

Abstract
Knowledge of the genetic architecture of pathogen infectivity and host resistance is essential for a mechanistic understanding of coevolutionary processes, yet the genetic basis of these interacting traits remains unknown for most host–pathogen systems. We used a comparative genomic approach to explore the genetic basis of infectivity in Pasteuria ramosa, a Gram-positive bacterial pathogen of planktonic crustaceans that has been established as a model for studies of Red Queen host–pathogen coevolution. We sequenced the genomes of a geographically, phenotypically, and genetically diverse collection of P. ramosa strains and performed a genome-wide association study to identify genetic correlates of infection phenotype. We found multiple polymorphisms within a single gene, Pcl7, that correlate perfectly with one common and widespread infection phenotype. We then confirmed this perfect association via Sanger sequencing in a large and diverse sample set of P. ramosa clones. Pcl7 codes for a collagen-like protein, a class of adhesion proteins known or suspected to be involved in the infection mechanisms of a number of important bacterial pathogens. Consistent with expectations under Red Queen coevolution, sequence variation of Pcl7 shows evidence of balancing selection, including extraordinarily high diversity and absence of geographic structure. Based on structural homology with a collagen-like protein of Bacillus anthracis, we propose a hypothesis for the structure of Pcl7 and the physical location of the phenotype-associated polymorphisms. Our results offer strong evidence for a gene governing infectivity and provide a molecular basis for further study of Red Queen dynamics in this model host–pathogen system.
Keywords: Daphnia magna; Pasteuria ramosa; Red Queen; host–pathogen coevolution; balancing selection; GWAS; pathogenicity, collagen-like protein
Introduction
The abundance and persistence of heritable variation in natural populations is often cited as a puzzling characteristic of life. Genetic drift and the most intuitive forms of natural selection tend to erode diversity over time, yet genetic variation seems the rule, not the exception, in most natural populations (Lewontin and Hubby 1966; Charlesworth et al. 2016). One of the most powerful mechanisms thought to be responsible for the observed variation is negative frequency-dependent selection (NFDS; Ayala and Campbell 1974; Clarke 1979), which posits that the fitness of a variant is inversely proportional to the frequency of that variant in the population. A specific form of NFDS is Red Queen coevolution, whereby pathogens and hosts continually evolve unique mechanisms of infection and defense, respectively (Jaenike 1978; Hamilton 1980; Salathe et al. 2008). Under this model, pathogens, which often have the faster inherent rate of evolution, adapt rapidly to exploit common host phenotypes. Uncommon host phenotypes thus enjoy a selective advantage and increase in frequency until they become common. This, in turn, selects for pathogens capable of infecting the newly common host phenotype. This model can result in cycling frequencies of host and pathogen genetic variants. Apart from its importance as an engine of diversification, Red Queen coevolution is one of the best explanations for the evolution and widespread occurrence of sexual reproduction (Hamilton 1980), as the attendant recombinatorial diversity is thought to provide longer-lived hosts a means of keeping up with their faster evolving pathogens.
Theories of evolutionary mechanism are predicated, either implicitly or explicitly, on an underlying model of the genetic basis of the traits involved, and this is especially true for the Red Queen model. This form of rapid antagonistic coevolution requires that interactions between host and pathogen are strong and highly specific (Jaenike 1978; Hamilton 1980; Ebert 2018). This, in turn, requires a simple genetic architecture, whereby pathogen infectivity and host resistance are conferred by specific alleles at a small number of loci, and successful infection can only occur if alleles at the interacting host and pathogen loci form a competent match. These fundamental genetic details have typically been assumed or inferred from phenotypic patterns, but the genetic basis of interaction is not known for most host–pathogen associations (but see Samson et al. 2013; Koskella and Brockhurst 2014). Identifying the specific coevolving genetic loci thus represents the next frontier in understanding host–pathogen coevolution (Ebert 2018).
One well-studied host–pathogen association for which progress toward a mechanistic understanding has been made is the Daphnia magna–Pasteuria ramosa system. Daphnia magna is a planktonic crustacean that inhabits bodies of lentic freshwater across Eurasia, North Africa, and North America, and the bacterium P. ramosa is one of its primary pathogens. This system has become a model for host–pathogen coevolution, in large part because its natural ecology has been well studied, and it exhibits characteristic features of Red Queen dynamics (reviewed in Ebert 2008; Ebert et al. 2016). The fitness consequences of interaction are substantial for both host and pathogen (Ebert et al. 2004), resulting in strong reciprocal antagonistic selection. And infection exhibits high genotypic specificity (Carius et al. 2001; Luijckx et al. 2011), whereby the outcome of interaction is binary, with certain host and pathogen genotype combinations resulting in infection and others resulting in complete resistance. Moreover, patterns of infectivity and resistance in natural populations are consistent with predictions of Red Queen theory, with pathogens best adapted to contemporary hosts (Decaestecker et al. 2007). More recently, studies of the genetic architecture of host resistance have identified a genetic locus that is strongly associated with resistance of D. magna to P. ramosa (Routtu and Ebert 2015; Bento et al. 2017, 2020), and breeding experiments provide evidence for at least two other closely linked loci in epistatic interaction with the first (Luijckx et al. 2013; Metzger et al. 2016; Ameline et al. 2020). However, no such genetic evidence has been available for the pathogen, leaving half the story untold.
Here, we identify a genetic locus explaining a widespread polymorphic infectivity phenotype in P. ramosa. We used a geographically broad collection of P. ramosa field isolates to refine and select a genetically and phenotypically diverse sample of distinct P. ramosa clones. Using the full genomic sequence of 24 of these clones, we conducted a genome-wide association study (GWAS) to identify polymorphisms associated with the infection phenotype. We found a cluster of polymorphisms within a single gene for the collagen-like protein (CLP), Pcl7, which corresponds perfectly with infection phenotype. In a second step, we confirmed this pattern with an additional independent validation sample of 32 P. ramosa clones. Phylogenetic and geographic analyses are consistent with the hypothesis that the infectivity phenotype is preserved as a stable polymorphism over a large geographic area, most likely maintained by long-term balancing selection. These findings support the hypothesis that Red Queen coevolution can maintain a pool of standing genetic variation at infectivity loci in pathogens, as it does for resistance loci in hosts. Studies of a homologous protein in the closely related genus Bacillus provide a basis for inference on the structure and function of Pcl7 and suggest hypotheses for the mechanistic significance of the phenotype-associated polymorphisms. Our results conclusively establish the genetic basis of a natural polymorphism for infectivity in this model pathogen. Together with recent advances in our understanding of the genetic basis of resistance in the host, our study is one of the first to provide an explanation of the genetic basis of Red Queen coevolution.
Results
Genome Assembly, SNP Calling, and Descriptive Statistics
The reference-assisted, de novo genome assemblies of the 24 individual P. ramosa clones had an average length of ∼1.34 Mb (min. = 1.01 Mb, max. = 1.53 Mb), a mean contig number of 552 (min. = 387, max. = 1,007), a mean maximum contig length of 48.2 kb (min. = 21.7 kb, max. = 86.7 kb), and a mean n50 of 6.2 kb (min. = 2.6 kb, max. = 8.9 kb). These individual contig assemblies were all successfully scaffolded, resulting in a final assembly of the same approximate length as the reference C1 strain. However, individual assemblies differed in the fraction containing gaps. The average number of gaps per assembly was 482 (min. = 353, max. = 607), with an average proportion of gaps being 25% of the genome length (min. = 14%, max. = 51%).
In addition to these de novo assemblies, we also mapped individual reads to the reference genome from the C1 P. ramosa strain. The average read depth per site was 116X (min. = 33X, max. = 426X). We identified a total of 11,193 single nucleotide polymorphisms (SNPs) across the 24 sequenced P. ramosa clones, with a mean number of 2,517 SNPs per genome (min. = 11, max. = 4,019). The amount of missingness per individual had an average of 9% (min. < 1%, max. = 17%). Mean genome-wide nucleotide diversity (π), based on 100-bp nonoverlapping windows, was 0.0052284 ± 0.006881613 SD.
