

Abstract
Thermoresponsive poly(N-isopropylacrylamide) (PNIPAAm) hydrogels are widely studied smart materials, particularly for biomedical applications, but are limited by their mechanical strength. In this study, double network (DN) hydrogels were prepared with an asymmetric crosslink design and inclusion of an electrostatic co-monomer, 2-acrylamido-2-methylpropane sulfonic acid (AMPS). These P(NIPAAm-co-AMPS)/PNIPAAm DN hydrogels were sequentially formed with a tightly crosslinked 1st network comprised of variable levels of AMPS (100 : 0 to 25 : 75 wt% ratio of NIPAAm:AMPS) and a loosely crosslinked 2nd network comprised of PNIPAAm. The impact of AMPS content in the 1st network on the volume phase transition temperature (VPTT), morphology, deswelling–reswelling kinetics and mechanical properties was evaluated. Without substantially altering the VPTT of conventional PNIPAAm hydrogels but with improving thermosensitivity, the DN hydrogel formed with 25 : 75 wt% of NIPAAm:AMPS achieved exceptional strength, high modulus and high %strain at break.
Introduction
In response to temperature change, thermoresponsive hydrogels reversibly switch from a water-swollen, hydrophilic state to a deswollen, hydrophobic state.1,2 Thermoresponsive hydrogels are prepared by crosslinking polymers that exhibit a lower critical solubility temperature (LCST) such as poly(N-isopropyl-acrylamide (PNIPAAm; LCST, ~32 °C).3 Thermal modulation of PNIPAAm hydrogels above and below its volume phase transition temperature (VPTT, ~33 to 35 °C) causes its reversible deswelling and reswelling, respectively.4,5 Because of its convenient VPTT, PNIPAAm hydrogels are particularly useful as smart materials for various biological applications,6 including: microfluidic actuation,7-9 separation,10,11 controlled drug delivery,12-14 cell sheet tissue engineering,15,16 anti-fouling coatings17-19 and “self-cleaning” membranes for implanted biosensors.20-23
Unfortunately, conventional PNIPAAm hydrogels prepared via copolymerization of N-isopropylacrylamide (NIPAAm) and a crosslinker such as N,N′-methylenebisacrylamide (BIS) exhibit relatively poor mechanical properties in the swollen state.24,25 Improving the mechanical strength as well as stiffness (i.e. modulus) of PNIPAAm hydrogels would enhance their utility. However, this must not be achieved to the detriment of thermosensitivity (i.e. rate and extent of deswelling and reswelling) which is already limited for crosslinked PNIPAAm hydrogels.24,26 This presents a significant challenge. For instance, the mechanical properties of PNIPAAm hydrogels are often compromised by strategies used to enhance the thermosensitivity (without altering the VPTT), including: comb-type networks,27-29 heterogeneous morphologies,30-33 poration34-36 or open channel structures.37
Interpenetrating polymer networks (IPNs) are comprised of two crosslinked networks held together by inter-network entanglements.38 Although PNIPAAm IPNs exhibited enhanced mechanical properties, their swelling was decreased.39 Compared to conventional single network (SN) hydrogels or even IPN hydrogels, double network (DN) hydrogels display enhanced mechanical properties as well as a high degree of swelling.40-42 A class of IPNs, DN hydrogels are comprised of a neutral and a polyelectrolyte network with one network more highly crosslinked than the other. Several DN hydrogel systems have been reported but none are thermoresponsive. The most extensively studied DN hydrogel system is that reported by Gong and co-workers consisting of a tightly crosslinked, ionizable 1st network comprised of poly(2-acrylamido-2-methylpropane sulfonic acid) (PAMPS) and a sparsely crosslinked, neutral 2nd network comprised of poly(acrylamide) (PAAm) (i.e. PAMPS/PAAm).43 Other DN hydrogels include PAMPS/poly(N,N′-dimethylacrylamide) (PDMAAm)44 and modified hyaluronan (HA)/PDMAAm.45 Frank and co-workers have reported DN hydrogels based on neutral poly(ethylene oxide) (PEO) as the tightly crosslinked 1st network and ionizable poly(acrylic acid) (PAA) as the loosely crosslinked 2nd network (i.e. PEO/PAA DN).46 Recently, Frank and co-workers reported a pH- and temperature “dual responsive” PNIPAAm/PAA hydrogel system which is an IPN due to the constant level of crosslinking in the 1st and 2nd networks.47
Of late, we have reported the first thermoresponsive PNIPAAm DN hydrogels.48 These were comprised of a tightly crosslinked PNIPAAm 1st network and a loosely crosslinked PNIPAAm 2nd network. Optionally, colloidal polysiloxane nanoparticles (~50 and 200 nm diameters) were incorporated during formation of the 1st or 2nd network to form nanocomposite DN hydrogels. Compared to the conventional PNIPAAm SN hydrogel, the PNIPAAm DN hydrogel exhibited similar swelling but improved strength and modulus. Inclusion of nanoparticles improved thermosensitivity without altering the VPTT but did not significantly improve mechanical properties relative to the DN hydrogel.
