-producing bacteria in the gut microbiota. Blautia has beneficial roles in glucose metabolism and obesity-associated inflammation[34,35] and are always observed at a lower relative abundance in type 2 diabetes patients than healthy controls[36,37]. Furthermore, recent reports indicated a correlation of Blautia abundance with altered glucose tolerance[38,39]. Although few studies have focused on the composition of the intestinal microbiome in patients with ICP, a study examining primary sclerosing cholangitis found an elevated relative abundance of Blautia when bile release into the small intestine was inhibited[40].
Interestingly, unlike other enteric bacterial isolates, such as Roseburia and Faecalibacterium, another butyrate-producing species, E. hallii, can produce butyrate from lactate and acetate at low pH levels[41]. Treatment with active E. hallii was discovered to improve fecal butyrate concentrations and to alter bile acid metabolism[35]. We also observed a decreased abundance of Bifidobacteria in the guts of ICP patients. Bifidobacteria is one of the major bacterial genera that make up the gastrointestinal tract microbiota in mammals and is also the most common bacteria in the infant gut microbiome[42]. It was reported that functional BSH exists in Bifidobacteria species[8]. Bile acid deconjugation implemented by bacteria with BSH activity can prevent the reuptake of bile acids from the small intestine. In fact, there is a close correlation between BSH and increased resistance to bile toxicity[8]. Our results suggested that alterations in the bile acid levels in ICP patients have an impact on the constituent of the gut microbiota. Our results suggest that aberrant bile acid levels have an effect on the intestinal microbiome, which may cause deterioration of the condition in ICP patients.
Our study found that Bilophila was enriched in the guts of ICP patients (Figure 4). In general, the abundance of B. wadsworthia is very low in healthy individuals but significantly elevated under pathological conditions. B. wadsworthia is a δ-Proteobacterium closely related to Desulfovibrio spp. Unlike sulfate-reducing bacteria using inorganic sulfate, B. wadsworthia can respire organic sulfonates, such as taurine[43,44]. B. wadsworthia has been isolated from clinical samples of patients with a variety of infectious diseases, including hidradenitis suppurativa, osteomyelitis, cholecystitis, vestibular mastitis and soft tissue abscess[45,46]. Devkota et al[47] found that the constituent of the gut microbiota significantly changed in response to a diet rich in saturated fat (milk extract), and an originally low abundance of B. wadsworthia was enriched. Diet promotes the secretion of tauro-conjugated bile acids thereby increasing the sulfur levels available to B. wadsworthia[47,48]. Alternatively, we also found that the relative abundance of Escherichia/Shigella is elevated in ICP patients, which may promote the colonization and growth of B. wadsworthia.
The enrichment of Parabacteroides is also consistent with previously reported results. Narushima et al[49] reported deoxycholic acid formation in gnotobiotic mice colonized with human fecal isolates, including Bacteroides uniformis, Bacteroides vulgatus, Parabacteroides distasonis, taurine-respiring B. wadsworthia and bile acid 7α-dehydroxylating Clostridium hylemonae and Clostridium hiranonis. Concomitantly, the KEGG analysis results revealed that the taurine and hypotaurine metabolism pathways were enriched in ICP patients (Figure 1). Furthermore, correlation analysis revealed a positive correlation between Parabacteroides and a variety of secondary bile acids (Figure 5). Parabacteroides has been reported to attenuate obesity and metabolic dysfunction by producing succinate and secondary bile acids[50]. This effect may be due to the increased bile acid content in the guts of ICP patients, which results in an elevated relative abundance of Parabacteroides. Collectively, these results suggest that dysregulated secretion of bile acids can affect the composition of the intestinal microbiome, which may further disrupt immune balance and trigger disease.