Spore Adhesion Assays
The 24 genome-sequenced P. ramosa clones (supplementary table 1, Supplementary Material online) were screened for their ability to adhere to and thus infect six different D. magna host clones (supplementary table 2, Supplementary Material online). Two of the host clones, DE-K1-IINB1 (from Germany) and CH-H-159 (from Switzerland), were not infected by any of the P. ramosa clones and were thus omitted from subsequent analysis, as they produced no variation in infection phenotype. Two other host clones, HU-HO-2 (from Hungary) and CH-H-67 (from Switzerland), were infected by the same six P. ramosa clones resulting in identical P. ramosa infection phenotypes. The Finnish host clones FI-XINB3 and FI-KELA-39-9 each produced their own distinct infection phenotype and were infected by four and six P. ramosa clones, respectively. The resulting three P. ramosa infection phenotypes (relative to HU-HO-2/CH-H-67, FI-XINB3, and FI-KELA-39-9) were each compared with genome-wide sequence polymorphisms via GWAS to identify loci of significant correlation between genotype and phenotype.
Genome-Wide Association Study
The GWAS “terminal” tests of the 24 genome-sequenced P. ramosa clones identified 21 SNPs whose variation segregated perfectly according to the HU-HO-2/CH-H-67 infection phenotype, (table 1, figs. 1 and 2A; supplementary figs. 1 and 2, Supplementary Material online). These phenotype-associated polymorphisms all fall within the Pcl7 gene, one of 37 known CLPs in the P. ramosa genome (Mouton et al. 2009; McElroy et al. 2011). Across the sequenced genomes, Pcl7, together with multiple other Pcl genes represented a disproportionately large fraction of the highest diversity regions. Among genome regions in the top 99th diversity percentile, Pcl genes accounted for 91% (48/53 annotated 100-bp windows, fig. 2B and supplementary table 3, Supplementary Material online). This includes four consecutive 100-bp windows within the Pcl7 gene as well as regions within 15 other Pcl genes.
Table 1.
Details of Infection Phenotype-Associated Polymorphisms within Pcl7.
| Nucleotide Position | Base: Infectious | Base: Noninfectious | Amino Acid: Infectious | Amino Acid: Noninfectious |
|---|---|---|---|---|
| 678 | T (one G exception: P37+) | C/G | G (synonymous) | G (synonymous) |
| 689 | A (one G exception: P37+) | G/T | Y (one W exception: P37+) | G/F/W |
| 690 | T (one G exception: P37+) | G/C | ||
| 691 | T | G (one T exception: P1032−) | S (one A exception: P37+) | A/V (one S exception: P1032−) |
| 693 | G | A | ||
| 691 | G | T/C | A/E | Y/H |
| 695 | C (one A exception: P37+) | A | ||
| 696 | A | T | ||
| 702 | T (one A exception: P37+) | A | G (synonymous) | G (synonymous) |
| 703 | G | A | G/V | S |
| 705 | A (one C exception: P37+) | T/C | ||
| 707 | G (one C exception: P37+) | A/C | G/A | E/S |
| 708 | T (one A exception: P37+) | A | ||
| 710 | C | G/T | A/T | G/V |
| 713 | G (one T exception: P37+) | C/T | G/F | A/V |
| 714 | A (one C exception: P37+) | C | ||
| 716 | C (one A exception: P37+) | A | A (one Y exception: P37+) | Y/E |
| 728 | C | T | A | V |
| 829 | G (one T exception: P1006+) | T | A (one S exception: P1006+) | L/S |
| 832 | T | C/G | S | P/G/A |
| 841 | G (one C exception: P37+) | C | G (one L exception: P37+) | L/R |
Note: Nucleotide and corresponding amino acid positions are based on translation-based alignment (supplementary fig. 2, supplementary material online). Gray shading indicates positions where infection phenotype is not perfectly correlated with nucleotide sequence, protein sequence, or both.
Fig. 1.

Map of the Western Palearctic with Pasteuria ramosa sampling localities indicated. Infection phenotypes are shown in red (positive attachment, infects host clone HU-HO-2) and blue (no attachment, does not infect host clone HU-HO-2).
Fig. 2.
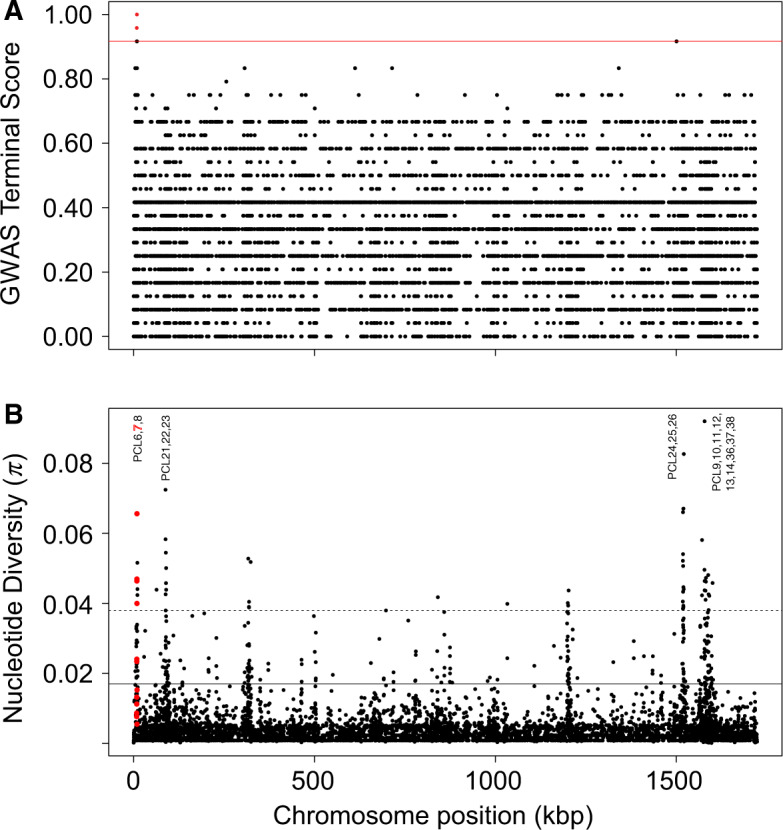
Genome scans of the 24 genome-sequenced Pasteuria ramosa clones. (A) Manhattan plot of the GWAS terminal test examining association between infection phenotype (relative to host clone HU-HO-2) and genetic polymorphisms across the tips of the P. ramosa phylogenetic tree. Red horizontal line indicates the significance threshold above which points indicate significant associations. Red points indicate significant polymorphisms, all located within the Pcl7 gene. (B) Nucleotide diversity (π). Each dot represents the average value of a given summary in a nonoverlapping window of 100 bp. Red dots represent bins that occur within the Pcl7 coding sequence. The horizontal solid black line delimits the 95% quantile, and the horizontal dotted black line delimits the 99% quantile. Text labels indicate closely spaced clusters of other Pcl genes that were also found to exhibit high levels of nucleotide diversity.
The two other tested infection phenotypes (relative to host clones FI-XINB3 and FI-KELA-39-9) produced no significant GWAS hits, and the “simultaneous” and “subsequent” tests produced no significant hits with respect to any phenotype. The positive GWAS tests provided the basis for additional Sanger sequencing of Pcl7 to validate these results.
Sanger Sequencing of Pcl7
Based on the Sanger sequences of all 56 clones in the P. ramosa diversity panel (24 genome-sequenced clones plus 32 additional clones), there were 157 variable sites (SNPs plus indels) across the 1,017-bp alignment of Pcl7 coding sequence (mean sequence length = 990.9 bp), equating to a variable site every 8.3 bases on average (supplementary fig. 2, Supplementary Material online). By comparison, the noncoding flanking sequence was less variable, with a variable site every 13.5 bases on average (39 variable sites across 526 bp). The variable sites within the coding sequence of Pcl7 resulted in 61 variable amino acids across the 339-residue alignment (mean sequence length = 330.3 residues).
Of the 21 perfectly phenotype-associated SNPs identified by GWAS, seven SNPs remained perfectly correlated with infection phenotype across the expanded sample set, and the remaining 14 were correlated with infection phenotype for all but one P. ramosa clone (table 1 and supplementary fig. 2, Supplementary Material online). The seven perfectly phenotype-associated polymorphisms correspond to a cluster of seven segregating amino acid substitutions across 47 residues, most of which represent substantial differences in size, hydrophobicity, and/or charge (table 1 and supplementary fig. 2, Supplementary Material online). Taken together, the cluster of segregating polymorphisms defines ten distinct haplotypes in the region of segregating polymorphisms (supplementary fig. 3, Supplementary Material online). The segregation of Pcl7 polymorphism by infection phenotype is also illustrated by a phylogeny of the entire gene sequence (fig. 3A), as all clones with a positive HU-HO-2/CH-H-67 infection phenotype form a monophyletic clade, despite the fact that these clones do not form a clade in the whole-genome phylogeny (fig. 3B).
Fig. 3.

