

Abstract
The GTPase of immunity-associated proteins (GIMAPs) are a family of genes believed to contribute to lymphocyte development, signaling, and apoptosis, thus playing an important role in immune system homeostasis. While models of gene derangement have been described in both mice and immortalized cell lines, human examples of these diseases remain exceptionally rare. In this manuscript we describe the first documented human cases of a homozygous deleterious GIMAP6 variant in the GIMAP6 gene and their subsequent clinical and immunological phenotype. In order to interrogate the patients’ immune defect, we performed whole-exome sequencing, western blot, flow cytometry analysis, lymphocyte activation and proliferation studies, cytokine release assays, and apoptosis studies. We found two siblings with a predicted deleterious homozygous variant in the GIMAP6 gene with no expression of GIMAP6 protein on western blot. Patients demonstrated accelerated apoptosis, but largely normal lymphocyte subpopulations, activation and proliferation and cytokine release. There appears to be a spectrum of clinical features associated with deficiency of GIMAP6 protein, with one patient suffering lymphopenia and recurrent sinopulmonary infections, and the other clinically asymptomatic. Biallelic variants in the GIMAP6 gene have now been shown to demonstrate disease in humans. The absence of GIMAP6 protein is associated with a spectrum of clinical manifestations and much remains to be learnt about the pathogenic mechanisms underlying this disease. We suggest that biallelic variants in the gene for GIMAP6 should be considered in children with lymphopenia and recurrent sinopulmonary infections.
Subject terms: Immunogenetics, Immunological deficiency syndromes
Introduction
The introduction of widespread next-generation sequencing (NGS) into clinical practice has fundamentally altered the practice of several medical subspecialties, perhaps none more so than immunology. NGS has enabled the discovery of new immunological diseases and mechanisms, providing ever greater understanding of immune system function in health and disease. This has also brought many challenges, as variants in genes of unclear significance are found, with a subsequent need to validate whether these variants are indeed the cause of a patient’s clinical and immunological phenotype [1].
The GIMAP (Guanosine triphosphatase (GTPase) of Immunity-Associated Proteins) proteins are a family of seven human genes and one possible pseudogene, all encoded at chromosome 7q36.1. Initially discovered in the late 1990s and referred to as immune-associated nucleotides (IANs), they appear to share a similar GTPase-binding motif and play a role in lymphocyte development and survival [2–7]. Expression of GIMAP family proteins fluctuates with the different stages of lymphocyte development, and in response to immune system challenge [2, 3, 5]. While some members of the GIMAP family (e.g. GIMAP4) appear to accelerate apoptosis, others (e.g. GIMAP5) appear to inhibit apoptosis [8]. There is also evidence to suggest some GIMAP proteins demonstrate decreased expression in cancerous tissue, particularly the apoptosis-inhibiting GIMAPs [9, 10].
Despite their potential widespread role, most GIMAP studies have been performed in mice or immortalized cell lines [2, 8, 11], and much about their function remains unknown. Human cases of GIMAP dysfunction are exceptionally rare, and in this paper we describe the first documented human cases of a homozygous deleterious GIMAP6 variant.
Materials and methods: patients, genetic investigations and validation of the variant
We collected data from the medical records of two siblings (P1 and P2) later diagnosed with homozygous variants in GIMAP6. The details of all genetic and immunological investigations can be found in the online repository of this article (Figs. E1–E4 and Tables E1–E4). All experiments to elucidate the validity and immunological impact of their genetic variants were performed after obtaining informed consent from the patients’ parents, and were approved by the Helsinki Committee of the Hadassah University Medical Center Institutional Review Board.
Our experiments were modeled on those of Ho and Tsai [8] who in 2017 showed that knock out of GIMAP6 in Jurkat immortalized cell lines lead to increased apoptosis, and that transfection of GIMAP6 into the Huh-7 cell lines (a liver cell line which does not express GIMAP6) lead to protection from apoptosis. When GIMAP6 was subsequently knocked out of the transfected Huh-7 cells, their sensitivity to an apoptosis-inducing agent (okadaic acid) was restored [8]. We have not repeated this knock in/knockout aspect of their study but have sought to determine whether the phenotypic features of GIMAP6-deficient cell lines are replicated in human beings lacking GIMAP6 protein expression.
