

A novel circRNA withholds the microRNA assembly line by arresting the manufacturing foreman in the nucleus of a cancer cell.
Abstract
The assortment of cellular microRNAs (“microRNAome”) is a vital readout of cellular homeostasis, but the mechanisms that regulate the microRNAome are poorly understood. The microRNAome of glioblastoma is substantially down-regulated in comparison to the normal brain. Here, we find malfunction of the posttranscriptional maturation of the glioblastoma microRNAome and link it to aberrant nuclear localization of DICER, the major enzymatic complex responsible for microRNA maturation. Analysis of DICER’s nuclear interactome reveals the presence of an RNA binding protein, RBM3, and of a circular RNA, circ2082, within the complex. Targeting of this complex by knockdown of circ2082 results in the restoration of cytosolic localization of DICER and widespread derepression of the microRNAome, leading to transcriptome-wide rearrangements that mitigate the tumorigenicity of glioblastoma cells in vitro and in vivo with correlation to favorable outcomes in patients with glioblastoma. These findings uncover the mechanistic foundation of microRNAome deregulation in malignant cells.
INTRODUCTION
MicroRNAs, short noncoding RNA (ncRNA) molecules that control the expression/activity of a multitude of protein-coding genes, populate the human genome with high frequency. Despite the ever-growing number of putative microRNAs, the number of high-confidence, validated loci has been recently approximated to be ~2300 (1). The number of putative protein-coding mRNAs that can be targeted via microRNA complementarity has been estimated at ~18,000, a number that approximates that of the protein-coding transcriptome (2). Quantitatively, this would imply that most of the cell transcriptome, and thus its proteome, are under microRNA surveillance.
The biogenesis and maturation of microRNAs have been thoroughly investigated since their discovery in the 1990s. MicroRNA genes embedded either within introns of protein-coding genes (intronic) or between them (intergenic) are transcribed first into long primary transcripts (pri-microRNAs) that are then processed in the nucleus into ~80-nucleotide (nt) precursors (pre-microRNAs) forming hairpin-like structures. Pre-microRNAs are transported into the cytoplasm where an enzymatic complex consisting of Endoribonuclease Dicer (DICER, encoded by the gene DICER1) and its cofactors cleave them further into short ~20-nt mature microRNAs. These mature microRNAs can then regulate mRNA expression by loading onto the RNA-induced silencing complex (RISC) protein complex that serves as an mRNA target seeker based on the complementarity between a 6- and 8-nt-long sequence within the microRNA known as the “seed” and its target site located usually within the 3′ untranslated region of mRNA (3). This interaction results in the cleavage of mRNA by RISC and/or less efficient translation at ribosomes, resulting in suppressed expression of the targeted gene. Although DICER-independent microRNAs exist (4), the vast majority of microRNAs rely on DICER for their processing, thus making the entire pathway vulnerable to malfunction if DICER activity is targeted.
The assortment of microRNAs expressed in the cell at any given time, the microRNAome, varies considerably between tissues, cell types, developmental and pathological stages, and upon response to stressors or stimuli. microRNAs, because of their ability to fine-tune scores of genes, have been recognized as master guardians of cell fate decisions and terminal differentiation (5). As malignant transformation can be perceived as a faulty execution of cell fate choices or as dedifferentiation that went awry at cellular backstops, the discovery that the microRNAome of the cancer cell is very much different than that of its cell-of-origin counterpart (6) was, perhaps with the benefit of hindsight, not particularly unexpected.
Cancer-specific signatures exist in most, if not nearly all, molecular readouts (transcriptomes, proteomes, metabolomes, etc.) of the cell. What makes the cancer microRNAome unique when compared to other classes of molecules (including other ncRNAs) is the persistent pattern of the deregulation. In essence, most microRNAs are suppressed, while relatively few are overexpressed (6). One possible explanation is that most microRNAs impose programs of terminal differentiation, and tumor cells evade these programs by microRNAome suppression (6).
Possible mechanistic explanations for this observation of cancer-specific global suppression of the microRNAome are few. Chromosomal anomalies and altered epigenetic landscapes, both common in malignancies, would result in changes that would be more localized across the genome or would affect other players of the transcriptome similarly. Transcriptional deregulation (again, frequent in transformed cells), on the other hand, functions mainly on a “case-by-case” fashion and thus cannot adequately explain this sweeping suppression of microRNAs. These considerations winnow the field of potential mechanistic culprits. Could disorders in the processing/maturation of microRNAs, primarily controlled by a single canonical pathway, be the wrongdoer in the observed cancer-specific transcriptome-wide quelling of the microRNAome?
Cancer stem-like cells (CSCs) that have characteristics associated with normal stem cells (e.g., expression of stem cell markers, the capacity for self-renewal, and long-term proliferation), but can form tumors (are tumorigenic), have now been described in most tumors. Their tumorigenic functions are especially relevant in the case of aggressive, poorly differentiated, and CSC-rich brain tumors, such as glioblastomas. These tumors are highly heterogeneous in their diverse transcriptomes of cell subpopulations and the whole spectrum of driver mutations, epigenomes, transcriptomes, and phenotypes (7). The current classification of glioblastoma heterogeneity is defined on the basis of the transcriptome of protein-coding genes and consists of three major subtypes: classical, proneural, and mesenchymal (or four in other classification) (8). Tumor-derived, glioblastoma CSCs (GSCs) can also be classified into the same three broad categories in the in vitro culture (9). This picture is muddled, although, by the temporal evolution of tumors, the coexistence of multiple subtypes within individual tumors, and the multidirectional subtype transitions that happen in response to therapy (8, 10–12). Also, microenvironmental factors (hypoxia, acidity, and nutrient flux) and intercellular communication (e.g., by the exchange of extracellular vesicles) further confound glioblastoma’s subtype classification (11, 13).
The activation of transcription, although indispensable, is one of many steps during which cells control the processing of genetic information into functional RNAs. Other well-studied steps in the processing of genetic information comprise alternative promoter choice, splicing, editing, and 3′ end formation—all of which end up generating the astonishingly diverse transcriptomes that include the well-regulated and complex output of ncRNAs from genic and intergenic regions of the genome. The role of these ncRNAs in numerous physiological and pathological processes has been well documented. Numerous classes of functional ncRNAs have been found, including microRNAs, long ncRNAs (lncRNAs), and, most recently, circular RNAs (circRNAs) (14, 15).
Over the years, circRNAs have mostly been disregarded as either transcriptional noise caused by malfunctioning splicing or rare curiosities with no meaningful footprint. A relatively recent demonstration of their widespread and persistent presence within eukaryotic transcriptomes along with numerous examples of cell type– or developmental stage–specific expression patterns suggestive of regulation has opened new insights into the intricacies of the ncRNA universe, including those of circRNAs that are particularly abundant in the human brain (15, 16). CircRNAs originate from linear transcripts via various mechanisms (15), and their hallmarks are a unique splice junction site, which mediates circularization, resulting in a covalently closed “head-to-tail” structure, along with the lack of 5′-3′ polarity and polyadenylated tail, as well as a low probability for encoding protein. CircRNAs have been shown to be long lived in vivo when compared to their linear counterparts, given that the bulk of RNA turnover involves exonucleases (17). CircRNAs have been shown to act as microRNA sponges (15, 18), but their function remains largely unknown because only a handful contain microRNA target sites. Increasingly, circRNAs are being implicated in numerous cancers; however, the functional relevance of the vast majority is yet to be found (19).
While rearrangements of microRNAome in pathologies, including cancer, were observed almost two decades ago, unexpectedly, little is known on their mechanistic nature. Numerous deregulations of microRNA expression have been linked to genetic and epigenetic irregularities, altered transcriptional activity, and some missteps in the maturation process. However, the overarching explanations of widespread suppression of microRNAome in cancer cells are still lacking. This gap has prompted us to analyze these processes, using a panel of patient-derived, subtype-characterized GSCs that are particularly tumorigenic and therapy-resistant as a model, and led to uncovering faulty precursor-to-mature microRNA transition as a widespread circumstance brought about by unusual subcellular distribution of DICER complex in the nucleus in these cells. Follow-up scrutiny of nuclear DICER interactome revealed the presence of RNA-protein complex formed by DICER with RNA binding motif protein 3 (RBM3) and circRNA denoted as circ2082. The knowledge about the role of circRNAs in shaping phenotypes of glioblastoma is virtually nonexistent. Thus, we performed the analysis of the circRNAome, which revealed subtype-specific signatures and pointed out to circ2082 as one of the most up-regulated circRNAs in GSCs. The disruption of the found complex by circ2082 antisense oligonucleotide (ASO) led to the cytosolic release of DICER, the resumption of microRNA processing resulting in the restoration of premalignant microRNAome, and, in consequence, mitigated tumorigenicity in vitro and in vivo. Close inspection of ensuing transcriptome signatures uncovered circRNA/microRNA-dependent effectors that can predict the outcomes of patients with glioblastoma. These findings thus revealed the mechanistic foundation of microRNAome deregulation in the malignant cell and provided new insight into the intricacies of the ncRNA universe.
RESULTS
The expression of the glioblastoma microRNAome is suppressed
Our efforts initially focused on assessing the extent of microRNAome rearrangements in several settings relevant to glioblastoma. These included analysis of (i) The Cancer Genome Atlas (TCGA) database, (ii) collected tumor tissue paired with matching (i.e., collected from the same individual) adjacent brain tissue devoid of gross pathology, and (iii) the selection of subtype-characterized GSCs and nonmalignant neural progenitor/stem cells (NPCs). As an additional dimension of this global analysis, we selected several microRNAs that have been identified in glioblastoma as either well-known tumor suppressors and oncogenes or are subtype-predictive and scrutinized the expression of both their precursor and mature forms. These include miR-124 and miR-1 [low expression in all GSCs (20, 21)] and miR-128 [particularly low expression in the mesenchymal (M) GSC subtype (11), as well as pan-glioblastoma highly expressed miR-21 (22), and miR-10b highly expressed in the proneural (P) GSC subtype (23), and the M GSC–specific miR-31 (24)]. These microRNAs are well characterized on the transcriptional level, the phenotypic consequences of their deregulation are well defined, and their mRNA targets are convincingly verified; thus, they can serve as indices for levels of expression in glioblastoma.
