

Abstract
This research presents two case studies in which a change in the disinfectant from free chlorine to chloramine caused an increase in lead corrosion. In both systems, the predominantly tetravalent lead (PbO2) scales destabilized as a result of disinfectant change. Orthophosphate corrosion control was used in both systems, and the effect of this treatment chemical on the destabilized PbO2 scales was examined. The absence of chemical reactivity between PbO2 and phosphorus is well known, and this research confirms that phosphorus does not interact with the legacy PbO2 scales. Instead, phosphorus and calcium were found to permeate through the destabilized PbO2 material and react with divalent lead [Pb(II)] at the surface of a basal litharge (PbO) layer. This reaction precipitated a crystalline lead phosphate in both systems, which could not be specifically identified by any known powder diffraction files. Further analysis suggested that the compound formed was not the typically modeled hydroxypyromorphite but rather a calcium-substituted hydroxypyromorphite. During scale formation, calcium is frequently bound to the Pb(II) phosphate crystal lattice structure, causing measurable crystal lattice distortion in powder X-ray diffraction patterns. The results of this study illustrate the longevity of legacy scales and how disequilibrium compounds persist long after treatment changes have been made.
Keywords: disinfectant change, legacy PbO2 scales, lead corrosion, hydroxypyromorphite, phosphate treatment
Graphical Abstract
INTRODUCTION
In large drinking water systems across the United States, which are estimated to contain millions of lead service lines (LSLs), the past 50 years have brought about many treatment changes that have greatly impacted parameters related to the corrosivity of the water, notably the pH, alkalinity, disinfection type, and the amount of disinfectant residual.1 Any time there is a change to water treatment, because of regulations or other reasons, there is a potential to impact corrosion.2–9
By 1996, several analyses of lead pipes from different US water systems had revealed that the tetravalent lead [Pb(IV)] oxide phase plattnerite (PbO2) could form,10 and the first descriptions of fully uniform deposits of PbO2 were described in 2001.11 When properly maintained, PbO2 solids are known to be highly insoluble contributing less lead to drinking water than the other known Pb(II) corrosion solids.11–14 The reductive dissolution of PbO2 by a drop in the oxidation–reduction potential (ORP) is predictable from a simple visual examination of potential–pH diagrams published by many authors.15–18 Schock and Lytle14 also emphasized that PbO2 reductive dissolution was thermodynamically favorable through a pH drop at the lead scale surface, which could be caused by any of several different treatment changes or localized pH changes from microbial activity such as nitrification. Subsequently, reductive dissolution of PbO2 scales by a pH drop has been described in both laboratory experiments19 and a municipal water system.20 The extent of the pH shift needed to cause the destabilization and dissolution of PbO2 would depend on where the system water was plotted on the equilibrium potential–pH diagram.21 As a result of the well-known lead release episode in Washington, DC, in the early 2000s,22–26 there was a considerable increase in research activity that looked at the feasibility of formation of PbO2 under viable drinking water conditions12,27 along with the reductive dissolution aspect of PbO2 destabilization caused by lowering the ORP by changing the secondary disinfectant from free chorine to chloramine at a neutral to basic pH14,27–33 or potentially by other water constituents, such as natural organic matter (NOM).34
In water systems with Pb(II) pipe scales, an established approach to mitigating lead release is to utilize orthophosphate to react with the Pb(II) constituents present in the scales or at plumbing material surfaces to form rather insoluble lead orthophosphate solids.35–40 Implicit to the computations and discussion of controlling divalent lead release is the reaction pathway of the pre-existing divalent lead corrosion byproduct scale, by which the Pb(II) carbonates, typically PbCO3 (cerussite) or Pb3(CO3)2(OH)2 (hydrocerussite), are converted in situ to any of several Pb(II) orthophosphates.35 In contrast, there are no thermodynamic constants or established chemical reactions reported for the direct interaction of orthophosphate, carbonate, or bicarbonate ions with PbO2 or Pb(IV) aqueous species under natural or drinking water conditions.
This research examines two case studies of pipe-scale evolution resulting from the interaction of destabilized PbO2 scales and phosphate treatment. These two drinking water distribution systems underwent destabilization of the existing PbO2 scales as a result of changing their drinking water disinfectant from free chlorine to chloramine. One system had dosed phosphoric acid for approximately 9 years prior to the switch, while the other system instituted phosphoric acid dosing once elevated lead levels were observed after the disinfectant switch. This analysis of exhumed LSLs provides insights into how phosphate was incorporated into the scales from these two complex drinking water systems. Mechanistic understanding is also supported by a limited scale characterization from a third water system.
MATERIALS AND METHODS
System Information and Water Quality.
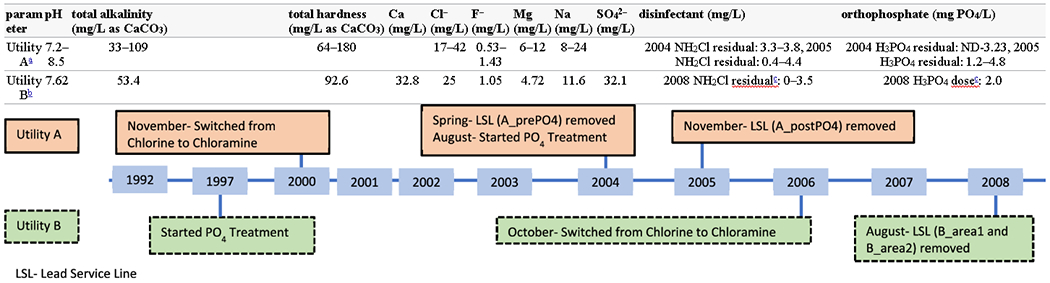
Utility A is a community water system that uses surface water in the Eastern United States, with conventional alum coagulation treatment, free chlorine for secondary disinfection and biofilm control, and pH adjustment with lime for corrosion control. The selected system operating and water quality parameters are summarized in Table 1 and Supporting Information Table S1. Seasonal variability in the source water is responsible for the wide range of pHs measured within the distribution system (Table 1). As a result of concerns about elevated disinfection byproducts, a switch was made to chloramine beginning November 1, 2000 in order to reduce total trihalomethanes (TTHMs). While this change was effective in lowering the TTHMs, a significant increase in lead levels was observed. Utility A’s lead 90th percentiles went from 0.012 mg/L in 2000 to 0.075 mg/L in 2002 (Supporting Information Table S2). In an effort to reduce the lead concentrations, a full-system orthophosphate treatment was instituted in August of 2004 with an initial target entry point residual of 3.2 to 3.5 mg PO4/L. Multiple LSLs were harvested from Utility A over a span of 4 years after the utility’s disinfection switch, each representing a specific known snapshot in time post-treatment change. These specimens represent distribution system conditions prior to orthophosphate addition, and at various intervals of postphosphate treatment.
Table 1.
Selected Water Quality Parameters for Utilities A and B Covering the Time Period in Which LSLs Were Removed and the Corresponding Timeline of Treatment Changes, 1992–2008
 |
Taken from Utility A’s 2004 and 2005 Consumer Confidence Reports (CCRs) unless otherwise noted.
2008 data posted on Utility B’s website unless otherwise noted.
Taken from Utility B’s 2008 CCR.
Utility B is also a surface water treatment system located in an Eastern United States community. This system utilized free chlorine as a secondary disinfectant, orthophosphate for corrosion control, and a variety of aluminum-based coagulants. In response to the Environmental Protection Agency’s (EPA) 1991 Lead and Copper Rule (LCR), the utility underwent lead and copper sampling in 1992, which yielded a Pb 90th percentile of 0.028 mg/L (Supporting Information Table S2). As a result, beginning in the spring of 1997, Utility B started feeding orthophosphate at a dose of 3.5 mg PO4/L to control Pb corrosion. An additional treatment change was carried out in October 2006 when the system switched disinfectants, from free chlorine to chloramine. Other details regarding system operation and water quality are included in Table 1 and Supporting Information Table S3. Only one LSL was provided from Utility B, and it was removed approximately 2 years (22 months) after the change in disinfectant.