The variant in GIMAP6 detected in P1 and P2 has been submitted to the ClinVar database, and the accession number is SCV001364378.
Results
Clinical features
P1 is an eight-year-old girl from Gaza, the fourth of five children born to consanguineous (first cousin) parents (Fig. E1). She was well until the age of five when she developed recurrent purulent otitis media and a chronic wet cough. A recent computer tomography (CT) scan of her chest demonstrates several small nodules in the right upper and middle lobe, but no evidence of bronchiectasis. She has mild hepatosplenomegaly and is lymphopenic, with an overall lymphocyte count that hovers around 1.2 × 109/L but has, on occasion, fallen to 0.7 × 109/L. Her older brother, P2, is asymptomatic.
Genetic findings
Homozygous variants in nine different genes were found in P1, with the c.257G>A, p.(Trp86Ter) variant in GIMAP6 the only candidate that could potentially explain her clinical phenotype (Table E1 of the Supplementary Materials). She was also found to carry two known pathogenic variants in the MEFV gene; however, these variants were both inherited from the same parent and P1 displays no clinical features of FMF.
Sanger sequencing of the GIMAP6 variant was performed for all members of the immediate family, all of whom were heterozygous for this variant except for P1’s older brother, P2, who is also homozygous (Fig. E1A of the Supplementary Materials). As P2 is clinically well, with no history of immune system disease, we extended his genetic analysis to WES. Of the nine genes with potentially disease-causing variants found in P1, segregation of the siblings was found in the genes for TAF5L, EPAS1, and TG; none of which were thought to be relevant for reasons elaborated in the Supplementary Materials linked to this manuscript.
Immunological phenotype
Both P1 and P2 demonstrated no expression of GIMAP6 protein on western blot analysis (Fig. 1). Lymphocyte subpopulations were largely in line with age-matched controls (Table E2), and lymphocyte activation, proliferation, and cytokine release assays were similar to those of healthy donors (HD) (Figs. E2–E4). We noted a slight increase in CD21lo B cells in P1, and both patients displayed elevated levels of double-negative αβ T cells.
Fig. 1. Immunoblot for GIMAP6 protein expression in PBMCs of the patients and control subjects.
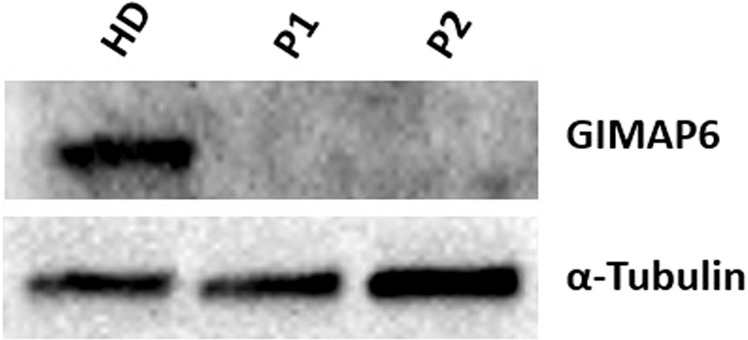
Cell lysates were prepared from P1 and P2 or healthy donor (HD) PBMCs. Cell lysates were separated through 4–20% gradient SDS-PAGE, and immunoblotted for GIMAP6 protein expression. α-Tubulin was used as a loading control.
Figure 2 demonstrates the accelerated apoptosis of both patients’ peripheral blood mononuclear cells (PBMCs), CD4+, and CD8+ cells. PBMCs of both P1 and P2 displayed baseline as well as post-stimulation increase in caspase-3 activity when compared to HD, indicative of increased apoptosis, and P1 displayed more apoptosis activity than P2. In assessing the CD4+ compartment, unstimulated PBMCs from P1 and P2 demonstrated similar levels of active caspase-3 cleavage at 0, 3, and 16 h of incubation, which was increased when compared to HD CD4+ cells. In response to stimulation with anti-CD3/CD28, at all time points both P1 and P2 demonstrated increased caspase-3 activity when compared to HD. Similarly, the CD8+ T cells of P1 and P2 demonstrated increased caspase-3 activity when compared to HD in media (unstimulated), anti-CD3/CD28 and PHA, with cells of P1 again demonstrating increased caspase-3 activity when compared to the cells of P2. This was true at all time points of the assay.