In this work, we report the formation of P(NIPAAm-co-AMPS)/PNIPAAm DNs to achieve improved mechanical properties versus PNIPAAm SN or DN hydrogels but with a concomitant improvement in thermosensitivity. These DNs are comprised of tightly crosslinked, ionized 1st network [P(NIPAAm-co-AMPS)] and a loosely crosslinked, interpenetrating 2nd network [PNIPAAm]. While P(NIPAAm-co-AMPS) SN hydrogels have been previously reported their mechanical properties were not investigated.49 AMPS is a strong electrolyte whose sulfonate groups completely dissociate over a wide pH range.50,51 In aqueous solution, polymers based on AMPS exhibit a marked coil expansion in aqueous solutions and networks are expanded.52 This behavior is attributed to the strong electrostatic repulsive forces which give rise to an increase in osmotic pressure.50 As a result, AMPS-containing SN hydrogels such as P(AAm-co-AMPS) exhibit enhanced swelling with increased AMPS content.50-53 In addition, P(AAm-co-AMPS) DN hydrogels exhibit notably high strength as well as modulus values.40,43,54,55 Thus, in this work, AMPS was utilized to impart these properties to this PNIPAAm-based DN system.
Herein, P(NIPAAm-co-AMPS)/PNIPAAm DN networks were formed with a tightly crosslinked 1st network comprised of variable levels of AMPS (100 : 0 to 25 : 75 wt% ratio of NIPAAm:AMPS) and a loosely crosslinked 2nd network comprised of PNIPAAm. The VPTT, morphology, equilibrium swelling, deswelling–reswelling kinetics and mechanical properties were evaluated.
Experimental
Materials
N-Isopropylacrylamide (NIPAAm, 97%) was obtained from Aldrich. 2-Acrylamido-2-methylpropane sulfonic acid (AMPS, 97%) and N,N′-methylenebisacrylamide (BIS, 99%) were purchased from ACROS. 1-[4-(2-Hydroxyethoxy)-phenyl]-2-hydroxy-2-methyl-1-propane-1-one (Irgacure 2959) was purchased from BASF.
Preparation of single network (SN) hydrogels
SN hydrogels (including those that serve as the 1st network of DN hydrogels) were prepared via in situ photocure of aqueous precursor solutions containing NIPAAm monomer, AMPS monomer (optional), BIS crosslinker, Irgacure-2959 photo-initiator and DI water (Fig. 1a). In a 50 mL round bottom (rb) flask equipped with a Teflon-covered stir bar, NIPAAm/AMPS (total weight equal to 1.0 g), BIS (0.04 g), and Irgacure (0.08 g) were dissolved in DI water (7.0 mL). The wt% ratio of NIPAAm to AMPS was systematically varied (Table 1).
Fig. 1.

Schematic depiction of P(NIPAAm-co-AMPS)/PNIPAAm double network (DN) hydrogels. Sequential formation of (a) 1st network (i.e. formation of single network, SN) and (b) subsequent formation of 2nd network (i.e. formation of DN). Photograph of DN hydrogels: (c) discs punched from a sheet immediately following crosslinking of 2nd network and (d) after subsequently soaking in DI water for 48 h. Per Table 1, DN hydrogels are denoted as “DN-X%” where X% equals the wt% of AMPS in the 1st network.
Table 1.
Composition, VPTT and mechanical properties of SN and DN hydrogels
| 1st networka |
2nd networkb |
VPTT |
Compressive properties |
|||||
|---|---|---|---|---|---|---|---|---|
| Notation | wt% ratio NIPAAm:AMPS |
mol% ratio NIPAAm:AMPS |
wt% ratio NIPAAm:AMPS |
To (°C) | Tmax (°C) | Modulus (MPa) | UCS (MPa) | %Strain at break |
| SN-0% | 100 : 0 | 100 : 0 | 100 : 0 | 32.0 | 33.9 | 0.081 ± 0.014 | 0.144 ± 0.012 | 57 ± 1 |
| DN-0% | 100 : 0 | 100 : 0 | 100 : 0 | 32.4 | 34.0 | 0.188 ± 0.007 | 0.452 ± 0.056 | 52 ± 3 |
| DN-5% | 95 : 5 | 97 : 3 | 100 : 0 | 31.6 | 33.4 | 0.192 ± 0.024 | 0.259 ± 0.029 | 40 ± 2 |
| DN-10% | 90 : 10 | 94 : 6 | 100 : 0 | 31.5 | 33.1 | 0.277 ± 0.060 | 0.563 ± 0.026 | 52 ± 7 |
| DN-25% | 75 : 25 | 85 : 15 | 100 : 0 | 30.9 | 32.8 | 0.341 ± 0.048 | 0.784 ± 0.004 | 46 ± 1 |
| DN-50% | 50 : 50 | 65 : 35 | 100 : 0 | 31.5 | 33.0 | 0.311 ± 0.035 | 2.532 ± 0.314 | 73 ± 5 |
| DN-60% | 40 : 60 | 55 : 45 | 100 : 0 | 31.5 | 33.0 | 0.303 ± 0.005 | 3.128 ± 0.545 | 71 ± 5 |
| DN-75% | 25 : 75 | 38 : 62 | 100 : 0 | 31.6 | 32.9 | 0.085 ± 0.018 | 17.50 ± 2.986 | 95 ± 2 |
wt% BIS: 4%.