Analysis of all these datasets revealed vastly different but consistently suppressed expression of the mature microRNAome in glioblastoma tissue/GSCs when compared to brain/NPCs, with some subtype-specific distribution in GSCs (Fig. 1, A to C, and table S1). Also, as expected, the expression of tumor suppressor microRNAs was low and oncogenic microRNA high in tumor/tumor-derived cells, with some distinct subtype characteristics [e.g., miR-128 was particularly low in the M GSCs, while miR-10b and miR-31 were elevated in the P GSCs and M GSCs, respectively] (Fig. 1, D to F, and fig. S1, A to F). However, levels of selected precursors did not display notably different patterns between malignant and nonmalignant material (Fig. 1, G to I, and fig. S1, A to F). Global principal components analysis (PCA) in the TCGA glioblastoma dataset confirmed the clear separation between groups using mature microRNAs but not their precursors (Fig. 1, J and K). These findings were thus the first clue that the deregulation of microRNA processing/maturation machinery may be the primary culprit for the observed widespread suppression of the glioblastoma microRNAome.
Fig. 1. Mature, but not precursor, microRNAome is suppressed in glioblastomas and GSCs.
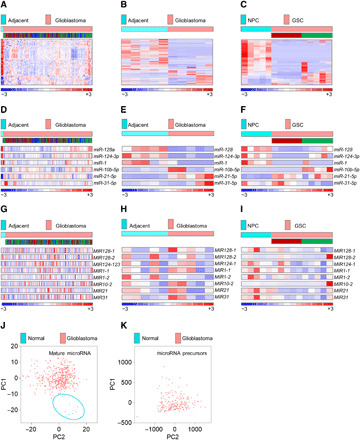
(A) Mature microRNAome in patients with glioblastoma (n = 490) versus healthy individuals (n = 11), based on 535 microRNAs in the TCGA database. The subtype identity: red, mesenchymal; blue, classical; green, proneural. (B and C) Mature microRNAome in glioblastoma tissue versus adjacent, nonpathological brain (n = 5 each) (B), and in GSCs (n = 5 per subtype) versus NPCs (n = 5) (C) based on 581 and 605 of 799 microRNAs by nCounter NanoString platform. GSC’s subtype identity is shown (red, mesenchymal; green, proneural). (D) Selected mature microRNAs in patients with glioblastoma (n = 490) versus healthy individuals (n = 11), based on the TCGA database. For the subtype identity, see (A). (E and F) Selected mature microRNAs in glioblastoma tissue versus adjacent, nonpathological brain (n = 5 each) (E), and in GSCs (n = 5 per subtype) versus NPCs (n = 5) (F), based on nCounter NanoString platform. For GSC’s subtype identity, see (B) and (C). (G) Selected microRNA precursors in patients with glioblastoma (n = 195) versus healthy individuals (n = 10), based on the TCGA database. For the subtype identity, see (A). (H and I) Selected microRNA precursors in glioblastoma tissue versus adjacent, nonpathological brain (n = 5 each) (H), and in GSCs (n = 5 per subtype) versus NPCs (n = 5) (I), based on custom NanoString precursor platform. For GSC’s subtype identity, see (B) and (C). (J and K) Mature microRNAome but not microRNA precursors distinguish between patients with glioblastoma and healthy individuals. PCA of mature (J) and precursors (K) microRNA in patients with glioblastoma (red dots, n = 490 and n = 205) versus healthy individuals (blue dots, n = 10) [(J) and (K), respectively].
DICER localizes to the nucleus in glioblastoma cells instead of the cytosol
To explore this further, we analyzed the expression of genes/proteins forming the major enzymatic complexes responsible for the transition between primary, precursor, and mature forms of microRNA, DROSHA and DICER. Several proteins compose the enzymatic core of DICER complex, with the most crucial cofactor of DICER being RISC-loading complex subunit TAR (HIV) RNA binding protein 2 (TARBP2), which increases the rate of RNA substrate recognition by DICER and the stability of DICER/RNA substrate complexes (25). Analysis of the TCGA database showed no significant difference in the expression of DROSHA and DICER between a normal brain and glioblastoma, but the expression of TARBP2 was markedly elevated in tumor tissue (fig. S2A). However, none of these genes/proteins correlated with the survival of patients with glioblastoma, suggesting that differences in their expression levels are unlikely to be that relevant to glioblastoma outcome (fig. S2B). However, database querying (https://subcellbarcode.org) suggested that in cancer cell lines (including glioblastoma), DICER and TARBP2 are mostly nuclear (fig. S2C), while the canonical DICER step involving the maturation of microRNA takes place in the cytosol. When we compared the distribution of DICER across the cellular compartments of GSCs and NPCs, the difference became apparent: While in NPCs, DICER was, as expected, predominantly cytosolic (less than 20% in the nucleus), in M and P GSCs, it was decidedly nuclear (70 to 80% based on densitometry analysis) (Fig. 2A). To confirm this distribution difference in vivo, we analyzed the subcellular distribution of DICER in GSC-originated xenografts in mice brains. As shown in Fig. 2B, DICER was predominantly nuclear in GSCs [green fluorescent protein (GFP)–positive], while in cells from surrounding tissue (GFP-negative), it was primarily cytosolic. These results clearly demonstrated that the change in subcellular localization of DICER is particular to GSCs.
Fig. 2. DICER is retained in the nucleus of GSCs, forming a complex with RBM3 protein.
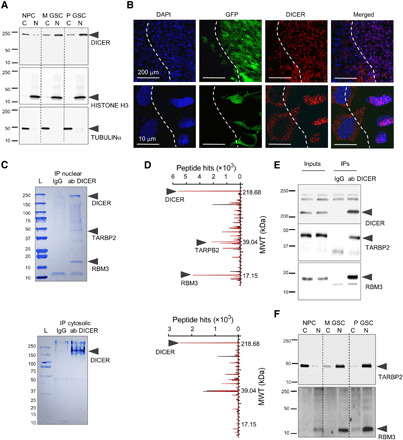
(A) Representative Western blotting of DICER in cytosolic (C) and nuclear (N) fractions from NPCs and GSCs. (B) Representative images of GFP-positive M GSC–originated intracranial xenograft sections 10 days after implantation with DAPI (blue), GFP (green), and DICER immunohistochemistry (red) are shown, and white dashed line denotes approximate boundary between xenograft and the brain. (C and D) Representative Coomassie blue staining (C) and mass spectroscopy peaks of DICER-associated proteins (D) using GSC nuclear (top) and cytosolic (bottom) extract IP with either IgG (black peaks) or DICER antibodies (red peaks) (n = 3). Molecular weight (MWT) in kDa is indicated. (E) Representative Western blots of nuclear lysate inputs and IP obtained by IgG and DICER antibodies. (F) Representative Western blotting of cytosolic (C) and nuclear (N) fractions from NPCs and GSCs. Loading and fraction purity controls are shown in (A).
RBM3 is an interacting partner of nuclear but not cytosolic DICER
The above finding prompted us to elucidate the mechanism for this change in subcellular localization by analyzing the interactome of nuclear DICER. Immunoprecipitation (IP) of DICER from the nuclear fraction of GSCs allowed the identification of interacting proteins and RNAs. Among proteins in the immunoprecipitate, we identified, predictably, DICER and its canonical interaction partner TARBP2 (25). In addition, RBM3 was found to be the most prominent nuclear DICER-interacting protein, but no binding was detected between cytosolic DICER and RBM3 or TARBP2 (Fig. 2, C and D, and table S2). RBM3 is a highly conserved protein engaged in the biosynthesis of different RNA species [including microRNAs (26)], and it is believed to function as a proto-oncogene associated with tumor progression and metastasis (27). Although RBM3 is strongly expressed in glioblastoma, its expression does not correlate with patient survival (fig. S2, A, B, and D). All these interactions were further confirmed by IP and Western blotting (Fig. 2E). In addition, the subcellular distribution of TARBP2 mirrors that of DICER, and RBM3 is strictly nuclear as expected from the data query (Fig. 2F and fig. S2C). These findings thus suggested that RBM3 is a previously identified protein interacting partner of the nuclear but not cytosolic DICER complex.
Circ2082, a circRNA that is highly expressed in cancer, binds to RMB3 and is part of the nuclear DICER complex
Having found RBM3 as a protein binding to nuclear DICER, we then analyzed nuclear DICER’s RNA interactome. We detected a population of small RNAs whose size corresponded to that of precursor microRNAs and one prominent, specific band (fig. S3A). Sequencing of the complementary DNA (cDNA) obtained from the immunoprecipitate revealed that it was a fragment located close to the 3′-end of MALAT1, an lncRNA with well-recognized protumorigenic function in multiple malignancies, including glioblastoma (28). Although quantitative polymerase chain reaction (qPCR) confirmed the enrichment of identified fragment in a nuclear immunoprecipitate of DICER, this approach failed to detect a larger fragment corresponding to MALAT1 (Fig. 3A). This and the fact that the PCR product did not disappear upon ribonuclease (RNase) treatment (Fig. 3A) led us to the hypothesis that this fragment may be a circRNA originating from the MALAT1 transcript. Circularization-predicting software (http://circbase.org) revealed that there was a high probability for a circRNA arising from this region of linear MALAT1 (fig. S3B, top). On the basis of this, we designed both divergent and convergent primers to distinguish between linear and circular forms of this transcript and to detect the junction site (Fig. 3B and fig. S3B, bottom). CircBase annotated this circRNA as hsa_circ_0002082 (chr11:65271199-65272066), and we abbreviated it as circ2082. Using circular-specific primers, we demonstrated enrichment of circ2082 in the nuclear DICER immunoprecipitate (Fig. 3C). To test the interaction between circ2082 and its protein partners, we used RNA antisense purification (RAP) assay followed by mass spectrometry and confirmed the presence of RBM3 in the circ2082 complex (Fig. 3, D to F, and table S2). In a set of reciprocal approach experiments, we used a fusion GFP-RBM3 protein to circumvent the lack of IP-grade RBM3 antibodies (Fig. 3G), and we detected circ2082 in the GFP immunoprecipitate (Fig. 3, H and I). Last, we performed a series of circ2082 protein partner knockdown experiments (fig. S3, C and D) followed by RAP assay to assess the interdependency of proteins as binding partners to circ2082. The knockdown of neither protein affected the levels of circ2082 (fig. S3E). While RBM3 was readily detectable in pull-down material regardless of the presence of DICER, the DICER itself was present only with RBM3 intact (fig. S3F), suggesting that only RBM3 binds directly to circ2082. These results provided proof for the existence of nuclear RNA/protein complex consisting of proteins indispensable in microRNA maturation (DICER), RNA biogenesis/processing (RBM3), and a noncoding circRNA (circ2082) originating from a notorious noncoding oncogene, MALAT1.
