

Abstract
Pathogenic mechanisms involved in the autoimmune disorder systemic lupus erythematosus (SLE) remain elusive. However, dysbiosis of the gut microbiota may be involved in disease pathogenesis. We demonstrate that the dysbiotic gut microbiota of lupus-prone mice induced autoimmunity when transferred into non-autoimmune germfree wildtype mice. An increase in kynurenine, a metabolite of tryptophan metabolism, was observed in lupus-prone mice, which decreased after antibiotic treatment. Low dietary tryptophan prevented autoimmune pathology, whereas high dietary tryptophan exacerbated disease. Further, a reduction in dietary tryptophan resulted in changes in gut microbial taxa in lupus-prone mice and the congenic normal control mice. The interplay of gut microbial dysbiosis, tryptophan metabolism and host genetic susceptibility suggested that aberrant tryptophan metabolism in lupus-susceptible mice could be one of the mechanisms contributing to autoimmune activation in this disease.
One Sentence Summary:
Gut microbiota dysbiosis induces lupus-like autoimmunity in mice through altered tryptophan metabolism
Accessible Summary:
Interplay of genes, microbiome, and tryptophan controls autoimmunity
Alterations in gut microbiota may contribute to autoimmunity through bacterial-mediated immune dysregulations. Tryptophan, an essential amino acid, is critically involved in kynurenine biosynthesis. High kynurenine is observed in patients with lupus and lupus-prone mice. Choi et al. how that gut bacterial dysbiosis found in lupus-prone mice dysregulates tryptophan metabolism, which, in turn, exacerbates the disease progression. They also demonstrate that autoimmunity can be mitigated by reducing dietary tryptophan. Thus, they propose a model in which genetic susceptibility induces autoimmunity, which leads to gut bacterial dysbiosis and an increased tryptophan metabolism that enhances lupus pathogenesis in the host.
INTRODUCTION
Dysbiosis of the gut microbiota may contribute to autoimmune disease (1), including systemic lupus erythematosus (SLE) in human (2–4) and a lupus-like autoimmune disease in mice (3, 5–10). The derivation of gnotobiotic lupus-prone mice demonstrates that gut bacteria are not necessary for lupus development (11). However, microbial from the gut to peripheral organs induces autoimmune phenotypes in the (NZW x BSXB)F1 and B6.TLR7 transgenic mouse models of lupus (7, 9). Further, cross-reactivity with commensal orthologues of the human lupus-associated autoantigen Ro60 contributes to autoimmunity and the induction of anti-RNA autoantibodies (12). We have used a congenic lupus-prone mouse model, B6.Sle1.Sle2.Sle3 (dubbed TC for triple congenic mice) sharing over 95% genome identity with control C57BL/6 (B6) mice (13). In this model, phenotypic differences, including differences in the gut microbiota and metabolites, between the TC and control B6 mice are caused by the 5% of the genome responsible for lupus susceptibility (14). The autoantibodies induced in TC mice are dominated by DNA reactivity, which may arise from distinct mechanisms of autoimmune activation instead of autoantibodies directed against RNA.
We have demonstrated differences in gut microbial dysbiosis in TC mice, contributing to autoimmune activation when feces were transferred to germfree (GF) mice or to specific pathogen-free (SPF) B6 mice. Further, TC mice showed no evidence of altered intestinal permeability resulting in bacterial translocation, which has been implicated in lupus pathogenesis. Here, we investigated whether changing the availability of bacterial-associated metabolites could mitigate autoimmune activation. Specifically, our focus centered on tryptophan and its metabolites, which differ in the feces of TC mice compared to B6 mice. Tryptophan is an essential amino acid used for the biosynthesis of key compounds, including 5-hydroxytryptamine, also called serotonin,) and kynurenine. The gut microbiota is critically involved in dietary tryptophan metabolism (15), which has several biological pathways that can be modified (16, 17). Relevant to lupus, bacterial-associated tryptophan metabolism orchestrates immunoregulatory effects (18–20) via the generation of aryl hydrocarbon receptor (AhR) ligands (19), or short-chain fatty acids (20). High kynurenine and low serotonin have been found in the sera (21, 22) and blood cells of individuals with lupus (23). Furthermore, tryptophan metabolites were predictive of the response of a lupus patient cohort to therapy (23). It has been proposed that increased expression of indoleamine-pyrrole 2,3-dioxygenase 1 (IDO1) produces kynurenine as a consequence of high interferon (IFN) activity observed in individuals with lupus (24). Alternatively, high oxidation found in peripheral blood cells of lupus patients may promote kynurenine production and activate the mechanistic target of rapamycin (mTOR), a master regulator of the elevated cellular metabolism observed in lupus (23). In this study, we investigate whether gut dysbiosis contributes to aberrant tryptophan metabolism in lupus-prone mice, and whether the interplay between gut microbes, tryptophan metabolism and genetic susceptibility could potentially drive systemic autoimmunity.
RESULTS
Translocation of the gut microbiota does not initiate autoimmunity in TC mice
Although dysfunctional intestinal barrier integrity has been documented in three lupus mouse models (6, 7, 9) and in some individuals with SLE (4, 7), intestinal inflammation has not been thoroughly examined. Here, we demonstrate that the colons of aged TC mice were longer than the age-matched B6 control mice and showed sub-clinical inflammation (P < 0.001), as well as increased numbers of CD45+ sub-epithelial infiltrating immune cells (Fig. 1A–B). Further, some TC mice were positive for fecal blood, indicating severe gut inflammation, whereas all B6 mice were negative for fecal blood (Fig. 1C). Intestinal barrier integrity was investigated by FITC-dextran gavage of mice, and by measuring serum endotoxin and the expression of tight junction genes (Fig. 1D–F). Results did not indicate disruption of the intestinal barrier in the late stage of the lupus-like disease in TC mice. Moreover, gut bacterial translocation to the liver or mesenteric lymph nodes was as found in less than half of aged TC mice and less than 10% of young TC mice (Fig. 1G and H). The frequency and amount of bacterial translocation were higher in aged TC mice compared to B6 mice (P < 0.05). However, it was not correlated with either the frequency of follicular helper T (Tfh) cells (Fig. 1I), or serum anti-dsDNA IgG (fig. S1A), two key characteristics of human SLE. Furthermore, no anaerobic bacterial growth was detected in the livers of TC mice, which was documented in (NZW x BSXB)F1 and B6.TLR7 transgenic mice (7, 9). Sequence analysis demonstrated that all TC gut microbial cultures contained Staphylococcus xylosus or other related Staphylococcus species (fig. S1B and Fig. 1J). S. xylosus was found in B6 but not in TC mouse feces (Fig. 1K), potentially suggesting a different intestinal location of this bacterial species between the two mouse strains.
Fig. 1. TC mice show gut inflammation but limited bacterial translocation from the gut to the periphery.

(A) Shown are representative whole colon sections (top panels, scale bars, 7 mm) and proximal colon sections () stained with H&E (middle panels, scale bars 300 μm) and with anti-CD45 antibody (bottom panels, scale bars 300 μm) from) from B6 and TC mice. (B) Colon pathology scores, ranked from 1 for the mice with the lowest inflammation to 29 for the mice the highest inflammation, with medians are shown. (C) Shown is the frequency of fecal blood in B6 and TC feces (sample size is indicated in center). (D) Shown is serum FITC-dextran in B6 and TC mice after gavage of animals with FITC-dextran to detect gut barrier integrity. (E) Shown are serum endotoxin concentrations in TC and B6 mice. (F) Expression of tight junction genes (Cldn2, Zo-1, Ocln, Jam) in the duodenum and colon of TC and B6 mice are shown. (G) The frequency of TC and B6 mouse liver and mesenteric lymph node (MLN) cultures that were positive for bacteria are presented. (H) CFU counts in liver and mesenteric lymph node cultures from B6 and TC mice, either at 2-5 months (young, Y) or 6-8 (older, O) months of age are shown. The sample size for each column is indicated. (I) Shown is a Pearson’s correlation between the frequency of splenic Tfh cells and the sum of liver and mesenteric lymph node CFUs (Pearson’s correlation R2 = 0.02, p = 0.58). (J) CFU numbers in TC liver and mesenteric lymph node cultures that were negative or positive for Staphylococcus xylosus (Sx − or Sx +, respectively) are shown. (K) S. xylosus operational taxonomic unit (OTU) counts in 16S rDNA sequences in B6 and TC mouse feces. (L) Bacterial 16S rDNA sequences in B6 and TC mouse feces are depicted. On the left, the taxonomic cladogram shows the phylogenetic distribution of differentially enriched taxa between the two mouse strains. Fecal mouse B6-enriched taxa are shown in blue and fecal mouse TC-enriched taxa are shown in red. On the right, a bubble plot of linear discriminant analysis (LDA) scores reveals the most differentially abundant taxa between the two mouse strains. Only taxa meeting the criteria (LDA score > 2 and p < 0.05) are shown. Fecal B6-enriched taxa are represented with positive LDA scores and fecal TC-enriched taxa with negative LDA scores. Data analyzed were from 2 independent experiments. Mice were 6 - 12-months old, except in (H). Statistical analysis for panels B-K: Each symbol represents a mouse; bars show means and standard error of the mean (SEM). Statistical tests used were Mann-Whitney tests in panels B,D,E and J, Fisher’s exact test in panels C and G, and t test in panel F. * P < 0.05, ** P < 0.01, *** P < 0.001.
Aged TC mice exhibit gut microbial dysbiosis and lupus-associated phenotypes
16S rDNA sequencing demonstrated different microbial communities in the feces of TC and B6 mice (fig. S2A). Whereas there were no changes in the relative bacterial abundance (fig. S2B–D) and alpha diversity indices (fig. S2D), marked alterations were observed in several taxa, including Prevotellaceae, Paraprevotella and Lactobacillus (Fig. 1L), mirroring the changes reported in B6.TLR7 transgenic and imiquimod-treated B6 mice (9).
To determine whether differences in the gut microbiota contribute to autoimmune pathogenesis, TC mice were treated with antibiotics, which reduced lymphoid tissue expansion (fig. S3A) and delayed the onset of anti-dsDNA IgG production (fig. S3B). Whereas total IgG was not affected, antibiotic treatment decreased serum anti-dsDNA IgG relative to total IgG (fig. S3C–D). Antibiotic treatment also reduced proinflammatory CD4+ T cells and the frequency of effector memory CD4+ T cells, Tfh cells (fig. S3E–F), and CD4+ T cells producing IFNγ, IL-17A, or IL-10, but increased the follicular regulatory T cell (Tfr) to Tfh cell ratio (fig. S3G–J). Finally, antibiotic treatment increased the frequency of Foxp3+ Treg cells and diminished their effector to naïve T cell ratio (fig. S3K–L).