wt% BIS: 0.2%.
Hydrogel sheets were prepared by pipetting the precursor solution into a rectangular mold formed by sandwiching poly-carbonate spacers (1.5 mm thick) between two clamped glass microscope slides. The mold was submerged in an ice water bath (~7 °C) and subjected to UV light (UV-transilluminator, 6 mW cm−2, 365 nm) for 30 min. After removal from the mold, the hydrogel sheet was rinsed with DI water and then soaked in DI water for 2 days with daily water changes to remove impurities. Specimens used for analyses were taken from hydrogel sheets following soaking.
Preparation of double network (DN) hydrogels
The designated SN hydrogel (i.e. the 1st network) was soaked in a solution of NIPAAm (6.0 g), BIS (0.012 g), Irgacure-2959 (0.24 g), DI water (21 mL) for 24 h. The hydrogel sheet was then transferred to a rectangular mold (2.3 mm thick), photocured and purified as above (Fig. 1b). Specimens used for analyses were taken from hydrogel sheets following soaking.
Extent of crosslinking
The percentage of uncrosslinked material (i.e. %extractables) in select hydrogels was determined by weight loss following soaking in dichloromethane (CH2Cl2). For a given hydrogel, three hydrogel discs (13 mm diameter, 1.5 mm thickness) were punched from a single hydrogel sheet with a die and dried in a vacuum oven [30 in. Hg, room temperature (RT), 24 h] and weighed. Each dried disc was soaked in 10 mL of CH2Cl2 for 24 h and weighed after similarly drying in a vacuum oven. The % extractables was calculated as the average weight difference of the extracted versus unextracted weight divided by the unextracted weight.
Volume phase transition temperature (VPTT)
The VPTT of swollen hydrogels was determined by differential scanning calorimetry (DSC, TA Instruments Q100). Water-swollen hydrogels were blotted with a Kim Wipe and a small piece sealed in a hermetic pan. After cooling to −50 °C, the temperature was increased to 50 °C at a rate of 3 °C min−1 for 2 cycles. The resulting endothermic phase transition peak was characterized by the initial temperature at which the endotherm starts (To) and the peak temperature of the endotherm (Tmax). Reported data are from the 2nd cycle.
Morphology
Hydrogel discs (13 mm diameter) were punched from a hydrogel sheet. Discs were submerged in liquid nitrogen for 1 min and immediately freeze-dried (Labconco Freezone 2.5) over-night. Cross-sections were prepared by cutting with a clean razor blade. Specimen cross-sections were subjected to Ptsputter coating and viewed with a field emission scanning electron microscope (FEI Quanta 600 FE-SEM) at accelerated electron energy of 10 keV.
Kinetic deswelling
Three discs (13 mm diameter) were prepared as above. Each disc was placed in a sealed vial containing 20 mL DI water, immersed in a water bath for 24 h at 22 °C to reach equilibrium (Ws) and quickly transferred into a 50 °C water bath. At 10, 20, 40, 80, 120 and 180 min, each disc was removed, blotted with a Kim Wipe, immediately weighed (Wt) and returned to the vial for subsequent measurements. After 180 min, the discs were dried in a vacuum oven (30 in. Hg, 60 °C, 24 h) and weighed (Wd). Water retention (WR) is defined as: WR = (Wt – Wd)/Ws.
Kinetic reswelling
Three discs (13 mm diameter) were prepared as above. Each disc was placed in an open vial, dried in a vacuum oven (30 in. Hg, 60 °C, 24 h) and weighed (Wd). To each vial was added 20 mL DI water and the sealed vial immersed in a water bath at 22 °C. At 10, 20, 40, 80, 120, 200, 320, 450 and 640 min, each disc was removed, blotted with a Kim Wipe and weighed (Wt). Kinetic reswelling ratio is defined as: SR = Wt/Wd.