Fig. 3. DICER/RBM3 forms complex with circ2082.
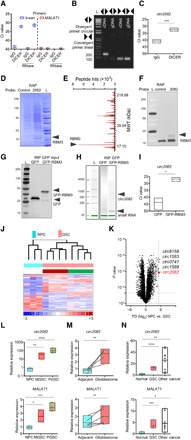
(A) RIP of nuclear lysates with DICER antibodies followed by RNA isolation, RNase treatment, and qPCR (n = 3) using indicated primers. (B) Agarose gel of PCR products using cDNA or genomic DNA (gDNA) and indicated primers. (C) RIP of nuclear extracts from GSC using IgG and DICER antibodies, qPCR analysis with means ± SD. (D to F) Coomassie blue staining (D), mass spectroscopy peaks of circ2082-associated proteins (E), and Western blot (F) using GSC protein extracts upon RAP with control or anti-circ2082 probes (n = 3). (G to I) RIP of lysates from GSC transfected with GFP or GFP-RBM3 vector. Protein inputs analyzed by Western blot (G), RNA profile by Agilent Bioanalyzer (H), and circ2082 enrichment in RIP by qPCR with means ± SD (I). (J) Heatmap with a hierarchical clustering for NPCs (n = 3) and GSCs (n = 4 per subtype), (n = 1270 of 12,659 detectable, P < 0.05). Subtype identity: red, mesenchymal; green, proneural. (K) Volcano plot for NPCs (n = 3) and GSCs (n = 10) and the top five most up-regulated circRNAs. Dashed lines indicate P value and fold cutoffs. (L to N) qPCR analysis of circ2082 (top) and linear MALAT1 (bottom) with means ± SD in NPCs versus M GSCs (50.1- and 2.5-fold, respectively) and NPCs versus P GSCs (150.2- and 3.8-fold, respectively) (n = 4 per group) (L); adjacent brain versus glioblastoma (2.3- and 1.4-fold paired, respectively), matching lines identify pairs (n = 5) (M); and NPCs versus GSCs (12.8- and 3.9-fold, respectively), and NPC versus other cancers (27.1- and 4.6-fold, respectively) (nonmalignant cells, n = 6; GSCs, n = 3 per subtype; other cancers, n = 9) (N); P values: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
This discovery prompted us to take a closer look at the circRNAome of GSCs and NPCs. To this end, we used the human circRNA array. We detected 12,659 of 13,617 probes on Arraystar human circRNA array [confirming their abundance in the human brain (16)] in both types of cells. Of these, 478 were significantly down-regulated in GSCs, while almost twice as many, 792, were significantly up-regulated (Fig. 3J). Circ2082 was among the top circRNAs up-regulated in GSCs (Fig. 3K). The analysis of the expression of circ2082 and its transcript of origin, MALAT1, revealed several interesting details. First, although both transcripts were high in GSCs and neither of them was subtype specific, circ2082 was more profoundly up-regulated in GSCs than its linear parental transcript (Fig. 3L, ~50- to 100-fold versus ~3-fold). Second, the degree of overexpression of circ2082 in glioblastoma in comparison to adjacent matched brain tissue was again higher than that of linear MALAT1 (Fig. 3M). Last, both transcripts were elevated to various degrees in cancers other than glioblastoma, again more strongly in the case of circ2082 (Fig. 3N).
As glioblastoma is notorious for its invasiveness, we cannot exclude the possibility that seemingly healthy tissue can contain a fraction of tumor cells. However, the compromised purity of the sample can be perceived as somewhat ambiguous as the stromal component often infiltrates a bulk tumor tissue. Also, notably, the matching pairs of samples were harvested from the same individual, effectively nullifying high patient-to-patient heterogeneity. Last, as both circ2082 and MALAT1 are notably lower in the normal brain, it implies that the significance we detected would only become stronger if we have dealt with pure populations. Cell assays using cancer and nonmalignant cells substantiated our bulk tissue findings. These results strongly suggested that while both MALAT1 and its circularized fragment, circ2082, are overexpressed in malignant cells, the circular form is, in most cases, present at higher levels.
Circ2082 knockdown leads to normalization of DICER localization to the cytosol with reestablishment of microRNAome homeostasis
Circularization generates a unique sequence with no homology in the entire human genome, allowing precise targeting of circ2082 via ASO, which leaves the linear parental transcript intact (Fig. 4, A and B). Conversely, small interfering RNA (siRNA)–mediated knockdown of MALAT1 effectively removes both linear and circular transcripts, as the former is the source of origin for the latter (fig. S4A). Both transcripts are almost exclusively nuclear (fig. S4B), which we already demonstrated for linear MALAT1 (13). Having the ability to silence circ2082 effectively and specifically, we attempted to characterize the molecular and phenotypic footprint of the knockdown. To this end, we analyzed changes to the protein-coding transcriptome and microRNAome in circ2082 knockdown GSCs. Notably, the effect of circ2082 knockdown on the protein-coding transcriptome, albeit substantial, was cell type–dependent (Fig. 4C and fig. S4C). Conversely, the impact of circ2082 expression on the microRNAome was so potent that knockdown cells from both subtypes clustered together, apart from their parental cells (Fig. 4D and table S1). Detailed analysis of microRNAome effect revealed the pattern that was common for both subtypes: widespread derepression of numerous weakly expressed microRNAs and inhibition of a relatively few that were strongly expressed in control GSCs (Fig. 4E and table S1). The analysis of specific microRNAs with known function or subtype-predictive ones (same as in Fig. 1 and fig. S1) confirmed this—the expression of tumor-suppressive miR-128, miR-124, and miR-1 was unblocked regardless of subtype. Simultaneously, the expression of oncogenic miR-21, P GSC–specific oncogenic miR-10b, and M GSC–specific miR-31 were suppressed (fig. S4D). The levels of their precursors were not altered upon circ2082 knockdown (Fig. 4F, fig. S4E, and table S1), suggesting again that unblocked processing rather than transcriptional change was responsible for the observed phenomenon. Last, we were able to demonstrate that knockdown of circ2082 resulted in cytosolic relocalization of the DICER complex, while RBM3 was and remained strictly nuclear (Fig. 4, G and H, and fig. S4F). These results showed that nuclear retention of the DICER complex, a molecular consequence of high levels of circ2082 in GSCs, leads to the blockade of mature microRNAome.
Fig. 4. The knockdown of circ2082 results in a widespread derepression of GSC microRNAome.
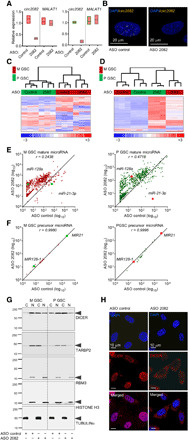
(A) qPCR analysis for circ2082 or linear MALAT1 in GSCs with means ± SD. GSC (green, proneural; red, mesenchymal; n = 3 per subtype) were transfected with ASO control or circ2082. (B) Representative FISH images of the circ2082 in GSC transfected with ASO control or circ2082 with a fluorescently labeled junction probe and nuclei stained with DAPI (n = 4). (C) Heatmap with a hierarchical clustering for GSCs transfected with ASO control or circ2082 (n = 3 each subtype) based on the most variable mRNA transcripts (n = 3740 of 31,555, P < 0.05). GSC’s subtype identity: red, mesenchymal; green, proneural. (D) Heatmap with an unsupervised hierarchical clustering for GSCs transfected with ASO control or circ2082 (n = 3 per subtype) based on levels of 756 mature microRNAs. GSC’s subtype identity: red, mesenchymal; green, proneural. (E) Scatterplots for GSCs (M GSC: red, left; P GSC: green, right) transfected with ASO control or circ2082 (n = 3 each) based on levels of 756 mature microRNAs. (F) Scatterplots for GSCs (M GSC: red, left; P GSC: green, right) transfected with ASO control or circ2082 (n = 3 each) based on levels of 22 precursor. (G) Representative Western blotting of cytosolic (C) and nuclear (N) fractions of GSCs (n = 4) transfected with ASO control or circ2082. (H) Representative images of cultured GSC (n = 3) transfected with ASO control or circ2082. DAPI staining (blue) and DICER immunohistochemistry staining (red) analysis were performed.