Sample Preparation and Analytical Techniques.
Details regarding sample preparation of the LSLs received from both utilities are discussed in the Methods section of the Supporting Information and have been described in previous publications.13,14,41–46 For powder X-ray diffraction (PXRD), depending on the volume of sample available, samples were either top-loaded or mounted using an amyl acetate slurry onto silicon or quartz zero-background sample holders. Samples were analyzed using a PANalytical X’Pert Pro theta-/theta powder diffractometer using Cu Kα radiation generated at 1.8 kW (45 kV, 40mA) and an X’celerator real time multiple strip detector. Samples were spun at 1 revolution/s to improve particle statistics. Patterns were collected in a continuous scan mode, from 5 to 89.994° 2θ at a scan speed of 0.035556°/s, with data binned into 0.0167113° steps. Diffraction patterns were analyzed using MDI Jade version 9 software (Livermore, CA) and the 2020 ICDD PDF-4+ database (Newtown Square, PA).
Representative sections of each of the LSLs were set in epoxy and prepared for scanning electron microscopy and energy-dispersive spectroscopy (SEM–EDS). These cross-sectioned regions were examined with a JEOL JEM 6490LV SEM at an accelerating voltage of 15 kV. EDS elemental analysis was performed using an Oxford X-act silicon drift detector (Concord, MA). Additional information regarding the SEM–EDS analysis is included in the Methods section of the Supporting Information.
Crystal Lattice Distortion Computations.
Fourteen phases were identified from the ICDD PDF 4+ 2020 database as being part of the solid solution series that exists between hydroxypyromorphite [Pb5(PO4)3OH] and hydroxyapatite [Ca5(PO4)3OH], both members of the hexagonal crystal system (Supporting Information Table S4). These phases include a variety of naturally occurring and synthesized materials with various proportions of Pb and Ca, with both directly measured and theoretically calculated powder diffraction patterns. The molar concentration of Ca was determined from the chemical formula for each phase ranging from 0 to 5, along with the d-spacing of the 99–100% intensity peak (Supporting Information Table S4). With this information, the empirical relationship Vegard’s law was applied to evaluate the correlation between lattice d-spacing and Ca content. Vegard’s law states that the lattice parameter of a particular compound in the solid solution series is a linear combination of the two components if both components are of the same crystal structure.47 A linear relationship was established with an r2 value of 0.9799 for the 14 phases (Supporting Information Figure S1), and the equation of the line was used to calculate the empirical molar concentration for Ca in each of the scale samples that displayed a peak with a d-spacing between 2.8 and 2.9 Å corresponding to the (211) crystal lattice plane. The chemical formula (Cax,Pb5–x) (PO4)3OH was then assumed to encompass all compounds that would form along the solid-solution series, and the molar concentration of Ca was used to calculate the weight percent of Ca, P, Pb, and O that would be expected for each scale sample.
RESULTS AND DISCUSSION
Utility A Scale Mineralogy.
Two characteristic LSLs from Utility A were analyzed for this case study. Both were removed after the disinfectant switch, but one is representative of the timeframe prior to orthophosphate treatment (A_prePO4) and the other was exposed to approximately 15 months of orthophosphate (A_postPO4). A detailed description of the scale layers and photographs of each pipe are included in the section LSL Scale Descriptions of the Supporting Information (Figure S2). PXRD results from pipe A_prePO4 showed that the two layers sampled from the pipe were composed of crystalline lead minerals. At the pipe scale–drinking water interface, the uppermost scale layer (L1) was mainly composed of plattnerite (β-PbO2) with minor hydrocerussite [Pb3(CO3)2(OH)2] and a trace of litharge (PbO). The layer directly against the lead pipe wall (L2) mainly comprised litharge with minor hydrocerussite and β-PbO2, along with metallic lead (artifact of the sampling process from the lead pipe wall) (Supporting Information Table S5).
A distinct difference in the mineralogy of the scale materials was observed after 15 months of orthophosphate treatment (Supporting Information Table S5 and Figure S3). A_postPO4 L1 comprised a minor portion of the scale and was mainly composed of α-PbO2 with some amorphous content. L2 also contained mainly β-PbO2 with a trace amount of hydrocerussite. Evidence indicating the presence of phosphate appeared within L3. While the mineralogy of L3 was predominantly the PbO2 compounds, β-PbO2 and scrutinyite (α-PbO2), a moderate amount of lead phosphates and hydrocerussite were also noted. However, the lead phosphate identified was poorly crystalline, leading to the broad humps visible in the PXRD pattern (Supporting Information Figure S3). A trace amount of lead phosphates and β-PbO2 occurred in layer L4a with the predominant mineral being litharge, along with a minor amount of hydrocerussite. L4b comprised the layer directly against the lead pipe wall and was composed of mainly litharge with a trace of hydrocerussite and some metallic lead incorporated from the pipe wall during sampling.
Utility B Scale Mineralogy.
The scale in the pipe from Utility B was observed to be nonuniform along the length of the pipe received and was partitioned for description and sampling as B_area1 and B_area2 (Figure 1). Descriptions of each scale layer removed from these two areas of pipe are included in the LSL scale description section of the Supporting Information and shown in Figure S4. PXRD results for B_area1 scale layers are shown in Figure 2A (Supporting Information Table S5). L1 is made up of poorly crystalline or X-ray amorphous phases, with β-PbO2 present likely from the small amounts of L2 material that were observed clinging to the base of L1 during sampling. L2 is predominately composed of β-PbO2, while L3 is composed of lead phosphate phases with a minor amount of β-PbO2. Directly against the Pb pipe wall, L4 mainly comprised litharge, and although β-PbO2 and lead phosphate phases were also detected in L4, these likely represent sampling carryover from L3. The elemental lead detected in L4 is contamination from the lead pipe wall.
Figure 1.

Macrophotograph of the received LSL from Utility B with the locations of area 1 and area 2 scales, scale bar in mm.
Figure 2.