The pathogenic potential of TC gut microbial dysbiosis was directly assessed by fecal transfer from TC mice into GF B6 mice. TC fecal transfer resulted in elevated serum anti-dsDNA IgG and IgM, as well as total IgG, in transplanted GF B6 mice (Fig. 2A–C). TC fecal transfer also induced anti-nuclear autoantibodies with the prototypical homogenous nuclear staining pattern (Fig. 2D). Next, we investigated whether TC fecal transfer increased IgA, given that higher amounts of fecal IgA have been found in patients with SLE (4). TC mice produced higher serum and fecal IgA compared to B6 control mice, and TC fecal transfer induced more IgA in GF B6 recipient mice than did B6 fecal transfer (P < 0.05, Fig. 2E). TC fecal transfer also promoted the formation of more lymphoid nodules harboring B and T cells in the colon compared to B6 fecal transfer (P < 0.05, Fig. 2F). Finally, TC fecal transfer elicited immune activation in the mesenteric lymph node of GF B6 recipient mice, with higher numbers of germinal center B cells, plasma cells and Tfh cells (Fig. 2G–I). The frequency of Tfh cells was also increased with TC fecal transfer and, conversely, TC fecal transfer reduced the Tfr to Tfh ratio as compared to B6 fecal transfer (P < 0.05, Fig. 2I). Notably, Tfh cell expansion is a characteristic of lupus (28), wherein the frequency of circulating Tfh cells (26) and the Tfh/Tfr ratio (30) positively correlate with disease activity.
Fig. 2. Fecal microbiota contributes to autoimmune activation in TC mice.
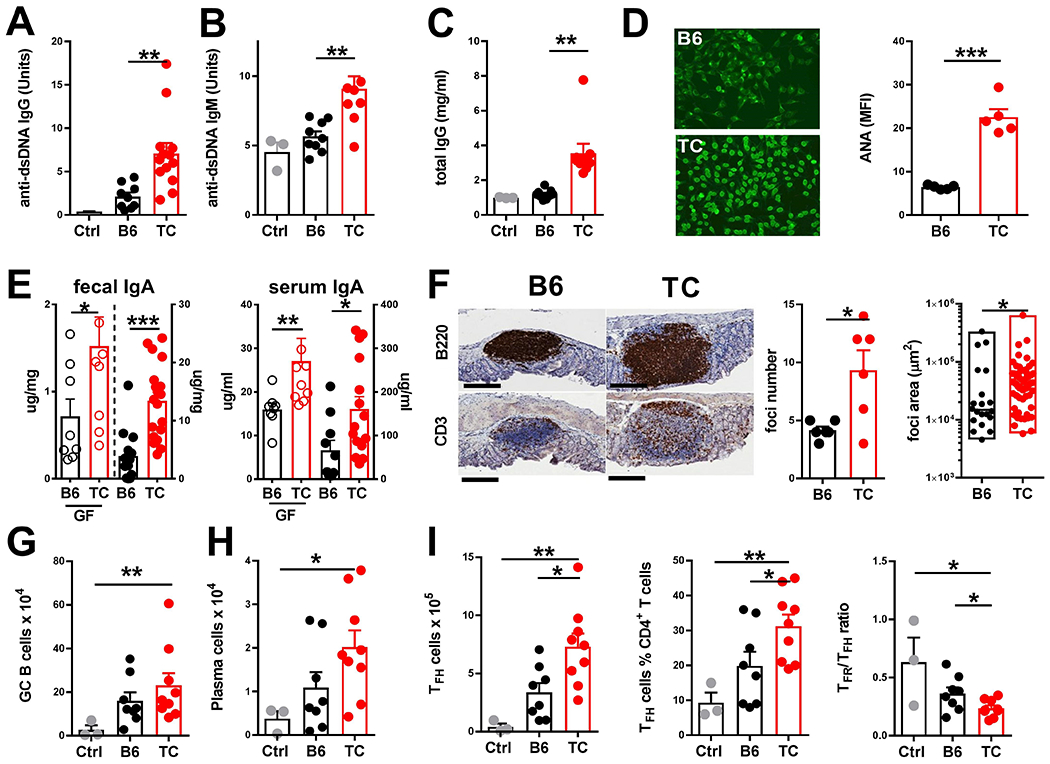
GF B6 mice were gavaged with feces from anti-dsDNA IgG-positive TC mice, age-matched B6 mice, or were gavaged with PBS (Ctrl). Immune phenotypes were evaluated 4 weeks later. Serum anti-dsDNA IgG (A) and IgM (B), total IgG (C) and anti-nuclear autoantibodies (ANA) were quantified by anti-IgG by FITC staining (mean fluorescence intensity, MFI) with representative images (20X magnification) of cells stained with B6 or TC sera shown on the left (D). (E) Fecal and serum IgA was measured in GF mice gavaged with feces from B6 or TC mice (left axes) or in the B6 and TC mice used as fecal donors (right axes). (F) Representative B220 and CD3 staining in sections of colonic lymphoid tissue from B6 and TC mice are shown (scale bars, 200 μm). Number of lymphoid foci per colon is shown for6 mice for the B6 and TCTC strain from two separate cohorts; surface area of individual foci (min-max boxes with median) is presented (n = 20 B6, n=57 TC). Numbers of germinal center B cells (G) and plasma cells (H) are shown in mesenteric lymph nodes of B6 and TC mice. (I) Numbers and frequencies of mesenteric lymph node Tfh cells, as well as ratio of Tfr / Tfh cells is shown for B6 and TC mice. Data are presented as means and SEM of data pooled from three cohorts undergoing fecal transfer, with each symbol representing one mouse, except in (F). Comparisons were done using t tests, except for panel F where a Mann-Whitney test was used. * P < 0.05, ** P < 0.01, *** P < 0.001.
We further investigated whether gut microbial dysbiosis emerged in TC mice at an early age. Gut microbiota composition differed in TC and B6 mice at 1 month of age, but these differences were reduced in mice at 2 months of age (fig. S4A). Linear discriminant analysis of the gut microbiota of young TC and B6 mice showed little overlap with the bacterial taxa seen in the gut microbiota of adult mice (Fig. 1L). Further, pair-wise analyses suggested that the abundance of Bifidobacterium and Allobaculum increased in B6 and TC mice at 1-2 months of age (fig. S4C), and the relative abundance of Bifidobacteria was lower in TC compared to B6 mice (fig. S4B, D). Importantly, there were no differences between strains at 1-2 months of age in the genera of Prevotella/Paraprevotella, or Lactobacillus (fig. S4D), suggesting that these taxa may differentially populate the gut of TC mice after they develop autoimmunity. To determine whether the proinflammatory phenotype of TC gut microbiota existed prior to disease development, we colonized GF B6 recipient mice with feces isolated from 1-month-old B6 or TC mice. Young TC fecal transfer did not induce anti-dsDNA antibodies nor increase total IgG, IgM, or IgA (fig. S5A–B), or result in germinal center activation in the mesenteric lymph nodes (fig. S5C).
To test whether B and T cells contributed to the induction of a proinflammatory gut microbiota, GF TC and B6 mice were colonized with feces of TC.Rag1−/− or B6.Rag1−/− mice, or with control feces from TC or B6 mice. Fecal transfer from TC.Rag1−/− mice induced intermediate amounts of anti-dsDNA IgG and lower amounts of anti-dsDNA IgM, total IgG and IgA, as well as mesenteric lymph node activation, compared to TC fecal transfer (fig. S5D–G). However, TC.Rag1−/− and TC fecal transfer supported a similar expansion of Tfh relative to Tfr cells (fig. S5G). Moreover, transferring TC.Rag1−/− feces resulted in higher anti-dsDNA IgM and total IgA compared to B6.Rag1−/− fecal transfer (fig. S5D–E). Collectively, these data demonstrate that TC fecal transfer induced autoantibodies and local inflammation in the gut-associated lymphoid tissue that skewed CD4+ T cells toward Tfh cells. These autoimmune phenotypes were only fully induced by fecal transfer from mice with ongoing autoimmune manifestations.
We next assessed the ability of lateral gut microbiota transmission to mitigate autoimmunity by co-housing of B6 and TC mice. After 6 months of co-housing TC mice with B6 mice, TC mice produced less anti-dsDNA IgG and anti-nuclear autoantibodies than did control non co-housed TC mice, and cytoplasmic anti-nuclear autoantibody staining was increased in co-housed TC mice compared to the typical nuclear pattern seen in control non co-housed TC mice (P < 0.05, Fig. 3A and B). However, sharing microbiota between TC and B6 mice was not sufficient to augment production of autoantibodies in co-housed B6 mice (P > 0.05, Fig. 3A). Serum total IgA showed intermediate abundance in co-housed B6 mice compared to control B6 mice that were not co-housed (P < 0.05, Fig. 3C). There was no difference in glomerulonephritis severity between co-housed and control TC mice (Fig. 3D). However, co-housed B6 mice exhibited an increased glomerular cellularity and mesangial expansion (fig. S6), leading to higher pathology scores compared to control non co-housed B6 mice (Fig. 3D). Colonic pathology was less severe in co-housed than in control TC mice (P < 0.05), and more severe in co-housed than control B6 mice (P < 0.01, Fig. 3E and fig. S6). Additionally, renal and colonic pathology were positively correlated across the four groups (Fig. 3F), suggesting a link between the immune activation in these two organs and the dysbiotic gut microbiota. Sharing the gut microbiota between TC and B6 mice also affected immunophenotypes. The large number of splenic plasma cells of TC mice (28) was lower in co-housed compared to control TC mice (P < 0.05, Fig. 3G). Moreover, the variance in plasma cell numbers in co-housed B6 mice was higher than in control B6 mice, suggesting a variable response to the TC gut microbiota. Additionally, exposure to TC gut microbiota increased the CD69+ CD4+ T cell and T effector cell frequencies in co-housed B6 mice (P < 0.05, Fig. 3H–I). Finally, sharing B6 gut microbiota expanded the frequency of Tfr relative to Tfh cells in co-housed TC mice, reduced in B6 mice sharing TC mouse gut microbiota relative to control B6 mice (Fig. 3J).
Fig. 3. Horizontal transmission reveals interactions between lupus susceptibility genes and the gut microbiota of TC mice.