Circ2082 knockdown has anticancer effects
The efficient and specific knockdown of circ2082 allowed precise characterization of its phenotypic footprint in GSCs. In vitro, both limiting dilution assay and neurosphere formation assays demonstrated the strong anti-GSC effect of circ2082 knockdown (Fig. 5, A and B, and fig. S5A), underlining pan-glioblastoma effects of circ2082, as its knockdown affected equally both tested GSC subtypes, regardless of their very different transcriptomes. For testing in vivo effects of circ2082, GSCs were pretreated with ASOs and implanted into the brains of athymic mice. Similar to in vitro observations, ensuing tumors were significantly smaller when M GSCs were treated with ASO circ2082 compared to scrambled control ones (fig. S5, B and C). The analysis of tumor volume in P GSC–originated tumors was not feasible as these do not form nodular tumors but are very diffused, as we demonstrated previously (11). We also observed significant survival benefits of circ2082 knockdown. Mice bearing very aggressive M GSC–originated tumors showed an increase in median survival from 12 to 17 days, i.e., more than 40% of the post-implantation time, upon circ2082 knockdown. Implantation of P GSC–originated tumors pretreated with ASO circ2082 did not result in mortality up to 300 days, suggesting the inhibitory effect of the knockdown during the tumor initiation phase (Fig. 5, C and D). These results indicated that transcriptome deregulated by circ2082 in both subtypes might be critical for the observed phenotypes. Thus, to discover the culprits, we analyzed the fraction of the transcriptome deregulated in both subtypes (Fig. 5E). Although these 377 genes were significantly deregulated in both subtypes, they had no power to overcome the clustering of knockdown cells with their parental counterparts (Fig. 5F). Thus, to set up a circ2082-dependent signature into a broader clinical context, we queried these genes with the TCGA glioblastoma patient dataset. It allowed us to filter out six genes annotated with hazard ratios from Cox analysis, which were sufficient to cluster patients’ samples accordingly with either up- or down-regulation of these genes upon circ2082 knockdown (Fig. 5G). These clusters had significant power to predict the outcome using Kaplan-Meier estimator survival analysis (Fig. 5H). These results thus identified and described the action of the effectors of circ2082-dependent phenotype in GSCs; yet, the role of microRNAome deregulation in these processes remained unsubstantiated. So instead of further scrutiny of the transcriptome itself, we revisited microRNAome analysis to verify whether direct targets of selected microRNAs provide sufficient evidence for the explanation of a circ2082-dependent effect.
Fig. 5. The knockdown of circ2082 mitigates tumorigenicity of GSCs.
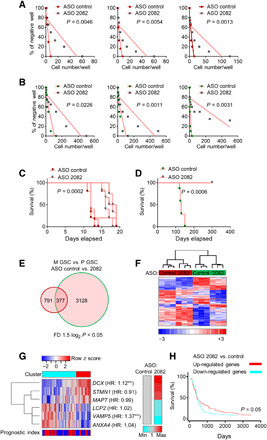
(A and B) Representative sphere frequency assays using linear regression plot. GSCs (n = 3 per subtype) were transfected with ASO control or circ2082. P values are indicated. (C and D) Kaplan-Meier curves of mice intracranially injected (n = 5 per group) with GSCs (red, mesenchymal; green, proneural) transfected with ASO control or circ2082. P values are indicated. (E and F) GeneVenn analysis (E) and heatmap with hierarchical clustering (F) for GSCs transfected with ASO control or circ2082 (n = 3 per subtype) based on the most variable mRNA transcripts in both subtypes (n = 377 of the total 31,555), fold difference (FD) 1.5 log2, and P < 0.05 are shown. GSC’s subtype identity: red, mesenchymal; green, proneural. (G) Heatmap of the most varied genes (n = 6) in GSC with circ2082 knockdown with color annotations according to profile similarity and prognostic index stratification cluster based on TCGA database (left) and average relative expression based on qPCR analysis (n = 3) as a color bar (right). Molecule names are annotated with hazard ratios (HR) from Cox analysis; **, hazard ratios (HRs) with P ≤ 0.05. (H) Kaplan-Meier curve survival analysis based on six gene signatures in the TCGA database stratified according to their cluster membership [see (G)].
Clinical significance of circ2082 knockdown
As a proof of concept, we first aimed to identify genes correlated with the expression of microRNAs selected previously as deregulated in glioblastoma and being circ2082-dependent (see description of Figs. 1 and 5). To show a correlation of expression between genes and microRNAs, we revisited the TCGA glioblastoma dataset. It revealed a correlation signature of ~1700 genes that were either positively or negatively associated with microRNAs (Fig. 6A and table S3) and stratified the samples into two clusters: miR-128, miR-124, and miR-1 (red cluster) and miR-10b, miR-31, and miR-21 (blue cluster) with the power of outcome prediction (Fig. 6B). However, we had already demonstrated that the subtype identity remained the dominant force in this clustering (Fig. 5, E and F). To circumvent this dominance in the analysis of microRNA-mediated circ2082-dependent effect, we thus supervised the analysis of previously identified microRNA clusters’ mRNA targets. To this end, we separately scrutinized the expression of genes (from an initially selected set of ~1700) after circ2082 knockdown in M GSCs only, in P GSCs only, and in both subtypes. On the basis of these results, we selected one previously validated, significantly altered mRNA target for each of six proof-of-concept microRNAs in all three scenarios to experimentally evaluate the effect of circ2082 knockdown–dependent derepression of microRNAome (Fig. 6, C to E). These included the following: in M GSC circ2082 knockdown cells, GLI2, a down-regulated target of miR-124 (29), and JARID2, an up-regulated target of miR-31 (30); in P GSC circ2082 knockdown cells, CCNG1, a down-regulated target of miR-128 (31), and SART3, an up-regulated target of miR-10b (32); and, in both GSC circ2082 knockdown cells, MET, a down-regulated target of miR-1 (21), and PDCD4, an up-regulated target of miR-21 (33). We compared their expression upon (i) circ2082 knockdown, (ii) exogenous overexpression of depleted microRNAs, and (iii) antisense-mediated knockdown of highly expressed microRNAs. The apparent expression pattern revealed that the genes repressed after circ2082 knockdown were also suppressed in response to exogenous overexpression of their respective microRNAs in both GSC subtypes (Fig. 6D). On the contrary, genes maintained after circ2082 knockdown were derepressed in response to the inhibition of their “master” microRNAs only in the scenario when these microRNAs and mRNAs were expressed in control cells (Fig. 6E). These results suggested that the derepression of microRNAome by circ2082 knockdown had a strong cell type–independent effect, while the circ2082 knockdown–associated repression of oncogenic microRNAs was strictly cell type specific. Although the one-by-one analysis of microRNA/mRNA pairs presented above was useful as a proof of concept, it is unlikely that these few tandems are responsible for the observed far-reaching phenotype, as few of them are significantly outcome-predictive (fig. S6, A to C). Thus last, to delineate a circ2082-dependent microRNA-associated signature in the clinical context, we queried microRNA correlation genes with the TCGA glioblastoma patient dataset. Genes deregulated in P GSCs, M GSCs, and all GSCs together (fig. S6, D to F) were filtered to keep the most varied molecules among the list annotated with hazard ratios from Cox analysis. All three analyses had power to predict the outcome using Kaplan-Meier estimator survival analysis (Fig. 6F). These results thus identified and clarified the circ2082-dependent phenotype in GSCs in the context of circ2082-implemented microRNAome surveillance and deregulation.
Fig. 6. The circ2082-dependent footprint is mediated via the rearrangements of the microRNAome.
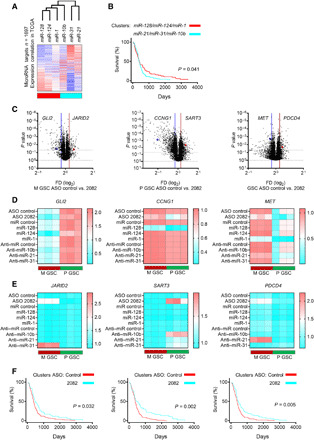
(A) Heatmap displaying the clustering of expression correlation of genes (n = 1697) and selected microRNAs using the TCGA database. The correlation value (blue, negative and red, positive) and annotations of profile similarity (bottom bar) are shown. (B) Kaplan-Meier curve survival analysis based on genes associated with six microRNA in the TCGA database and stratified according to their cluster membership [see (A)]. (C) Volcano plots of genes [n = 1697, see (A)] for M GSCs (n = 3, left), P GSCs (n = 3, middle), and all GSCs (n = 6, right) transfected with ASO control or circ2082 are shown. The selected deregulated targets are shown. Lines indicate a P value and fold difference cutoffs. (D and E) qPCR analysis for indicated mRNAs upon the treatment by ASO circ2082 or tumor-suppressive microRNAs (D) and ASO circ2082 or oncogenic anti-microRNAs (E) in GSCs (n = 3 per subtype). (F) Kaplan-Meier curve survival analysis using TCGA database and stratified according to their cluster membership (see fig. S6, D to F) after circ2082 knockdown in M GSCs (left), P GSCs (middle), and all GSCs (right).
DISCUSSION
CSC-rich tumors such as glioblastoma are more aggressive than more differentiated ones, e.g., meningioma. Although differentiation lineage programs exist in CSCs, groups of genes act as brakes to avoid setting these programs in motion. So, releasing those brakes should dampen their aggressiveness (34). This switch requires sweeping changes in gene expression, so microRNAs that simultaneously repress multiple factors from certain pathways/developmental programs are efficient regulators of cell fates. Thus, it can be argued that microRNA suppression that is common in cancer cells reflects their undifferentiated status (35). We showed previously that the widespread loss of microRNAs in GSCs enhanced their “stemness” (24) and that the restoration of premalignant microRNA landscape favors more differentiated and a less neoplastic milieu (23). So, the mechanism of the initial loss of microRNAs is in the research crosshairs now, with a focus on the disruption of microRNA maturation. However, the mechanisms of cancer cell type–specific alterations of microRNAome are unknown.
