

Abstract
We closely characterized 7-Dimethylamino-4-trifluromethylcoumarin (Coumarin 152, C152), a substrate metabolized by multiple P450 species, to establish a new fluorogenic probe for the studies of functional integration in the cytochrome P450 ensemble,
Scanning fluorescence spectroscopy and LC/MS-MS were used to characterize the products of N-demethylation of C152 and optimize their fluorometric detection. The metabolism of C152 by the individual P450 species was characterized using the microsomes containing cDNA-expressed enzymes. C152 metabolism in human liver microsomes (HLM) was studied in a preparation with quantified content of eleven P450 species
C152 is metabolized by CYP2B6, CYP3A4, CYP3A5, CYP2C19, CYP1A2, CYP2C9, and CYP2C8 listed in the order of decreasing turnover. The affinities exhibited by CYP3A4, CYP2C9, and CYP2C8 were lower than those characteristic to the other enzymes.
The presumption of additivity suggests the participation of CYP3A4, CYP2B6, and CYP2C19 to be 84, 8, and 0.2%, respectively. Contrary to this prediction, inhibitory analysis identified CYP2C19 as the principal C152-metabolizing enzyme.
We thoroughly characterize C152 for the studies of drug metabolism in HLM and demonstrate the limitations of the proportional projection approach by providing an example, where the involvement of individual P450 species cannot be predicted from their content.
Keywords: Cytochrome P450, Coumarin 152, fluorogenic substrates, human liver microsomes, drug metabolism, inhibition, CYP2C19, CYP3A4
Introduction
The functional properties of the human drug-metabolizing system are primarily determined by the composition of the cytochrome P450 ensemble responsible for the metabolism of over 75% of all marketed drugs and new drug candidates (Rendic and Guengerich 2010; Wienkers and Heath 2005). A functional versatility of the cytochrome P450 ensemble is achieved through the presence of over a dozen of cytochrome P450 species differing in their substrate specificity. Although the premise that the properties of this ensemble represent a simple aggregate of the properties of the constituting P450 enzymes continues to be the cornerstone of a rational analysis of the routes of drug metabolism, its validity became essentially compromised. There are numerous indications of important nonadditivity of the properties of the individual cytochrome P450 species constituting the human drug-metabolizing ensemble (Fang et al. 2018a; Fang et al. 2018b; Gao et al. 2016; Volpe 2019; Zhang et al. 2016).
One of the major sources of nonadditivity is represented by the consequences of protein-protein interactions of cytochrome P450, including the formation of complexes of multiple P450 species and their interactions with potential regulatory proteins (Davydov 2011; Davydov 2016; Reed and Backes 2016; Reed and Backes 2017; Ryu et al. 2017). Further uncovering the manifestations of this nonadditivity and elucidation of its mechanistic grounds represent the task of vital importance for the biochemistry of drug metabolism.
The success of further studies in this direction depends on an appropriate selection of probe substrates. Most strategies of in vitro studies of drug metabolism relied on the use of isoform-specific P450 substrates used for probing the specific activity of each particular P450 species considered in isolation. However, there are only a few known examples of drugs metabolized by a single P450 enzyme (Spaggiari et al. 2014; Yuan et al. 2002). Most of the widely used pharmaceuticals exhibit similar affinities and comparable turnovers with multiple P450 species. The studies on the effect of the composition of the P450 pool on the routes of metabolism of polyspecific substrates may provide information of vital importance for avoiding erroneous attribution of the pathways of drug metabolism and uncovering the principles of integration of the individual P450 functionalities.
Important advantages for in vitro studies of drug metabolism are provided by the use of fluorogenic substrates that offers a cost-effective and expeditious alternative to laborious chromatographic techniques. Our search for fluorogenic substrates for the studies of functional integration in the human cytochrome P450 ensemble brought into our view a study of Santosh Kumar (Kumar 2007), which introduces 7-dimethylamino-4-trifluoromethyl coumarin (Coumarin 152, C152) as a fluorogenic substrate preferentially metabolized by multiple CYP2C enzymes. Unfortunately, the experimental design of this study suffers from several important flaws that did not allow the author to characterize the metabolism of C152 well enough to enable practical use of this substrate in further research. In particular, the above-cited study failed to take into account the background fluorescence of the substrate, did not address a possibility of subsequent formation of two products of C152 N-demethylation and did not characterize the fluorescent properties of the products.
Here we address the limitations of the above study, characterize and quantify the metabolites of C152 with scanning fluorescence spectroscopy, and liquid chromatography with tandem mass spectrometry (LC-MS/MS), and probe the ability of major P450 species to metabolize C152. Our studies demonstrated that C152 exhibits similarly high affinity and comparable turnover numbers with CYP2B6, CYP3A4, CYP2C19, and CYP1A2. Examining C152 metabolism in pooled HLM preparation with the quantified content of 11 major drug-metabolizing P450s, we observed a severe nonadditivity in the functionalities of the P450 species involved in its metabolism. C152 may, therefore, serve as a valuable probe in the studies of rerouting the pathways of metabolism of P450 substrates in response to changes in the composition of the P450 pool.
Material and Methods
Pooled human liver microsomes and their characterization with mass spectrometry.
The HLM sample used in this study was the InVitroCYP™ M-class 50-donor mixed gender pooled HLM preparations lot LBA obtained from BioIVT Corporation (Baltimore, MD). The composition of the cytochrome P450 ensemble in this preparation was characterized by mass-spectrometric analysis with a triple quadrupole mass spectrometer using the method of multiple reaction monitoring (MRM), as described previously (Davydova et al. 2019). Content of NADPH-cytochrome P450 reductase and cytochromes P450 1A2, 2A6, 2B6, 2D6, 2C8, 2C9, 2C18, 2C19, 2E1, 3A4 and 3A5 was determined based on the concentration of protein-specific tryptic peptides in trypsin-digested samples with the use of the stable isotope-labeled internal standards (SIS) (Davydova and others 2019). For each protein, one standard peptide with three transitions was used. The peptides, which sequences are provided in Table 2, were arranged into one Selected Reaction Monitoring (SRM) assay.
Table 2.
Composition of the cytochrome P450 ensemble in HLM preparation used in this study*
| Cytochrome P450 Species | Tryptic peptide used in the assay | Content, pmol/mg protein | Specific fraction, %a |
|---|---|---|---|
| CYP1A2 | ASGNLIPQEK | 12.6 ± 2.4 | 5.4 ± 0.3 |
| CYP2A6 | GTGGANIDPTFFLSR | 46.8 ± 6.7 | 22 ± 2 |
| CYP2B6 | GYGVIFANGNR | 2.7 ± 0.7 | 1.2 ± 0.1 |
| CYP2C8 | GLGIISSNGK | 2.6 ± 3.6 | 0.66 ± 0.92 |
| CYP2C9 | GIFPLAER | 33.9 ± 7.1 | 16.5 ± 0.3 |
| CYP2C18 | IAENFAYIK | 0.50 ± 0.06 | 0.30 ± 0.06 |
| CYP2C19 | ICVGEGLAR | 0.52 ± 0.12 | 0.26 ± 0.07 |
| CYP2D6 | LLDLAQEGLK | 1.3 ± 0.5 | 0.50 ± 0.18 |
| CYP2E1 | GIIFNNGPTWK | 26.0 ± 6.8 | 12 ± 1 |
| CYP3A4 | LSLGGLLQPEKPVVLK | 109 ± 54 | 41 ± 10 |
| CYP3A5 | DVEINGVFIPK | 1.7 ± 1.6 | 0.5 ± 0.4 |
| Total of all species | 217.7 ± 56.4 | 53 ± 14 | |
| P450 content by UV/VIS spectroscopy | 404.5 ± 94.9 | ||
| CPR (by LC-MS/MS) | NPFLAAVTTNR | 34.3 ± 8.1 | 55 ± 13 |
| CPR content by specific activity | 63.0 ± 12.2 |
The values given in the table correspond to the averages of 11 individual measurements, except for CYP2C8, which was determined in 2 individual assays. The“±” values are the confidence intervals calculated for p=0.05.
The values of the specific content of P450 species are the averages of the values calculated for each individual dataset. The fractions that are shown for the total P450 content and the content of CPR represent the ratios of the amounts detected with LS-MS/MS to the amount of the protein determined with the absorbance spectroscopy (P450) and activity (CPR) assays.
Microsomes containing recombinant human cytochromes P450.
All microsomal preparations containing individual P450 enzymes (Supersomes™) were the products of BD Gentest, now a part of Corning Life Sciences (Tewksbury, MA). In the present study we used the preparations containing CYP2B6 (SS(2B6), lot 31487), CYP1A2 (SS(1A2), lot 69375), CYP2C8 (SS(2C8), lot 8239002); CYP2C9 (SS(2C9), lot 41274), CYP2C19 (SS(2C19), lot 73445) and CYP3A4 (SS(3A4), lot 35933). All those preparations contained human NADPH-cytochrome P450 reductase (CPR) and cytochrome b5, except for SS(1A2), which had no cytochrome b5 co-expressed.
Characterization of the content of NADPH-cytochrome P450 reductase and cytochromes P450 in HLM.
The concentration of NADPH-cytochrome P450 reductase in microsomal membranes was determined based on the rate of NADPH-dependent reduction of cytochrome c, as described earlier (Davydova and others 2019). The total concentration of cytochromes P450 in HLM was determined with a variant of “oxidized CO versus reduced CO difference spectrum” method (Davydova and others 2019). Mass-spectrometry measurements of P450 content were performed using the equipment of “Human Proteome” Core Facilities of the Institute of Biomedical Chemistry.
Fluorometric assays of C152 metabolism.
The rate of demethylation of C152 was measured with a real-time continuous fluorometric assay using a Cary Eclipse fluorometer (Agilent Technologies, Santa Clara, CA, USA) or a custom-modified PTI QM-1 fluorometer (Photon Technology International, New Brunswick, NJ (Davydov et al. 2017). In the experiments with Cary Eclipse, the excitation was performed with a monochromatic light centered at 405 nm with 5 nm bandwidth. In the case of PTI QM-1, the excitation light centered at 405 nm was emitted by a CPS405 collimated laser diode module (Thorlabs Inc, Newton, NJ). In the single-wavelength assays, the emission wavelength was set at 500 nm with a 20 nm slit. The rate of formation of the fluorescent products was estimated by determining the slope of the linear part of the kinetic curve recorded over 3 – 5 min. In the experiments with scanning fluorimetry, the spectra of fluorescence in 450 – 650 nm region were recorded with 15 – 45 s time interval. The resulting series of spectra were analyzed with Principal Component Analysis.
All kinetic assays were performed in 0.1 M Na-HEPES buffer, pH 7.4, containing 60 mM KCl. In the case of the use of a Cary Eclipse instrument, the total volume of the incubation mixture was equal to 300μl, and a 5 × 5 mm quartz cell was used. In the experiments with PTI QM-1 fluorometer, we used a 3 × 3 mm quartz cell, and the volume of the sample was equal to 60μl. With both instruments, the kinetic assays were carried out at continuous stirring, and the temperature was maintained at 30 °C with a circulating water bath. An aliquot of 15–20 mM stock solution of C152 in acetone was added to attain the desired concentration in the range of 0.1 – 30 μM. The reaction was initiated by addition of 20 mM solution of NADPH to the concentration of 200 μM.
Determination of the rate of formation of the demethylated product with LC-MS.
The amount of formed product of N-demethylation of C152, 7-amino-4-trifluoromethyl coumarin (Coumarin 151, C151), was quantified with LC-MS/MS. The probes containing 5 – 20 pmol of cytochrome P450 in the volume of 60 μl were prepared in the same buffer as described for the fluorimetric assays. C152 was added as a 0.5 mM or 4 mM solution in acetone. The reaction was started by adding NADPH to the final concentration of 500 μM and carried on for 5 min at 30 °C in a shaking water bath. Quenching of the reaction was achieved by an addition of 240 μl of isopropanol containing a known concentration of phenacetin as an internal standard and followed by centrifugation at 9,300 g for 10 min.
