

Abstract
Introduction
Allopregnanolone is an endogenous neurosteroid with the potential to be a novel regenerative therapeutic for Alzheimer's disease (AD). Foundations of mechanistic understanding and well‐established preclinical safety efficacy make it a viable candidate.
Methods
A randomized, double‐blinded, placebo‐controlled, single and multiple ascending dose trial was conducted. Intravenous allopregnanolone or placebo was administered once‐per‐week for 12 weeks with a 1‐month follow‐up. Participants with early AD (mild cognitive impairment due to AD or mild AD), a Mini‐Mental State Examination score of 20–26 inclusive, and age ≥55 years were randomized (6:2 to three allopregnanolone dosing cohorts or one placebo cohort). Primary endpoint was safety and tolerability. Secondary endpoints included pharmacokinetic (PK) parameters and maximally tolerated dose (MTD). Exploratory endpoints included cognitive and imaging biomarkers.
Results
A total of 24 participants completed the trial. Allopregnanolone was safe and well tolerated in all study participants. No differences were observed between treatment arms in the occurrence and severity of adverse events (AE). Most common AE were mild to moderate in severity and included rash (n = 4 [22%]) and fatigue (n = 3 [17%]). A single non‐serious AE, dizziness, was attributable to treatment. There was one serious AE not related to treatment. Pharmacokinetics indicated a predictable linear dose‐response in plasma concentration of allopregnanolone after intravenous administration over 30 minutes. The maximum plasma concentrations for the 2 mg, 4 mg, 6 mg, and 10 mg dosages were 14.53 ng/mL (+/−7.31), 42.05 ng/mL (+/−14.55), 60.07 ng/mL (+/−12.8), and 137.48 ng/mL (+/−38.69), respectively. The MTD was established based on evidence of allopregnanolone‐induced mild sedation at the highest doses; a sex difference in the threshold for sedation was observed (males 10 mg; females 14 mg). No adverse outcomes on cognition or magnetic resonance imaging–based imaging outcomes were evident.
Conclusions
Allopregnanolone was well tolerated and safe across all doses in persons with early AD. Safety, MTD, and PK profiles support advancement of allopregnanolone as a regenerative therapeutic for AD to a phase 2 efficacy trial.
Trial registration
ClinicalTrials.gov‐NCT02221622
Keywords: allopregnanolone, Alzheimer’s disease, maximally tolerated dose, neurogenesis, pharmacokinetics, phase 1 clinical trial, regenerative therapeutic, translational research
1. INTRODUCTION
Thus far no interventions have demonstrated meaningful therapeutic efficacy to treat Alzheimer's disease (AD) resulting in a 99.6% clinical trial failure rate with several accelerating disease progression. 1 , 2 , 3 , 4 Current thinking in the field supports the complexity of AD pathophysiology, which has enabled a more diverse therapeutic pipeline. 1 , 5
An innovative approach is to target the regenerative system of the brain while simultaneously activating systems that reduce burden of AD pathology. 6 , 7 , 8 Allopregnanolone (Allo), 3α‐hydroxy‐5α‐pregnan‐20‐one, is an endogenous pregnane neurosteroid and a reduced metabolite of progesterone. 9 , 10 Unlike its precursor, Allo is inactive at nuclear progesterone receptors and instead promotes neurogenesis through activation of GABAA receptor complex on neural stem cells. 6 , 9 , 10 , 11 , 12 , 13 , 14 , 15 Allo is a first‐in‐class regenerative therapeutic aimed at treating AD, with a strong foundation of human safety exposure. 11 , 16 , 17 , 18 , 19 , 20 , 21 , 22 Intravenous infusion of Allo has been proven safe and tolerable in healthy adults, 16 both adults and children with super‐refractory status‐epilepticus, 19 , 23 men with fragile‐X associated tremor/ataxia syndrome (FXTAS), 18 , 24 and women with postpartum depression. 25 , 26 , 27 The most common side effect induced by high doses of Allo is sedation/somnolence, which has been previously reported 16 , 17 , 18 , 25 , 26 , 27 and is an expected effect due to its known positive allosteric modulation of GABAA receptors. FXTAS patients demonstrated improvement in executive functioning, episodic memory, and learning after treatment with Allo. 18 In patients with postpartum depression, Allo (brexanolone) was superior to placebo in improvement of depressive symptoms. 25 , 26 , 27
Both females and males are exposed to Allo during fetal development and throughout their lifetime. During their reproductive years, women are chronically exposed to Allo. During pregnancy, blood concentration of Allo is highest during the third trimester reaching, on average, 157 nmol/l (50 ng/mL), which is not associated with adverse effects and is safe for mother and fetus. 9 , 28 , 29 , 30 In the aged and degenerated brain, Allo content is diminished, and both the pool of neural stem cells and proliferative capacity are markedly reduced. 9 , 31 , 32
The primary objective of the phase 1b/2a trial was to assess the safety and tolerability of Allo in persons with early AD after a single‐dose administration and after chronic exposure over a 12‐week period of once‐per‐week dosing. Pharmacokinetic (PK) profile and maximally tolerated dose (MTD) were established, and cognitive, imaging, and blood‐based biomarkers explored. This is the first report describing the weekly administration of Allo in this patient population.
2. METHODS
2.1. Trial design
This was a randomized, double‐blinded, placebo‐controlled, multiple ascending dose study of 12 weeks’ duration in persons with early AD, defined as mild cognitive impairment (MCI) due to AD or mild AD. The study was comprised of three dosing cohorts of Allo: 2 mg, 4 mg, and 6–18 mg cohort. After trial commencement, adjustments to the dosing regimen were done as described in Section 2.4. The primary endpoint was the occurrence of adverse events (AE) and serious adverse events (SAE) in participants treated with Allo compared to placebo. Secondary endpoints included the MTD and pharmacokinetic parameters of Allo administered intravenously for 30 minutes. Exploratory endpoints included cognitive assessments, imaging, and blood‐based biomarkers.
The study was approved by the US Food and Drug Administration and the University of Southern California (USC) Institutional Review Board and was registered with clinicaltrials.gov (NCT02221622). All participants provided written informed consent prior to participation.
2.2. Participants
Eligible participants were at least 55 years of age, met criteria for MCI due to AD or probable AD, 33 , 34 had a Mini‐Mental State Examination (MMSE) 35 score ≥20 at screening, and provided informed consent.