Dynamic mechanical analysis (DMA)
Five discs (13 mm diameter) were prepared as above. DMA of the discs was performed in the compression mode with a dynamic mechanical analyzer (TA Instruments Q800) equipped with parallel-plate compression clamp with a diameter of 40 mm (bottom) and 15 mm (top). The swollen disc was blotted with a Kim Wipe, clamped between the parallel plates and silicone oil placed around the exposed hydrogel edge to prevent dehydration. Following equilibration below the VPTT at 25 °C (5 min), the specimens were tested in a multi-frequency-strain mode (1 to 25 Hz).
Compression tests
Three discs (6 mm diameter) were punched from a single sheet with a die. Compressive tests were performed with an Instron 3340 at RT. A swollen disc (6 mm diameter) was blotted with a Kim Wipe and clamped between the parallel plates with an initial pre-load force of ~0.5 N. Compressive strain was applied at a rate of 1 mm min−1 until the disc fractured. The following parameters were determined: (1) compressive modulus (E); (2) ultimate compressive strength (UCS), and (3) %strain at break. The modulus was obtained from the slope of the stress–strain curve from 0–10% strain.44
Results and discussion
Preparation of SN and DN hydrogels
Conventional PNIPAAm SN hydrogels (SN-0%) were formed by photopolymerization of aqueous solutions containing NIPAAm and BIS crosslinker (4 wt% based on NIPAAm weight) (Table 1). SN-0% served as a conventional PNIPAAm SN control for all analyses. In the case DN hydrogel fabrication, the 1st network (i.e. SN) was likewise formed but with the monomer being a combination of a varying ratio of NIPAAm:AMPS (100 : 0 to 25 : 75 wt%). Subsequent soaking of these 1st networks in the 2nd network solution comprised of NIPAAm and BIS crosslinker (0.2 wt% based on NIPAAm weight) yielded the corresponding DN hydrogels (Table 1, Fig. 1a and b). Per Table 1, DN hydrogels are denoted as “DN-X%” where X% equals the wt% of AMPS in the 1st network. DN-0% served as a PNIPAAM DN control for all analyses. The remaining DN hydrogels are comprised of a highly crosslinked, P(NIPAAm-co-AMPS) 1st network of varying amounts of ionized sulfonate groups and an interpenetrating, loosely crosslinked, neutral PNIPAAM 2nd network.
For P(NIPAAm-co-AMPS)/PNIPAAm DN hydrogels, a 25 : 75 wt% ratio of NIPAAm:AMPS was selected as the maximum level of AMPS in the 1st network. This was based on our observation that 1st networks (i.e. SNs) prepared with higher levels of AMPS produced poorly cured hydrogels. We verified that the 1st network used to eventually form DN-75% (i.e. NIPAAm:AMPS = 25 : 75 wt%) contained low levels of uncrosslinked material (2% extractables) (Table S1†). However, 1st networks prepared with 15 : 85 and 5 : 95 wt% of NIPAAm:AMPS indeed produced higher %extractables (4 and 32%, respectively).
All hydrogels were photocured at low temperatures (Tprep < 20 °C) to obtain a homogeneous rather than a heterogeneous morphology.56,57 Immediately following cure, hydrogel discs were somewhat iridescent due to the presence of unreacted NIPAAm monomer (Fig. 1c). However, following soaking in DI water to remove impurities, all hydrogels were rather transparent which is consistent with homogeneous hydrogels (Fig. 1d). Notably, the extent to which the freshly cured hydrogel discs swelled (i.e. increased in diameter) after soaking in DI water dramatically increased with higher levels of AMPS. This is attributed to the greater expansion of the 1st network due to a rise in electrostatic repulsive forces with AMPS levels which produces an increase in osmotic pressure.50
VPTT
When heated above its VPTT, thermoresponsive PNIPAAm hydrogels deswell due to breaking of hydrogen bonds of surrounding water molecules and subsequent increase in hydrophobic interactions. As recorded by DSC, an endothermic peak accompanies this process and VPTT is defined in terms of the onset (To) and the maximum temperature (Tmax) of the endothermic peak.58,59 As expected for “PNIPAAm-only” hydrogels (i.e. SN-0% and DN-0%), there was not a significant change in the VPTT (Table 1, Fig. 2). Incorporation of a hydrophilic, anionic comonomer is generally associated with an increase in the VPTT.60 Indeed, for P(NIPAAm-co-AMPS) SN hydrogels, VPTT was observed to increase with AMPS content.49 We likewise observed this effect for select P(NIPAAm-co-AMPS) SN (i.e. 1st networks) (Fig. S1†). However, for our P(NIPAAm-co-AMPS)/PNIPAAm DN hydrogels, the VPTT was slightly decreased (~1 °C for Tmax) versus that of SN-0% and DN-0%, irrespective of AMPS content in the 1st network. Thus, the “PNIPAAm-only” 2nd network exhibits at PNIPAAm hydrogel-like VPTT despite the presence of a P(NIPAAm-co-AMPS) 1st network. The slightly lower VPTT values are attributed to the PNIPAAm 2nd networks' lower crosslink density.61
Fig. 2.