This study investigated the consequences of ICP with respect to the serum metabolome and gut microbiota for the first time. Our data suggest that ICP can cause significant changes in the serum metabolome and gut microbiota. ICP-induced changes in the relative abundance of bile acid-related compounds in the serum may have an effect on the gut microbiota, which in turn may have a detrimental effect on health. These findings suggest a mechanism of ICP that is associated with the microbiota via changes in serum metabolites. An obvious limitation of this study is that its sample size was small, which causes the analysis of the differences in the gut microbiota to lack statistical significance. Our future studies will collect more samples to examine the changes in the gut microbiota of ICP patients. We will also conduct intervention studies using prebiotics, probiotics and synbiotics to promote the establishment of beneficial microbiota and investigate whether it can have a positive impact on the health of ICP patients. In addition, based on the analysis of 16S rRNA sequences, the reduction in microbial diversity simply demonstrates an unbalanced intestinal ecosystem but does not provide us with more detailed information on the species and functions of certain microorganisms. Therefore, large-scale metagenomics and functional studies are needed to investigate the role of gut microbes in the molecular pathogenesis of ICP.
CONCLUSION
In summary, our study suggested that ICP patients had an altered serum metabolome and gut microbiota as evidenced by a decrease in SCFA-producing bacteria and an increase in bile acid metabolism-related bacteria. The serum metabolome was significantly correlated with the gut microbiota, indicating that the gut microbiota plays an important role in the occurrence and development of ICP. Although the mechanism by which ICP affects the serum metabolome and gut microbiota of patients remains unclear, our findings suggest that the intestinal microbiome can be used as a therapeutic target to provide new strategies for the diagnosis and treatment of ICP.
ARTICLE HIGHLIGHTS
Research background
Intrahepatic cholestasis in pregnancy (ICP) is the most common liver disease during pregnancy, and its cause and course of progression are still poorly understood.
Research motivation
Women with ICP have a significantly increased risk of preterm birth, stillbirth and admission to the neonatal unit for treatment. At present, there is no specific drug for the treatment of ICP in clinical practice.
Research objectives
We speculate that changes in serum metabolism caused by ICP may influence the intestinal tract circumstance and thus the intestinal microbiome. The gut microbiota may represent a mechanism by which ICP affects the health of pregnant women and fetuses. Our goal is to find the novel therapeutic strategies of ICP in the field of the gut microbiota.
Research methods
The serum nontarget metabolomes from each group were determined. Amplification of the 16S rRNA V3-V4 region was performed using fecal samples from the disease and healthy groups. By comparing the differences in the microbiota and metabolite compositions between the two groups, the relationship between the gut microbiota and serum metabolites was also investigated.
Research results
ICP patients have critical differences in the intestinal microflora composition (beta diversity) from that in healthy controls. At the genus level, most of the bacteria depleted in ICP are able to produce short-chain fatty acids (e.g., Faecalibacterium, Blautia and Eubacterium hallii), while the bacteria enriched in ICP are associated with bile acid metabolism (e.g., Parabacteroides and Bilophila). Our results also showed that specific genera were associated with the serum metabolome.
Research conclusions
The serum metabolome was significantly correlated with the gut microbiota, indicating that the gut microbiota plays an important role in the occurrence and development of ICP. Our findings suggest that the intestinal microbiome can be used as a therapeutic target to provide new strategies for the diagnosis and treatment of ICP.
Research perspectives
Our group will study gut microbiota and metabolome changes before and after drug treatment to provide a new regimen for ICP treatment. In the future we will also conduct intervention studies using prebiotics, probiotics and synbiotics to promote the establishment of beneficial microbiota and investigate whether it can have a positive impact on the health of ICP patients.
ACKNOWLEDGEMENTS
We would like to express our appreciation to the Department of Obstetrics, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine for help with patient recruitment and sample collection.
Footnotes
Institutional review board statement: This study was approved by the Ethics Committee of Shanghai First Maternity and Infant Health Hospital (KS2035).
Informed consent statement: All study participants or their legal guardian provided informed written consent about personal and medical data collection prior to study enrolment.
