

Abstract
AIMS:
Brain mural cells (BMC), smooth muscle cells and pericytes, interact closely with endothelial cells and modulate numerous cerebrovascular functions. A loss of BMC function is suspected to play a role in the pathophysiology of Alzheimer’s Disease (AD).
METHODS:
BMC markers, namely smooth muscle alpha actin (α-SMA) for smooth muscle cells, as well as platelet-derived growth factor receptor β (PDGFRβ) and aminopeptidase N (ANPEP or CD13) for pericytes, were assessed by Western immunoblotting in microvessel extracts from the parietal cortex of 60 participants of the Religious Orders study, with ages at death ranging from 75 to 98 years old.
RESULTS:
Participants clinically diagnosed with AD had lower vascular levels of α-SMA, PDGFRβ and CD13. These reductions were correlated with lower cognitive scores for global cognition, episodic and semantic memory, perceptual speed and visuospatial ability. In addition, α-SMA, PDGFRβ and CD13 were negatively correlated with vascular Aβ40 concentrations. Vascular levels of BMC markers were also inversely correlated with cleaved insoluble phosphorylated transactive response DNA binding protein 43 (TDP-43) (25 kDa) and positively correlated with cleaved soluble phosphorylated TDP-43 (35 kDa) in cortical homogenates, suggesting strong association between BMC loss and cleaved phosphorylated TDP-43 aggregation.
CONCLUSIONS:
The results of this study highlight a loss of BMC in AD. The associations between a-SMA, PDGFRβ and CD13 vascular levels with cognitive scores, TDP-43 aggregation and cerebrovascular accumulation of Aβ in the parietal cortex suggest that BMC loss contributes to both AD symptoms and pathology, further strengthening the link between cerebrovascular defects and dementia.
Keywords: brain mural cells, smooth muscle cells, pericytes, Alzheimer’s Disease, cognitive impairment, TDP-43, cerebrovasculature
Introduction
Defects in key aspects of cerebrovascular function are inseparable from the classical pathophysiological pathways of Alzheimer’s Disease (AD) [1–4]. It has been consistently shown, for instance, that patients with AD display reduced brain glucose uptake as well as decreased cerebral blood flow [5, 6]. Post-mortem analyses show a thickening of the basement membrane [7–9] and a high prevalence of cerebral amyloid angiopathy (CAA) in AD [10, 11]. Accordingly, we recently reported that patients with both AD and CAA had the highest vascular concentrations of Aβ peptides, with lower levels of efflux transporters and degrading enzymes as well as higher levels of its precursor cleaving enzyme (BACE1) in brain microvascular cells as possible underlying mechanisms [2].
Brain mural cells (BMC) are in close contact with brain endothelial cells along blood vessels and play a key role in the cerebrovascular function, including the regulation of BBB function and cerebral blood flow, as well as angiogenesis and neuroinflammatory responses [12–20]. BMC are subdivided in two main cell types, namely smooth muscle cells (SMC) and pericytes, which can be differentiated using various markers, such as smooth muscle alpha actin (α-SMA) for SMC and platelet-derived growth factor receptor β (PDGFRβ), aminopeptidase N (ANPEP or CD13) and neural/glial antigen 2 (NG2) for pericytes. However, the definition of BMC subtypes is still ambiguous, mostly because BMC can have heterogeneous phenotypes in terms of morphology and protein expression [21–26]. Indeed, imaging analyses in transgenic mice showed that arterioles were surrounded by smooth muscle cells while the capillaries were covered by pericytes, with distinct morphologies observed along the capillary bed [22, 23]. Hybrid cells, expressing both SMC and pericyte markers, were also visualized at the arteriolar end of the capillary bed, leading to their classification as either SMC [24] or pericytes [22], the latter being in accordance with the initial description of pericytes by Zimmermann [27].
Various studies have reported changes in SMC in AD. For instance, it has been shown that, compared to controls, endothelial coverage of α-SMA staining on cortical vessels is diminished in AD patients [28, 29] but increased in non-demented subjects with high plaque burden [30], leading the authors to hypothesize that higher α-SMA levels at the preclinical stage could be a mechanism to counteract vascular dysfunction in AD. Similar conclusions were drawn in the APPSweDI model (harbouring the Swedish, Dutch and Iowa mutations on the APP gene) following the observation that both mRNA and protein levels of α-SMA were increased in microvessels of 12-month-old mice [31]. In contrast, analyses in Tg2576 mice, another mouse model of Aβ neuropathology, showed that SMC first become disorganized, evidenced by a dysfunctional vasodilation response, then reduced in number with the concomitant progression of AD neuropathology [32].
Concerning pericytes, experiments in APPsw mice showed that genetic invalidation of one Pdgfrb allele (APPsw/0/Pdgfrb+/−) leads to accelerated vascular accumulation of Aβ peptides compared to age-matched APPsw/0/Pdgfrb+/+ mice [33], suggesting a role for pericytes in Aβ clearance. Beside an aggravated cognitive deficit, these double transgenic mice also exhibited tau pathology and early neuronal loss, both being uncharacteristic features of most APP models [33]. Human post-mortem studies probing BMC are more limited. Immunofluorescence studies on small number of samples report either a decrease or no change in pericyte number and endothelial coverage in the cortex and hippocampus of AD patients [34, 35]. A more recent study showed lower concentrations of ELISA-detected PDGFRβ in total precuneus homogenates from AD cases compared to controls [36]. The cerebrospinal fluid (CSF) has also been used to estimate changes in central pericytes, showing for instance increased concentrations of the cleaved, soluble form of PDGFβ in the CSF of individuals with mild cognitive impairment [37, 38] or with a clinical or biomarker-based diagnosis of AD [39]. Altogether, these studies suggest that BMC deficiency plays a pivotal role in the pathophysiology of AD [28, 29, 34–36, 40].
Taking advantage of our recently validated method to isolate microvessels from frozen human brain material [2], we sought to directly probe BMC by measuring levels of SMC and pericyte markers in microvessel samples from a cohort of study volunteers divided in three groups based on detailed ante mortem assessment of cognitive function: no cognitive impairment (NCI), mild cognitive impairment (MCI) or AD. Brain samples from the cohort used here have underwent an extensive biochemical characterization not only for Aβ and tau, but also for transactive response DNA-binding protein 43 (TDP-43) and vascular pathologies [2, 41, 42]. We thus interrogated whether the extent of BMC pathology in the parietal cortex was related to neuropathological markers, vascular Aβ levels and cognitive decline.
Methods
Human samples: Religious Orders Study (Rush Alzheimer’s Disease Center)
Parietal cortex samples were obtained from participants in the Religious Orders Study, a longitudinal clinical and pathological study of aging and dementia from which extensive amounts of clinical and neuropathological data were available [43]. Each participant enrolled without known dementia and underwent annual uniform structured clinical evaluations until death. Briefly, dementia and AD diagnosis required evidence of meaningful decline in cognitive function and impairment in at least 2 domains of cognition, one of which was episodic memory, based on the results of 21 cognitive performance tests and their review by a clinical neuropsychologist and expert clinician [44]. “MCI” refers to participants with cognitive impairment as assessed by the neuropsychologist but without a diagnosis of dementia, as determined by the clinician [45]. “NCI” refers to participants with no cognitive impairment [46]. A global measure of cognition along with five cognitive domains (episodic, semantic and working memory, perceptual speed and visual-spatial ability) were generated from 19 cognitive performance tests [47]. Each subject was also interviewed about their current prescription medication usage, such as antihypertensive and diabetes medications, in the last two weeks prior to its follow-up as reported [48, 49]. At death, a neurologist, blinded to all post-mortem data, reviewed select clinical data and rendered a summary diagnostic opinion regarding the clinical diagnosis at the time of death. Participants thus received a clinical diagnosis of MCI (n = 20) or AD (n = 20) or NCI (n = 20), as previously described [50]. The neuropathological assessment was performed using the ABC scoring method found in the revised National Institute of Aging - Alzheimer’s Association (NIA-AA) guidelines for the neuropathological diagnosis of AD [51]. Each case was given, by examiners blinded to all clinical data, a score for three different neuropathological parameters: A) Thal score assessing phases of Aβ plaque accumulation [52], B) Braak score assessing neurofibrillary tangle pathology [53], and C) CERAD score assessing neuritic plaque pathology [54]. These scores were then combined to obtain an ABC score, reported as AX, BX, CX with X ranging from 0 to 3 for each parameter [51]. Neuritic plaques, diffuse plaques, and neurofibrillary tangles in the parietal cortex were counted following Bielschowsky silver impregnation, as previously described. Each ABC score was converted into one of four levels of AD neuropathological changes, not, low, intermediate or high. According to the chart found in the revised NIA-AA guidelines, intermediate or high levels of AD neuropathological changes are consistent with a neuropathological diagnosis of AD, while no or a low level of AD neuropathological changes are not [51]. Therefore, when group comparisons were based on the neuropathological diagnosis, individuals with intermediate or high levels of AD neuropathological changes were classified as AD while participants with no or a low level of AD neuropathological changes were classified as Controls. In addition, the presence of cerebral amyloid angiopathy in parenchymal vessels of the parietal cortex, and cerebral macroinfarcts and microinfarcts was determined during neuropathological evaluations, as described previously [2, 55]. Table 1 summarizes clinical, neuropathological and biochemical data of participants grouped by the clinical diagnosis.
