

Abstract
Mediastinal involvement is considered essential for the diagnosis of primary mediastinal large B-cell lymphoma (PMBL). However, we have observed cases of diffuse large B-cell lymphoma (DLBCL) with features of PMBL but without detectable mediastinal involvement. The goal was to assess our previously established gene expression profiling (GEP) signature for PMBL in classifying these cases. In a large series of DLBCL cases, we identified 24 cases with a GEP signature of PMBL, including 9 cases with a submission diagnosis of DLBCL consistent with PMBL (G-PMBL-P) and 15 cases with a submission diagnosis of DLBCL. The pathology reviewers agreed with the diagnosis in the 9 G-PMBL-P cases. Among the other 15 DLBCL cases, 11 were considered to be PMBL or DLBCL consistent with PMBL, 3 were considered to be DLBCL, and 1 case was a gray-zone lymphoma with features intermediate between DLBCL and classical Hodgkin lymphoma. All 9 G-PMBL-P and 9 of the 15 DLBCL cases (G-PMBL-M) had demonstrated mediastinal involvement at presentation. Interestingly, 6 of the 15 DLBCL cases (G-PMBL-NM) had no clinical or radiologic evidence of mediastinal involvement. The 3 subgroups of PMBL had otherwise similar clinical characteristics, and there were no significant differences in overall survival. Genetic alterations of CIITA and PDL1/2 were detected in 26% and 40% of cases, respectively, including 1 G-PMBL-NM case with gain of PDL1/2. In conclusion, PMBL can present as a nonmediastinal tumor without evidence of mediastinal involvement, and GEP offers a more precise diagnosis of PMBL.
Keywords: primary mediastinal large B-cell lymphoma, gene expression profiling, CIITA, PDL1/2
Primary mediastinal large B-cell lymphoma (PMBL) is a distinct subtype of diffuse large B-cell lymphoma (DLBCL) that is thought to arise from thymic medullary B cells and is recognized on the basis of its distinctive clinical, pathologic, and molecular features.1 PMBL is usually seen in young adults with a primary mediastinal mass and has a better prognosis compared with nonmediastinal DLBCL.2–4 Gene expression profiling (GEP) has suggested a relationship between PMBL and classical Hodgkin lymphoma,5,6 and this is further supported by shared genetic abnormalities.7–9 The typical PMBL is characterized by compartmentalizing fibrosis and medium-sized to large B cells with pale cytoplasm, variable positivity for CD30 and CD23, and the frequent absence of surface immunoglobulin. However, it is sometimes difficult to render a definitive diagnosis of PMBL, as it may overlap with systemic DLBCL with secondary mediastinal involvement and some cases of classical Hodgkin lymphoma.10
By definition, mediastinal involvement has been considered essential for the diagnosis of PMBL. The mediastinal mass is usually bulky and may extend locally into adjacent thoracic structures. Occasionally, PMBL disseminates to extranodal sites such as the kidney and the brain.11 Nodal or extranodal DLBCL without mediastinal involvement or with predominantly nonmediastinal disease is generally diagnosed as DLBCL-not otherwise specified (NOS). However, we have recently observed several DLBCL cases with pathologic features of PMBL but without detectable mediastinal involvement. Conversely, there are cases with both mediastinal and nonmediastinal disease, but the pathologic features are not typical of PMBL. It is unclear whether such cases should be classified as PMBL or DLBCL, which is important in terms of clinical management and prognostication.
Translocations involving the MHC class II transactivator (CIITA), as well as gains/amplification of chromosome 9p24.1 including the gene loci of JAK2 and programmed cell death protein 1 ligands (PDL1 and PDL2), have been detected frequently in cases of PMBL.8,9,12–14 Interestingly, the PDL genes are frequent partners in CIITA translocation. Translocations involving PDL1 and PDL2, some of which are independent of CIITA have also been described in PMBL.15 In contrast, these genetic alterations are rare in DLBCL.5,16,17 CIITA is an essential transactivator of MHC class II expression,18 and CIITA gene fusions result in downregulation of surface MHC class II expression. Thus, overexpression of PD-1 ligands can result from gene amplification or translocation15,19 and leads to inhibition of effector T-cell activation in the tumor microenvironment. Therefore, it has been proposed that the pathogenesis of PMBL involves immune escape of malignant B cells by downregulation of MHC class II molecules secondary to CIITA gene fusions and upregulation of PD-1 ligands with induction of T-cell anergy.20
Previously, we derived a robust GEP signature for PMBL that can distinguish it from other types of DLBCL.5 We used this signature to examine its utility in diagnosing PMBL in biopsies of nonmediastinal sites and, in particular, in cases without clinical or radiologic evidence of mediastinal involvement. We also examined the frequencies of CIITA translocation and 9p24.1 rearrangements or copy number variations in these cases.
MATERIALS AND METHODS
Patients
The biopsies of DLBCL cases before treatment were collected from multiple institutions.21,22 The cases with a submission diagnosis of PMBL were excluded. Clinical data were available on all patients. Pathology review of the cases was performed by a Leukemia/Lymphoma Molecular Profiling Project panel of expert pathologists. If the tumor had the histologic features of PMBL including medium-sized to large-sized cells with abundant pale cytoplasm in the background of compartmentalized alveolar fibrosis and variable CD30 and CD23 expression, a diagnosis of PMBL or consistent with PMBL was rendered. The cases without these features were classified as DLBCL-NOS. A final consensus diagnosis for all discrepant cases was reached by discussing the cases over a multihead microscope. This study was approved by the various contributing centers’ Institutional Review Boards.