This study was conducted at USC from June 2015 to February 2018 and consisted of a 4‐week screening period, 12‐week treatment period, and 1‐month follow‐up. Recruitment was conducted in three separate stages, one for each cohort. Recruitment for cohorts two and three began after all participants in the preceding cohort had completed the study and the data and safety monitoring board (DSMB) approved the continuation of the study.
Participants who met eligibility requirements were randomly assigned to Allo or placebo in a 6:2 allocation ratio. Computer‐generated random allocation was programmed by the study statistician. Randomization was stratified on sex to ensure balance across cohorts.
The sample size of six participants per active dose cohort and six total placebo participants was selected to identify major safety and tolerability signals related to dose. In terms of estimation of PK and other continuously measured parameters, selected sample sizes allowed estimation with a 95% confidence interval with limits of ±0.8 standard deviation (SD). Confidence intervals on mean group differences allowed interval limits of ±1 SD.
HIGHLIGHTS
Targeting the regenerative system of the brain while simultaneously activating systems that reduce burden of pathology is an innovative therapeutic approach in treating Alzheimer's disease (AD).
Allopregnanolone is a first‐in‐class regenerative therapeutic with a strong foundation of human safety.
Allopregnanolone was safe and well tolerated when administered intravenously in an early AD cohort.
Pharmacokinetics and maximally tolerated dose were established for a regenerative dosing regimen in early AD.
Outcomes support advancement of allopregnanolone to later stages of clinical development.
RESEARCH IN CONTEXT
Systematic Review: Allopregnanolone is a pleiotropic regenerative agent that promotes neurogenesis, restores cognitive function, and reduces neural pathology in a preclinical Alzheimer's disease (AD) model. Abundant data support the safety of allopregnanolone in animals and humans. Translational research indicates the potential of allopregnanolone to promote regeneration of neural pathways affected by AD to restore brain function.
Interpretation: Allopregnanolone administered intravenously over 12 weeks in patients with early AD was safe and well tolerated at every dose. Our findings are consistent with published preclinical translational data and are the first published clinical data in an AD study population.
Future directions: Outcomes of this Phase 1b/2a clinical trial will guide dose selection and overall design of a randomized‐controlled phase 2 efficacy trial.
2.3. Interventions and study drug
Allopregnanolone or placebo were administered via an intravenous (IV) infusion once weekly for 12 consecutive weeks. The infusion lasted 30 minutes and was administered with a syringe pump. Volume infused varied by dose, ranging from approximately 0.3 to 3 mL. Allo was formulated as a clear aqueous solution packaged in an IV bag with non‐Di(2‐Ethylhexyl)Phthalate fluid path (Baxter Healthcare Corporation, Deerfield, Illinois, USA). The final product included Allo and sulfobutylether‐β‐cyclodextrin (Dexolve, Cyclolabs, Budapest, Hungary) in 0.9% sodium chloride (NaCl). Placebo solution (0.9% NaCl for injection, USP) was matched in color and fill volume, and packaged in the exact same manner to maintain the study blind. All products were manufactured and packaged at the University of California Davis Good Manufacturing Practices Laboratory (Sacramento, California, USA).
Dose selection of 2, 4, and 6 ascending to 18 mg was determined by pre‐clinical dose‐response analyses for neurogenesis, an IV bridging study in 3xTgAD mice, prior clinical studies, physiologically based PK modeling, and simulations of pharmacodynamic responses using available pre‐clinical and clinical data of non‐sedative to mildly sedative doses. 11 , 21 Based on observations of a regenerative effect of Allo at sub‐sedative doses versus suppression of neurogenesis at sedative doses, 6 , 27 , 36 we targeted a sub‐sedative dose and therefore used mild sedation as a clinical indicator for the MTD of Allo.
2.4. Multiple ascending dose adjustment
The first and second cohort received 2 mg and 4 mg of IV Allo, respectively, for 12 weeks. Symptoms of sedation were monitored during the infusions using a combination of a validated self‐administered visual analogue scale, the Mood Rating Scale (MRS), 13 and a clinician administered questionnaire, the Stanford Sleepiness Scale (SSS). 37 These assessments showed no symptoms of sedation in either the 2 mg or 4 mg cohorts. Consequently, a dose escalation regimen was implemented for the participants in the third cohort to ensure that a sedative dose would be identified. Dose escalation for the last cohort occurred in the following manner: 6 mg–10 mg–14 mg–18 mg. If any participant met the criteria for qualified sedation at any dose, higher doses were not tested for that participant and the lower dose was evaluated during the next infusion. Participants continued at a sub‐sedative maintenance dose for the remainder of the study. Additionally, if any of the pre‐established critical laboratory criteria were met within a dose cohort, dose escalation was temporarily suspended until safety data across all participants and dose cohorts was thoroughly evaluated.
2.5. Safety assessments
The primary objective of the trial was to evaluate the safety and tolerability of a weekly administration of Allo for 12 weeks in persons with early AD. Weekly safety assessments included vital signs, treatment emergent adverse events (TEAEs), AEs, SAEs, and suicidal ideation as per the Columbia‐suicide severity scale. TEAE were defined as an AE occurring during the treatment window (24 hours post‐infusion). Clinical laboratory measurements were done at screening, weeks 5 and 9 of treatment, and at end‐of‐study (week 13). Physical and neurological examinations were performed at screening, and 1‐week and 1‐month post‐treatment. All participants underwent magnetic resonance imaging (MRI) to assess amyloid‐related imaging abnormalities (ARIA) related to vasogenic edema (ARIA‐E) or microhemorrhages and hemosiderosis (ARIA‐H). 38 Both MRI and electrocardiograms were performed at baseline and end‐of‐study.
Safety data from the intent‐to‐treat study population was analyzed by the DSMB during and at the end of the study. Safety data from per‐protocol study population is reported and analyzed here. The incidence and severity of TEAEs was tabulated for participants randomized to each dose and compared to placebo using Fisher's exact tests. Summary and descriptive statistics were used to further detail the safety assessment data that were measured on a continuous scale. Mean values on these continuous safety measures were compared by randomized group using mixed effects models, with randomized group and visit (follow‐up) as fixed effects and patient as a random effect. For these safety analyses, the primary comparisons of interest were differences in each dose group relative to placebo.