Phylogenies of the 24 genome-sequenced Pasteuria ramosa clones based on (A) the full coding sequence of the Pcl7 gene and (B) the full genome sequence, excluding Pcl7. Red branch tips indicate clones that were infectious to host phenotype HU-HO-2/CH-H-67, and black branch tips indicate noninfectious clones.
Structural Inference
Searches of Pcl7 translated sequences against a database of known protein structures identified multiple potential structural homologs spanning part or all of the C-terminal half of the polypeptide. The most significant homolog identified was BclA, a CLP that is the major surface protein on the exosporium of bacteria in the genus Bacillus (UniProt: Q83WB0; Sylvestre et al. 2002; Todd et al. 2003), a close relative of Pasteuria (Charles et al. 2005; Schmidt et al. 2008). Bacterial CLPs are unified by the characteristic presence of an internal collagen-like region (CLR) composed of Gly–Xaa–Yaa repeats that can combine with other collagen-like polypeptides in hetero- and homotrimers to form triple helices. On either side of the CLR sit the N-terminal (NTD) and C-terminal (CTD) domains. The crystal structure of the CTD homotrimer of BclA has been solved by X-ray diffraction, revealing a globular jelly-roll topology composed of 12 β-strands that pack together to stabilize the core of the structure, with intervening loops between the β-strands forming the proximal and distal ends of the structure (Réty et al. 2005). Structural models of Pcl7 using BclA as a template align up to 90% of the CTD, including all of the key segregating sequence polymorphisms (fig. 4 and supplementary fig. 4, Supplementary Material online). Similar structural alignments were obtained regardless of which Pcl7 query sequence was used. Alignment quality across the entire sequence was variable (p = 7.4 × 10−6, Sequence similarity = 29%, Global Model Quality Estimation Score = 0.16), yet interspersed throughout were regions of high quality. The portions of the Pcl7 CTD that align best with BclA encompass the majority of the structurally important β-strands (fig. 4 and supplementary fig. 4, Supplementary Material online). These structural homologies, along with the phylogenetic proximity and biological similarities of Pasteuria and Bacillus, suggest that Pcl7 may possess a similar overall topology to BclA.
Fig. 4.

(A) Structural model of C-terminal globular domain of Pcl7 homotrimer, based on alignment with BclA. The collagen-like triple-helix domain and the N-terminus (not shown) would descend from the bottom center of the structure shown. The backbone is illustrated as a ribbon diagram with the transparent protein surface overlaid. Balls along the backbone of one of the three identical monomers indicate the locations of polymorphic residues whose variation segregates perfectly by infection phenotype. Balls and any corresponding surface-exposed area are highlighted according to their coordinates in the protein alignment: Red for the cluster of polymorphisms at residues 232–243, yellow for the polymorphism at residue 278, blue for the segregating glycosylation site at residues 308–310. Polymorphisms are named according to the residue(s) of the infectious haplotypes, the coordinate in the alignment, and the residue(s) of the noninfectious haplotypes. All polymorphisms are surface exposed, except residues 238 and 278. (B) The structure has been rotated forward 90° to show the top view.
Glycosylation
Based on the presence of specific target sequences as well as the predicted surface accessibility of those sequences, we identified 15 potential N-linked glycosites across all translated sequences of Pcl7. There were no potential O-linked glycosites identified in any of the Pcl7 sequences. Fourteen of the 15 predicted N-glycosites in Pcl7 were polymorphic among P. ramosa clones, and one of the sites on a surface-exposed loop of the CTD (NAS, nucleotide alignment positions 922–929; fig. 4 and supplementary fig. 2, Supplementary Material online) was predicted for all P. ramosa clones that were infectious to HU-HO-2/CH-H-67 but was not predicted for any of the noninfectious clones. Interestingly, the NAS target sequence was present in some noninfectious clones (7 out of 40) but was not identified as surface accessible in these sequences. Thus, although this predicted structural/functional feature of Pcl7 segregates perfectly with infection phenotype, it does not correlate precisely with an underlying sequence polymorphism.
A BLAST search (Altschul et al. 1997) of translated Pcl7 sequences against a database of verified glycoproteins (ProUGP) identified two strong hits, BclA and BclB (BclA, EMBL ID: AF246294, E = 3 × 10−49, 92/170 identities; BclB, EMBL ID: AE016879, E = 6 × 10−44, 96/170 identities), both CLPs from the genus Bacillus that are known to be highly glycosylated spore surface proteins (Charlton et al. 1999; Todd et al. 2003; Waller et al. 2005). This result corroborates the identification of BclA as a structural homolog of Pcl7 and suggests that, in addition to nucleotide/peptide polymorphisms, variation in glycosylation status may play a role in determining the infection phenotype of P. ramosa.
Discussion
Our study aimed to explore the molecular genetic basis of infectivity in P. ramosa, a bacterial pathogen of planktonic crustaceans that has become a model organism for studies of host–pathogen coevolution via NFDS. We performed a GWAS to investigate correlations between infection phenotype and genomic variation. We identified a single, highly diverse gene with a number of polymorphisms that correlate perfectly with an infection phenotype in all 56 P. ramosa clones surveyed.
The phenotype-associated gene, Pcl7, is a member of a class known as prokaryotic collagens or CLPs. Collagens are an abundant, widespread, and versatile class of proteins whose defining feature is a region of repetitive Gly–Xaa–Yaa peptide sequence that can combine in homo- or heterotrimers to form a supercoiled triple helix (Brodsky and Ramshaw 1997). Although collagens were originally thought to occur only in multicellular animals, they are now known to occur in all three domains of life, including a range of bacterial taxa (Rasmussen et al. 2003; Yu et al. 2014). CLPs have been implicated in the pathogenicity of a number of bacteria. For example, Clostridium difficile, a notorious gut pathogen, has at least three CLPs (Pizarro-Guajardo et al. 2014) that have been implicated in both infectivity and virulence (Phetcharaburanin et al. 2014). Streptococcus pyogenes, a clinically important agent of human skin infections, is known to have at least two CLPs that are anchored on the cell surface and bind to a number of host proteins (reviewed in Lukomski et al. 2017). Burkholderia mallei and B. pseudomallei, significant pathogens of the respiratory tracts of humans and livestock and known agents of biological weaponry, possess at least 13 CLPs that are thought to be involved in pathogenesis and multidrug resistance (Bachert et al. 2015). And a CLP of Legionella pneumophila, the causative agent of Legionnaire’s disease, was shown to be involved in the adhesion to and infection of host cells (Vandersmissen et al. 2010).
CLPs have also been previously suggested as possible candidates for infectivity of P. ramosa. Mouton et al. (2009) first identified a CLP in P. ramosa, Pcl1a, based on differential 2D gel electrophoresis patterns between two pathogen isolates with different infection phenotypes. Subsequently, McElroy et al. (2011) discovered at least 37 distinct Pcl genes across the P. ramosa genome and identified sequence polymorphisms at some of these genes. However, these studies were based on very few P. ramosa isolates. Our independent identification of a Pcl gene, using a de novo unbiased GWAS approach with a large and diverse sample, strongly corroborates the hypothesis that at least one CLP is involved in mediating the specificity of infection in P. ramosa.
Structural Inference
The sequence of CLPs can be divided into three domains: an internal repetitive CLR that forms the triple helix, flanked on either side by an NTD and a CTD. In our study, all infection phenotype-associated polymorphisms within Pcl7 were located in the CTD (fig. 4), a section of the protein that was found to have structural homology with the CTD of BclA, the principle CLP of Bacillus anthracis. In B. anthracis, which is the causative agent of anthrax and a close relative of Pasteuria (Charles et al. 2005; Schmidt et al. 2008), homotrimers of BclA compose the filaments of a hair-like nap that covers the outermost surface of the exosporium (Sylvestre et al. 2002, 2003). The NTD of BclA is known to provide the anchor point of the filament to the exosporium; the CLR composes the fibrous stalk, and the globular CTD is oriented outward forming the most distal tip of the filament (Boydston et al. 2005). These filaments have been implicated in the interaction of B. anthracis with host cells during infection (Bozue et al. 2007; Oliva et al. 2008, 2009).
