

Abstract
Protein-protein interactions (PPIs) play a critical role in fundamental biological processes. Competitive inhibition of these interfaces requires compounds that can access discontinuous binding epitopes along a large, shallow binding surface area. Conformationally-defined protein surface mimics present a viable route to target these interactions. However, the development of minimal protein mimics that engage intracellular targets with high affinity remains a major challenge because mimicry of a portion of the binding interface is often associated with the loss of critical binding interactions. Covalent targeting provides an attractive approach to overcome the loss of non-covalent contacts but have the inherent risk of dominating non-covalent contacts and increasing the likelihood of non-selective binding. Here, we report the iterative design of a proteolytically-stable α3β chimeric helix mimic that covalently targets oncogenic G12C Ras as a model system. We explored several electrophiles to optimize preferential alkylation with the desired C12 on Ras. The designed lead peptide modulates nucleotide exchange, inhibits activation of the Ras-mediated signaling cascade, and is selectively toxic towards mutant G12C Ras cancer cells. The relatively high frequency of acquired cysteines as missense mutations in cancer and other diseases suggests that covalent peptides may offer an untapped therapeutic approach for targeting aberrant protein interactions.
Graphical Abstract
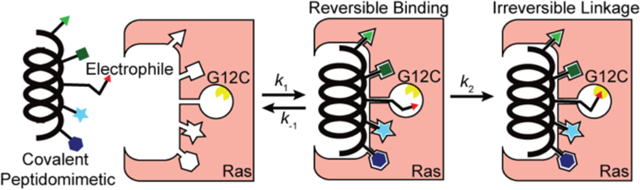
INTRODUCTION
Activation of the small GTPase Ras is a tightly regulated process that involves GDP release and GTP binding, and is catalyzed predominantly by the guanine nucleotide exchange factor (GEF) Son of Sevenless (Sos).1 When bound to GTP, Ras adopts a conformation that enables interaction with effector proteins with high affinity.2 Ras deactivation occurs upon the intrinsic hydrolysis of GTP to GDP, which is stimulated by GTPase activating proteins (GAPs).3 Aberrant activation of Ras can arise by various mechanisms that deregulate these processes including somatic or germline mutations in genes encoding GEFs, GAPs, receptor tyrosine kinases (RTKs), and Ras itself.4 G12C is an activating K-Ras mutation in approximately 10–20% of all Ras-driven cancers. Shokat, Wells, and coworkers have described covalent small molecules, which inhibit nucleotide exchange and Ras signaling.5 The lead compound was discovered using a fragment screening assay to access a pocket near the Ras Switch I/II regions and allosterically inhibit nucleotide exchange (Figure 1). Advanced derivatives of this lead compound have also been described.6, 7
Figure 1.
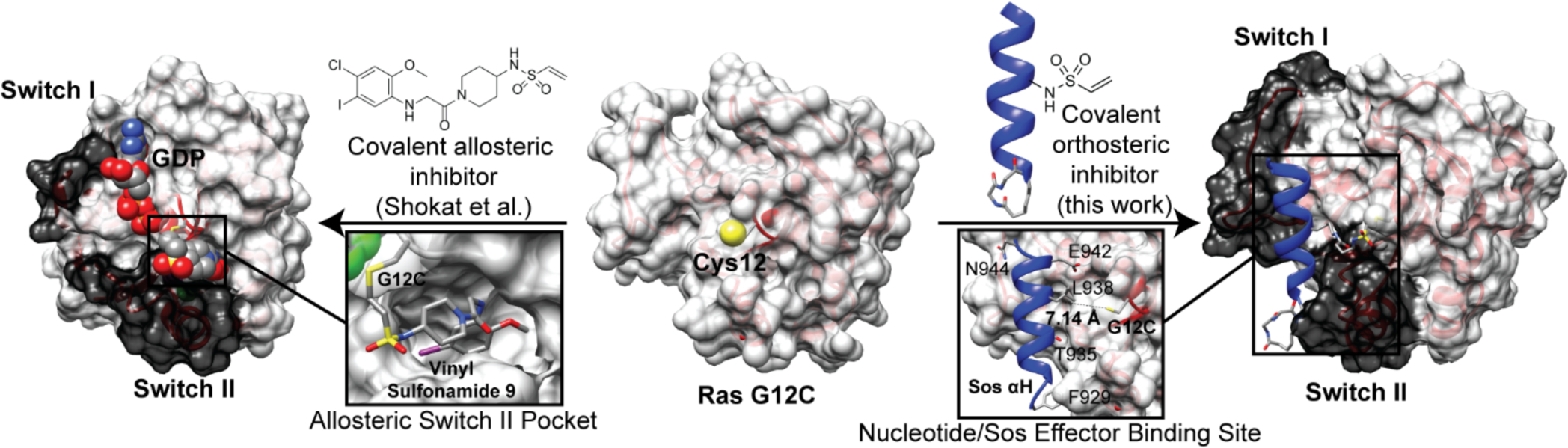
Potential routes for covalent inhibition of G12C Ras. (Left) Shokat et al. recently revealed a small molecule that occupies an allosteric pocket and inhibits Ras signaling (PDB: 4LYH). (Right) We aimed to develop a covalent orthosteric ligand by mimicking the contact helix from Sos (PDB: 1NVW)
Sos catalyzes nucleotide exchange by binding Ras at its conformationally dynamic Switch I/II surface and inserting a helical hairpin into the nucleotide binding region.2 Mimics of the Sos helical hairpin would be expected to compete with Sos binding and inhibit its GEF activity. Based on this hypothesis, we previously developed ligands that mimic the contact αH helix of Sos.8 The stabilized peptide helix (HBSSOS) was shown to bind Ras on the Switch surface and inhibit Ras activation.8 A shortcoming of this Sos mimicry approach is that Ras and Sos are representative of weak PPIs with a KD of 28 μM in the absence of membrane localization.9 The Sos αH helix mimic also binds Ras with mid-micromolar affinity,8 but we postulated that this orthosteric ligand may serve as a scaffold for covalent targeting of the mutant G12C Ras (Figure 1).
Specific targeting by covalent ligands requires that noncovalent interactions drive molecular recognition before the warhead reacts with the receptor.10 The potential for specific protein targeting is reduced if the native binding affinity of the non-covalent portion is not high.11 Although many classes of covalent inhibitors have been described, and their use as drug candidates is accelerating,12, 13 the best candidates feature high affinity non-covalent components.11, 14 It remains to be determined if protein surface mimics, which often bind the target with weak affinity, can be tuned to achieve selective covalent labeling. The extensive Ras–Sos PPI captures this challenge for inhibitor design,15, 16 and HBSSOS represents a case study for covalent peptidomimetic design: namely, can a rationally designed weak inhibitor be converted into a potent and specific cellular reagent with an appropriately reactive warhead?
Only a few examples of covalent inhibitors derived from protein surface mimicry have been described. de Araujo et al. developed a constrained peptide based on the pro-apoptotic BIM BH3 helix to covalently target Cys55 of Bcl2A1.17 Huhn et al. also employed a stapled BH3 peptide for inhibiting BFL-1, a Bcl2 homolog.18 Both groups utilized acrylamide-based warheads against the nucleophilic residue at relatively short distances (3.6–3.9 Å). The proximity of the nucleophilic cysteine peptide allow accommodation for the relatively slow and chemoselective covalent labeling of the acrylamide group. Targeting of Ras by Sos mimics is likely to be more challenging both because of the lower cognate affinity for the non-covalent complex formation and distance the reactive nucleophile (C12) on Ras resides from the nearest Sos residue (>7 Å) (PDB: 1NVW). This distance necessitates a long linker from the peptide backbone, increasing the likelihood for non-specific alkylations with other nucleophiles.
Excluding the C-terminal membrane-targeting region, H-Ras contains three native cysteine residues beyond the G12C mutation, providing built-in controls to probe the specificity of electrophilic Sos mimics. We developed a series of covalent ligands to correlate the reactivity of the electrophile with specific labeling of cysteine-12 of Ras. The overall workflow is depicted in Figure 2. We began our studies by designing a conformationally-defined Sos mimic to engage Ras at its binding surface. This constrained peptide was then converted to a covalent Sos mimic for initial testing and warhead optimization. Further improvements to the lead compound required a proteolytically stable main chain consisting of an α3β chimeric sequence. These studies resulted in a proteolytically stable, cell-permeable covalent Sos derivative, which is selectively toxic towards G12C Ras over wild-type (WT) and G12V Ras cell lines.