Gene Expression Profiling
Of the 24 cases with a GEP signature of PMBL in this study, 13 were selected from a set of DLBCL cases using a 46 gene Bayesian predictor on the basis of Lymphochip DNA microarray.5,21 The remaining 11 cases were selected from another set of DLBCL cases22 by a version of this model that had been modified to be compatible with the Affymetrix U133 + 2 platform (Affymetrix, Santa Clara, CA). The modification was performed as follows: from the previous set of 188 DLBCL samples and 34 PMBL samples21 analyzed using a U133 + 2 platform, a linear predictor score for each sample was calculated by summing log transformed gene expression of each of the 46 genes and multiplied each by the t statistic for differential expression between the DLBCL and PMBL samples. The mean and variance of these scores within the DLBCL and PMBL subgroups was then calculated and used to generate a Bayesian probability that a given score was associated with a PMBL sample. The cases with a Bayesian probability score of > 90% in the direction of PMBL were declared to be PMBL.
Fluorescence In Situ Hybridization
Interphase fluorescence in situ hybridization (FISH) analysis for detection of CIITA (16p13.13) and PDL1/PDL2 (9p24.1) rearrangements, gains, and amplifications was performed on formalin-fixed, paraffin-embedded sections as previously described.19 In brief, we used in-house bacterial artificial chromosome break-apart, dual-color probes for CIITA and PDL1/PDL2. For the purpose of this study, the cutoff value for break-apart and copy number gains/amplifications was set at > 5%. A minimum of 200 interphase cells were scored per case. Copy number gains were defined as 3 or 4 signals and amplifications as > 4 signals per probe. Slides were analyzed using a Carl Zeiss Axio Imager Z2 microscope equipped with a Plan Apochromat × 100/1.4 oil objective. The images were acquired using the Cool Cube Digital and Metasystems software (version 5.5.1).
Immunohistochemistry
PDL2 immunostaining was performed on formalin-fixed, paraffin-embedded tissue sections. After deparaffinization and antigen retrieval, automated immunohistochemical staining for PDL2 was performed on the Biocare Intellipath FLX autostainer. Sections were blocked for peroxidase activity and nonspecific binding with peroxidase-1 and background sniper (Biocare, Concord, CA). PDL2 mouse monoclonal antibody (clone 366C.9E5, kindly provided by Dr Gordon J. Freeman, Dana-Farber Cancer Institute, Boston, MA) was used at a concentration of 0.07 μg/mL in Renaissance background reducing diluent for 30 minutes at room temperature. Slides were then incubated with MACH 2 mouse-HRP polymer for 30 minutes at room temperature and were developed using intelliPATH FLX DAB chromogen for 5 minutes at room temperature. Reactivity for PDL2 was determined and scored on the basis of the percentage of tumor cells showing positive staining (0% to 100%, in 10% increments) and the intensity of positive staining (0 = negative, 1 + = weak, 2+ = moderate, 3 + = strong). A case was scored as positive if > 20% of the tumor cells stained positive with an intensity of at least 1 +. The PDL2 immunohistochemical stains were interpreted without knowledge of the FISH results.
Statistical Analysis
The Fisher exact test was used to compare patient characteristics between groups. The Wilcoxon rank sum was used to test the age distribution between groups. The Kaplan-Meier method was used to estimate overall survival (OS) and event-free survival (EFS) distributions. OS was defined as the time from diagnosis to death or last follow-up. EFS was defined as the time from diagnosis to relapse, progression, or death. Patients who were alive and relapse-free/progression-free at last follow-up were treated as censored. The log rank test was used to compare survival distributions between groups. P-values ≤ 0.05 were considered to be statistically significant.
RESULTS
Clinical Features of Cases With a PMBL GEP Signature
We previously have used GEP to develop a robust molecular predictor that precisely distinguishes PMBL from DLBCL of the germinal center B-cell–like and activated B-cell–like subtypes.5 We applied this PMBL predictor to a large group of DLBCL patients with GEP data. A total 24 cases showed a PMBL GEP signature, including 9 cases with a submission diagnosis of DLBCL consistent with PMBL (referred to as G-PMBL-P) and 15 cases with a submission diagnosis of DLBCL.
The initial biopsy slides of these 24 cases, including hematoxylin and eosin stains and immunostains, were reviewed by a pathology review panel, and a consensus diagnosis was reached for each case. The reviewers agreed with the diagnosis of the 9 G-PMBL-P cases. Of the 15 DLBCL cases, 11 were diagnosed as DLBCL consistent with PMBL, 3 were called DLBCL, and 1 case was diagnosed as a gray-zone lymphoma (B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and classical Hodgkin lymphoma). The characteristics of the 24 patients with a PMBL GEP signature on initial presentation are listed in Table 1. The patients were young (median age, 31 y), and the bone marrow was negative at initial presentation in all patients. As expected, the G-PMBL-P cases were predominantly young female individuals (median age, 29 y) with localized mediastinal disease. In contrast, the 15 DLBCL patients were older (median age, 45 y), had an equal sex distribution, and had a higher incidence of advanced-stage disease. However, such differences in age, sex distribution, and incidence of advanced-stage disease did not reach statistical significance between groups (Table 1).