Similar filaments (AKA microfibers, fimbriae, or pili) are also a conspicuous anatomical feature of Pasteuria spores (fig. 5; Davies 2009; Duneau et al. 2011). In their inert resting state, P. ramosa spores are encased within a durable outer exosporium (fig. 5A). Ingestion by any Daphnid host triggers the spore to indiscriminately shed its exosporium, releasing the endospore within (fig. 5B). The endospore is the active infectious stage, which either adheres to susceptible hosts or fails to adhere to resistant hosts in the key binary step of the infection process. The endospore has an overall shape that has been likened to a sombrero or a fried egg, and it has regions that are densely covered by filaments very similar in appearance to the hair-like nap of Bacillus spores (fig. 5C and D). It has been hypothesized that these filaments are the specific anatomical structures involved in adhesion of Pasteuria to its host (Davies 2009; Duneau et al. 2011). Although the molecular composition of the spore surface filaments of Pasteuria has not yet been determined, proteome sequencing of the endospore has identified an abundance of CLPs, including Pcl7 (Fredericksen M, unpublished data), indicating that this protein is expressed. If the Pcl7 protein in Pasteuria composes filaments on the surface of the endospore similar to the BclA filaments on the exosporium of Bacillus, which seems a reasonable hypothesis in light of the phylogenetic proximity of the taxa and the structural homology of the proteins, the surface-exposed segregating polymorphisms would be located at the absolute distal tip of the filament, one of the first physical points of contact between pathogen and host. Given that these polymorphisms result in a total of seven amino acid changes across a short stretch of sequence, most of which represent substantial differences in size, hydrophobicity, or charge, it is likely that they are consequential for the structure and function of the CTD of Pcl7.
Fig. 5.

Micrographs of Pasteuria ramosa spores, modified from Duneau et al. (2011), with permission. (A) Scanning electron micrograph of inactive spore with exosporium intact. (B) Transmission electron micrograph of spore “activation,” showing the endospore being released from the exosporium (ex). (C) Transmission electron micrograph of exosporium, showing the densely packed filaments (fil) on the surface. (D) Scanning electron micrograph of “activated” spores with densely packed surface filaments clearly visible.
In addition to sequence polymorphisms, we also identified a predicted N-glycosylation site in the CTD of Pcl7 (fig. 4) whose presence or absence correlates perfectly with infection phenotype. Glycans have been linked to infectivity in a wide variety of pathogens (Nothaft and Szymanski 2013; Rodrigues et al. 2015; Varki 2017), and BclA, the putative structural homolog of Pcl7, is also known to be highly glycosylated (Sylvestre et al. 2002; Maes et al. 2016). The infection phenotype-associated variation in a predicted glycosylation site, together with variation in amino acid sequence, located at the absolute distal tip of a putative spore surface protein, provide a highly plausible molecular basis for the strong genotypic specificity observed in host attachment and infection of P. ramosa. This hypothesized structure and function for Pcl7 is consistent with numerous other known examples of bacterial adhesins, which are typically organized as thread-like cell surface structures with the specific adhesive component at the distal tip (reviewed in Klemm and Schembri 2000).
Genomic and Geographic Patterns of Balancing Selection
Patterns of genetic diversity across the genome and across geographic space can help elucidate the demographic and selective processes that produced them. Because the sample set of this study aimed to maximize the evenness of known phenotypic diversity, it was not random and thus is not suited for rigorous quantitative population genetic analysis. Nonetheless, several strong and noteworthy patterns are evident, and these are unlikely to be artifacts of sampling design. First, the nucleotide diversity of Pcl7 was exceptionally high relative to the rest of the genome (fig. 2B), which is consistent with the action of balancing selection on this locus (Nielsen 2001; Charlesworth 2006; Nygaard et al. 2010; Ochola et al. 2010; Ebert and Fields 2020). Second, there was no discernable geographic pattern in the distribution of infection phenotypes or their corresponding Pcl7 haplotypes; despite the fact that Pcl7 haplotypes were segregated phylogenetically (fig. 3), infective haplotypes are more or less evenly spread across the entire sampling range (fig. 1). In contrast, the genome-wide population genetic structure of P. ramosa has been shown to follow a pattern of isolation by distance (IBD) across its range (Andras et al. 2018), a pattern that we also observed in the genome-wide SNP data of our 24 fully sequenced clones, as well as microsatellite data of our expanded diversity panel (data not shown). A similar disparity has been observed for the D. magna host, which shows a genome-wide pattern of IBD but no spatial structure of resistance phenotypes or genotypes (Bourgeois Y, personal communication). NFDS between pathogens and their hosts is expected to create a rapidly fluctuating geographic mosaic of genotypes that are involved in the interaction (Kaltz and Shykoff 1998; Thompson 2005; Tellier and Brown 2011), and genes under balancing selection are expected to have higher effective migration rates than neutral parts of the genome (Schierup et al. 2000; Muirhead 2001; Leducq et al. 2011; Ebert and Fields 2020). These processes will tend to obscure the signatures of slower and weaker demographic processes evident elsewhere across the genome. Thus, the unusually high diversity and lack of spatial patterning observed at Pcl7 supports the hypothesis that this infection phenotype-associated locus is undergoing Red Queen coevolution with the host.
The observed patterns of diversity at Pcl7 are also noteworthy because the source of variation for adaptive evolution is a topic of ongoing investigation (Barrett and Schluter 2008; Peter et al. 2012). For multicellular organisms, evidence suggests that selection acts predominantly on standing genetic variation (Jones et al. 2012; Peter et al. 2012; Reid et al. 2016; Lai et al. 2019). However, it is not clear whether the same is true for rapidly evolving unicellular organisms. Microparasites typically have very large population sizes, high mutation rates, and strongly structured populations (Woolhouse et al. 2002). Adaptive evolution at loci interacting with the host might therefore be expected to draw primarily upon de novo mutation, rather than standing genetic variation (Peter et al. 2012; Charlesworth et al. 2017). For P. ramosa, we observed the broad geographic distribution of a closely related clade of infection-associated haplotypes. This pattern suggests that the infection-positive phenotype did not arise primarily through independent novel mutations, but rather that the causative haplotype has persisted for some time as standing variation, and, aided by selection, has dispersed more broadly than neutral parts of the genome. An interesting exception to this pattern is seen in clone P37 from Israel, which is both a geographic and a genetic outlier (fig. 1 and supplementary fig. 5, Supplementary Material online). This clone has a positive infection phenotype, but its Pcl7 gene sequence is not a part of the monophyletic infection-positive clade and is quite divergent from all other sampled isolates. Despite this substantial disparity though, P37 shares with other infection-positive clones the seven perfectly segregating SNPs in the Pcl7 gene (table 1). Thus, the large majority of observed infection-associated Pcl7 haplotypes share a common origin, although a single rare outlier may have arisen independently through de novo mutation or recombination. Overall, these results indicate that balancing selection can maintain standing diversity in pathogens, as it does in their hosts, and this diversity can be geographically widespread and stable over time.
The patterns of variation observed at Pcl7 correlate with just one of the three infection phenotypes surveyed and do not therefore capture the global infectivity of a given P. ramosa clone. In fact, universally infectious phenotypes have never been observed for P. ramosa despite extensive sampling and study, and patterns of infection specificity are much more consistent with a matching-allele model of host–pathogen interaction and the associated trade-offs among different infection phenotypes (Luijckx et al. 2013; Ebert et al. 2016; Metzger et al. 2016; Bento et al. 2017). As evidence of this point, all of the clones with positive infection phenotypes in our GWAS analysis were unable to infect at least one of the other two host clones tested (supplementary table 1, Supplementary Material online). There are certainly other unscored infection phenotypes in our diversity panel as well as many unsampled infection phenotypes across the species’ range. Accordingly, there are likely other genes in the P. ramosa genome that are also involved in determining the specificity of infection. It has been previously speculated that the remarkable intragenomic diversity of the Pcl family, comprised at least 37 different genes, has been driven by coevolution with the host and is thus likely involved in infectivity (McElroy et al. 2011). Our results specifically confirm the involvement of one particular Pcl gene and also highlight patterns of interest across the broader Pcl gene family. In addition to Pcl7, a number of other Pcl genes were observed to have unusually high nucleotide diversity (fig. 2B and supplementary table 3, Supplementary Material online). Such metrics are commonly used as principle guiding criteria in the search for pathogenicity loci (reviewed in: Weedall and Conway 2010; Llaurens et al. 2017). It is quite possible that the abundant polymorphisms observed at other Pcl loci govern the specific patterns of other infection phenotypes. Given that collagen-like surface proteins can be composed of heterotrimers as well as homotrimers, it is also possible that the high diversity observed across the Pcl gene family could produce additional variation in infection phenotype through combinations of variants from different loci. The involvement of other Pcl genes in P. ramosa infectivity would also be consistent with our understanding of the genetic basis of resistance in the D. magna host, which includes at least three different loci (Metzger et al. 2016; Bento et al. 2017; Ameline et al. 2020; Bento et al. 2020). As the P. ramosa diversity panel is expanded and other infection phenotypes are described, the other identified highly polymorphic loci, many of which are Pcl genes, will serve as candidates of special interest in the effort to fully describe the genetics of pathogenicity.