Conflict-of-interest statement: The authors have no conflicts of interest to declare.
STROBE statement: The manuscript has been prepared and revised according to the STROBE statement.
Manuscript source: Unsolicited manuscript
Peer-review started: September 9, 2020
First decision: September 29, 2020
Article in press: November 4, 2020
Specialty type: Obstetrics and gynecology
Country/Territory of origin: China
Peer-review report’s scientific quality classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Marasco G S-Editor: Zhang H L-Editor: Filipodia P-Editor: Liu JH
Contributor Information
Guo-Hua Li, Department of Reproductive Immunology, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai 200040, China.
Shi-Jia Huang, Department of Obstetrics, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai 200040, China.
Xiang Li, Department of Obstetrics, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai 200040, China.
Xiao-Song Liu, Department of Obstetrics, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai 200040, China.
Qiao-Ling Du, Department of Obstetrics, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai 200040, China. qldu2004@126.com.
Data sharing statement
The raw sequencing data have been submitted to the NCBI Sequence Read Archive under accession number PRJNA657645.
References
- 1.Wood AM, Livingston EG, Hughes BL, Kuller JA. Intrahepatic Cholestasis of Pregnancy: A Review of Diagnosis and Management. Obstet Gynecol Surv. 2018;73:103–109. doi: 10.1097/OGX.0000000000000524. [DOI] [PubMed] [Google Scholar]
- 2.Lee NM, Brady CW. Liver disease in pregnancy. World J Gastroenterol. 2009;15:897–906. doi: 10.3748/wjg.15.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geenes V, Chappell LC, Seed PT, Steer PJ, Knight M, Williamson C. Association of severe intrahepatic cholestasis of pregnancy with adverse pregnancy outcomes: a prospective population-based case-control study. Hepatology. 2014;59:1482–1491. doi: 10.1002/hep.26617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brouwers L, Koster MP, Page-Christiaens GC, Kemperman H, Boon J, Evers IM, Bogte A, Oudijk MA. Intrahepatic cholestasis of pregnancy: maternal and fetal outcomes associated with elevated bile acid levels. Am J Obstet Gynecol 2015; 212: 100.e1-100. :e7. doi: 10.1016/j.ajog.2014.07.026. [DOI] [PubMed] [Google Scholar]
- 5.Zecca E, De Luca D, Marras M, Caruso A, Bernardini T, Romagnoli C. Intrahepatic cholestasis of pregnancy and neonatal respiratory distress syndrome. Pediatrics . 2006;117:1669–1672. doi: 10.1542/peds.2005-1801. [DOI] [PubMed] [Google Scholar]
- 6.Wahlström A, Sayin SI, Marschall HU, Bäckhed F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016;24:41–50. doi: 10.1016/j.cmet.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 7.de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013;17:657–669. doi: 10.1016/j.cmet.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci USA. 2008;105:13580–13585. doi: 10.1073/pnas.0804437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jia W, Xie G, Jia W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Rev Gastroenterol Hepatol. 2018;15:111–128. doi: 10.1038/nrgastro.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall HU, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17:225–235. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Zheng X, Huang F, Zhao A, Lei S, Zhang Y, Xie G, Chen T, Qu C, Rajani C, Dong B, Li D, Jia W. Bile acid is a significant host factor shaping the gut microbiome of diet-induced obese mice. BMC Biol. 2017;15:120. doi: 10.1186/s12915-017-0462-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Tang R, Leung PSC, Gershwin ME, Ma X. Bile acids and intestinal microbiota in autoimmune cholestatic liver diseases. Autoimmun Rev. 2017;16:885–896. doi: 10.1016/j.autrev.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Pei LY, Ke YS, Zhao HH, Wang L, Jia C, Liu WZ, Fu QH, Shi MN, Cui J, Li SC. Role of colonic microbiota in the pathogenesis of ulcerative colitis. BMC Gastroenterol. 2019;19:10. doi: 10.1186/s12876-019-0930-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Magro DO, Santos A, Guadagnini D, de Godoy FM, Silva SHM, Lemos WJF, Vitulo N, Torriani S, Pinheiro LV, Martinez CAR, Saad MJA, Coy CSR. Remission in Crohn's disease is accompanied by alterations in the gut microbiota and mucins production. Sci Rep. 2019;9:13263. doi: 10.1038/s41598-019-49893-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xue AJ, Miao SJ, Sun H, Qiu XX, Wang SN, Wang L, Ye ZQ, Zheng CF, Huang ZH, Wang YH, Huang Y. Intestinal dysbiosis in pediatric Crohn's disease patients with IL10RA mutations. World J Gastroenterol. 2020;26:3098–3109. doi: 10.3748/wjg.v26.i22.3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Biase AR, Marasco G, Ravaioli F, Dajti E, Colecchia L, Righi B, D'Amico V, Festi D, Iughetti L, Colecchia A. Gut microbiota signatures and clinical manifestations in celiac disease children at onset: a pilot study. J Gastroenterol Hepatol. 2020 doi: 10.1111/jgh.15183. [DOI] [PubMed] [Google Scholar]
- 17.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oksanen J, Blanchet F, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin P, O’Hara R, Simpson G, Solymos P. Vegan: Community Ecology Package. R package 2019; version 2. :5–6. [Google Scholar]
- 19.Gómez-Rubio V. ggplot2 - Elegant Graphics for Data Analysis (2nd Edition). J Statist Software 2017; 77. [Google Scholar]
- 20.Liaw A, Wiener M. Classification and regression by randomForest. R news . 2002;2:18–22. [Google Scholar]
- 21.Smith DD, Rood KM. Intrahepatic Cholestasis of Pregnancy. Clin Obstet Gynecol . 2020;63:134–151. doi: 10.1097/GRF.0000000000000495. [DOI] [PubMed] [Google Scholar]
- 22.Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Bäckhed HK, Gonzalez A, Werner JJ, Angenent LT, Knight R, Bäckhed F, Isolauri E, Salminen S, Ley RE. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell . 2012;150:470–480. doi: 10.1016/j.cell.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miquel S, Martín R, Bridonneau C, Robert V, Sokol H, Bermúdez-Humarán LG, Thomas M, Langella P. Ecology and metabolism of the beneficial intestinal commensal bacterium Faecalibacterium prausnitzii. Gut Microbes. 2014;5:146–151. doi: 10.4161/gmic.27651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miquel S, Martín R, Rossi O, Bermúdez-Humarán LG, Chatel JM, Sokol H, Thomas M, Wells JM, Langella P. Faecalibacterium prausnitzii and human intestinal health. Curr Opin Microbiol. 2013;16:255–261. doi: 10.1016/j.mib.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 25.Quévrain E, Maubert MA, Michon C, Chain F, Marquant R, Tailhades J, Miquel S, Carlier L, Bermúdez-Humarán LG, Pigneur B, Lequin O, Kharrat P, Thomas G, Rainteau D, Aubry C, Breyner N, Afonso C, Lavielle S, Grill JP, Chassaing G, Chatel JM, Trugnan G, Xavier R, Langella P, Sokol H, Seksik P. Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn's disease. Gut. 2016;65:415–425. doi: 10.1136/gutjnl-2014-307649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miquel S, Leclerc M, Martin R, Chain F, Lenoir M, Raguideau S, Hudault S, Bridonneau C, Northen T, Bowen B, Bermúdez-Humarán LG, Sokol H, Thomas M, Langella P. Identification of metabolic signatures linked to anti-inflammatory effects of Faecalibacterium prausnitzii. mBio. 2015;6:e00300–15. doi: 10.1128/mBio.00300-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martín R, Miquel S, Chain F, Natividad JM, Jury J, Lu J, Sokol H, Theodorou V, Bercik P, Verdu EF, Langella P, Bermúdez-Humarán LG. Faecalibacterium prausnitzii prevents physiological damages in a chronic low-grade inflammation murine model. BMC Microbiol. 2015;15:67. doi: 10.1186/s12866-015-0400-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, Grangette C, Vasquez N, Pochart P, Trugnan G, Thomas G, Blottière HM, Doré J, Marteau P, Seksik P, Langella P. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leonel AJ, Alvarez-Leite JI. Butyrate: implications for intestinal function. Curr Opin Clin Nutr Metab Care. 2012;15:474–479. doi: 10.1097/MCO.0b013e32835665fa. [DOI] [PubMed] [Google Scholar]
- 30.Eeckhaut V, Machiels K, Perrier C, Romero C, Maes S, Flahou B, Steppe M, Haesebrouck F, Sas B, Ducatelle R, Vermeire S, Van Immerseel F. Butyricicoccus pullicaecorum in inflammatory bowel disease. Gut. 2013;62:1745–1752. doi: 10.1136/gutjnl-2012-303611. [DOI] [PubMed] [Google Scholar]
- 31.Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. 2016;7:189–200. doi: 10.1080/19490976.2015.1134082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russell WR, Hoyles L, Flint HJ, Dumas ME. Colonic bacterial metabolites and human health. Curr Opin Microbiol. 2013;16:246–254. doi: 10.1016/j.mib.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 33.Terpstra ML, Sinnige MJ, Hugenholtz F, Peters-Sengers H, Remmerswaal EB, Geerlings SE, Bemelman FJ. Butyrate production in patients with end-stage renal disease. Int J Nephrol Renovasc Dis. 2019;12:87–101. doi: 10.2147/IJNRD.S200297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Q, Xiao X, Li M, Yu M, Ping F, Zheng J, Wang T, Wang X. Vildagliptin increases butyrate-producing bacteria in the gut of diabetic rats. PLoS One. 2017;12:e0184735. doi: 10.1371/journal.pone.0184735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Udayappan S, Manneras-Holm L, Chaplin-Scott A, Belzer C, Herrema H, Dallinga-Thie GM, Duncan SH, Stroes ESG, Groen AK, Flint HJ, Backhed F, de Vos WM, Nieuwdorp M. Oral treatment with Eubacterium hallii improves insulin sensitivity in db/db mice. NPJ Biofilms Microbiomes. 2016;2:16009. doi: 10.1038/npjbiofilms.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inoue R, Ohue-Kitano R, Tsukahara T, Tanaka M, Masuda S, Inoue T, Yamakage H, Kusakabe T, Hasegawa K, Shimatsu A, Satoh-Asahara N. Prediction of functional profiles of gut microbiota from 16S rRNA metagenomic data provides a more robust evaluation of gut dysbiosis occurring in Japanese type 2 diabetic patients. J Clin Biochem Nutr. 2017;61:217–221. doi: 10.3164/jcbn.17-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park SK, Kim MS, Bae JW. Blautia faecis sp. nov., isolated from human faeces. Int J Syst Evol Microbiol. 2013;63:599–603. doi: 10.1099/ijs.0.036541-0. [DOI] [PubMed] [Google Scholar]