Figure 2.
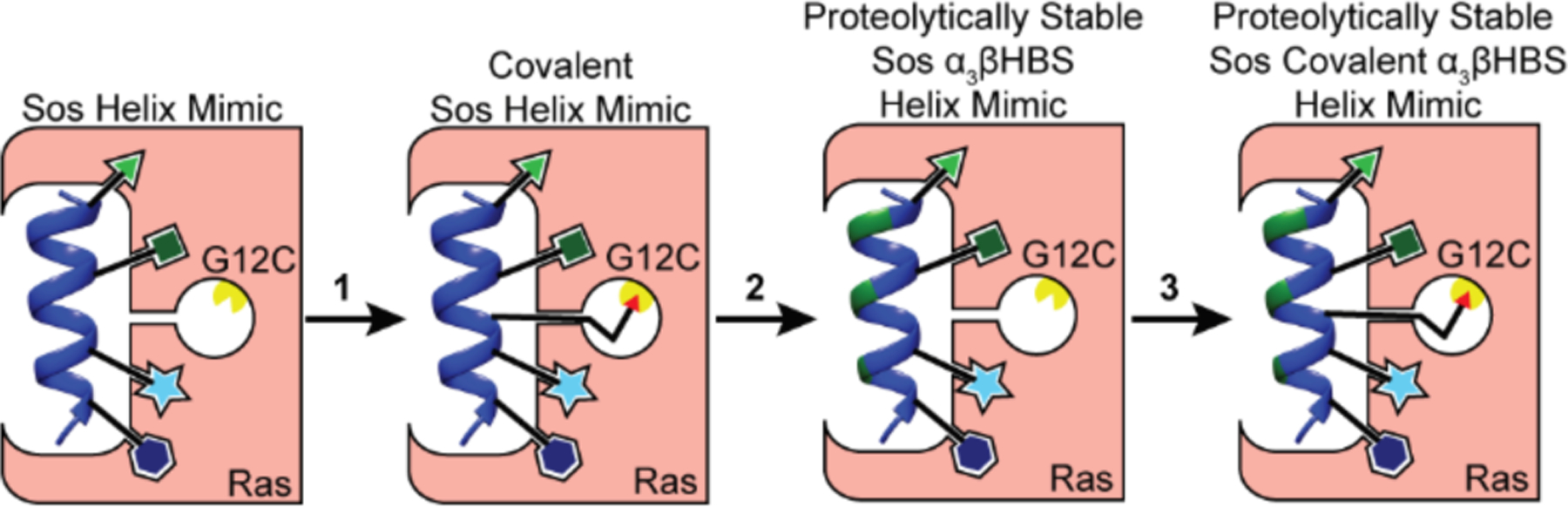
General workflow for covalent inhibition of G12C Ras: (1) Electrophilic warhead optimization, (2) incorporation of β-residues to enhance proteolytic stability, and (3) addition of optimal electrophile to generate lead compound. Green shading in helix ribbon denotes β-residues.
RESULTS AND DISCUSSION
Development of Proteolytically Resistant Sos αH Mimics.
The high-resolution structure of Ras in complex with Sos depicts a large interacting surface area encompassing >3600 Å2, making it particularly difficult to disrupt with small molecules.2 The critical αH helix of Sos forms several direct contacts with the GTP-binding Switch I and Switch II regions of Ras, making it an attractive target and basis for helix mimicry.19 Computational alanine scanning data on this helix supports the experimental observation that four residues (F929, T235, E942 and N944) are essential for binding, with residues F929 and N944 making critical contacts to Ras (Table S1). Based on our structural and computational analyses, we designed HBSSOS as a ligand for Ras. The design required extensive peptide engineering to obtain a water-soluble, stable α-helix that engages Ras.8
To mimic Sos αH, we generated a helical peptide utilizing the hydrogen bond surrogate (HBS) approach, which creates a macrocycle at the N-terminus to nucleate an α-helix (HBSSOS).20 The HBS method involves the replacement of an N-terminal backbone hydrogen bond with an isosteric, covalent carbon-carbon bond (Figure 3A). The resulting preorganized HBS macrocycle encourages the initial nucleation and subsequent propagation of a helical conformation in the remaining peptide.21 Several strategies for helix mimicry have been employed to modulate a diverse variety of protein-protein interactions.22–26 The unique advantage of the HBS strategy is that in contrast to the side-chain helix stapling method,27 it replaces a main-chain hydrogen bond for helix stabilization and allows all side chain residues to remain available for molecular recognition.28
Figure 3.
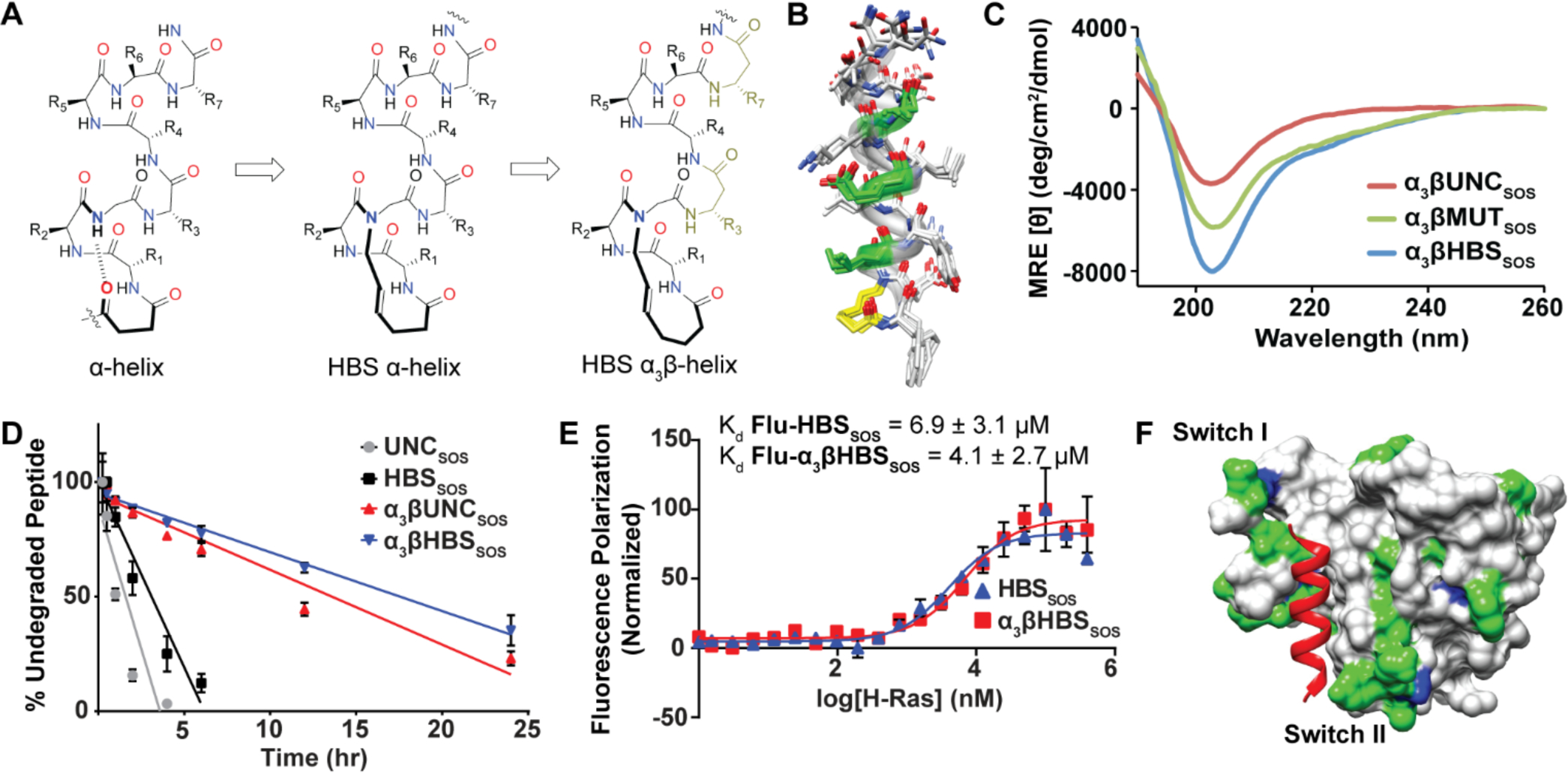
(A) Incorporation of β3-residues within each turn of the HBS helix. (B) Ensemble of the 10 lowest energy structures derived from 2D NMR spectroscopy of α3βHBSSOS (yellow) with β3-residues in green. (C) CD spectra of α3βHBSSOS, α3βMUTSOS, and α3βUNCSOS. (D) Proteolytic degradation of HBS peptides by trypsin over 24 hours. Error bars are mean ± STD of biological triplicates. (E) Fluorescence polarization binding assays of Flu-HBSSOS and Flu-α3βHBSSOS for WT H-Ras. (F) Ribbon view of the 1H-15N-Ras-α3βHBSSOS interaction titrated with increasing amounts of peptide. Residues are colored according to mean chemical shift change: minimal (gray), moderate (green), and significant (blue)
HBSSOS was shown to bind recombinant H-Ras with micromolar affinity, within range of full-length Sos.8 1H-15N-HSQC NMR titration experiments with the peptide revealed concentration-dependent chemical shifts for Ras residues corresponding to the binding cleft of the native Sos – namely, the flexible Ras switch I and II regions.8 HBSSOS was also shown to modulate Ras activation and ERK signaling in a concentration-dependent manner.8 These earlier results provide a firm basis for the generation of an orthosteric covalent ligand for the oncogenic G12C Ras isoform.