Conclusions and Future Directions
The substantial body of research on the P. ramosa–D. magna system has led to valuable insights into the ecology and evolution of host–pathogen interactions. Yet our understanding of this system, like nearly all other natural host–pathogen associations, has heretofore been limited to the level of the phenotype. Here we extend our knowledge of this model system to the molecular level, providing strong evidence that a single CLP serves as a basis of infectivity in P. ramosa. Our findings join recent studies on the genetic basis of resistance in the host to confirm that this host–pathogen system evolves according to NFDS based on an underlying matching-allele architecture. Future studies of Pcl7 will explore the structure and functional significance of this protein through targeted biochemical and immunochemical manipulation in vivo to test the hypotheses proposed here. The continued expansion of the P. ramosa diversity panel will extend the investigation into the molecular bases of pathogenicity to new infection phenotypes and likely new genetic loci. In addition, studies of natural populations will be essential to connect spatial and temporal variation in genotypes and phenotypes with population dynamics. Together, these efforts will help produce a complete picture of Red Queen coevolution in this model host–pathogen system, from broad ecological and evolutionary patterns, through phenotypes, down to the fundamental molecular basis of the interaction.
Materials and Methods
Study Organism
Pasteuria ramosa is an endospore-forming, Gram-positive bacterium of the Bacillus–Clostridium clade that is an obligate pathogen of freshwater planktonic crustaceans, primarily D. magna (Ebert et al. 1996). Pasteuria ramosa disperses and infects its hosts via the environmental endospore stage, which resides primarily in the sediments of ponds and lakes. Daphnia ingest these spores during filter feeding, and, if the host and pathogen genotypes form a competent match, P. ramosa spores adhere to the host esophagus, after which they penetrate the body wall and grow vegetatively in the host’s hemolymph. Though infected hosts can live for about 50 days, their reproduction is halted shortly after infection, as energy is diverted to pathogen growth. Thus, the fitness consequences of infection are severe, and selection for resistance is commensurately strong (Ebert et al. 2016). Pasteuria ramosa sporulates within the host, and when infected hosts die, they fall to the benthos and decompose, releasing millions of spores to the pond sediment to begin the infection cycle again.
Pasteuria ramosa can be isolated from natural populations in the form of live infections or spore-laden sediments. It can be passaged repeatedly in susceptible hosts, and spores can be harvested, cleaned, and kept indefinitely for experiments and analysis. However, P. ramosa cannot be maintained in pure culture, and it is therefore not currently amenable to genetic manipulation. Thus, all genetic analyses of P. ramosa must be performed on either the nonpurified vegetative stage isolated directly from an infected host or endospores that can be isolated from the host and purified.
Collection and Preparation of the Pasteuria ramosa Diversity Panel
Nearly all of the P. ramosa clones included in this study were isolated via experimental infection from pond sediment samples as a part of a previously reported population genetic survey (Andras et al. 2018). For this survey, P. ramosa isolates were genotyped at 12 polymorphic microsatellite loci, which allowed us to determine the diversity of haplotypes in each isolate and to identify which isolates were genetically distinct. From this much larger sample set, we used four criteria to select a subset of clones to compose the P. ramosa diversity panel. We chose 1) genetically distinct isolates 2) that appeared to be monoclonal or nearly so 3) from as broad a geographic range as possible 4) representing an even sample of phenotypes based on the D. magna clone(s) they were known to infect. To verify the monoclonal status of these isolates, or, in some cases, to reduce their original diversity to a single clone, the initial infections that were isolated from the sediment were serially passaged three additional times at low spore dosage (as in Luijckx et al. 2011) using the same D. magna host clone with which they were originally isolated. After each passage, the resulting isolate was genotyped again. Isolates that had a distinct monoclonal haplotype for at least the last two sequential passages were classified as clones and included in the P. ramosa diversity panel. We selected 21 distinct clones from 17 localities, along with three previously isolated and characterized laboratory clones for whole-genome sequencing. After a GWAS analysis of these 24 genomes produced a candidate locus for infectivity (see below), we produced an independent sample of another 32 genetically distinct P. ramosa clones from 21 localities for targeted Sanger sequencing of the candidate locus. In total, the P. ramosa diversity panel employed in this study includes 56 (24 + 32) genetically distinct clones from 28 geographic locations spanning 4,000 km across Europe, Western Asia, and the Near East (supplementary table 1, Supplementary Material online and fig. 1).
Infection Phenotyping
To determine the infection phenotype of clones in the P. ramosa diversity panel, we used a previously published assay that measures the ability of spores to adhere to the host foregut (Duneau et al. 2011). This is an essential and binary step in the infection process—spore attachment results in successful infection, whereas the inability of spores to attach indicates host resistance (Ebert et al. 2016). We chose six different genotypes of D. magna for testing, based on known differences in their resistance phenotype (supplementary table 2, Supplementary Material online). Clonal offspring from each of the six D. magna genotypes were individually exposed to 104 fluorescently labeled spores of each P. ramosa clone, and spore attachment to the host foregut was ascertained with a fluorescence microscope. Twelve replicate clonal offspring were tested for each D. magna genotype × P. ramosa clone combination, and combinations were scored as a positive match when at least 10 of 12 replicates showed clear adhesion.
DNA Extraction and Whole-Genome Sequencing
Approximately 200 million mature spores of each P. ramosa clone were harvested from ten infected D. magna individuals 6 weeks after the third serial passage infection. Pooled infected D. magna were suspended in 1 ml of sterile water and crushed with a plastic pestle, and the resulting solution was passed through a sterile 40-μm cell strainer. Pasteuria ramosa spores were pelleted via centrifugation for 3 min at 16 RCF, and the supernatant was removed. Spores were washed via resuspension/centrifugation twice more with 1 ml distilled water. Contaminating bacteria and D. magna tissue in the spore samples was digested with lysozyme (1 h at 37 °C in 1 ml TES buffer with 5 μl of 250 U/μl Epicentre Ready-Lyse lysozyme), followed by proteinase K (12 h at 56 °C, 16 μl of 20 mg/ml proteinase K). These conditions were determined in advance to not lyse the P. ramosa spores. The spores were then washed three additional times with 1 ml sterile water, and contaminating DNA was removed by digesting with DNase (30 min at 37 °C in 400 μl reaction buffer with 3 μl of 1 U/μl Promega RQ1 DNase). DNase digestion was terminated with 40 μl stop solution, and spores were washed three additional times with 1 ml sterile water. The cleaned spores were then lysed via bead beating and enzymatic digestion, and DNA was extracted according to previously published protocols (Andras and Ebert 2013). Because previous testing had revealed consistently low yields, all P. ramosa DNA extracts were whole-genome amplified using a Qiagen Repli-g Mini Kit according to the manufacturer’s protocols. Whole-genome amplifications were sequenced via Illumina MiSeq PE 250 bp using the Nextera XT DNA preparation kit. Sequencing was performed at the Department of Biosystems Science and Engineering, ETH-Zurich in Basel, Switzerland.
Genome Assembly, SNP Calling, and Descriptive Statistics
Read quality was assessed using FastQC v.0.10.1 (Andrews 2010). Trimmomatic was used to remove Illumina adapters as well as to trim low-quality sequences (Bolger et al. 2014). In order to generate reference-assisted assemblies of individual P. ramosa isolates, we used the Assembly by Reduced Complexity, or ARC, pipeline (Hunter et al. 2015). Briefly, ARC works by mapping a set of reads against a reference, extraction of mapped reads, and de novo assembly of mapped reads, a process which is iterated until further improvement of assembly completeness cannot be made. Within the ARC pipeline, we used Bowtie2 (Langmead and Salzberg 2012; Langmead et al. 2019) for mapping and the assembler SPAdes v. 3.8 (Nurk et al. 2013). For initial read mapping, we used a previously assembled, single scaffold reference sequence and annotation of the C1 clone of P. ramosa (Paljakka M, Fields PD, Ebert D, in preparation; NCBI; genome accession number: QBIE00000000). In order to improve the contiguity of the resultant ARC-based assemblies, we used the Ragout scaffolder (Kolmogorov et al. 2014, 2018), once again using the C1 assembly as a reference.