Table 1.
Clinical Characteristics of 24 Patients With a PMBL GEP Signature
| n (%) |
|||||
|---|---|---|---|---|---|
| All Patients N = 24 (100%) |
G-PMBL-P N = 9 (38%) |
G-PMBL-M N = 9 (100%) |
G-PMBL-NM N = 6 (100%) |
P* | |
| Age (y) | |||||
| Median | 31 | 29 | 44.5 | 46 | 0.24 |
| Range | 18-90 | 20-45 | 18-90 | 19-85 | |
| Sex | |||||
| Male | 7 (32) | 2 (22) | 2 (29) | 3 (50) | 0.54 |
| Female | 15 (68) | 7 (78) | 5 (71) | 3 (50) | |
| LDH | |||||
| Normal | 3 (16) | 0 (0) | 1 (14) | 2 (33) | 0.48 |
| Increased | 16 (84) | 6 (100) | 6 (84) | 4 (67) | |
| B symptoms (+) | 8 (38) | 2 (25) | 3 (43) | 3 (50) | 0.63 |
| ECOG performance status | |||||
| 0-1 | 17 (89) | 7 (100) | 5 (83) | 5 (83) | 0.51 |
| ≥ 2 | 2 (11) | 0 (0) | 1 (17) | 1 (17) | |
| Extranodal sites | |||||
| > 1 | 6 (29) | 4 (50) | 0 (0) | 2 (33) | 0.11 |
| Ann Arbor stage | |||||
| I-II | 10 (45) | 5 (63) | 2 (25) | 3 (50) | 0.43 |
| III-IV | 12 (55) | 3 (37) | 6 (75) | 3 (50) | |
| International Prognostic Index | |||||
| 0-2 | 12 (63) | 5 (83) | 4 (57) | 3 (50) | 0.59 |
| 3-5 | 7 (37) | 1 (17) | 3 (43) | 3 (50) | |
Patient characteristics were compared between the groups with the Fisher exact test or the Wilcoxon rank sum test. Not all features were available for all patients. LDH indicates lactate dehydrogenase.
The most frequent site of involvement at presentation and relapse was the mediastinum, followed by lymph nodes and lung (Table 2). Extrathoracic sites were uncommon. The majority of the patients were treated with CHOP or CHOP-equivalent chemotherapy with or without rituximab (Table 3).
TABLE 2.
Sites of PMBL at Presentation and Relapse
| Presentation N = 24 (%) |
Relapse N = 11 (%) |
|
|---|---|---|
| Mediastinum | 18 (75) | 4 (36) |
| Lymph nodes | 6 (25) | 3 (27) |
| Intrathoracic sites | ||
| Lung | 3 (13) | 2 (18) |
| Chest wall/pleura | 2 (8) | 0 (0) |
| Extrathoracic sites | ||
| Parotid gland | 1 (4) | 0 (0) |
| Gastrointestinal tract | 2 (8) | 1 (9) |
| Kidney | 1 (4) | 0 (0) |
| Adrenal gland | 1 (4) | 0 (0) |
| Breast | 1 (4) | 0 (0) |
| Cervical soft tissue | 1 (4) | 0 (0) |
| Intra-abdominal/pelvic | 1 (4) | 1 (9) |
| Bone | 0 (0) | 1 (9) |
| Bone marrow | 0 (0) | 1 (9) |
TABLE 3.
Distribution of Treatment for PMBL
| CHOP/CHOP Equivalent |
ProMACE | |
|---|---|---|
| All (n = 21) | ||
| With rituximab | 9 | 0 |
| Without rituximab | 11 | 1 |
| G-PMBL-P (n = 8) | ||
| With rituximab | 2 | 0 |
| Without rituximab | 5 | 1 |
| G-PMBL-M and G-PMBL-NM (n = 13) | ||
| With rituximab | 7 | 0 |
| Without rituximab | 6 | 0 |
CHOP indicates cyclophosphamide, doxorubicin, vincristine, prednisone; ProMACE, prednisone, methotrexate, doxorubicin, cyclophosphamide, etoposide.
Patients Without Evident Mediastinal Disease
All 9 G-PMBL-P cases and 9 of 15 DLBCL cases (referred to as G-PMBL-M) involved the mediastinum at initial presentation. Interestingly, 6 of 15 DLBCL cases had no radiologic evidence of mediastinal involvement by chest computed tomography scans (n = 5) or no documentation of mediastinal involvement in the clinical notes (n = 1) (referred to as G-PMBL-NM) (Table 4). There were no significant differences in the clinical characteristics of the 3 groups. The majority of the 6 G-PMBL-NM cases (cases 2 to 5) had morphologic features of PMBL, whereas the remaining 2 cases were classified as gray-zone lymphoma (case 1) or DLBCL (case 6) (Table 4). The primary site(s) at presentation included both lymph nodes (n = 3), parotid gland (n = 1), pelvis (n = 1), and kidney/adrenal gland (n = 1) (Table 4).
TABLE 4.