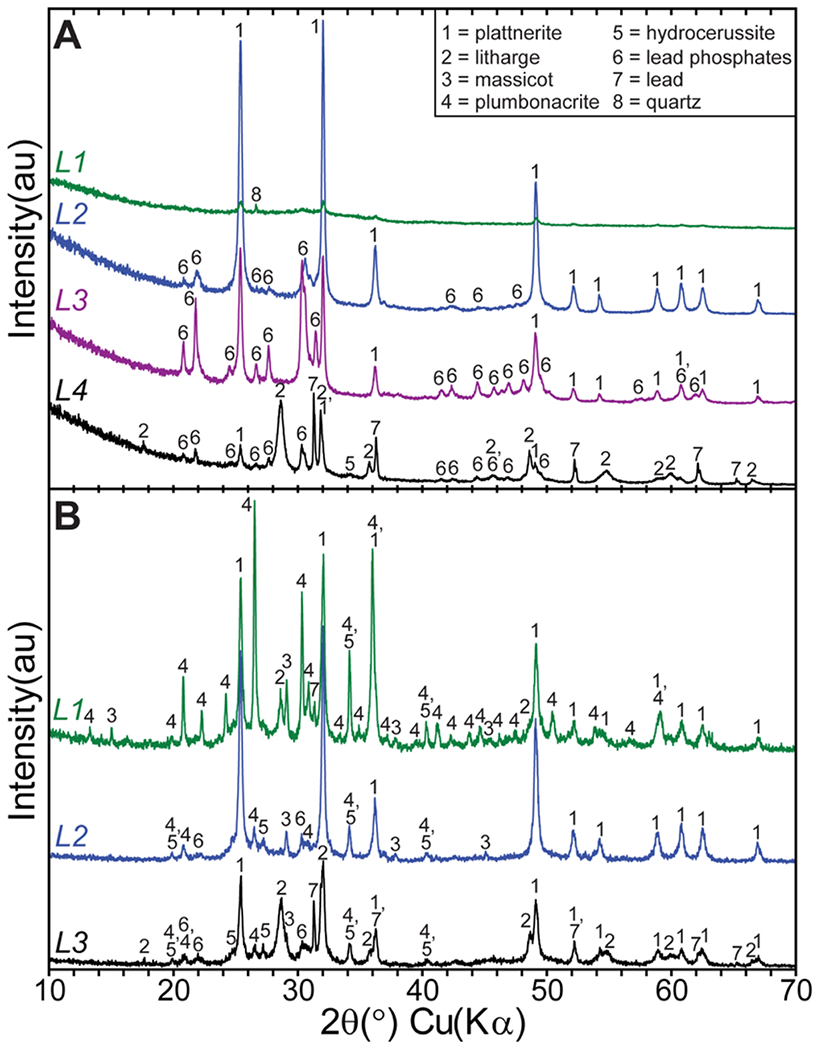
Utility B PXRD patterns collected from (A) pipe B_area1 scale layers and (B) pipe B_area2 scale layers. Numbered phases indicate the location of diffraction lines for the individual compounds identified.
Area 2 in the LSL from Utility B begins approximately 3 inches away from the center of the wiped joint in area 1 and continues for the rest of the length of the received LSL (Figure 1 and Supporting Information Figure S4). PXRD results for B_area2 scale layers are shown in Figure 2B (Supporting Information Table S5). In this area, the surface (L1) layer is predominately composed of plumbonacrite [Pb5O-(OH)2(CO3)3] and β-PbO2. Minor amounts of both Pb(II) oxides, litharge and massicot, were also detected. The latter phases are likely from the irregular local patches exposed on the scale surface (Supporting Information Figure S4C). L2 is predominantly β-PbO2 with minor amounts of plumbonacrite, hydrocerussite, and lead phosphate phases. L3 is primarily composed of litharge, with β-PbO2 and subordinate amounts of plumbonacrite, hydrocerussite, and lead phosphate phases being likely carried over during sampling from the overlying layer.
Overall Mineralogy.
The PXRD results established the predominance of PbO2 compounds within both utilities’ scales. The PbO2 compounds among all the scales sampled were observed to occupy at least 50 μm thickness of the scale material. In some cases, the PbO2 compounds were in direct contact with flowing drinking water, and in others, the PbO2 extended down to lower layers of the scale. In addition, each of the LSLs was also observed to contain a litharge layer directly against the Pb pipe wall that ranged between ~20 and 170 in thickness.
In a recent study by Bae et al.,48 a laboratory-created PbO2 LSL scale was examined for lead release and scale composition after a simulated disinfectant switch when orthophosphate had been predosed for 14 weeks. The study acknowledged that it did not capture all the complexities present in real-world LSL scales, one of which is easily contrasted by this current work as the PbO2 scale created and tested by Bae et al.48 was relatively pure and had a thickness of ~15 μm. In addition to the increased PbO2 thickness observed in the two case studies presented in this paper, multiple lead phases were identified among the scales studied in Utilities A and B with all sampled layers containing at least two different lead phases. The presence of multiple lead phases forming in real-world LSLs is not uncommon, and over the years, the authors have found very few systems in which the scales are found to contain only one or two pure lead phases.42,46,49
Numerous observations of what could be termed “mature” PbO2 scales from multiple utilities have been made in the authors’ laboratory, and the interpretations of these scales have been refined over time.13 Though there are multiple ways in which PbO2 manifests itself in the many corrosion scales investigated from North American water systems,39,41 the form common to these stable and mature PbO2 scales appears to be analogous to the “duplex” conductor scale structure that has been frequently observed in a passivated copper pipe.50–52 In these, a conductive Cu(I) solid Cu2O (cuprite) layer bonds to the copper pipe and is overlain continuously by a conductive layer of the stable Cu(II) form, which is typically either CuO (tenorite) or Cu2(OH)2CO3 (malachite) depending on the pipe age and water chemistry. This upper Cu (II) scale layer can alter reversibly, as the water/scale interface redox conditions change. In mature PbO2 scales, there is a continuous compact scale, with essentially pure α-PbO (litharge) bonded to the lead pipe at the base, blending into an upper scale layer of pure β-PbO2 (plattnerite) resulting in an ORP gradient from the pipe to the scale/water interface. This interface is in direct contact with the primary strong electron-acceptor residual in water, typically the free chlorine species. Though there is some variation across different systems, the litharge layer is often considerably thicker than the plattnerite layer. Occasionally, some depositional “crustal” material, such as aluminum, iron, or manganese oxyhydroxides, or amorphous calcareous material, may be present on top of the plattnerite layer.
This dual valence state (duplex) scale is inherently unstable in all but extremely high pH drinking waters. Thus, without the persistent presence of a strong oxidant residual, reductive dissolution is inevitable. When the scale/water interface pH or ORP is reduced for a prolonged period of time, these conductive scales begin to decompose at both scale boundaries (scale-water interface and pipe wall). The electron sources for reductive dissolution vary depending on the location within the scale and include a combination of water (as noted from the inception of computations of Pourbaix and other potential–pH diagrams) and other electron donor species (reductants) that could be present in water, such as (but not exclusive to) NOM.14–18,34
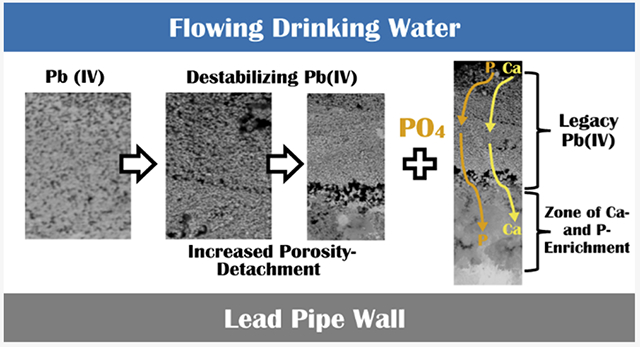
As divalent lead ions or aqueous complexes are formed, they can react with the inorganic carbonate species in water, or orthophosphate ions if present, to form lead solids of much higher solubility than PbO2. Neither carbonate nor phosphate will react with either tetravalent lead aqueous or solid species, so that while these species will start permeating the increasingly porous (dissolving) PbO2 scale, they will not accumulate through precipitation reactions until they contact the litharge underlayer. There, the carbonate and phosphate will be able to spread and develop a conversion reaction front of the lead oxide scale to a carbonate or phosphate-based divalent lead scale. Over time, this will undermine the remaining PbO2 layer and begin to cause separation, breaking the electrochemical continuity of the duplex oxide scale. Though it is not currently possible to observe this phenomenon evolving in the pipes in real time, it seems possible to generally infer at a conceptual level, the sequence of scale evolution events from pipe scale samples, water quality information, and treatment history. From these observations, hypotheses have been developed on the mechanisms of decomposition and potential implications for water systems over time.