2.6. Pharmacokinetic assessment
A 24‐hour PK profile was established after a single administration of Allo at week 1, and at week 12 after repeated weekly dosing. Plasma samples were collected before and after the start of the infusion at the following time points: 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, and 24 hours. Allo concentration was measured using tandem quadrupole mass spectrometer Waters Acquity ultra high‐performance liquid chromatography, 18 with a quantification range of 2.5 ng/mL to 1500 ng/mL.
Plasma PK parameters were derived using Phoenix WinNonlin (version 8.0). 18 The following parameters were derived using non‐compartmental analysis of the plasma concentration‐time profiles for each participant: tmax (time to reach maximum plasma concentration), Cmax (maximum plasma concentration), AUC0‐last (area under the plasma concentration‐time curve from time zero to time of the last measured concentration above the limit of quantification), t½ (terminal elimination half‐life).
A two‐compartment model fit was used for calculating the following parameter estimates: CL (clearance), CL2 (intercompartmental distribution), V1 (central compartment volume), and V2 (peripheral compartment volume). Missing data were not imputed. For plotting purposes only, if values were below the limit of quantitation (BQL), they were set to lower limit of quantitation (LLOQ)/2 (1.25 ng/mL).
2.7. Exploratory assessments
2.7.1. Cognitive assessment
The MMSE and Clinical Dementia Rating (CDR) were administered at screening only. The following tests were administered at baseline, weeks 5 and 9 of treatment, and at end‐of‐study (week 13): Alzheimer's Disease Assessment Scale‐Cognition (ADAS‐Cog)14, Montreal Cognitive Assessment (MoCA), and Cogstate Brief Battery.
2.7.2. Imaging biomarkers
MRI scanning of the brain without IV contrast was performed at baseline and end‐of‐study using a 3T whole‐body scanner (General Electric Signa HDxt). The imaging protocol used was a modification of the Alzheimer's Disease Neuroimaging Initiative 3T MRI protocol. T1 weighed images were acquired and volumetric analysis was done using Freesurfer 6.0 longitudinal version. Automated segmentation was used to calculate hippocampal volumes. Scans were also evaluated using Corlnsights MRI to assess overall baseline characteristics.
3. RESULTS
3.1. Study population
Overall, 47 participants were screened and of these, 26 participants were randomized (Figure A.1 in supporting information). One participant from the placebo arm discontinued the intervention and dropped out of the study for reasons not related to treatment. One participant from the Allo arm was removed from the study after randomization due to a pre‐existing medical condition. Both participants were subsequently replaced to ensure equal numbers in each cohort. Patient retention for the study was 96.2%. Demographic and baseline characteristics are summarized in Table A.1 in supporting information.
3.2. Safety and tolerability
Overall, Allo IV administration was safe and well tolerated by all participants. AEs were reported by 83% of participants in the placebo arm and 61% of participants in the Allo arm (Table 1). No differences in incidence and severity, nor differences in frequency of AEs within MedDRA classifications were detected across treatment groups (Table 1). No participants discontinued treatment due to AEs. The most frequently reported AEs (≥2 participants) and SAEs, irrespective of causality, are summarized in Table 2. One SAE (rectal hemorrhage) occurred in the Allo 2 mg cohort but was determined to be unrelated to study medication, and the participant completed the study without interruption. Of the total reported AEs, only one (2%) was determined to be “possibly related” at the time of assessment. That participant experienced dizziness, which was reported to have occurred within 24 hours after the infusion. Overall, there were no clinically significant changes in echocardiograms, physical exams, or clinical laboratory assessments. Additionally, imaging analyses did not detect any ARIA‐E or ‐H across cohorts (Table 2).
TABLE 1.
Adverse events (AE) by treatment cohort. A) Number of participants reporting categorized a AE; B) Number of participants with ARIA; C) Number of AEs distributed by severity
| Placebo | Allo 2 mg | Allo 4 mg | Allo 6–18 mg | ||
|---|---|---|---|---|---|
| (N = 6) | (N = 6) | (N = 6) | (N = 6) | P‐value c | |
| A) Serious AE, N (%) b | |||||
| Gastrointestinal disorders | 0 | 1 (17) | 0 | 0 | 1.0 |
| AE, N (%) b | |||||
| Cardiac disorders | 0 | 0 | 0 | 1 (17) | 1.0 |
| Gastrointestinal disorders | 0 | 1 (17) | 1 (17) | 0 | 1.0 |
| General disorders and administration site conditions | 2 (33) | 1 (17) | 1 (17) | 2 (33) | 1.0 |
| Infections and infestations | 2 (33) | 1 (17) | 1 (17) | 1 (17) | 1.0 |
| Injury, poisoning, and procedural complications | 1 (17) | 0 | 0 | 0 | 1.0 |
| Metabolism and nutrition disorders | 0 | 0 | 1 (17) | 0 | 1.0 |
| Musculoskeletal and connective tissue disorders | 2 (33) | 0 | 0 | 0 | 0.22 |
| Nervous system disorders | 0 | 1 (17) | 1 (17) | 0 | 1.0 |
| Respiratory, thoracic, and mediastinal disorders | 0 | 1 (17) | 0 | 2 (33) | 0.57 |
| Skin and subcutaneous tissue disorders | 0 | 1 (17) | 2 (33) | 1 (17) | 0.88 |
| Surgical and medical procedures | 0 | 0 | 1 (17) | 0 | 1.0 |
| B) ARIA, N (%) b | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1.0 |
| C) AE severity, N (%) d | Placebo | Allo 2 mg | Allo 4 mg | Allo 6‐18 mg | P‐value e |
|---|---|---|---|---|---|
| Mild | 5 (62.5) | 6 (75) | 8 (53) | 6 (60) | 0.73 |
| Moderate | 3 (37.5) | 2 (25) | 6 (40) | 3 (30) | |
| Severe | 0 | 0 | 1 (7) | 1 (10) |
MedDRA classifications. Abbreviation: ARIA, amyloid related imaging abnormalities.
N (%) = no. of participants; Participants may have reported more than one event within a classification.
Fisher's exact test comparing proportion of participants reporting an event across treatment cohort.
N (%) = no. of events.
Fisher's exact test comparing distribution of the most severe AE reported by a participant across treatment cohorts.
TABLE 2.