Clinical Characteristics of 6 G-PMBL-NM Patients
| Consensus Diagnosis | Age/Sex | Stage | Abnormal LDH | Extranodal Sites | Primary Site | Treatment | Response | Outcome (Follow-up [mo]) |
|---|---|---|---|---|---|---|---|---|
| 1 DLBCL, c/w gray-zone lymphoma | 31/F | IV | + | 2 | Pelvic mass | CHOP-RT | CR | Dead (9) (COD: lymphoma) |
| 2 PMBL | 53/M | I | − | 0 | Cervical LN | R-CHOP-21 | CR | Alive (100) |
| 3 DLBCL, c/w PMBL | 85/F | II | + | 0 | Periaortic, inguinal LNs | R-CHOP-21 | PR | Dead (66) (COD: lymphoma) |
| 4 DLBCL, c/w PMBL | 39/F | IV | + | 4 | Kidney, adrenal gland, pancreas, small intestine | R-EPOCH | CR | Dead (34) (COD: lymphoma) |
| 5 DLBCL, c/w PMBL | 19/M | I | − | 1 | Parotid gland | R-CHOP-21 | CR | Alive (93) |
| 6 DLBCL | 82/M | III | + | 0 | Axillary LN | R-CHOP-21 | CR | Dead (64) (COD: lymphoma |
CHOP indicates cyclophosphamide, doxorubicin, vincristine, prednisone; COD, cause of death; CR, complete remission; EPOCH, etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin; LN, lymph node; PR, partial response; R, rituximab; RT, radiation therapy.
Fluorescence In Situ Hybridization
Although CIITA rearrangements and copy number variants/rearrangements of 9p24.1 are frequent in PMBL, both events are rare in DLBCL.5,15,19 A total of 21 of the 24 cases were available for the CIITA and PDL1/PDL2 FISH analyses. FISH tests failed in 2 cases for CIITA and in 1 case for PDL1/PDL2. Genetic alterations including rearrangements, gains, and amplifications of CIITA and PDL1/PDL2 were detected in 26.3% (5/19) and 40% (8/20) of the cases, respectively (Table 5 and Fig. 1). There were 3 cases with both a CIITA rearrangement and PDL1/PDL2 gain or amplification. A gain of PDL1/PDL2 was present in 1 of 5 cases without mediastinal involvement, whereas rearrangements of CIITA or PDL1/PDL2 were only detected in cases with mediastinal involvement (Table 5).
TABLE 5.
CIITA and PDL1/2 Rearrangements, Gains, and Amplifications in Patients With PMBL
| G-PMBL-P | G-PMBL-M | G-PMBL-NM | Total | |
|---|---|---|---|---|
| CIITA | N = 7 | N = 8 | N = 4 | 19 |
| Rearrangement | 2 | 2 | 0 | 4 |
| Gain | 0 | 1 | 0 | 1 |
| n (%) | 2/7 (29) | 3/8 (38) | 0/4 (0) | 5/19 (26) |
| PDL1/2 | N = 7 | N = 8 | N = 5 | 20 |
| Rearrangement | 2 | 0 | 0 | 2 |
| Gain | 3 | 0 | 1 | 4 |
| Amplification | 0 | 2 | 0 | 2 |
| n (%) | 5/7 (71) | 2/8 (25) | 1/5 (20) | 8/20 (40) |
|
CIITA rearrangement and PDL1/2 abnormality (n [%]) |
1/7 (14) | 2/8 (25) | 0/4 (0) | 3/19 (16) |
FIGURE 1.
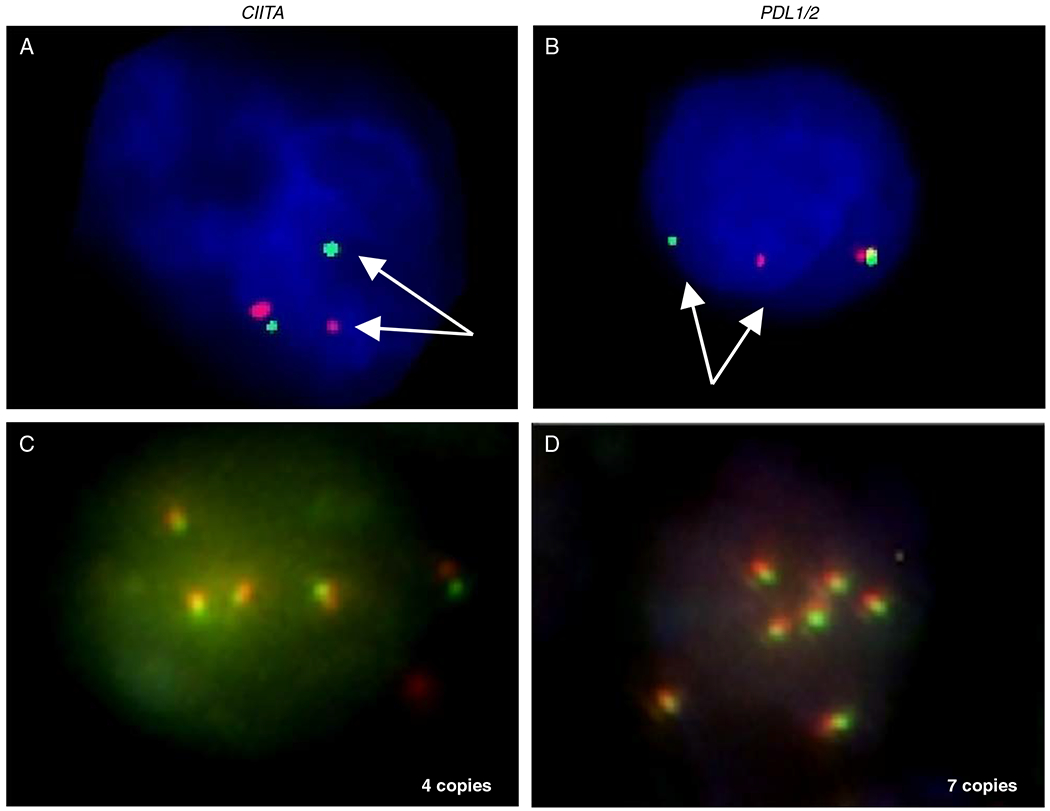
Representative interphase FISH analysis for detection of chromosomal rearrangement, gain, or amplification involving CIITA (A and C) and PDL1/2 (B and D). Detection of separate red and green signals indicates a chromosomal breakpoint affecting the CIITA locus at 16p13.13 (A) or the PDL1/2 locus at 9p24.1 (B). Multiple fused green and red signals indicate amplification of CIITA (C) and PDL1/2 (D).