Previously trimmed reads used for assembly were also mapped to the reference sequence of the C1 clone of P. ramosa using BWA-MEM (Li and Durbin 2009; Li 2013), and the resulting SAM alignment file was converted to a BAM file and coordinate-sorted using SAMtools (Li and Durbin 2009). Duplicate reads were marked using Picard tools (Picard Toolkit 2018). Variant calls were made using GATK HaplotypeCaller (McKenna et al. 2010). We used a haploid-aware version of VCFtools (Danecek et al. 2011) to calculate nucleotide diversity (π) in 100-bp, nonoverlapping bins across the genome.
A FASTA alignment was generated from the genome-wide biallelic SNPs using Variant Call Format Kit (Cook and Andersen 2017). After exclusion of all SNPs in the 1,017 bp coding region of the Pcl7 candidate gene (see details below), the resultant alignment was used to generate a genome-wide phylogenetic tree using BEAST2 (Bouckaert et al. 2019). Within BEAST2, we specified a single partition, a general time reversible substitution model (frequencies estimated), strict clock, and a coalescent constant population prior. The Markov chain Monte Carlo (MCMC) settings in BEAST2 included a chain length of 100 million iterations, with a sample drawn every 1,000 iterations. The convergence of the MCMC run resulting from BEAST2 was assessed using Tracer (Rambaut et al. 2018). All estimated parameters reached an effective sample size >1,000. We used the BEAST2 tool TreeAnnotator to remove the first 50% of the MCMC chain as burn-in and generate a maximum clade credibility tree with median node heights.
GWAS Analysis
To test for statistical correlations between genomic sequence polymorphisms and infection phenotypes, we used a GWAS approach on the 24 fully sequenced P. ramosa genomes. This relatively low initial sample size was a consequence of the practical challenge of isolating new P. ramosa clones from the environment. Although low sample sizes can limit the statistical power of GWAS approaches, these limitations are countered by other attributes of our study system. Namely, the infection phenotypes of interest are binary, suggesting that they are controlled by few genes of large effect. Also, the phenotypes show no environmental plasticity (Duneau et al. 2011) and can be assessed across clonal replicates, which allows phenotypes to be assigned with near perfect accuracy and without loss of statistical power due to noisy data.
We performed GWAS tests as implemented by the treeWAS R package (Collins and Didelot 2018). This method, designed specifically for use with bacteria, uses a phylogenetic approach to maintain high statistical power for association tests while controlling for the potentially confounding effects of population structure and recombination. The treeWAS method simultaneously tests for three different types of association (Collins and Didelot 2018). The “terminal test” examines association across only the tips of the phylogenetic tree, aiming to identify broad patterns of correlation. The “simultaneous test” uses parsimony to reconstruct ancestral states on a phylogeny and examine simultaneous changes in phenotypic and genotypic states throughout the phylogeny. The “subsequent test” measures the proportion of branches in the phylogeny for which the genotype and phenotype are in the same state. The software generates null distributions of simulated association scores to determine a specific significance threshold for each test, and any scores exceeding that threshold indicate a significant association. The authors of treeWAS emphasize that an association deemed significant by any one of these three tests is a viable candidate for further investigation.
For our GWAS analysis, a matrix of genome-wide binary SNP and indel data from the 24 genome-sequenced P. ramosa clones was compared individually with vectors of binary phenotype data (i.e., columns of binary data indicating whether or not individual P. ramosa clones adhered to a given host). Separate tests were performed for each infection phenotype using the default treeWAS parameters and Bonferroni-adjusted significance thresholds. Two of the six tested host clones (HU-HO-2 from Hungary and CH-H-67 from Switzerland) were infected by the exact same P. ramosa clones, so these GWAS tests were effectively identical. Two other host clones (DE-K1-IINB1 from Germany and CH-H-159 from Switzerland) were not infected by any of the 24 P. ramosa clones, so GWAS tests relative to these hosts were not possible, resulting in three distinct infection phenotypes that were tested.
Sanger Sequencing of Pcl7
The candidate-free GWAS analyses identified several phenotype-associated sequence polymorphisms in a single gene, Pcl7 (see Results for details). To expand the sample set and to verify the sequence polymorphisms using a candidate test, we used Sanger sequencing to examine a region encompassing the gene for Pcl7 in all 56 clones from the P. ramosa diversity panel (24 original genome-sequenced clones + 32 additional clones). Using sequence data from the whole-genome alignments, we designed primers to amplify 1,596 bp encompassing Pcl7 (forward: 5′-CCTTACCCAGGTGGCACAAT-3′; reverse: 5′-CCTTACCCAGGTGGCACAAT-3′). DNA was extracted from the 32 additional P. ramosa clones as described by Andras and Ebert (2013), and polymerase chain reaction (PCR) was performed using the Qiagen Taq PCR Master Mix Kit according to the manufacturer’s protocols (annealing temperature = 60°). Amplicons were sequenced in both the forward and reverse directions at Microsynth AG (Switzerland). Paired reads were assembled into continuous full-length sequences, and all sequences were aligned based on translation using ClustalW in GENIOUS PRIME. All polymorphisms were verified by manual inspection. Based on this alignment, we constructed a phylogenetic tree of Pcl7 following the same approach as the whole-genome tree.
Structural Inference/Glycosylation
To investigate possible consequences of the phenotype-associated polymorphisms, we compared the translated sequences of Pcl7 with protein databases of known structural and functional characteristics. First, we used SWISSMODEL (Waterhouse et al. 2018) and RaptorX (Källberg et al. 2012), which are server-based analysis suites that construct tertiary structural models of a polypeptide query sequence based on alignment to homologous proteins with solved structures. We also searched translated Pcl7 sequences for potential glycosylation sites using GLYCOPP v1.0 (Chauhan et al. 2012). This approach, which is specifically designed for prokaryotes, uses both local sequence identity and predicted surface accessibility to infer potential N- and O-glycosylation sites. We used a prediction model based on Average Surface Accessibility and Binary Profile of Patterns (ASA + BPP) and the default SVM threshold of 0. Finally, we used Blast search (Altschul et al. 1997) to compare translated Pcl7 sequences with a database of verified glycoproteins (ProUGP).
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was funded by the Swiss National Science Foundation, Grant number: 310030B_166677. The authors would like to thank Jean-Claude Walser for consultation on analysis and Jürgen Hottinger and Urs Stiefel for assistance in the laboratory.
Author Contributions
Conceptualization: J.P.A. and D.E.; Methodology: J.P.A. and P.D.F; Resources: J.P.A., M.F., and D.E; Investigation: J.P.A; Formal analysis: J.P.A., P.D.F, and L.D.P; Software: J.P.A. and P.D.F; Writing—original draft: J.P.A.; Writing review and editing: J.P.A., P.D.F., L.D.P, M.F., and D.E.; and Funding acquisition: D.E. and J.P.A.