Most frequently reported adverse events (AEs) and serious adverse events by treatment cohort
| Placebo | Allo 2 mg | Allo 4 mg | Allo 6–18 mg | |
|---|---|---|---|---|
| (N = 6) | (N = 6) | (N = 6) | (N = 6) | |
| Any AE, N (%) | 5 (83.3) | 3 (50) | 5 (83.3) | 3 (50) |
| AEs (≥ 2 participants) a , N (%) | ||||
| Dizziness | 0 (0) | 1 (16.7) | 1 (16.7) | 0 (0) |
| Fatigue | 1 (16.7) | 1(16.7) | 1 (16.7) | 1 (16.7) |
| Nasal congestion | 0 (0) | 0 (0) | 0 (0) | 2 (33.3) |
| Nasopharyngitis | 2 (33.3) | 0 (0) | 1 (5.6) | 0 (0) |
| Rash | 0 (0) | 1 (16.7) | 2 (33.3) | 1 (16.7) |
| Sinusitis | 1 (16.7) | 1 (16.7) | 0 (0) | 0 (0) |
| Serious AEs, N (%) | ||||
| Rectal hemorrhage | 0 (0) | 1(16.7) | 0 (0) | 0(0) |
AEs that were reported by two or more participants.
N (%) = no. of participants.
3.3. Pharmacokinetics
Allo PK parameters are summarized as mean values and standard deviations (Table 3). Mean plasma concentration‐time profiles are presented on a semilogarithmic scale (Figure 1). At study weeks 1 and 12, mean plasma values of Allo after the administration of 2 mg, 4 mg, and 6 mg exhibited a peak (Tmax) at approximately 30 minutes after the start of the infusion, which corresponded to the end of the infusion (Figure 1). After the peak, the mean concentration‐time profile decreased steadily, with all participants exhibiting concentrations close to, or below, the LLOQ (2.5 ng/mL) by the 4‐hour time point. All plasma concentrations at 24 hours remained BQL (Figure 1).
TABLE 3.
Pharmacokinetic parameters
| Single dose PK a | Allo 2 mg (N = 6) | Allo 4 mg (N = 5) | Allo 6 mg (N = 6) |
|---|---|---|---|
| TMax (h) | 0.42 (0.13) | 0.45 (0.11) | 0.46 (0.10) |
| CMax (ng/mL) | 14.53 (7.31) | 42.05 (14.55) | 60.07 (12.80) |
| AUClast (h*ng/mL) | 7.73 (3.38) | 24.35 (13.46) | 37.00 (11.58) |
| Multiple‐dose PK b | Allo 2 mg (N = 6) | Allo 4 mg (N = 5) | Allo 6 mg (N = 3) | Allo 10 mg (N = 3) |
|---|---|---|---|---|
| TMax (h) | 0.42 (0.13) | 0.50 (0) | 0.50 (0) | 0.42 (0.14) |
| CMax (ng/mL) | 25.85 (8.7) | 38.69 (5.9) | 58.47 (16.34) | 137.5 (38.69) |
| AUClast (h*ng/mL) | 13.21 (4.89) | 25.06 (7.02) | 34.26 (16.38) | 76.29 (26.27) |
| Two‐compartment estimates c | Allo 2 mg | Allo 4 mg | Allo 6 mg | Allo 10 mg |
|---|---|---|---|---|
| CL (L/h) | 134 | 122 | 137 | 115 |
| CL2 (L/h) 2 | 80 | 71 | 50 | 37 |
| V (L) 3 | 28 | 50 | 43 | 26 |
| V2 (L) 4 | 35 | 37 | 68 | 33 |
Single dose means (SD) PK parameters by dose cohort at treatment week 1.
Multiple dose (steady state) mean (SD) PK parameters by dose cohort at treatment week 12.
Clearance and volume of distribution obtained from modeling conducted for the mean data at each dose level. Abbreviations:CL, clearance; CL2, intercompartmental distribution; V, central compartment volume; V2, peripheral compartment volume.
FIGURE 1.
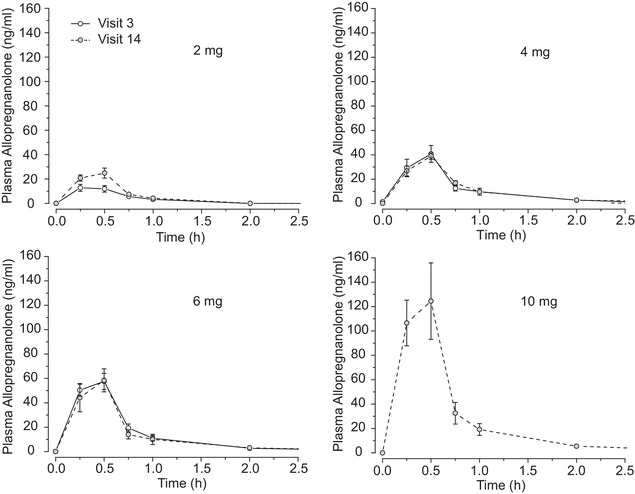
Semilogarithmic mean plasma concentration‐time profiles of Allopregnanolone doses. Allopregnanolone 2 mg, 4 mg, and 6 mg doses shown at visit 3 and visit 14; 10 mg dose shown at visit 14 only. Measurements in all cases after 2 hours were below limit of quantitation (4 hours, 6 hours, 8 hours, and 24 hours). Error bars indicate standard error of the mean
In dosing cohort three, all participants received 6 mg infusions at study week 1. All participants were individually dosed to their MTD, which was found to be 6 mg for the three male participants and 10 mg for the three female participants. Differences in exposure at study week 1 across three doses (2, 4, and 6 mg) show a less than dose‐proportional concentration response at the lower dose. Differences in exposure at study week 12 across four doses (2 mg, 4 mg, 6 mg, and 10 mg), demonstrates that Cmax was consistent with a linear dose‐concentration relationship. The AUC0‐last, data support a linear dose‐exposure relationship after repeated dosing over 12 weeks (Table 3).
For all doses, the apparent t1/2 occurred at approximately 30 minutes, at which sufficient data points were available to characterize the elimination (Figure 1). These data are consistent with the observed profiles in which drug levels are close to depletion after ≈5 half‐lives. The terminal elimination phase was consistent with first‐order kinetics in the dose range studied.
Two‐compartment PK parameter estimates obtained from modeling conducted for the mean data at each dose level demonstrated a clearance of 127 L/h, an intercompartmental distribution of 59 L/h, a central compartment volume of 36 L, and a peripheral compartment volume of 52 L (Table 3).