PDL2 Expression
Translocations, gains, and amplifications of PDL1/PDL2 can result in elevated PDL2 transcript levels.14,15 We next evaluated the correlation between FISH results and protein expression by immunohistochemistry for PDL2. Nine cases in total, including 1 case with PDL1/PDL2 rearrangement, 5 cases with a PDL1/PDL2 gain or amplification, and 3 cases without PDL1/PDL2 abnormalities, were available for PDL2 immunostaining. Positive PDL2 staining was detected in all 6 cases with PDL1/PDL2 genetic alterations, including the case without mediastinal involvement. These 6 cases showed membrane staining of PDL2 in at least 60% tumor cells with variable staining intensity, from 1 + to 3 + (Fig. 2). In the 3 cases without PDL1/PDL2 abnormalities by the FISH studies, 2 were negative but 1 was positive for PDL2 in 80% of tumor cells at 1+ intensity. This discordant case was a DLBCL with mediastinal involvement. These results indicate excellent correlation between FISH and immunohistochemistry studies, which confirm the findings reported recently.17,23 The discordant case suggests that other mechanisms of upregulation of PDL2 may also exist.17
FIGURE 2.
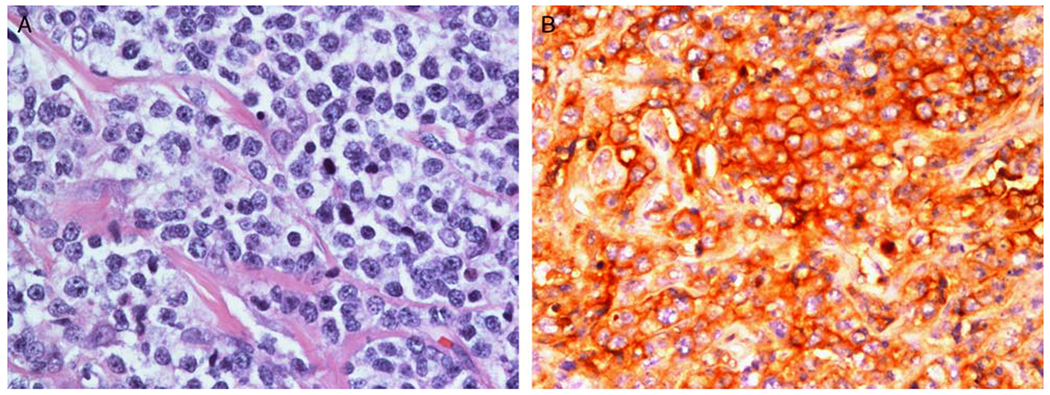
A representative G-PMBL-P case with PDL1/PDL2 genetic alterations showing large atypical cells with pale cytoplasm in a background of fibrosis (H&E stain) (A) and intense membranous staining for PDL2 (B).
Survival
Survival data were available for 22 patients, with a median OS of 5.7 years and a 5-year OS of 54.5%. Nine patients were alive at last contact with a median follow-up of 10.1 years (range, 2.9 to 26.0 y). The OS and EFS of the 3 subgroups or between the cases with or without mediastinal involvement were not significantly different (Fig. 3), but there was a trend of worse EFS for the G-PMBL-P group compared with the other 2 groups (P = 0.062). A significantly worse EFS for G-PMBL-P was observed compared with that of the 2 other groups combined (P = 0.018), possibly because more of the patients from G-PMBL-P were treated without rituximab. The OS and EFS were also compared between cases with or without CIITA and/or PDL1/PDL2 rearrangement. However, no significant difference was observed (Fig. 4).
FIGURE 3.