We began the current project by converting HBSSOS into a proteolytically stable helix mimic. In an earlier report, we investigated the proteolytic stability of HBS helices composed of α-amino acids, and found a direct correlation between helicity and proteolytic stability since proteases bind and cleave peptides in the extended conformation.29 However, the extent of proteolytic stability of HBS α-helices was found to be sequence and length dependent. Inspired by the ability of β-peptides and chimeric α/β-peptides to resist degradation,30 we sought to examine if insertion of β3-residues within an α-peptide chain could lead to more stable HBS helices which also retain their functional properties. We previously reported the design of HBS α3β motifs which contain one β3-residue per helical turn such that the heterogeneous sequence contains three α-residues followed by one β3-residue. Such α3β sequences have been characterized as suitable mimics of α-helices. The design of conformationally stable α-helix mimics from α3β-sequences required proper alignment of the hydrogen-bonding patterns and a 14-membered HBS macrocycle as opposed to the 13-membered ring HBS macrocycle in the helix composed of all α-residues (Figure 3A).31 Extensive NMR and circular dichroism studies on model sequences showed that the HBS α3β motif is conformationally stable and can bind to the desired protein receptors with similarly high affinity as HBS helices composed of purely α-amino acids.31
We applied the α3β design strategy to the HBSSOS helix to create α3βHBSSOS. β-residues were inserted along the non-interacting face of the Sos αH helix, based on the observation that α3β-sequences displayed higher binding affinity when non-binding residues were mutated to their β-analogues. As mentioned above, experimental and computational mutagenesis experiments show that F929, E942, and N944 of Sos are essential for complex formation. Helical faces incorporating these hot spot residues were left unaltered, and the non-binding residues at positions 932, 936, and 940 were replaced with the corresponding β-amino acid residues (α3βHBSSOS). For the initial biophysical characterization experiments, two control peptides were generated: 1) the unconstrained α3β sequence lacking the HBS macrocycle (α3βUNCSOS) and 2) the alanine mutant α3βHBS helix (α3βMUTSOS), where the hot spot residues for Ras binding (F929, E942, and N944) are replaced with alanine.
Structural and Biophysical Characterization of α3βHBSSOS.
To assess the conformational rigidity and helical propensity of α3βHBS of the Sos αH helix, we utilized a combination of 2D NMR Nuclear Overhauser effect (NOESY) and circular dichroism (CD) spectroscopies. NMR data concludes that α3βHBSSOS exhibits pronounced helical character. While both α3βHBSSOS and α3βUNCSOS possessed several short (NN(i,i+1)) and medium-range (αN(i,i+4)) NOEs, only the HBS sequence exhibited long-range (αN(i,i+4)) NOE contacts, which are indicative of a stable helical conformation. The resulting solution structures of the constrained and unconstrained α3β sequences were determined from NOESY cross-peaks and 3JNH-CαH coupling constants via a Monte-Carlo-based conformational search and energy minimization protocol. For α3βHBSSOS, the molecular dynamics calculations utilized a total of 92 NOE restraints (16 medium- and long-range, 33 sequential, and 43 intra-residue) and 15 angle restraints. No explicit hydrogen-bond restraints were used in these calculations. The final 10 lowest energy structures had no significant distance violations. The 20-conformer ensemble obtained for the peptide shows a backbone root mean squared deviation (RMSD) of 0.61 ± 0.39 Å. Overall, the NMR structure of α3βHBSSOS confirms that a well-defined conformation is accessed in this sequence (Figure 3B). In contrast, the calculated NMR structure of the unconstrained sequence (α3βUNCSOS) suggests that the sequence lacks helical stability without the HBS constraint (Figure S1). CD studies support the conclusions derived from NMR spectroscopy that α3βHBSSOS possess high helical character. The CD spectra trace of α3βHBSSOS shows a global minimum near 205 nm and maximum at 190 nm, consistent with previous reports for α3β helices.23, 31, 32 In contrast, the CD trace for α3βUNCSOS was less intense, indicating its lower helical content (Figure 3C). CD spectra indicate that α3βHBSSOS is more helical than α3βMUTSOS, likely due to the potential loss of an i to i +3 ionic interaction between K939 and E942 upon mutation of the latter to alanine.
Proteolytic Stability of α3βHBSSOS.
Enzymatic proteolysis is an important consideration when studying the effects of peptidic compounds in cellular models. By design, α3βHBS peptides are predicted to resist proteolytic degradation based on the stability of heterogeneous α/β unconstrained peptides.33 For our initial assays with trypsin, the rate of proteolytic cleavage was determined via an HPLC-based assay with tryptophan serving as the internal control standard (Figure 3D). The Sos sequence contains two trypsin cleavage sites following lysine and arginine, providing site-specific positions for determining the stability of the α and α3β sequences. Comparison of the tryptic digestion rates revealed a ~6-fold increase in half-life for α3βHBSSOS compared to HBSSOS (t1/2 values of 17.6 and 3.1 hours, respectively). Similarly, we observed an 8.5-fold improvement in half-life for α3βUNCSOS over αUNCSOS (t1/2 = 13.6 and 1.6 hours, respectively). We observed similarly high resistance to proteolytic degradation of α3βHBSSOS using 25% human serum (Figure S2A) and proteinase K, which targets aromatic and aliphatic residues (Figure S2B). Comparison of proteolysis rates illustrated a ~4-fold increase in half-life for α3βHBSSOS in serum and a 360-fold increase with proteinase K.
α3βHBSSOS Engages Ras Within the Nucleotide Binding Pocket.
We next determined the potential of α3βHBSSOS to bind Ras using a fluorescence polarization assay with purified recombinant HRas and fluorescein-labeled peptides.8 In nucleotide-free conditions, the α-peptide-based Flu-HBSSOS bound to wild-type H-Ras with an affinity of 6.9 ± 3.1 μM, which is comparable to previously reported values (Figure 3E). As expected based on prior results, Flu-α3βHBSSOS also bound with similar affinity (4.1 ± 2.7 μM). The dissociation constant of Flu-α3βHBSSOS for Ras is ~4-fold lower than the KD values obtained for the catalytic domain of Sos (SosCAT)9, indicating the potential utility of α3βHBS peptides as orthosteric inhibitors of the Ras-Sos interaction.
We further interrogated the α3βHBSSOS-Ras binding interaction with titration 1H-15N-HSQC NMR spectroscopy. The protein 15N chemical shift changes upon titration with ligand indicate residues potentially involved in binding. According to the crystal structure of the Ras-Sos complex (PDB: 1NVW),2 the native Sos αH helix binds between the flexible Switch I/II regions of Ras to mediate nucleotide entry and exit. Titration HSQC experiments with α3βHBSSOS revealed significant shifts in residues from both Ras Switch regions as well as the anti-parallel β-sheet linking them together (Figure 3F, S3–5). Overall, the NMR data provides strong support for the hypothesis that the designed derivative is engaging the desired Ras surface.
Design of Electrophilic HBS Peptides.