3.4. Pharmacodynamics and target engagement: sedation
No indicators of sedation were observed at the 2 mg and 4 mg doses of Allo as evaluated by the SSS and MRS. Scores consistent with increased levels of sleepiness were observed with both assessment scales in the higher dose cohort (6–18 mg). Mean post‐infusion MRS composite scores are shown in Table 4. MRS scores are 1 to 100, with lower scores indicating increased sedation. There was a statistically significant difference in post‐infusion MRS scores among dosing cohorts in alertness, and mental and physical sedation that was dose dependent (P = 0.006, 0.002 and 0.01, respectively). Overall, incidence of SSS scores consistent with sedation (≥7) was directly proportional to Allo dose. The highest rated SSS score for participants in cohort one (2 mg) was 2; the majority had a score of 1, which denotes a person who is “feeling active, vital, alert, or wide awake.” The highest rated SSS score in cohort two (4 mg) was 4, which indicates that a person is “somewhat foggy or let down.” For cohort three (6–18 mg) the highest rated SSS score was ≥7, defining a person with “sleep onset soon” or who is asleep.
TABLE 4.
Mood Rating Scale composite scores by treatment cohort a
| MRS score b | Placebo (N = 6) | Allo 2 mg (N = 6) | Allo 4 mg (N = 6) | Allo 6–18 mg (N = 6) | P‐value c |
|---|---|---|---|---|---|
| Alertness d | 75 (65–86) | 84 (73–94) | 67 (56–78) | 58 (48–69) | 0.006 |
| Mental sedation e | 76 (65–88) | 80 (69–91) | 66 (55–78) | 52 (41–63) | 0.002 |
| Physical sedation f | 74 (63–85) | 86 (75–97) | 69 (58–80) | 60 (49–71) | 0.01 |
Numbers in table are mean (95% confidence interval).
MRS range is 0–100 with lower scores indicating increased sedation.
Linear mixed model with random subject effect was used to compare mean MRS scores among treatment cohorts. P‐value represents overall difference across all treatment cohorts. Dunnett's method was used for multiple comparisons between each Allo cohort with placebo. The mean difference in mental sedation between Allo 6–18 mg and placebo is significant (P = 0.007). The difference in alertness between Allo 6–18 mg placebo is marginally significant (P = 0.07). All other P‐values >0.17.
Alertness is the average of scores for (a) alert/drowsy, (b) clear‐headed/muzzy, (c) quick‐witted/mentally slow, (d) attentive/dreamy, (e) strong/feeble, (f) well‐coordinated/clumsy, (g) energetic/lethargic, (h) proficient/incompetent, and (i) interested/bored.
Mental sedation is a subcomponent of Alertness (average of scores a, b, c, d).
Physical sedation is a subcomponent of Alertness (average of scores e, f, g, h).
Abbreviation: MRS, Mood Rating Scale.
In all female participants, no sedation was observed per the SSS and MRS at either the 6 mg or 10 mg but was observed at 14 mg and 18 mg. In contrast, male participants demonstrated sedation at doses ≥ 6 mg.
3.5. Exploratory endpoints
All 24 participants completed pre‐ and post‐treatment cognitive assessments. After 12 weeks of treatment, no statistically significant differences were observed amongst cohorts on the ADAS‐Cog14 total score, MoCA total score, and Cogstate Brief Battery composite score (Table 5), and no indicators of adverse impact on cognition was apparent.
TABLE 5.
Exploratory outcomes: comparison of change scores across treatment cohorts a
| Change score b | Placebo (N = 6) | Allo 2 mg (N = 6) | Allo 4 mg (N = 6) c | Allo 6–18 mg (N = 6) | P‐value |
|---|---|---|---|---|---|
| ADAS‐Cog14** | 0.2 (−4.9, 4.5) | −1.0 (−3.7, 5.6) | 0.5 (−5.1, 4.2) | 0.5 (−5.2, 4.2) | 0.96 |
| Cogstate composite d | 0.08 (−0.36, 0.53) | 0.56 (0.12,1.0) | 0.23 (−0.21, 0.67) | 0.19 (−0.26, 0.63) | 0.43 |
| MoCA total | −0.1 (−3.6, 3.3) | −1.1 (−4.4, 2.3) | −0.9 (−4.3, 2.5) | 1.9 (−1.5, 5.4) | 0.57 |
| Change in hippocampal volume | Placebo (N = 6) | Allo 2 mg (N = 5) | Allo 4 mg (N = 6) | Allo 6‐18 mg (N = 6) | P‐value |
|---|---|---|---|---|---|
| Left, mm3 | −150 (−275, −26) | −75 (−211, 60) | 28 (−96, 151) | −91 (−216, 34) | 0.230 |
| Right, mm3 | −80 (−172, 11) | −121 (−223, −20) | 36 (−56, 128) | −25 (−117, 67) | 0.124 |
| Total (L+R), mm3 | −231 (−424, −40) | −193 (−404, 17) | 61 (−130, 253) | −116 (−309, 77) | 0.156 |
Analysis of covariance on change in cognitive test scores and change in hippocampal volume (week 13 minus baseline), controlling for baseline scores and volume, respectively. Least squares means, 95% confidence intervals, and P‐values are shown in the table.
To facilitate comparisons across cognitive tests, ADAS‐Cog scores were reversed to a positive score. Thus, positive change indicates improvement in all tests.
One participant had a missing Cogstate ONB score at week 13; their week 12 score was carried forward for analysis.
Composite: Change in average z‐score (Detection test + Identification test + One back test + One card learning test).
Abbreviation: ADAS‐Cog14, Alzheimer's Disease Assessment Scale‐Cognition 14; MoCA, Montreal Cognitive Assessment.
Baseline and post‐treatment MRI imaging data were available for 23 subjects. Volumetric analysis hippocampal volume demonstrated no adverse outcome of Allo over the course of treatment. There was no statistically significant difference in hippocampal volumes among the four cohorts (Table 5). Analysis of change in left and right hippocampal volumes suggest a trend of decreased atrophy in Allo‐treated participants.
4. DISCUSSION
Safety and tolerability were the primary outcome measures in this first‐in‐human study of Allo in persons with early AD. As anticipated, both the single dose IV administration and the weekly dosing over the course of 12 weeks were safe and well tolerated in this study population. Safety outcomes are consistent with previously reported safety data in both animals and humans, 11 , 16 , 17 , 18 , 19 , 23 , 24 , 39 and from an open‐label study 25 and two randomized‐controlled trials 26 , 27 of Allo in the treatment of severe postpartum depression.