Table 1 –
Cohort characteristics
| Characteristics | NCI | MCI | AD | Statistical Analysis | P-value | |
|---|---|---|---|---|---|---|
| N | 20 | 20 | 20 | ___ | ||
| Men | 4/20 | 9/20 | 7/20 | C; Pearson test, χ2 = 2.85 | p = 0.024 | |
| Mean age at death | 87.1 (5.8) | 87.1 (5.2) | 87.3 (4.9) | A; F(groups)2,57 = 0.01 | p = 0.99 | |
| Mean education, years | 18.5 (3.6) | 18.6 (3.0) | 17.5 (3.0) | A; F(groups)2,57 = 0.69 | p = 0.51 | |
| Mean MMSE | 27.2 (1.8) | 25.5 (3.1) | 15.8 (7.9)¶ | A; F(groups)2,57 = 30.17 | p < 0.0001 | |
| Global cognition score | −0.03 (0.38) | −0.43 (0.45) | −1.66 (0.89)¶ | A; F(groups)2,56 = 38.03 | p < 0.0001 | |
| apoE ε4 allele carriage (%) | 30 | 30 | 35 | C; Pearson test, χ2 = 0.15 | p = 0.93 | |
| Thal amyloid score 0/1/2/3 (n) | 4/8/4/4 | 2/6/7/5 | 1/2/6/11 | |||
| Braak score 0/1/2/3 (n) | 0/3/17/0 | 0/1/18/1 | 0/3/7/10 | |||
| CERAD score 0/1/2/3 (n) | 7/3/8/2 | 7/3/6/4 | 1/1/6/12 | |||
| Neuropathological diagnosis Control/AD (n) | 11/9 | 8/12 | 3/17$ | C; Pearson test, χ2 = 7.03 | p = 0.0297 | |
| Parenchymal CAA stage in parietal cortex 0/1/2/3/4 (n) | 10/6/0/1/0 | 12/2/2/2/2 | 11/3/4/0/1 | C; Pearson test, χ2 = 10.49 | p = 0.23 | |
| Presence of chronic cortical macroinfarcts 0/1 (n) | 19/1 | 17/3 | 17/3 | C; Pearson test, χ2 = 1.29 | p = 0.52 | |
| Presence of chronic cortical microinfarcts 0/1 (n) | 17/3 | 17/3 | 17/3 | ___ | ||
| Usage of antihypertensive medication 0/1 (n) | 2/18 | 4/16 | 1/19 | C; Pearson test, χ2 = 2.26 | p = 0.32 | |
| Usage of diabetes medication 0/1 (n) | 15/5 | 18/2 | 15/5 | C; Pearson test, χ2 = 1.88 | p = 0.39 | |
| Cerebellar pH | 6.35 (0.34) | 6.32 (0.28) | 6.31 (0.46) | A; F(groups)2,57 = 0.07 | p = 0.93 | |
| Postmortem delay, hours | 7.1 (5.6) | 7.8 (5.2) | 7.8 (4.8) | A; F(groups)2,57 = 0.13 | p = 0.88 | |
| Diffuse plaque count in the parietal cortex | 8.7 (15.9) | 14.5 (13.2) | 19.6 (18.2) | A; F(groups)2,57 = 2.36 | p = 0.10 | |
| Neuritic plaque count in the parietal cortex | 6.3 (8.9) | 7.0 (7.7) | 18.0 (15.5)* | A; F(groups)2,57 = 6.81 | p = 0.0022 | |
| Neurofibrillary tangle count in the parietal cortex | 0.15 (0.49) | 0.25 (0.72) | 5.25 (11.10)& | A; F(groups)2,57 = 4.12 | p = 0.02 | |
| Insoluble Aβ40 (pg/mg tissue) | 4185 (6606) | 1094 (1931) | 2224 (3542) | A; F(groups)2,39 = 1.51 | p = 0.23 | |
| Insoluble Aβ42 (pg/mg tissue) | 622.9 (646.7) | 843.4 (934.3) | 2331 (4841) | A; F(groups)2,52 = 1.95 | p = 0.15 | |
| Insoluble phosphorylated tau (AD2; ROD) | 212.5 (763.2) | 1038 (2814) | 5164 (8650)& | A; F(groups)2,57 = 5.07 | p = 0.0094 | |
| Cyclophilin B in microvessel extracts (ROD) | 2.70 (0.82) | 2.95 (0.70) | 2.44 (0.76) | A; F(groups)2,53 = 2.01 | p = 0.13 | |
| CD31 levels in microvessel extracts (normalized ROD) | 0.49 (0.35) | 0.54 (0.38) | 0.38 (0.64) | A; F(groups)2,51 = 0.51 | p = 0.60 |
Participants were grouped according to the clinical diagnosis. The neuropathological diagnosis was established based on the ABC scoring method described in the revised NIA-AA guidelines (Montine et al., 2012). Parenchymal CAA stages in the parietal cortex were determined in the angular gyrus. Brain pH was measured in cerebellum extracts. Insoluble Aβ peptide concentrations in the parietal cortex were determined by ELISA and are expressed as picograms/milligram of tissue. Relative concentrations of insoluble phosphorylated tau in the parietal cortex were determined by Western immunoblotting using the AD2 antibody. Vascular levels of cyclophilin B and CD31 were measured by Western immunoblotting in microvessel-enriched extracts from the same parietal cortex samples as reported in our previous work (Bourassa et al., 2019b). Values are expressed as means (SD) unless specified otherwise. Statistical analysis: (A) one-way analysis of variance followed by a Tukey’s post hoc test,
p < 0.05 versus NCI and MCI,
p < 0.01 versus NCI and MCI,
p < 0.0001 versus NCI and MCI; (C) Contingency, Pearson test,
p < 0.05. CD31 data in microvessel extracts were normalized with cyclophilin B as loading control.
Abbreviations: AD, Alzheimer’s disease; C, contingency; CAA, cerebral amyloid angiopathy; CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; MCI, mild cognitive impairment; NCI, healthy controls with no cognitive impairment; ROD, relative optical density.
Isolation of human brain microvessels
Microvessel-enriched extracts from frozen human brain samples used in this study were described in our previous publication [2]. This method consists of a series of centrifugation steps, including one density gradient centrifugation with dextran, after which the tissue is filtered through a 20-μm nylon filter, generating two fractions: one enriched in cerebral microvessels, the other consisting of microvessel-depleted parenchymal cell populations. Our previous work and data shown in Figure 1 confirm the composition of each fraction using immunodetection of endothelial and neuronal markers [2]. Proteins of both fractions were extracted using a lysis buffer and supernatants were kept for Western immunoblotting analyses. Protein concentrations were all determined using the bicinchoninic acid assay. The detailed experimental procedure is described in the Supplementary material.
Figure 1: Mural cell markers are enriched in human and murine brain microvessel extracts.
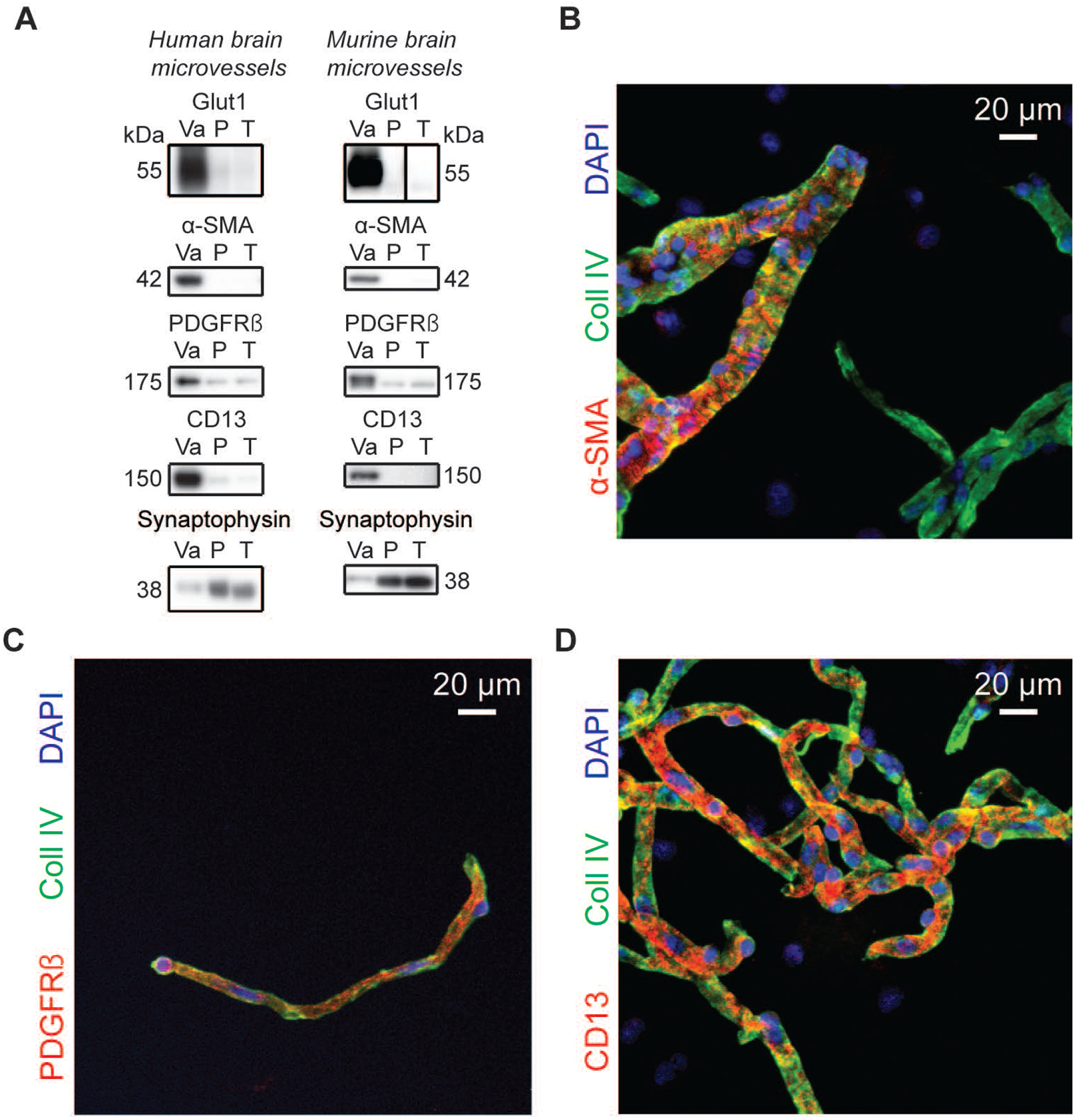
A) Western immunoblotting analyses on human and murine brain microvessel extracts show that the endothelial marker Glut1 as well as known mural cell markers are concentrated in the vascular fraction whereas synaptophysin, a neuronal marker, is enriched in the microvessel-depleted parenchymal fraction. Representative photo examples were taken from the same immunoblot experiment, and black vertical line was inserted to indicate nonconsecutive bands. The same amount (8 μg) of proteins per sample was loaded. B-D) Subsequent immunofluorescence assays on human brain microvessels showed that antibodies raised against α-SMA labeled larger vessels while capillary-like smaller vessels were stained by PDGFRβ and CD13 antibodies. Scale bar: 20 μm. Abbreviations: α-SMA, smooth muscle alpha actin; CD13, aminopeptidase N; Coll IV, type IV collagen; P, microvessel-depleted parenchymal fraction; PDGFRβ, platelet-derived growth factor receptor β; T, total homogenate; Va, vascular fraction, enriched in microvessels.
Immunofluorescence analysis of isolated human microvessels
Extracts from the vascular fraction were dried on Superfrost Plus slides and fixed using a 4% paraformaldehyde solution in PBS for 20 minutes at RT and then blocked with a 10% normal horse serum and 0.1% Triton X-100 solution in PBS for 1 hour at RT. For Aβ immunolabeling only, a pretreatment with 90% formic acid for 10 minutes was performed between the fixation and blockage steps. Then, samples were stained with goat anti-type IV collagen and with rabbit anti-smooth muscle alpha actin (Abcam, ab5694, 1:100), rabbit anti-PDGFRβ (Abcam, ab32570, 1:100), rabbit anti-CD13 (Proteintech, 14553–1-AP, 1:100) or mouse anti-Aβ40 (BioLegend, 805401, 1:100). Extracts were then incubated with secondary antibodies (donkey anti-goat Alexa Fluor 488, donkey anti-rabbit Alexa Fluor 555 or donkey anti-rabbit Alexa Fluor 647, and donkey anti-mouse Alexa Fluor 647). Finally, cell nuclei were counterstained with DAPI and slides were mounted with Mowiol mounting medium. Between each step, three washes of 5 minutes with PBS were performed. Images were taken using a laser scanning confocal microscope (Olympus IX81, FV1000; Ontario, Canada) and were acquired by sequential scanning using optimal z-separation at a magnification of 20X.
Protein fractionation from human parietal cortex
Each inferior parietal cortex sample (~ 100 mg) was sequentially centrifuged to generate a Tris-buffered saline (TBS)-soluble protein fraction containing soluble intracellular, nuclear and extracellular proteins, a detergent-soluble fraction containing membrane-bound proteins and a detergent-insoluble fraction (formic acid extract) containing insoluble aggregates, as previously reported [42]. The TBS-soluble fraction was used to probe for phosphorylated TDP-43 (Ser403/404) and total TDP-43, whereas the formic acid-soluble fraction was used to probe for phosphorylated TDP-43 (Ser409/410), total TDP-43, and phosphorylated tau (Ser396/404, clone AD2). The resulting whole homogenates of total soluble proteins (TBS- and detergent-soluble protein homogenates) were also used for comparison with microvessel-enriched extracts.