Carbon-based electrophiles, i.e. acrylamides, vinyl sulfonamides, and vinyl sulfones, were chosen as warheads due to their chemoselectivity for cysteine and the irreversible nature of the resulting covalent bond.5 These Michael acceptors have been previously employed as electrophiles in covalent inhibitors within clinical contexts.14 Ideally, the initial noncovalent binding interaction of the HBS peptide would allow presentation of the warhead to its intended target protein prior to covalent bond formation (Figure 4A). Computational analysis of the Ras-Sos complex indicates Leu938 has the proper orientation and closest proximity (7.1 Å) from Sos to Ras (PDB: 1NVW). We replaced this residue with lysine for further modification with α,β-unsaturated alkenes. HBS synthesis is described in Supporting Information. Figure 4B lists the series of compounds synthesized based on the HBS scaffold as part of our initial exploration. HBS 1 is a direct analog of the previously published HBSSOS compound with a lysine residue in place of Leu938.8 Addition of electrophiles to 1 yields a set of compounds for initial biochemical evaluations: acrylamides (2a-b), vinyl sulfonamides (3a-b), and vinyl sulfones (4a-b). The derivatives in each electrophile class differ in linker lengths.
Figure 4.
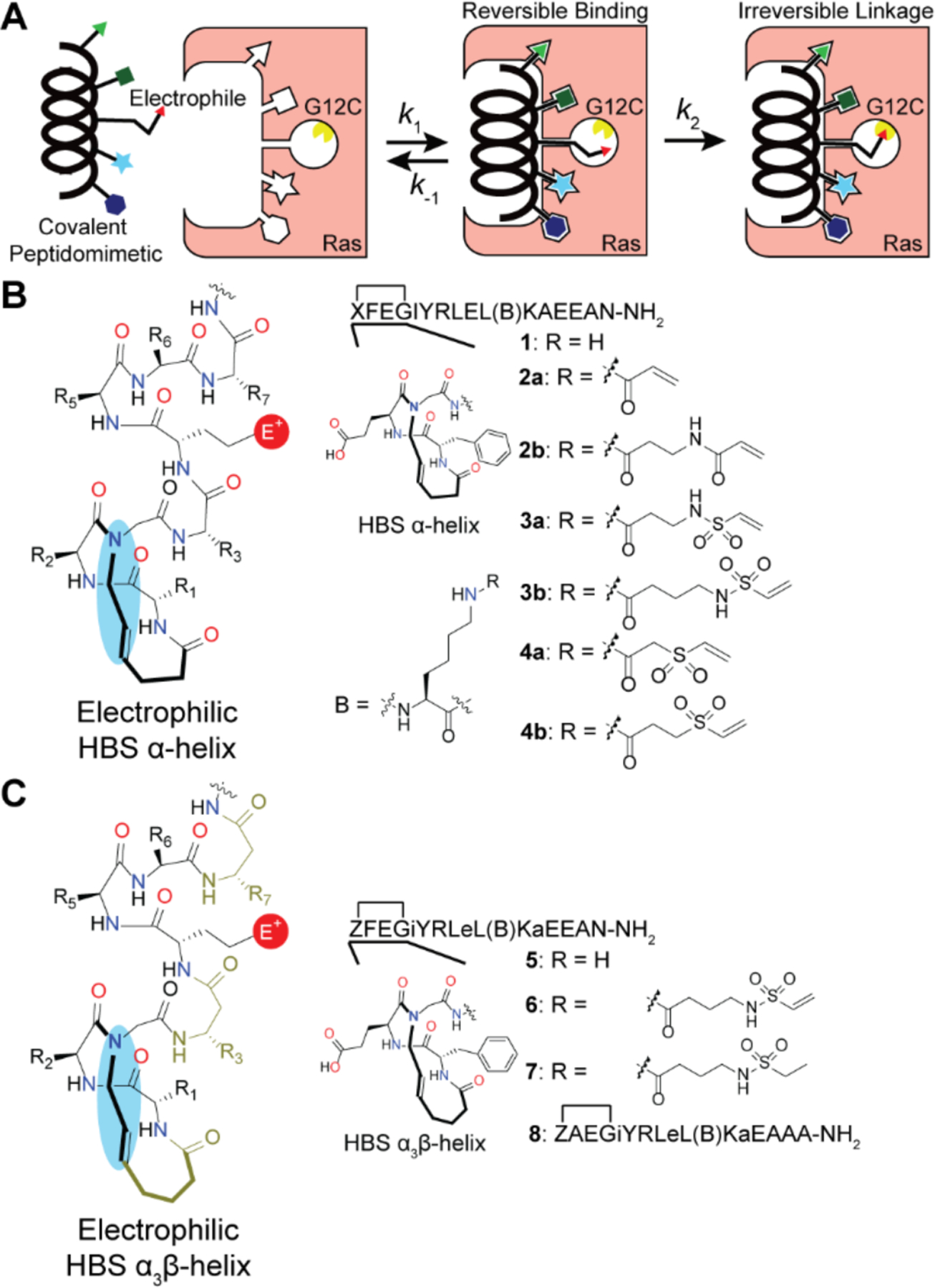
(A) Cartoon schematic depicting binding interactions between the covalent peptidomimetics and target protein (Ras G12C). (B) Covalent HBS designs. (C) Covalent α3βHBS designs. Lower case letters designate β3-residues.
The enhanced proteolytic stability offered by the α3β chimeric helix and its ability to specifically engage the Ras Switch regions with similar affinity as the natural sequence provide a strong basis for the transformation of this sequence into an electrophilic ligand for G12C Ras. Incorporation of the L938K mutation within the α3βHBSSOS sequence yielded the base peptide (5) for our second-generation covalent peptide inhibitors. Attachment of the most promising electrophile, vinyl sulfonamide, generated the proteolytically stable lead α3β sequence (6) (Figure 4C). The saturated warhead control (7) and an alanine mutant sequence with the vinyl sulfonamide group (8) were synthesized as controls.
Site-specific Labeling of Cys-12 With Covalent HBS.
The irreversible binding potency of the covalent peptides was evaluated by MALDI mass spectrometry. We calculated the area under the unlabeled and modified Ras peaks with the assumption that both proteins would have a similar ionization profile. The assay was standardized to 10 μM recombinant H-Ras G12C and an excess of peptide (50 eq.). A comparison of labeling efficiencies after 24 h incubation revealed a trend where higher peptide-Ras adduct formation was observed based on the reactivity of the electrophile. A summary of these results for %Ras labeling is illustrated in Figure 5A. The acrylamides 2a-b are not very reactive. The vinyl sulfones 4a-b are highly reactive and lead to alkylation of multiple cysteine residues on Ras. Both vinyl sulfonamides provide roughly 25% labeling of the desired cysteine alkylation and low non-specific alkylation of other cysteine thiols on Ras (vide infra). Based on the results, we chose 3b as our lead peptide for further evaluation. In general, minor changes in the linker lengths did not lead to substantial differences in the reactivity profiles of different electrophiles.
Figure 5.