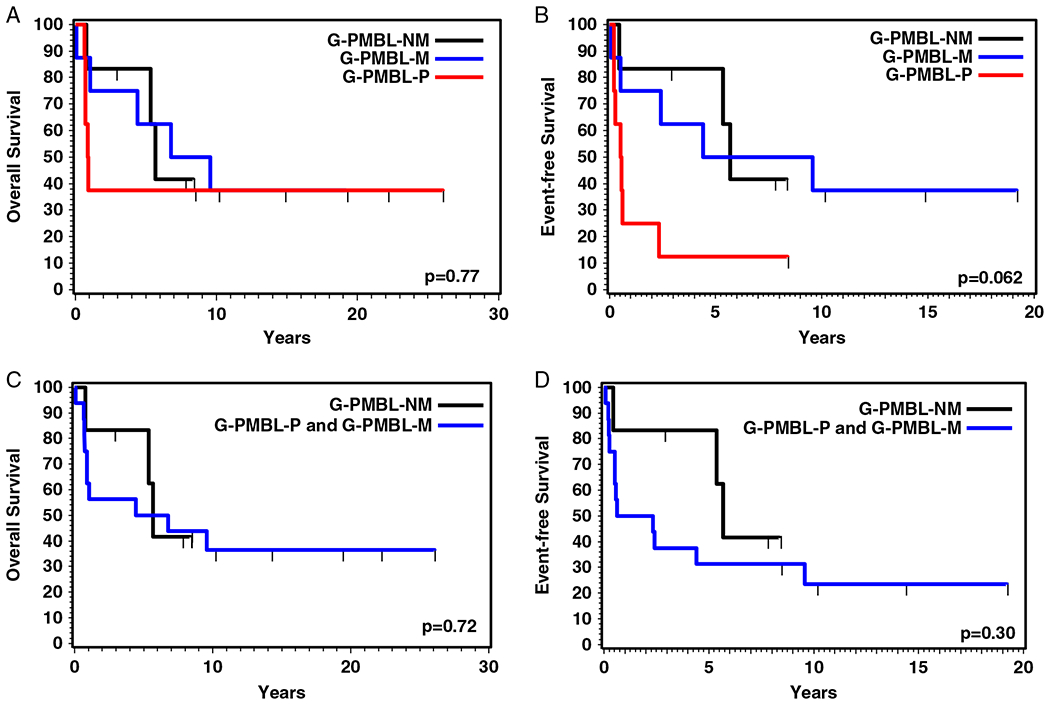
OS (A) and EFS (B) of patients with PMBL (G-PMBL-P), DLBCL with mediastinal involvement (G-PMBL-M), and DLBCL without mediastinal involvement (G-PMBL-NM). OS (C) and EFS (D) of patients with mediastinal involvement (G-PMBL-P and G-PMBL-M) and those without mediastinal involvement (G-PMBL-NM).
FIGURE 4.
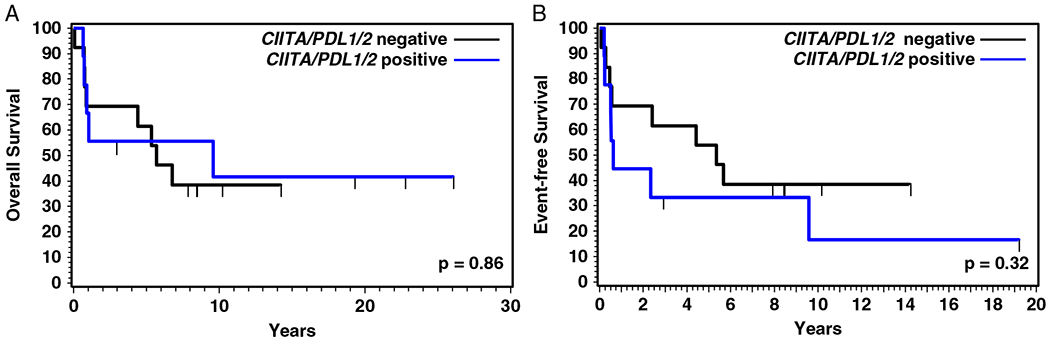
OS (A) and EFS (B) of patients with or without CIITA and/or PDL1/2 genetic alterations.
DISCUSSION
PMBL is known to disseminate to nodal and extranodal sites,11,24 including some unusual sites such as the kidney and adrenal gland, but bone marrow involvement is uncommon. The pathologic features that led pathologists to consider the cases as consistent with PMBL include medium-sized to large-sized cells with multilobated nuclei and abundant pale cytoplasm in the background of fibrosis, as well as variable CD30 and CD23 expression. However, the diagnosis of PMBL in nonmediastinal sites is extremely challenging, especially with limited tissue and atypical morphology. Even in cases with a mediastinal mass, separating PMBL from DLBCL with secondary mediastinal involvement may be challenging. The availability of a robust molecular signature will help resolve these diagnostic problems.
In this study, 24 cases classified as PMBL using a GEP signature were identified in a large series of DLBCL cases. Some of the cases were called DLBCL consistent with PMBL by the submitting pathologist on the basis of the morphology and the presence of mediastinal disease. The expert panel confirmed the diagnosis of G-PMBL-P and G-PMBL-M cases and also determined that the majority of G-PMBL-NM cases had features of PMBL. Even after the review, 4 cases were classified as DLBCL-NOS. Previous studies have also found cases initially classified as DLBCL that were actually PMBL by GEP and vice versa.5,6 In the study of Savage et al,6 6 of 176 cases diagnosed as DLBCL were reclassified as PMBL by GEP, and not all of the cases had mediastinal disease. Saarinen et al25 recently described a family with 3 siblings with PMBL and a cousin with extranodal DLBCL without mediastinal involvement. Interestingly, all 4 cases had a similar morphology and immunophenotype. All had an MLL missense mutation, suggesting that the DLBCL case might really be a PMBL but without mediastinal disease. For cases with atypical morphology or a nonmediastinal presentation, the molecular GEP signature could be a powerful tool to aid in the diagnosis.