References
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ.. 1997. Gapped Blast and PSI-Blast. Nucleic Acids Res. 25(17):3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameline C, Bourgeois Y, Voegtli F, Savola E, Andras JP, Engelstaeter J, Ebert D.. 2020. A two-locus system with strong epistasis underlies rapid parasite-mediated evolution of host resistance. BioRxiv. doi: 10.1101/2020.06.11.145391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andras JP, Ebert D.. 2013. A novel approach to parasite population genetics: experimental infection reveals geographic differentiation, recombination and host-mediated population structure in Pasteuria ramosa, a bacterial parasite of Daphnia. Mol Ecol. 22(4):972–986. [DOI] [PubMed] [Google Scholar]
- Andras JP, Fields PD, Ebert D.. 2018. Spatial population genetic structure of a bacterial parasite in close coevolution with its host. Mol Ecol. 27(6):1371–1384. [DOI] [PubMed] [Google Scholar]
- Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. Available from:https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. [Google Scholar]
- Ayala FJ, Campbell CA.. 1974. Frequency-dependent selection. Annu Rev Ecol Syst. 5(1):115–138. [Google Scholar]
- Bachert BA, Choi SJ, Snyder AK, Rio RVM, Durney BC, Holland LA, Amemiya K, Welkos SL, Bozue JA, Cote CK, et al. 2015. A unique set of the Burkholderia collagen-like proteins provides insight into pathogenesis, genome evolution and niche adaptation, and infection detection. PLoS One. 10(9):e0137578–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett R, Schluter D.. 2008. Adaptation from standing genetic variation. Trends Ecol Evol. 23(1):38–44. [DOI] [PubMed] [Google Scholar]
- Bento G., Fields PD., Duneau D., Ebert D. 2020. An alternative route of bacterial infection associated with a novelresistance locus in the Daphnia–Pasteuria host–parasite system. Heredity 1–11. https://doi.org/10.1038/s41437-020-0332-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bento G, Routtu J, Fields PD, Bourgeois Y, Du Pasquier L, Ebert D.. 2017. The genetic basis of resistance and matching-allele interactions of a host-parasite system: the Daphnia magna-Pasteuria ramosa model. PLoS Genet. 13(2):e1006596–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert R Vaughan TG Barido-Sottani J Duchêne S Fourment M Gavryushkina A Heled J Jones G Kühnert D De Maio N, . et al. 2019. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 15(4):e1006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boydston JA, Chen P, Steichen CT, Turnbough CL.. 2005. Orientation within the exosporium and structural stability of the collagen-like glycoprotein BclA of Bacillus anthracis. J Bacteriol. 187(15):5310–5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozue J, Moody KL, Cote CK, Stiles BG, Friedlander AM, Welkos SL, Hale ML.. 2007. Bacillus anthracis spores of the bclA mutant exhibit increased adherence to epithelial cells, fibroblasts, and endothelial cells but not to macrophages. Infect Immun. 75(9):4498–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky B, Ramshaw JA.. 1997. The collagen triple-helix structure. Matrix Biol. 15(8–9):545–554. [DOI] [PubMed] [Google Scholar]
- Carius HJ, Little TJ, Ebert D.. 2001. Genetic variation in a host‐parasite association: potential for coevolution and frequency‐dependent selection. Evolution 55(6):1136–1145. [DOI] [PubMed] [Google Scholar]
- Charles L, Carbone I, Davies KG, Bird D, Burke M, Kerry BR, Opperman CH.. 2005. Phylogenetic analysis of Pasteuria penetrans by use of multiple genetic loci. J Bacteriol. 187(16):5700–5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Charlesworth D, Coyne JA, Langley CH.. 2016. Hubby and Lewontin on protein variation in natural populations: when molecular genetics came to the rescue of population genetics. Genetics 203(4):1497–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth D. 2006. Balancing selection and its effects on sequences in nearby genome regions. PLoS Genet. 2(4):e64–e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth D, Barton NH, Charlesworth B.. 2017. The sources of adaptive variation. Proc R Soc B. 284(1855):20162864–20162812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton S, Moir AJ, Baillie L, Moir A.. 1999. Characterization of the exosporium of Bacillus cereus. J Appl Microbiol. 87(2):241–245. [DOI] [PubMed] [Google Scholar]
- Chauhan JS, Bhat AH, Raghava GPS, Rao A.. 2012. GlycoPP: a webserver for prediction of N- and O-glycosites in prokaryotic protein sequences. PLoS One. 7(7):e40155–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke BC. 1979. The evolution of genetic diversity. Proc R Soc B. 205:453–474. [DOI] [PubMed] [Google Scholar]
- Collins C, Didelot X.. 2018. A phylogenetic method to perform genome-wide association studies in microbes that accounts for population structure and recombination. PLoS Comput Biol. 14(2):e1005958–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DE, Andersen EC. 2017. VCF-kit: assorted utilities for the variant call format. Bioinformatics 33(10):1581–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 2011. The variant call format and VCFtools. Bioinformatics 27(15):2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies K. 2009. Understanding the interaction between an obligate hyperparasitic bacterium, Pasteuria penetrans and its obligate plant-parasitic nematode host, Meloidogyne spp. Adv Parasitol. 68:211–245. [DOI] [PubMed] [Google Scholar]
- Decaestecker E, Gaba S, Raeymaekers JAM, Stoks R, Van Kerckhoven L, Ebert D, De Meester L.. 2007. Host–parasite “Red Queen” dynamics archived in pond sediment. Nature 450(7171):870–873. [DOI] [PubMed] [Google Scholar]
- Duneau D, Luijckx P, Ben-Ami F, Laforsch C, Ebert D.. 2011. Resolving the infection process reveals striking differences in the contribution of environment, genetics and phylogeny to host-parasite interactions. BMC Biol. 9:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert D, Fields PD. Forthcoming 2020. Host-parasite coevolution and its genomic signature. Nat Rev Genet. [DOI] [PubMed] [Google Scholar]
- Ebert D. 2008. Host–parasite coevolution: insights from the Daphnia–parasite model system. Curr Opin Microbiol. 11(3):290–301. [DOI] [PubMed] [Google Scholar]
- Ebert D. 2018. Open questions: what are the genes underlying antagonistic coevolution? BMC Biol. 16(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert D, Carius HJ, Little T, Decaestecker E.. 2004. The evolution of virulence when parasites cause host castration and gigantism. Am Nat. 164(S5):S19–S32. [DOI] [PubMed] [Google Scholar]
- Ebert D, Duneau D, Hall MD, Luijckx P, Andras JP, Du Pasquier L, Ben-Ami F.. 2016. A population biology perspective on the stepwise infection process of the bacterial pathogen Pasteuria ramosa in Daphnia. Adv Parasitol. 91:265–310. [DOI] [PubMed] [Google Scholar]
- Ebert D, Rainey P, Embley TM, Scholz D.. 1996. Development, life cycle, ultrastructure and phylogenetic position of Pasteuria ramosa Metchnikoff 1888: rediscovery of an obligate endoparasite of Daphnia magna straus. Philos Trans R Soc B. 351:1689–1701. [Google Scholar]
- Hamilton WD. 1980. Sex versus non-sex versus parasite. Oikos 35(2):282–290. [Google Scholar]
- Hunter SS, Lyon RT, Sarver BAJ, Hardwick K, Forney LJ, Settles ML.. 2015. Assembly by reduced complexity (ARC): a hybrid approach for targeted assembly of homologous sequences. BioRxiv. doi:/10.1101/014662. [Google Scholar]
- Jaenike J. 1978. An hypothesis to account for the maintenance of sex within populations. Evol Theory. 3:191–194. [Google Scholar]
- Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J, Swofford R, Pirun M, Zody MC, White S, et al. 2012. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484(7392):55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Källberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J.. 2012. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 7(8):1511–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltz O, Shykoff JA.. 1998. Local adaptation in host–parasite systems. Heredity 81(4):361–370. [Google Scholar]
- Klemm P, Schembri MA.. 2000. Bacterial adhesins: function and structure. Int J Med Microbiol. 290(1):27–35. [DOI] [PubMed] [Google Scholar]
- Kolmogorov M, Armstrong J, Raney BJ, Streeter I, Dunn M, Yang F, Odom D, Flicek P, Keane TM, Thybert D, et al. 2018. Chromosome assembly of large and complex genomes using multiple references. Genome Res. 28(11):1720–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmogorov M, Raney B, Paten B, Pham S.. 2014. Ragout: a reference-assisted assembly tool for bacterial genomes. Bioinformatics 30(12):i302–i309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskella B, Brockhurst MA.. 2014. Bacteria–phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol Rev. 38(5):916–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y-T, Yeung CKL, Omland KE, Pang E-L, Hao Y, Liao B-Y, Cao H-F, Zhang B-W, Yeh C-F, Hung C-M, et al. 2019. Standing genetic variation as the predominant source for adaptation of a songbird. Proc Natl Acad Sci U S A. 116(6):2152–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL.. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9(4):357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Wilks C, Antonescu V, Charles R.. 2019. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 35(3):421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leducq J-B, Llaurens V, Castric V, Saumitou-Laprade P, Hardy OJ, Vekemans X.. 2011. Effect of balancing selection on spatial genetic structure within populations: theoretical investigations on the self-incompatibility locus and empirical studies in Arabidopsis halleri. Heredity 106(2):319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin RC, Hubby JL.. 1966. A molecular approach to the study of genic heterozygosity in natural populations. II. Amount of variation and degree of heterozygosity in natural populations of Drosophila pseudoobscura. Genetics 54:595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arxiv.org. [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llaurens V, Whibley A, Joron M.. 2017. Genetic architecture and balancing selection: the life and death of differentiated variants. Mol Ecol. 26(9):2430–2448. [DOI] [PubMed] [Google Scholar]