Isolation of murine brain microvessels
The 3xTg-AD mouse model used in this study has been described previously. Briefly, 3xTg-AD mice express mutant preselinin-1 (PS1M146V), APPswe, and tau (P301L) transgenes, leading to the progressive accumulation of Aβ plaques and neurofibrillary tangles [56–58]. Both 3xTg-AD and nontransgenic (NonTg) mouse lines are maintained in our animal facility. Mice aged 12 and 18 months were used in this study. In this model, the AD-like neuropathology becomes readily detectable at 12 months and is widespread after 18 months [57]. All mice had free access to standard laboratory food and water and were kept on a 12-hour light-dark cycle. All experiments were performed in accordance with the Canadian Council on Animal Care guidelines and were approved by the Laval University animal ethics committee. Brain microvessels from 3xTg-AD mice were generated with a protocol similar to the one used for human frozen samples, using a procedure reported in our previous work [59] and described in the Supplementary material.
Western blot
Protein homogenates from human and murine brain microvessel extracts were added to Laemmli’s loading buffer and heated 10 minutes at 70°C. TBS- and formic acid-soluble fractions from human parietal cortex were also added to Laemmli’s loading buffer and heated 5 minutes at 95°C. Equal amounts of proteins per sample (8 μg for both human and murine brain microvessel extracts and 15 μg for protein homogenates of human parietal cortex) were resolved on a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). All samples, loaded in a random order, were run on the same immunoblot experiment for quantification. Proteins were electroblotted on PVDF membranes, which were then blocked during 1h at RT with a PBS solution containing 5% non-fat dry milk, 0.5% BSA and 0.1% Tween 20. Membranes were then incubated overnight at 4°C with primary antibodies (microvessel extracts: rabbit anti-smooth muscle alpha actin, 1:2000; rabbit anti-PDGFRp, 1:2000; rabbit anti-CD13 1:2000; rabbit anti-cyclophilin B, Abcam, ab16045, 1:1000; TBS-soluble homogenates: rabbit anti-phosphoTDP-43 Ser403/404 antibody (Cosmo Bio Co., TIP-PTD-P05, 1:2000) and mouse anti-human TDP-43 (clone 2E2-D3), Abnova, H00023435-M01, 1:2000); formic acid-soluble homogenates: rabbit anti-phosphoTDP-43 Ser409/410 (Cosmo Bio Co., TIP-PTD-P02, 1:2000) and mouse anti-human TDP-43 (clone 2E2-D3), 1:2000). Membranes were then washed three times with PBS containing 0.1% Tween 20 and incubated during 1h at RT with the secondary antibody (goat anti-rabbit HRP or goat anti-mouse HRP, Jackson ImmunoResearch Laboratories, West Grove, PA; 1:50,000 in PBS containing 0.1% Tween 20 and 1% BSA). Membranes were probed with chemiluminescence reagent (Luminata Forte Western HRP substrate; Millipore) and imaged using the myECL imager system (Thermo Fisher Scientific). Densitometric analysis was performed using the mylmageAnalysis™ Software provided with the imaging system. Uncropped gels of all immunoblot assays are shown in the Supplementary material (Figures S2, S3 and S4).
Data and statistical analysis
When comparing two groups, an unpaired Student’s t-test was performed. When more than two groups were compared, parametric one-way ANOVA followed by Tukey’s multiple comparison tests or twoway ANOVA were used. Correlations with antemortem clinical scores were adjusted for the following covariates: sex, age at death, educational level and APOE genotype. For all data, statistical significance was set at p < 0.05. Individual data were excluded for technical reasons or if determined as an outlier. All statistical analyses were performed with Prism 6 (GraphPad, San Diego, CA, USA) or JMP (version 13; SAS Institute Inc., Cary, IL) software.
Results
Levels of mural cell markers are reduced in volunteers with AD and are correlated with cognitive performance
We first validated that mural cell markers were enriched in brain microvessel extracts used in this study. As represented in Figure 1A, α-SMA, PDGFRβ and CD13 were all highly concentrated in both human and murine brain microvessel fractions. Immunofluorescence analyses on human brain microvessels revealed that all mural cell markers colocalized with microvessels, labelled with type IV collagen, a marker of basement membrane. These analyses further showed that α-SMA immunoreactivity was observed on larger vessels (Figure 1B), while pericyte markers PDGFRβ and CD13 were detected mostly on smaller capillary-like vessels (Figure 1C, D).
Western blot analyses revealed that PDGFRP levels were significantly lower for participants with a clinical diagnosis of AD compared to both NCI (−76%) and MCI (−72%). Alpha (α)-SMA and CD13 levels were significantly lower in persons with clinical AD compared to those with MCI (−60% and −49% respectively), but not compared to NCI (−41% and −38% respectively) (Figure 2). Differences between NCI and MCI were not significant for all three mural cell markers investigated (Figure 2A–C). Additional analyses after grouping NCI and MCI further revealed that all three mural cell markers were significantly lower in the persons with clinical AD compared to non-demented individuals (not shown). By contrast, the levels of CD31, an endothelial marker, remained similar between groups (Table 1), as previously reported [2].
Figure 2: Mural cell markers are reduced in AD and are correlated with global cognitive performance.
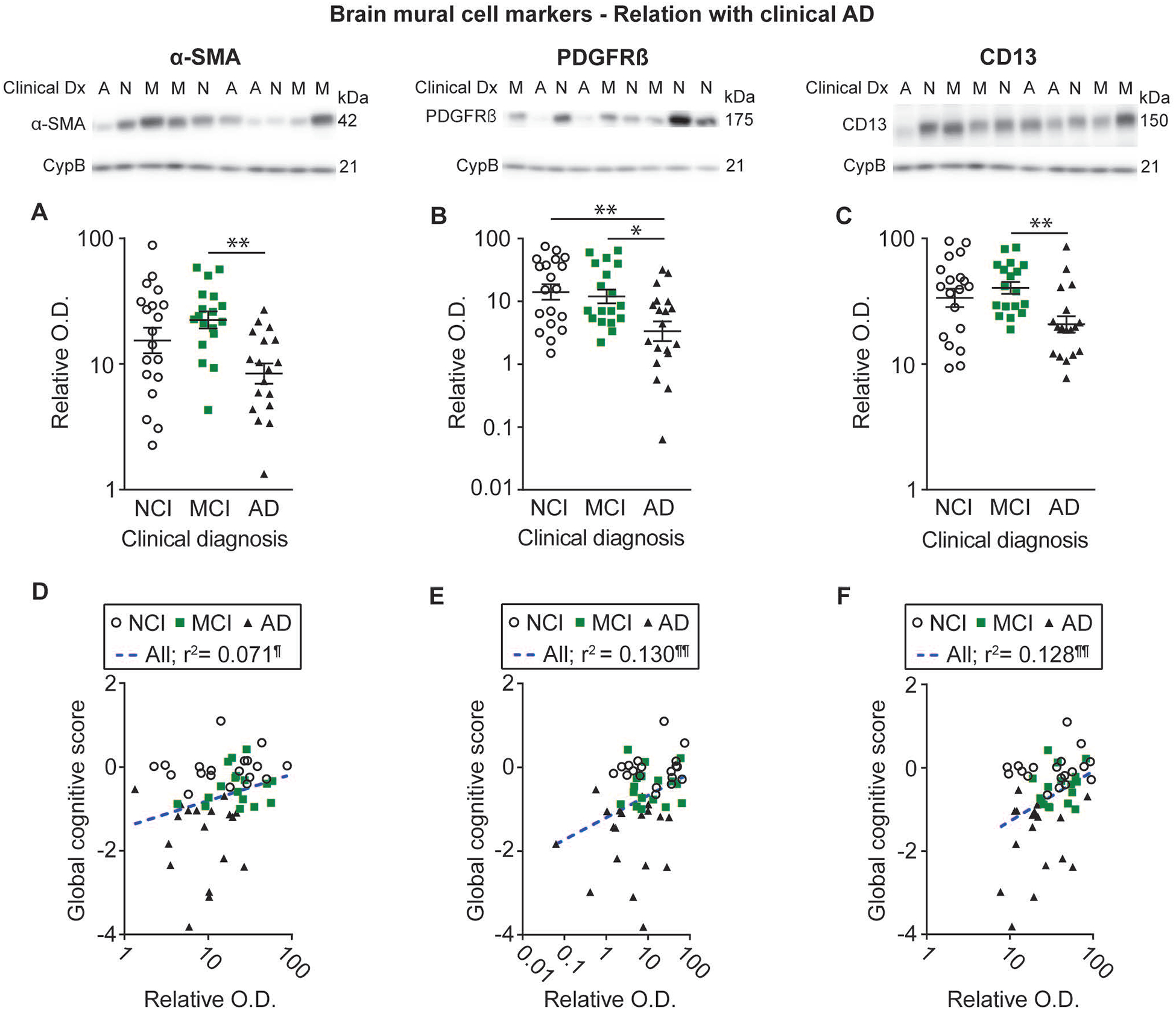
Levels of mural cell markers in human microvessel extracts were determined by Western immunoblotting. Data are represented as scatterplots, with relative optical density values indicated using a logarithmic scale. Representative photo examples illustrate consecutive bands, with cyclophilin B being shown as loading control. Uncropped gels of all immunoblot assays are shown in Figure S2. A-C) Subjects were grouped according to the clinical diagnosis. Alpha (α)-SMA, PDGFRβ and CD13 levels were all reduced in individuals clinically classified as AD compared to NCI or MCI. Horizontal lines indicate mean ± S.E.M. Statistical analysis: one-way analysis of variance followed by a Tukey’s post hoc test using log transformed relative optical density values, * p < 0.05; ** p < 0.01. D-F) Linear regression analyses further revealed that all markers were positively associated with global cognitive scores. Statistical analysis: Coefficients of determination (r2) computed after log transformation of relative optical density values for mural cell markers, ¶ p < 0.05; ¶¶ p < 0.01. Correlations were adjusted for the following covariates: sex, age at death, educational level and APOE genotype. Abbreviations: α-SMA, smooth muscle alpha actin; A-AD, Alzheimer’s Disease; CD13, aminopeptidase N; Clinical Dx, clinical diagnosis; CypB, cyclophilin B; M-MCI, mild cognitive impairment; N-NCI, healthy controls with no cognitive impairment; O.D., optical density; PDGFRβ, platelet-derived growth factor receptor β.
To determine whether levels of BMC markers were associated with antemortem cognitive performance, we performed linear regression analyses with various domain-specific cognitive scores, with adjustments for sex, age at death, educational level and APOE genotype. All three markers were positively associated with global cognitive scores (α-SMA: r2 = 0.071, p < 0.05; PDGFRβ: r2 = 0.130, p < 0.01; CD13: r2 = 0.128, p < 0.01), semantic memory (α-SMA: r2 = 0.086, p < 0.05; PDGFRβ: r2 = 0.141, p < 0.01; CD13: r2 = 0.149, p < 0.01) and perceptual speed (a-SMA: r2 = 0.075, p < 0.05; PDGFRβ: r2 = 0.169, p < 0.01; CD13: r2 = 0.081, p < 0.05) (Figure 2D–F and Table 2). Pericyte markers PDGFRβ (r2 = 0.098, p < 0.05) and CD13 (r2 = 0.114, p < 0.05) were also positively correlated with episodic memory, while a similar trend was noted for α-SMA (r2 = 0.070, p = 0.0507) (Table 2).