DSC thermograms of hydrogels.
Morphology
Hydrogel morphology was studied by SEM. Specimens were prepared with freeze-drying as this is known to preserve the structure and volume of swollen hydrogels even after all (or almost all) of the solvent is removed.62 The pore size of DN hydrogels increased substantially as the AMPS content was increased in the 1st network (Fig. 3). This observation is consistent with the corresponding increase in hydrogel dimension when allowed to swell in water (Fig. 1d).
Fig. 3.

SEM micrographs of hydrogels. All scale bars = 50 μm.
Kinetic deswelling/reswelling
For release and delivery applications, PNIPAAm-based hydrogels should exhibit a rapid and extensive response to thermal modulation (i.e. thermosensitivity). The thermosensitivity of hydrogels were determined by measuring the rate and extent to which they deswelled at 50 °C (>VPTT) (Fig. 4) and subsequently re-swelled at 22 °C (<VPTT) (Fig. 5).
Fig. 4.

Hydrogel deswelling kinetics at 50 °C (by mass).
Fig. 5.

Hydrogel reswelling kinetics at 22 °C (by mass).
DN-0% exhibited an enhanced rate and extent of deswelling versus SN-0% due to the former's asymmetrically crosslinked network structure and larger pore size which permitted a greater collapse (Fig. 4).48 Thermosensitivity was dramatically improved for DN-5%. However, as AMPS levels were further increased in the 1st network, deswelling systematically decreased. Thus, as AMPS levels were increased (beyond a 95 : 5 wt% ratio of NIPAAm:AMPS), the increase in electrostatic forces appeared to inhibit the ability of the hydrogel to collapse when heated above the VPTT. Still, DN-75% exhibited enhanced deswelling behavior versus SN-0% and DN-0%.
When the deswollen specimens were reswollen, DN-0% and SN-0% exhibited similar thermosensitivity. As AMPS levels were systematically increased in the 1st network, the P(NIPAAm-co-AMPS)/PNIPAAm DN hydrogels exhibited an increased rate and extent of reswelling (Fig. 5). In contrast to deswelling, increased electrostatic forces (i.e. increased AMPS) enhances electrostatic repulsive forces to facilitate re-expansion and reswelling of the network via an increase in osmotic pressure.50
Mechanical properties
The ability of PNIPAAm-hydrogels to withstand mechanical forces both in vitro and in vivo is critical to their utility. An increase in hydrogel swelling ratio is typically associated with an decrease in strength and modulus.63 Thus, the swelling ratio (SR) of the hydrogel specimen must be considered when evaluating measured mechanical properties. Here, the SR of specimens subjected to mechanical tests may be considered to be that recorded at time = 640 min (Fig. 5).
DMA was used to measure hydrogel stiffness in terms of the storage modulus (G′) as a function of frequency of the applied compressive strain (Fig. 6). Despite having a similar SR, G′ of DN-0% was nearly twice that of SN-0% due former's asymmetrically crosslinked network.59 G′ generally increased as the AMPS levels were increased in the 1st network up to ~50 : 50 wt % ratio of NIPAAm:AMPS (i.e. DN-50%). However, higher levels of AMPS produced a decrease in G′. Likewise, during quasistatic compression tests, the compressive modulus (E) was observed to increase with AMPS content in the 1st network before decreasing again (Table 1, Fig. 7). This modulus trend has also been previously observed for the P(AAm-co-AMPS) SN64 and P(DMAAm-co-AMPS) SN65 hydrogels. It has been proposed that, beyond a certain level of AMPS, the effective crosslink density of the network is decreased due to a high concentration of electrostatic forces (i.e. AMPS content) which produce a highly extended chain conformation.64 This reduction in the effective crosslink density leads to a decrease in modulus. Still, the E value of ~0.3 MPa for DN-25% and DN-50% is notable and similar to that observed for PAMPS/PAAm DN hydrogels.43
Fig. 6.