We have established that DICER, an enzyme responsible for microRNA maturation, predominantly localizes in the nucleus of CSCs, as it was suggested before for breast carcinoma (36), while in NPCs and other nonmalignant cells, it is localized in the cytosol (37). Thus, we asked what mechanism was responsible for this phenomenon and whether this change in localization was accountable for the observed defect in microRNA processing. To this end, we immunoprecipitated nuclear DICER from CSCs for the analysis of both its protein and RNA interactomes. Among proteins bound to nuclear DICER, its canonical interaction partner TRBP2 (38) and RNA binding protein RBM3 came into view. The interaction with RBM3, which was associated with microRNA biogenesis at the DICER step (26), and with small RNAs of a size corresponding to precursor microRNAs, further supported the idea of impaired microRNA maturation in CSCs as an underlying reason of suppressed microRNAome. Besides these, we also detected the presence of the circRNA, circ2082. It belongs to the relatively recently found ncRNA family of circRNAs implicated in multiple cellular processes (15, 39). Circ2082 originates from the well-established noncoding oncogenic lncRNA MALAT1 that promotes protumorigenic traits in numerous solid tumors, including glioblastoma (28, 40). We have tools that allow us to distinguish between the effects of linear and circular forms of any given transcript. Circularization brings together two sequences that are far apart in a linear molecule, thus creating a unique sequence not found in linear codification, allowing the design of ASO that targets exclusively circular form. Conversely, antisense targeting of linear transcripts removes both forms as linear structure serves as a template for circular one.
Circ2082 is one of the most highly expressed circRNAs regardless of the GSC subtype, but it is expressed at low levels in NPCs. In contrast, the full-length transcript (MALAT1) from which circ2082 derives is only moderately elevated in GSCs versus NPCs, suggesting distorted stoichiometry. circRNAs are reported to be more stable compared to their parental linear RNAs (17), and it is likely that while cicr2082 accumulates, MALAT1 gets rapidly degraded in cells, accounting for the observed differences. It is also plausible that circularization efficiency may play a role in this discrepancy, as circular transcripts emerge at the expense of linear ones. It allows us to speculate that the cancer cell can promote the circularization by yet-undetermined mechanisms, directing its transcriptional output toward increased circularization of specific transcripts or the overall enhancement of its circRNAome. It also lets us to hypothesize that at least part of the linear transcript’s footprint (either protein-coding or noncoding), both phenotypic and molecular, can be mediated through circRNA originating from such transcript.
The discovery of circRNA in the complex with DICER raises the obvious question: What does it do? Knockdown of strictly nuclear circ2082 shifted the expression of the protein-coding transcriptome in a cell-dependent manner, i.e., knockdown cells remained relatively similar to parental ones. However, its effect on the microRNAome was much more pronounced as knockdown cells clustered together but not with their parental cells, indicating far-reaching rearrangements of their microRNAome. The deregulation of microRNAs was not random—the ones repressed in CSCs were derepressed (miR-128 was among the most induced), while few abundant, oncogenic microRNAs (e.g., miR-21) were suppressed. It can be argued, although direct evidence is still lacking and will be the subject of follow-up studies, that derepression of a multitude of lowly expressed microRNAs puts a strain on the capacity of processing machinery, thus leading to relative suppression of previously highly expressed microRNAs.
On the basis of the published association between global down-regulation of microRNAs and cancer growth, we propose that the loss of the microRNAome by circ2082-dependent nuclear retention of DICER determines the CSCs’ molecular identity and their tumorigenic potential. Thus, the targeting of circ2082 reverses the cell fate of CSCs via facilitating the restoration of a premalignant microRNAome, and cytosolic release of the DICER complex seems to play a vital role in this phenomenon. Additional studies will elucidate whether all substantially enriched/depleted microRNAs are necessary to affect cellular fate or whether all these changes are directly circ2082 dependent. We hypothesize that an excellent candidate for a future study could be miR-451, known to be transcriptionally regulated and DICER independent while being relevant to glioblastoma phenotypes in response to microenvironmental cues (4, 41–43).
So, on the basis of our past discoveries on the association between global microRNAs down-regulation and cancer growth, we propose that loss of microRNAome by circ2082-dependent DICER nuclear retention determines CSC molecular identity and their tumorigenic potential. Thus, the targeting of circ2082 reverses cell fate in CSCs by cytoplasmic relocalization of DICER that mediates microRNAome correction.
Each mature cell that is poised to perform specialized tasks within the human body starts as a primordial cell. So, its journey to cellular adulthood is determined by binary decisions until it reaches its final destination. However, when these steps go awry, the stem cell may take a turn down the oncogenic path. To retract the wrong decision made by these primitive cells as they differentiate, it requires unblocking their differentiation potential. So, the modulation of microRNA biogenesis at posttranscriptional steps by ncRNA and RNA binding proteins is the pivotal point of regulatory control over the expression of microRNAs and the cellular processes they affect. However, the extent and conditions under which the microRNA pathway is amenable to regulation at posttranscriptional steps are poorly understood in cancer.
As widespread underexpression of mature microRNAs is often observed in solid tumors occurring in organs other than brain, the restoration of premalignant microRNA landscape can be thus highly beneficial in cancer cells regardless of the cell origin. circRNA, characterized by a unique circularization site, can be highly accurately targeted using ASO, even in the presence of its linear counterparts, which open possibility for smooth delivery [even systemic, as we showed previously (32)] into the brain.
CircRNAs are recognized as relatively new and promising candidates for biomarkers of pathologies because of their high stability and specificity of detection via junction site. However, as protein-coding transcriptome of glioblastoma is well characterized at both bulk and single-cell level, there are no data on the expression of circRNAs in a large cohort of TCGA database. Thus, we performed a transcriptome array in CSCs to assess circ2082-dependent footprint. Our data showed that genes differentially deregulated in circ2082 knockdown cells are potent effectors of glioblastoma progression, as reflected by the survival analysis. As this did not provide unquestionable proof of whether the unblocking of the microRNAome is functionally involved in the survival benefits, we performed the analysis of direct, validated targets of selected microRNAs that offered sufficient evidence for the circ2082-dependent microRNA engagement in this process.
We have achieved considerable progress in deconstructing ncRNA-driven cellular phenotypes and their molecular mechanism of action. However, our current knowledge has a considerable gap in understanding what are the mechanisms of microRNAome suppression during tumorigenesis and how vital is microRNA homeostasis in tipping the balance between differentiation and stemness. Our results strongly suggest the engagement of a member of the recently found class of circRNA (circ2082) in these processes. We argue that these interactions interfere with microRNA processing machinery. Thus, a fascinating picture of ncRNA networks in the very core of human brain physiology and pathology begins to emerge, bringing new opportunities for the expansion of our knowledge and future translational applications.
MATERIALS AND METHODS
Cell culture
GSCs and NPC were cultured as neurospheres under stem cell–enriching conditions using Neurobasal medium supplemented with 1% glutamine, 2% B27, and EGF and FGF (20 ng/ml) (epidermal growth factor and fibroblast growth factor, respectively) using ultralow attachment plates/flasks. The unique identity of cultured patient-derived cells was confirmed by short tandem repeat analysis. All GSCs used in this study are isocitrate dehydrogenase wild type (9). Mycoplasma testing was routinely performed by PCR. Mesenchymal and proneural subclass classification by gene expression profiling was described previously (13). Nonmalignant brain cells (n = 6) included neurons, two NPCs, two astrocytes, and brain microvasculature endothelial cells; other cancer cell lines (n = 9) included two thyroid carcinomas, leukemia, three breast carcinomas, melanoma, and two lung carcinomas.
Cell transfection
Lipofectamine 2000 was used for all transfections. For transfection, 1 μg of plasmid or 10 pg of oligo was added in 500 μl of a medium, followed by addition of 6 μl of Lipofectamine 2000 in 500 μl of medium and incubated for 5 min. The two mixtures were pooled and incubated further for 10 min at room temperature. The respective transfection mixture was then added to the six-well ultralow attachment plate with 0.5 × 106 cells. Cells were incubated at 37°C for 18 hours in a 5% CO2 by 72 hours and then harvested by centrifugation (5 min/1000 rpm at 4°C).
Plasmids
cDNA of human RBM3 was cloned into Eco RI and Xho I sites of the pCDH-EF1-copGFP vector.
Cell cytoplasmic/nuclear fractionation
Cytoplasmic and nuclear fractions were isolated via mild lysis and centrifugation using the Nuclear/Cytosol Fractionation Kit (Biovision, Milpitas, CA). Briefly, 2 × 106 cells were treated with trypsin-EDTA (Gibco) and resuspended with cytosol extraction buffer A. Cytosol extraction buffer B was added to the suspension followed by centrifugation to obtain cytosolic protein/RNA containing supernatant, whereas the pellet was further processed for nuclear proteins/RNAs.
Protein purification
Total cell protein content was isolated via extraction for 30 min in ice-cold lysis buffer containing 50 mM tris-Cl (pH 7.5), 100 mM NaCl, 1% Triton X-100, 1 mM dithiothreitol (DTT), 1 mM EDTA, 1 mM EGTA, 2 mM Na3VO4, 50 mM glycerophosphate, and a protease inhibitor cocktail (GE Healthcare, Piscataway, NJ), followed by centrifugation (15 min/13,000 rpm at 4°C).
Quantitative PCR
Total RNA was extracted using a standard TRIzol protocol (Invitrogen). The RNA quantity and quality were measured using a NanoDrop 2000 (Thermo Fisher Scientific) and analyzed using a Bioanalyzer 2100. Gel images are provided for visualization of fragment sizing and distribution, as well as for a visual representation of the RNA ladder. For RNase R treatment, 1 μg of total RNA was incubated for 30 min at 37°C with or without 2.5 U of RNase R (Epicentre Technologies, Madison, WI). For mRNA analysis, 3 μg of total RNA was treated with deoxyribonuclease (Promega) for 2 hours to remove genomic DNA. For microRNA analysis, 10 ng of total RNA was used. Reverse transcription was performed using random hexamers and iScript (Bio-Rad), and qPCR was performed using TaqMan or SYBR Green master mix (Applied Biosystems).