Table 2 –
Linear regressions between brain mural cell markers in isolated human brain microvessels and antemortem cognitive evaluation for each domain
| Global cognitive score | Episodic memory | Semantic memory | Perceptual speed | Working memory | Visuospatial ability | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| r2 | r2 | r2 | r2 | r2 | r2 | ||||||
| α-SMA | 0.071¶ | 0.070 | 0.086¶ | 0.075¶ | 0.041 | 0.001 | |||||
| PDGFRβ | 0.130¶¶ | 0.098¶ | 0.141¶¶ | 0.169¶¶ | 0.022 | 0.015 | |||||
| CD13 | 0.128¶¶ | 0.114¶ | 0.149¶¶ | 0.081¶ | 0.023 | 0.091¶ |
Alpha (α)-SMA, PDGFRβ and CD13 were measured by Western immunoblotting. Relative optical density values were log transformed to normalize distribution for statistical analyses. Correlations were adjusted for the following covariates: sex, age at death, educational level and APOE genotype. Linear regressions were performed to obtain coefficients of determination (r2).
p < 0.05 and
p < 0.01.
Abbreviations: α-SMA, smooth muscle alpha actin; CD13, aminopeptidase N; PDGFRβ, platelet-derived growth factor receptor β.
No significant differences in BMC markers were detected between volunteers according to their neuropathological diagnosis of AD or apoE4 carriage
We further compared the volunteers included in this study based on their neuropathological diagnosis as either AD or no AD, determined using the revised NIA-AA criteria [51]. Moreover, given that it has been previously shown that markers of mural cells were influenced by apoE4 carriage [36, 40], individuals rated as having an AD were subdivided in two groups based on apoE4 carriage, while the only two apoE4 carriers in the control group were excluded from this analysis for statistical reasons. No significant difference was noted between groups (Figure 3A–C). Nevertheless, linear regression analyses revealed that vascular levels of both α-SMA and PDGFRβ were negatively associated with the number of neuritic plaques in the parietal cortex (Figure 3D–F and Table S1). Moreover, α-SMA was also inversely correlated with insoluble Aβ42 concentrations in the whole parietal cortex (Figure 3G–I and Table S1). However, no significant association of BMC markers was found with tangle counts and insoluble phosphorylated tau in the parietal cortex (Figure 3J–L and Table S1).
Figure 3: Alzheimer’s Disease neuropathology and apoE4 carriage are not associated with the loss of mural cell markers.
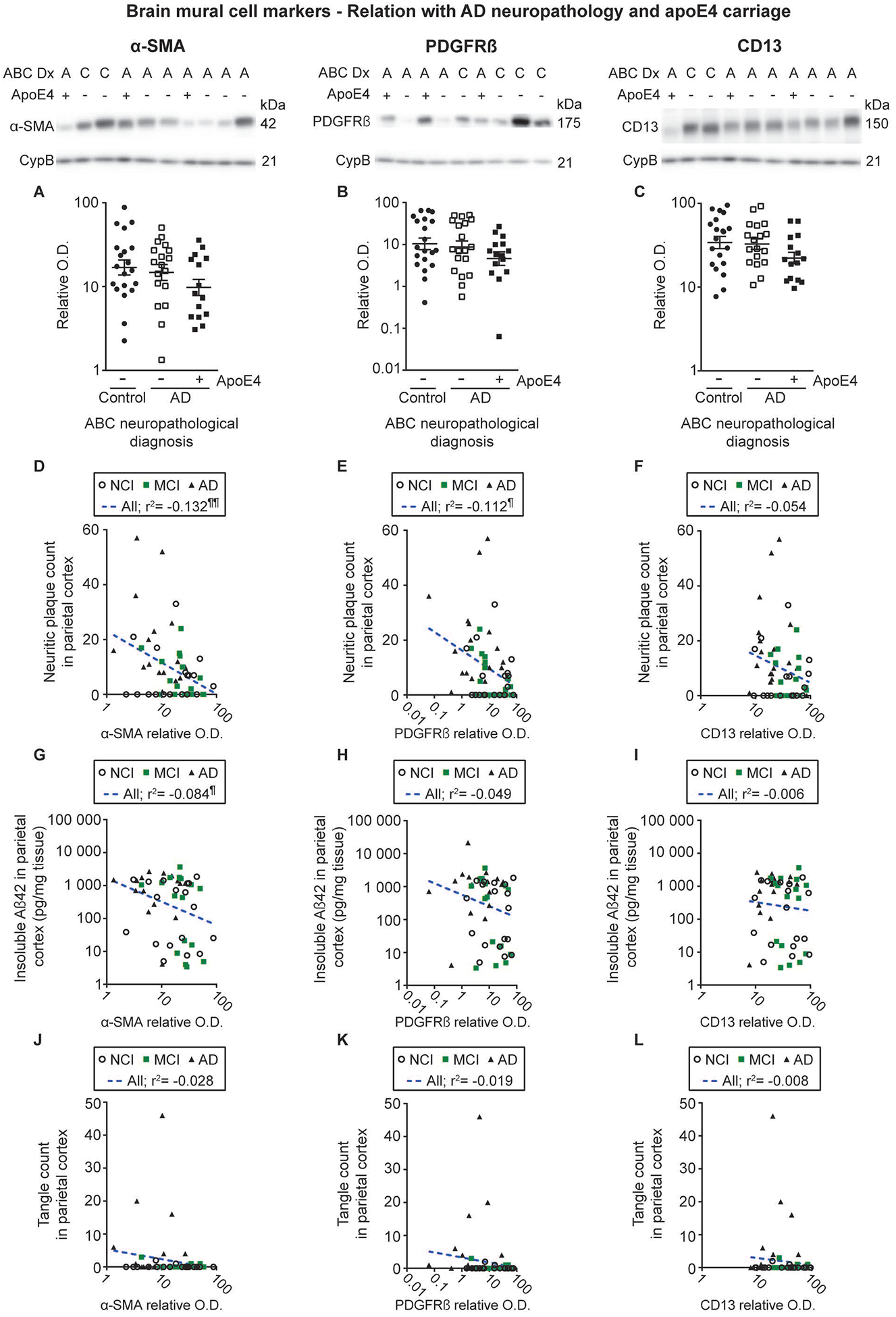
Levels of mural cell markers in human microvessel extracts were determined by Western immunoblotting. Data are represented as scatterplots, with relative optical density values indicated using a logarithmic scale. Representative photo examples illustrate consecutive bands, with cyclophilin B being shown as loading control. Uncropped gels of all immunoblot assays are shown in Figure S2. A-C) Subjects were grouped according to the neuropathological diagnosis, the AD group being further subdivided based on apoE4 carriage. For all mural cell markers investigated, no significant difference was observed in participants with an autopsy-confirmed AD neuropathology compared to controls, without any additional effect from apoE4 carriage. Statistical analysis: one-way analysis of variance followed by a Tukey’s post hoc test using log transformed relative optical density values for mural cell markers, p > 0.05. D-L) Vascular α-SMA and PDGFRβ levels were negatively associated with neuritic plaque counts in the parietal cortex, while only a trend was observed for CD13. Vascular levels of α-SMA but not PDGFRβ and CD13, were also negatively associated with the concentrations of insoluble cortical Aβ42. No significant association was observed between mural cell markers and tangle counts in the parietal cortex. Statistical analysis: Coefficients of determination (r2) computed after log transformation of relative optical density values for mural cell markers and insoluble cortical Aβ concentrations, ¶ p < 0.05; ¶¶ p < 0.01. Abbreviations: (-), apoE4 non carrier; (+), apoE4 carrier; α-SMA, smooth muscle alpha actin; A-AD, Alzheimer’s Disease; ABC Dx, ABC neuropathological diagnosis; CD13, aminopeptidase N; CypB, cyclophilin B; O.D., optical density; PDGFRβ, platelet-derived growth factor receptor β.
Cerebrovascular accumulation of Aβ is inversely correlated with levels of mural cell markers
The utilization of microvessel extracts gives us the possibility to perform a direct assessment of the accumulation of Aβ40 and Aβ42 specifically in the vascular compartment isolated from the same persons [2]. Experiments conducted in APPsw mice showed that a genetic ablation of one Pdgfrb allele aggravates cerebrovascular Aβ accumulation [33], suggesting a role for mural cells in CAA. Interestingly, pericyte markers PDGFRβ (r2 = −0.109, p < 0.05) and CD13 (r2 = −0.197, p < 0.001) were negatively correlated with microvascular Aβ40 concentrations, whereas a strong trend was found for α-SMA (r2 = −0.052, p = 0.1012) (Figure 4A–C). Conversely, no significant association was noted between vascular Aβ42 and all mural cell markers tested (Figure 4D–F). We further performed immunofluorescence experiments to evaluate whether mural cell markers colocalized with Aβ peptides. Although both Aβ40 and α-SMA were detected on large vessels, only scarce evidence of colocalization, visible in orange, was found between these markers, indicating that most of the Aβ40 appears to accumulate predominantly in vascular segments devoid of α-SMA immunoreactivity (Figure 4G). Aβ40 immunoreactivity was also observed adjacent to type IV collagen and α-SMA staining, in agreement with previous evidence of an accumulation of vascular Aβ in the basement membrane surrounding SMC [60]. As pericyte are mostly on smaller capillary-like vessels, almost no colocalization was found between CD13 and Aβ40 (Figure 4H). Finally, no significant differences in BMC markers were detected between volunteers according to the parenchymal CAA stage in the parietal cortex or the presence or not of chronic cortical infarcts (Figure S1).
Figure 4: Levels of mural cell markers are inversely correlated with Aβ concentrations in human microvessel extracts.