Categories of adverse events were highly variable and did not differ significantly between placebo and Allo‐treated cohorts. No participants discontinued treatment due to any AE. Most frequent reported AEs in the Allo‐treated cohorts were rash (22%), fatigue (17%), and dizziness (11%). All instances of rash and fatigue were deemed not related to the study intervention because the events did not occur within 24 hours of the infusion, based on the study drug's short half‐life. Additionally, it was confirmed that skin conditions existed prior to study enrollment. The only treatment‐emergent AE was dizziness reported by a participant in the 4 mg cohort. This AE occurred within 24 hours of the infusion and was likely related to Allo. Dizziness is a common AE of sedative drugs and has previously been reported as a frequent AE at high doses of Allo. 26 Nasopharyngitis and musculoskeletal disorders were the only AEs reported with greater frequency in the placebo group compared to the Allo treated groups. These AEs were associated with specific episodes of the common cold and pre‐existing osteoarthritis. Importantly, Allo did not induce indicators of ARIA. Unlike therapeutics designed to remove amyloid beta (Aβ) plaques from the brain, which are associated with ARIA, 40 the mechanisms by which Allo reduces Aβ load in the brain are related to its reduced generation. 6
Sedation was dose dependent (Table 4) and the threshold dose was sex specific. In the multiple ascending dose cohort (Allo 6–18 mg) all female participants received two 18 mg infusions, which resulted in marked sedation. Subsequently the dose was deescalated back to 10 mg, which resulted in no sedation. In contrast, two of three male participants demonstrated mild sedation at 6 mg and all three participants demonstrated sedation at 10 mg. One participant was escalated up to 14 mg for two infusions, which resulted in marked sedation; this participant was deescalated back to 6 mg for the remainder of the treatment period. Based on these results, the MTD was determined to be 10 mg for females and 6 mg for males.
The dosing regimen is a critical factor for the neuro‐regenerative effect of Allo, which occurs at nanomolar concentrations and is suppressed at higher doses. 6 , 9 , 12 Allo exhibits an inverted U‐shaped dose‐response which at low concentrations results in promotion of neural stem cell regeneration and at high concentrations suppresses proliferation, which protects against unchecked cell division. 6 , 9 , 12 Previous preclinical data indicated that Allo administered once per week was maximally efficacious for increasing neurogenesis and markers of white matter generation while simultaneously reducing multiple indicators of AD pathology. 11 , 12 , 41 , 42 Conversely, frequent or continuous dosing regimens, similar to the one used to treat postpartum depression, 25 do not promote, and likely suppress, neurogenesis. 43 , 44 , 45 Mode of administration will also influence absorption and PK. 12
Blood levels of Allo reached a dose‐dependent Cmax after both a single dose and 12 weeks of weekly infusions. The Cmax levels obtained from the 4 to 6 mg doses are consistent with the serum levels that occur during the third trimester of pregnancy, which range from 40 to 50 ng/mL. 29 These results are comparable to those reported in an open label trial of Allo in men with FXTAS, in which participants received the exact same dosing and treatment regimen. 18 A previous study reported the same steady‐state plasma concentration of approximately 50 ng/mL in a postpartum cohort after 12 hours of continuous infusion with dosing up to 48 hours before a graduated decline in dose over the next 12 hours. 25
This trial was powered for safety and PK, thus given the small sample size and duration of treatment, the efficacy of Allo could not be determined. By design, assessments of cognition and imaging biomarkers were exploratory. Results indicated that Allo did not alter cognition, which can be interpreted as having no adverse effects on cognition over the course of 3 months of once‐per‐week treatment. Likewise, Allo exerted no adverse outcome on total hippocampal MRI volumetric structure over the course of 3 months. Analysis of change in hippocampal volume suggested a potential signal in the 4 mg Allo dose that will be investigated further in an appropriately powered phase 2 trial as a surrogate marker of regeneration. 46 ‐49 Moreover, noted differences in hippocampal volume appeared to be differentially affected by apolipoprotein E (APOE) ε4 genotype. Analyses of other MRI structural volumes, resting state functional MRI (fMRI), and diffusion tensor imaging, as well as other exploratory outcomes, are ongoing and will be published in detail separately. Overall, exploratory outcome measures support safety outcomes by demonstrating that Allo infusions were not detrimental to cognition or imaging biomarkers.
5. CONCLUSIONS
Allopregnanolone is a first in class regenerative therapeutic for early AD that targets endogenous neural stem cells and disease‐modifying mechanisms. The results of this phase 1b/2a clinical trial demonstrated that Allo was well tolerated and safe in an AD study population. Pharmacokinetic profile and MTD obtained will guide dose selection for a randomized‐controlled phase 2 trial to investigate the long‐term safety and efficacy of this novel therapeutic for AD.
CONFLICTS OF INTEREST
This study was funded by the National Institute on Aging (NIA) and the Alzheimer's Drug Discovery Foundation (ADDF).
Dr. Hernandez reports no disclosures. Dr. Solinsky reports no disclosures. Dr. Mack reports no disclosures. Mrs. Kono reports no disclosures. Dr. Rodgers reports no disclosures. Dr. Wu and Dr. Mollo are paid consultants and co‐founders of TOMO Pharmacometrics, which was contracted to conduct processing and pharmacokinetic analysis. Mrs. Lopez reports no disclosures. Dr. Pawluczyk reports no disclosures. Mr. Bauer reports no disclosures. Dr. Matthews is a paid consultant and CEO of ADM Diagnostics, Inc. Dr. Shi reports no competing interests. Dr. Law reports no competing interests. Dr. Rogawski is principal investigator on research grants to the University of California, Davis, from NINDS (U54NS079202, R25NS099170, U01NS112102), CURE – Citizens United for Research in Epilepsy, and Mallinckrodt Pharmaceuticals; has served as a consultant to Eisai, West Therapeutics Development, Xenon Pharmaceuticals, Supernus Pharmaceuticals, Aquestive Therapeutics, and H. Lundbeck A/S; is a member of the board of directors of Epalex; is a member of the scientific advisory boards of Zynerba Pharmaceuticals, Marinus Pharmaceuticals, and OB Pharmaceuticals; and is named as an inventor of patents and patent applications assigned to the Regents of the University of California. Dr. Schneider reports grants from Biogen, Roche/Genentech, Eli Lilly and Company, Novartis, and Biohaven; personal fees from Merck, Eli Lilly and Company, and Roche/Genentech for serving on the data and safety monitoring boards; personal fees from Takeda for serving as a consultant and on an adjudication committee; and consulting fees from AC Immune, Avraham Pharmaceuticals, Boehringer Ingelheim, Cognition Therapeutics, Cotexyme, Eisai, Neurim Pharmaceuticals, Neuronix, Tau RX, Toyama, Abbott, and vTv Therapeutics outside the submitted work. Dr. Brinton holds patent US8969329B2 for allopregnanolone for the treatment of neurodegenerative diseases.