An LC-20AD series high-performance liquid chromatography system (Shimadzu, Columbia, MD) fitted with an HTC PAL autosampler (LEAP Technologies, Carrboro, NC) was used to perform chromatography on a Kinetex® reverse-phase column (100 × 2.1 mm, Phenomenex, Torrance, CA). Chromatographic separation was performed using the binary method at the flow rate of 0.2 ml/min. The separation was initiated by 0.3 min flow at 10% mobile phase B. This was then increased to 70% and 95% until 1.5 and 6 min, respectively. The flow of mobile phase B was gradually decreased and equilibrated to initial flow conditions for 1 min. The quantification of the metabolite was conducted using an API 4000 Q-Trap tandem mass spectrometry system manufactured by Applied Biosystems/MDS Sciex (Foster City, CA) using a Turbo Spray (ESI) source operating in positive ion mode. The mass spectrometer parameters were set at curtain gas, 20; collision gas, medium; ion spray voltage, 5500; ion source gas 1, 60; ion source gas 2, 40; temperature, 400, declustering potential, 70; entrance potential, 10; collision energy, 35; collision cell exit potential, 15. The analyte (C151) and the internal standard (phenacetin) were detected using multiple reactions monitoring (MRM) mode by monitoring the m/z transition from 230.16 to 157.2 and 180.200 to 110.1, respectively. The determination of the product amounts was achieved with a calibration curve ranging from 0 to 2000 nM concentration of C151.
Enzyme inhibition experiments.
Inhibition of C152 metabolism by ketoconazole (KCZ), clotrimazole (CTZ), 4-(4-Chlorobenzyl)pyridine (CBP), and (S)-(+)-N-3-benzylnirvanol (BNVL) was probed with a real-time single wavelength fluorimetric assay described above. The incubation mixture containing 30 μM C152 and 0.01–0.05 μM of Supersomes (by P450 content) or 0.1 – 0.2 mg/ml of HLM (by protein content) was placed into a fluorimetric cell and thermostated at 30° C. The reaction was initiated by adding NADPH to the concentration of 200 μM. The reaction traces were linear for at least 500 sec in the absence of inhibitor. The inhibition experiments were performed by consecutive additions of aliquots of 1 – 10 mM stock solution of inhibitor in acetone. In these experiments, the reaction was initiated in the absence of the inhibitor and monitored for 100–150 s before its first addition. After recording a representative linear trace (100–150 s), the inhibitor concentration was increased, and the monitoring continued for another 100–150 s. One incubation mixture was used to measure the reaction rates at 2–4 increasing concentrations of inhibitor. The duration of one inhibition run did not exceed 500 s. The slope of each segment of the recorded trace was determined by linear approximation, and the fractional inhibition at each inhibitor concentration was determined from the ratio of the slope of the respective segment to the slope of the segment recorded at no inhibitor added. The resulting dependence of the fractional inhibition on the inhibitor concentration was fitted to a hyperbolic equation (Krohn and Link 2003):
| (Eq. 1) |
In this equation, FI and vi are the fractional inhibition and the reaction velocity observed at inhibitor concentration [I], v0 is the reaction rate at no inhibitor added, and Amax is the maximal amplitude of inhibition. IC50 is the concentration of inhibitor at which the inhibition reaches 50% of its maximal amplitude (Amax). In the canonical case of competitive inhibition, the value of IC50 is a function of the inhibition constant (Ki), the concentration of the probe substrate ([S]) and the Michaelis constant (KM) that characterizes its affinity to the enzyme (Cheng and Prusoff 1973):
| (Eq. 2) |
Data analysis.
Analysis of series of spectra obtained in the assays with scanning fluorescence spectroscopy was done with the method of Principal Component Analysis (PCA) (Davydov et al. 1995; Halaka et al. 1985), which was used as a method of global analysis that minimizes the contribution of incidental variation in the shape of the spectra. To quantify the amount of formed products, we used a linear least-squares fitting of the spectra of the first and second principal components by a combination of the prototypical spectra of emission of two major products of N-demethylation of C152 – 7-methylamino-4-trifluoromethyl coumarin (desmethyl-C152, DC152) and C151. These spectral standards were acquired as described under Results. All data analyses were performed using the SpectraLab software (Davydov and others 1995; Davydov et al. 2016), which is available for download at http://cyp3a4.chem.wsu.edu/spectralab.html.
Results
General characterization of the changes in fluorescence of C152 associated with its metabolism.
The structure of Coumarin 152 and two major products of its metabolism by cytochromes P450, 7-Methylamino-4-trifluoromethylcoumarin (Desmethyl-Coumarin 152, DC152) and amino-4-trifluoromethylcoumarin (Coumarin 151, C151), are shown in Figure 1. The fluorescence properties of C152 and C151 are well studied. The spectrum of fluorescence of C152 features a diffuse asymmetric band with the maximum positioned at 504–515 nm, depending on the polarity of the solvent (Brackmann 2000; Nad et al. 2003). The band of fluorescence of C151 is more compact and exhibits a maximum positioned at 480–485 nm (Brackmann 2000; Nad and Pal 2001). In contrast, the mono-demethylated product of C152 metabolism (DC152) is not commercially available, and its fluorescent properties are not characterized. However, by analogy with a closely similar fluorophore, 7-ethylamino-4-trifluoromethyl coumarin (Coumarin 500), we may expect the maximum of fluorescence of DC152 to be positioned at ≥500 nm (Brackmann 2000; Nad and Pal 2003).
Figure 1.
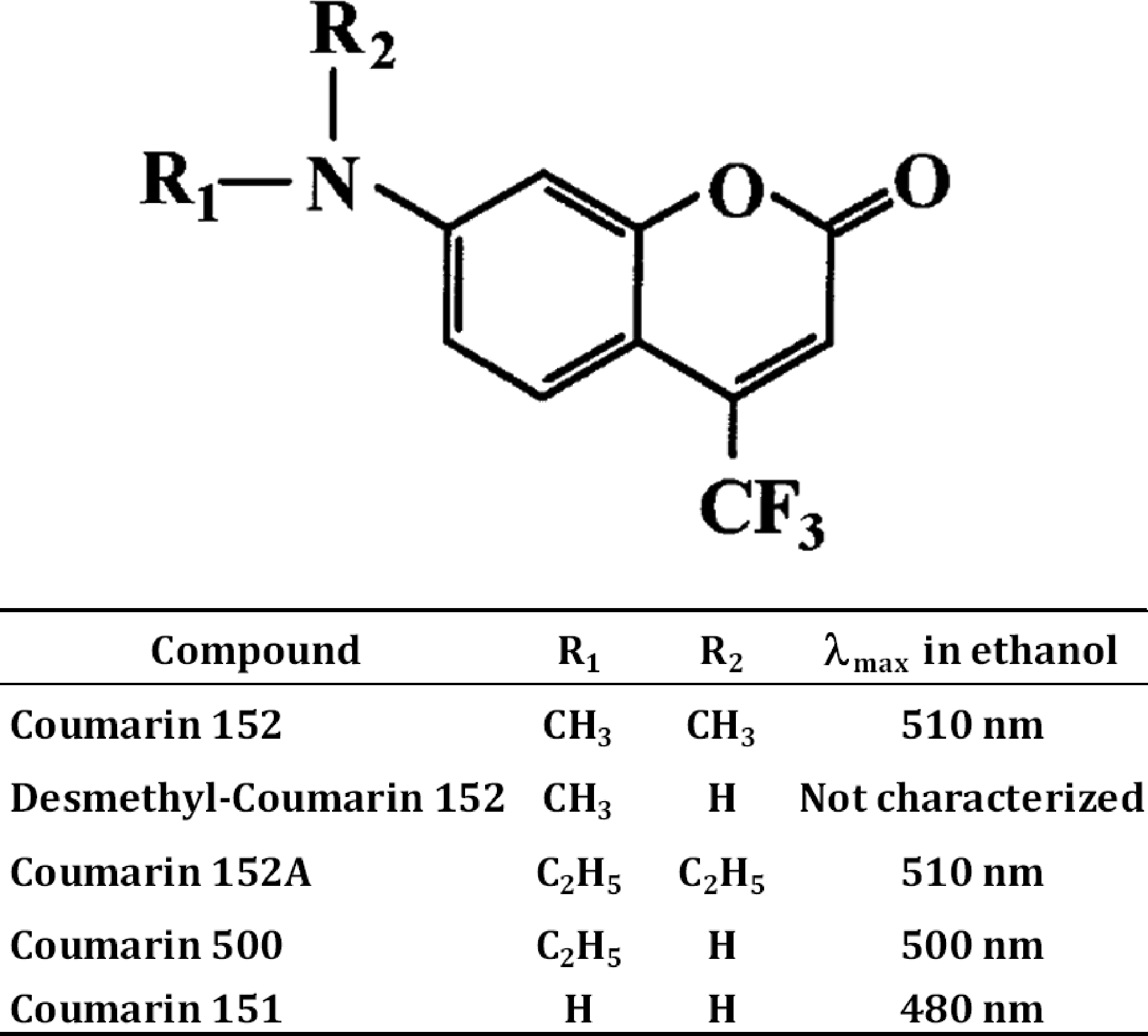
Structure and fluorescence properties of Coumarin 152, its metabolites, and related compounds.
Our experiments with scanning fluorescence spectroscopy demonstrated that the incubation of C152 with HLM in the presence of NADPH results in a substantial increase in fluorescence with the maximum positioned at ~500 nm (Figure 2a). We can, therefore, infer that C151 does not represent the major product of HLM-catalyzed metabolism of C152. The predominant formation of DC152, the mono-demethylated product, is anticipated instead.
Figure 2.
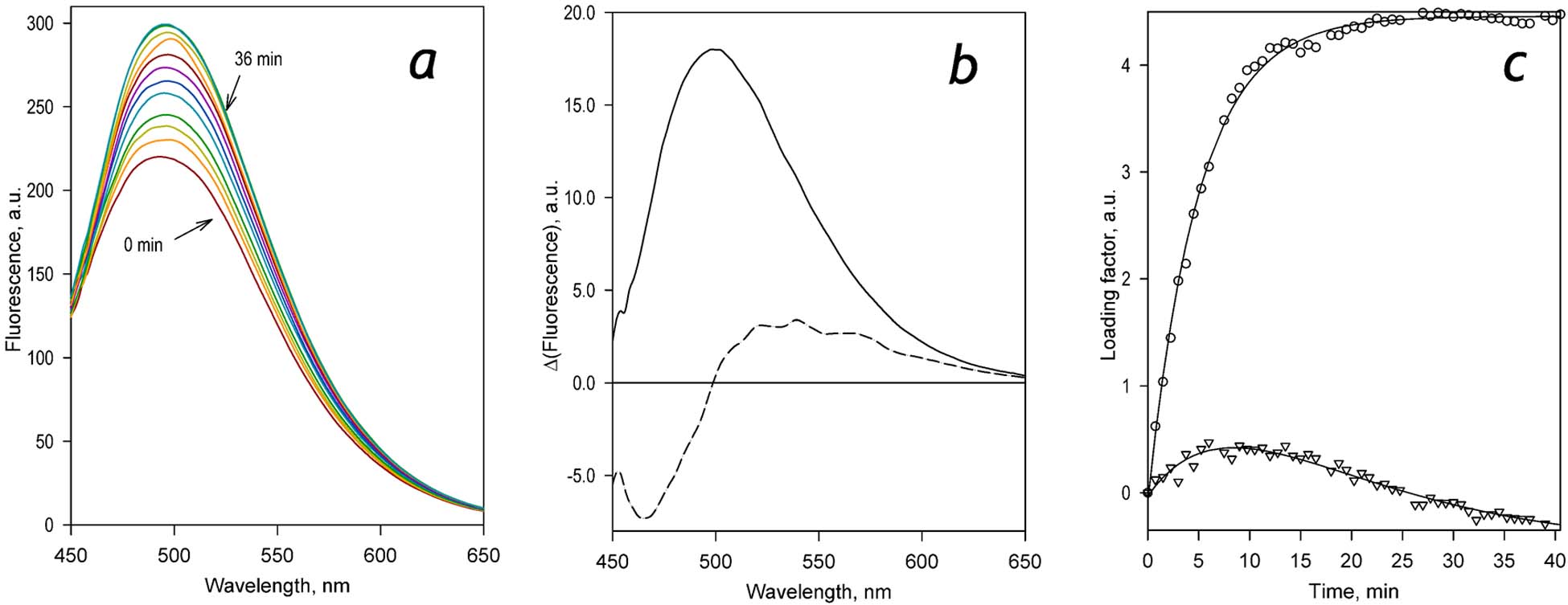
Changes in fluorescence resulting from the metabolism of C152 by HLM. Panel a shows a series of spectra of fluorescence recorded at 0, 0.75, 1.5, 1.25, 3.75, 4.5, 6, 12, 24, and 36 min after adding NADPH to the reaction mixture containing HLM (0.076 μM by the content of CPR) and 29 μM C152. Panels b and c show respectively the spectra and the loading factors of the first (solid line, circles) and the second (dashed line, triangles) principal components derived from the application of PCA to the series of spectra shown in panel a. The lines shown in panel c represent approximations of the time dependencies of the loading factors with a bi-exponential equation. These approximations are displayed solely to emphasize the general trend of the observed changes and have no interpretational value.