B6 and TC mice were housed from weaning either with littermates (shown as B6 and TC on the graphs) or with age-matched mice from the other strain (cohoused, shown as CoH on the graphs). Immune phenotypes were analyzed in 7-month-old B6 or TC mice. (A) Shown is the concentration in arbitrary units of serums anti-dsDNA IgG in control or cohoused B6 and TC mice. (B) Frequency of nuclear localization of anti-nuclear autoantibody staining in the sera of cohoused and control TC mice is shown; the sample size is shown in the middle. Representative images (20X magnification) of nuclear and cytoplasmic staining by a control TC and a cohouse TC serum are on the right. Shown are renal (D) and colon (E) pathology ranks, as well as correlation between the two (F), for control and cohoused B6 and TC mice. (G) Numbers of splenic plasma cells for control and cohoused B6 and TC mice are shown. Frequencies of splenic CD69+CD4+ T cells (H), and CD62L−CD44+ Tem cells (I) are shown for control and cohoused B6 and TC mice. (J) Splenic Tfr/Tfh ratio is shown for control and cohoused B6 and TC mice. Data are means and SEM pooled from three cohorts, with each symbol representing one mouse (D-F). For simplicity, comparisons between control B6 and TC mouse results are not shown (all p < 0.05). Comparisons between cohoused B6 and TC mouse groups were done with t tests (* P < 0.05, ** P < 0.01, *** P < 0.001), Fisher exact test (B), Pearson r correlation test (F); and analysis of variance, vvv P < 0.001 (G). (K) Heatmap of differentially expressed fecal metabolites between cohoused B6 and TC mice was analyzed comparing strains regardless of housing conditions. Each column represents one mouse. (L) Enriched KEGG pathways in fecal metabolites between TC and B6 mice are shown with the p values and the number of metabolites represented in each pathway. The size of each bubble represents the number of metabolites differentially expressed for each pathway with a scale on the lower right (set size).
Metabolites from TC mice indicate an altered tryptophan metabolism
To elucidate the mechanisms through which the TC mouse gut microbiota could activate autoimmunity, we performed an unbiased metabolic screen of the feces of separately housed, or co-housed B6 and TC mice. A clear separation was found between B6 and TC mouse fecal metabolites, with co-housed B6 and TC mice falling in between these two groups (Fig. 3K and fig. S7A–B). Analysis of variance showed that mouse strain was the dominant factor, although co-housing improved the significance (P < 0.05) for 30 metabolites (Fig. 3K, fig. S7B and Table S1). Purine, pyrimidine and amino acid synthesis pathways differed between the mouse strains (Fig. 3L). These pathways have been associated with CD4+ T cell dysregulation in TC mice and in patients with SLE (23, 32). Thus, these data suggested that some of the fecal metabolites observed in TC mice could contribute to immune activation.
We focused on the tryptophan pathway, which was found to be different in the TC and B6 fecal metabolite analysis (Fig. 3L). Species of Prevotella, which were expanded in TC feces (Fig. 1L), are known to metabolize tryptophan into indole (30). We observed a lower abundance of serum tryptophan and serotonin, and higher kynurenine in TC mouse serum samples (P < 0.05, Fig. 4A), as previously reported for SLE patients (21, 23). Abundance of fecal tryptophan was similar between B6 and TC mice, but fecal kynurenine and serotonin showed the same skewed distribution as found in the sera of these two mouse strains (P < 0.05, Fig. 4A). Consistent with the involvement of gut bacteria in processing dietary tryptophan (15), antibiotic-treated TC and B6 mice demonstrated increased fecal tryptophan (P < 0.05). However, antibiotics decreased serum kynurenine and the kynurenine/serotonin ratio in TC (P < 0.01) but not in B6 mice (Fig. 4B). We compared the expression of enzymes converting tryptophan to kynurenine in B6 and TC mouse tissues. IDO1 expression either transcriptionally (fig. S8A) or translationally (fig. S8B) was similar, or lower, in dendritic cells (DCs) of TC mice compared to B6 mice. Furthermore, gut Ido1 and liver tryptophan 2, 3-dioxygenase 2 (Tdo2) expression was similar in the two mouse strains (fig. S8C and D). Overall, these data suggest that the endogenous production of kynurenine is not altered in TC mice, but that tryptophan is metabolized differently by the gut microbiota of TC compared toto B6 mice.
Fig. 4. Tryptophan metabolism modulates autoimmune phenotypes in TC mice.

(A) Concentrations of tryptophan (Trp), kynurenine (Kyn) and serotonin (5HT) in feces and serum of 6-month-old B6 and TC mice are shown. (B) Shown is the quantitation of (F) fecal tryptophan, (S) serum kynurenine and the kynurenine/serotonin ratio in B6 and TC mice treated with antibiotic combination therapy (ampicillin, metronidazole, neomycin and vancomycin, AMNV) for 5 months or untreated. (C) Body weight loss of B6 and TC mice fed with tryptophan-deficient chow is shown (n = 5 per group). Shown are serum anti-dsDNA IgG concentrations in arbitrary units in TC mice fed chow with the indicated amount of tryptophan (%) after 4 months of treatment (D) and as a time-course during the treatment (E). (F) Correlation between serum kynurenine and anti-dsDNA IgG concentration in TC and B6 mice fed with chow containing 0.08 to 1.19 % tryptophan (n = 3 -- 5/group). (G) Renal pathology indicated as glomerulonephritis (GN) ranked scores from the lowest at 1 to the most severe at 24 in TC mice fed with variable amounts of tryptophan in their chow is shown. Frequencies of CD69+CD4+ cells (H), Ki67+ TAct cells (I), Tfh cells (J) and Ki67+ Tfh cells (K) in spleen are shown. (L) Shown is proliferation of effector T cells in vitro in the presence of Treg cells isolated from TC mice fed chow with different amounts of tryptophan (0.08%, low, or1.19% high) n = 6/group). (M) Shown is the frequency of splenic germinal center B cells of TC mice fed chow with the indicated amounts of tryptophan (n = 5-10/group). Data are presented as means and SEM and are compared with Dunnetťs multiple comparison tests (panels A, B, D G-K), 2-way ANOVA (C and E), Pearson’s correlation (F and L), or Fisher Exact test (G). In panel D, medians are compared with the Mann-Whitney Test. * P < 0.05, ** P < 0.01, *** P < 0.001.
Dietary tryptophan modulates autoimmune phenotypes in TC mice
To directly test the effect of tryptophan on autoimmune phenotypes, B6 and TC mice were fed synthetic chows that differed only in their tryptophan content starting at 6 weeks of age for 4 months. When fed the tryptophan-deficient chow, TC mice lost more than 20% of their body weight during the first week (Fig. 4C), whereas B6 mice showed a moderate weight loss (31), suggesting different tryptophan utilization by these two mouse strains. To compensate for the weight loss in TC mice, all mice fed the tryptophan-deficient chow were switched to a 0.19% tryptophan chow on weekends, resulting in an overall 0.08% dietary tryptophan (tryptophan low). Serum tryptophan, kynurenine and serotonin increased with dietary tryptophan in a dose-dependent manner (fig. S9A–C). However, serum tryptophan was consistently lower in TC than in B6 mice receiving the same amount of dietary tryptophan (fig. S9A). Conversely, kynurenine was higher, and serotonin was lower in TC mouse sera at the two higher dietary tryptophan doses (fig. S9B–C), further suggesting a different utilization of dietary tryptophan by these two mouse strains. We assessed whether changes in dietary tryptophan altered tryptophan metabolism through endogenous enzymes. High dietary tryptophan increased IDO1 expression in DCs of TC mice compared to low dietary tryptophan (fig. S8E), whereas there was no effect on liver Tdo2 expression (fig. S8F). Given that IDO1 in DCs and TDO in liver are the largest endogenous enzyme contributors to systemic kynurenine production, it seems unlikely that these enzymes alone contributed to differential distribution of tryptophan-associated metabolites observed between TC and B6 mice.
Low dietary tryptophan for 4 months resulted in a greater body weight loss in TC compared to B6 mice (fig. S9D), and limited lymphoid tissue expansion in TC mice, but not in B6 mice (fig. S9E–F). Low dietary tryptophan prevented the development of anti-dsDNA IgG compared to either 0.3 or 1.19% tryptophan (P < 0.05, Fig. 4D), consistently during the 4 months of treatment (P < 0.01, Fig. 4E). Serum anti-dsDNA IgG was equivalent in TC mice fed standard chow and synthetic chow with the same 0.3% tryptophan content (fig. S9G), indicating that tryptophan may serve as a dietary determinant for autoimmunity. Serum anti-dsDNA IgG and tryptophan were positively correlated in TC mice. However, anti-dsDNA IgG was observed only in TC and not in B6 mice fed similar amounts of tryptophan (fig. S9H). In contrast, anti-dsDNA IgG was found solely in TC mice with more serum kynurenine than B6 mice (Fig. 4F), suggesting that elevated kynurenine may contribute to autoimmunity in TC mice. High dietary tryptophan increased anti-dsDNA IgG in B6 mice; however, such an increase was lower than in TC mice (fig. S9I) indicating that high dietary tryptophan was not sufficient to trigger autoimmunity in the absence of Sle susceptibility genes. Importantly, the kidneys of TC mice fed low tryptophan were not distinguishable from those of aged-matched B6 mice, with less renal inflammation than TC mice fed high tryptophan (P < 0.01, Fig. 4G).
Pronounced differences were observed for splenic CD4+ T cell phenotypes in both TC and B6 mice fed with tryptophan low and high chows. High dietary tryptophan decreased the frequency of CD4+ T cells (fig. S10A and fig. S11A) and increased that of proliferative activated CD69+ CD4+ T cells in both mouse strains (Fig. 4H and I, and fig. S11B–C), as well as CD44+ CD62L− CD4+ T effector cells in TC mice (fig. S10B). Additionally, high dietary tryptophan increased the proliferation of TC mouse CD4+ T cells (fig. S10C), which are more metabolically active compared to those of B6 control mice (32, 35). High dietary tryptophan enhanced Tfh cell frequency in both TC (P < 0.05, Fig. 4J) and B6 mice (fig. S11D), which corresponded to an increased Tfh cell proliferation in TC mice (P < 0.001, Fig. 4K). Conversely, low dietary tryptophan increased the Tfr/Tfh ratio in TC mice (fig. S10D). Low dietary tryptophan increased CD25 expression on TC and B6 Treg cells (fig. S10E and fig. S11E), suggesting that tryptophan metabolism may contribute to expansion of CD25low Treg cells, which has been previously described for individuals with SLE (33). Consistent with this finding, low dietary tryptophan increased Treg suppressive function (P < 0.01, Fig. 4L). The suppressive capacity of Treg cells from TC mice fed low tryptophan was improved and similar to that of Treg cells from B6 mice, which were not affected by dietary tryptophan (fig. S11F). Further, low dietary tryptophan decreased the frequency of Th17 cells, with a stronger effect on the mesenteric lymph nodes, but no effect on IFNγ+CD4+ T cells (fig. S10F and G). The effect of low dietary tryptophan on mTORC1 activation varied, demonstrating an increase in pS6 but a decrease in p4E-BP1 (fig. S10H and I), which is consistent with the effect of kynurenine on Jurkat T cells (23). Finally, dietary tryptophan demonstrated little effect on total B cells in either mouse strain (fig. S10J and fig. S11G). In contrast, high dietary tryptophan increased the frequency of germinal center B cells in TC but not in B6 mice (P < 0.01, Fig. 4M and fig. S11H). However, plasma cells were increased in B6 but not in TC mice (fig. S10K and fig. S11I), suggesting that germinal center B cells from TC mice may possess an intrinsically higher response to either tryptophan metabolites, the expansion of Tfh cells (37), or both. This effect was not extended to the stage of plasma cells that is less T-cell dependent. Thus, tryptophan metabolites manipulated through dietary tryptophan could modulate T cell activation and effector functions in both autoimmune and control mice.