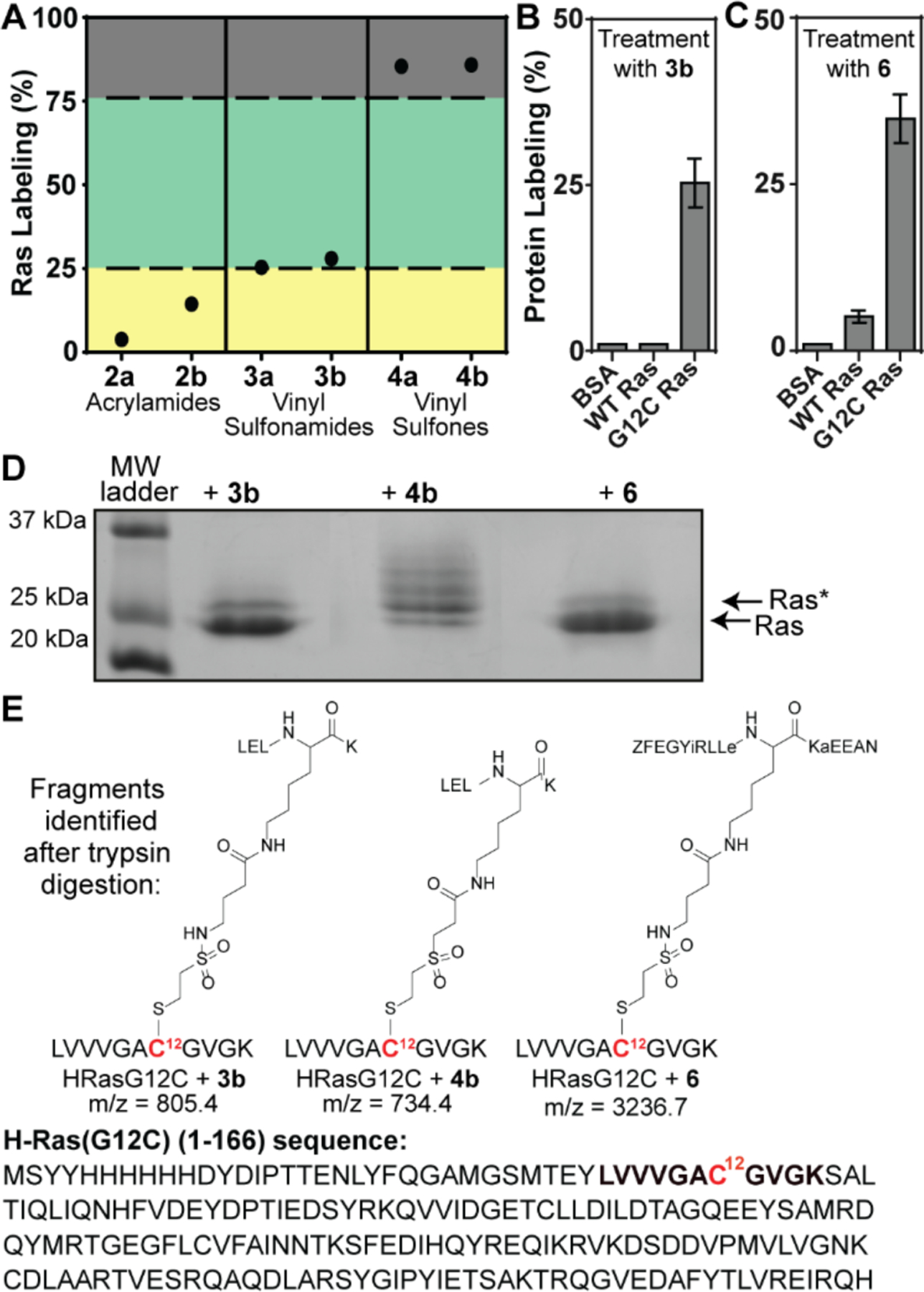
(A) Scatter plot outlining differences in overall reactivity of the three electrophilic moieties: yellow (low), green (medium), and red (high). (B) Covalent labeling of 3b to BSA, WT H-Ras, and H-Ras G12C. (C) Covalent labeling of 6 to BSA, WT H-Ras, and HRas G12C. (D) Gel shift assay with H-Ras G12C labeled with 3b, 4b or 6. Ras and Ras* refer to the unmodified and monoalkylated protein bands, respectively. (E) Fragment structures identified from MS analysis of trypsin-digested monolabeled Ras gel band. Error bars are mean ± SD of at least two biological replicates.
Selective engagement of the target residue requires careful balancing of electrophile reactivity and stability. The vinyl sulfonamide group offers a balance between potency and specificity, especially when considering the relatively modest reversible binding affinity of the native sequence and medium- range distance to the target nucleophile. Proper binding of GDP/GTP within the nucleotide binding site of Ras requires a crucial magnesium ion that coordinates to various neighboring residues.34 Addition of ethylenediaminetetraacetic acid (EDTA) to nucleotide-bound Ras sequesters Mg2+ and predisposes Ras towards its nucleotide-free state. Without EDTA, Ras remains tightly bound to its nucleotide ligand and does not expose the C12 residue to the electrophilic peptides. The recombinant H-Ras G12C sequence contains four cysteine residues that can potentially be targeted by 3b: C12, C51, C80, and C118. While C51 and C80 are buried deep within the hydrophobic core, C12 becomes more accessible to the Sos-derived peptides in the nucleotide-free state upon unfurling of the highly flexible Switch regions of Ras.35 C118 remains amenable to labeling in either conformation and is a useful specificity control for our covalent peptidomimetics.
Treatment with 3b results in the modification of H-Ras G12C but not bovine serum albumin (BSA) and WT Ras (Figure 5B). WT Ras shows no labeling although it contains multiple free cysteines (C51, C80, and C118). BSA contains a single free cysteine residue (along with several disulfide groups) and is also unreactive toward 3b. The specificity of 3b for G12C Ras over wt-Ras highlights the suitability of this compound as a lead. MALDI spectra of samples of Ras G12C incubated with 6 revealed robust labeling of Ras at a similar level as the all-α variant (Figure 5C). Comparison of reactivity of 6 with G12C Ras and wt-Ras shows that this compound is also highly specific for the mutant Ras isoform. As a specificity control, we employed BSA, containing only a single available cysteine residue, and observed no detectable levels of covalent crosslinking with 6.
We next probed the biochemical reactivity of the covalent peptides with gel electrophoresis as a complement to MALDI MS. Gel shift assays confirm the relative differences in reactivity between the vinyl sulfonamides and vinyl sulfones. Incubation of H-Ras G12C with 3b yielded a single covalent adduct band in addition to the unlabeled Ras band (Figure 5D). In contrast, treatment with the vinyl sulfone-based 4b resulted in multiple bands, indicating non-specific labeling of the other three free cysteine residues. The exact labeling site of covalent peptides was determined from MS/MS analysis of trypsin-digested Ras fragments. Monolabeled bands for both 3b and 4b correspond to peptides attached to C12 (Figure 5E).
H-Ras G12C and 6 showed a single band within the gel-shift assay, indicating a monolabeled species (Figure 5D). MS analysis of trypsin-treated monolabeled samples revealed molecular ion peaks consistent with the digested protein fragment covalently attached to 6 at the C12 position. With the proteolytic stability of α3βHBSSOS sequence and specific targeting of C12, we observed the Ras fragment linked to fully undigested 6 at the desired Ras residue (Figure 5E).
Inhibition of Nucleotide Exchange In Vitro.
The intrinsic rate of nucleotide exchange by Ras proteins is extremely low. Sos and other GEFs facilitate Ras activation by disrupting the Ras-nucleotide complex and stabilizing the nucleotide-free conformation.2 Nucleotide-free Ras rebinds GTP and then dissociates from Sos. Inhibitors of Ras are, therefore, judged on their ability to block nucleotide exchange. However, K-Ras mutants are thought to exist in a permanently active (GTP- bound) state in cancer cells, which implies that drugs targeting the nucleotide-free or GDP-bound Ras states would be ineffective as these states do not exist in the context of mutant Ras. Recent studies have challenged this classical understanding of the role of Sos in Ras activation and revealed that K-Ras G12C mutants retain nucleotide hydrolysis activity and can cycle between the active and inactive states. In support of this mechanism, covalent allosteric ligands that trap GDP-bound Ras have been shown to downregulate tumor burden in mouse models harboring the G12C mutation.6 Accumulating evidence also indicates that the tumorigenic potential of oncogenic Ras is dependent upon both Sos catalytic activity and expression of the WT Ras isoforms.36
To assess the nucleotide exchange inhibitory activity of 6, we measured its effect on Sos-mediated nucleotide exchange in vitro.5, 6 We conducted Sos-catalyzed exchange reactions with the fluorescent 2’-deoxy-3’-O-(N-methylanthraniloyl) (mant)-GDP with and without 6 in the presence of excess unlabeled GDP, as described.5 The general process involved initial labeling of recombinant H-Ras G12C (1 μM) with a specified amount of covalent peptide, incubation with excess mant-GDP, and exposure to SosCAT in the presence of excess GDP (Figure 6A). α3βHBS 6 led to a concentration-dependent inhibition of nucleotide exchange (Figure 6B–C). Assessment of nucleotide association in a time-dependent fashion revealed a 4-fold enhancement in binding, which was predicated on the formation of the covalent bond (Figure S6). The inhibitory activity of 6 is significant when compared to the negative controls: 10 equivalents of 6 lead to >50% decrease in Ras binding to mant- GDP, whereas 50 equivalents of the control α3βHBS peptide sequences 7 and 8 failed to lead to notable inhibition.
Figure 6.