It has been postulated that PMBL originates from a thymic B cell.1 However, the presence of PMBL cases without mediastinal disease raises the question regarding the origin of PMBL. One possibility is that such cases arise from ectopic thymus. Ectopic thymic tissue is usually embedded along the embryologic path of descent, which spans the angle of the mandible to the superior mediastinum, including sites such as the thyroid, trachea, and esophagus. Although there are reports of ectopic tissue that occur elsewhere in the body,26,27 the anatomic sites of ectopic thymus cannot explain all of the nonmediastinal sites involved by PMBL in our study, arguing against this idea. Another possibility is that there is a small, undetectable lymphoma in the thymus of these cases, but this is difficult to substantiate. The other possibility is that these cases share an origin from the B cell that also gives rise to classical Hodgkin lymphoma. This cell has a propensity to migrate into the thymus and some other organs, which may also explain the peculiarity of some disease sites. Our observation of 1 case with gray-zone morphology but the GEP of PMBL is highly interesting and supports these latter hypotheses.
Several studies have shown that CD30-positive DLBCLs tend to have a favorable outcome.28,29 Although PMBL was excluded from these studies on the basis of morphology and mediastinal involvement, our study demonstrated that PMBL can present as a nonmediastinal lesion with minimal or no evidence of a mediastinal mass. GEP is more accurate to definitively exclude these cases from such studies. Therefore, a subset of the CD30-positive DLBCL could be PMBL, which may partially account for the better prognosis of these tumors.
CIITA rearrangement and chromosomal gain of 9p24.1 (PDL1/PDL2) are the 2 most frequent genetic alterations in PMBL and can be seen in 38% and 63% of the cases, respectively.14,19,30 Recent studies have shown that PDL1/PDL2 is rearranged at a frequency of 20% in PMBL.15 Our FISH results for CIITA and PDL1/PDL2 rearrangements, and chromosomal gain/amplification of PDL1/PDL2, showed a lower frequency than reported for PMBL. The genetic abnormalities involving CIITA and PDL1/PDL2 were more frequently identified in the cases with mediastinal disease than those without mediastinal disease. A prior study of gray-zone lymphoma showed that cases with mediastinal disease more frequently exhibited alterations in chromosome 9p24.1, whereas alterations involving CIITA were more common in those without mediastinal disease.31 It is unclear whether the frequency of CIITA and PDL1/PDL2 gene rearrangement in PMBL is associated with mediastinal involvement. As some of our results may be limited by the small sample size in our study, large-scale studies are desirable to further address these issues. Steidl et al19 found that the presence of a CIITA rearrangement significantly correlated with a shorter disease-specific survival. Because of the small sample size, we combined the cases with CIITA and/or PDL1/PDL2 alterations and compared them with the cases without either genetic alteration. However, no significant difference in OS and EFS was observed between these 2 groups in our study.
Whether PMBL cases without apparent mediastinal involvement form a different subset with older age, more equal sex distribution, and lower incidence of CIITA and PDL1/PDL2 abnormalities will require future studies of a large group of patients.
In summary, we identified 24 cases with a PMBL GEP signature, and 6 of these 24 cases had no evidence of mediastinal disease. Our results demonstrate that PMBL can present as a nonmediastinal tumor without the clinical suspicion of PMBL. The molecular diagnosis of PMBL using a gene expression signature may offer a more precise diagnosis of such cases than currently available methods. Although such cases are not common, awareness and accurate diagnosis will better direct the appropriate clinical management for the patients and predict prognosis.
ACKNOWLEDGMENTS
The authors wish to thank Kate Milne for the PDL2 immunostaining and Dr Gordon J. Freeman for providing the PDL2 antibody. We also thank Susana Ben-Neriah for excellent technical support performing the FISH studies.
Partly supported by National Institutes of Health grant U01CA157581 to W.C.C.
Footnotes
Conflicts of Interest and Source of Funding: The authors have disclosed that they have no significant relationships with, or financial interest in, any commercial companies pertaining to this article.