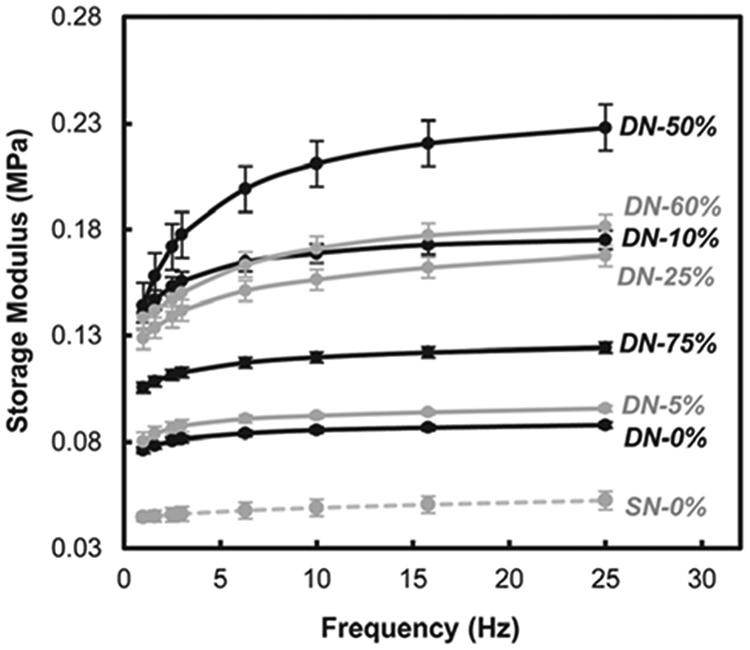
Storage modulus (G′) of hydrogels.
Fig. 7.

Compressive modulus (E) of hydrogels.
Compression tests also were also used to measure hydrogel ultimate compressive strength (UCS) (Table 1, Fig. 8). UCS of DN-0% was over three times greater than that of SN-0% due former's asymmetrically crosslinked network.59 For DN-5%, UCS decreased somewhat. However, as AMPS levels in the 1st network was increased from 90 : 10 to 40 : 60 wt% (NIPAAm:AMPS), UCS steadily increased. Most noteworthy was the exceptional UCS (~17.5 MPa) achieved by DN-75%. This is particularly significant given its SR of ~11.5 (i.e. a weight increase of 11.5× versus the dry state) (Fig. 5). The UCS of DN-75% is similar to that of PAMPS/PAAm DN hydrogels (~17.2 MPa) whose strength has been attributed a 1st network which, by itself, is quite brittle, but whose crack propagation is prevented by a loosely crosslinked 2nd network which effectively dissipates stress.40,43,66 The %strain at break of DN-75% (95%) was also the highest for this series of hydrogels.
Fig. 8.

Ultimate compressive strength (UCS) of hydrogels.
Conclusions
To summarize, P(NIPAAm-co-AMPS)/PNIPAAm DN hydrogels were produced from a tightly crosslinked 1st network comprised of variable levels of AMPS (100 : 0 to 25 : 75 wt% ratio NIPAAm:AMPS) and a loosely crosslinked 2nd network comprised of PNIPAAm. For all of these DN hydrogels, their VPTT was only slightly decreased (~1 °C) versus the “all PNIPAAm” SN-0% and DN-0%. Thus, the 2nd PNIPAAm network effectively dominated the VPTT process. As the AMPS content increased in the 1st network, the pore size of the DN hydrogels increased substantially and produced enhanced water uptake and swollen gel dimensions. All P(NIPAAm-co-AMPS)/PNIPAAm DN hydrogels exhibited enhanced thermosensitivity versus SN-0% and DN-0%. While increased electrostatic repulsive forces with AMPS content resulted in a systematic reduction in the extent and rate of deswelling, re-swelling behavior was enhanced. Consistent with that observed for other polyelectrolyte networks, the modulus of P(NIPAAm-co-AMPS)/PNIPAAm DN hydrogels initially increased with AMPS content but then decreased. UCS steadily increased with AMPS content, reaching an impressive 17.5 MPa for DN-75% (i.e. 25 : 75 wt% NIPAAm:AMPS in the 1st network). The combined properties of the ultra-strong DN-75% (UCS = 17.5 MPa), including a high modulus (E = 0.085 MPa) and strain at break (95%), distinguishes it from conventional thermoresponsive PNIPAAm SN hydrogels.
Supplementary Material
Acknowledgements
Funding from NSF/CBET (0854462) is gratefully acknowledged.