Fig. 2. Stimulation-induced apoptosis.

PBMC apoptosis in response to anti-CD3/CD28 and PHA stimulation. PBMCs from healthydonor (HD, continuous line), P1 (dashed line), and P2 (dotted line) were analyzed for intracellular activated Caspase-3 expression following exposure to anti-CD3/CD28 or PHA for 0, 3, 16, and 25 h. PBMCs (left panels) gated on CD4+ (middle panels) and CD8+ (right panels) were analyzed for cleaved Caspase-3 expression. Significant values were indicated as follows: *p < 0.05; **p < 0.01; ***p < 0.005.
Discussion
GIMAP6 is a member of the “GTPase of Immunity-Associated Proteins” family of proteins, which consists of seven human genes and one pseudogene, all encoded at chromosome 7q36.1 [4]. This is a complicated family of proteins united by a common GTPase-binding motif and predominantly expressed in hematopoietic cells, particularly lymphoid cells [6, 12, 13]. The positioning of the GIMAP genes relative to one another along chromosome 7 is conserved among all vertebrates and some higher plants, which suggests conservation throughout evolution despite species divergence [3, 6, 8]. Given the centrality of GTP binding to cellular functioning, this is perhaps not surprising [4, 5]. The role of the GIMAP proteins appears strongly associated with lymphocyte apoptosis, particularly at key junctures during lymphocyte development, selection, and survival [3, 7, 12]. Balanced lymphocyte death and survival is vital to a healthy immune system and the expression and interaction of various GIMAPs is closely linked with pro- and anti-apoptotic factors in concert with cell survival and death signals supplied via the T cell receptor (TCR), cytokines, and various other cell surface receptors [2, 3]. Despite this seemingly central role, knockout models of GIMAP deficiencies performed in mice have in some instances shown no impact on lymphocyte development, and much about their function remains unknown [7].
GIMAP1 appears to be important in early T lymphocyte development and is highly expressed in double-positive lymphocytes [2]; it has also been shown in mouse models that GIMAP1 protein deficiency is associated with a severe decrease in peripheral T and B lymphocytes [8]. GIMAP2 has no known role in either mice or humans [3] and GIMAP3 is sometimes referred to as a pseudogene [3, 4, 8], although there is some suggestion that it is transcribed with GIMAP5 in a redundant manner during the early stages of thymocyte development, at least in a mouse model [2, 3, 11]. GIMAP4 has been found, in mice models, to accelerate apoptosis [3, 7, 9, 13]. GIMAP7 has no known functional role [3] and GIMAP8 is reported to be an apoptosis inhibitor, protecting NIH 3T3 and CHO-K1 cells from anisomycin-induced apoptosis [8].
GIMAP5 is the best characterized of the GIMAP family, and the only member with a described human case with homozygous variants in the GIMAP5 gene. A 2018 paper by Patterson et al. [12] described GIMAP5 protein as being localized to multi-vesicular bodies and lysosomes, and acting to inhibit GSK3β. GSK3β normally acts to phosphorylate a range of substrates like c-Myc, Nuclear Factor of Activated T Cells 1 (NFATc1), Mcl-1, and β-catenin, all of which are required for lymphocyte differentiation, proliferation, and are involved in apoptosis. In the absence of GIMAP5 protein, GSK3β is not inhibited and these processes are impaired. The patient described in this paper suffered from splenomegaly, hepatomegaly, and lymphopenia affecting CD4+, CD8+, and NK cells with impaired lymphocyte proliferation. Knockout cell lines and the patient’s own cells were treated with lithium chloride, itself a GSK3β inhibitor, which rescued the phenotype in vitro. In vivo treatment with lithium chloride was not reported, but the potential for treatment with a readily available targeted therapy is certainly promising [12]. A rat model of GIMAP5 knockout demonstrated increased activation of nuclear factor kappa B (NFKβ), with a subsequent Th1–Th2 imbalance and the development of autoimmunity [2, 14], and in another study Jurkat cells were protected from the effects of okadaic acid and γ-irradiation by the presence of GIMAP5 protein [8].