In all three samples in this study (A_postPO4, B_area1, and B_area2) phosphorus and calcium were found to have migrated through the remnant surface layer containing PbO2 and reacted along the interface at the point of contact with the underlying PbO layer. Eventually, the formation of Pb(II) phosphate phases at depth can provide an additional physicochemical wedge to further degrade the upper PbO2 layer from both the top and bottom, making it more vulnerable to both dissolution in and physical release into water. The mineralogy of the LSLs in this study confirmed that divalent lead phosphate phases had formed in the lower layers of the scales which had been exposed to orthophosphate (A_postPO4, B_area1, and B_area2). To further characterize these changes and examine the layer relationships in more detail, epoxy cross sections were analyzed from each pipe.
SEM Line Scans–Elemental Analysis.
The cross sections taken from Utility A LSLs provide a chronology that shows the effects of a disinfectant change and where the newly added phosphate treatment was being incorporated into the pre-existing scale. Prior to the phosphate addition pipe A_prePO4, the scale is predominately composed of Pb, which exhibits a gradual increase in concentration with increasing scale depth (Figure 3A). Elemental analysis of prephosphate L1 scales indicated the presence of varying amounts of Al and Fe, a minor amount of Ca, with no evidence of P (P results were lower than the detection limit) (Supporting Information Table S6). This corresponds well with the PXRD which identified A_prePO4 L1 as consisting mainly of β-PbO2 with minor amounts of hydrocerussite. A few cracks are visible within the PbO2 textured area of L1 (sandy appearance, mainly constituting the upper portion of the crosssection backscatter image). This potentially indicates a loss of integrity of the PbO2 material as this LSL was removed approximately 3.5 years after Utility A lowered the ORP of their system by switching disinfectants from free chlorine to chloramine. However, an LSL from before the disinfectant switch was not available for analysis, therefore, the original starting conditions of this pipe are unknown. The minor amounts of hydrocerussite indicated in the PXRD patterns are also visible in the cross section as the generally vertical blade structures in the middle to lower half of the backscatter image.
Figure 3.

Light microscopy cross-sectional views coupled with backscatter SEM images overlain with EDS elemental line data for two cross sections from Utility A. White boxes in the light microscopy images show the area of the cross section where the backscatter image was captured, and scale layers are notated along the left-hand side. Light microscopy scale bars = 100 μm; backscatter microscopy scale bars = 60 μm. (A) Pipe A_prePO4 and (B) Pipe A_postPO4.
Figure 3B shows a backscatter SEM image overlain with EDS line scan data from the cross section of A_postPO4. This LSL was removed approximately 5 years after the disinfectant switch and 1 year after the orthophosphate treatment was started. While L1 is not visible in the cross section, this layer was found to be up to 100 μm thick, containing mainly PbO2 with some amorphous component(s). Porosity and loss of scale integrity are easily visible in the backscatter image of this cross section, particularly at the boundary between the PbO2-dominated L2 and the lead phosphate containing L3. Some of the porosity could have been accentuated during sample preparation. However, given the changes in disinfection prior to removal and the observed increase in Pb release to drinking water, the porosity is likely of natural formation resulting from the dissolution and removal of PbO2 from the scale. The EDS line scan of this cross section shows that phosphorus migrates through L2, which is mainly composed of PbO2 and is only found within L3 and the upper portion of L4a. This corroborates the PXRD results for the layers sampled from A_postPO4. Additionally, Ca and, to some extent, Al also follow this trend of bypassing L2 and increasing slightly in the areas where P resides. This lack of interaction between PbO2 and P has been noted previously by Giani et al.53 and Lytle et al.54 Elemental analyses provided in the Supporting Information Table S6 show that after orthophosphate addition, Ca and P accumulated not only in the lower scale layers, as indicated by the cross-sectional analysis, but also in L1. Furthermore, the L1 material sampled from nine additional LSLs from Utility A also showed an enrichment of Al, Ca, Fe, and P. This indicates that the uppermost layer is potentially serving as a sink for P and Ca, impeding the migration of these elements to the lower Pb-rich scale layers. A material similar to L1 that was enriched in Ca, Al, Fe, and P was also observed as a particulate in the distribution system and was the cause of consumer cloudy water complaints after the switch to orthophosphate.55
For Utility B, Figure 4 shows backscatter SEM micrographs overlain with EDS elemental line scan data across cross sections of undisturbed scale layers. The increased detail visible in the Figure 4B cross section shows that B_area2 L2 consists of two sublayers comparable to layers L2 and L3 of B_area1 (Figure 4A and Supporting Information Table S5). Scales from sample B_area2 (Figure 4B) exhibit considerably more porosity than those of B_area1 (Figure 4A). This porosity may have been enhanced slightly during the epoxy sample preparation but is much more likely a natural formation feature. The scale in area 1 shows a relatively intact PbO2 layer under which Ca and P have combined with Pb. The thickness of B_area1 L1 (400–500 μm) after the disinfectant switch may be acting as a barrier and protecting the underlying PbO2-rich L2 from the rapid dissolution PbO2 in B_area2 is experiencing. Similar to A_postPO4, P and Ca were found to be enriched in a zone deep within the scale unassociated with PbO2 (L3 in Figure 4A; the lowest part of L2 in 4B), and to a lesser degree in a thin (~5 μm) band on top of layer L2 (Figure 4B).
Figure 4.

Light microscopy cross-sectional views coupled with backscatter SEM images overlain with EDS elemental line data for two cross sections from Utility B. White boxes in the light microscopy images show the area of the cross section where the backscatter image was captured, and scale layers are notated along the left-hand side. Light microscopy scale bars = 100 μm; backscatter microscopy scale bars = 100 μm. (A) Pipe B_area1 and (B) pipe B_area2.
These cross sections illustrate that the disturbance caused by the disinfectant switch likely increased the ability of Ca and P to migrate through the scale material; however, the Ca- and P- rich layers at depth within these scales had likely been forming all along, as evidenced by Figure 4A. Formation of a Ca–Pb phosphate layer below a legacy PbO2 layer has been confirmed in another water system (Utility C) that has not undergone a disinfectant change (Supporting Information Figures S5 and S6). Additionally, Figure 4B shows another potential cooccurring process with the formation of discrete crystals of the Pb(II) carbonate plumbonacrite in L1 (Supporting Information Table S5). Pb(II) carbonates were not present in area 1, and one hypothesis is that as PbO2 dissolved in area 2, some of the Pb may have reprecipitated at the scale-water surface as plumbonacrite. Notably, although this pipe was removed from the system approximately 2 years after the disinfectant change and was exposed to orthophosphate, L1 in pipe B_area2 does not contain phosphorus or any crystalline Ca–Pb phosphates (Figure 4B and Supporting Information Table S5).
Identification of Lead Phosphate.
The combination of PXRD and SEM-EDS data confirmed the presence of lead phosphate minerals in the scales of both utilities, once the primary PbO2 material had been destabilized to form Pb(II) and the orthophosphate had time to migrate and concentrate within the scale. Lead phosphates were identified at depth within the scale materials of A_postPO4, B_area1 and B_area2; however, no individual powder diffraction file (PDF) card of over 400,000 entries in the ICDD PDF-4+ (2020) database exactly matched the peaks these materials displayed.
For example, the PXRD pattern from pipe A_postPO4 L3 shows a broad peak at d-spacing = 2.90 Å (Figure 5, dashed line). The layer is known to contain P, Ca, and Pb, as evidenced by the SEM–EDS analysis of the cross section (Figure 3B). The presumed lead phosphate peak falls amidst lines corresponding to the (211) lattice plane of a series of compounds within the solid solution between hydroxypyromorphite (Pb end member) and hydroxyapatite (Ca end member). Starting from hydroxypyromorphite, as more Ca is incorporated into the structure, the crystal lattice spacing decreases, resulting in an increasingly compact structure (Supporting Information Figure S7). This sliding chemical formula alters the d-spacings, and thus, the PXRD patterns that were generated. The result is variable crystal lattice distortion due to the differing amounts of incorporated Ca, which is visible in samples from both utilities. Note that this analysis focused on the (211) lattice plane because it is the most intense peak and is thus the easiest to consistently identify if the phase occurs in a mixture and/or as a minor component of the sample. However, other lattice planes (e.g., Figure 5, d = 4.03 Å line) also shift with varying Ca and Pb.