Next, we investigated whether the beneficial effects of low dietary tryptophan could be achieved via inhibiting IDO1 production of kynurenine by treating TC mice with 1-methyl tryptophan (1MT), while they were fed low or high dietary tryptophan chow for 6 weeks. As expected, 1MT treatment reduced serum kynurenine (fig. S8G), but it showed no effect on splenic weight, anti-dsDNA IgG, or renal pathology (fig. S8I–J). IDO1 inhibition had a complex effect on TC mouse CD4+ T cells that was independent from that of dietary tryptophan. 1MT treatment expanded CD69+ and IFN-gamma+CD4+ T cells, and increased pS6 expression, suggesting mTORC1 activation (fig. S8K–M). 1MT decreased p4E-BP1 expression, another well-established mTORC1 target, as well as the frequency of Th17 cells and increased the relative expression of CD25 on Treg cells over the other two IL-2R chains, suggesting an enhanced suppressive capacity (fig. S8N–Q). assessed whether increasing amounts of either tryptophan or kynurenine could mitigate the activation of CD4+ T cell effector functions in vitro. Exogenous tryptophan or kynurenine treatment enhanced Th1 cell polarization in both B6 and TC mice (fig. S12A–B) and resulted in a modest Treg cell expansion in B6 but not TC mice (fig. S12 C–D). Further, the highest amount of kynurenine led to a lower Treg cell polarization in TC mice compared to B6 mice (fig S12D). These results suggest that tryptophan and kynurenine availability may modulate T cell functions.
Given that tryptophan metabolites serve as AhR ligands (35), we compared AhR expression in TC and B6 mice. AhR expression was the highest in Treg and Tfr cells in both mouse strains (fig. S10L–M). In early disease, AhR expression was higher in TC than in B6 Treg and Tfr cells, but no difference was observed later in disease. We then investigated the preventive effect of low dietary tryptophan fed toto TC mice 6 weeks starting in early disease development when the majority of mice produced anti-dsDNA IgG, and then later, when the majority of mice exhibited renal pathology. At both disease stages, low dietary tryptophan reduced anti-dsDNA IgG (fig S13A–E). Early treatment also mitigated renal pathology (fig. S13B); however, it had no effect on the kidneys of older mice. In early disease, low tryptophan also decreased germinal center B cell frequency, however, there was a trend for reduced Tfh cells (fig. S13C–D). In late disease, low tryptophan reduced T effector cells, Tfh cell and plasma cell frequency, as well as CD4+ T cell proliferation (fig. S13F–I).
Calorie restriction is known to delay the development of lupus in mice (39, 40). To investigate the impact of weight loss on the protective effect of a low tryptophan diet, we compared circulating adiponectin in mice fed with low or high tryptophan chows. Higher amounts of adiponectin were found in mice fed a low tryptophan diet (fig. S14A). Interestingly, adiponectin was less abundant in TC than in B6 mice with a high correlation with body weight (fig. S14B). Studies have reported increased adiponectin in patients with SLE, but adiponectin deficiency protects several murine models against the disease (41).. Thus, we submitted TC mice fed with control (0.3%) or enriched (0.48%) tryptophan8 diets to a 20% calorie restriction and compared them to mice fed ad libitum with 0.08 or 0.3 % tryptophan diets without calorie restriction. Body weight loss in calorie-restricted mice was similar to that of TC mice fed a low tryptophan diet (fig. S14C). However, calorie-restricted mice exhibited lower serum tryptophan and kynurenine than did the control mice fed a 0.3% tryptophan diet (fig. S14D–E). Nonetheless, calorie-restricted mice generated anti-dsDNA IgG, showed renal pathology (fig. S14–G) as well as immune activation (fig. S14H–J) similar to that of control TC mice fed 0.3% tryptophan diet ad libitum. To further assess whether low dietary tryptophan resulted in immune suppression, we compared the response to immunization with a protein conjugated to nitrophenyl (NP) in mice on low or high tryptophan chows. As previously shown (35, 42), TC mice produced lower amounts of anti-NP antibodies than did B6 mice. However, similar production of high and low affinity anti-NP IgG1 (fig. S14K–M), as well as NP-specific germinal center B cells and splenic plasma (fig. S14O–P) were observed in TC mice fed either diet. Further, CD4+ T cells of TC mice fed low or high tryptophan diets responded similarly to alloantigen in a mixed lymphocyte reaction (fig. S14Q). Collectively, these results suggest that weight loss may not be a major contributor to the protective effect of low dietary tryptophan on autoimmune development in TC mice.
Variations in tryptophan metabolites modulate the fecal microbiota of TC mice
The impact of variations in dietary tryptophan on the gut microbiota was evaluated in TC and B6 mice. In both mouse strains, dietary tryptophan changed the gut microbiota such that tryptophan low and high fed groups showed separate clusters of bacterial species (fig. S15A and E). The distribution of major phyla was affected by dietary tryptophan in a manner that was different among the two mouse strains (fig. S15C–D and G–H), but the diversity indices were not affected (fig. S15B and F). Tryptophan deficiency expanded specific bacterial families in B6 mice (Fig. 5A). Further, higher relative abundance of Paraprevotella was observed in B6 mice fed a high tryptophan-supplemented diet (fig. S15D). Analyzing the gut microbiota of TC mice fed a high tryptophan diet showed increased Paraprevotella, Lactobacillus and Prevotellaceae (Fig. 5B), as observed in TC mice compared to B6 mice (Fig. 1L).
Fig. 5. Dietary tryptophan impacts TC gut microbiota composition and diversity.

16S rDNA sequence analyses of fecal bacteria from B6 and TC mice fed tryptophan high (Trp plus) or tryptophan low (Trp minus) chows are presented. Taxonomic cladograms and bubble plots of enriched taxa in feces of B6 mice fed tryptophan-low compared to tryptophan-high chow (A) and TC mice fed tryptophan-high compared to tryptophan-low chow (B) are shown. Only taxa meeting the criteria (LDA score > 2 and p < 0.05) are shown. Data are from three independent experiments (n = 5/group). (C-I) GF B6 mice were gavaged with feces of TC mice maintained on a low or high tryptophan chow for 4 months, or were gavaged with PBS as a control and were analyzed 3 weeks later. Shown are mesenteric lymph node cell numbers (C),),serum anti-dsDNA IgM concentrations (D), number of Tfh cells in mesenteric lymph nodes (E), number of Th17 cells in mesenteric lymph nodes (F), number of germinal center B cells (G), and plasma cells (H). CD25 expression (MFI) on Treg cells in mesenteric lymph nodes are shown (I). Data are presented as means and SEM and are compared with t tests. * P < 0.05, ** P < 0.01 (n= 5-10/group).
To test whether the gut microbial changes induced by dietary tryptophan exerted functional consequences, we performed fecal transfers from TC mice fed with low or high tryptophan chow into GF B6 mice. Three weeks later, TC mice fed with a high tryptophan diet showed an increase in cell number in the mesenteric lymph nodes (P < 0.01, Fig. 5C) and in serum anti-dsDNA IgM (P < 0.05, Fig. 5D). These mice also showed expanded numbers of Tfh, Th17, germinal center B cells and plasma cells (P < 0.05, Fig. 5E–H). TC mice fed a low tryptophan diet also showed increased CD25 expression on Treg cells (P < 0.05, Fig. 5I).
Finally, to further shed light on the gut microbes altered in TC mice, we performed metagenomic analysis of the gut microbiomes of B6 and TC mice fed different tryptophan diets (Table S3). Data demonstrated alterations in gut microbial consortia between the two mouse strains as well as between TC mice fed a low or high tryptophan diet. Lactobacillus spp. and Bacteroides dorei increased in abundance in TC mice fed a high tryptophan diet (Fig. 6A–B), and Lactobacillus species (e.g., L. reuteri, L. Johnsonii) represented the major contributing taxa in most of the bacterial families profiled (Fig. 6D–G). Importantly, although Prevotella spp. per se was not increased in our metagenomic analysis, numerous Prevotella-associated gene families such as amino acid permeases were enriched in TC mice fed a high tryptophan diet (Fig. 6D, E and G). Further, metabolic pathway analyses revealed a decrease in anaerobic reductive tricarboxylic acid cycle components but elevated L-citrulline biosynthesis enzymes in the gut microbiota of TC compared to B6 mice (Fig. 6C, G). In the TC mouse gut microbiota, there was also an increase in glucose catabolic pathway components, glucose uptake and dihydrolipoamide S-acetyltransferase involved in pyruvate metabolism (Fig. 6 E–F). Many gene families involved in amino acid metabolism (e.g., aspartate ligase) were enriched in TC compared to B6 mice (Fig. 6 D–G). Interestingly, although altered tryptophan biosynthesis was not pronounced in our analysis, tryptophan metabolism via kynurenine showed an elevated trend in the gut microbiota of TC mice compared to B6 mice (Fig. 6G). Finally, genes associated with nucleoside metabolism, including purine operon repressors controlling purine biosynthesis, were upregulated in TC mice compared to B6 mice (Fig. 6E–G). Further, amino acid and nucleoside pathways were also observed to be increased in TC mice in our metabolomic analyses (Fig. 3L).
Figure 6. Alterations in the gut microbiota potentially modify TC mouse metabolism.

Shown is shotgun sequencing for rDNA sequences in the feces of B6 and TC mice fed a control diet (n = 4/group), and TC mice fed a low tryptophan diet (TC.Trp minus) or high tryptophan diet (TC.Trp plus, n = 5/group). (A) LEfSe analyses of fecal taxa comparisons between B6 and TC groups are shown. (B) Enrichment analyses of fecal taxa comparisons of low and high tryptophan diet- TC mice are shown. (C-G) Linear discriminant analysis effect size (LEfSe) analyses of metabolic pathways and sub-level gene families in fecal samples from lupus-prone mice (either TC or TC.Trp plus) compared to those from control groups (either B6 or TC.Trp minus) are shown. (C) BioCyc metabolic pathway analysis of fecal samples are shown comparing either the B6 and TC mouse strain on control diet or the TC mice on high and low tryptophan diet. ?? (D-F) The two columns of the left show linear discriminant analysis scores of annotated gene families recategorized by EC number (D), Pfam (E) and KEGG pathways (F), comparing B6 to TC microbiota, and low to high tryptophan TC microbiota. The columns on the right indicate the dominant bacterial species to which the majority of the sequencing reads mapped to the gene families found in the microbiota of TC mice fed low or high tryptophan. (*) indicates annotations in either the Prevotella or Alloprevotella genus. (G) Shown are selected bar plots indicating genus-stratified profiles of metabolic pathways and gene families. The top of each set of stacked bars indicates the total stratified abundance of the pathway within a single fecal sample. Species and “unclassified” stratifications are linearly scaled within the total bar height.