- Luijckx P, Ben-Ami F, Mouton L, Du Pasquier L, Ebert D.. 2011. Cloning of the unculturable parasite Pasteuria ramosa and its Daphnia host reveals extreme genotype-genotype interactions. Ecol Lett. 14(2):125–131. [DOI] [PubMed] [Google Scholar]
- Luijckx P, Fienberg H, Duneau D, Ebert D.. 2013. A matching-allele model explains host resistance to parasites. Curr Biol. 23(12):1085–1088. [DOI] [PubMed] [Google Scholar]
- Lukomski S, Bachert BA, Squeglia F, Berisio R.. 2017. Collagen-like proteins of pathogenic streptococci. Mol Microbiol. 103(6):919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes E, Krzewinski F, Garenaux E, Lequette Y, Coddeville B, Trivelli X, Ronse A, Faille C, Guerardel Y.. 2016. Glycosylation of BclA glycoprotein from Bacillus cereus and Bacillus anthracis exosporium is domain-specific. J Biol Chem. 291(18):9666–9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElroy K, Mouton L, Du Pasquier L, Qi W, Ebert D.. 2011. Characterisation of a large family of polymorphic collagen-like proteins in the endospore-forming bacterium Pasteuria ramosa. Res Microbiol. 162(7):701–714. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20(9):1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger C, Luijckx P, Bento G, Mariadassou M, Ebert D.. 2016. The Red Queen lives: epistasis between linked resistance loci. Evolution 70(2):480–487. [DOI] [PubMed] [Google Scholar]
- Mouton L, Traunecker E, McElroy K, Du Pasquier L, Ebert D.. 2009. Identification of a polymorphic collagen-like protein in the crustacean bacteria Pasteuria ramosa. Res Microbiol. 160(10):792–799. [DOI] [PubMed] [Google Scholar]
- Muirhead CA. 2001. Consequences of population structure on genes under balancing selection. Evolution 55(8):1532–1541. [DOI] [PubMed] [Google Scholar]
- Nielsen R. 2001. Statistical tests of selective neutrality in the age of genomics. Heredity 86(6):641–647. [DOI] [PubMed] [Google Scholar]
- Nothaft H, Szymanski CM.. 2013. Bacterial protein N-glycosylation: new perspectives and applications. J Biol Chem. 288(10):6912–6920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski AD, Pyshkin A, Sirotkin A, Sirotkin Y, et al. 2013. Assembling single-cell genomes and mini-metagenomes from highly chimeric MDA products. J Comput Biol. 20(10):714–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard S, Braunstein A, Malsen G, Van Dongen S, Gardner PP, Krogh A, Otto TD, Pain A, Berriman M, McAuliffe J, et al. 2010. Long- and Short-term selective forces on Malaria parasite genomes. PLoS Genet. 6(9):e1001099–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochola LI, Tetteh KKA, Stewart LB, Riitho V, Marsh K, Conway DJ.. 2010. Allele frequency-based and polymorphism-versus-divergence indices of balancing selection in a new filtered set of polymorphic genes in Plasmodium falciparum. Mol Biol Evol. 27(10):2344–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva C, Turnbough CL, Kearney JF.. 2009. CD14-Mac-1 interactions in Bacillus anthracis spore internalization by macrophages. Proc Natl Acad Sci U S A. 106(33):13957–13962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva CR, Swiecki MK, Griguer CE, Lisanby MW, Bullard DC, Turnbough CL, Kearney JF.. 2008. The integrin Mac-1 (CR3) mediates internalization and directs Bacillus anthracis spores into professional phagocytes. Proc Natl Acad Sci U S A. 105(4):1261–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter BM, Huerta-Sanchez E, Nielsen R.. 2012. Distinguishing between selective sweeps from standing variation and from a de novo mutation. PLoS Genet. 8(10):e1003011–e1003014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phetcharaburanin J, Hong HA, Colenutt C, Bianconi I, Sempere L, Permpoonpattana P, Smith K, Dembek M, Tan S, Brisson M-C, et al. 2014. The spore-associated protein BclA1 affects the susceptibility of animals to colonization and infection by Clostridium difficile. Mol Microbiol. 92(5):1025–1038. [DOI] [PubMed] [Google Scholar]
- Pizarro-Guajardo M, Olguín-Araneda V, Barra-Carrasco J, Brito-Silva C, Sarker MR, Paredes-Sabja D.. 2014. Characterization of the collagen-like exosporium protein, BclA1, of Clostridium difficile spores. Anaerobe 25:18–30. [DOI] [PubMed] [Google Scholar]
- Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. 2018. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst Biol. 67(5):901–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen M, Jacobsson M, Björck L.. 2003. Genome-based identification and analysis of collagen-related structural motifs in bacterial and viral proteins. J Biol Chem. 278(34):32313–32316. [DOI] [PubMed] [Google Scholar]
- Reid NM, Proestou DA, Clark BW, Warren WC, Colbourne JK, Shaw JR, Karchner SI, Hahn ME, Nacci D, Oleksiak MF, et al. 2016. The genomic landscape of rapid repeated evolutionary adaptation to toxic pollution in wild fish. Science 354(6317):1305–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Réty S, Salamitou S, Garcia-Verdugo I, Hulmes DJS, Le Hégarat F, Chaby R, Lewit-Bentley A.. 2005. The crystal structure of the Bacillus anthracis spore surface protein BclA shows remarkable similarity to mammalian proteins. J Biol Chem. 280(52):43073–43078. [DOI] [PubMed] [Google Scholar]
- Rodrigues JA, Acosta-Serrano A, Aebi M, Ferguson MAJ, Routier FH, Schiller I, Soares S, Spencer D, Titz A, Wilson IBH, et al. 2015. Parasite glycobiology: a bittersweet symphony. PLoS Pathog. 11(11):e1005169–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routtu J, Ebert D.. 2015. Genetic architecture of resistance in Daphnia hosts against two species of host-specific parasites. Heredity 114(2):241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salathe M, Kouyos R, Bonhoeffer S.. 2008. The state of affairs in the kingdom of the Red Queen. Trends Ecol Evol. 23(8):439–445. [DOI] [PubMed] [Google Scholar]
- Samson JE, Magadán AH, Sabri M, Moineau S.. 2013. Revenge of the phages: defeating bacterial defences. Nat Rev Microbiol. 11(10):675–687. [DOI] [PubMed] [Google Scholar]
- Schierup MH, Vekemans X, Charlesworth D.. 2000. The effect of subdivision on variation at multi-allelic loci under balancing selection. Genet Res. 76(1):51–62. [DOI] [PubMed] [Google Scholar]
- Schmidt LM, Mouton L, Nong G, Ebert D, Preston JF.. 2008. Genetic and immunological comparison of the cladoceran parasite Pasteuria ramosa with the nematode parasite Pasteuria penetrans. Appl Environ Microbiol. 74(1):259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvestre P, Couture-Tosi E, Mock M.. 2002. A collagen-like surface glycoprotein is a structural component of the Bacillus anthracis exosporium. Mol Microbiol. 45(1):169–178. [DOI] [PubMed] [Google Scholar]
- Sylvestre P, Couture-Tosi E, Mock M.. 2003. Polymorphism in the collagen-like region of the Bacillus anthracis BclA protein leads to variation in exosporium filament length. J Bacteriol. 185(5):1555–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellier A, Brown J.. 2011. Spatial heterogeneity, frequency-dependent selection and polymorphism in host-parasite interactions. BMC Evol Biol. 11(1):319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JN. 2005. Coevolution: the geographic mosaic of coevolutionary arms races. Curr Biol. 15(24):R992–R994. [DOI] [PubMed] [Google Scholar]
- Todd SJ, Moir AJG, Johnson MJ, Moir A.. 2003. Genes of Bacillus cereus and Bacillus anthracis encoding proteins of the exosporium. J Bacteriol. 185(11):3373–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandersmissen L, De Buck E, Saels V, Coil DA, Anné J.. 2010. A Legionella pneumophila collagen-like protein encoded by a gene with a variable number of tandem repeats is involved in the adherence and invasion of host cells. FEMS Microbiol Lett. 306(2):168–176. [DOI] [PubMed] [Google Scholar]
- Varki A. 2017. Biological roles of glycans. Glycobiology 27(1):3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller LN, Stump MJ, Fox KF, Harley WM, Fox A, Stewart GC, Shahgholi M.. 2005. Identification of a second collagen-like glycoprotein produced by Bacillus anthracis and demonstration of associated spore-specific sugars. J Bacteriol. 187(13):4592–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46(W1):W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weedall GD, Conway DJ.. 2010. Detecting signatures of balancing selection to identify targets of anti-parasite immunity. Trends Parasitol. 26(7):363–369. [DOI] [PubMed] [Google Scholar]
- Woolhouse MEJ, Webster JP, Domingo E, Charlesworth B, Levin BR.. 2002. Biological and biomedical implications of the co-evolution of pathogens and their hosts. Nat Genet. 32(4):569–577. [DOI] [PubMed] [Google Scholar]
- Yu Z, An B, Ramshaw JAM, Brodsky B.. 2014. Bacterial collagen-like proteins that form triple-helical structures. J Struct Biol. 186(3):451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.