While the two patients we have described are the first documented human cases with homozygous GIMAP6 variants, GIMAP6 deficiency has been studied both in vitro and in vivo. A 2017 paper by Ho and Tsai [8] used Jurkat cell lines to demonstrate that GIMAP6 is anti-apoptotic and predominantly found in T lymphocytes. Knock down of GIMAP6 by short hairpin-RNA leads to increased, and more rapid, cell death upon exposure to hydrogen peroxide, FasL, and okadaic acid. It also lead to more rapid T cell activation following stimulation by PMA and ionomycin, and the authors showed for the first time that GIMAP6 can function as an ATPase (the only member of the GIMAP family shown to do so) [8]. In 2018, Pascall et al. [15, 16] established a knockout mouse model of GIMAP6 deficiency and showed that these mice suffered a 50–70% reduction in the numbers of circulating CD4+ and CD8+ lymphocytes as well as reduced autophagy.
The experiments we performed to investigate the lymphocyte function of our two patients were largely based upon those of Ho and Tsai [8], to determine if this cellular phenotype was replicated in human cases of GIMAP6 protein deficiency. The most impressive finding from our patients is that both demonstrated an increased susceptibility to apoptosis, with increased caspase-3 activity compared to HD at all time points upon exposure of CD4 and CD8 lymphocytes to anti-CD3/CD28 (this was also true upon exposure to PHA for P1, but not for P2). CD4 and CD8 lymphocytes of both P1 and P2 also demonstrated increased baseline levels of FAS receptor (CD95) and HLA-DR expression, but activation in response to anti-CD3/CD28 and PHA was comparable to control for both patients, as was proliferative ability and cytokine expression. The significance of this heightened immune activation and its correlation to the patients’ clinical findings is unclear, particularly given that P2 has no outward features of immune system dysfunction. Both patients displayed elevated levels of double-negative αβ T cells and P1 expressed higher levels of CD21lo B lymphocytes, phenomena which are known to be associated with autoimmunity, particularly in autoimmune lymphoproliferative disease [17] and common variable immune deficiency [18].
Interestingly, T cell function was only slightly affected in P1 and P2, and they both demonstrated broadly normal lymphocyte subpopulations, with only minor deviations from the normal range for children their age. Given that the clinical phenotype of P2 is milder than P1, it is not surprising that P1’s cellular phenotype is more severely affected than that of her older brother. It remains unclear, however, why a discrepancy exists in both clinical and cellular phenotype between these two siblings despite both inheriting the same variants and demonstrating complete absence of the GIMAP6 protein. Heterozygous variants in MEFV have been described as disease modifiers in conditions such as systemic lupus erythematosus, however attributing causality in P1’s case would be purely speculative [19]. It is also unclear exactly how P1’s increased predisposition to apoptosis explains her clinical phenotype which, although mild, has resulted in lymphopenia, recurrent acute otitis media, lung disease, and hepatosplenomegaly; however, these features are noted in other immune defects associated with apoptosis [17]. While P1 does not require replacement of intravenous immunoglobulin, prophylactic antibiotic therapy, or a hematopoietic stem cell transplant at this stage, her future clinical course is difficult to predict. In line with the role of the GIMAP genes in either accelerating or preventing apoptosis, links have emerged between variants in GIMAP genes and various malignancies, including leukemia, hepatocellular carcinoma, and lung cancer [4, 9, 20, 21]. Thus, we have adopted a “watch and wait” approach with reviews every 3–6 months and standard immunological blood tests at each review and early introduction of antibiotics to treat any signs of infection.
One weakness of this study is that we have not repeated the aforementioned experiments in the heterozygous members of this family. Unfortunately, we were not able to obtain blood samples from them due to restricted movements of patients and families over the Gaza–Israeli border, particularly due to the coronavirus pandemic.
These cases again underscore the utility of WES in modern clinical immunology. Although P1 displays a relatively mild clinical phenotype (and P2 is asymptomatic), a genetic variant has been found that explains her symptoms. In addition to ensuring more prompt introduction of anti-microbial therapy and increased malignancy surveillance, the diagnosis of homozygous GIMAP6 variants has extended our understanding of the GIMAP family of genes and the clinical spectrum of disease associated with them. Much remains to be learnt about the mechanisms linking the genetic variant with the clinical features. We suspect there may be some redundancy protecting against excessive apoptosis, with other genes playing similar roles to GIMAP6. There may also be gene–gene interactions or epigenetic changes that explain the differences between the two siblings, as well as differences in microbial exposure between them.