DISCUSSION
The potential immunomodulatory effects of an altered gut microbiota have been readily documented, however, the causative effects of either gut bacterial dysbiosis or the presence of specific bacteria on autoimmunity are still elusive. The absence of commensal bacteria in the gut does not exert consistent effects on the development of autoimmune pathology in mouse models of spontaneous lupus (11). However, antibiotic treatment prevents or delays disease manifestations in lupus-prone mice with different genetic backgrounds (7, 9, 27, 43). Here, we confirm this finding in TC mice (13). Furthermore, alterations in the composition of the gut microbiota have been reported in independent cohorts of patients with SLE (3, 44, 45) and in multiple mouse models of lupus (3, 5–10). We confirm this finding in TC relative to B6 mice, and because these two mouse strains are congenic for most of their genome, the differential distribution of bacterial communities in this model is thus tightly associated with SLE susceptibility genes. We observed no tangible evidence for reduced gut bacterial diversity in TC mice, but did observe a differential distribution of bacterial taxa, with a marked abundance of Prevotella, Paraprevotella or Lactobacillus genera. Importantly, these bacterial taxa exhibit an expansion in the gut microbiota of individuals with rheumatoid arthritis (44) and in the oral microbiota of individuals with SLE (47) and in lupus prone B6.TLR7 transgenic mice (9). Further, we show that alterations in the TC mouse gut microbiota may involve functional consequences. Indeed, transfer of fecal microbiota from aged TC into GF B6 mice induced anti-dsDNA IgG antibodies, as well as the expansion of germinal center B cells and Tfh cells, which are phenotypes implicated in lupus disease progression. Interestingly, lateral transmission of the fecal microbiota among SPF B6 and TC mice also mitigated the same lupus-associated phenotypes. Thus, these results suggest that an altered gut microbiota may contribute to lupus pathogenesis. The inability of gut bacteria from young TC mice to induce an autoimmune phenotype in GF B6 mice suggests that gut microbial dysbiosis does not trigger but rather may amplify the autoimmune phenotype. This observation is also supported by the inability of gut bacteria derived from TC.Rag1−/− mice to induce autoimmunity in GF B6 mice. Furthermore, horizontal bacterial transfer experiments demonstrated that the most severe autoimmune phenotype developed in mice expressing lupus susceptibility genes and having a dysbiotic gut microbiota. Finally, antibiotic treatment only delayed the development of autoimmunity in TC mice, indicating that expression of susceptibility genes eventually led to autoimmune activation even though there were major changes in the gut microbiota. Overall, our results suggest a model in which lupus susceptibility genes triggered autoimmune activation in TC mice that potentially promoted gut dysbiosis, which then reinforced autoimmune activation.
Several mechanisms may account for the ability of bacteria to induce autoimmunity associated with lupus. Accordingly, commensal bacteria expressing orthologues of human Ro60 autoantigens are commonly found in patients with SLE, and Ro60-specific T cells can be activated and trigger anti-Ro60 antibodies in GF mice (12), supporting molecular mimicry as a potential mechanism of autoimmunity. Further, impaired gut barrier integrity is also documented in several murine models of lupus (3, 7, 9) and in some patients with SLE (3, 7), and the translocation of Enterococcus gallinarium (7) or Lactobacillus reuteri (9) from the gut to the periphery results in systemic immune activation GF B6 mice.in ?? In the present investigation, our evaluation of the intestine of TC mice found colonic inflammation that developed in late disease, potentially correlating with the severity of renal pathology, suggesting that these two types of tissue injury may be functionally linked. However, a combination of commonly used assays showed no clear evidence of deteriorated gut barrier integrity in TC mice, although some aged TC mice did exhibit minimal Staphylococcusus in peripheral organs. There was, however, no correlation between the presence or abundance of these translocated bacteria and any autoimmune manifestation in these TC mice. Additionally, no bacterial translocation was observed in TC mice before they produced autoantibodies. Overall, these results do not demonstrate any tangible bacterial translocation in the periphery that may have critically contributed to autoimmune activation leading to the emergence of lupus in the TC mice.
Microbial-associated metabolites serve as a major source of immunomodulation (46). Individuals with SLE present a distinctive gut microbiota-derived metabolite signature (49, 50), but the consequences of these findings have not been investigated. Thus, here, we elucidated whether metabolites could account for the autoimmune activation that may have been induced by gut microbiota dysbiosis in TC mice. Among the metabolites observed in TC mouse feces compared to control mouse feces, the tryptophan pathway was of particular interest because an altered distribution of tryptophan-associated metabolites has been consistently observed by other groups in the serum and immune cells from patients with SLE often in correlation with disease activity (21–23, 51–54) and in the feces of patients with SLE (48). Moreover, bacterial and host metabolites synthesized from dietary tryptophan are known to contribute to immune modulation and microbiota-host communication (16, 55, 56). We showed increased kynurenine and reduced serotonin in the sera and feces of TC mice. Elevated kynurenine has been associated with IDO1 activation induced by IFNs in patients with SLE (24). Whereas expression of both type I and II IFNs is high in TC mice (37, 57), our results do not support a contribution of IDO1 to the altered tryptophan metabolism in these animals. Further, treatment with an IDO1 inhibitor did not change the autoimmune outcomes in TC mice, contrary to the disease acceleration reported in lupus-prone MRL/lpr mice treated with the same inhibitor (58). Our results support a gut microbial contribution to tryptophan metabolism as antibiotic treatment decreased kynurenine in TC mice. TC mice also responded with acute weight loss to tryptophan deficiency, suggesting that these mice may process this essential amino acid differently, possibly because they specifically harbor gut microbes that catabolize tryptophan. In support of this notion, some species of Prevotella, Paraprevotella, or Lactobacillus, were expanded in the TC gut microbiota and differentially catabolized tryptophan (33, 56, 59), resulting in the generation of indole derivatives that may have negatively impacted TC mouse immune cells.
We showed that reduced dietary tryptophan exerted a protective effect on lupus development in TC mice and that anti-dsDNA IgG correlated with serum kynurenine in cohorts of TC mice exposed to variable amounts of dietary tryptophan. Further, variations in dietary tryptophan affected B6 and TC mouse gut microbiotas differently. Notably, tryptophan supplementation was associated with the expansion of Prevotella, Paraprevotella, or Lactobacillus species in TC mouse feces compared to B6 mice fed standard chow. Accordingly, fecal transfers from TC mice exposed to high dietary tryptophan enhanced inflammatory phenotypes in GF B6 mice compared to fecal transfers from TC mice fed low dietary tryptophan. An alteration of the gut microbiota by tryptophan supplementation has been reported in piglets, with, as in TC mice, an expansion of Prevotella species (60). Interestingly, tryptophan supplementation in these animals increased the expression of genes involved in gut epithelial integrity, which may be related, at least in part, to the absence of a “leaky gut” phenotype in TC mice. Overall, these results suggest a functional link between tryptophan metabolism, gut microbial dysbiosis and autoimmune activation in TC mice (fig. S17).
There are several limitations to this study. Although we have clearly linked gut microbial dysbiosis to altered tryptophan metabolism in the lupus-prone TC mice, we are currently not certain whether an altered tryptophan metabolism and gut microbial dysbiosis are also linked to patients with SLE. Further, we have not tested whether altering dietary tryptophan would be beneficial in patients with SLE. Mechanistically, we have not identified the bacterial species responsible for the differential processing of dietary tryptophan and tryptophan metabolites that may have induced the autoimmune activation in TC mice. Moreover, although we have documented phenotypic changes in the lymphocytes of TC mice in response to variations in tryptophan availability, we have not yet identified the molecular machinery by which this occurs.
It is also unknown which of the altered tryptophan metabolites generated by the TC mouse gut microbiota may mitigate the development of autoimmune pathogenesis. Kynurenine and AhR ligands have been associated with immunosuppression (46),, however, kynurenine activates mTORC1 in human T cells (23). As mTORC1 activation is a critical pathway in lupus pathogenesis, including in TC mice (32, 35), and its targeting with rapamycin demonstrates beneficial therapeutic effects in patients with SLE (61), mTOR activation may be one of the mechanisms through which elevated kynurenine may enhance immune activation in TC mice. Variations in dietary tryptophan modulate mTORC1 activation in a complex manner that requires future investigation. Tryptophan metabolism integrates both mammalian host and gut bacteria pathways (16). Although not as well documented as the production by IDO1, bacterial production of kynurenine has been documented in Pseudomonas aeruginosa (62), which may represent a functional association between tryptophan metabolism, gut microbial dysbiosis and autoimmune activation in TC mice. Our in vitro results suggest that tryptophan and kynurenine may directly alter T cell functions. Additionally, tryptophan bacterial-associated metabolites other than kynurenine may also trigger autoimmune activation in TC mice. Collectively, our investigation here demonstrates that modifying tryptophan metabolism through the gut microbiota may alter autoimmune pathogenesis. A recent study has reported that a restriction of dietary tryptophan impaired encephalitogenic T cell responses in a mouse model of multiple sclerosis, most likely through an altered gut microbiota (63). Thus, tryptophan availability may regulate autoreactive pathogenic CD4+ T cells in a variety of autoimmune settings.
MATERIAL AND METHODS
Study design
This study aimed to elucidate the role of gut microbial dysbiosis in the TCTC mouse model of lupus-like disease compared to congenic B6 control mice, and to investigate possible mechanisms by which gut microbial dysbiosis could affect the development of autoimmune activation. We examined gut barrier integrity by a combination of approaches. We compared the distribution of fecal microbial communities between young and aged TC and B6 mice fed with control chow, as well as animals fed high or low tryptophan chow using 16S rDNA and shotgun metagenomic sequence analyses. The contribution of gut microbial dysbiosis to autoimmune phenotypes was examined by treating mice with a cocktail of antibiotics, by fecal gavage from TC mice into GF B6 mice and by horizontal transfer between co-housed TC and B6 mice. Fecal metabolites were compared between B6 and TC mice with an untargeted screen using LC-HRMS/MS with a focus on tryptophan metabolism. The abundance of tryptophan, kynurenine and serotonin was measured in serum and feces by LC-HRMS/MS and HLPC. The contribution of tryptophan to autoimmune activation was assessed by feeding TC and B6 mice synthetic chows with low and high percentages of tryptophan for various durations (6 weeks to 6 months). The contribution of weight loss presented by TC mice fed with a low tryptophan diet was evaluated by subjecting mice to calorie restriction, as well as by assessing the response of TC mice on a low tryptophan diet to immunization with a protein antigen. The contribution of endogenous enzymes to the high amounts of kynurenine found in TC mice was assessed by measuring IDO1 expression using q-RT-PCR and flow cytometry. TDO2DO expression was measured by qRT-PCR in the liver. In addition, TC mice were treated with the IOD1 inhibitor 1MT. Finally, the contribution of gut bacteria to the autoimmune activation induced by dietary tryptophan was evaluated by fecal gavages from TC mice fed either low or high tryptophan into GF B6 mice. At the end of the treatments, spleens and mesenteric lymph nodes were collected for immunophenotyping by flow cytometry focused on CD4+ T cells and B cells, and kidneys and colons were evaluated for pathology in a blinded fashion. Autoantibodies and total immunoglobulin were measured by ELISA in serum and feces. All experiments were conducted according to protocols approved by the University of Florida Institutional Animal Care and Usage Committee. All mice are assigned randomly to treatment groups and no data was excluded from analysis.