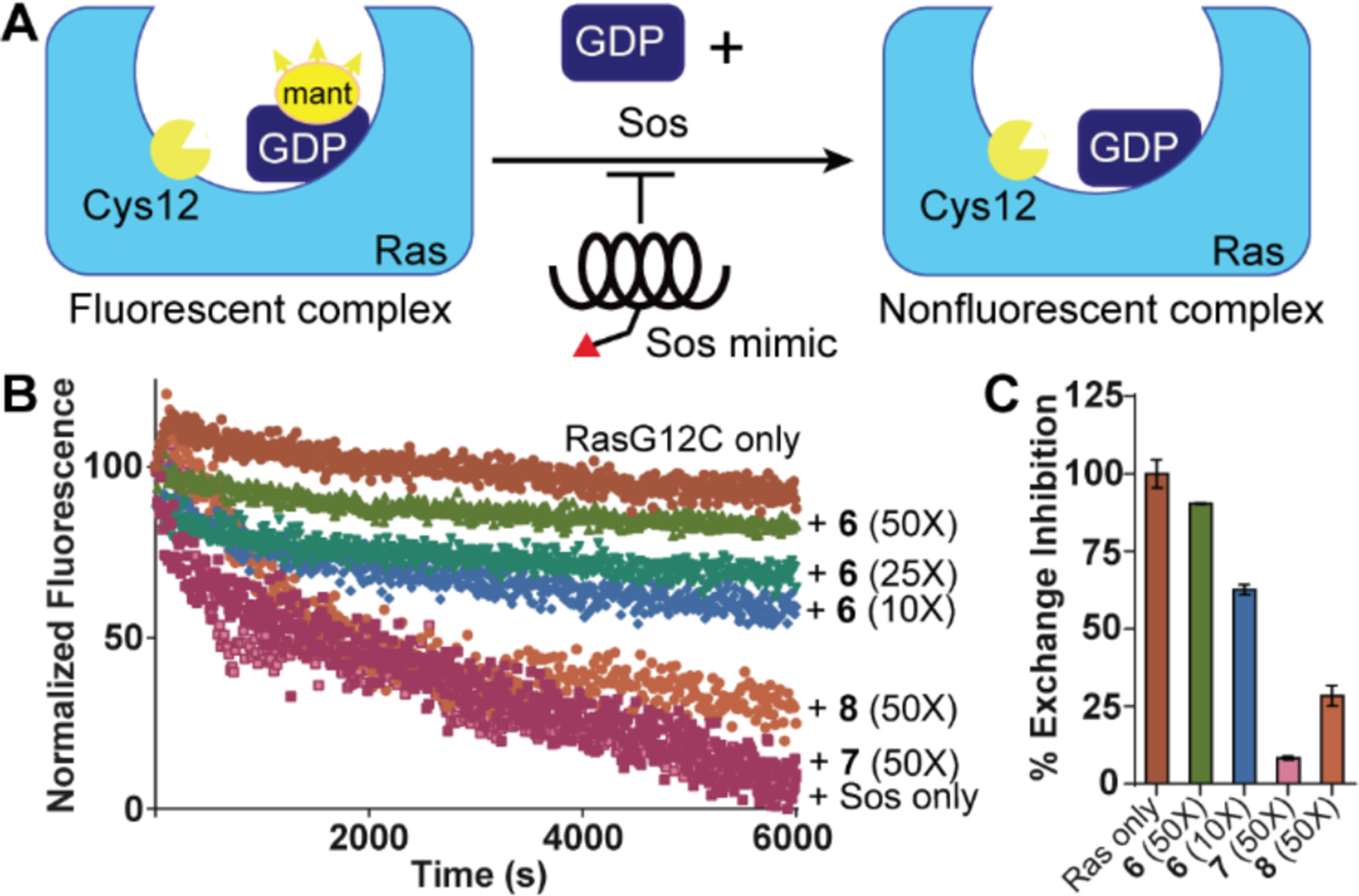
(A) Nucleotide exchange inhibition by covalent peptidomimetics with a fluorescent nucleotide analog (mant-GDP) as a probe. (B) Sos-mediated nucleotide exchange assays with H-Ras G12C and increasing concentrations of covalent α3βHBS 5. (C) Bar graph compares the efficacy of 5 and control peptides 6 and 7. Error bars are mean ± SD of biological duplicates.
Cellular Uptake of α3βHBSSOS.
The in vitro results suggest that the designed compounds may modulate cellular Ras signaling. Effective cellular modulation requires that the peptides are efficiently taken up into the cell. Live cell fluorescence microscopy indicated significant cellular uptake of Flu-α3βHBSSOS into the cytosol (Figure 7A). We further assessed cellular uptake of the peptides in H358 cells incubated with 1 μM fluorescein-labeled Flu-HBSSOS and Flu-α3βHBSSOS for 4 hours and quantified the uptake via flow cytometry (Figure 7B). These experiments indicate that the concentration of Flu-α3βHBSSOS in live cells is nearly four-fold higher than the all α-residue variant, potentially due to the higher proteolytic stability of α3β helix.
Figure 7.
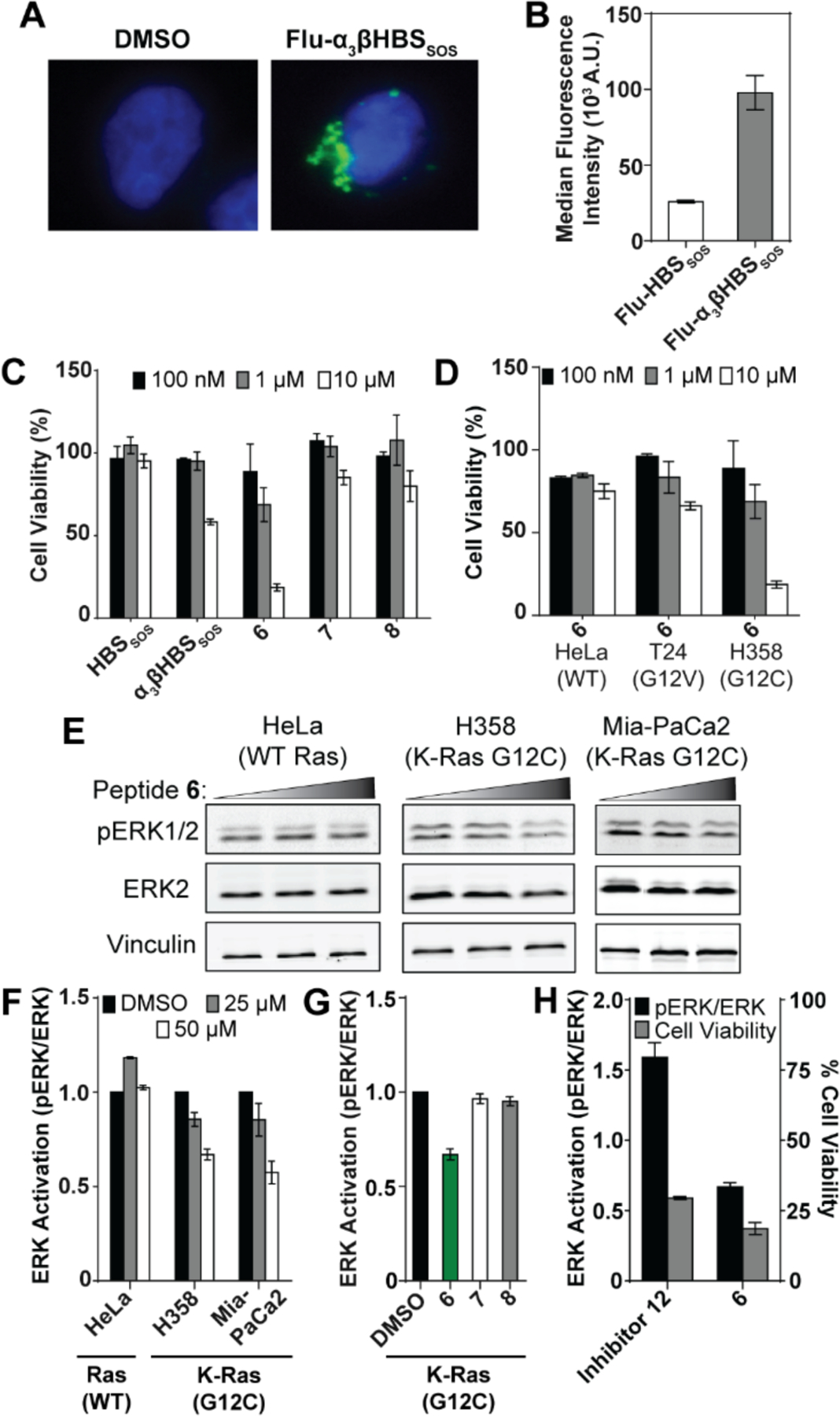
(A) Live cell fluorescence imaging of DAPI-treated H358 cells incubated with Flu-α3βHBSSOS for 4 hours at 40x magnification. (B) Relative median fluorescence intensities in H358 cells after 4-hour incubation with Flu-HBSSOS and Flu-α3βHBSSOS. (C) MTT cell viability in H358 lung cancer cells treated with α3βHBSSOS, α3βHBSSOS, and 4–7 for 72 hours. (D) Comparison of cell viability between HeLa (WT Ras), T24 (H-Ras G12V), and H358 (K-Ras G12C) cells treated with 5. (E-F) ERK activation in HeLa, H358, and Mia-PaCa2 (K-Ras G12C) cells post-treatment with 5. (G) ERK activation in H358 cells post-treatment with our lead compound 5 and control peptides 6–7. (H) Comparison of ERK activation and corresponding cell viability for Inhibitor 12 and 5 in H358 cells. Error bars are mean ± SD of at least two biological replicates.