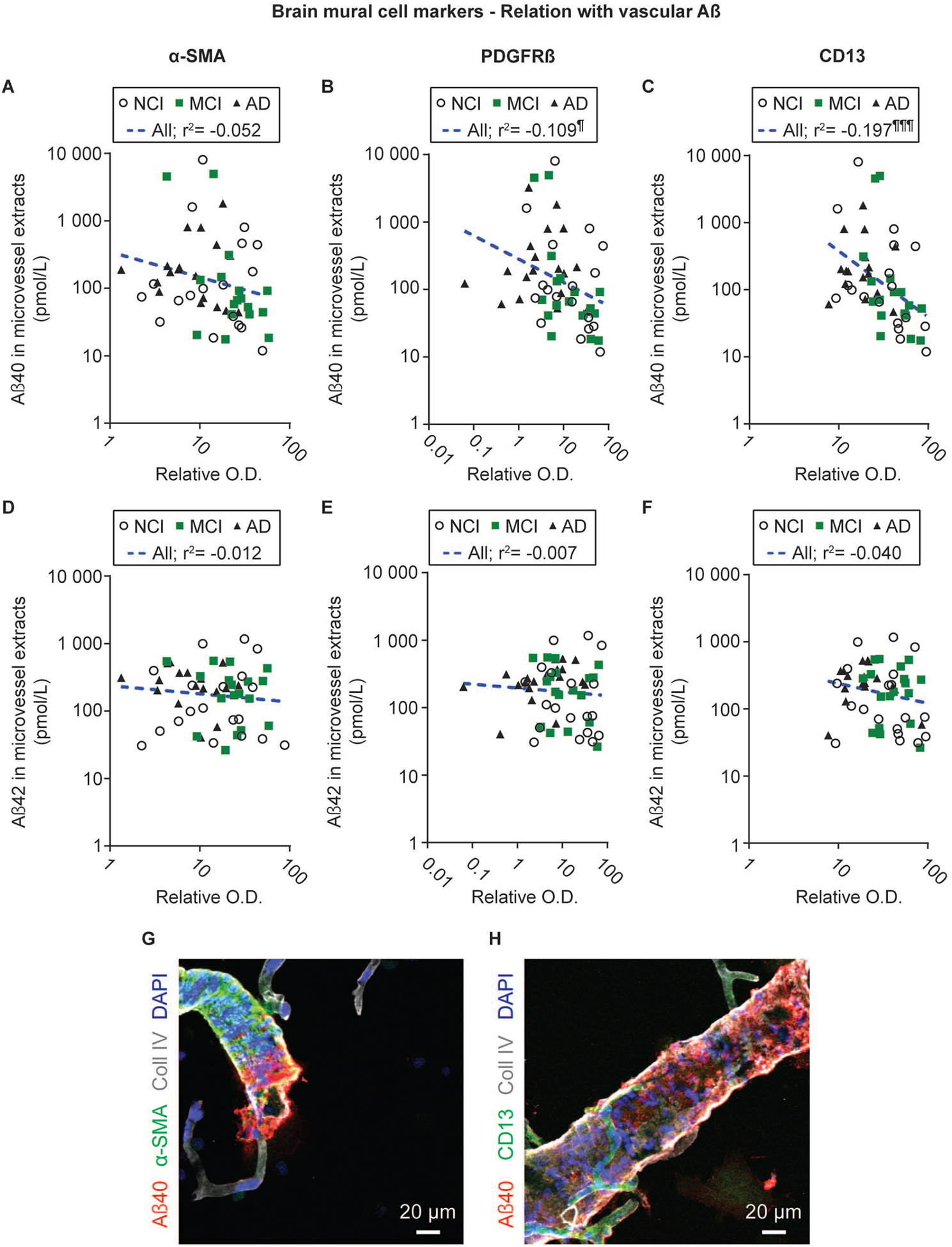
Aβ concentrations used for linear regression analyses were measured by ELISA and previously published [2]. PDGFRβ and CD13 levels were inversely correlated to vascular Aβ40 concentrations, while a strong trend was noted for α-SMA (A-C). On the contrary, no correlation was observed with vascular Aβ42 (D-F). Statistical analysis: Coefficients of determination (r2) computed after log transformation of both vascular Aβ peptide concentrations and relative optical density values for mural cell markers, ¶¶ p < 0.01. Aβ40 and α-SMA immunoreactivities were detected on larger vessels, with only a scarce colocalization (visible in orange), while CD13-positive signal was found on smaller, capillary-like vessels (G-H). Abbreviations: α-SMA, smooth muscle alpha actin; AD, Alzheimer’s Disease; CD13, aminopeptidase N; Coll IV, type IV collagen; O.D., optical density; PDGFRβ, platelet-derived growth factor receptor β.
Levels of CD13, but not PDGFRβ or α-SMA, are reduced in brain microvessels from old 3xTg-AD mice
To determine whether changes in BMC are a consequence of AD neuropathology, we applied the same methodology in brain samples from old 3xTg-AD mice. This model is transgenically designed to develop plaque and tangle neuropathology with age, but it also develops cerebrovascular deficits such as reduced glucose and docosahexaenoic acid uptake, reduced cerebrovascular volume, and a thickening of the basement membrane [59, 61–63]. Immunoblotting experiments in microvessel-enriched extracts showed lower CD13 levels in old 3xTg-AD mice compared to age-matched NonTg mice (−33% for mice aged 12 months and −21% for mice aged 18 months, respectively), whereas α-SMA and PDGFRβ remained unchanged (Figure 5A–C).
Figure 5: CD13 levels are reduced in brain microvessels from old 3xTg-AD mice.
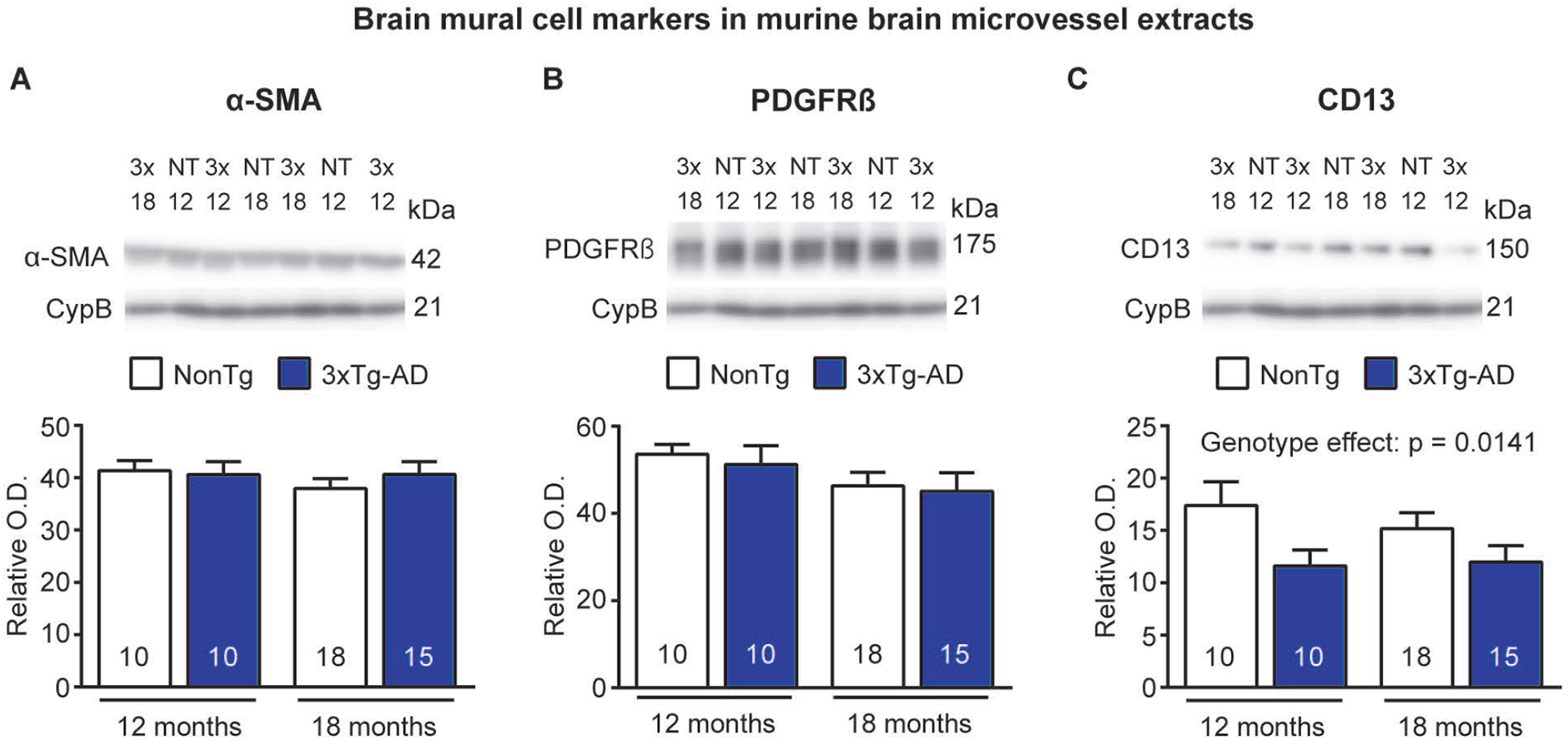
A-C) Levels of mural cell markers in microvessel extracts from NonTg and 3xTg-AD mice aged 12 and 18 months were determined by Western immunoblotting. Representative photo examples illustrate consecutive bands, with cyclophilin B being shown as loading control. Uncropped gels of all immunoblot assays are shown in Figure S3. CD13 levels are decreased in old 3xTg-AD mice compared to age-matched NonTg mice. No difference was noted for α-SMA and PDGFRβ. Statistical analysis: two-way analysis of variance. Data are represented as bar graphs indicating mean ± S.E.M. Sample size for each group is indicated in the graph bars. Abbreviations: 3x-3xTg-AD, triple transgenic mice; α-SMA, smooth muscle alpha actin; CD13, aminopeptidase N; CypB,cyclophilin B; NT-NonTg, nontransgenic mice; O.D., optical density; PDGFRβ, platelet-derived growth factor receptor β.
Loss of mural cell markers in microvessels is associated with cortical TDP-43 pathology
The brains of up to 57% of AD patients contain post-mortem transactive response DNA-binding protein 43 (TDP-43) neuropathology [11, 42, 64–66]. We thus examined whether differences in mural cell markers in vascular samples we observed in clinical AD were related to TDP-43 pathology assessed in TBS-soluble and formic acid-soluble protein homogenates, with an emphasis on its cleaved, phosphorylated forms given their putative role in the toxicity of TDP-43 [67–70]. As represented in Figure 6B and Table S2, cleaved phosphorylated forms of TDP-43 were less concentrated in TBS-soluble extracts from volunteers with AD. In contrast, levels of cleaved phosphorylated TDP-43 were higher in the formic acid-soluble fractions containing insoluble proteins from AD subjects (Figure 6E and Table S2), suggesting a conversion of pTDP-43 into its insoluble form in AD. Despite similar trends, no significant changes were observed for their full-length counterparts in the same fractions (Figure 6C, F and Table S2). Linear regression analyses, adjusted for age at death, showed that a-SMA (soluble cleaved phosphorylated TDP-43 / insoluble cleaved phosphorylated TDP-43: r2 = 0.076, p < 0.05 / r2 = −0.178, p < 0.01) and PDGFRβ (soluble cleaved phosphorylated TDP-43 / insoluble cleaved phosphorylated TDP-43: r2 = 0.258, p < 0.0001 / r2 = −0.423, p < 0.0001) levels were strongly related with cleaved, phosphorylated TDP-43 concentrations, while only non-significant trends were observed for CD13 (soluble cleaved phosphorylated TDP-43/insoluble cleaved phosphorylated TDP-43: r2 = 0.028, p > 0.05 / r2 = −0.047, p > 0.05) (Figure 6G–L). The association was particularly strong between cortical pTDP-43 and vascular PDGFRβ, but opposite for the soluble and insoluble species, suggesting that cleavage and aggregation of the 25-kDa fragment may be deleterious for pericytes. Analyses also revealed that the two pericyte markers investigated (PDGFRβ: r2 = −0.165, p < 0.01; CD13: r2 = −0.098, p < 0.05), but not α-SMA (r2 = −0.019, p > 0.05), were inversely associated with total insoluble TDP-43 levels (Figure 6M–O).
Figure 6: Mural cell marker levels are associated with cortical TDP-43 pathology in human volunteers.