The application of the PCA procedure to the series of spectra shown in Figure 2a allowed us to resolve two principal components (PC) that represent the spectral signature of C152 metabolism (Figure 2b). Together these two PCs cover over 99.9% of the changes in fluorescence observed during incubations. The higher-order principal components represented mainly the spectral jitter and did not reveal any spectral features characteristic to the fluorescence of the substrate or the products of its demethylation. While the first PC features a single asymmetric band with the maximum at 500 nm (Figure 2b, solid line), the second PC exhibits a trough centered around 470 nm, crosses the zero line at ~500 nm and displays a maximum positioned at 536 nm (Figure 2b, dashed line).
The two principal components exhibit different kinetics of their loading factors during the incubation. Both of them show a nearly linear increase during the initial 5 – 8 min. However, the increase in the contribution of the first PC reaches its maximum and stabilizes after ~20 min. In contrast, the contribution of the second PC starts to decrease linearly after 10 min of incubation (Figure 2c). This behavior is consistent with the sequential formation of the two products of C152 demethylation – DC152 (mono-demethylated product) and C151 (bis-demethylated product). While the spectrum of the first principal component apparently corresponds to a sum of the spectra of the two products with the predominant contribution of DC152, the second PC represents a difference between the spectra of emission of the mono- and bis-demethylated products (DC152 and C151 respectively). This observation is consistent with the expected positioning of the maxima of fluorescence of C151 and DC152 discussed above. According to this initial analysis of the experiment presented in Figure 2, the formation of DC152 comes to an end after 20 min of incubation. In contrast, the successive formation of the bis-demethylated product continues almost linearly for another 30 min of incubation.
Multiple experiments carried out with HLM in the setup exemplified by Figure 2 at concentrations of C152 varying from 10 to 40 μM all yielded similar results. In all cases, the first principal component exhibited a single band with a maximum positioned at 500–502 nm, and the second PC showed a combination of two oppositely directed bands that crosses the zero line at ~500nm. According to these results, the spectra of the two products of C152 metabolism have an isosbestic point at 500 nm. Therefore the changes in fluorescence at this wavelength may be used for monitoring the overall process of C152 demethylation in a simple single-wavelength assay.
Obtaining spectral standards of fluorescence of C152 and C151 and the setup of the fluorimetric assay of C152 metabolism
Monitoring the increase in fluorescence at 500 nm, the isosbestic point of the spectra of changes in fluorescence caused by the formation of the two successive products of demethylation, makes the single-wavelength assay specific for the first step of metabolism – the formation of mono-demethylated product (DC152). To interpret the observed changes in terms of the molar amount of the formed DC152, we determined the spectra of fluorescence of 1μM C151 and C152 (Figure 3, the spectra shown in solid lines). The difference between the normalized spectra of fluorescence of C151 and C152 obtained in this way (Figure 3, dashed line) was used as the prototypical spectrum of the changes expected upon conversion of 1μM C152 to C151. The value of the difference between the intensities of fluorescence of C151 and C152 at 500 nm was used as a calibration coefficient for calculating the rates of formation of DC152 from the results of the single wavelength fluorimetric assays.
Figure 3.
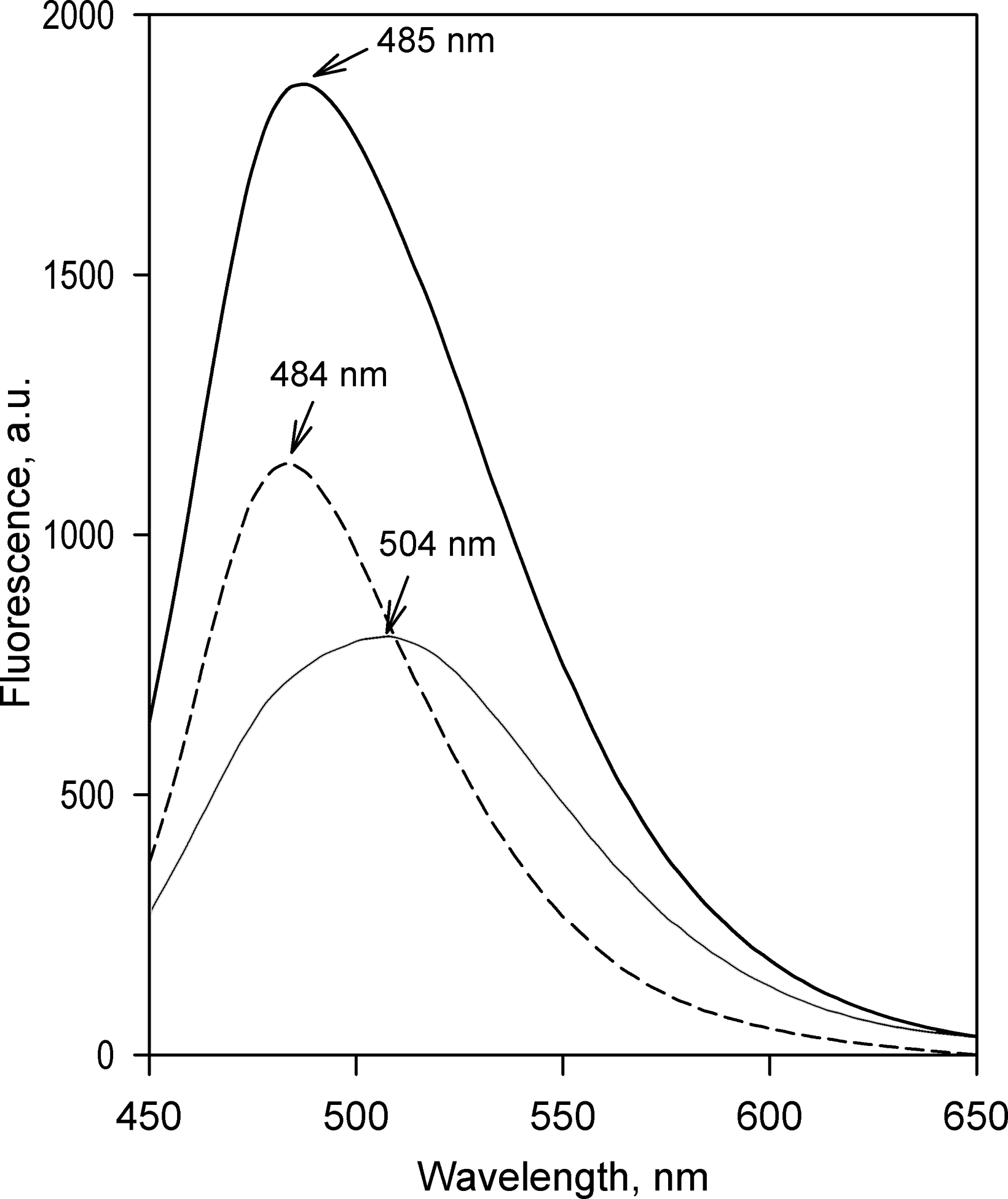
Fluorescence of Coumarin 152 and Coumarin 151. The graph shows the spectra of emission of 1 μM C152 (solid line) and C151 (thin solid line) in 0.1 M Na-Hepes buffer, pH 7.4, containing 60 mM KCl in the presence of HLM-2 (0.33 mg protein/ml). The dashed line shows the difference between the spectra of emission of C152 and C151.
Probing the ability of recombinant human P450 species to metabolize C152
To characterize the ability of individual human cytochrome P450 species to metabolize C152, we investigated its metabolism by recombinant human CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and CYP3A5 using commercial preparations of Supersomes™ containing the respective enzymes. Three of the ten probed cytochrome P450 species - CYP2A6, CYP2D6, and CYP2E1 – showed no detectable activity with C152. Results obtained in the studies with other P450 species are summarized in Table 1 and illustrated in Figure 4a.
Table 1.
Kinetic parameters of metabolism of Coumarin 152 by recombinant SupersomesTM and HLM*
| Microsomal preparation | C152 metabolism (fluorimetric assay) | Formation of C151 (LC-MS) | Fraction of C151 formed,% | |||||
|---|---|---|---|---|---|---|---|---|
| KM or S50, μMa | Na | Vmax, min−1b | KM or S50, μMa | Na | Vmax, min−1b | LC-MS | Fluorimetry | |
| SS(1A2) | 2.9±1.7 | N/A | 5.0±1.1 | 1.7±0.6 | N/A | 0.98±0.12 | 20±2 | 26±7 |
| SS(2B6) | 1.8±0.2 | N/A | 49±11 | 1.0±0.4 | N/A | 1.8±0.4 | 4.0±0.8 | 7.3±3.4 |
| SS(2C8) | 12±6 | 2.6±0.3 | 2.6±0.8 | N/Dc | N/D | N/D | ||
| SS(2C9) | 29±8 | N/A | 3.9±1.2 | N/D | N/D | N/D | ||
| SS(2C19) | 5.7±1.1 | N/A | 7.8±3.4 | 6.7±1.5 | N/A | 0.43±0.03 | 5.6±0.4 | 3.2±2.4 |
| SS(3A4) | 4.5±1.2 | 1.6±0.2 | 17±3 | 6.4±1.1 | 1.4±0.4 | 0.94±0.41 | 5.4±2.4 | 2.5±0.3 |
| SS(3A5) | 19±9 | N/A | 15±4 | N/D | N/D | N/D | ||
| HLM | 4.5±1.5 | 1.8±0.3 | 58±24 | 3.0±0.5 | N/A | 1.3±0.2 | 2.4±0.4 | 5.7±2.0 |
The estimates given in the table correspond to the averages of 2–6 individual measurements. The“±” values are the confidence intervals calculated for p=0.05.
Parameters given in the table were obtained from the fitting of the substrate dependencies of the reaction rate with the Michaelis-Menten or Hill equation. S50 and N are the concentration of half-saturation with the substrate and the Hill coefficient. In the instances when the Michaelis-Menten equation was used, the Hill coefficient is marked as not applicable (N/A).