Footnotes
Electronic supplementary information (ESI) available: Table S1 and Fig. S1. See DOI: 10.1039/c3sm27226e
Notes and references
- 1.Wu XS, Hoffman AS and Yager P, J. Polym. Sci., Part A: Polym. Chem, 1992, 30, 2121–2129. [Google Scholar]
- 2.Liu F and Urban MW, Prog. Polym. Sci, 2010, 35, 3–23. [Google Scholar]
- 3.Schild HG, Prog. Polym. Sci, 1992, 17, 163–249. [Google Scholar]
- 4.Hoffman AS, Afrassiabi A and Dong LC, J. Controlled Release, 1986, 4, 213–222. [Google Scholar]
- 5.Zhang J, Pelton R and Deng Y, Langmuir, 1995, 11, 2301–2302. [Google Scholar]
- 6.Chaterji S, Kwon K and Park K, Prog. Polym. Sci, 2007, 32, 1083–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eddington DT and Beebe DJ, Adv. Drug Delivery Rev, 2004, 56, 199–210. [DOI] [PubMed] [Google Scholar]
- 8.Harmon ME, Tang M and Frank CW, Polymer, 2003, 44, 4547–4556. [Google Scholar]
- 9.Li Z, He Q, Ma D and Chen H, Anal. Chim. Acta, 2010, 665, 107–112. [DOI] [PubMed] [Google Scholar]
- 10.Freitas RFS and Cussler EL, Sep. Sci. Technol, 1987, 22, 911–919. [Google Scholar]
- 11.Zhiming L, Qiaohong H, Dan M, Hengwu C and Soper SA, Anal. Chem, 2010, 82, 10030–10036. [DOI] [PubMed] [Google Scholar]
- 12.Chilkoti A, Dreher MR, Meyer DE and Raucher D, Adv. Drug Delivery Rev, 2002, 54, 613–630. [DOI] [PubMed] [Google Scholar]
- 13.Nakayama M, Drug Delivery Syst., 2008, 23, 627–636. [Google Scholar]
- 14.Qiu Y and Park K, Adv. Drug Delivery Rev, 2001, 53, 321–329. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi J and Okano T, Sci. Technol. Adv. Mater, 2010, 11, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamato M, Akiyama Y, Kobayashi J, Yang J, Kikuchi A and Okano T, Prog. Polym. Sci, 2007, 32, 1123–1133. [Google Scholar]
- 17.Callwaert M, Rouxhet PG and Boulange-Petermann L, J. Adhesion Sci. Technol, 2005, 19, 765–781. [Google Scholar]
- 18.Cunliffe D, Smart CA, Tsibouklis J, Young S, Alexander C and Vulfson EN, Biotechnol. Lett, 2000, 22, 141–145. [Google Scholar]
- 19.Ista L and Lopez G, J. Ind. Microbiol. Biotechnol, 1998, 20, 121–125. [Google Scholar]
- 20.Chen J, Yoshida M, Maekawa Y and Tsubokawa N, Polymer, 2001, 42, 9361–9365. [Google Scholar]
- 21.Gant R, Abraham A, Hou Y, Grunlan MA and Cote GL, Acta Biomater., 2010, 6, 2903–2910. [DOI] [PubMed] [Google Scholar]
- 22.Gant R, Hou Y, Grunlan MA and Cote GL, J. Biomed. Mater. Res., Part A, 2009, 90, 695–701. [DOI] [PubMed] [Google Scholar]
- 23.Guenther M, Gerlach G, Kuckling D, Kretschmer K, Corten C, Weber J, Sorber J, Suchaneck G and Arndt K-F, Proc. SPIE, 2006, 6167, 61670T/61671–61670T/61611. [Google Scholar]
- 24.Zhang X-Z, Xu X-D, Cheng S-X and Zhuo R-X, Soft Matter, 2008, 4, 385–391. [DOI] [PubMed] [Google Scholar]
- 25.Haraguchi K and Li H-J, Macromolecules, 2006, 39, 1898–1905. [Google Scholar]
- 26.Yoshida R, Uchida K, Kaneko Y, Sakai K, Kikuchi A, Sakurai Y and Okano T, Nature, 1995, 374, 240–242. [Google Scholar]
- 27.Liu Q, Zhang P, Qing A, Lan Y, Shi J and Lu M, Polymer, 2006, 47, 6963–6969. [Google Scholar]
- 28.Matsuura T, Sugiyama M, Annaka M, Hara Y and Okano T, Polymer, 2003, 44, 4405–4409. [Google Scholar]
- 29.Yoshida R, Uchida K, Kaneko Y, Sakurai Y and Okano T, Nature, 1995, 374, 240–242. [Google Scholar]
- 30.Xue W, Champ S, Huglin MB and Jones TGJ, Eur. Polym. J, 2004, 40, 703–712. [Google Scholar]
- 31.Yan Q and Hoffman AS, Polymer, 1995, 36, 887–889. [Google Scholar]
- 32.Zhang X-H, Yang Y-Y and Chung T-S, J. Colloid Interface Sci, 2002, 246, 105–111. [DOI] [PubMed] [Google Scholar]