Mice and treatments
TC and TC.Rag1−/− strains were used (13, 37) along with strains purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and maintained at the University of Florida: (NZW x B6.MPJ.Yaa) F1 male mice, C57BL/6J (B6), B6.Rag1−/−, B6.SJL-Ptprca Pepcb/BoyJ (B6.SJL), B6(C)-H2-Ab1bm12/KhEgJ (Bm12) and B6.129P2-Tcrbtm1Mom/J (B6.TCRβ KO) mice. B6.Ido1−/− mice were kindly provided by Dr. Lei Jin (Dept. of Medicine, University of Florida). Germ free (GF) B6 mice were produced by the University of Florida Animal Care Services. Only female mice were used in this study, except for the (NZW x B6.MPJ.Yaa) F1 and their controls, which were housed with mice from the same strain unless indicated otherwise, in the same SPF room. “Aged” or late disease stage mice are defined as 6 to 9-month-old, when TC mice present a full autoimmune activation with high production of autoantibodies. TC.Rag1−/− mice were used at 5 to 6 months of age, due to the heavy mortality in older mice from that strain. All age-matched groups within an experiment were tested simultaneously. In the fecal transfer experiments, 2 to 3-month-old GF mice were acclimated in SPF static cages for 3 d before the transfers to separate cages according to the strain of origin of the fecal donor. Feces were collected from B6 and TC mice as detailed for each experiment, as well as from 5-month-old B6.Rag1−/− and TC.Rag1−/− mice. Pooled fecal pellets were diluted in PBS (1 pellet / 150 ul) and 200 ul of fecal slurry was gavaged in each GF recipients. GF controls received PBS only. Phenotypes were evaluated one month after transfer. In the co-housing experiments, TC and B6 mice age-matched within 2 days were housed from weaning to 7 months of age either with the same strain, or mixed with other strain (3 TC : 2 B6 and 2 TC : 3 B6, without any significant difference between the two configurations). Mice were provided with autoclaved reversed osmosis drinking water, and unless indicated, fed with irradiated Envigo 7912 standard chow at lib that contains 0.3% tryptophan. Tryptophan-modified synthetic chows that differ only by their tryptophan content (0, 0.19, 0.3, or 1.19%, A11022501-04, Research Diet) were fed to mice for the indicated periods of time. To replenish tryptophan, tryptophan-deficient chow was switched to tryptophan (0.19) chow on Friday afternoons and switched back to tryptophan (0) chow on Monday mornings, corresponding to an approximate distribution of 60% tryptophan (0): 40% tryptophan (0.19), or an overall 0.08% dietary tryptophan. Overall food consumption was similar across chows, and higher in TC mice corresponding to a higher body weight than in age-matched B6 controls. Some mice were treated with 1-methyl tryptophan (1MT) in drinking water (2 mg/ml) in addition to the tryptophan-modified chows. For calorie restriction experiments, mice were fed 2 g/mouse/d (a 20% reduction from mice fed ad libitum) of either tryptophan 0.3 control chow or a 1:4 mixture of tryptophan 1.19 and tryptophan 0.3 chows. Some mice were treated with a cocktail of antibiotics (AMNV: ampicillin 0.5g/L, metronidazole 0.5g/L, neomycin 0.5g/L and vancomycin 0.25 g/L) from 6 weeks to 6 months of age. All experiments were conducted on at least 2 independent cohorts per group to avoid cage effects.
Renal and colon pathology
Kidneys and colon were processed as previously described (62) and PAS (kidneys) H&E (colon) -stained sections were ranked or scored on a 0-4 scale in a blinded fashion by a pathologist (BPC). The surface area of lymphoid foci in the colon was measured with Aperio ImageScope. Colon sections were also stained with antibodies to CD45 (BD Pharmingen 30F11, 1:25 dilution), B220 (BD Pharmingen RA3-6B2, 1:800) and CD3 (Serotec MCA1477, 1:1,000) by immunohistochemistry.
Microbiota analysis
Fecal samples were stored at −80°C. Part of the collected fecal samples were processed for microbiota analysis. Briefly, fecal DNA was isolated using ZymoBIOMICS DNA Mini Kit (Zymo Research, Irvine, CA). Fecal DNA samples were amplified by pairs of Miseq compatible primers, targeting the 16S rDNA V4-V5 regions as reported previously (64, 65). Amplicons were purified, normalized, pooled and sequenced on an Illumina Miseq with 2 x 250 bp pair-end reads. Filtered operational taxonomic units (OTUs) were rarefied to a depth of 21,234 sequences per sample, 16,451 sequences per sample, and 23,624 sequences per sample in the experiments comparing aged B6 to TC mice, tryptophan high compared to tryptophan low fed mice, and 1-2-month-old mice, respectively. Principle coordinate analysis (PCoA) was performed in QIIME (v1.9.1), as described previously (64, 65) and the data was plotted using Python (V2.7.14). Alpha diversity analyses, including Chao’s richness were also performed in QIIME. To identify significantly enriched microbial species, linear discriminant analysis effect size (LEfSe) analyses were performed (64, 65). To avoid batch effects, we set the batches as the subclass and the genotype or diet as the main class in the LEfSe analyses with the all-against-all strategy, which is stricter. Significant taxa were selected with default criteria (P < 0.05 by Kruskal-Wallis test; linear discriminant analysis (LDA) score > 2) and plotted in a cladogram based on their phylogenetic relationship.
To perform shotgun metagenomic sequencing, libraries were generated from 18 fecal samples using Nextseq500 in high-output mode (2x75 bp). Raw reads were quality-controlled using KneadData version 0.7.2 (http://huttenhower.sph.harvard.edu/kneaddata), whereupon low-quality bases (< 60 nt in length) were trimmed and discarded. Identified reads mapped against the mouse genome (mouse_C57BL_6NJ) using Bowtie 2 (65). As end-pairing relationships were not considered during HUMAnN2’s alignment steps, the paired-end reads were concatenated together as a single joint file. The average amount of data generated for each sample was 13.82 Gb, which met the industry standard (66). Furthermore, quality-filtered metagenomes were taxonomically profiled using MetaPhlAn2 version 2.6.0 via default parameters (67). Functional profiling was performed using HUManN2 version 2.8.1 in UniRef50 mode (68). Metagenomic reads were mapped to pangenomes of species identified during MetaPhlAn2 step. Coding sequences have been pre-annotated to their respective UniRef50 families. Unaligned reads were subjected to translated search against UniRef50 database with DIAMOND and the obtained hits were filtered by the alignment quality, sequence length and sequence coverage. This strategy resulted in an average of 47% translated alignment rate that was sufficient for further analyses (68). UniRef50 gene families were generated in reads/kilobase units (RPK). Pathway abundance and coverage tables were created by default. These gene family and pathway abundance tables were further renormalized to relative abundance and regrouped according to databases (e.g., KEGG, Pfam, EC numbers). Tables were further joined and dissected by different contrasts of interest (B6 versus TC; TC.trp.minus versus TC.trp.plus). Both HUManN2-associated module (68) and LEfSe (69) were applied for statistical analyses. The significant features were further subjected to HUManN2-barplot to visualize the stratification of microbial taxa in detail. The body of generated data, translated alignment rates and HUManN2-associate analyses is documented in table S3. The metagenomic raw FastQ sequencing files were deposited into the National Center for Biotechnology Information Sequence Read Archive and are available under the accession numbers SRP234839 or PRJNA593830. Additional processed data reported in this study are available upon request.
Metabolomic analysis
Feces were homogenized in 5 mM ammonium acetate at 20 mg/ml. Following homogenization, the sample was centrifuged at 20,000 rcf and 100 µL was transferred to a new tube. A 20 µL aliquot of isotopic standards were added for both calibration and profiling experiments. The internal standard solution consisted of tryptophan-13C11 (5 µg/mL), creatine-D3 (4 µg/mL), leucine-D10 (4 µg/mL), citric acid-13C6 (8 µg/mL), tyrosine-13C6 (4 µg/mL), phenyalanine-13C6 (4 µg/mL), serotonin-D4 (5 µg/mL), kynurenine-D5 (5 µg/mL), kynurenic acid-D5 (0.5 µg/mL), anthranilic acid-13C6 (0.8 µg/mL), xanthurenic acid-D4 (0.8 µg/mL). Next, 800 µL of a mixture of acetonitrile, methanol and acetone (8:1:1) was added followed by centrifugation to precipitate and pellet the proteins. The supernatant was transferred to a clean tube and dried under a gentle stream of nitrogen before reconstitution in 100 µL of 0.1% formic acid in water for metabolomic analysis. Global metabolomic analysis was performed on a Thermo Q-Exactive High Resolution Mass Spectrometer (HRMS) with a Dionex UHPLC and autosampler running in both positive and negative ionization as separate injections. Mass resolution is 35,000 at m/z 200 with mass accuracy of less than 5 ppm in positive mode and less than 10 ppm in negative mode. Separation was achieved on an Ace C18-PFP column (100 x 2.1 mm, 2 µm) with 0.1% formic acid in water as mobile phase A and acetonitrile as mobile phase B with a column temperature of 25°C. Flow rate was 350 µL/min with a total run time of 20 min (70, 71). Exact chromatography and additional method details can be found at www.metabolmicsworkbench.org. Metabolite identification of 234 named metabolites retained for subsequent analysis was performed through reference to metabolite library of 1,000 compounds, curated by running each standard at the Southeast Center for Integrated Metabolomics (SECIM, www.secim.ufl.edu). Feature alignment and curation were performed by MZmine (72) through an automated routine developed in-house. From the same type of global metabolite profiling from serum and fecal samples, we quantified 6 tryptophan metabolites (tryptophan, serotonin, kynurenine, kynurenic acid, xanthurenic acid, and anthranilic acid) through the addition of isotopically labeled versions of each target species and referenced to an external calibration curve covering the expected concentration range. The metabolomics data (including 234 named metabolites retained for subsequent analysis) were first log-2 transformed and then centered. They were further examined to ensure that the normality assumption was satisfied. The four groups (B6/TC strains, co-housed and housed separately) were first analyzed using principal component analysis, to obtain a global viewpoint of the data and the groups. Subsequently, for each of the 234 named metabolites, an analysis of variance model was fitted, with two main effects (factors), the strain factor and the co-housing factor, as well as an interaction between the two. Adjusted p-values for multiple comparisons, based on the Benjamini-Hochberg procedure, are given in Table S1. Subsequently, the named metabolites were mapped to 114 KEGG pathways that were then tested for enrichment across the B6 and TC strains. P-values were adjusted for multiple comparisons based on the Benjamini-Hochberg procedure.