Electrophilic α3βHBS Selectively Reduces Ras G12C Cell Viability.
Encouraged by the in vitro results, we probed the potential of the lead derivative in cellular models. We next tested the cellular potency of 6 and relevant control analogs in H358 (K-Ras G12C) lung cancer cells and HeLa (WT Ras) cervical cancer cells using the MTT assay. Peptide 6 exhibited concentration-dependent toxicity to cells expressing K-Ras G12C, while displaying no discernible effect in the control HeLa cell line (Figure 7C–D). The peptide was also minimally effective against T24 (H-Ras G12V) bladder cancer cells. In contrast, controls 7 and 8 showed no appreciable effect on cell viability. α3βHBSSOS is designed from the Ras-binding domain of Sos and should show a modest effect against WT Ras cell lines. Consistent with its design, α3βHBSSOS impacts viability of H358 cells at higher concentrations.8 Together, the cell viability studies demonstrate that the lead covalent peptide shows higher toxicity against cells bearing the nucleophilic C12 mutation.
Inhibition of Downstream Ras Signaling.
Ras is a key activator of various signal transduction pathways. To determine if the inhibitory effect of 6 on Ras can modulate cellular signaling, we probed the phosphorylation state of ERK, a well-documented downstream effector of the Ras signal transduction pathway and implicated in cell proliferation and differentiation.37 Serum-starved H358 and HeLa cells were treated with increasing concentrations of 6, and the lysates were blotted for phosphorylated ERK. As expected, 6 markedly reduced the extent of ERK activation in the Ras G12C cell line while having no effect in cells bearing WT Ras (Figure 7E–F). We also verified the effect of the lead covalent inhibitor 6 on another Ras G12C cell line (Mia-PaCa2) and again observed concentration-dependent inhibition of ERK phosphorylation. Control peptides 7 and 8 did not suppress ERK phosphorylation in H358 cells (Figure 7G, S7). α3βHBS 6 alone leads to a decrease in cell viability with a concomitant decrease in ERK activation. Surprisingly, the acrylamide-based small molecule ligand Inhibitor 125 elicited upregulation in ERK activation (Figure 7H, S8). The increase in ERK levels is likely attributable to a stress-induced feedback mechanism.38, 39
CONCLUSION
Mimics of protein secondary structures have emerged as an attractive class of compounds for such targets.40, 41 However, the approach faces a fundamental challenge: proteins often do not engage partner proteins with high affinity, and so mimics of protein interfaces are also weak binders. Various strategies that address this challenge have been devised. The most common approach relies on the introduction of non-native residues through selection42 or rational design to exploit unoccupied pockets on protein surfaces.43 Covalent targeting provides a classical drug discovery approach to gaining potency. Several classes of drugs that complex with the target through an irreversible interaction have been reported.12–14 The present study describes our efforts to engage Ras using a covalent orthosteric ligand derived from Sos.
Ras remains a significant challenge for drug discovery. The G12C mutation offers a reactive nucleophile near the guanine nucleotide binding site of Ras. The C12 residue has been targeted by covalent allosteric inhibitors of Ras as well as electrophilic-GTP mimics.44–46 The allosteric inhibitors can access a nearby crevice and react with cysteine; however, the buried C12 residue presents a challenge for orthosteric ligands that emerge from rational mimicry of protein partners. Significantly, Ras features several cysteine residues outside the nucleotide binding region, and the specificity of the ligand for C12 becomes an overriding requirement. Design of a covalent orthosteric ligand required proper selection of the electrophilic warhead. We began with a previously described stabilized Sos helix mimic. We tested different electrophiles and varied linker lengths to arrive at the optimized construct. This compound alkylated C12 with high selectivity in biochemical assays and supported our hypothesis that Sos-derived orthosteric ligands may be tuned to react with the desired nucleophilic residue. Encouraged by these findings, we probed the toxicity of the lead electrophilic helix against mutant K-Ras G12C-bearing cells as compared to cells expressing WT Ras, and found the lead derivative to be significantly more toxic to G12C cells as compared to cells bearing WT Ras or the G12V mutation. Finally, we assayed the potential of the compound to inhibit signaling and activation of downstream effectors. As expected, the control compounds do not show any activity.
Mutant Ras G12C is a model system for discovery of covalent ligands for protein surfaces. In contrast to its prevalence within the proteome, cysteine is one of the most commonly acquired missense mutations in human cancers.47 We have categorized PPIs that are mediated by high affinity secondary and tertiary structure motifs.48, 49 The high affinity motifs were defined as short segments that contain several residues which contribute to binding affinity as revealed by computational alanine mutagenesis studies. As part of our studies, we classified interfaces that feature a cysteine residue within 8 Å of a high affinity α-helix. These entries are listed in the Supporting Information (Table S2). The lessons derived from the current, and related examples of electrophilic peptides, provide a blueprint for rational design of peptidomimetics that can target PPIs featuring a native or mutated nucleophilic cysteine residue.
Supplementary Material
ACKNOWLEDGMENT
We dedicate this paper to S. Gellman on the occasion of his 60th Birthday. This work was supported by grants from NIH/NIGMS (R35GM130333) to P.S.A., NIH/NCI (R35CA210263/R01-CA055360) to D.B.-S. and NIH/NCI (T32CA009161) to A.D.H.
Footnotes
The Supporting Information is available free of charge on the ACS Publications website.