- 38.Zhou W, Sailani MR, Contrepois K, Zhou Y, Ahadi S, Leopold SR, Zhang MJ, Rao V, Avina M, Mishra T, Johnson J, Lee-McMullen B, Chen S, Metwally AA, Tran TDB, Nguyen H, Zhou X, Albright B, Hong BY, Petersen L, Bautista E, Hanson B, Chen L, Spakowicz D, Bahmani A, Salins D, Leopold B, Ashland M, Dagan-Rosenfeld O, Rego S, Limcaoco P, Colbert E, Allister C, Perelman D, Craig C, Wei E, Chaib H, Hornburg D, Dunn J, Liang L, Rose SMS, Kukurba K, Piening B, Rost H, Tse D, McLaughlin T, Sodergren E, Weinstock GM, Snyder M. Longitudinal multi-omics of host-microbe dynamics in prediabetes. Nature. 2019;569:663–671. doi: 10.1038/s41586-019-1236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Egshatyan L, Kashtanova D, Popenko A, Tkacheva O, Tyakht A, Alexeev D, Karamnova N, Kostryukova E, Babenko V, Vakhitova M, Boytsov S. Gut microbiota and diet in patients with different glucose tolerance. Endocr Connect. 2016;5:1–9. doi: 10.1530/EC-15-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Torres J, Bao X, Goel A, Colombel JF, Pekow J, Jabri B, Williams KM, Castillo A, Odin JA, Meckel K, Fasihuddin F, Peter I, Itzkowitz S, Hu J. The features of mucosa-associated microbiota in primary sclerosing cholangitis. Aliment Pharmacol Ther. 2016;43:790–801. doi: 10.1111/apt.13552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duncan SH, Louis P, Flint HJ. Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl Environ Microbiol. 2004;70:5810–5817. doi: 10.1128/AEM.70.10.5810-5817.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turroni F, Peano C, Pass DA, Foroni E, Severgnini M, Claesson MJ, Kerr C, Hourihane J, Murray D, Fuligni F, Gueimonde M, Margolles A, De Bellis G, O'Toole PW, van Sinderen D, Marchesi JR, Ventura M. Diversity of bifidobacteria within the infant gut microbiota. PLoS One. 2012;7:e36957. doi: 10.1371/journal.pone.0036957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peck SC, Denger K, Burrichter A, Irwin SM, Balskus EP, Schleheck D. A glycyl radical enzyme enables hydrogen sulfide production by the human intestinal bacterium Bilophila wadsworthia. Proc Natl Acad Sci USA. 2019;116:3171–3176. doi: 10.1073/pnas.1815661116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laue H, Denger K, Cook AM. Taurine reduction in anaerobic respiration of Bilophila wadsworthia RZATAU. Appl Environ Microbiol. 1997;63:2016–2021. doi: 10.1128/aem.63.5.2016-2021.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng Z, Long W, Hao B, Ding D, Ma X, Zhao L, Pang X. A human stool-derived Bilophila wadsworthia strain caused systemic inflammation in specific-pathogen-free mice. Gut Pathog. 2017;9:59. doi: 10.1186/s13099-017-0208-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baron EJ. Bilophila wadsworthia: a unique Gram-negative anaerobic rod. Anaerobe . 1997;3:83–86. doi: 10.1006/anae.1997.0075. [DOI] [PubMed] [Google Scholar]
- 47.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature. 2012;487:104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turnbaugh PJ. Microbiology: fat, bile and gut microbes. Nature. 2012;487:47–48. doi: 10.1038/487047a. [DOI] [PubMed] [Google Scholar]
- 49.Narushima S, Itoha K, Miyamoto Y, Park SH, Nagata K, Kuruma K, Uchida K. Deoxycholic acid formation in gnotobiotic mice associated with human intestinal bacteria. Lipids. 2006;41:835–843. doi: 10.1007/s11745-006-5038-1. [DOI] [PubMed] [Google Scholar]
- 50.Wang K, Liao M, Zhou N, Bao L, Ma K, Zheng Z, Wang Y, Liu C, Wang W, Wang J, Liu SJ, Liu H. Parabacteroides distasonis Alleviates Obesity and Metabolic Dysfunctions via Production of Succinate and Secondary Bile Acids. Cell Rep 2019; 26: 222-235. :e5. doi: 10.1016/j.celrep.2018.12.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw sequencing data have been submitted to the NCBI Sequence Read Archive under accession number PRJNA657645.