- 33.Zhang X-Z, Yang Y-Y and Chung T-S, Langmuir, 2002, 18, 2538–2542. [Google Scholar]
- 34.Serizawa T, Wakita K and Akashi M, Macromolecules, 2002, 35, 10–12. [Google Scholar]
- 35.Serizawa T, Wakita K, Kaneko T and Akashi M, J. Polym. Sci., Part A: Polym. Chem, 2002, 40, 4228–4235. [Google Scholar]
- 36.Serizawa T, Uemura M, Kaneko T and Akashi M, J. Polym. Sci., Part A: Polym. Chem, 2002, 40, 3542–3547. [Google Scholar]
- 37.Kaneko T, Asoh T-A and Akashi M, Macromol. Chem. Phys, 2005, 206, 566–574. [Google Scholar]
- 38.Suthar B, Xiao HX, Klempner D and Frisch KC, Polym. Adv. Technol, 1995, 7, 221–233. [Google Scholar]
- 39.Zhang X-Z, Wu D-Q and Chu C-C, Biomaterials, 2004, 25, 3793–3805. [DOI] [PubMed] [Google Scholar]
- 40.Gong JP, Soft Matter, 2010, 6, 2583–2590. [Google Scholar]
- 41.Myung D, Waters D, Wiseman M, Duhamel P-E, Noolandi J, Ta CN and Frank CW, Polym. Adv. Technol, 2008, 19, 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haque MA, Kurokawa T and Gong JP, Polymer, 2012, 53, 1805–1822. [Google Scholar]
- 43.Gong JP, Katsuyama Y, Kurokawa T and Osada Y, Adv. Mater, 2003, 15, 1155–1158. [Google Scholar]
- 44.Yasuda K, Gong JP, Katsuyama Y, Nakayama A, Tanabe Y, Kondo E, Ueno M and Osada Y, Biomaterials, 2005, 26, 4468–4475. [DOI] [PubMed] [Google Scholar]
- 45.Weng L, Gouldston A, Wu Y and Chen W, Biomaterials, 2008, 29, 2153–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Myung D, Koh W, Ko J, Hu Y, Carrasco M, Noolandi J, Ta CN and Frank CW, Polymer, 2007, 48, 5376–5387. [Google Scholar]
- 47.Kelmanovich SG, Parke-Houben R and Frank CW, Soft Matter, 2012, 8, 8137–8148. [Google Scholar]
- 48.Fei R, George JT, Park J and Grunlan MA, Soft Matter, 2012, 8, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turan E, Demirci S and Caykara T, J. Polym. Sci., Part B: Polym. Phys, 2008, 46, 1713–1724. [Google Scholar]
- 50.Travas-Sejdic J and Easteal A, Polym. Gels Networks, 1998, 5, 481–502. [Google Scholar]
- 51.Melekaslan D and Okay O, Polymer, 2000, 41, 5737–5747. [Google Scholar]
- 52.Liu X, Tong Z and Hu O, Macromolecules, 1995, 28, 3813–3817. [Google Scholar]
- 53.Okay O, Sariisik SB and Zor SD, J. Appl. Polym. Sci, 1998, 70, 567–575. [Google Scholar]
- 54.Na Y-H, Kurokawa T, Katsuyama Y, Tsukeshiba H, Gong JP, Osada Y, Okabe S, Karino T and Shibayama M, Macromolecules, 2004, 37, 5370–5374. [Google Scholar]
- 55.Nakajima T, Furukawa H, Gong JP, Lin EK and Wu W-L, Macromol. Symp, 2010, 291–292, 122–126. [Google Scholar]
- 56.Kayaman N, Kazan D, Erarslan A, Okay O and Baysal BM, J. Appl. Polym. Sci, 1998, 67, 805–814. [Google Scholar]
- 57.Rathjen CM, Park C-H, Goodrich PR and Walgenbach DD, Polym. Gels Networks, 1995, 3, 101–115. [Google Scholar]
- 58.Singh D, Knuckling D, Choudhary V, Adler H-J and Koul V, Polym. Adv. Technol, 2006, 17, 186–192. [Google Scholar]
- 59.Tanaka Y, Gong JP and Osada Y, Prog. Polym. Sci, 2005, 30, 1–9. [Google Scholar]
- 60.Feil H, Bae YH, Feijen J and Kim SW, Macromolecules, 1993, 26, 2496–2500. [Google Scholar]
- 61.Zhang X-Z, Wu D-Q and Chu C-H, J. Polym. Sci., Part B: Polym. Phys, 2003, 41, 582–593. [Google Scholar]
- 62.Bekiari V and Lianos P, Langmuir, 2006, 22, 8602–8606. [DOI] [PubMed] [Google Scholar]
- 63.Anseth KS, Bowman CN and Brannon-Peppas L, Biomaterials, 1995, 17, 1647–1657. [DOI] [PubMed] [Google Scholar]
- 64.Okay O and Durmaz S, Polymer, 2002, 43, 1215–1221. [Google Scholar]
- 65.Tong Z and Liu Z, Macromolecules, 1993, 26, 4964–4966. [Google Scholar]
- 66.Brown HR, Macromolecules, 2007, 40, 3815–3818. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.