Supporting information
Supplementary information
ACKNOWLEDGMENTS
We acknowledge and appreciate the exceptional contributions of the USC Alzheimer Disease Research Center (ADRC) leadership, Dr. Helena Chui, and staff; members of the Brinton laboratory team, Drs. Ronald Irwin, Eliza Bacon, and Maunil Desai, for their valuable contribution to the clinical trial; and Dr. Adam Raikes for providing imaging analysis support. To all the study participants and their families, we extend sincere appreciation for their commitment and dedication to the phase 1 clinical trial of allopregnanolone for Alzheimer's disease.
Hernandez GD, Solinsky CM, Mack WJ, et al. Safety, tolerability, and pharmacokinetics of allopregnanolone as a regenerative therapeutic for Alzheimer's disease: A single and multiple ascending dose phase 1b/2a clinical trial. Alzheimer's Dement. 2020;6:1–10. 10.1002/trc2.12107
REFERENCES
- 1. Cummings J, Aisen PS, Dubois B, et al. Drug development in Alzheimer's disease: the path to 2025. Alzheimers Res Ther. 2016;8:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hampel H, Vergallo A, Aguilar LF, et al. Precision pharmacology for Alzheimer's disease. Pharmacol Res. 2018;130:331–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cummings JL, Morstorf T, Zhong K. Alzheimer's disease drug‐development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6(4):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schneider LS, Mangialasche F, Andreasen N, et al. Clinical trials and late‐stage drug development for Alzheimer's disease: an appraisal from 1984 to 2014. J Intern Med. 2014;275(3):251–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer's disease drug development pipeline: 2019. Alzheimers Dement (N Y). 2019;5:272–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brinton RD. Neurosteroids as regenerative agents in the brain: therapeutic implications. Nat Rev Endocrinol. 2013;9(4):241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Diaz Brinton R, Ming Wang J. Therapeutic potential of neurogenesis for prevention and recovery from Alzheimer's disease: allopregnanolone as a proof of concept neurogenic agent. Curr Alzheimer Res. 2006;3(3):185–190. [DOI] [PubMed] [Google Scholar]
- 8. Brinton R, Wang J. Preclinical analyses of the therapeutic potential of allopregnanolone to promote neurogenesis in vitro and in vivo in transgenic mouse model of Alzheimer's disease. Curr Alzheimer Res. 2006;3(1):11–17. [DOI] [PubMed] [Google Scholar]
- 9. Wang JM. The neurosteroid allopregnanolone promotes proliferation of rodent and human neural progenitor cells and regulates cell‐cycle gene and protein expression. J Neurosci. 2005;25(19):4706–4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mellon SH. Neurosteroid regulation of central nervous system development. Pharmacol Ther. 2007;116(1):107–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Irwin RW, Wang JM, Chen S, Brinton RD. Neuroregenerative mechanisms of allopregnanolone in Alzheimer's disease. Front Endocrinol (Lausanne). 2011;2:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Irwin RW, Solinsky CM, Brinton RD. Frontiers in therapeutic development of allopregnanolone for Alzheimer's disease and other neurological disorders. Front Cell Neurosci. 2014;8(203). Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bond AJ, James DC, Lader MH. Sedative effects on physiological and psychological measures in anxious patients. Psychol Med. 1974;4(4):374–380. [DOI] [PubMed] [Google Scholar]
- 14. Gee KW, Bolger MB, Brinton RE, Coirini H, McEwen BS. Steroid modulation of the chloride ionophore in rat brain: structure‐activity requirements, regional dependence and mechanism of action. J Pharmacol Exp Ther. 1988;246(2):803–812. [PubMed] [Google Scholar]
- 15. Chen S, Wang T, Yao J, Brinton RD. Allopregnanolone promotes neuronal and oligodendrocyte differentiation in vitro and in vivo: therapeutic implication for Alzheimer's disease. Neurotherapeutics. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Timby E, Balgård M, Nyberg S, et al. Pharmacokinetic and behavioral effects of allopregnanolone in healthy women. Psychopharmacology (Berl). 2006;186(3):414–424. Art. no. 2006. [DOI] [PubMed] [Google Scholar]
- 17. Van Broekhoven F, Bäckström T, Van Luijtelaar G, Buitelaar JK, Smits P, Verkes RJ. Effects of allopregnanolone on sedation in men, and in women on oral contraceptives. Psychoneuroendocrinology. 2007;32(5):555–564. Clinical Trial Comparative Study Research Support, Non‐U.S. Govt. [DOI] [PubMed] [Google Scholar]
- 18. Wang JY, Trivedi AM, Carrillo NR, et al. Open‐Label allopregnanolone treatment of men with fragile X‐Associated Tremor/Ataxia Syndrome. Neurotherapeutics. 2017;14(4):1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vaitkevicius H, Husain AM, Rosenthal ES, et al. First‐in‐man allopregnanolone use in super‐refractory status epilepticus. Ann Clin Transl Neurol. 2017;4(6):411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Irwin RW, Brinton RD. Allopregnanolone as regenerative therapeutic for Alzheimer's disease: translational development and clinical promise. Prog Neurobiol. 2014;113:40–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Irwin RW, Solinsky CM, Loya CM, et al. Allopregnanolone preclinical acute pharmacokinetic and pharmacodynamic studies to predict tolerability and efficacy for Alzheimer's disease. PLoS One. 2015;10(6):e0128313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hernandez GD, Brinton RD. Allopregnanolone as a Therapeutic to Regenerate the Degenerated Brain In: Brinton R D, Genazzani A R, Simoncini T, and Stevenson J C, eds. Sex Steroids' Effects on Brain, Heart and Vessels: Volume 6: Frontiers in Gynecological Endocrinology. Cham: Springer International Publishing; 2019:111–123. [Google Scholar]