Figure 5.
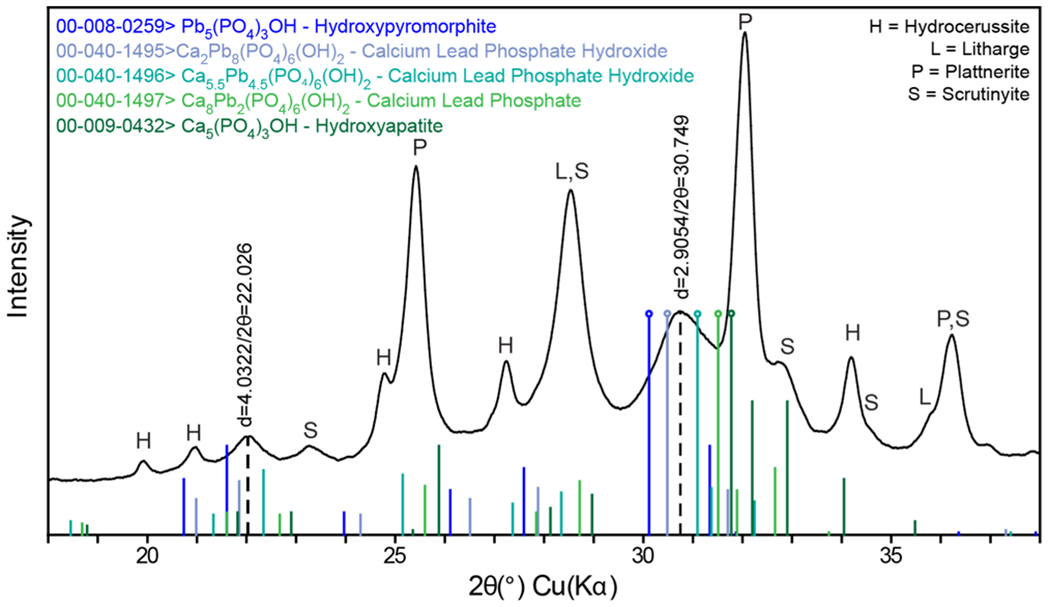
PXRD pattern from pipe A_postPO4 L3 overlain with peak positions for five Pb-, Ca-, and Ca–Pb-phosphates, with most intense lines marked by an open circle. The position of the peak corresponding to the (211) lattice plane of A_postPO4 is noted by a dashed line, d-spacing = 2.90 Å. Another lattice plane associated with Ca–Pb-phosphates is noted by the dashed line at d-spacing = 4.03 Å.
Vegard’s law was applied (Supporting Information Figure S1) in order to better characterize the proportions of Ca and Pb in the lead phosphates occurring in the scale samples. Weight percents for Ca, Pb, P, and O were calculated for each sample based on the d-spacing of the (211) peak (Table 2). Other trace elements such as Cl or F (common substitutions with OH− in apatites and pyromorphites) were not detected in the EDS analysis. These theoretical calculations were then compared to an independent line of evidence using measured EDS values taken from pipe: A_postPO4 L3, B_area1 L3, and B_area2 L2/L3 cross sections (Table 2). The measured Ca weight percents were all within 1–2 weight percent of the theoretically calculated value, O within 2–3 weight percent, P within 3–4 weight percent, and Pb within 4–7 weight percent. The measured Pb results displayed the most variation from the theoretically calculated value, with higher concentrations observed than calculated. However, this can be explained by the presence of other Pb minerals co-mingled with (Cax, Pb5-x) (PO4)3OH, as evidenced by the PXRD results (Supporting Information Table S5). In both utilities, one to five other lead phases were identified by PXRD to be in the same layer as (Cax, Pb5-x) (PO4)3OH, thus potentially contributing to higher lead concentrations observed in the EDS analysis.
Table 2.
Theoretical and Measured Elemental Concentrations for the Scale Samples Containing Suspected Calcium Lead Phosphates
| sample/layer | A_postPO4 L3 | B_area1 L3 | B_area2 L2 | B_area2 L3 |
|---|---|---|---|---|
| d-spacing_(211) (Å) | 2.9054 | 2.9450 | 2.9164 | 2.9194 |
| Ca (mol) | 1.8 | 0.6 | 1.5 | 1.4 |
| calculated Ca (wt %) | 7.0 | 1.8 | 5.4 | 4.9 |
| measured Ca (wt %) EDS | 5.6 | 2.2 | 5.3 | 5.3 |
| calculated Pb (wt %) | 63.9 | 73.9 | 67.1 | 67.9 |
| measured Pb (wt %) EDS | 70.0 | 79.8 | 73.1 | 73.1 |
| calculated P (wt %) | 9.0 | 7.5 | 8.5 | 8.4 |
| measured P (wt %) EDS | 5.3 | 4.2 | 4.9 | 4.9 |
| calculated O (wt %) | 20.1 | 16.7 | 19.0 | 18.7 |
| measured O (wt %) EDS | 17.1 | 14.5 | 16.6 | 16.6 |
The results from these two utilities agree with the findings of Hopwood et al., 201656 that the theoretical lead phosphate solid, hydroxypyromorphite, typically used in Pb solubility calculations for systems that use orthophosphate treatment, was not identified in these real-world LSLs. Instead, the identification of variable formulated lead apatites better fits the data and no currently available reference pattern was found to match the data collected from these two utilities. Based on the Ca molar concentrations determined from the PXRD patterns, pipe A_postPO4 and pipe B_area2 contain similar calcium lead phosphate solids, with the estimated formulas Ca1.8, Pb3.2(PO4)3OH and Ca1.5, Pb3.5(PO4)3OH, respectively. The calcium lead phosphate in pipe B_area1 has a formula with slightly less Ca: Ca0.6, Pb4.4(PO4)3OH. This further suggests that the thicker L1 deposit on pipe B_area1 may act as a diffusion barrier, and in addition to precluding the rapid dissolution of PbO2, it also precludes some of the diffusion of Ca and P down into the plane of lead phosphate formation.
Effect of Treatment Changes.
These two utilities can serve as useful case studies to demonstrate the effects of treatment changes and how the sequence of those changes impact active distribution system LSL scales. Both water systems were likely primarily PbO2 systems in advance of their treatment changes, made a disinfectant change, instituted an orthophosphate treatment, and experienced a lead action level exceedance. Although the sequence of events for these two utilities was different, the changes manifested in the scales had some remarkably similar signatures that could serve as a word of caution for other utilities contemplating similar changes in treatment.
In both systems, the disinfectant change yielded an almost immediate lead action level exceedance. For Utility A, that exceedance persisted for a total of 5 years, and only after approximately 1.5 years of an orthophosphate treatment did 90th percentile lead levels drop to around 10 ppb. This prolonged period of lead release can be explained through an examination of the scales. The uppermost scale layer observed in A_prePO4 and A_postPO4 was found to be composed of PbO2 compounds which begin to dissolve when water quality parameters such as the ORP change.28,35 This chronology of cross sections also provides an explanation as to why a delay was observed between the addition of orthophosphate and seeing a reduction in 90th percentile lead levels within the distribution system. EDS line scans and PXRD demonstrated that P completely bypassed the PbO2-rich layer and instead established itself in lower layers of the scale material, thus leaving the dissolving PbO2 directly accessible to flowing drinking water in some of the LSLs. As noted by Tesfai et al.55 and identified in other LSLs analyzed from this system, a Ca-, Al-, P-, and Fe-rich precipitate was formed in the distribution system. As the material accumulated on the surface, this deposit potentially created a diffusion barrier between the dissolving PbO2 and the flowing drinking water.