Thus, in this manuscript we have described the first human cases of a deleterious homozygous GIMAP6 variant and shown that patients exhibit increased apoptosis, but a relatively mild clinical phenotype in one sibling and no clinical features in her older brother. GIMAP6 appears to play only a minor role in lymphocyte activation and proliferation in humans, but we will continue to monitor both P1 and P2 for development of additional disease. This study once again emphasizes the utility of next-generation sequencing in extending our understanding of primary immune deficiency [22–24].
Supplementary information
Supplementary materials with active track changes to indicate changes made since last review
Acknowledgements
We would like to thank our patient and her family for allowing us to publish this manuscript. We would also like to thank our departmental nursing and administrative staff for their assistance in caring for this patient. We would also like to thank Professor Zeev Rotstein, Director of The Hadassah Medical Center, for his support of our department and our patients.
Funding
BS’s position is supported by the Australian Government Research Training Program Scholarship and Hadassah, Australia. This work was supported by the Deutsche Forschungsgemeinschaft (Discovery and Evaluation of new Combined Immunodeficiency Disease Entities (DECIDE); grant DFG WA 1597/4-2) and the ERA-Net ERARE Consortium EURO-CID.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Bella Shadur, Nathalie Asherie, Shlomit Kfir-Erenfeld
These authors jointly supervised this work: Hagar Mor-Shaked, Polina Stepensky
Supplementary information
The online version of this article (10.1038/s41431-020-00773-x) contains supplementary material, which is available to authorized users.
References
- 1.Casanova J, Conley M, Seligman S, Abel L, Notarangelo L. Guidelines for genetic studies in single patients: lessons from primary immunodeficiencies. J Exp Med. 2014;211:2137–49. doi: 10.1084/jem.20140520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ciucci T, Bosselut R. Gimap and T cells: a matter of life or death. Eur J Immunol. 2014;44:348–51. doi: 10.1002/eji.201344375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nitta T, Takahama Y. The lymphocyte guard-IANs: regulation of lymphocyte survival by IAN/GIMAP family proteins. Trends Immunol. 2006;28:58–65. doi: 10.1016/j.it.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Krucken J, Schroetel R, Muller I, Saidani N, Marinovski P, Benten P, et al. Comparative analysis of the human gimap gene cluster encoding a novel GTPase family. Gene. 2004;341:291–304. doi: 10.1016/j.gene.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Filen S, Lahesmaa R. GIMAP proteins in T-lymphocytes. J Signal Transduct. 2010;10:268589. 10.1155/2010/268589. [DOI] [PMC free article] [PubMed]
- 6.Poirier G, Anderson G, Huvar A, Wagaman P, Shuttleworth J, Jenkinson E, et al. Immune-associated nucleotide-1 (IAN-1) is a thymic selection marker and defines a novel gene family conserved in plants. J Immunol. 1999;163:4960–9. [PubMed] [Google Scholar]
- 7.Saunders A, Webb L, Janas M, Hutchings A, Pascall J, Carter C, et al. Putative GTPase GIMAP1 is critical for the development of mature B and T lymphocytes. Blood. 2010;115:3249–57. doi: 10.1182/blood-2009-08-237586. [DOI] [PubMed] [Google Scholar]
- 8.Ho C, Tsai S. Functional and biochemical characterization of a T cell-associated anti-apoptotic protein, GIMAP6. J Biol Chem. 2017;292:9305–19. doi: 10.1074/jbc.M116.768689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shiao Y, Chang Y, Liu Y, Li J, Su J, Liu K, et al. Dysregulation of GIMAP genes in non-small cell lung cancer. Lung Cancer. 2008;62:287–94. doi: 10.1016/j.lungcan.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 10.Dalberg U, Markholst H, Hornum L. Both Gimap5 and the diabetogenic BBDP allele of Gimap5 induce apoptosis in T cells. Int Immunol. 2007;19:447–53. doi: 10.1093/intimm/dxm009. [DOI] [PubMed] [Google Scholar]