Synthesis, characterization, biological, biochemical, and spectroscopy data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Chardin P, Camonis JH, Gale NW, van Aelst L, Schlessinger J, Wigler MH, and Bar-Sagi D (1993) Human Sos1: a guanine nucleotide exchange factor for Ras that binds to GRB2, Science 260, 1338–1343. [DOI] [PubMed] [Google Scholar]
- (2).Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, and Kuriyan J (1998) The structural basis of the activation of Ras by Sos, Nature 394, 337–343. [DOI] [PubMed] [Google Scholar]
- (3).Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, and Wittinghofer A (1997) The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants, Science 277, 333–338. [DOI] [PubMed] [Google Scholar]
- (4).Smith MJ, Neel BG, and Ikura M (2013) NMR-based functional profiling of RASopathies and oncogenic RAS mutations, Proc. Natl. Acad. Sci. U. S. A 110, 4574–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ostrem JM, Peters U, Sos ML, Wells JA, and Shokat KM (2013) K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions, Nature 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, Chen Y, Babbar A, Firdaus SJ, Darjania L, Feng J, Chen JH, Li S, Li S, Long YO, Thach C, Liu Y, Zarieh A, Ely T, Kucharski JM, Kessler LV, Wu T, Yu K, Wang Y, Yao Y, Deng X, Zarrinkar PP, Brehmer D, Dhanak D, Lorenzi MV, Hu-Lowe D, Patricelli MP, Ren P, and Liu Y (2018) Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor, Cell 172, 578–589 e517. [DOI] [PubMed] [Google Scholar]
- (7).Lito P, Solomon M, Li L-S, Hansen R, and Rosen N (2016) Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism, Science 351, 604–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Patgiri A, Yadav KK, Arora PS, and Bar-Sagi D (2011) An orthosteric inhibitor of the Ras-Sos interaction, Nat. Chem. Biol 7, 585–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sondermann H, Soisson SM, Boykevisch S, Yang SS, Bar-Sagi D, and Kuriyan J (2004) Structural analysis of autoinhibition in the Ras activator Son of sevenless, Cell 119, 393–405. [DOI] [PubMed] [Google Scholar]
- (10).Lagoutte R, Patouret R, and Winssinger N (2017) Covalent inhibitors: an opportunity for rational target selectivity, Curr. Opin. Chem. Biol 39, 54–63. [DOI] [PubMed] [Google Scholar]
- (11).De Cesco S, Kurian J, Dufresne C, Mittermaier AK, and Moitessier N (2017) Covalent inhibitors design and discovery, Eur. J. Med. Chem 138, 96–114. [DOI] [PubMed] [Google Scholar]
- (12).Singh J, Petter RC, Baillie TA, and Whitty A (2011) The resurgence of covalent drugs, Nat. Rev. Drug Discov 10, 307–317. [DOI] [PubMed] [Google Scholar]
- (13).Lonsdale R, and Ward RA (2018) Structure-based design of targeted covalent inhibitors, Chem. Soc. Rev 47, 3816–3830. [DOI] [PubMed] [Google Scholar]
- (14).Baillie TA (2016) Targeted Covalent Inhibitors for Drug Design, Angew. Chem. Int. Ed. Engl 55, 13408–13421. [DOI] [PubMed] [Google Scholar]
- (15).Smith MC, and Gestwicki JE (2012) Features of protein-protein interactions that translate into potent inhibitors: topology, surface area and affinity, Expert Rev. Mol. Med 14, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Scott DE, Bayly AR, Abell C, and Skidmore J (2016) Small molecules, big targets: drug discovery faces the protein-protein interaction challenge, Nat. Rev. Drug Discov 15, 533–550. [DOI] [PubMed] [Google Scholar]
- (17).de Araujo AD, Lim J, Good AC, Skerlj RT, and Fairlie DP (2017) Electrophilic Helical Peptides That Bond Covalently, Irreversibly, and Selectively in a Protein-Protein Interaction Site, ACS Med. Chem. Lett 8, 22–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Huhn AJ, Guerra RM, Harvey EP, Bird GH, and Walensky LD (2016) Selective Covalent Targeting of Anti-Apoptotic BFL-1 by Cysteine-Reactive Stapled Peptide Inhibitors, Cell Chem Biol 23, 1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, Bar-Sagi D, and Kuriyan J (2003) Structural evidence for feedback activation by Ras.GTP of the Ras-specific nucleotide exchange factor SOS, Cell 112, 685–695. [DOI] [PubMed] [Google Scholar]
- (20).Patgiri A, Jochim AL, and Arora PS (2008) A hydrogen bond surrogate approach for stabilization of short peptide sequences in alpha-helical conformation, Acc. Chem. Res 41, 1289–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zimm BH, and Bragg JK (1959) Theory of the Phase Transition between Helix and Random Coil in Polypeptide Chains, J. Chem. Phys 31, 526–535. [Google Scholar]
- (22).Milroy L-G, Grossmann TN, Hennig S, Brunsveld L, and Ottmann C (2014) Modulators of Protein–Protein Interactions, Chem. Rev 114, 4695–4748. [DOI] [PubMed] [Google Scholar]
- (23).Horne WS, Johnson LM, Ketas TJ, Klasse PJ, Lu M, Moore JP, and Gellman SH (2009) Structural and biological mimicry of protein surface recognition by alpha/beta-peptide foldamers, Proc. Natl. Acad. Sci. U. S. A 106, 14751–14756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Azzarito V, Long K, Murphy NS, and Wilson AJ (2013) Inhibition of alpha-helix-mediated protein-protein interactions using designed molecules, Nat. Chem 5, 161–173. [DOI] [PubMed] [Google Scholar]
- (25).Verdine GL, and Hilinski GJ (2012) Stapled Peptides for Intracellular Drug Targets, Methods Enzymol. 503, 3–33. [DOI] [PubMed] [Google Scholar]
- (26).Walensky LD, and Bird GH (2014) Hydrocarbon-Stapled Peptides: Principles, Practice, and Progress, J. Med. Chem [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yang B, Liu D, and Huang Z (2004) Synthesis and helical structure of lactam bridged BH3 peptides derived from pro-apoptotic Bcl-2 family proteins, Bioorg. Med. Chem. Lett 14, 1403–1406. [DOI] [PubMed] [Google Scholar]
- (28).Miller SE, Thomson PF, and Arora PS (2014) Synthesis of hydrogen-bond surrogate alpha-helices as inhibitors of protein-protein interactions, Curr. Protoc. Chem. Biol 6, 101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tyndall JD, Nall T, and Fairlie DP (2005) Proteases universally recognize beta strands in their active sites, Chem. Rev 105, 973–999. [DOI] [PubMed] [Google Scholar]
- (30).Seebach D, and Gardiner J (2008) Beta-peptidic peptidomimetics, Acc. Chem. Res 41, 1366–1375. [DOI] [PubMed] [Google Scholar]
- (31).Patgiri A, Joy ST, and Arora PS (2012) Nucleation effects in peptide foldamers, J. Am. Chem. Soc 134, 11495–11502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Boersma MD, Haase HS, Peterson-Kaufman KJ, Lee EF, Clarke OB, Colman PM, Smith BJ, Horne WS, Fairlie WD, and Gellman SH (2012) Evaluation of diverse alpha/beta-backbone patterns for functional alpha-helix mimicry: analogues of the Bim BH3 domain, J. Am. Chem. Soc 134, 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Cheloha RW, Maeda A, Dean T, Gardella TJ, and Gellman SH (2014) Backbone modification of a polypeptide drug alters duration of action in vivo, Nat. Biotechnol 32, 653–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Farnsworth CL, and Feig LA (1991) Dominant inhibitory mutations in the Mg(2+)-binding site of RasH prevent its activation by GTP, Mol. Cell. Biol 11, 4822–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wong KA, Russo A, Wang X, Chen YJ, Lavie A, and O’Bryan JP (2012) A new dimension to Ras function: a novel role for nucleotide-free Ras in Class II phosphatidylinositol 3-kinase beta (PI3KC2beta) regulation, PLoS One 7, e45360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Jeng HH, Taylor LJ, and Bar-Sagi D (2012) Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis, Nat Commun 3, 1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Garcia-Gomez R, Bustelo XR, and Crespo P (2018) Protein-Protein Interactions: Emerging Oncotargets in the RAS-ERK Pathway, Trends Cancer 4, 616–633. [DOI] [PubMed] [Google Scholar]
- (38).Oh YT, Deng JS, Yue P, and Sun SY (2016) Paradoxical activation of MEK/ERK signaling induced by B-Raf inhibition enhances DR5 expression and DR5 activation-induced apoptosis in Ras-mutant cancer cells, Sci. Rep 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Mendoza MC, Er EE, and Blenis J (2011) The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation, Trends Biochem. Sci 36, 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sawyer N, Watkins AM, and Arora PS (2017) Protein Domain Mimics as Modulators of Protein–Protein Interactions, Acc. Chem. Res 50, 1313–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Milroy LG, Grossmann TN, Hennig S, Brunsveld L, and Ottmann C (2014) Modulators of protein-protein interactions, Chem. Rev 114, 4695–4748. [DOI] [PubMed] [Google Scholar]
- (42).Wuo MG, and Arora PS (2018) Engineered protein scaffolds as leads for synthetic inhibitors of protein–protein interactions, Curr. Opin. Chem. Biol 44, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Rooklin D, Modell AE, Li H, Berdan V, Arora PS, and Zhang Y (2017) Targeting Unoccupied Surfaces on Protein–Protein Interfaces, J. Am. Chem. Soc 139, 15560–15563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Lim SM, Westover KD, Ficarro SB, Harrison RA, Choi HG, Pacold ME, Carrasco M, Hunter J, Kim ND, Xie T, Sim T, Janne PA, Meyerson M, Marto JA, Engen JR, and Gray NS (2014) Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor, Angew. Chem. Int. Ed. Engl 53, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zeng M, Lu J, Li L, Feru F, Quan C, Gero TW, Ficarro SB, Xiong Y, Ambrogio C, Paranal RM, Catalano M, Shao J, Wong KK, Marto JA, Fischer ES, Janne PA, Scott DA, Westover KD, and Gray NS (2017) Potent and Selective Covalent Quinazoline Inhibitors of KRAS G12C, Cell Chem Biol 24, 1005–1016 e1003. [DOI] [PubMed] [Google Scholar]
- (46).Lito P, Solomon M, Li LS, Hansen R, and Rosen N (2016) Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism, Science 351, 604–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Visscher M, Arkin MR, and Dansen TB (2016) Covalent targeting of acquired cysteines in cancer, Curr. Opin. Chem. Biol 30, 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Watkins AM, Wuo MG, and Arora PS (2015) Protein-Protein Interactions Mediated by Helical Tertiary Structure Motifs, J. Am. Chem. Soc 137, 11622–11630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Watkins AM, and Arora PS (2014) Anatomy of beta-strands at protein-protein interfaces, ACS Chem. Biol 9, 1747–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.