Utility B presented a different sequence of events in which the utility had implemented an orthophosphate treatment for approximately 9 years prior to switching disinfectants. After the switch, the system experienced a lead action exceedance for one quarter in 2007 and then returned to below the action level. A time series of cross sections was not available for this utility; however, the cross sections from the two different areas on the LSL received show a similar pattern to Utility A. Both P and Ca are present at depth within the scale, bypassing a majority of the PbO2-dominated layers and not creating a surface barrier of lead phosphates between the scale and flowing drinking water. In comparison to Utility A, it does appear that the 9 years of orthophosphate addition did reduce the length of Utility B’s lead action exceedance, but it did not eliminate the increase in water lead levels caused by the switch. Other factors, such as the precipitation of plumbonacrite at the surface (in pipe B_area2) or the presence of an amorphous deposit (in pipe B_area1) may have played a role in abbreviating the high lead release episode. However, the full extent of the lead release and its progression over time cannot be truly known from only the first draw 1 L sample, which rarely contain water that has stagnated within the LSL. Such samples are generally more representative of lead release within each individual house’s premise plumbing in close proximity to the sampled tap.
State of Disequilibrium.
The scales from Utilities A and B clearly show chemical reaction boundaries for solid-phase conversion occurring deep below the water contact surface, as well as evidence of simultaneous deposition on the uppermost scale surface. These surface deposits, although not necessarily contain the lower solubility Ca–Pb phosphates, can still have a strongly mitigating influence on lead release, particularly, if they also protect the underlying layers and help to maintain the physical stability of the scale material.
In both systems, the scale structure displayed a large degree of partial reaction and partial mineral conversion, even after over a year of consistent treatment once the disinfectant was changed and orthophosphate was present. A porous PbO2 zone also notably persists in both systems, emphasizing that, for many years, a state of disequilibrium governed the pipe scale mineralogy. This disequilibrium allowed for both dissolved and particulate lead release within the systems, dissolved lead via the dissolution of PbO2, and particulate release because of a weakened and porous scale structure. Fieldwork conducted with Utility A by other researchers identified the presence of lead particulates in drinking water tap samples, specifically PbO2 particulates.57,58 The A_postPO4 sample and additional LSLs received from Utility A (not detailed in this study) showed localized sloughing of outer scale layers (PbO2) above a detachment surface at the boundary between the PbO2 and Ca–Pb phosphate layers. These LSLs also show legacy-destabilized PbO2 and only partial conversion of the scale material to Ca-substituted hydroxypyromorphite even 4 years after the addition of orthophosphate to the system. This research reaffirms the fact that orthophosphate does not directly react with stable or remnant PbO2 scale layers.
The issue of prolonged destabilized PbO2 particulate release has been confirmed in a system analogous to Utilities A and B, although Utility C has not undergone a change in the disinfectant type as the source of chemical instability. PXRD results from a LSL scale analysis (Supporting Information Figure S5) illustrate a similar scale structure with a Ca–Pb phosphate layer below a legacy PbO2 layer (Supporting Information Figure S6). A physical investigation of a residence in Utility C revealed the presence of particulates within the premise plumbing. These particulates, which had collected in faucet aerators and a toilet tank, were also analyzed by PXRD and found to contain β-PbO2, displaying a similar pattern to the PXRD patterns collected from the Utility C LSL scale materials (Figure 6).
Figure 6.
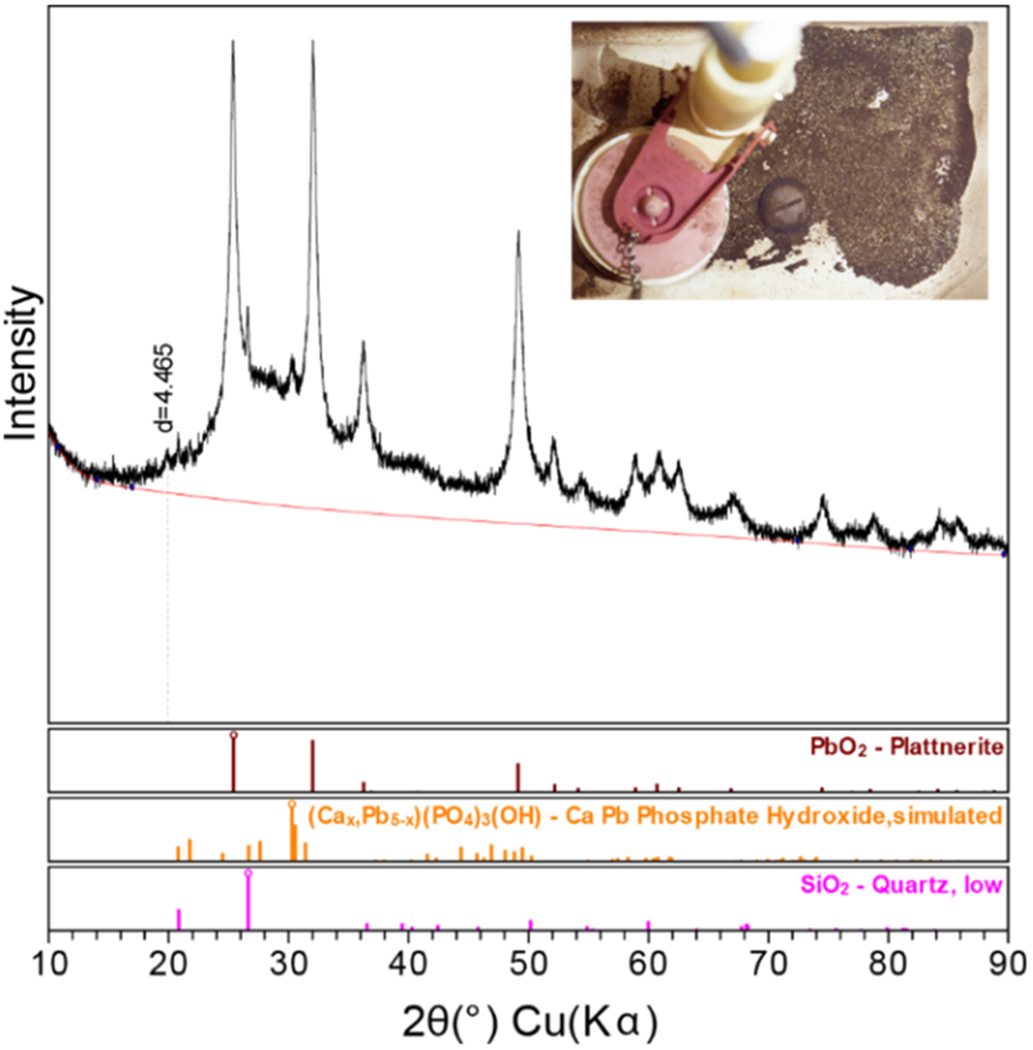
Utility C PXRD pattern and identified crystalline phases from sediment collected in a residential toilet tank (upper right-hand image). The peak at a d-spacing of 4.465 Å is likely attributable to Pb(II) carbonate.