The values of Vmax given in the table are expressed as moles of C152 metabolized, or moles of C151 formed per mol of enzyme per minute. In the case of HLM, these values are normalized per concentration of CPR, while in the case of Supersomes, the normalization was made on the concentration of P450.
N/D – not determined
Figure 4.
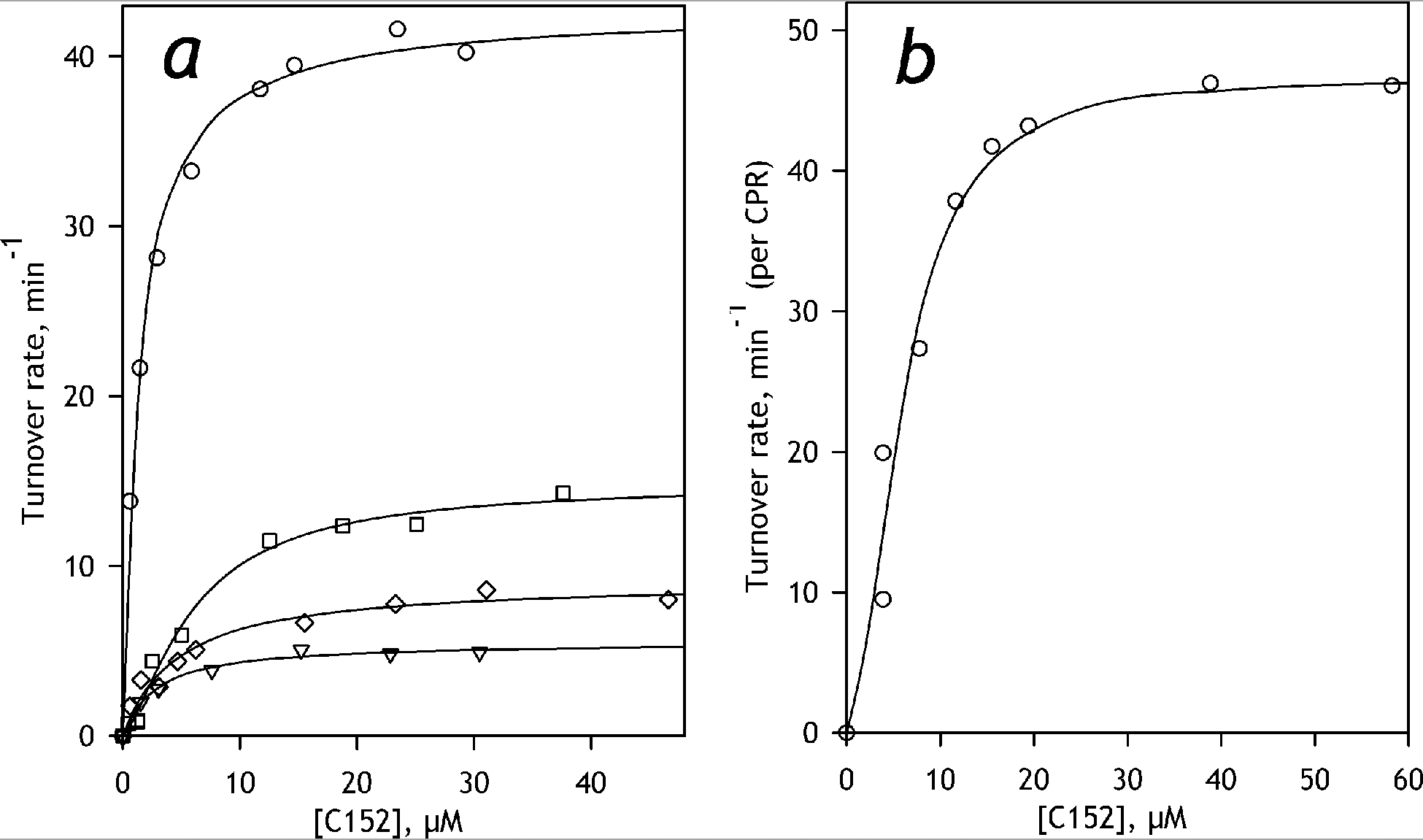
Dependencies of the rate of C152 metabolism on substrate concentration. Panel a shows the representative datasets obtained with SS(2B6) (circles), SS(3A4) (squares), SS(1A2) (triangles) and SS(2C19) (diamonds). The results obtained with HLM are exemplified in panel b. The solid lines represent the results of fitting of the datasets to the Hill (HLM and SS(3A4)) or Michaelis-Menten (SS(2B6), SS(1A2), and SS(2C19)) equations.
The highest rate of metabolism of C152 was exhibited by CYP2B6, for which the turnover numbers with C152 are among the highest observable with drug-metabolizing P450 enzymes with any substrates. A considerably slower, but still quite high rate of turnover was observed with CYP3A4 and CYP3A5, which were followed by CYP2C19. Much lower rates of turnover were exhibited by CYP1A2, CYP2C9, and CYP2C8.
For the majority of the probed P450 species, the dependencies of the rate of C152 turnover on substrate concentration could be adequately approximated with a hyperbolic (Michaelis-Menten) equation (Figure 4a). The two exceptions are CYP3A4 and CYP2C8, which reveal a distinct positive homotropic cooperativity that is frequently observed with these enzymes (Davydov and Halpert 2008; Kaspera et al. 2011; Roberts et al. 2011; Schoch et al. 2008). Four of the seven enzymes capable of metabolism of C152 - CYP2B6, CYP3A4, CYP2C19, and CYP1A2 - exhibited quite similar and rather high affinity to this substrate characterized with the KM or S50 values in the range of 1.8 – 6 μM (Table 1). KM values displayed by CYP2C9 and CYP3A5, as well as the S50 value specific to CYP2C8, were considerably higher (Table 1).
In addition to studying the metabolism of C152 with the fluorimetric assay, we also investigated the rate of formation of C151, the bis-demethylated product, with LC-MS/MS. As seen from the results of these experiments (Table 1), the values of KM or S50 exhibited by probed P450 species are close to those found in the fluorimetric assays. However, the rates of formation of C151 were significantly slower than the overall rates of C152 metabolism. The fraction of the formed C151 over the total amount of C152 metabolized varies in the range of 4 – 20%, depending on the P450 species examined (Table 1). The fraction of C152 converted to C151 by CYP1A2 (20%) was the highest among all P450 species compared, while CYP2B6 displayed the lowest (4%) fraction of C151 formed.
Metabolism of C152 by HLM
The dependencies of the rate of C152 turnover by HLM on the substrate concentration are exemplified in Figure 4b. It should be noted that the rate of metabolism of C152 in HLM in this study was normalized on the concentration of CPR in the microsomal membrane (see Materials and Methods). Given a high excess of cytochromes P450 over CPR in HLM, this normalization was considered as the most appropriate approach for comparing the rates of metabolism in the HLM preparations with different compositions of the cytochrome P450 ensemble.
Dependencies of the rate of metabolism of C152 by HLM reveal positive homotropic cooperativity and could be best approximated with the Hill equation with the Hill coefficient of 1.8 ± 0.3 (Figure 4b). The value of S50 exhibited by HLM was close to the values of KM or S50 obtained with SS(3A4) and SS(2C19) (Table 1). Similar to what was found in our studies with Supersomes, the rate of formation of C151 by HLM was radically lower than the rate of C152 turnover (Table 1). According to LC-MS/MS measurements, only around 2.4% of the total amount of C152 metabolized by HLM in the initial phase of reaction is converted to C151.
Resolving the kinetics of the two steps of C152 N-demethylation with scanning fluorimetry.
As discussed above, the spectra of fluorescence of C151 and DC152 differ considerably as to the position of the maximum of fluorescence. This difference, together with different kinetics of accumulation of these two products of C152 N-demethylation, gives rise to two distinct principal components resolved from the series of spectra of fluorescence recorded in the process of C152 metabolism by HLM (Figure 2). Experiments with scanning fluorimetry carried out with Supersomes containing individual P450 yielded very similar results and allowed to resolve two types of spectral changes (two principal components of the observed changes) with different kinetic behavior.
The general features of the spectra of the two PC obtained with Supersomes were similar to those described above for the experiments with HLM (Figure 2). However, while the second PC reveal no differences between the results obtained with HLM and different preparations of Supersomes, the spectra of the first PC obtained with SS(1A2), SS(2B6), SS(2C19) and SS(3A4) exhibit some variation in the position and the width of the band. This difference, which is illustrated in Figure 5 for the pair of SS(1A2) and SS(2B6), is commensurate with the difference between the P450 species in the fraction of C151 formed in C152 metabolism. Thus, CYP1A2, which was found to be the most efficient in the catalysis of the second demethylation step (Table 1), is characterized with the spectrum of the first PC centered at 494 nm, while the maximal changes in fluorescence observed with CYP2B6 were observed at 504 nm, close to the expected position of the maximum of fluorescence of DC152 (Figure 5).
Figure 5.
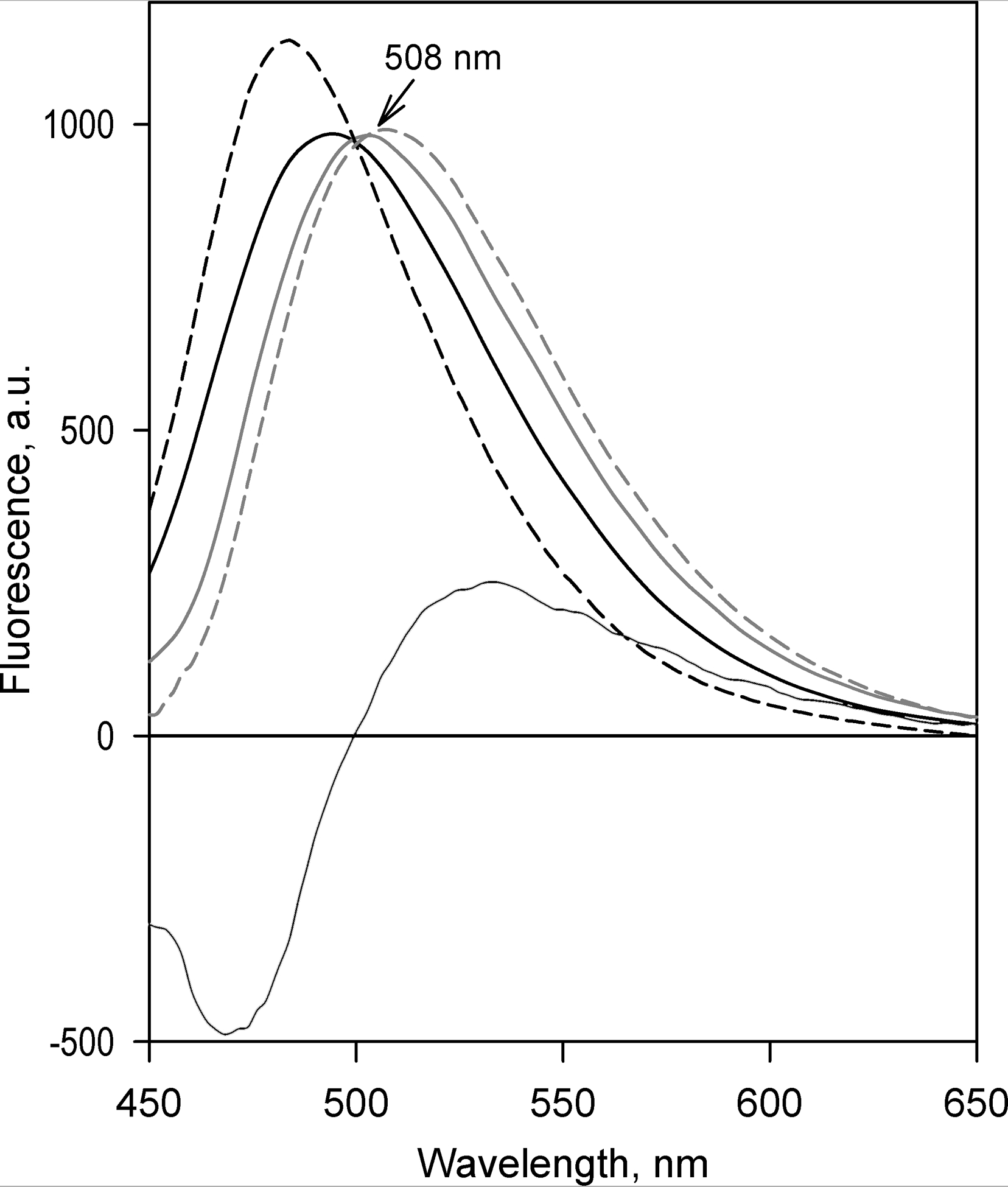
Spectral properties of fluorescent products of C152 metabolism explored with Principal Component Analysis. The solid lines show the spectra of the first principal component obtained with SS(1A2) (black) and SS(2B6) (gray). Thin solid line indicates the spectrum of the second principal component averaged over the results obtained with all probed preparations of Supersomes. The dashed gray line shows the prototypical spectrum of the changes in fluorescence, reflecting the conversion of C152 into DC152. The dashed black line represents the difference in the spectra of fluorescence of C151 and C152 (see Figure 3).