- 11.Yano K, Carter C, Yoshida N, Abe T, Yamada A, Nitta T,, et al. Gimap3 and Gimap5 cooperate to maintain T-cell numbers in the mouse. Eur J Immunol. 2014;44:561–72. doi: 10.1002/eji.201343750. [DOI] [PubMed] [Google Scholar]
- 12.Patterson A, Endale M, Lampe K, Aksoylar H, Flagg A, Woodgett J, et al. Gimap5-dependent inactivation of GSK3β is required for CD4+ T cell homeostasis and prevention of immune pathology. Nat Commun. 2018;9:430. 10.1038/s41467-018-02897-7. [DOI] [PMC free article] [PubMed]
- 13.Schwefel D, Arasu B, Marino S, Lamprecht B, Kochert K, Rosenbaum E, et al. Structural Insights into the mechanism of GTPase activation in the GIMAP family. Structure. 2013;21:550–9. doi: 10.1016/j.str.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 14.Hellquist A, Zucchelli M, Kivinen K, Saarialho-Kere U, Koskenmies S, Widen E, et al. The human GIMAP5 gene has a common polyadenylation polymorphism increasing risk to systemic lupus erythematosus. J Med Genet. 2007;44:314–21. doi: 10.1136/jmg.2006.046185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pascall J, Webb L, Eskelinen E, Innocentin S, Attaf-Bouabdallah N, Butcher G. GIMAP6 is required for T cell maintenance and efficient autophagy in mice. PLoS ONE. 2018;13:e0196504. 10.1371/journal.pone.0196504. eCollection 2018. [DOI] [PMC free article] [PubMed]
- 16.Pascall J, Rotondo S, Mukadam A, Oxley D, Webster J, Walker S, et al. The Immune system GTPase GIMAP6 interacts with the Atg8 homologue GABARAPL2 and is recruited to autophagosomes. PLoS ONE. 2013;8e77782. 10.1371/journal.pone.0077782. eCollection 2013. [DOI] [PMC free article] [PubMed]
- 17.Fisher G, Rosenberg F, Straus S, Dale J, Middleton L, Lin A, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–46.. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 18.Rakhmanov M, Keller B, Gutenberger S, Foerster C, Hoenig M, Driessen G, et al. Circulating CD21low B cells in common variable immunodeficiency resemble tissue homing, innate-like B cells. Proc Natl Acad Sci USA. 2009;106:13451–6. doi: 10.1073/pnas.0901984106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shinar Y, Kosach E, Langevitz P, Zandman-Goddard G, Pauzner R, Rabinovch E, et al. Familial Mediterranean Fever gene (MEFV) mutations as a modifier of systemic lupus erythematosus. Lupus. 2012;21:993–8. doi: 10.1177/0961203312441048. [DOI] [PubMed] [Google Scholar]
- 20.Huang Z, Zhang W, Gao C, Ji B, Chi X, Zheng W, et al. Dysregulation of GTPase IMAP family members in hepatocellular cancer. Mol Med Rep. 2016;14:4119–23. doi: 10.3892/mmr.2016.5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanati N, Iancu O, Wu G, Jacobs J, McWeeney S. Network-based predictors of progression in head and neck squamous cell carcinoma. Front Genet. 2018;9:183. 10.3389/fgene.2018.00183. eCollection 2018. [DOI] [PMC free article] [PubMed]
- 22.Wang D, Zhang S, Liu B. TAF5L functions as transcriptional coactivator of MITF involved in the ummune response of the clam Meretrix petechialis. Fish Shellfish Immunol. 2020;98:1017–23. doi: 10.1016/j.fsi.2019.11.039. [DOI] [PubMed] [Google Scholar]
- 23.Schelker RC, Herr W, Grassinger J. A new exon 12 mutation in the EPAS1 gene possibly associated with erythrocytosis. Eur J Haematol. 2019;103:64–6. doi: 10.1111/ejh.13241. [DOI] [PubMed] [Google Scholar]
- 24.Narumi S, Muroya K, Asakura Y, Aachi M, Hasegawa T. Molecular basis of thyroid dyshormonogenesis: genetic screening in population-based Japanese patients. J Clin Endocrinol Metab. 2011;96:E1838–42. doi: 10.1210/jc.2011-1573. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials with active track changes to indicate changes made since last review