The results from Utility C suggest that even if a phosphate-rich layer exists above a remnant PbO2 layer, there is a real and ongoing particulate risk in a system that has not experienced a recent treatment or water quality change. Scale instability, whether caused by a single or a combination of chemical or physical factors, can be a long-term (years to decades), ongoing source of particulate lead to drinking water. This is due in part to the thickness of real-world PbO2 scales. The case studies presented in this article illustrate that the PbO2 portion of the scale comprises a thickness of at least 50 μm on the pipe wall. When this material is destabilized, the result is not an immediate sloughing and removal of the PbO2 scale. Instead, because of the thickness of the initial PbO2 scale, it takes time for the material to be removed via dissolution and particulate release. The erratic and long-lasting nature of the release, which may be caused by chemical or hydraulic changes not observed or controlled by the consumers, and the high lead content of the usually invisible particulates, make it a potential ongoing lead exposure risk that is difficult to predict or quantify.
Supplementary Material
ACKNOWLEDGMENTS
All scale metal analyses were conducted by the United States Geological Survey’s Mineral Resource Surveys Program under Interagency Agreement number DW14999901 under the direction of Dr. Stephen A. Wilson. The authors also acknowledge pipe sampling and analysis assistance by Meghan Welch, Hannah Luebbers, Rachel Copeland, and Mallikarjuna Nadagouda.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.est.0c03027.
Water utility treatment, lead 90th percentiles, X-ray diffraction patterns, methods, LSL scale descriptions, and graphics related to the Ca-substituted lead phosphate (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.est.0c03027.
The authors declare no competing financial interest.
This document has been reviewed in accordance with U.S. Environmental Protection Agency policy and approved for publication. The views expressed in this article are those of the author(s) and do not necessarily represent the views or policies of the U.S. Environmental Protection Agency. Any mention of trade names, manufacturers, or products does not imply an endorsement by the United States Government or the U.S. Environmental Protection Agency. EPA and its employees do not endorse any commercial products, services, or enterprises.
Contributor Information
Michael K. DeSantis, Center for Environmental Solutions and Emergency Response, Water Infrastructure Division, U.S. Environmental Protection Agency, Cincinnati, Ohio 45268, United States.
Michael R. Schock, Center for Environmental Solutions and Emergency Response, Water Infrastructure Division, U.S. Environmental Protection Agency, Cincinnati, Ohio 45268, United States.
Jennifer Tully, Center for Environmental Solutions and Emergency Response, Water Infrastructure Division, U.S. Environmental Protection Agency, Cincinnati, Ohio 45268, United States.
Christina Bennett-Stamper, Center for Environmental Solutions and Emergency Response, Water Infrastructure Division, U.S. Environmental Protection Agency, Cincinnati, Ohio 45268, United States.
REFERENCES
- (1).USEPA. Economic Analysis for the Proposed Lead and Copper Rule Revisions, 2019. Supporting and Related Material in Regulations.gov Docket: EPA-HQ-OW-2017-0300. [Google Scholar]
- (2).USEPA. National Primary Drinking Water Regulations; Total Coliforms (Including Fecal Coliforms and E. Coli). 54 Fed. Reg 124 (June 29, 1989), 27544–27568. [Google Scholar]
- (3).USEPA. National Primary Drinking Water Regulations: Filtration, Disinfection; Turbidity, Giardia lamblia, Viruses, Legionella, and Heterotrophic Bacteria. 54 Fed. Reg 124 (June 29, 1989), 27486–27541. [Google Scholar]
- (4).USEPA. National Primary Drinking Water Regulations: Disinfectants and Disinfection Byproducts. 63 Fed. Reg 241 (December 16, 1998), 69390–69476. [Google Scholar]
- (5).USEPA. National Primary Drinking Water Regulations: Interim Enhanced Surface Water Treatment. 63 Fed. Reg 241 (December 16, 1998), 69478–69521. [Google Scholar]
- (6).USEPA. National Primary Drinking Water Regulations: Long Term 1 Enhanced Surface Water Treatment Rule. 67 Fed. Reg 9 (January 14, 2002), 1812–1844. [PubMed] [Google Scholar]
- (7).USEPA, National Primary Drinking Water Regulations: Stage 2 Disinfectants and Disinfection Byproducts Rule. 71 Fed. Reg 2 (January 4, 2006), 388–493. [Google Scholar]
- (8).CDC. History of Drinking Water Regulations, https://www.cdc.gov/healthywater/surveillance/drinking-water-reg-history.html (accessed January 30, 2020).
- (9).USEPA. National Primary Drinking Water Regulations: Lead and Copper. 56 Fed. Reg. 110 (June 7, 1991), 26460–26564. [Google Scholar]
- (10).Schock MR Corrosion Inhibitor Applications in Drinking Water Treatment: Conforming to the Lead and Copper Rule. NACE International Annual Conference and Exposition: Denver, CO, 1996. [Google Scholar]
- (11).Schock MR; Harmon SM; Swertfeger J; Lohmann R Tetravalent Lead: A Hitherto Unrecognized Control of Tap Water Lead Contamination. AWWA Water Quality Technology Conference: Nashville, TN, November 11–15, 2001. [Google Scholar]
- (12).Lytle DA; Schock MR The Formation of Pb(IV) Oxides in Chlorinated Water. J. Am. Water Works Assoc. 2005, 97, 102–114. [Google Scholar]
- (13).Triantafyllidou S; Schock MR; DeSantis MK; White C Low contribution of PbO2-coated lead service lines to water lead contamination at the tap. Environ. Sci. Technol. 2015, 49, 3746–3754. [DOI] [PubMed] [Google Scholar]
- (14).Schock MR; Lytle DA Internal Corrosion and Deposition Control, 6th ed.; McGraw-Hill, Inc: New York, 2011. [Google Scholar]
- (15).Schock MR Response of Lead Solubility to Dissolved Carbonate in Drinking-Water. J. Am. Water Works Assoc. 1980, 72, 695–704. [Google Scholar]
- (16).Schock MR Response of Lead Solubility to Dissolved Carbonate in Drinking Water (erratum). J. Am. Water Works Assoc 1981, 73, 36. [Google Scholar]
- (17).Pourbaix M; De Zoubov N; Vanleuenhaghe C; Van Rysselberghe P Section 17.5. Lead. Atlas of Electrochemical Equilibria in Aqueous Solutions; National Association of Corrosion Engineers: Houston, TX, 1966; pp 485–492. [Google Scholar]
- (18).Garrels RM; Christ CL Solutions, Minerals and Equilibria; Freeman, Cooper and Company: San Francisco, California, 1965. [Google Scholar]
- (19).Xie Y; Wang Y; Singhal V; Giammar DE Effects of pH and Carbonate Concentration on Dissolution Rates of the Lead Corrosion Product PbO2. Environ. Sci. Technol. 2010, 44, 1093–1099. [DOI] [PubMed] [Google Scholar]
- (20).Lytle D; Schock M; Formal C; Bennett-Stamper C; Harmon S; Nadagouda M; DeSantis M; Tully J; Pham M Lead Particle Size Fractionation and Identification in Newark, New Jersey’s Drinking Water. Environ. Sci. Technol. 2020, Accepted paper, DOI: 10.1021/acs.est.0c03797 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Copeland A; Lytle DA Measuring the oxidation-reduction potential of important oxidants in drinking water. J. AWWA 2014, 106, E10–E20. [Google Scholar]