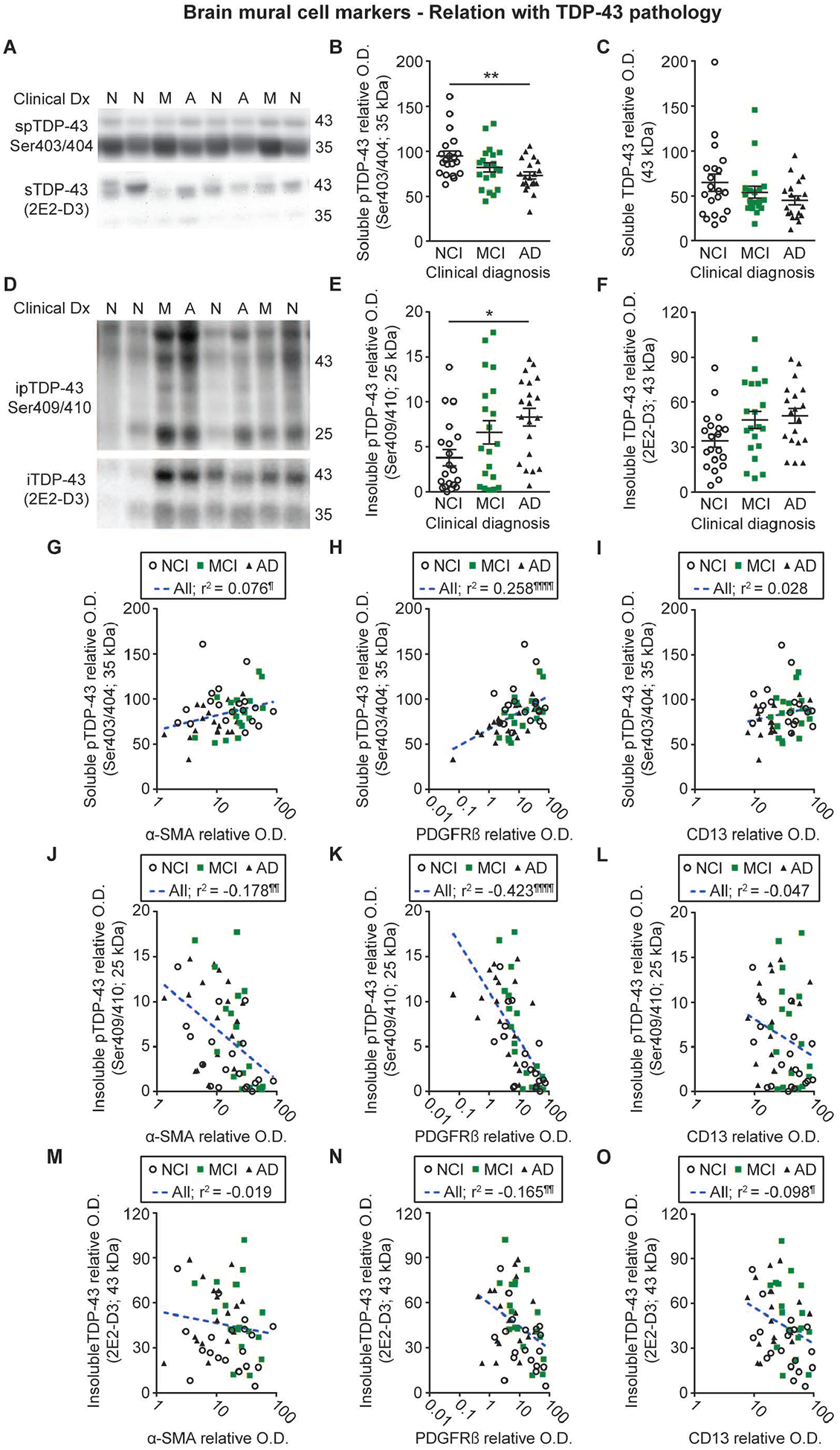
Levels of soluble and insoluble phosphorylated as well as total insoluble TDP-43 were determined by Western immunoblotting in TBS-soluble and formic acid-soluble protein homogenates from the parietal cortex of the same series of donors. Representative photo examples illustrate consecutive bands. Uncropped gels of all immunoblot assays are shown in Figure S4. A-C) Soluble phosphorylated TDP-43 levels were reduced in volunteers clinically classified as AD whereas total soluble TDP-43 levels remained similar between groups. D-F) Insoluble phosphorylated TDP-43 levels were increased in participants with clinical AD while a strong upward trend was noted for total insoluble TDP-43 in the same group. Values in the MCI group were intermediate as compared to the AD and NCI groups. Statistical analysis: one-way analysis of variance followed by a Tukey’s post hoc test, * p < 0.05; ** p 0.01. G-O) Vascular α-SMA and PDGFRβ levels were respectively positively and negatively associated with TBS- and formic acid-soluble phosphorylated TDP-43, while only non-significant trends were observed for CD13. Vascular levels of PDGFRβ and CD13, but not α-SMA, were also negatively associated with total insoluble TDP-43. Statistical analysis: Coefficients of determination (r2) adjusted for age at death and computed after log transformation of relative optical density values for mural cell markers, ¶ p < 0.05; ¶¶ p < 0.01; ¶¶¶ p < 0.001; ¶¶¶¶ p < 0.0001. Abbreviations: α-SMA, smooth muscle alpha actin; A-AD, Alzheimer’s Disease; CD13, aminopeptidase N; Clinical Dx, clinical diagnosis; ipTDP43, insoluble phosphorylated TDP-43; iTDP43, insoluble TDP-43; M-MCI, mild cognitive impairment; N-NCI, healthy controls with no cognitive impairment; O.D., optical density; PDGFRβ, platelet-derived growth factor receptor β; spTDP43, TBS-soluble phosphorylated TDP-43.
Discussion
The present study sought to investigate the extent of vascular mural cell pathology in AD using brain extracts enriched in microvessels generated from human parietal cortex samples. Altogether, our results suggest that a loss of smooth muscle cells and pericytes occurs in the parietal cortex in individuals with clinical AD, correlating with cognitive decline and cerebrovascular accumulation of Aβ. In addition, regression analysis of our data highlight that such loss of mural cells is strongly associated with TDP-43 pathology, weakly associated with cortical Aβ content, but not at all with tau neuropathology.
Lower BMC levels in microvessel extracts are associated with cognitive impairment and a diagnosis of AD.
Data from the present work indicate that α-SMA, PDGFRβ and CD13 levels were lower in microvessel extracts from individuals with lower cognitive scores or with a clinical diagnosis of AD. This could be explained by either a loss of cells or a decreased expression. The latter is a less likely interpretation since all three markers were decreased. On the other hand, a loss of mural cells is in agreement with previous reports showing a reduced coverage of cortical and hippocampal microvessels by smooth muscle cells and pericytes in post-mortem AD brains, using immunohistochemistry [28] and immunofluorescence [34] paradigms. However, while PDGFRβ- and CD13-positive pericyte coverage of cortical capillaries was reduced by a similar magnitude [34], the present immunoblot approach allowed us to determine that the magnitude of reduction for PDGFRβ was twice more marked than for CD13 in the parietal cortex in AD. Contrasting with the decrease of α-SMA, PDGFRβ and CD13 levels, no massive degeneration of the cerebral endothelium was observed, thus suggesting a rather specific loss of BMC. This is in line with a previous study showing similar von Willebrand factor levels in the precuneus of AD patients despite reduced PDGFRβ concentrations [36].
Recent studies reported that subjects clinically diagnosed with AD as well as subjects at preclinical stages had higher CSF concentrations of the cleaved, soluble form of PDGFRβ [37–39], this elevation being interpreted as resulting from pericyte degeneration [14, 37–39]. While this previous work is consistent with the present results, the increase in CSF PDGFRβ is already detectable at the MCI stage, which was not the case here. Notably, we observed a strong association between mural cell loss and impaired cognitive performance. Indeed, all three mural cell markers investigated were not only positively correlated with global cognition, but also with several specific cognitive domains including episodic memory, a domain predominantly affected in AD [71, 72]. Whether mural cell loss plays a causal role in cognitive changes cannot be ascertained from these associative studies, but genetic induction of pericyte deficiency in a mouse of AD was shown to result in cognitive decline [33].
A weaker association between BMC and classical AD neuropathology.
Contrasting with cognition, vascular BMC showed a more complex relationship with Aβ and tau neuropathology. On the one hand, conflicting results were reported following analyses of cleaved, soluble PDGFRβ (sPDGFRβ) concentrations in the CSF. For instance, Nation and colleagues showed that CSF levels of sPDGFRβ were unrelated to CSF measurements of Ap42 and phosphorylated tau [38], while another recent study rather found that sPDGFRβ content in the CSF was associated with CSF concentrations of phosphorylated tau, but not Aβ42 [39]. On the other hand, in agreement with anterior post-mortem measurements in whole brain homogenates [36], we noted that PDGFRβ levels in vascular extracts were negatively correlated to the number of neuritic plaques in the parietal cortex, an association even stronger for α-SMA. Conversely, no significant association was found with neurofibrillary tangle counts in the same brain region for all brain mural cell markers, illustrating an apparent dichotomy between CSF and neuropathological findings. Nevertheless, based on these observations, one could have expected significant variations for BMC markers in volunteers with a neuropathological diagnosis of AD. Yet, unlike in previous studies [28, 34, 39], BMC markers levels were not statistically different when subjects were distinguished according to the neuropathological diagnosis, determined using the ABC scoring method. These discrepancies can possibly be explained by differences in the neuropathological definition of the control group as anterior reports only included in their control group participants that were given low Braak (0 or I) and CERAD (absent or sparse neuritic plaques) scores whereas, in this work, a small number of subjects with high Braak or CERAD scores were included, in line with the NIA-AA guidelines [51].
Measurements of BMC markers in brain microvessels from 3xTg-AD mice argue against a major causal role of Aβ and tau pathologies in the loss of mural cell markers. In 3xTg-AD mice aged 12 and 18 months, only CD13 was reduced compared to age-matched NonTg mice, which is in accordance with previous findings showing a reduced number of CD13-positive cell bodies but similar PDGFRβ levels in cortical vessels of APPsw mice compared to controls [33]. Also using APPsw mice, Christie and colleagues reported that a loss of SMC surrounding leptomeningeal vessels occurs with the progression of cerebrovascular Aβ pathology [32]. These discrepancies with our findings are possibly due to the variable extent of CAA pathology found in each of these models [73, 74] and the study material (leptomeningeal versus parenchymal vessels). In addition, the lack of concordance between the two pericyte markers CD13 and PDGFRβ in murine microvessel extracts from different AD mouse models suggests a decreased expression of this marker in BMC rather than a loss of BMC, contrasting with the observations in human microvascular extracts. However, it is worth noting that the 3xTg-AD mouse develops a less extensive AD pathology than what is found in human post-mortem AD patients, and perhaps insufficient to trigger a widespread mural cell loss.
The relation between pericyte markers and microvascular Aβ tells a different story, as we observed a relatively strong inverse association between levels of PDGFRβ and CD13 with Ap40 in human brain microvessels. This is probably because Aβ40 is the predominant Aβ species found clustered onto microvessels in CAA, whereas Aβ42 is rather predominant in neuritic plaques [75–77]. Such observation is to some extent in agreement with a previous work reporting that APP-overexpressing mice invalidated for one Pdgfrb allele displayed a five-fold increase in Aβ immunoreactivity in cortical arteries compared to age-matched APPsw mice [33]. Since pericytes are not located on larger vessels where Aβ peptides predominantly accumulate, as shown in our immunofluorescence analysis, this suggests a role for pericytes in Aβ clearance mostly on smaller vessels. Of note, the interstitial cerebral fluid and its solutes, including Aβ peptides, are thought to be drained out of the brain into the basement membranes of capillaries and then along those of arteries [78]. Therefore, an impaired clearance of Aβ peptides by pericytes at the level of capillaries could also contribute to the vascular accumulation of Aβ. The spontaneous contraction of SMC, termed vasomotion, is thought to be a major driving force of the intramural periarterial drainage (IPAD) pathway of the brain [79, 80]. A loss of SMC, as suggested by data gathered in this study, could thus lead to a deficiency of this clearance mechanism and a subsequent accumulation of Aβ in the vessel walls. Nonetheless, the loss of mural cells evidenced in this study is most likely associated with vascular abnormalities not limited to Aβ accumulation.
A strong association between BMC and TDP-43.