REFERENCES
- 1.Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: Internation Agency for Research on Cancer (IARC); 2008:358–360. [Google Scholar]
- 2.Cazals-Hatem D, Lepage E, Brice P, et al. Primary mediastinal large B-cell lymphoma. A clinicopathologic study of 141 cases compared with 916 nonmediastinal large B-cell lymphomas, a GELA (“Groupe d’Etude des Lymphomes de l’Adulte”) study. Am J Surg Pathol. 1996;20:877–888. [DOI] [PubMed] [Google Scholar]
- 3.Savage KJ, Al-Rajhi N, Voss N, et al. Favorable outcome of primary mediastinal large B-cell lymphoma in a single institution: the British Columbia experience. Ann Oncol. 2006;17:123–130. [DOI] [PubMed] [Google Scholar]
- 4.Zinzani PL, Martelli M, Bertini M, et al. Induction chemotherapy strategies for primary mediastinal large B-cell lymphoma with sclerosis: a retrospective multinational study on 426 previously untreated patients. Haematologica. 2002;87:1258–1264. [PubMed] [Google Scholar]
- 5.Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 2003;198:851–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Savage KJ, Monti S, Kutok JL, et al. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood. 2003;102:3871–3879. [DOI] [PubMed] [Google Scholar]
- 7.Joos S, Kupper M, Ohl S, et al. Genomic imbalances including amplification of the tyrosine kinase gene JAK2 in CD30 + Hodgkin cells. Cancer Res. 2000;60:549–552. [PubMed] [Google Scholar]
- 8.Joos S, Otano-Joos MI, Ziegler S, et al. Primary mediastinal (thymic) B-cell lymphoma is characterized by gains of chromosomal material including 9p and amplification of the REL gene. Blood. 1996;87:1571–1578. [PubMed] [Google Scholar]
- 9.Bentz M, Barth TF, Bruderlein S, et al. Gain of chromosome arm 9p is characteristic of primary mediastinal B-cell lymphoma (MBL): comprehensive molecular cytogenetic analysis and presentation of a novel MBL cell line. Genes Chromosomes Cancer. 2001;30: 393–401. [DOI] [PubMed] [Google Scholar]
- 10.Traverse-Glehen A, Pittaluga S, Gaulard P, et al. Mediastinal gray zone lymphoma: the missing link between classic Hodgkin’s lymphoma and mediastinal large B-cell lymphoma. Am J Surg Pathol. 2005;29:1411–1421. [DOI] [PubMed] [Google Scholar]
- 11.Bishop PC, Wilson WH, Pearson D, et al. CNS involvement in primary mediastinal large B-cell lymphoma. J Clin Oncol. 1999;17: 2479–2485. [DOI] [PubMed] [Google Scholar]
- 12.Wessendorf S, Barth TF, Viardot A, et al. Further delineation of chromosomal consensus regions in primary mediastinal B-cell lymphomas: an analysis of 37 tumor samples using high-resolution genomic profiling (array-CGH). Leukemia. 2007;21:2463–2469. [DOI] [PubMed] [Google Scholar]
- 13.Oschlies I, Burkhardt B, Salaverria I, et al. Clinical, pathological and genetic features of primary mediastinal large B-cell lymphomas and mediastinal gray zone lymphomas in children. Haematologica. 2011;96:262–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Green MR, Monti S, Rodig SJ, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116:3268–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Twa DD, Chan FC, Ben-Neriah S, et al. Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood. 2014;123:2062–2065. [DOI] [PubMed] [Google Scholar]
- 16.Steidl C, Connors JM, Gascoyne RD. Molecular pathogenesis of Hodgkin’s lymphoma: increasing evidence of the importance of the microenvironment. J Clin Oncol. 2011;29:1812–1826. [DOI] [PubMed] [Google Scholar]
- 17.Twa DD, Mottok A, Chan FC, et al. Recurrent genomic rearrangements in primary testicular lymphoma. J Pathol. 2015. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 18.Silacci P, Mottet A, Steimle V, et al. Developmental extinction of major histocompatibility complex class II gene expression in plasmocytes is mediated by silencing of the transactivator gene CIITA. J Exp Med. 1994;180:1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steidl C, Shah SP, Woolcock BW, et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature. 2011;471:377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steidl C, Gascoyne RD. The molecular pathogenesis of primary mediastinal large B-cell lymphoma. Blood. 2011;118:2659–2669. [DOI] [PubMed] [Google Scholar]
- 21.Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–1947. [DOI] [PubMed] [Google Scholar]
- 22.Lenz G, Wright G, Dave SS, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008;359:2313–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi M, Roemer MG, Chapuy B, et al. Expression of programmed cell death 1 ligand 2 (PD-L2) is a distinguishing feature of primary mediastinal (thymic) large B-cell lymphoma and associated with PDCD1LG2 copy gain. Am J Surg Pathol. 2014;38:1715–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lazzarino M, Orlandi E, Paulli M, et al. Treatment outcome and prognostic factors for primary mediastinal (thymic) B-cell lymphoma: a multicenter study of 106 patients. J Clin Oncol. 1997;15:1646–1653. [DOI] [PubMed] [Google Scholar]
- 25.Saarinen S, Kaasinen E, Karjalainen-Lindsberg ML, et al. Primary mediastinal large B-cell lymphoma segregating in a family: exome sequencing identifies MLL as a candidate predisposition gene. Blood. 2013;121:3428–3430. [DOI] [PubMed] [Google Scholar]
- 26.Okamura JM, Barr RJ. Cutaneous lymphoepithelial neoplasms. Adv Dermatol. 1997;12:277–294. discussion 295. [PubMed] [Google Scholar]
- 27.Hiraumi H, Tabuchi K, Kitajiri S. Dermal thymus: case report and review of the literature. Am J Otolaryngol. 2001;22:294–296. [DOI] [PubMed] [Google Scholar]
- 28.Hu S, Xu-Monette ZY, Balasubramanyam A, et al. CD30 expression defines a novel subgroup of diffuse large B-cell lymphoma with favorable prognosis and distinct gene expression signature: a report from the International DLBCL Rituximab-CHOP Consortium Program Study. Blood. 2013;121:2715–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maes B, Anastasopoulou A, Kluin-Nelemans JC, et al. Among diffuse large B-cell lymphomas, T-cell-rich/histiocyte-rich BCL and CD30+ anaplastic B-cell subtypes exhibit distinct clinical features. Ann Oncol. 2001;12:853–858. [DOI] [PubMed] [Google Scholar]
- 30.Rui L, Emre NC, Kruhlak MJ, et al. Cooperative epigenetic modulation by cancer amplicon genes. Cancer Cell. 2010;18:590–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eberle FC, Salaverria I, Steidl C, et al. Gray zone lymphoma: chromosomal aberrations with immunophenotypic and clinical correlations. Mod Pathol. 2011;24:1586–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]