- (22).Renner R Plumbing the depths of D.C.’s drinking water crisis. Environ. Sci. Technol. 2004, 38, 224A–227A. [DOI] [PubMed] [Google Scholar]
- (23).Renner R Leading to lead. Sci. Am. 2004, 291, 22–24. [DOI] [PubMed] [Google Scholar]
- (24).Renner R Chloramine’s effect on lead in drinking water. Environ. Sci. Technol. 2006, 40, 3129–3130. [PubMed] [Google Scholar]
- (25).Renner R Mapping out lead’s legacy. Environ. Sci. Technol. 2009, 43, 1655–1658. [DOI] [PubMed] [Google Scholar]
- (26).Renner R Out of Plumb When Water Treatment Causes Lead Contamination. Environ. Health Perspect. 2009, 117, A542–A547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Edwards M; Dudi A Role of chlorine and chloramine in corrosion of lead-bearing plumbing materials. J. Am. Water Works Assoc. 2004, 96, 69–81. [Google Scholar]
- (28).Switzer JA; Rajasekharan VV; Boonsalee S; Kulp EA; Bohannan EW Evidence that monochloramine disinfectant could lead to elevated Pb levels in drinking water. Environ. Sci. Technol. 2006, 40, 3384–3387. [DOI] [PubMed] [Google Scholar]
- (29).Xie Y; Wang Y; Giammar DE Impact of chlorine disinfectants on dissolution of the lead corrosion product PbO2. Environ. Sci. Technol. 2010, 44, 7082–7088. [DOI] [PubMed] [Google Scholar]
- (30).Lin Y-P; Valentine RL Release of Pb(II) from monochloramine-mediated reduction of lead oxide (PbO2). Environ. Sci. Technol. 2008, 42, 9137–9143. [DOI] [PubMed] [Google Scholar]
- (31).Lin Y-P; Valentine RL Reduction of lead oxide (PbO2) and release of Pb(II) in mixtures of natural organic matter, free chlorine and monochloramine. Environ. Sci. Technol. 2009, 43, 3872–3877. [DOI] [PubMed] [Google Scholar]
- (32).Rajasekharan VV; Clark BN; Boonsalee S; Switzer JA Electrochemistry of free chlorine and monochloramine and its relevance to the presence of Pb in drinking water. Environ. Sci. Technol. 2007, 41, 4252–4257. [DOI] [PubMed] [Google Scholar]
- (33).Schock MR; Giani R Oxidant/Disinfectant Chemistry and Impacts on Lead Corrosion. Sunday Workshop, AWWA Water Quality Technology Conference: San Antonio, TX, November 14, 2004. [Google Scholar]
- (34).Dryer DJ; Korshin GV Investigation of the reduction of lead dioxide by natural organic matter. Environ. Sci. Technol. 2007, 41, 5510–5514. [DOI] [PubMed] [Google Scholar]
- (35).Schock MR; Wagner I; Oliphant R The Corrosion and Solubility of Lead in Drinking Water Internal Corrosion of Water Distribution Systems, 2nd ed.; AWWA Research Foundation/TZW: Denver, CO, 1996; pp 131–230. [Google Scholar]
- (36).Edwards M; McNeill LS Effect of phosphate inhibitors on lead release from pipes. J. Am. Water Works Assoc. 2002, 94, 79–90. [Google Scholar]
- (37).Schock MR Understanding Corrosion Control Strategies for Lead. J. Am. Water Works Assoc. 1989, 81, 88–100. [Google Scholar]
- (38).Schock MR; Clement JA Lead and Copper Control with Non-Zinc Orthophosphate. J. N. Engl. Water Work. Assoc. 1998, 112, 20–42. [Google Scholar]
- (39).Sheiham I; Jackson PJ The Scientific Basis for Control of Lead in Drinking Water by Water Treatment. J. Inst. Water Eng. Sci. 1981, 35, 491–515. [Google Scholar]
- (40).Gregory R; Jackson PJ Central Water Treatment to Reduce Lead Solubility. Proceedings AWWA Annual Conference: Dallas, TX, June 10–14, 1984. [Google Scholar]
- (41).Schock MR; Scheckel KG; DeSantis MK; Gerke TL Mode of Occurrence, Treatment and Monitoring Significance of Tetravalent Lead. AWWA Water Quality Technology Conference: Quebec City, Canada, November 6–10, 2005. [Google Scholar]
- (42).Desantis MK; Schock MR Ground Truthing the ‘Conventional Wisdom’ of Lead Corrosion Control Using Mineralogical Analysis. 2014 AWWA Water Quality Technology Conference: New Orleans, LA, November 16–20, 2014. [Google Scholar]
- (43).DeSantis MK; Schock MR; Bennett-Stamper C Incorporation of Phosphate in Destabilized PbO2 Scales. AWWA Water Quality Technology Conference: Seattle, WA, November 15–19, 2012. [Google Scholar]
- (44).Desantis MK; Welch MM; Schock MR Mineralogical Evidence of Galvanic Corrosion in Domestic Drinking Water Pipes. AWWA Water Quality Technology Conference: Seattle, WA, November 15–19, 2009. [Google Scholar]
- (45).Schock MR; Cantor AF; Triantafyllidou S; Desantis MK; Scheckel KG Importance of pipe deposits to Lead and Copper Rule compliance. J. Am. Water Works Assoc. 2014, 106, E336–E349. [Google Scholar]
- (46).Wasserstrom LW; Miller SA; Triantafyllidou S; Desantis MK; Schock MR Scale Formation Under Blended Phosphate Treatment for a Utility With Lead Pipes. J. Am. Water Works Assoc. 2017, 109, E464–E478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Denton AR; Ashcroft NW Vegard’s law. Phys. Rev. A 1991, 43, 3161–3164. [DOI] [PubMed] [Google Scholar]
- (48).Bae Y; Pasteris JD; Giammar DE The Ability of Phosphate To Prevent Lead Release from Pipe Scale When Switching from Free Chlorine to Monochloramine. Environ. Sci. Technol. 2020, 54, 879–888. [DOI] [PubMed] [Google Scholar]
- (49).Tully J; DeSantis MK; Schock MR Water quality–pipe deposit relationships in Midwestern lead pipes. AWWA Water Sci. 2019, 1, e1127. [PMC free article] [PubMed] [Google Scholar]
- (50).Ives DJG; Rawson AE Copper Corrosion. J. Electrochem. Soc. 1962, 109, 458. [Google Scholar]
- (51).Ferguson JL; von Franquá O; Schock MR Corrosion of Copper in Potable Water Systems Internal Corrosion of Water Distribution Systems, 2nd ed.; AWWA Research Foundation/DVGW-TZW: Denver, CO, 1996; pp 231–268. [Google Scholar]
- (52).Werner W; Groß H-J; Sontheimer H Corrosion of Copper Pipes in Drinking Water Installations. Translation of: gwf-Wasser/Abwasser 1994, 135, 1–15. [Google Scholar]
- (53).Giani R; Donnelly M; Ngantcha T The Effects of Changing Between Chloramine and Chlorine Disinfectants on Lead Leaching. AWWA Water Quality Technology Conference: Quebec City, Canada, November 6–10, 2005. [Google Scholar]
- (54).Lytle DA; Schock MR; Scheckel K The inhibition of Pb(IV) oxide formation in chlorinated water by orthophosphate. Environ. Sci. Technol. 2009, 43, 6624–6631. [DOI] [PubMed] [Google Scholar]
- (55).Tesfai F; Pierre C; Reiber S; Giani R; Donnelly M Precipitate formation in the distribution system following addition of orthophosphate. AWWA Water Quality Technology Conference: Denver, CO, November 5–9, 2006. [Google Scholar]
- (56).Hopwood JD; Derrick GR; Brown DR; Newman CD; Haley J; Kershaw R; Collinge M The Identification and Synthesis of Lead Apatite Minerals Formed in Lead Water Pipes. J. Chem. 2016, 2016, 1–11. [Google Scholar]
- (57).Triantafyllidou S; Parks J; Edwards M Lead Particles in Potable Water. J. Am. Water Works Assoc. 2007, 99, 107–117. [Google Scholar]
- (58).Triantafyllidou S; Edwards M Lead (Pb) in Tap Water and in Blood: Implications for Lead Exposure in the United States. Crit. Rev. Environ. Sci. Technol 2012, 42, 1297–1352. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.