Clinicopathological studies highlight that TDP-43 pathology occurs in an important subset of AD patients, estimated between 14 and 57 % [11, 42, 64–66]. However, the relative pathogenicity of TDP-43 depends on the exact cellular localization, full-length versus cleaved form and phosphorylation status [42, 81–83]. Involved in several steps of RNA metabolism, TDP-43 is naturally shuttled between the nucleus and the cytoplasm [84]. Through a yet unknown mechanism, TDP-43 is cleared out of the nucleus and accumulates in the cytoplasm where it is cleaved and phosphorylated, such modifications exacerbating its aggregation [67–70]. The present analyses confirmed that soluble and insoluble phosphorylated TDP-43 are respectively decreased and increased in AD, with this translocation into cytoplasmic aggregates correlating best with cognitive function [42].
Full-length TDP-43, and its cleaved, phosphorylated form found in the insoluble extracts as well as lower amounts of its soluble counterpart, were all associated with reduced levels of mural cell markers in human brain microvessel extracts, with higher coefficients of determination than cortical Aβ42 and neuritic plaque burden. To our knowledge, this is the first study linking the reduction of mural cell markers and specific aspects of TDP-43 pathology in the same cohort of AD patients, our biochemical approach providing the possibility to distinguish between specific fragments. Here, strong and opposite correlations were found with cleaved phosphorylated TDP-43 species while weaker associations were noted with its parent form. Such observation suggests that brain mural cell loss is more related to the transition from a soluble to an insoluble state of cleaved phosphorylated TDP-43 than the full-length protein, consistent with anterior findings showing that such a biochemical process has pathological implications [67–70]. Imaging analyses in patients with the behavioural variant of frontotemporal dementia (bvFTD) or amyotrophic lateral sclerosis (ALS) showed that a hypoperfusion was observed in brain regions matching the progression of TDP-43 pathology in these diseases and correlating with cognitive and behavioural impairments [85]. Likewise, a hypoperfusion is also noted in AD, but is thought to occur several years before the onset of clinical symptoms [5, 86, 87]. In the subset of AD patients displaying post-mortem TDP-43 pathology, it is thus conceivable that hypoperfusion may be among the initial stressors triggering the development of this proteinopathy and that the neuronal dysfunction consecutive to this pathology could subsequently lead to alterations in the neurovascular unit, causing the loss of mural cells. Experiments in mice deficient in mural cells or modelling TDP-43 pathology would be of interest to better understand the link between this pathology and cerebrovascular alterations, including IPAD failure and the loss of mural cell markers. Emerging biomarkers for TDP-43 may also be useful tools to identify the patients that are more likely to display cerebrovascular alterations [88–90].
Loss of BMC: a late event in AD
Our results suggest that the loss of BMC is a late event in the AD continuum as individuals with MCI displayed similar levels of BMC markers than controls. Very limited changes of BMC markers in brain microvessels of 3xTg-AD mice also argue against an early event occurring before the apparition of an overblown AD neuropathology. Based on the data presented in this study, one potential explanation could be that TDP-43 pathology, generally thought to be a secondary neuropathology in AD, is required for this cell loss to occur. The accumulation of cleaved, soluble PDGFRβ observed in the CSF of participants rated as MCI [38] could thus be a reflection of an increased shedding of the receptor, an insult pericytes may be able to recover from at early stages [91, 92], but not in later stages, leading to mural cell loss. Alternatively, it is also possible that markers expressed by BMC other than those investigated in this work may be modulated at earlier stages. Lysyl oxidase appears as a potential candidate as this enzyme is implicated in the formation and modulation of the extracellular matrix, and alterations of its protein level or activity could in turn affect Aβ accumulation and deposition in vessel walls as well as vascular stiffening and remodelling [93–95].
Conclusion:
These data unveil a path in which a loss of BMC occurs in the parietal cortex along with the progression of the cognitive symptoms of AD. Besides being related to the accumulation of Aβ, particularly in microvessels, the loss of BMC is more strongly associated with the conversion of phosphorylated TDP-43 from a soluble to an insoluble cleaved form. This is supportive of a putative role for mural cell alterations in the pathophysiology of AD.
Supplementary Material
Acknowledgments
The authors are indebted to the nuns, priests and brothers from the Catholic clergy participating in the Religious Orders Study.
Funding
Funding was provided by the Canadian Institutes of Health Research (CIHR) to F.C (MOP 125930). The study was supported in part by P30AG10161 and R01AG15819 (D.A.B). F.C is a Fonds de recherche du Québec - Santé (FRQ-S) senior research scholar. P.B held scholarships from the Reseau quebecois de recherche sur le medicament (RQRM), Fondation du CHU de Quebec and a joined scholarship from the FRQ-S and the Alzheimer Society of Canada (ASC) and now holds a scholarship from the CIHR.
Footnotes
Competing interests
The authors report no competing interests.
Ethical approval
All procedures performed with volunteers included in this study were in accordance with the ethical standards of the institutional ethics committees. Informed consent was obtained from all individual participants included in this study.
References
- 1.Erickson MA, Banks WA. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J Cereb Blood Flow Metab 2013; 33: 1500–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bourassa P, Tremblay C, Schneider JA, Bennett DA, Calon F. Beta-amyloid pathology in human brain microvessel extracts from the parietal cortex: relation with cerebral amyloid angiopathy and Alzheimer’s disease. Acta Neuropathol 2019; 137: 801–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sweeney MD, Montagne A, Sagare AP, Nation DA, Schneider LS, Chui HC, et al. Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimers Dement 2019; 15: 158–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sweeney MD, Kisler K, Montagne A, Toga AW, Zlokovic BV. The role of brain vasculature in neurodegenerative disorders. Nat Neurosci 2018; 21: 1318–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson NA, Jahng GH, Weiner MW, Miller BL, Chui HC, Jagust WJ, et al. Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: initial experience. Radiology 2005; 234: 851–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piert M, Koeppe RA, Giordani B, Berent S, Kuhl DE. Diminished glucose transport and phosphorylation in Alzheimer’s disease determined by dynamic FDG-PET. J Nucl Med 1996; 37: 2018. [PubMed] [Google Scholar]
- 7.Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol 2001; 64: 575–611. [DOI] [PubMed] [Google Scholar]
- 8.Kalaria RN, Pax AB. Increased collagen content of cerebral microvessels in Alzheimer’s disease. Brain Res 1995; 705: 349–52. [DOI] [PubMed] [Google Scholar]
- 9.Lepelletier FX, Mann DM, Robinson AC, Pinteaux E, Boutin H. Early changes in extracellular matrix in Alzheimer’s disease. Neuropathol ApplNeurobiol 2017; 43: 167–82. [DOI] [PubMed] [Google Scholar]
- 10.Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol 2011; 69: 320–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kapasi A, DeCarli C, Schneider JA. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol 2017; 134: 171–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood-brain barrier. Nature 2010; 468: 557–61. [DOI] [PubMed] [Google Scholar]
- 13.Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010; 68: 409–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sweeney MD, Ayyadurai S, Zlokovic BV. Pericytes of the neurovascular unit: key functions and signaling pathways. NatNeurosci 2016; 19: 771–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kisler K, Nelson AR, Rege SV, Ramanathan A, Wang Y, Ahuja A, et al. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat Neurosci 2017; 20: 406–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014; 508: 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mishra A, Reynolds JP, Chen Y, Gourine AV, Rusakov DA, Attwell D. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat Neurosci 2016; 19: 1619–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olson LE, Soriano P. PDGFRbeta signaling regulates mural cell plasticity and inhibits fat development. Dev Cell 2011; 20: 815–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S, Voisin MB, Larbi KY, Dangerfield J, Scheiermann C, Tran M, et al. Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J Exp Med 2006; 203: 1519–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Voisin MB, Probstl D, Nourshargh S. Venular basement membranes ubiquitously express matrix protein low-expression regions: characterization in multiple tissues and remodeling during inflammation. Am J Pathol 2010; 176: 482–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Attwell D, Mishra A, Hall CN, O’Farrell FM, Dalkara T. What is a pericyte? J Cereb Blood Flow Metab 2016; 36: 451–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hartmann DA, Underly RG, Grant RI, Watson AN, Lindner V, Shih AY. Pericyte structure and distribution in the cerebral cortex revealed by high-resolution imaging of transgenic mice. Neurophotonics 2015; 2: 041402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grant RI, Hartmann DA, Underly RG, Berthiaume AA, Bhat NR, Shih AY. Organizational hierarchy and structural diversity of microvascular pericytes in adult mouse cortex. J Cereb Blood Flow Metab 2017: 271678X17732229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hill RA, Tong L, Yuan P, Murikinati S, Gupta S, Grutzendler J. Regional Blood Flow in the Normal and Ischemic Brain Is Controlled by Arteriolar Smooth Muscle Cell Contractility and Not by Capillary Pericytes. Neuron 2015; 87: 95–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nehls V, Drenckhahn D. Heterogeneity of microvascular pericytes for smooth muscle type alpha-actin. J Cell Biol 1991; 113: 147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bandopadhyay R, Orte C, Lawrenson JG, Reid AR, De Silva S, Allt G. Contractile proteins in pericytes at the blood-brain and blood-retinal barriers. J Neurocytol 2001; 30: 35–44. [DOI] [PubMed] [Google Scholar]
- 27.Krueger M, Bechmann I. CNS pericytes: concepts, misconceptions, and a way out. Glia 2010; 58: 1–10. [DOI] [PubMed] [Google Scholar]
- 28.Ervin JF, Pannell C, Szymanski M, Welsh-Bohmer K, Schmechel DE, Hulette CM. Vascular smooth muscle actin is reduced in Alzheimer disease brain: a quantitative analysis. J Neuropathol Exp Neurol 2004; 63: 735–41. [DOI] [PubMed] [Google Scholar]
- 29.Stopa EG, Butala P, Salloway S, Johanson CE, Gonzalez L, Tavares R, et al. Cerebral cortical arteriolar angiopathy, vascular beta-amyloid, smooth muscle actin, Braak stage, and APOE genotype. Stroke 2008; 39: 814–21. [DOI] [PubMed] [Google Scholar]