Amplification was performed using the StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA), and the software determined Ct thresholds. Expression was quantified using the ΔΔCt method using 18S ribosomal RNA (for mRNA/circRNA) or U6 small nuclear RNA (for microRNA or nuclear RNA fraction) as reference genes. PCR products were cloned into pGEM-T Easy, and different clones were picked for Sanger sequencing. Probes and primers are listed in table S4.
Immunoprecipitation
Cleared whole cell or cell fraction protein lysates were incubated at 4°C for 3 hours with the appropriate antibody precoupled to protein A/G plus agarose beads (sc-2003, Santa Cruz Biotechnology). The beads were washed twice with extraction buffer, twice with extraction buffer containing 0.5 M LiCl, and twice with assay buffer [40 mM tris-Cl (pH 7.5), 0.1 mM EDTA, 5 mM MgCl2, and 2 mM DTT].
RNA IP
Cells (control) or transfected with pCDH-EF1-copGFP/pCDH-EF1-copGFP-RBM3 vectors were ultraviolet (UV) cross-linked at 400 mJ/cm2. Cells or nuclei were collected with RNA immunoprecipitation (RIP) buffer [100 mM NaCl, 20 mM tris-Cl (pH 8.0), 0.5 mM EDTA, 0.5% NP-40, 0.1% Na-deoxycholate, 0.5 mM DTT, RNasin (100 U/ml), protease, and phosphatase inhibitors]. One-tenth of each cell or fraction lysate was used for RNA extraction using TRIzol reagent (Invitrogen), and the rest was incubated with anti-DICER or immunoglobulin G (IgG) or GFP antibodies coupled with protein A/G plus agarose beads (sc-2003, Santa Cruz Biotechnology) overnight at 4°C. Protein/RNA complexes were washed three times with RIP buffer and three times with high-salt buffer (1 M NaCl modified RIP buffer). Samples were then treated with proteinase K (Invitrogen), and RNA was extracted using TRIzol. The qPCR was performed as described above.
RNA antisense purification
GSC (2 × 106) (control or with siRNA-mediated knockdown of DICER or RBM3) were UV cross-linked at 400 mJ/cm2, and cell lysates (500 μg) were subjected to pull-down using 3 μg of biotin-labeled circ2082 probe (see table S4) and streptavidin beads at room temperature for 2 hours. The reaction was washed three times with RIP buffer and three times with high-salt buffer (1 M NaCl modified RIP buffer). RNA was then digested using RNase A, and bound proteins were analyzed by immunoblotting.
Western blotting
Proteins were separated by SDS–polyacrylamide gel electrophoresis, transferred to a polyvinylidene fluoride membrane (Immobilon-P, EMD Millipore) by liquid transfer, and the Western blots were probed using the appropriate primary antibodies (1:1000) followed by alkaline phosphatase secondary antibodies (1:5000). The signals were detected using a chemiluminescence system (Thermo Fisher Scientific), followed by Gel Dock system (Bio-Rad) imaging.
Single-molecule fluorescence in situ hybridization
Single-molecule fluorescence in situ hybridization (FISH) was performed on GSCs grown on glass coverslips according to the following protocol. Cells were washed twice in phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde (Electron Microscopy Sciences) in PBS for 10 min, then washed in PBS, and stored in 70% ethanol for ≥2 hours at 4°C. Coverslips were equilibrated for ≥2 min in washing buffer (10% formamide, 2× SSC) and probing using custom probes (44) labeled with Alexa Fluor (see table S4) diluted to 25 nM in hybridization solution [10% formamide, 2× SSC, and dextran sulfate (100 mg/ml)] in a humidifying chamber at 37°C overnight. The excess probe was washed for 30 min in washing buffer containing DAPI (4′,6-diamidino-2-phenylindole; 100 ng/ml) and 5 min in washing buffer to remove the excess of DAPI. Nikon Eclipse Ti microscope was used for signal localization and imaging.
Immunofluorescence
Paraformaldehyde-fixed, paraffin-embedded specimens were cut into 5- to 10-μm-thick sections and mounted on chromogelatin-precoated slides. After paraffin removal in xylene for 30 min, tissue was hydrated in decreasing grades (98 to 50%) of ethyl alcohol. Antigen retrieval was achieved by incubation in a sodium citrate buffer (pH 6) and boiled for 20 min. Nonspecific antigens were blocked using 10% normal rabbit serum (NRS) (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA). Incubation with primary antibodies and monoclonal mouse anti-DICER1 (1:150; Thermo Fisher Scientific) was performed overnight at 4°C. Afterward, slides were washed in tris-buffered saline buffer [50 mM tris-HCl (pH 7.4) and 150 mM NaCl] and incubated for 1 hour with secondary antibodies rabbit and anti-mouse conjugated with Alexa Fluor 594 (1:5000; Thermo Fisher Scientific). Negative control sections were stained with primary antibodies replaced with either 10% NRS (Abcam) or mouse IgG1 (1:150; Abcam). Last, sections were mounted in UltraCruz Mounting Medium (Santa Cruz Biotechnology) containing DAPI.
In vitro cell assay
For spheroid formation, GSCs were dissociated to single cells and plated at 500 cells per well in a 96-well plate in 100 μl of supplemented Neurobasal medium. Size and number of spheroids were quantified after 96 hours using ImageJ, and spheroid volume was calculated. For limited dilution spheroid assay, single-cell suspensions were plated in ultralow attachment 96-well plates at different concentrations (from 1 to 500 cells per well) in 0.1 ml of supplemented Neurobasal medium. Cultures were left undisturbed for 7 days. After incubation, spheres were imaged using a Nikon Eclipse Ti microscope, and the percentage of wells not containing spheres for each cell concentration was calculated and plotted against the number of cells per well.
Human tissue processing
The collection of the human operative specimens was performed in accordance with the Brigham and Women’s Hospital/Dana Farber Cancer Institute Institutional Review Board protocol (no. 10-417) and after obtaining informed consent. Frozen surgical specimens with histopathology-confirmed glioblastoma or normal brains were obtained through the Department of Neuropathology at the Dana Farber Cancer Institute.
Animal studies
Animal experiments were performed using 6- to 8-week-old immunodeficient athymic mice (FoxN1 nu/nu, Envigo, South Easton, MA), in compliance with all relevant ethical regulations applied to the use of small rodents, and with approval by the Institutional Animal Care and Use Committees at the Brigham and Women’s Hospital and Harvard Medical School (HMS) (no. 2016N000384). For intracranial tumor implantation, a stereotactic frame (Kopf) was used to inoculate each animal in the right striatum with M GSC (5000 cells per point, n = 3) or GSC admixed with ASO control or circ2082. Mice were euthanized and perfused 6 days after surgery (for tumor immunohistochemistry and tumor volume analysis) or when they reached their predetermined end points (for survival analysis).
RNA cloning and sequencing
For the analysis of DICER-bound RNAs, we performed RIP procedure as described above, using anti-DICER and anti-IgG antibodies. Isolated RNA was treated with RNase T1 at a final concentration of 1 U/μl and incubated at 22°C for 15 min. Reverse transcriptase iScript (Bio-Rad) reactions with random hexamers were followed by TOPO cloning (Invitrogen) and sequencing of clones.
Gene microarray
Transcriptome expression analysis was performed on total RNA extracted from GSCs (n = 10) transfected with ASO control or circ2082. Array Star Inc. performed RNA labeling and array hybridization. Briefly, total RNA from each sample was linearly amplified and labeled with Cy3-UTP (uridine 5′-triphosphate). The labeled antisense RNAs (cRNAs) were purified using an RNAeasy mini kit (Qiagen). The concentration and specific activity of the labeled cRNAs (picomol of Cy3 per microgram of cRNA) were measured by a NanoDrop ND-1000. The labeled cRNA (1 μg each) was fragmented by adding 11 μl of 10× blocking agent and 2.2 μl of 25× fragmentation buffer, then heated at 60°C for 30 min, and lastly, 55 μl of 2× GE Hybridization Buffer was added to dilute the labeled cRNA. Hybridization solution (100 μl) was dispensed into the gasket slide and assembled to the gene expression microarray slide. The slides were incubated for 17 hours at 65°C in an Agilent hybridization oven. Agilent Feature Extraction software (version 11.0.1.1) was used to analyze acquired array images. Quantile normalization and subsequent data processing were performed using the GeneSpring GX v12.1 software (Agilent Technologies).
circRNA microarray
CircRNA expression analysis was performed on total RNA extracted from GSCs (n = 10) and NPCs (n = 4). Array Star Inc. performed RNA labeling and array hybridization. Briefly, total RNA was digested with RNase R (Epicentre, Madison, WI, USA) to remove linear RNA and to enrich circRNAs. The remaining RNAs were amplified and transcribed into fluorescent cRNA using a random priming method (Arraystar Super RNA Labeling Kit; Arraystar) and hybridized onto the Arraystar circRNA Array (8 × 15 K, Arraystar) (Rockville, MD, USA). After washing, the arrays were scanned by the Agilent Scanner G2505C. Agilent Feature Extraction software (version 11.0.1.1) was used to analyze acquired array images.
Array data analysis
After quantile normalization of the raw data, genes that had flags in at least 3 of 12 samples as detected (“all targets value”) were chosen for further data analysis. The raw expression intensities were log2-transformed and normalized by quantile normalization. Differential analysis between groups was performed by t test. The cutoffs were P ≤ 0.05 and fold change was ≥2.0. Normality was assumed for log2-transformed normalized intensity values across samples per gene. Genes (>90%) in our dataset passed the Shapiro-Wilk normality test. Differentially expressed transcripts with statistical significance were identified through volcano plot filtering (GraphPad Prism). Hierarchical clustering was performed using the R software (version 2.15).
nCounter assay
nCounter in-solution hybridization method (NanoString) technology and service were used to analyze mature and precursor microRNAs in cells (GSC, n = 10 and NPC, n = 5), glioblastoma tissue specimens (n = 10 from five donors), and GSC transfected with control or anti-circ2082 antisense oligo. Sample preparation and procedure were performed according to the manufacturer’s instructions. Briefly, 100 ng of RNA for solution-phase hybridization between the target (mature or precursor) microRNA and reporter-capture probe pairs and excess probes were removed, and the probe/target complexes were aligned and immobilized in the nCounter cartridge (24 samples × 800 probes), which was then placed in a digital analyzer for image acquisition and data processing. For data analysis, positive and negative corrections, as well as a sample content normalization to the raw data, were applied as per the manufacturer’s guidelines.