Serum kynurenine (Kyn) and tryptophan (Trp) were also quantified by a high-performance liquid chromatography (HPLC) system (Agilent 1220 Infinity II, Agilent Technologies) equipped with an automated sampler, a gradient pump and a VWD detector. Briefly, 50 ul of serum was mixed with 6.5 ul 40% periodic acid, followed by vortex-mixing for 1 min and centrifugation at 17,000 g for 10 min. Subsequently, the supernatants were passed through a 0.22 um filter (Millex), and 20 ul of the samples was injected into the HPLC system. Separations were carried out using a mobile phase consisting of 15 mM aqueous zinc acetate (pH 4.0) and acetonitrile (96:4, v/v) in a ZORBAX Eclipse AAA column (3.5 μm, 3 × 150 mm), with a flow rate of 1.0 ml/min. The eluates were monitored by the programmed wavelength detection setting at 365 nm (kynurenine) and 404 nm (tryptophan, excitation at 254 nm). The data were acquired and processed using Agilent ChemStation software. Concentrations of serum kynurenine and tryptophan were calculated based on the standard curves using serial dilutions of standards (Sigma).
Cellular assays
For the Treg suppression assays, CD4+CD25− effector T cells were isolated from B6.SJL mice and labeled with Cell Trace Violet (CTV, Life Technologies). DCs were isolated from B6 spleens with CD11c microbeads and served as antigen-presenting cells. CD4+CD25+ Treg cells were isolated from B6 and TC mice fed tryptophan low or high chow for 6 - 10 weeks. DCs (104) and effector T cells (6.6 x 104) were incubated with Treg cells at a 1:1 to 1:8 Teff:Treg ratio in the presence of soluble anti-CD3 antibody (1 mg/ml) for 3 days. The proliferation of CD45.1+ effector T cells was determined by CTV and the proliferation index was calculated by the FlowJo software. Two independent cohorts were performed for TC mice and one for B6 mice. The mixed lymphocyte reaction was adapted from an assay previously described (73). Briefly, CD4+ T cells purified from TC mice fed with tryptophan low or high chows for 6 weeks were labeled with CTV and mixed with H-2b mismatched CD4-depleted splenocytes from Bm12 mice at a 1:1 ratio in complete RPMI 1640 media for 4 days. Controls included TC CD4+ T cells alone and Bm12 CD4+ T cells stimulated from TCRβ KO mice. Proliferation was assessed by Ki-67 staining on CTV-positive cells. For T cell polarization assays, total splenic CD4+ T cells were incubated in the presence of anti-CD3/CD28 antibodies under Th1 (IL-12 (10 ng/mL) and anti-IL-4 (10 ug/mL)) or Treg (TGF-β (3 ng/mL), IL-2 (50 ng/mL), anti-IFNγ (300 nm.), and anti-IL-4 (10 ug/mL)) polarizing conditions with L-Tryptophan or L-Kynurenine (both from Sigma) in complete McCoy’s 5A media, which has a 15 mM tryptophan concentration, for 96 h. IFN-gamma production and Foxp3 expression were assessed by flow cytometry.
Flow cytometry
Flow cytometry was performed on splenic and mesenteric lymph node cells as previously described (74) with the gating strategy shown in fig. S17. Fluorochrome-conjugated antibodies against the following markers were purchased from BD Biosciences, eBiosciences, or BioLegend: AhR (4MEJJ), BCL6 (K112-91), B220 (RA3-6B2), CD3e (145-2C11), CD4 (RM4-5), CD11c (HL3), CD19 (1D3), CD25 (7D4), CD44 (IM7), CD62L (MEL-14), CD69 (H1.2F3), CD95 (15A7), CD122 (TM-β1), CD132 (TUGm2), CD138 (281-2), CXCR5 (2G8), Foxp3 (FJK-16s), GL-7 (GL7), Ido1 (mIDO-48), p4E-BP1 (236B4), pS6 (D57.2.2E), PD-1 (J43), IFNγ (XMG1.2), IL-10 (JES5-16E3), IL-17A (TC11-18h10.1), I-Ab (AF6-120.1), and PSGL-1 (2PH1). 4-Hydroxy-3-nitrophenylacetyl (NP)-Phycoerythrin (Biosearch Technology) was used to analyze NP-positive germinal center B cells and plasma cells. Tfh cells were gated as CD4+PD-1hiCXCR5+Bcl-6+Foxp3−, Tfr cells as CD4+PD-1hiCXCR5+Bcl-6+Foxp3+, Treg cells as CD4+Foxp3+. Naïve and effector memory CD4+ T cells were gated as CD4+CD44−CD62L+ and CD4+CD44+CD62L−, respectively. Germinal center B cells were gated as B220+GL7+Fas+, and plasma cells as CD138+IgD−. Cell proliferation was measured with Ki-67 (SolA15) intracellular staining. To analyze intracellular IFN-gamma and IL-17A production, spleen cells were treated with a leukocyte activation cocktail (LKA, BD Biosciences) for 5 h and fixed with the Fixation/Permeabilization kit (eBioscience) after anti-CD4 antibody staining.
Antibody and adiponectin measurements
Serum anti-ssDNA IgG and IgM, anti-dsDNA IgG and IgM were quantified by ELISA on sera diluted 1:100 as previously described (29). Anti-dsDNA IgA was quantified in sera diluted 1:100 using the same method with goat anti- mouse IgA-AP (Thermo Fisher) diluted 1:1000. Serum IgM and IgG were measured with sera diluted 1:50,000. Total IgA was measured using the IgA Ready-Set-Go ELISA kit (eBioscience) on feces diluted 1:100, and serum diluted 1:10,000. Anti-nuclear autoantibodies were visualized on Hep-2 slides with serum diluted 1:40 reacted with anti-mouse IgG FITC, which was quantified as mean fluorescence intensity (MFI) with ImageJ. The production of anti-NP IgG1 was measured as previously described (32) in the serum of mice immunized with NP-keyhole limpet hemocyanin in alum, boosted at day 14, and euthanized 7 days later. Adiponectin was measured in sera collected 2 h into the dark cycle at a 1:2,000 dilution according to the manufacturer’s instructions (R&D systems).
Statistical Analysis
Statistical analyses were performed using the GraphPad Prism 6.0 software. Unless indicated, data were normally distributed, and graphs show means and standard deviations of the mean (SEM) for each group. Unless indicated, results were compared with 2-tailed tests with a minimal level of significance set at P < 0.05. Bonferroni corrections were applied for multiple comparisons.
Supplementary Material
fig. S1. Bacterial cultures from liver and mesenteric lymph nodes of TC mice.
fig. S2. Alteration of bacterial communities in feces of TC mice compared to B6 mice.
fig. S3. Antibiotic treatment delays autoimmune activation in TC mice.
fig. S4. Distinct signatures of fecal microbial consortia in young TC mice compared to B6 mice.
fig. S5. Fecal microbiota from either young or lymphocyte-deficient TC mouse donors failed to induce autoimmune activation.
fig. S6. Representative kidney and colon tissue sections showing histology.
fig. S7. TC and B6 mice have a distinct fecal metabolome.
fig. S8. TC and B6 mice present similar expression of kynurenine producing enzymes.
fig. S9. Differential tryptophan metabolism in TC and B6 mice in response to changes in dietary tryptophan.
fig. S10. Dietary tryptophan regulates immune activation in lupus-prone TC mice.
fig. S11. Dietary Tryptophan regulates T cell activation in non-autoimmune B6 mice.
fig. S12. Tryptophan and kynurenine mitigate CD4+ T cell polarization into Th1 and Treg cells.in
Fig. S13. Short-term variations in dietary tryptophan change autoimmunity in lupus-prone TC mice.
fig. S14. Effects of low dietary tryptophan and weight loss on immune phenotypes in TC mice.
fig. S15. Tryptophan supplementation or deficiency perturbs gut bacterial communities.
fig. S16. Proposed model linking genetic susceptibility, autoimmunity, gut microbial dysbiosis and tryptophan metabolism in TC mice.
fig. S17. Representative FACS gating strategy showing gating for different lymphocyte populations
table S1. ANOVA-results comparing fecal metabolites by mouse strain and cohousing conditions
table S2. Primer sequence for tight junction gene expression and house-keeping gene controls
table S3. Metagenomics analysis
Acknowledgments:
We thank members of the Morel laboratory for helpful discussions as well as Antonio La Cava, UCLA, for helpful discussions, the Department of Pathology Molecular Pathology Core for technical expertise with histology.
Funding: This research was supported by R21 AI122338 and R01 AI143313 to LM and in part by NIH R01 DK109560 to MM.
Footnotes
Competing interests: All authors have no competing interests.
Data and materials availability: All data associated with this study are presented in the main paper or in the supplementary materials. The metagenomic raw FastQ files are available under the accession numbers SRP234839 or PRJNA593830 at the NCBI Sequence Read Archive.