- 30.Hulette CM, Ervin JF, Edmonds Y, Antoine S, Stewart N, Szymanski MH, et al. Cerebrovascular smooth muscle actin is increased in nondemented subjects with frequent senile plaques at autopsy: implications for the pathogenesis of Alzheimer disease. J Neuropathol Exp Neurol 2009; 68: 417–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hutter-Schmid B, Humpel C. Alpha-Smooth Muscle Actin mRNA and Protein Are Increased in Isolated Brain Vessel Extracts of Alzheimer Mice. Pharmacology 2016; 98: 251–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Christie R, Yamada M, Moskowitz M, Hyman B. Structural and functional disruption of vascular smooth muscle cells in a transgenic mouse model of amyloid angiopathy. Am JPathol 2001; 158: 1065–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A, et al. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun 2013; 4: 2932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Sengillo JD, Winkler EA, Walker CT, Sullivan JS, Johnson M, Zlokovic BV. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol 2013; 23:303–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schultz N, Brannstrom K, Byman E, Moussaud S, Nielsen HM, Netherlands Brain B, et al. Amyloid-beta 1–40 is associated with alterations in NG2+ pericyte population ex vivo and in vitro. Aging Cell 2018; 17: e12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miners JS, Schulz I, Love S. Differing associations between Abeta accumulation, hypoperfusion, blood-brain barrier dysfunction and loss of PDGFRB pericyte marker in the precuneus and parietal white matter in Alzheimer’s disease. J Cereb Blood Flow Metab 2018; 38: 103–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015; 85: 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nation DA, Sweeney MD, Montagne A, Sagare AP, D’Orazio LM, Pachicano M, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med 2019; 25: 2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miners JS, Kehoe PG, Love S, Zetterberg H, Blennow K. CSF evidence of pericyte damage in Alzheimer’s disease in associated with markers of blood-brain barrier dysfunction and disease pathology. Alzheimers Res Ther 2019; 11: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, Winkler EA, et al. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab 2016; 36: 216–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tremblay C, Francois A, Delay C, Freland L, Vandal M, Bennett DA, et al. Association of Neuropathological Markers in the Parietal Cortex With Antemortem Cognitive Function in Persons With Mild Cognitive Impairment and Alzheimer Disease. JNeuropatholExp Neurol 2017; 76: 70–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tremblay C, St-Amour I, Schneider J, Bennett DA, Calon F. Accumulation of transactive response DNA binding protein 43 in mild cognitive impairment and Alzheimer disease. J Neuropathol Exp Neurol 2011; 70: 788–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious Orders Study and Rush Memory and Aging Project. JAlzheimers Dis 2018; 64: S161–S89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bennett DA, Schneider JA, Aggarwal NT, Arvanitakis Z, Shah RC, Kelly JF, et al. Decision rules guiding the clinical diagnosis of Alzheimer’s disease in two community-based cohort studies compared to standard practice in a clinic-based cohort study. Neuroepidemiology 2006; 27: 169–76. [DOI] [PubMed] [Google Scholar]
- 45.Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, et al. Natural history of mild cognitive impairment in older persons. Neurology 2002; 59: 198–205. [DOI] [PubMed] [Google Scholar]
- 46.Bennett DA, Wilson RS, Boyle PA, Buchman AS, Schneider JA. Relation of neuropathology to cognition in persons without cognitive impairment. Ann Neurol 2012; 72: 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilson RS, Beckett LA, Barnes LL, Schneider JA, Bach J, Evans DA, et al. Individual differences in rates of change in cognitive abilities of older persons. Psychol Aging 2002; 17: 179–93. [PubMed] [Google Scholar]
- 48.Arvanitakis Z, Grodstein F, Bienias JL, Schneider JA, Wilson RS, Kelly JF, et al. Relation of NSAIDs to incident AD, change in cognitive function, and AD pathology. Neurology 2008; 70: 2219–25. [DOI] [PubMed] [Google Scholar]
- 49.Arvanitakis Z, Schneider JA, Wilson RS, Bienias JL, Kelly JF, Evans DA, et al. Statins, incident Alzheimer disease, change in cognitive function, and neuropathology. Neurology 2008; 70: 1795–802. [DOI] [PubMed] [Google Scholar]
- 50.Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006; 66: 1837–44. [DOI] [PubMed] [Google Scholar]
- 51.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 2012; 123: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002; 58: 1791–800. [DOI] [PubMed] [Google Scholar]
- 53.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82: 239–59. [DOI] [PubMed] [Google Scholar]
- 54.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991; 41: 479–86. [DOI] [PubMed] [Google Scholar]
- 55.Arvanitakis Z, Leurgans SE, Barnes LL, Bennett DA, Schneider JA. Microinfarct pathology, dementia, and cognitive systems. Stroke 2011; 42: 722–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 2003; 39: 409–21. [DOI] [PubMed] [Google Scholar]
- 57.Bories C, Guitton MJ, Julien C, Tremblay C, Vandal M, Msaid M, et al. Sex-dependent alterations in social behaviour and cortical synaptic activity coincide at different ages in a model of Alzheimer’s disease. PLoS One 2012; 7: e46111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dal-Pan A, Dudonne S, Bourassa P, Bourdoulous M, Tremblay C, Desjardins Y, et al. Cognitive-Enhancing Effects of a Polyphenols-Rich Extract from Fruits without Changes in Neuropathology in an Animal Model of Alzheimer’s Disease. JAlzheimers Dis 2017; 55: 115–35. [DOI] [PubMed] [Google Scholar]
- 59.Bourassa P, Alata W, Tremblay C, Paris-Robidas S, Calon F. Transferrin Receptor-Mediated Uptake at the Blood-Brain Barrier Is Not Impaired by Alzheimer’s Disease Neuropathology. Mol Pharm 2019; 16: 583–94. [DOI] [PubMed] [Google Scholar]
- 60.Keable A, Fenna K, Yuen HM, Johnston DA, Smyth NR, Smith C, et al. Deposition of amyloid beta in the walls of human leptomeningeal arteries in realtion to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim Biophys Acta 2016; 1862: 1037–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bourasset F, Ouellet M, Tremblay C, Julien C, Do TM, Oddo S, et al. Reduction of the cerebrovascular volume in a transgenic mouse model of Alzheimer’s disease. Neuropharmacology 2009; 56:808–13. [DOI] [PubMed] [Google Scholar]
- 62.Do TM, Alata W, Dodacki A, Traversy MT, Chacun H, Pradier L, et al. Altered cerebral vascular volumes and solute transport at the blood-brain barriers of two transgenic mouse models of Alzheimer’s disease. Neuropharmacology 2014; 81: 311–7. [DOI] [PubMed] [Google Scholar]
- 63.Calon F Omega-3 polyunsaturated fatty acids in Alzheimer’s disease: key questions and partial answers. Curr Alzheimer Res 2011; 8: 470–8. [DOI] [PubMed] [Google Scholar]
- 64.Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 2007; 61: 435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Uryu K, Nakashima-Yasuda H, Forman MS, Kwong LK, Clark CM, Grossman M, et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. JNeuropatholExp Neurol 2008; 67: 555–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol 2014; 127: 811–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A 2009; 106: 7607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang YJ, Gendron TF, Xu YF, Ko LW, Yen SH, Petrucelli L. Phosphorylation regulates proteasomal-mediated degradation and solubility of TAR DNA binding protein-43 C-terminal fragments. MolNeurodegener 2010; 5: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prasad A, Bharathi V, Sivalingam V, Girdhar A, Patel BK. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front Mol Neurosci 2019; 12: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gao J, Wang L, Huntley ML, Perry G, Wang X. Pathomechanisms of TDP-43 in neurodegeneration. JNeurochem 2018; 146: 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weintraub S, Wicklund AH, Salmon DP. The neuropsychological profile of Alzheimer disease. Cold Spring Harb Perspect Med 2012; 2: a006171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tarawneh R, Holtzman DM. The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb Perspect Med 2012; 2: a006148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oddo S, Caccamo A, Cheng D, LaFerla FM. Genetically altering Abeta distribution from the brain to the vasculature ameliorates tau pathology. Brain Pathol 2009; 19: 421–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Klohs J, Rudin M, Shimshek DR, Beckmann N. Imaging of cerebrovascular pathology in animal models of Alzheimer’s disease. Front Aging Neurosci 2014; 6: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kakuda N, Miyasaka T, Iwasaki N, Nirasawa T, Wada-Kakuda S, Takahashi-Fujigasaki J, et al. Distinct deposition of amyloid-beta species in brains with Alzheimer’s disease pathology visualized with MALDI imaging mass spectrometry. Acta Neuropathol Commun 2017; 5: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gravina SA, Ho L, Eckman CB, Long KE, Otvos L Jr., Younkin LH, et al. Amyloid beta protein (A beta) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43). J Biol Chem 1995; 270: 7013–6. [DOI] [PubMed] [Google Scholar]
- 77.Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM. Progression of cerebral amyloid angiopathy: accumulation of amyloid-beta40 in affected vessels. J Neuropathol Exp Neurol 1998; 57: 353–9. [DOI] [PubMed] [Google Scholar]
- 78.Weller RO, Djuanda E, Yow HY, Carare RO. Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol 2009; 117: 1–14. [DOI] [PubMed] [Google Scholar]
- 79.Aldea R, Weller RO, Wilcock DM, Carare RO, Richardson G. Cerebrovascular smooth muscle cells as the drivers of intramural periarterial drainage of the brain. Front Aging Neurosci 2019; 11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.van Veluw SJ, Hou SS, Calvo-Rodriguez M, Arbel-Ornath M, Snyder AC, Frosch MP, et al. Vasomotion as a driving force for paravascular clearance in the awake mouse brain. Neuron 2020; 105: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic- predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rohn TT. Caspase-cleaved TAR DNA-binding protein-43 is a major pathological finding in Alzheimer’s disease. Brain Res 2008; 1228: 189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.James BD, Wilson RS, Boyle PA, Trojanowski JQ, Bennett DA, Schneider JA. TDP-43 stage, mixed pathologies, and clinical Alzheimer’s-type dementia. Brain 2016; 139: 2983–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ayala YM, Zago P, D’Ambrogio A, Xu YF, Petrucelli L, Buratti E, et al. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci 2008; 121: 3778–85. [DOI] [PubMed] [Google Scholar]
- 85.Ferraro PM, Jester C, Olm CA, Placek K, Agosta F, Elman L, et al. Perfusion alterations converge with patterns of pathological spread in transactive response DNA-binding protein 43 proteinopathies. Neurobiol Aging 2018; 68: 85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Perez JM, Evans AC, Alzheimer’s Disease Neuroimaging I. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun 2016; 7: 11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Binnewijzend MA, Benedictus MR, Kuijer JP, van der Flier WM, Teunissen CE, Prins ND, et al. Cerebral perfusion in the predementia stages of Alzheimer’s disease. Eur Radiol 2016; 26: 506–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Foulds P, McAuley E, Gibbons L, Davidson Y, Pickering-Brown SM, Neary D, et al. TDP-43 protein in plasma may index TDP-43 brain pathology in Alzheimer’s disease and frontotemporal lobar degeneration. Acta Neuropathol 2008; 116: 141–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kasai T, Tokuda T, Ishigami N, Sasayama H, Foulds P, Mitchell DJ, et al. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol 2009; 117: 55–62. [DOI] [PubMed] [Google Scholar]
- 90.Suarez-Calvet M, Dols-Icardo O, Llado A, Sanchez-Valle R, Hernandez I, Amer G, et al. Plasma phosphorylated TDP-43 levels are elevated in patients with frontotemporal dementia carrying a C9orf72 repeat expansion or a GRN mutation. J Neurol Neurosurg Psychiatry 2014; 85: 684–91. [DOI] [PubMed] [Google Scholar]
- 91.Berthiaume AA, Grant RI, McDowell KP, Underly RG, Hartmann DA, Levy M, et al. Dynamic Remodeling of Pericytes In Vivo Maintains Capillary Coverage in the Adult Mouse Brain. Cell Rep 2018; 22: 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Berthiaume AA, Shih AY. Sharpening the tools for pericyte research. Nat Neurosci 2019; 22: 1041–3. [DOI] [PubMed] [Google Scholar]
- 93.Wilhelmus MM, Bol JG, van Duinen SG, Drukarch B. Extracellular matrix modulator lysyl oxidase colocalizes with amyloid-beta pathology in Alzheimer’s disease and hereditary cerebral hemorrhage with amyloidosis—Dutch type. Exp Gerontol 2013; 48, 109–14. [DOI] [PubMed] [Google Scholar]
- 94.Martinez-Revelles S, Garcia-Redondo AB, Avendano MS, Varona S, Palao T, Orriols M, et al. Lysyl oxidase induces vascular oxidase stress and contributes to arterial stiffness and abnormal elastin structure in hypertension: role of p38MAPK. AntioxidRedox Signal 2017; 27: 379–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Varona S, Orriols M, Galan M, Guadall A, Canes L, Aguilo S, et al. Lysyl oxidase (LOX) limits VSMC proliferation and neointimal thickening through its extracellular enzymatic activity. Sci Rep 2018; 8: 13258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.