- 23. Broomall E, Natale JE, Grimason M, et al. Pediatric super‐refractory status epilepticus treated with allopregnanolone. Ann Neurol. 2014l;76(6):911–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Napoli E, Schneider A, Wang JYi, et al. Allopregnanolone treatment improves plasma metabolomic profile associated with GABA metabolism in fragile X‐Associated Tremor/Ataxia syndrome: a pilot study. Mol Neurobiol. 2019;56(5):3702–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kanes SJ, Colquhoun H, Doherty J, et al. Open‐label, proof‐of‐concept study of brexanolone in the treatment of severe postpartum depression. Hum Psychopharmacol. 2017;32(2):e2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kanes S, Colquhoun H, Gunduz‐Bruce H, et al. Brexanolone (SAGE‐547 injection) in post‐partum depression: a randomised controlled trial. Lancet North Am Ed. 2017;390(10093):480–489. [DOI] [PubMed] [Google Scholar]
- 27. Meltzer‐Brody S, Colquhoun H, Riesenberg R, et al. Brexanolone injection in post‐partum depression: two multicentre, double‐blind, randomised, placebo‐controlled, phase 3 trials. Lancet. 2018;392(10152):1058–1070. [DOI] [PubMed] [Google Scholar]
- 28. Dombroski RA, Casey ML, Macdonald PC. 5‐Alpha‐dihydroprogesterone formation in human placenta from 5alpha‐pregnan‐3beta/alpha‐ol‐20‐ones and 5‐pregnan‐3beta‐yl‐20‐one sulfate. J Steroid Biochem Mol Biol. 1997;63(1‐3):155–163. [DOI] [PubMed] [Google Scholar]
- 29. Luisi S, Petraglia F, Benedetto C, et al. Serum allopregnanolone levels in pregnant women: changes during pregnancy, at delivery, and in hypertensive patients. J Clin Endocrinol Metab. 2000;85(7):2429–2433. [DOI] [PubMed] [Google Scholar]
- 30. Gago N, El‐Etr M, Sananès N, et al. 3α, 5α‐tetrahydroprogesterone (allopregnanolone) and γ‐aminobutyric acid: autocrine/paracrine interactions in the control of neonatal PSA‐NCAM+ progenitor proliferation. J Neurosci Res. 2004;78(6):770–783. [DOI] [PubMed] [Google Scholar]
- 31. Bernardi F, Salvestroni C, Casarosa E, et al. Aging is associated with changes in allopregnanolone concentrations in brain, endocrine glands and serum in male rats. Eur J Endocrinol. 1998;138(3):316–321. [DOI] [PubMed] [Google Scholar]
- 32. Weill‐Engerer Sé, David J‐P, Sazdovitch Vé, et al. Neurosteroid quantification in human brain regions: comparison between Alzheimer's and nondemented patients. J Clin Endocrinol Metab. 2002;87(11):5138–5143. [DOI] [PubMed] [Google Scholar]
- 33. Mckhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Albert MS, Dekosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):270–279. Consensus Development Conference, NIH Research Support, Non‐U.S. Govt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Folstein MF, Folstein SE, Mchugh PR. Mini‐mental state. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198. [DOI] [PubMed] [Google Scholar]
- 36. Chen S, Wang JM, Irwin RW, Yao J, Liu L, Brinton RD. Allopregnanolone promotes regeneration and reduces β‐amyloid burden in a preclinical model of Alzheimer's disease. PLoS One. 2011;6(8):e24293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hoddes E, Zarcone V, Smythe H, Phillips R, Dement WC. Quantification of sleepiness: a new approach. Psychophysiology. 1973;10(4):431–436. [DOI] [PubMed] [Google Scholar]
- 38. Sperling RA, Jack CR, Black SE, et al. Amyloid‐related imaging abnormalities in amyloid‐modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7(4):367–385. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang JM, Singh C, Liu L, et al. Allopregnanolone reverses neurogenic and cognitive deficits in mouse model of Alzheimer's disease. Proc Natl Acad Sci. 2010;107(14):6498–6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sperling R, Salloway S, Brooks DJ, et al. Amyloid‐related imaging abnormalities in patients with Alzheimer's disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 2012;11(3):241–249. Research Support, Non‐U.S. Govt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen S, Wang JM, Irwin RW, Yao J, Liu L, Brinton RD. Allopregnanolone promotes regeneration and reduces beta‐amyloid burden in a preclinical model of Alzheimer's disease. PLoS One. 2011;6(8):e24293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Singh C, Liu L, Wang JM, et al. Allopregnanolone restores hippocampal‐dependent learning and memory and neural progenitor survival in aging 3xTgAD and nonTg mice. Neurobiol Aging. 2012;33(8):1493–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reddy DS, Rogawski MA. Neurosteroid replacement therapy for catamenial epilepsy. Neurotherapeutics. 2009;6(2):392–401. Research Support, N.I.H., Extramural Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lan NC, Gee KW. Neuroactive steroid actions at the GABAA receptor. Horm Behav. 1994;28(4):537–544. Review. [DOI] [PubMed] [Google Scholar]
- 45. Kask K, Bäckström T, Nilsson L‐G, Sundström‐Poromaa I. Allopregnanolone impairs episodic memory in healthy women. Psychopharmacology (Berl). 2008;199(2):161–168. Art. no. 2008. [DOI] [PubMed] [Google Scholar]
- 46. Schuff N, Woerner N, Boreta L, et al. MRI of hippocampal volume loss in early Alzheimer's disease in relation to ApoE genotype and biomarkers. Brain. 2009;132(4):1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Veitch DP, Weiner MW, Aisen PS, et al. Understanding disease progression and improving Alzheimer's disease clinical trials: recent highlights from the Alzheimer's Disease Neuroimaging Initiative. Alzheimers Dement. 2019;15(1):106–152. [DOI] [PubMed] [Google Scholar]
- 48. Horgusluoglu‐Moloch E, Nho K, Risacher SL, et al. Targeted neurogenesis pathway‐based gene analysis identifies ADORA2A associated with hippocampal volume in mild cognitive impairment and Alzheimer's disease. Neurobiol Aging. 2017;60:92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Horgusluoglu‐Moloch E, Risacher SL, Crane PK, et al. Genome‐wide association analysis of hippocampal volume identifies enrichment of neurogenesis‐related pathways. Sci Rep. 2019;9(1):14498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information