Mass spectrometry–based proteomics
Proteomics analysis was performed on immunoprecipitated nuclear and cytoplasmic lysates from GSCs (n = 4) with DICER antibodies. The Taplin Mass Spectrometry facility performed mass spectrometry at HMS. Briefly, protein bands were excised from colloidal blue–stained gels (Thermo Fisher Scientific), treated with DTT and iodoacetamide to alkylate the cysteines before in-gel digestion using modified trypsin (Promega; sequencing grade). The resulting peptides from the whole line were analyzed by nano–liquid chromatography tandem mass spectrometry (UltiMate 3000 coupled to LTQ-Orbitrap Velos, Thermo Fisher Scientific) using a 25-min gradient. Peptides and proteins were identified using Mascot (Matrix Science) and filtered using IRMa software.
TCGA data analysis
The collection of the data from TCGA (TCGA Research, 2008) was compliant with all applicable laws, regulations, and policies for the protection of human subjects, and necessary ethical approvals were obtained. Experimental and clinical data were downloaded (https://tcga-data.nci.nih.gov/), as described in TCGA Research Network. For analysis of mature microRNA, precursor microRNA, and gene expression in glioblastoma, we used normalization of data and aggregation at the feature level as designated by the TCGA glioblastoma the “Level3.” Transcript expression data have been generated from experiments on three different platforms: Affymetrix HT HG U133A (10 + 488 patient samples × 12,042 features). Data were analyzed using free available portals (GlioVis; GBM-BioDP; Betastasis) as a resource for accessing and displaying interactive views of TCGA data associated with glioblastoma.
Displaying a summary of experimental data associated with selected genes
The samples (columns on the heatmap) are annotated in two ways: First, according to cluster membership (the optimal number of clusters was determined using NbClust); second, by inspecting the status of a prognostic index (which was computed by weight-averaging the gene expressions with the regression coefficients of a multigene Cox proportional hazards model). The gene names are annotated with their respective hazard ratios in a multigene Cox proportional hazards model. When search results involve more than 50 genes, we filter them by keeping the 50 genes whose expression is the most varied among the samples.
Performing gene survival analysis
The Kaplan-Meier survival curve compare samples stratified according to gene expression levels. The default options stratified samples into two groups: those with expression levels smaller than the median over the subgroup and those with expression levels higher than the median. For gene searches that result in multiple hits, we analyzed how the expression profiles affect survival. We performed two types of survival analyses: First, the optimum clusters were selected by the stratification of the samples according to the heatmap cluster membership (see the first annotation bar), where the optimal number of clusters is picked out algorithmically using silhouette width index. Next, we used a Kaplan-Meier model to analyze the differences in survival between groups using a log-rank statistic. These analyses were performed using the “NbClust” package in R.
Displaying heatmap clustering of gene and microRNA expression data correlation
For selected multiple hits of microRNAs, we present a heatmap of the correlation between the expression of genes and microRNAs. Each cell of the heatmap represents how the expression of the gene in the row and the microRNA in that column are correlated, and it is annotated with the correlation value. A pairwise Pearson correlation analysis was performed for the selected six microRNAs. The results were displayed as a heatmap using hierarchical clustering analysis using the average linkage distance metric.
Quantification and statistical analysis
Graphs (scatterplots, box plots, PCA) were generated, and statistical analyses were performed using GraphPad Prism 7. Statistical parameters, including the value of n, statistical test, and statistical significance (P value), are reported in the figures and their legends. For studies involving mouse tissues, replicates refer to samples derived from different mice. For studies involving cell culture, replicates refer to technical (transfection) or biological (cells/tissues obtained from a different patient) replicates. No statistical methods were used to predetermine the sample size. Statistical tests were selected on the basis of the desired comparison. Unpaired two-tailed t tests were used to assess significance when comparing data between two variances. One-way analysis of variance (ANOVA) was used to determine significance when comparing data between ≥3 variances; significant ANOVA results were followed by post hoc testing either comparing every mean with every other mean (Tukey’s multiple-comparison test) or comparing every mean to the wild-type mean (Dunnett’s multiple-comparison test). For the differential expression of global measurements (platforms), the DESeq2 software generated adjusted P values using the Wald test with the Benjamini-Hochberg procedure to correct for multiple hypothesis testing. The Mann-Whitney test was used to compare cumulative distributions of gene fold changes between two gene sets.
Supplementary Material
Acknowledgments
Funding: This research was funded by the National Cancer Institute 1R01 CA176203-01A1 (to J.G.), K08NS101091, and R01NS11614 (to P.P.); and the National Science Center Poland (2018/29/B/NZ1/01016) (to A.B.). Author contributions: Conceptualization: A.B. and J.G. Methodology: A.B. and A.K.R. Investigation: A.B., A.K.R., and P.P. Formal analysis: A.B. and E.S. Validation: A.K.R. Data curation: A.B. and K.K. Writing (original draft): J.G. and A.B. Writing (review and editing): E.A.C. and B.P. Funding acquisition: J.G. and E.A.C. Resources: I.N. Supervision: J.G. Competing interests: A.B., J.G., and E.A.C. are inventors on a provisional patent related to this work filed by Brigham and Women’s Hospital (no. 63/091,251, filed 13 October 2020). The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The accession number for the array data reported in this paper is GEO: GSE146440, GSE146463. Further information and requests for resources and reagents (13, 45–52) should be directed to and will be fulfilled by the lead contact, A.B., at abronisz@bwh.harvard.edu.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabc0221/DC1
REFERENCES AND NOTES
- 1.Alles J., Fehlmann T., Fischer U., Backes C., Galata V., Minet M., Hart M., Abu-Halima M., Grässer F. A., Lenhof H.-P., Keller A., Meese E., An estimate of the total number of true human miRNAs. Nucleic Acids Res. 47, 3353–3364 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helwak A., Kudla G., Dudnakova T., Tollervey D., Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153, 654–665 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lewis B. P., Shih I.-h., Jones-Rhoades M. W., Bartel D. P., Burge C. B., Prediction of mammalian microRNA targets. Cell 115, 787–798 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Cheloufi S., Dos Santos C. O., Chong M. M., Hannon G. J., A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 465, 584–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ivey K. N., Srivastava D., MicroRNAs as regulators of differentiation and cell fate decisions. Cell Stem Cell 7, 36–41 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Lu J., Getz G., Miska E. A., Alvarez-Saavedra E., Lamb J., Peck D., Sweet-Cordero A., Ebert B. L., Mak R. H., Ferrando A. A., Downing J. R., Jacks T., Horvitz H. R., Golub T. R., MicroRNA expression profiles classify human cancers. Nature 435, 834–838 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Brown D. V., Stylli S. S., Kaye A. H., Mantamadiotis T., Multilayered heterogeneity of glioblastoma stem cells: Biological and clinical significance. Adv. Exp. Med. Biol. 1139, 1–21 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Neftel C., Laffy J., Filbin M. G., Hara T., Shore M. E., Rahme G. J., Richman A. R., Silverbush D., Shaw M. L., Hebert C. M., Dewitt J., Gritsch S., Perez E. M., Castro L. N. G., Lan X., Druck N., Rodman C., Dionne D., Kaplan A., Bertalan M. S., Small J., Pelton K., Becker S., Bonal D., Nguyen Q.-D., Servis R. L., Fung J. M., Mylvaganam R., Mayr L., Gojo J., Haberler C., Geyeregger R., Czech T., Slavc I., Nahed B. V., Curry W. T., Carter B. S., Wakimoto H., Brastianos P. K., Batchelor T. T., Stemmer-Rachamimov A., Martinez-Lage M., Frosch M. P., Stamenkovic I., Riggi N., Rheinbay E., Monje M., Rozenblatt-Rosen O., Cahill D. P., Patel A. P., Hunter T., Verma I. M., Ligon K. L., Louis D. N., Regev A., Bernstein B. E., Tirosh I., Suvà M. L., An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 835–849.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mao P., Joshi K., Li J., Kim S.-H., Li P., Santana-Santos L., Luthra S., Chandran U. R., Benos P. V., Smith L., Wang M., Hu B., Cheng S.-Y., Sobol R. W., Nakano I., Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. U.S.A. 110, 8644–8649 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patel A. P., Tirosh I., Trombetta J. J., Shalek A. K., Gillespie S. M., Wakimoto H., Cahill D. P., Nahed B. V., Curry W. T., Martuza R. L., Louis D. N., Rozenblatt-Rosen O., Suvà M. L., Regev A., Bernstein B. E., Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rooj A. K., Ricklefs F., Mineo M., Nakano I., Chiocca E. A., Bronisz A., Godlewski J., MicroRNA-mediated dynamic bidirectional shift between the subclasses of glioblastoma stem-like cells. Cell Rep. 19, 2026–2032 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J., Lee I.-H., Cho H. J., Park C.-K., Jung Y.-S., Kim Y., Nam S. H., Kim B. S., Johnson M. D., Kong D.-S., Seol H. J., Lee J.-I., Joo K. M., Yoon Y., Park W.-Y., Lee J., Park P. J., Nam D.