We may, therefore, assume that the spectrum of the first PC obtained with each particular P450 species represents a sum of the spectra corresponding to the formation of C151 and DC152 taken in the proportion determined by the partitioning of C151 in the products of C152 metabolism. The other way round, the spectrum of the second PC may be interpreted as a difference between the spectra of fluorescence of C151 and DC152. Using these inferences in combination with the estimates for the partitioning of C151 in the products of C152 metabolism determined with LC-MS/MS (Table 1), we may obtain an approximation of the spectrum of the increase in fluorescence caused by the conversion of C152 to DC152. The respective spectral standard obtained by averaging the results of calculations performed for each probed P450 species is shown in Figure 5 in a dashed red line.
The combination of the newly obtained spectral standard of DC152 with the experimentally determined spectrum representing the conversion of C152 into C151 (Figure 5, dashed black line) can now be used for quantitative analysis of the series of spectra recorded in the process of metabolism of C152. Approximation of the spectra of the first two PC with the set of these prototypical spectra allows for resolving the kinetics of accumulation of two subsequent products of C152 metabolism, as illustrated in Figure 6 for the case of SS(1A2).
Figure 6.
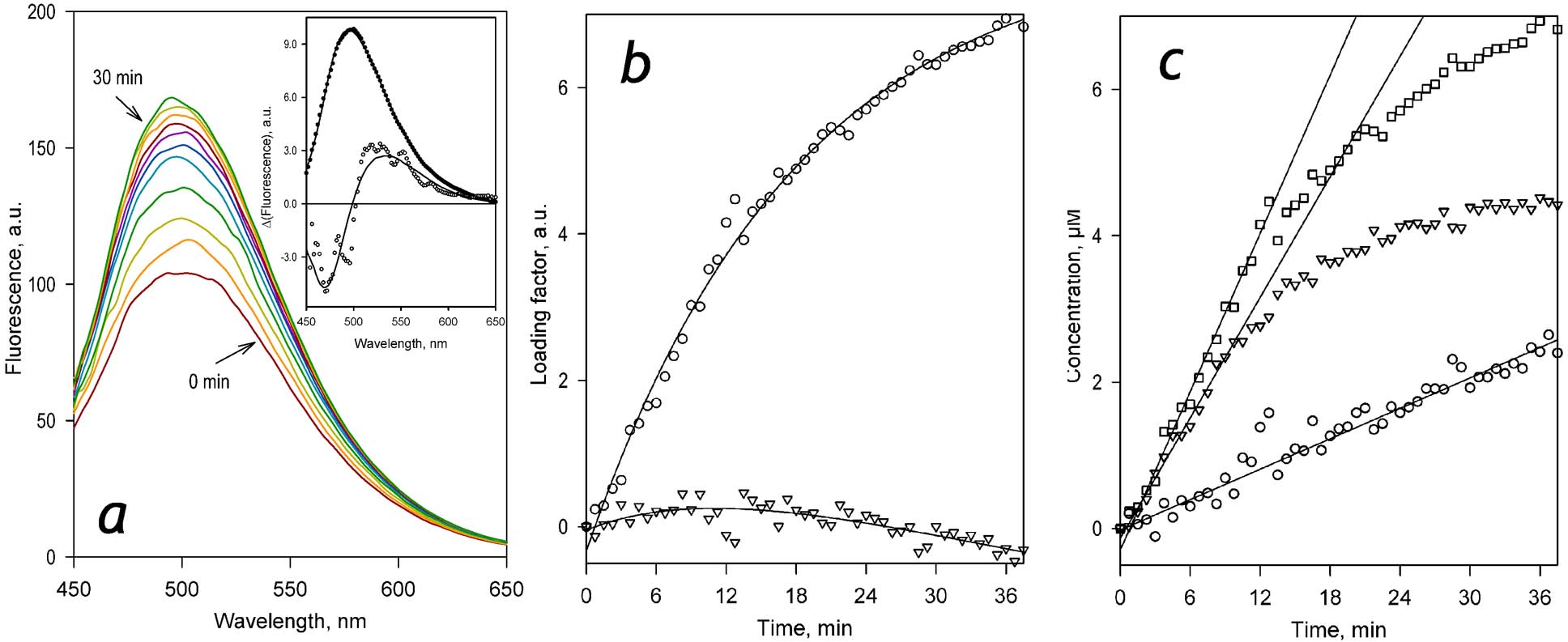
Analysis of the kinetics of accumulation of two sequential products of C152 metabolism with scanning fluorimetry and PCA. Panel a shows a series of spectra of fluorescence recorded in 3 min intervals during the incubation of SS(1A2) (0.076 μM P450) with 29 μM C152 in the presence of NADPH. The inset shows the spectra of the first (filled circles) and the second (open circles) principal components derived with PCA. Solid lines represent the approximations of these spectra with the prototypical spectra of fluorescence of DC152 and C151 (see Figure 5). Panel b shows the time dependencies of the respective loading factors. Solid lines shown in this panel represent the approximations of the time dependencies of the loading factors with a bi-exponential equation. These approximations are presented solely to emphasize the general trend of the observed changes and have no interpretational value. Panel c shows the kinetic curves of the changes in the concentrations of DC152 (triangles), C151 (circles), and the total of the two products (squares) obtained from the results of PCA. Solid lines illustrate the initial velocities of product accumulation derived from the approximation of the initial fragments of the kinetic curves with a first-order polynomial.
As seen from the kinetic traces shown in Figure 6c, the initial steady-state segment of where the kinetic traces of accumulation of both DC152 and C152 remain linear lasts for approximately 10 min. After that time, the rate of formation of DC152 starts to decrease and almost nullifies after 30 min of incubation. At the same time, the production of DC152 continues at a constant rate. Initial proportionality of the velocities of DC152 and C151 formation may indicate that the fraction of C151 formed in this initial phase reflects the probability of a sequential two-step demethylation that takes place without dissociation of the enzyme-substrate complex.
Importantly, the maximal level of formation of DC152 (~4 μM) is much lower than the initial concentration of the substrate (28 μM). Therefore, the inhibition of the production of DC152 after the initial linear phase cannot be explained by substrate depletion and may instead reflect the inhibition of turnover of C152 due to its competition with the product of mono-demethylation (DC152).
Similar regularities were also observed with other probed recombinant P450 and HLM, although the fraction of formed C151 and the maximal amplitude of the first demethylation step (formation of DC152) varies considerably between different P450 species (Figure 7). The fraction of the production of C151 in the total rate of C152 metabolism determined with scanning fluorimetry was in all cases in a good agreement with that estimated from LC-MS/MS experiments (Table 1). Both methods point CYP1A2 as the most efficient catalyst of the second demethylation step (20–25% of C151 formed), while with the other probed recombinant P450 species (CYP2B6, CYP2C19, and CYP3A4) and HLM the fraction of the formed C151 was much lower (see Table 1).
Figure 7.
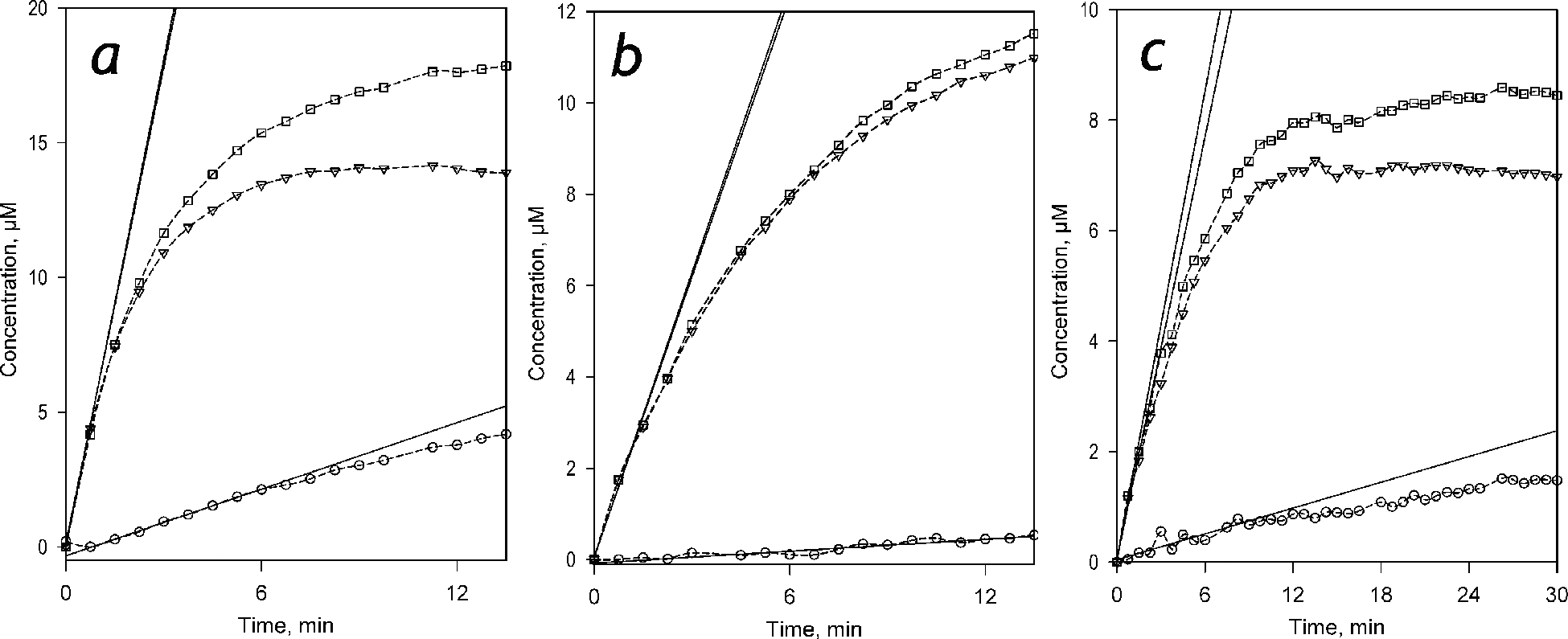
Kinetics of accumulation of the products of C152 metabolism derived from the application of PCA to the results of experiments with scanning fluorimetry. Panels a, b and c represent the results obtained with SS(2B6), SS(3A4), and HLM, respectively. The concentrations of Supersomes and HLM in the incubation mixture were equal to 0.076 μM P450 and 1.5 mg protein/ml (or 0.076 μM CPR), respectively. The plots show the kinetic curves of the changes in the concentrations of DC152 (triangles), C151 (circles), and the total of the two products (squares) obtained from the results of PCA. Solid lines illustrate the initial velocities of product accumulation derived from the approximation of the initial fragments of the kinetic curves with the first or second-order polynomial.