REFERENCES AND NOTES
- 1.Clemente JC, Manasson J, Scher JU, The role of the gut microbiome in systemic inflammatory disease. BMJ 360, j5145 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hevia A et al. , Intestinal dysbiosis associated with systemic lupus erythematosus. mBio 5, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo XM et al. , Gut microbiota in human systemic lupus erythematosus and a mouse model of lupus. Appl Environ Microbiol 84, e02288–02217 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azzouz D et al. , Lupus nephritis is linked to disease-activity associated expansions and immunity to a gut commensal. Ann Rheum Dis 78, 947–956 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H, Liao X, Sparks JB, Luo XM, Dynamics of gut microbiota in autoimmune lupus. Appl Environ Microbiol 80, 7551–7560 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mu Q et al. , Control of lupus nephritis by changes of gut microbiota. Microbiome 5, 73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manfredo Vieira S et al. , Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science 359, 1156–1161 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaudreau MC, Johnson BM, Gudi R, Al-Gadban MM, Vasu C, Gender bias in lupus: does immune response initiated in the gut mucosa have a role? Clin Exp Immunol 180, 393–407 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zegarra-Ruiz DF et al. , A diet-sensitive commensal lactobacillus strain mediates TLR7-dependent systemic autoimmunity. Cell Host Microbe 25, 113–127 e116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma Y et al. , Gut microbiota promote the inflammatory response in the pathogenesis of systemic lupus erythematosus. Molecular medicine 25, 35 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vieira S, Pagovich O, Kriegel M, Diet, microbiota and autoimmune diseases. Lupus 23, 518–526 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greiling TM et al. , Commensal orthologs of the human autoantigen Ro60 as triggers of autoimmunity in lupus. Sci Transl Med 10, pii: eaan2306 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morel L et al. , Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc Natl Acad Sci USA 97, 6670–6675 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morel L, Mapping lupus susceptibility genes in the NZM2410 mouse model. Adv Immunol 115, 113–139 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Lamas B, Natividad JM, Sokol H, Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol 11, 1024–1038 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Cervenka I, Agudelo LZ, Ruas JL, Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 357, pii: eaaf9794 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Gao J et al. , Impact of the gut microbiota on intestinal immunity mediated by tryptophan metabolism. Front Cell Infect Microbiol 8, 13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rothhammer V et al. , Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med 22, 586–597 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cervantes-Barragan L et al. , Lactobacillus reuteri induces gut intraepithelial CD4+CD8alphaalpha+ T cells. Science 357, 806–810 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dodd D et al. , A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 551, 648–652 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bengtsson AA et al. , Metabolic profiling of systemic lupus erythematosus and comparison with primary Sjogren’s syndrome and systemic sclerosis. PloS one 11, e0159384 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akesson K et al. , Kynurenine pathway is altered in patients with SLE and associated with severe fatigue. Lupus Sci Med 5, e000254 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perl A et al. , Comprehensive metabolome analyses reveal N-acetylcysteine-responsive accumulation of kynurenine in systemic lupus erythematosus: implications for activation of the mechanistic target of rapamycin. Metabolomics 11, 1157–1174 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lood C et al. , Type I interferon-mediated skewing of the serotonin synthesis is associated with severe disease in systemic lupus erythematosus. PloS one 10, e0125109 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Craft JE, Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol 8, 337–347 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi JY et al. , Circulating follicular helper-like T cells in systemic lupus erythematosus: association with disease activity. Arthritis Rheumatol 67, 988–999 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu B et al. , The ratio of circulating follicular T helper cell to follicular T regulatory cell is correlated with disease activity in systemic lupus erythematosus. Clin Immunol 183, 46–53 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erickson LD, Lin LL, Duan B, Morel L, Noelle RJ, A genetic lesion that arrests plasma cell homing to the bone marrow. Proc Natl Acad Sci USA 100, 12905–12910 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin Y et al. , Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med 7, 274ra218 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sasaki-Imamura T, Yoshida Y, Suwabe K, Yoshimura F, Kato H, Molecular basis of indole production catalyzed by tryptophanase in the genus Prevotella. FEMS Microbiol Lett 322, 51–59 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Browne CA, Clarke G, Dinan TG, Cryan JF, An effective dietary method for chronic tryptophan depletion in two mouse strains illuminates a role for 5-HT in nesting behaviour. Neuropharmacology 62, 1903–1915 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Choi SC et al. , Inhibition of glucose metabolism selectively targets autoreactive follicular helper T cells. Nat Commun 9, 4369 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horwitz D, Identity of mysterious CD4+CD25-Foxp3+ cells in systemic lupus erythematosus. Arthritis Research & Therapy 12, 101 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi SC et al. , Relative contributions of B cells and dendritic cells from lupus-prone mice to CD4(+) T cell polarization. J Immunol 200, 3087–3099 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gutierrez-Vazquez C, Quintana FJ, Regulation of the immune response by the Aryl Hydrocarbon Receptor. Immunity 48, 19–33 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson BC, Gajjar A, Kubo C, Good RA, Calories versus protein in onset of renal disease in NZB x NZW mice. Proc Natl Acad Sci USA 83, 5659–5662 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mizutani H et al. , Calorie restriction prevents the occlusive coronary vascular disease of autoimmune (NZW x BXSB)F1 mice. Proc Natl Acad Sci USA 91, 4402–4406 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li HM et al. , Emerging role of adipokines in systemic lupus erythematosus. Immunol Res 64, 820–830 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Niu H, Sobel ES, Morel L, Defective B-cell response to T-dependent immunization in lupus-prone mice. Eur J Immunol 38, 3028–3040 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mu Q et al. , Antibiotics ameliorate lupus-like symptoms in mice. Sci Rep 7, 13675 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jain S et al. , Associations of autoimmunity, immunodeficiency, lymphomagenesis, and gut microbiota in mice with knockins for a pathogenic autoantibody. Amer J Pathol 187, 2020–2033 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hevia A et al. , Intestinal dysbiosis associated with systemic lupus erythematosus. MBio 5, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Azzouz D et al. , Lupus nephritis is linked to disease-activity associated expansions and immunity to a gut commensal. Ann Rheum Dis, annrheumdis-2018-214856 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scher JU et al. , Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife 2, e01202 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Correa JD et al. , Subgingival microbiota dysbiosis in systemic lupus erythematosus: association with periodontal status. Microbiome 5, 34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ost KS, Round JL, Communication between the microbiota and mammalian immunity. Annu Rev Microbiol 72, 399–422 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rojo D et al. , Ranking the impact of human health disorders on gut metabolism: systemic lupus erythematosus and obesity as study cases. Sci Rep 5, 8310 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Q et al. , Fecal metabolomics and potential biomarkers for systemic lupus erythematosus. Front Immunol 10, 976 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mandel EH, Appleton HD, Tryptophan metabolism. Results of studies in discoid lupus erythematosus. Arch Dermatol 94, 358–360 (1966). [DOI] [PubMed] [Google Scholar]
- 50.Widner B et al. , Degradation of tryptophan in patients with systemic lupus erythematosus. Adv Exp Med Biol 467, 571–577 (1999). [DOI] [PubMed] [Google Scholar]
- 51.Widner B et al. , Enhanced tryptophan degradation in systemic lupus erythematosus. Immunobiol 201, 621–630 (2000). [DOI] [PubMed] [Google Scholar]
- 52.Pertovaara M et al. , Indoleamine 2,3-dioxygenase activity is increased in patients with systemic lupus erythematosus and predicts disease activation in the sunny season. Clin Exp Immunol 150, 274–278 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Agus A, Planchais J, Sokol H, Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe 23, 716–724 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Roager HM, Licht TR, Microbial tryptophan catabolites in health and disease. Nat Commun 9, 3294 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sang A et al. , Dysregulated cytokine production by dendritic cells modulates B cell responses in the NZM2410 mouse model of lupus. PloS one 9, e102151 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ravishankar B et al. , Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenase. Proc Natl Acad Sci USA 109, 3909–3914 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zelante T et al. , Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39, 372–385 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Liang H et al. , Dietary L-tryptophan modulates the structural and functional composition of the intestinal microbiome in weaned piglets. Front Microbiol 9, 1736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lai ZW et al. , Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet 391, 1186–1196 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bortolotti P et al. , Tryptophan catabolism in Pseudomonas aeruginosa and potential for inter-kingdom relationship. BMC Microbiol 16, 137 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sonner JK et al. , Dietary tryptophan links encephalogenicity of autoreactive T cells with gut microbial ecology. Nat Commun 10, 4877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cuda CM, Zeumer L, Sobel ES, Croker BP, Morel L, Murine lupus susceptibility locus Sle1a requires the expression of two sub-loci to induce inflammatory T cells. Genes Immun. 11, 542–553 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Colliou N et al. , Commensal Propionibacterium strain UF1 mitigates intestinal inflammation via Th17 cell regulation. J Clin Invest 127, 3970–3986 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ge Y et al. , Neonatal intestinal immune regulation by the commensal bacterium, P. UF1. Mucosal Immunol 12, 434–444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Quince C, Walker AW, Simpson JT, Loman NJ, Segata N, Shotgun metagenomics, from sampling to analysis. Nat Biotechnol 35, 833–844 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Segata N et al. , Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods 9, 811–814 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Franzosa EA et al. , Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods 15, 962–968 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Segata N et al. , Metagenomic biomarker discovery and explanation. Genome Biol 12, R60 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu H, Garrett TJ, Tayyari F, Gu L, Profiling the metabolome changes caused by cranberry procyanidins in plasma of female rats using (1) H NMR and UHPLC-Q-Orbitrap-HRMS global metabolomics approaches. Mol Nut Food Res 59, 2107–2118 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ulmer CZ, Yost RA, Chen J, Mathews CE, Garrett TJ, Liquid chromatography-mass spectrometry metabolic and lipidomic sample preparation workflow for suspension-cultured mammalian cells using jurkat t lymphocyte cells. J Proteom Bioinfor 08, 126–132 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pluskal T, Castillo S, Villar-Briones A, Oresic M, MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC bioinformatics 11, 395 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li W et al. , Targeting T cell activation and lupus autoimmune phenotypes by inhibiting glucose transporters. Front Immunol 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Choi SC et al. , The lupus susceptibility gene Pbx1 regulates the balance between follicular helper T cell and regulatory T cell differentiation. J Immunol 197, 458–469 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
fig. S1. Bacterial cultures from liver and mesenteric lymph nodes of TC mice.
fig. S2. Alteration of bacterial communities in feces of TC mice compared to B6 mice.
fig. S3. Antibiotic treatment delays autoimmune activation in TC mice.
fig. S4. Distinct signatures of fecal microbial consortia in young TC mice compared to B6 mice.
fig. S5. Fecal microbiota from either young or lymphocyte-deficient TC mouse donors failed to induce autoimmune activation.
fig. S6. Representative kidney and colon tissue sections showing histology.
fig. S7. TC and B6 mice have a distinct fecal metabolome.
fig. S8. TC and B6 mice present similar expression of kynurenine producing enzymes.
fig. S9. Differential tryptophan metabolism in TC and B6 mice in response to changes in dietary tryptophan.
fig. S10. Dietary tryptophan regulates immune activation in lupus-prone TC mice.
fig. S11. Dietary Tryptophan regulates T cell activation in non-autoimmune B6 mice.
fig. S12. Tryptophan and kynurenine mitigate CD4+ T cell polarization into Th1 and Treg cells.in
Fig. S13. Short-term variations in dietary tryptophan change autoimmunity in lupus-prone TC mice.
fig. S14. Effects of low dietary tryptophan and weight loss on immune phenotypes in TC mice.
fig. S15. Tryptophan supplementation or deficiency perturbs gut bacterial communities.
fig. S16. Proposed model linking genetic susceptibility, autoimmunity, gut microbial dysbiosis and tryptophan metabolism in TC mice.
fig. S17. Representative FACS gating strategy showing gating for different lymphocyte populations
table S1. ANOVA-results comparing fecal metabolites by mouse strain and cohousing conditions
table S2. Primer sequence for tight junction gene expression and house-keeping gene controls
table S3. Metagenomics analysis