-H., Spatiotemporal evolution of the primary glioblastoma genome. Cancer Cell 28, 318–328 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Mineo M., Ricklefs F., Rooj A. K., Lyons S. M., Ivanov P., Ansari K. I., Nakano I., Chiocca E. A., Godlewski J., Bronisz A., The long non-coding RNA HIF1A-AS2 facilitates the maintenance of mesenchymal glioblastoma stem-like cells in hypoxic niches. Cell Rep. 15, 2500–2509 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleaveland B., Shi C. Y., Stefano J., Bartel D. P., A network of noncoding regulatory RNAs acts in the mammalian brain. Cell 174, 350–362.e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., Maier L., Mackowiak S. D., Gregersen L. H., Munschauer M., Loewer A., Ziebold U., Landthaler M., Kocks C., le Noble F., Rajewsky N., Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333–338 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Rybak-Wolf A., Stottmeister C., Glažar P., Jens M., Pino N., Giusti S., Hanan M., Behm M., Bartok O., Ashwal-Fluss R., Herzog M., Schreyer L., Papavasileiou P., Ivanov A., Öhman M., Refojo D., Kadener S., Rajewsky N., Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol. Cell 58, 870–885 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Enuka Y., Lauriola M., Feldman M. E., Sas-Chen A., Ulitsky I., Yarden Y., Circular RNAs are long-lived and display only minimal early alterations in response to a growth factor. Nucleic Acids Res. 44, 1370–1383 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansen T. B., Jensen T. I., Clausen B. H., Bramsen J. B., Finsen B., Damgaard C. K., Kjems J., Natural RNA circles function as efficient microRNA sponges. Nature 495, 384–388 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Kristensen L. S., Hansen T. B., Venø M. T., Kjems J., Circular RNAs in cancer: Opportunities and challenges in the field. Oncogene 37, 555–565 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silber J., Lim D. A., Petritsch C., Persson A. I., Maunakea A. K., Yu M., Vandenberg S. R., Ginzinger D. G., James C. D., Costello J. F., Bergers G., Weiss W. A., Alvarez-Buylla A., Hodgson J. G., miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 6, 14 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bronisz A., Wang Y., Nowicki M. O., Peruzzi P., Ansari K., Ogawa D., Balaj L., De Rienzo G., Mineo M., Nakano I., Ostrowski M. C., Hochberg F., Weissleder R., Lawler S. E., Chiocca E. A., Godlewski J., Extracellular vesicles modulate the glioblastoma microenvironment via a tumor suppression signaling network directed by miR-1. Cancer Res. 74, 738–750 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papagiannakopoulos T., Shapiro A., Kosik K. S., MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res. 68, 8164–8172 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Bhaskaran V., Nowicki M. O., Idriss M., Jimenez M. A., Lugli G., Hayes J. L., Mahmoud A. B., Zane R. E., Passaro C., Ligon K. L., Haas-Kogan D., Bronisz A., Godlewski J., Lawler S. E., Chiocca E. A., Peruzzi P., The functional synergism of microRNA clustering provides therapeutically relevant epigenetic interference in glioblastoma. Nat. Commun. 10, 442 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Godlewski J., Ferrer-Luna R., Rooj A. K., Mineo M., Ricklefs F., Takeda Y. S., Nowicki M. O., Salińska E., Nakano I., Lee H., Weissleder R., Beroukhim R., Chiocca E. A., Bronisz A., MicroRNA signatures and molecular subtypes of glioblastoma: The role of extracellular transfer. Stem Cell Rep. 8, 1497–1505 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurzynska-Kokorniak A., Koralewska N., Pokornowska M., Urbanowicz A., Tworak A., Mickiewicz A., Figlerowicz M., The many faces of Dicer: The complexity of the mechanisms regulating Dicer gene expression and enzyme activities. Nucleic Acids Res. 43, 4365–4380 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pilotte J., Dupont-Versteegden E. E., Vanderklish P. W., Widespread regulation of miRNA biogenesis at the Dicer step by the cold-inducible RNA-binding protein, RBM3. PLOS ONE 6, e28446 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou R. B., Lu X.-L., Zhang C.-Y., Yin D.-C., RNA binding motif protein 3: A potential biomarker in cancer and therapeutic target in neuroprotection. Oncotarget 8, 22235–22250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voce D. J., Bernal G. M., Wu L., Crawley C. D., Zhang W., Mansour N. M., Cahill K. E., Szymura S. J., Uppal A., Raleigh D. R., Spretz R., Nunez L., Larsen G., Khodarev N. N., Weichselbaum R. R., Yamini B., Temozolomide treatment induces lncRNA MALAT1 in an NF-κB and p53 codependent manner in glioblastoma. Cancer Res. 79, 2536–2548 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu L., Liu H., Yan Z., Sun Z., Luo S., Lu Q., Inhibition of the Hedgehog signaling pathway suppresses cell proliferation by regulating the Gli2/miR-124/AURKA axis in human glioma cells. Int. J. Oncol. 50, 1868–1878 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Xue Y., Ouyang K., Huang J., Zhou Y., Ouyang H., Li H., Wang G., Wu Q., Wei C., Bi Y., Jiang L., Cai Z., Sun H., Zhang K., Zhang Y., Chen J., Fu X.-D., Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell 152, 82–96 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li M., Fu W., Wo L., Shu X., Liu F., Li C., miR-128 and its target genes in tumorigenesis and metastasis. Exp. Cell Res. 319, 3059–3064 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Teplyuk N. M., Uhlmann E. J., Gabriely G., Volfovsky N., Wang Y., Teng J., Karmali P., Marcusson E., Peter M., Mohan A., Kraytsberg Y., Cialic R., Chiocca E. A., Godlewski J., Tannous B., Krichevsky A. M., Therapeutic potential of targeting microRNA-10b in established intracranial glioblastoma: First steps toward the clinic. EMBO Mol. Med. 8, 268–287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang M., Song T., Yang L., Chen R., Wu L., Yang Z., Fang J., Nestin and CD133: Valuable stem cell-specific markers for determining clinical outcome of glioma patients. J. Exp. Clin. Cancer Res. 27, 85 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Thé H., Differentiation therapy revisited. Nat. Rev. Cancer 18, 117–127 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Anastasiadou E., Jacob L. S., Slack F. J., Non-coding RNA networks in cancer. Nat. Rev. Cancer 18, 5–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Passon N., Gerometta A., Puppin C., Lavarone E., Puglisi F., Tell G., di Loreto C., Damante G., Expression of dicer and drosha in triple-negative breast cancer. J. Clin. Pathol. 65, 320–326 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Much C., Auchynnikava T., Pavlinic D., Buness A., Rappsilber J., Benes V., Allshire R., O’Carroll D., Endogenous mouse dicer is an exclusively cytoplasmic protein. PLOS Genet. 12, e1006095 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Treiber T., Treiber N., Meister G., Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 20, 5–20 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Verduci L., Strano S., Yarden Y., Blandino G., The circRNA-microRNA code: Emerging implications for cancer diagnosis and treatment. Mol. Oncol. 13, 669–680 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao M., Wang S., Li Q., Ji Q., Guo P., Liu X., MALAT1: A long non-coding RNA highly associated with human cancers. Oncol. Lett. 16, 19–26 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ogawa D., Ansari K., Nowicki M. O., Salińska E., Bronisz A., Godlewski J., MicroRNA-451 inhibits migration of glioblastoma while making it more susceptible to conventional therapy. Noncoding RNA 5, 25 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ansari K. I., Ogawa D., Rooj A. K., Lawler S. E., Krichevsky A. M., Johnson M. D., Chiocca E. A., Bronisz A., Godlewski J., Glucose-based regulation of miR-451/AMPK signaling depends on the OCT1 transcription factor. Cell Rep. 11, 902–909 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Godlewski J., Nowicki M. O., Bronisz A., Nuovo G., Palatini J., De Lay M., Van Brocklyn J., Ostrowski M. C., Chiocca E. A., Lawler S. E., MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol. Cell 37, 620–632 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zirkel A., Papantonis A., Detecting circular RNAs by RNA fluorescence in situ hybridization. Methods Mol. Biol. 1724, 69–75 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Godlewski J., Nowicki M. O., Bronisz A., Williams S., Otsuki A., Nuovo G., Raychaudhury A., Newton H. B., Chiocca E. A., Lawler S., Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 68, 9125–9130 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Meyer K., Ferraiuolo L., Miranda C. J., Likhite S., Elroy S. M., Renusch S., Ditsworth D., Lagier-Tourenne C., Smith R. A., Ravits J., Burghes A. H., Shaw P. J., Cleveland D. W., Kolb S. J., Kaspar B. K., Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc. Natl. Acad. Sci. U.S.A. 111, 829–832 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schneider C. A., Rasband W. S., Eliceiri K. W., NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bowman R. L., Wang Q., Carro A., Verhaak R. G. W., Squatrito M., GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 19, 139–141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Celiku O., Johnson S., Zhao S., Camphausen K., Shankavaram U., Visualizing molecular profiles of glioblastoma with GBM-BioDP. PLOS ONE 9, e101239 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Y., Wang H., Tan C., Yan Y., Shen J., Huang Q., Xu T., Lin J., Chen J., Expression of amyloid precursor-like protein 2 (APLP2) in glioblastoma is associated with patient prognosis. Folia Neuropathol. 56, 30–38 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Orre L. M., Vesterlund M., Pan Y., Arslan T., Zhu Y., Woodbridge A. F., Frings O., Fredlund E., Lehtiö J., SubCellBarCode: Proteome-wide mapping of protein localization and relocalization. Mol. Cell 73, 166–182.e7 (2019). [DOI] [PubMed] [Google Scholar]
- 52.Verhaak R. G. W., Hoadley K. A., Purdom E., Wang V., Qi Y., Wilkerson M. D., Miller C. R., Ding L., Golub T., Mesirov J. P., Alexe G., Lawrence M., O’Kelly M., Tamayo P., Weir B. A., Gabriel S., Winckler W., Gupta S., Jakkula L., Feiler H. S., Hodgson J. G., James C. D., Sarkaria J. N., Brennan C., Kahn A., Spellman P. T., Wilson R. K., Speed T. P., Gray J. W., Meyerson M., Getz G., Perou C. M., Hayes D. N.; Cancer Genome Atlas Research Network , Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabc0221/DC1