Characterization of the composition of the cytochrome P450 pool in HLM and estimation of the apparent role of the individual P450 species in C152 metabolism
The preparation of the HLM used in this study was characterized by determining the content of NADPH-cytochrome P450 reductase and 11 major cytochrome P450 species, namely CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C18, CYP2C19, CYP2E1, CYP3A4, and CYP3A5, with mass-spectrometry. The total content of cytochromes P450 and the concentration of NADPH-cytochrome reductase (CPR) were also quantified with absorbance spectroscopy and determination of the rate of NADPH-dependent reduction of cytochrome c, respectively.
The degree of coverage of the P450 concentration determined by absorbance spectroscopy by the total of P450 contents determined by LC-MS/MS varied from 31 to 107% in 11 individual LC-MS assays with the mean coverage value of 53±14%. Similarly, the content of CPR determined by LC-MS ranged from 31 to 95% from the content estimated from the specific activity of the enzyme with the mean coverage value of 55 ± 15%. The variations in the recovery of P450 and CPR content always occurred in a strict parallel and may be explained by differences in the depth of proteolysis. These variations had no significant effect on the fractional content of the individual P450 species determined from the results of each replicate taken individually. The results of our analysis of the composition of the P450 pool in HLM are summarized in Table 2.
The fractional content of P450 species metabolizing C152 (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP3A4, and CYP3A5) estimated by LC-MS/MS in combination with their specific turnover numbers and affinities determined with recombinant enzymes (Table 1) allowed us to evaluate their expected fractional contribution into C152 metabolism in HLM. In these calculations, we followed the “total normalized rate” (TNR) approach proposed by Rodrigues (Rodrigues 1999) and further developed by others (Chen et al. 2011; Venkatakrishnan et al. 2000; Venkatakrishnan et al. 2001). This approach is based on a proportional projection of the presumed activities of the individual P450 enzymes to their ensemble with known composition. Normalized rates of C152 metabolism were calculated by multiplying the specific rates by the fractional content of the individual P450 species in the total of seven C152-metabolizing enzymes. The value of expected participation was calculated from the ratio of the respective normalized rate to the sum of all seven normalized rates. Results of our projections calculated for saturating C152 concentration and the concentration of 30 μM, which is near-saturating for the four major C152-metabolizing P450 species (CYP2B6, CYP3A4, CYP2C19, and CYP1A2), are presented in Table 3. As seen from these values, the primary involvement in C152 metabolism may be expected for CYP3A4, which is projected to be responsible for over 80% of the substrate turnover. The role of the secondary importance is predicted for CYP2B6 and CYP2C9, which were calculated to be responsible for around 7% of C152 each at substrate saturation. The expected involvement of CYP2C9, however, decreases to 4% at 30 μM C152 due to the lower affinity of this enzyme to the substrate.
Table 3.
Calculation of the projected involvement of P450 species in the metabolism of C152 in HLM.
| P450 species | Fraction in the P450 poola | Fraction in the total of seven C152-metabolizing P450 species | At saturation of all P450 species | At 30 μM C152 | ||||
|---|---|---|---|---|---|---|---|---|
| Specific rate, min−1b | Normalized rate, min−1c | Expected participation, %d | Specific rate, min−1e | Normalized rate, min−1. | Expected participation, %d | |||
| CYP1A2 | 0.055 | 0.083 | 5.0 | 0.412 | 3.10 | 4.5 | 0.375 | 3.08 |
| CYP2B6 | 0.012 | 0.019 | 49.3 | 0.914 | 6.89 | 46.6 | 0.863 | 7.10 |
| CYP2C8 | 0.007 | 0.010 | 2.2 | 0.022 | 0.17 | 1.9 | 0.020 | 0.16 |
| CYP2C9 | 0.166 | 0.251 | 3.9 | 0.981 | 7.39 | 2.0 | 0.496 | 4.08 |
| CYP2C19 | 0.003 | 0.004 | 7.8 | 0.031 | 0.24 | 6.5 | 0.026 | 0.22 |
| CYP3A4 | 0.413 | 0.625 | 17.3 | 10.8 | 81.3 | 16.5 | 10.3 | 84.75 |
| CYP3A5 | 0.005 | 0.008 | 15.3 | 0.120 | 0.91 | 9.5 | 0.074 | 0.61 |
The fractions of individual P450 species in the total pool of P450 species under analysis as shown in Table 2.
Vmax values determined with the recombinant enzymes in Supersomes, as shown in Table 1.
Normalized rates of C152 metabolism are calculated by multiplying the specific rates by the fractional content of the individual P450 species in the total of seven C152-metabolizing enzymes.
The value of expected participation is calculated from the ratio of the respective normalized rate to the sum of all seven normalized rates. This parameter is equivalent to the “% TNR” (Rodrigues 1999).
The values of the reaction rate at 30 μM C152 are calculated from the parameters of Hill or Michaelis equation for each particular P450 species shown in Table 1.
Determination of involvement of individual P450 species in C152 metabolism by inhibitory analysis
To probe the validity of the above projections, we performed an inhibitory analysis to examine the actual participation of individual P450 species in C152 metabolism in HLM. Analyzing the parameters of substrate dependencies of the rate of C152 metabolism by recombinant P450 enzymes and HLM (Table 1) suggests CYP2B6, CYP3A4, CYP2C9, and CYP1A2 as possible catalysts. The KM or S50 values exhibited by CYP2C9, CYP2C8, and CYP3A5 are much higher than the S50 value observed with HLM that makes their substantial involvement unlikely. From the other side, the low yield of C151 as a product of C152 demethylation by HLM contradicts any significant role of CYP1A2, the most potent catalyst of the second demethylation step. Therefore, our analysis was focused on probing the role of CYP3A4, CYP2C19 and CYP2B6 with the use of their specific inhibitors - clotrimazole and ketoconazole (CYP3A4), 4-(4-chlorobenzyl)pyridine (CYP2B6) and (+)-N-3-benzylnirvanol (CYP2C19). The results of theses experiments are summarized in Table 4.
Table 4.
Inhibition of Coumarin 152 metabolism in recombinant Supersomes and HLM*
| Micro-somes | KCZ | CTZ | CBP | BNVL | ||||
|---|---|---|---|---|---|---|---|---|
| IC50, μM | Inhi-bition, % | IC50, μM | Inhi-bition, % | IC50, μM | Inhi-bition, % | IC50, μM | Inhi-bition, % | |
| SS(2B6) | 9.5±2.8 (0.03) | 96±7 | 1.23±0.24 (0.02) | 100 | 0.038±0.016 (<10−3) | 96±2 | 8.5±2.3 (<10−5) | 81±7 |
| SS(2C19) | 2.2±1.1 (0.52) | 84±9 | 0.20±0.11 (0.62) | 100 | 215±118 (0.87) | 87±7 | 0.48±0.12 (0.65) | 89±14 |
| SS3A4 | 1.9±0.7 (0.82) | 84±6 | 0.015±0.007 (0.002) | 97±5 | 225±73 (0.99) | 92±8 | 27±3 (<10−7) | 59±10 |
| HLM | 1.9±0.2 | 89±7 | 0.24±0.05 | 71±2 | 225±30 | 88±12 | 0.51±0.03 | 57±10 |
The estimates given in the table correspond to the averages of 2–5 individual measurements. The“±” values are the confidence intervals calculated for p=0.05. The values given in parentheses represent the results of the Students’ T-test for the hypothesis of equality of the respective IC50 values to those observed with HLM. The instances where the probability that this hypothesis is valid is higher than 0.5 are highlighted in bold.
Ketoconazole (KCZ) is widely used as a specific inhibitor of CYP3A enzymes (Greenblatt et al. 2011; Pelkonen et al. 2008). According to Greenblatt and co-authors (Greenblatt et al. 2010), the geometric mean of Ki estimates for CYP3A4/KCZ pair found in literature is equal to 0.1 μM (90% confidence interval: 0.07 to 0.15 μM). The IC50 value derived from our experiments with inhibition of C152 metabolism by recombinant CYP3A4 is equal to 1.9 μM (Table 4), which corresponds to the Ki value of 0.21 μM. Therefore, while being at the high edge of the values observed with other probe substrates, our estimate of Ki is in a satisfactory agreement with the known data on CYP3A4 inhibition by KTZ.
However, besides inhibiting C152 metabolism by CYP3A4, KTZ revealed a high potency to inhibit the metabolism of this substrate by CYP2C19 (IC50=2.2 μM. Ki =0.4 μM, Table 4). Therefore, to discriminate between the metabolic roles of CYP2C19 and CYP3A4, we complemented the experiments with KCZ with studying the effect of clotrimazole (CTZ), which is reported to be a more potent and selective inhibitor of CYP3A4 than KCZ (Zhang et al. 2002). Results of our experiments with inhibition of C152 demethylation in Supersomes by CTZ confirmed a much higher affinity of this inhibitor to CYP3A4 as compared to both CYP2B6 and CYP2C19.
KCZ inhibition experiments with HLM yielded IC50 value similar to that observed with SS(3A4) and SS(2C19), but much lower than specific to CYP2B6. Inhibition experiments with CTZ are illustrated in Figure 8a. These results deny any noticeable role of CYP3A4 in C152 metabolism and suggest CYP2C19 as the principal P450 enzyme responsible for it.
Figure 8.
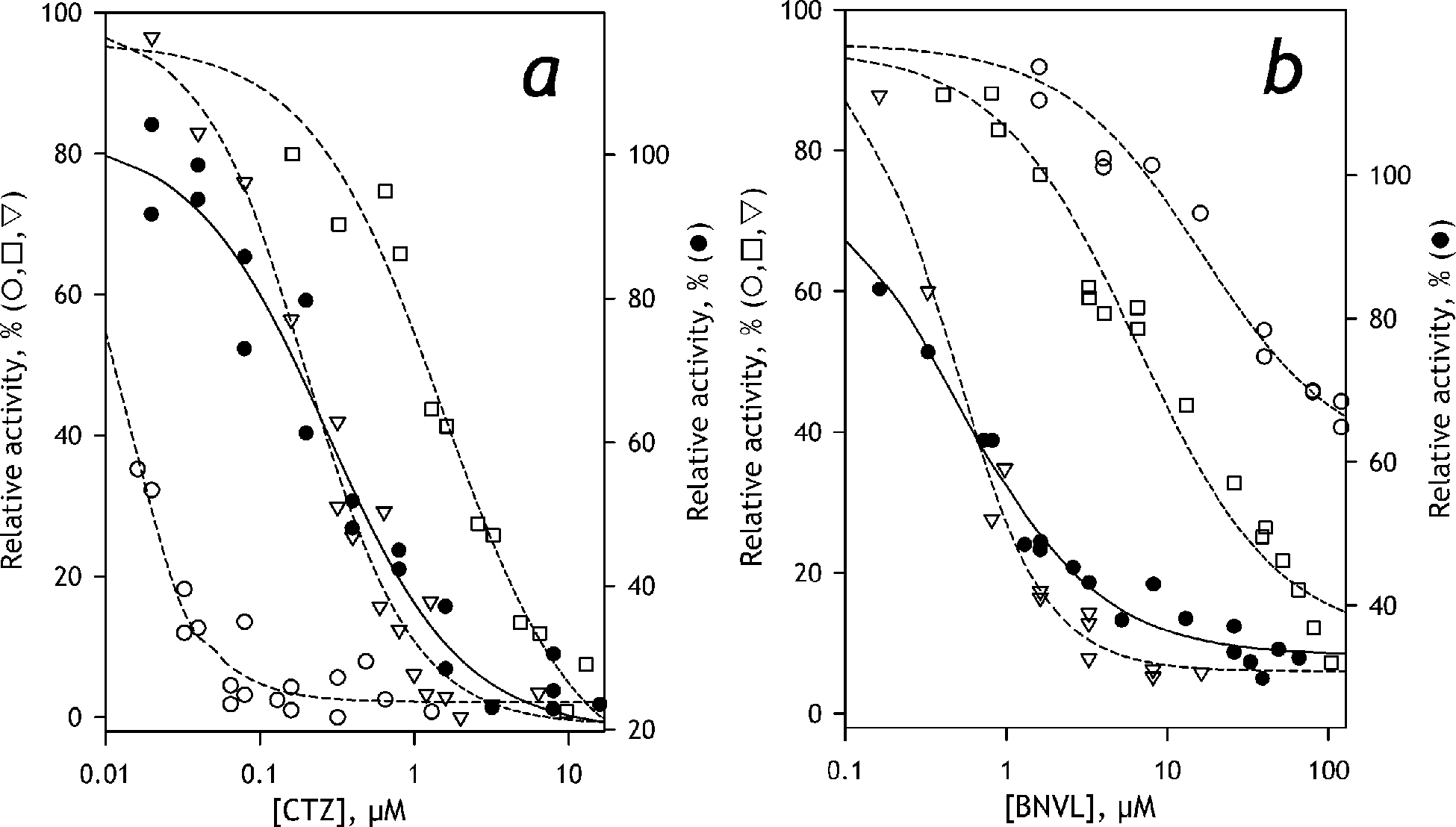
Inhibition of C152 demethylation by clotrimazole (a) and (+)-N-3-benzylnirvanol (b). The results obtained with SS(3A4), SS(2C19), and SS(2B6) are shown in open circles, triangles, and squares, respectively. The results obtained with HLM are shown in closed circles. The datasets exemplified in the plots represent combinations of 2–4 individual experiments. The lines (dashed for Supersomes and solid for HLM) show the approximations of the datasets with Eq. 1. The ordinates for the datasets obtained with HLM are shifted down for better clarity of the graphs
To probe this inference further, we used (+)-N-3-benzylnirvanol (BNVL) as a selective inhibitor of CYP2C19 (Suzuki et al. 2002; Walsky and Obach 2003). The results of these experiments are shown in Figure 8b. In compliance with the previous reports, our experiments with recombinant P450 enzymes demonstrated a much higher affinity of this inhibitor to CYP2C19, as compared to CYP3A4 and CYP2B6 (Figure 8b, Table 4). As illustrated in Figure 8b, BNVL efficiently inhibits C152 metabolism by HLM with IC50 similar to that specific to SS(2C19). Although these results convincingly demonstrate the primary role of CYP2C19, the fact that the maximal depth of BNVL inhibition in HLM did not exceed 60% (Table 4), suggest some involvement of other P450 species.
Probing the role of CYP2B6, the enzyme exhibiting the tightest affinity and the highest turnover number with C152, we used 4-(4-chlorobenzyl)pyridine as the selective inhibitor (Korhonen et al. 2007). Probing this inhibitor with recombinant CYP2B6, CYP2C19, and CYP3A4 enzymes, we confirmed its high affinity to CYP2B6. Experiments with HLM yielded the IC50 value much higher than it is observed with CYP2B6 and similar to that is characteristic to CYP2C19 and CYP3A4 (Table 4). Therefore, any sizable contribution of CYP2B6 into the metabolism of C152 is unlikely.
In summary, our inhibition experiments did not confirm the estimates based on the proportional projection of the properties of the individual P450 enzyme (Table 3). Despite the low content of CYP2C19 in HLM and its projected involvement in C152 metabolism of only 0.24%, this enzyme appears to be responsible for the significant part of C152 turnover in HLM.
Discussion
C152 as a fluorogenic substrate for human drug-metabolizing cytochromes P450
In this study, we thoroughly characterize C152 as a fluorogenic substrate for human drug-metabolizing cytochromes P450 and demonstrate its value as a model of drugs with broad isoform selectivity in the studies of functional integration in cytochrome P450 ensemble in HLM. C152 and its bis-demethylated metabolite, Coumarin 151 (C151), are commercially available chemicals, which are widely used as laser dyes. Similar to that observed with the Nile Red, another dimethylamine P450 substrate (Lampe et al. 2008), the metabolism of C152 occurs through two sequential N-demethylation steps. A combination of scanning fluorescence spectroscopy with determining the rate of the formation of C151 with LC-MS/MS allowed us to discriminate between the two major products with different fluorescent properties – mono-demethylated DC152 and bis-demethylated C151. We demonstrate that in the initial linear phase of the reaction, DC152 represents the predominant product of C152 metabolism in both HLM and model microsomes containing recombinant P450 enzymes.
Establishing a protocol for monitoring C152-demethylation activity, we demonstrated that the increase in fluorescence intensity at 500 nm with excitation at 405 nm is characteristic of the formation of DC152. This approach may be used in both real-time setup with continuous monitoring or in single-point assays. The only difference of the protocol of measurements with C152 from most other fluorimetric assays of P450 activity is that the determination of the molar amounts of the formed product requires a “double calibration.” Due to a significant background fluorescence of C152 and its changes upon penetration into the membrane, the fluorescence intensity of both C152 and C151 must be determined in the conditions of the activity assays (i.e., in the presence of the microsomal preparations) and the difference between these intensities should be used as a scaling factor.
Our results suggest an essential revision to the conclusions drawn from the early study of Santosh Kumar (Kumar 2007). In this study, which was performed in reconstituted systems with purified proteins, the rate of C152 turnover by CYP2C9 and CYP2C19 was reported to be much higher as compared to the rates observed with human CYP2C8, CYP2B6, CYP3A4, CYP3A5, and CYP2E1. In contrast, our results obtained in microsomes containing recombinant enzymes demonstrate that the highest rate of C152 turnover is exhibited by CYP2B6, which is followed by CYP3A4, CYP3A5, CYP2C19, CYP1A2, CYP2C9 and CYP2C8 listed in the order of decreasing activity. Our study demonstrates that, despite significant differences in the rate of C152 turnover, CYP2B6, CYP1A2, CYP2C19, and CYP3A4 displayed similarly high affinities to this substrate.
According to our results, C152 may be used as a high affinity and high turnover fluorogenic P450 substrate for the studies with HLM. Despite the lack of specificity of C152 for CYP2C enzymes, which was suggested by the earlier study (Kumar 2007), this fluorogenic probe may be useful in studying the functional interrelationship between P450 enzymes in HLM as an example of a substrate that has equally high binding affinities to most major drug-metabolizing P450 species.
Severe nonadditivity in the properties of the individual members of the cytochrome P450 ensemble revealed with C152
To probe the validity of the commonly used “proportional projection” or “total normalized rate” practice (Brian et al. 2016; Rodrigues 1999) in assigning the role of individual P450 species in the metabolism of drugs with broad isoform selectivity, we determined the composition of the ensemble of 11 major drug-metabolizing P450 enzymes in the HLM preparation used in this study. The general picture revealed in this analysis well agrees with the established knowledge on the isoform distribution in the P450 pool (Rodrigues 1999; Shimada et al. 1994; Zhang and others 2016). A notable difference, however, is the amount of CYP2A6 (22%), which was found to be considerably higher than it is usually presumed (~4–13% (Rodrigues 1999; Shimada and others 1994). This observation agrees well with the recent data of Wang and co-authors, who reported CYP2A6 as the most abundant P450 species based on the proteomic analysis of 109 individual HLM samples (Wang et al. 2020).
Calculating the projected involvement of each of the C152-metabolizing P450 enzymes in observed turnover with this substrate in HLM, we predicted the principal role of CYP3A4 in C152 metabolism (Table 3). Although the rate of N-demethylation of C152 exhibited by CYP2B6 is higher than that is specific to CYP3A4, the activity of the latter enzyme was projected to predominate due to its high abundance in HLM. Although the rate of C152 metabolism by CYP2C9 is low, this enzyme was also expected to be responsible for 4–7% of the overall turnover due to its substantial amount in HLM. The role of other C152-metabolizing P450 enzymes was predicted to be quite insignificant (Table 3).
To our surprise, the results of the inhibitory analysis did not confirm these expectations. The experiments with BNVL, a specific inhibitor of CYP2C19, demonstrate a dominant role of this enzyme in C152 turnover in HLM. In contrast, no significant contribution from CYP3A4 and CYP2B6 was revealed in the studies with their specific inhibitors (Table 4).
These results provide a spectacular example of nonadditivity in the properties of individual P450 species constituting the multienzyme system of drug metabolism in HLM. Predominance of CYP2C19 in the metabolism of C152 demonstrates a fundamental inefficiency of the “proportional projection” approach for identifying the role of individual P450 species in the metabolism of drugs with broad isoform selectivity. It provides a convincing illustration of a tight integration of the individual functionalities of multiple cytochrome P450 species constituting the drug-metabolizing ensemble.
Our observations are in good agreement with the concept of functional integration of multiple P450 species via their protein-protein interactions and formation of heteromeric P450 complexes in the microsomal membrane (Davydov 2011; Davydov 2016; Davydova and others 2019). According to this concept, a significant part of the cytochrome P450 pool in HLM is deposited in the inactive (“latent”) positions in heteromeric complexes of multiple P450 species. Availability of a particular cytochrome P450 for interaction with the reductase and catalytic activity is thought to be a complex function of the preferences of individual species for occupying the “active” and “latent” positions, the composition of the P450 pool and the presence of selective substrates of individual P450 species. These selective substrates are presumed to recruit their specific metabolizers for efficient catalysis (Davydov 2011; Davydov 2016; Davydova and others 2019).
Conclusion
In summary, our results demonstrate that C152 may be used as a high affinity and high turnover fluorogenic P450 substrate for in vitro studies of human drug metabolism. Despite the lack of specificity of C152 for CYP2C enzymes, which was suggested by the earlier study (Kumar 2007), this fluorogenic probe may be useful in studying the functional interrelationship between P450 enzymes as an example of a substrate that has equally high binding affinities to several major drug-metabolizing P450 species. Furthermore, an easy and reliable method for real-time monitoring of the formation of two sequential products of C152 N-demethylation with scanning fluorimetry introduced in our study provides a useful approach for in-depth investigation of kinetic mechanisms of P450-supported N-demethylation of dialkylamines.
Another, and, perhaps, more important aspect of our study is in providing a striking example where the specific activity of each of the P450 enzymes in HLM with a given substrate cannot be directly predicted from their rates of turnover measured with the individual enzymes, but rather represents a complex function of the overall composition of the P450 pool. Our results demonstrate a high informational value of the studies of the metabolism of polyspecific cytochrome P450 substrates in HLM and their utility in revealing the functional interrelationships in the cytochrome P450 ensemble. This study points out Coumarin 152 as a useful fluorogenic probe substrate for this kind of study.
Acknowledgments
This research was supported by the National Institute On Alcohol Abuse And Alcoholism of NlH under Award Number R21AA024548. The authors are grateful to Jeffrey P. Jones (WSU) for research support, assistance in obtaining and analyzing LC-MS/MS data, and continuous interest in this study. The authors also acknowledge the support from The State Academies of Sciences of the Russian Federation provided for the analysis of HLM samples with mass-spectroscopy within the framework of the Fundamental Scientific Research Program for 2013-2020.
References
- Brackmann U, (2000). Lambdachrome® Laser Dyes, 3rd Edition Goettingen, Germany: Lambda Physik AG; · [Google Scholar]
- Brian W, Tremaine LM, Arefayene M, de Kanter R, Evers R, Guo YY, Kalabus J, Lin W, Loi CM, Xiao GQ. (2016). Assessment of drug metabolism enzyme and transporter pharmacogenetics in drug discovery and early development: perspectives of the I-PWG. Pharmacogenomics, 17, 615–31. [DOI] [PubMed] [Google Scholar]
- Chen YA, Liu LL, Khanh N, Fretland AJ. (2011). Utility of Intersystem Extrapolation Factors in Early Reaction Phenotyping and the Quantitative Extrapolation of Human Liver Microsomal Intrinsic Clearance Using Recombinant Cytochromes P450. Drug Metab. Disp, 39, 373–82. [DOI] [PubMed] [Google Scholar]
- Cheng YC, Prusoff WH. (1973). Relationship between the inhibition constant and the concentration of inhibitor which causes 50 percent inhibition of an enzymatic reaction. Biochem Pharmacol, 22, 3099–108. [DOI] [PubMed] [Google Scholar]
- Davydov DR. (2011). Microsomal monooxygenase as a multienzyme system: the role of P450-P450 interactions. Expert Opin. Drug Metab. Toxicol, 7, 543–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov DR. (2016). Molecular Organization of the Microsomal Oxidative System: a New Connotation for an Old Term. Biochemistry Moscow-Supplement Series B-Biomedical Chemistry, 10, 10–21. [Google Scholar]
- Davydov DR, Davydova NY, Rodgers JT, Rushmore TH, Jones JP. (2017). Toward a systems approach to the human cytochrome P450 ensemble: interactions between CYP2D6 and CYP2E1 and their functional consequences. Biochem. J, 474, 3523–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov DR, Deprez E, Hui Bon Hoa G, Knyushko TV, Kuznetsova GP, Koen YM, Archakov AI. (1995). High-pressure-induced transitions in microsomal cytochrome P450 2B4 in solution - evidence for conformational inhomogeneity in the oligomers. Arch. Biochem. Biophys, 320, 330–44. [DOI] [PubMed] [Google Scholar]
- Davydov DR, Halpert JR. (2008). Allosteric P450 mechanisms: multiple binding sites, multiple conformers or both? Expert Opin. Drug Metab. Toxicol, 4, 1523–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov DR, Yang ZY, Davydova N, Halpert JR, Hubbell WL. (2016). Conformational mobility in cytochrome P450 3A4 explored by pressure-perturbation EPR spectroscopy. Biophys. J, 110, 1485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydova NY, Dangi B, Maldonado MA, Vavilov NE, Zgoda VG, Davydov DR. (2019). Toward a systems approach to cytochrome P450 ensemble: interactions of CYP2E1 with other P450 species and their impact on CYP1A2. Biochem. J [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Gao J, Wang T, Tian X, Gao N, Zhou J, Zhang HF, Wen Q, Jin H, Xing YR and others (2018a). Intraindividual Variation and Correlation of Cytochrome P450 Activities in Human Liver Microsomes. Molecular Pharmaceutics, 15, 5312–8. [DOI] [PubMed] [Google Scholar]
- Fang Y, Gao N, Tian X, Zhou J, Zhang HF, Gao J, He XP, Wen Q, Jia LJ, Jin H and others (2018b). Effect of P450 Oxidoreductase Polymorphisms on the Metabolic Activities of Ten Cytochrome P450s Varied by Polymorphic CYP Genotypes in Human Liver Microsomes. Cellular Physiology and Biochemistry, 47, 1604–16. [DOI] [PubMed] [Google Scholar]
- Gao N, Tian X, Fang Y, Zhou J, Zhang HF, Wen Q, Jia LJ, Gao J, Sun B, Wei JY and others (2016). Gene polymorphisms and contents of cytochrome P450s have only limited effects on metabolic activities in human liver microsomes. European Journal of Pharmaceutical Sciences, 92, 86–97. [DOI] [PubMed] [Google Scholar]
- Greenblatt DJ, Venkatakrishnan K, Harmatz JS, Parent SJ, von Moltke LL. (2010). Sources of variability in ketoconazole inhibition of human cytochrome P450 3A in vitro. Xenobiotica, 40, 713–20. [DOI] [PubMed] [Google Scholar]
- Greenblatt DJ, Zhao Y, Venkatakrishnan K, Duan SX, Harmatz JS, Parent SJ, Court MH, von Moltke LL. (2011). Mechanism of cytochrome P450–3A inhibition by ketoconazole. Journal of Pharmacy and Pharmacology, 63, 214–21. [DOI] [PubMed] [Google Scholar]
- Halaka FG, Babcock GT, Dye JL. (1985). The use of principal component analysis to resolve the spectra and kinetics of cytochrome-c oxidase reduction by 5,10-dihydro-5-methyl phenazine. Biophys. J, 48, 209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspera R, Naraharisetti SB, Evangelista EA, Marciante KD, Psaty BM, Totah RA. (2011). Drug metabolism by CYP2C8.3 is determined by substrate dependent interactions with cytochrome P450 reductase and cytochrome b5. Biochem Pharmacol, 82, 681–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korhonen LE, Turpeinen M, Rahnasto M, Wittekindt C, Poso A, Pelkonen O, Raunio H, Juvonen RO. (2007). New potent and selective cytochrome P450 2B6 (CYP2B6) inhibitors based on three-dimensional quantitative structure-activity relationship (3D-QSAR) analysis. British Journal of Pharmacology, 150, 932–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krohn KA, Link JM. (2003). Interpreting enzyme and receptor kinetics: keeping it simple, but not too simple. Nucl. Med. Biol, 30, 819–26. [DOI] [PubMed] [Google Scholar]
- Kumar S (2007). Identification of a novel laser dye substrate of mammalian cytochromes P450: Application in rapid kinetic analysis, inhibitor screening, and directed evolution. J. Biomol. Screen, 12, 677–82. [DOI] [PubMed] [Google Scholar]
- Lampe JN, Fernandez C, Nath A, Atkins WM. (2008). Nile red is a fluorescent allosteric substrate of cytochrome P450 3A4. Biochemistry, 47, 509–16. [DOI] [PubMed] [Google Scholar]
- Nad S, Kumbhakar M, Pal H. (2003). Photophysical properties of coumarin-152 and coumarin-481 dyes: Unusual behavior in nonpolar and in higher polarity solvents. J. Phys. Chem. A, 107, 4808–16. [Google Scholar]
- Nad S, Pal H. (2001). Unusual photophysical properties of coumarin-151. J. Phys. Chem. A, 105, 1097–106. [Google Scholar]
- Nad S, Pal H. (2003). Photophysical properties of coumarin-500 (C500): Unusual behavior in nonpolar solvents. J. Phys. Chem. A, 107, 501–7. [Google Scholar]
- Pelkonen O, Turpeinen M, Hakkola J, Honkakoski P, Hukkanen J, Raunio H. (2008). Inhibition and induction of human cytochrome P450 enzymes: current status. Arch. Toxicol, 82, 667–715. [DOI] [PubMed] [Google Scholar]
- Reed J, Backes W. (2016). The functional effects of physical interactions involving cytochromes P450: putative mechanisms of action and the extent of these effects in biological membranes. Drug Metab Rev. , 48, 453–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JR, Backes WL. (2017). Physical Studies of P450-P450 Interactions: Predicting Quaternary Structures of P450 Complexes in Membranes from Their X-ray Crystal Structures. Front. Pharmacol, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendic S, Guengerich FP. (2010). Update Information on Drug Metabolism Systems-2009, Part II. Summary of Information on the Effects of Diseases and Environmental Factors on Human Cytochrome P450 (CYP) Enzymes and Transporters. Curr. Drug Metab, 11, 4–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AG, Yang J, Halpert JR, Nelson SD, Thummel KT, Atkins WM. (2011). The structural basis for homotropic and heterotropic cooperativity of midazolam metabolism by human cytochrome P450 3A4. Biochemistry, 50, 10804–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues AD. (1999). Integrated cytochrome P450 reaction phenotyping - Attempting to bridge the gap between cDNA-expressed cytochromes P450 and native human liver microsomes. Biochem Pharmacol, 57, 465–80. [DOI] [PubMed] [Google Scholar]
- Ryu CS, Klein K, Zanger UM. (2017). Membrane Associated Progesterone Receptors: Promiscuous Proteins with Pleiotropic Functions - Focus on Interactions with Cytochromes P450. Front. Pharmacol, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch GA, Yano JK, Sansen S, Dansette PM, Stout CD, Johnson EF. (2008). Determinants of cytochrome P450 2C8 substrate binding - Structures of complexes with montelukast, troglitazone, felodipine, and 9-cis-retinoic acid. J. Biol. Chem, 283, 17227–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. (1994). Interindividual variations in human liver cytochrome-P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic-chemicals - studies with liver-microsomes of 30 japanese and 30 caucasians. J. Pharm. Exp. Ther, 270, 414–23. [PubMed] [Google Scholar]
- Spaggiari D, Geiser L, Daali Y, Rudaz S. (2014). A cocktail approach for assessing the in vitro activity of human cytochrome P450s: an overview of current methodologies. J Pharm Biomed Anal. , 101, 221–37. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Kneller MB, Haining RL, Trager WF, Rettie AE. (2002). (+)-N-3-benzyl-nirvanol and (−)-N-3-benzyl-phenobarbital: New potent and selective in vitro inhibitors of CYP2C19. Drug Metab. Disp, 30, 235–9. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan K, Von Moltke LL, Court MH, Harmatz JS, Crespi CL, Greenblatt DJ. (2000). Comparison between cytochrome P450 (CYP) content and relative activity approaches to scaling from cDNA-expressed CYPs to human liver microsomes: Ratios of accessory proteins as sources of discrepancies between the approaches. Drug Metab. Disp, 28, 1493–504. [PubMed] [Google Scholar]
- Venkatakrishnan K, von Moltke LL, Greenblatt DJ. (2001). Human drug metabolism and the cytochromes P450: Application and relevance of in vitro models. Journal of Clinical Pharmacology, 41, 1149–79. [DOI] [PubMed] [Google Scholar]
- Volpe DA. (2019). Interindividual Variability in Drug Metabolizing Enzymes. Curr. Drug Metab, 20, 1041–3. [DOI] [PubMed] [Google Scholar]
- Walsky RL, Obach RS. (2003). Verification of the selectivity of (+)N-3-benzylnirvanol as a CYP2C19 inhibitor. Drug Metab. Disp, 31, 343-. [DOI] [PubMed] [Google Scholar]
- Wang X, He B, Shi J, Li Q, Zhu H-J. (2020). Comparative Proteomics Analysis of Human Liver Microsomes and S9 Fractions. Drug Metab. Disp, 48, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienkers LC, Heath TG. (2005). Predicting in vivo drug interactions from in vitro drug discovery data. Nature Reviews Drug Discovery, 4, 825–33. [DOI] [PubMed] [Google Scholar]
- Yuan R, Madani S, Wei XX, Reynolds K, Huang SM. (2002). Evaluation of cytochrome P450 probe substrates commonly used by the pharmaceutical industry to study in vitro drug interactions. Drug Metab. Disp, 30, 1311–9. [DOI] [PubMed] [Google Scholar]
- Zhang HF, Wang HH, Gao N, Wei JY, Tian X, Zhao Y, Fang Y, Zhou J, Wen Q, Gao J and others (2016). Physiological content and intrinsic activities of 10 cytochrome P450 isoforms in human normal liver microsomes. J. Pharm. Exp. Ther, 358, 83–93. [DOI] [PubMed] [Google Scholar]
- Zhang WJ, Ramamoorthy Y, Kilicarslan T, Nolte H, Tyndale RF, Sellers EM. (2002). Inhibition of cytochromes P450 by antifungal imidazole derivatives. Drug Metab. Disp, 30, 314–8. [DOI] [PubMed] [Google Scholar]