

The S-adenosylhomocysteine (SAH) hydrolyzing enzyme AHCY functionally links the circadian clock to methionine metabolism.
Abstract
Circadian gene expression driven by transcription activators CLOCK and BMAL1 is intimately associated with dynamic chromatin remodeling. However, how cellular metabolism directs circadian chromatin remodeling is virtually unexplored. We report that the S-adenosylhomocysteine (SAH) hydrolyzing enzyme adenosylhomocysteinase (AHCY) cyclically associates to CLOCK-BMAL1 at chromatin sites and promotes circadian transcriptional activity. SAH is a potent feedback inhibitor of S-adenosylmethionine (SAM)–dependent methyltransferases, and timely hydrolysis of SAH by AHCY is critical to sustain methylation reactions. We show that AHCY is essential for cyclic H3K4 trimethylation, genome-wide recruitment of BMAL1 to chromatin, and subsequent circadian transcription. Depletion or targeted pharmacological inhibition of AHCY in mammalian cells markedly decreases the amplitude of circadian gene expression. In mice, pharmacological inhibition of AHCY in the hypothalamus alters circadian locomotor activity and rhythmic transcription within the suprachiasmatic nucleus. These results reveal a previously unappreciated connection between cellular metabolism, chromatin dynamics, and circadian regulation.
INTRODUCTION
The circadian clock is an endogenous, time-keeping system that directs multiple metabolic and physiological functions required for homeostasis (1, 2). Circadian transcription is driven by core transcription factors CLOCK and BMAL1, which heterodimerize and drive expression of a large number of clock-controlled genes (CCGs) by binding to E-box sequences within their promoters (3). Circadian transcription is associated with dynamic changes in chromatin structure (4, 5), including acetylation and methylation of a number of histone residues and nuclear spatial remodeling (6–8). In addition, CLOCK-BMAL1 associate with various remodelers that contribute to circadian chromatin dynamics and transcription (9–12).
Chromatin-modifying enzymes use cellular metabolites as substrates, thereby coupling metabolic pathways to epigenetic modifications and gene regulation (13–15). A notable feature of the metabolic-chromatin axis is the translocation of some metabolic enzymes into the nucleus and their contribution to localized availability of metabolites involved in epigenetic regulation (16). A key metabolic pathway able to influence gene expression through epigenetic mechanisms is the one-carbon cycle, as its intermediate metabolites, S-adenosyl methionine (SAM) and S-adenosyl homocysteine (SAH), activate and inhibit, respectively, the activity of enzymes that drive DNA and histone methylation (17). Biosynthesis of SAM is catalyzed by methionine adenosyltransferases (MATs) from methionine and adenosine triphosphate (ATP). As a by-product of methylation reactions, methyltransferases release the intermediate metabolite, SAH, which is further hydrolyzed by S-adenosyl-l-homocysteine hydrolase (AHCY) (18). Because SAH is a potent inhibitor of most methyltransferases (19, 20), efficient hydrolysis of SAH by AHCY is essential to sustain methylation reactions.
Metabolic control of chromatin dynamics is critical for a variety of biological processes ranging from cell fate and development to cancer pathophysiology and neural plasticity (17, 21, 22). However, the molecular pathways coupling cellular metabolism to circadian chromatin transitions are virtually unexplored.
Here, we show that AHCY controls circadian gene transcription by interacting with the core clock regulator BMAL1. The BMAL1-AHCY association occurs at chromatin, promoting rhythmic H3K4 trimethylation (H3K4me3) and cyclic BMAL1 recruitment to target genes. Our findings reveal a previously unidentified regulatory circuit of circadian control dictated by the direct interaction between a core clock component and a metabolic enzyme, illustrating the intimate connection between circadian rhythms and metabolic pathways.
RESULTS
BMAL1 interacts with AHCY in the nucleus
In search of fresh insights into circadian transcriptional regulation, we performed a mass spectrometry (MS) analysis of BMAL1 nuclear interactors. Endogenous BMAL1 was immunoprecipitated from nuclear fractions of mouse liver harvested at the peak (zeitgeber time, ZT8) and trough (ZT20) of circadian transcriptional activity, and coimmunoprecipitated proteins were analyzed by MS. Our analysis specifically identified CLOCK, the dimerization partner of BMAL1 (23), among the top enriched proteins (fold enrichment > log2; P < 0.05; Fig. 1, A and B, and data file S1). In addition, two enzymes of the methionine cycle, MAT1a and AHCY, were identified as putative BMAL1-interacting proteins (Fig. 1, A and B). Because of the technical limitations of immunoprecipitating endogenous BMAL1, we were unable to detect PER1/2 and CRY1/2 at ZT20 (24, 25).
Fig. 1. AHCY forms a complex with BMAL1.
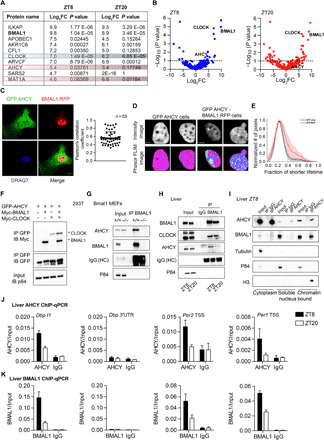
(A) Table showing the top BMAL1-interacting proteins in the nucleus ranked by log2 fold change (FC) value at ZT8. (B) Volcano plot of MS analysis of BMAL1-interacting proteins in the nucleus. The x axis indicates log2 ratio of normalized intensity (iBAQ) of proteins found in BMAL1 to IgG (n = 4). (C) AHCY colocalization with BMAL1 in the nucleus. GFP-AHCY and RFP-BMAL1 were imaged by confocal microscopy in MEF cells. The Pearson’s R coefficient indicates the extent of colocalization of GFP-AHCY and RFP-BMAL1 (n = 53). Scale bar, 10 μm. (D) Intensity (GFP channel) and phasor mapped fluorescence lifetime images (FLIM) of GFP-AHCY and GFP-AHCY/BMAL1-RFP MEF cell nucleus. Two representative cells are shown (n > 30 cells). Scale bar, 1 μm. (E) Histogram of the fractional intensity distributions of the donor (GFP) in the nucleus. The increasing fractional intensity of the shorter lifetime (FRET) component reflects the presence of FRET (red plot). The two average plots are separated by P < 0.001 (Kolmogorov-Smirnov test). (F) Co-IP experiment using 293T cells transiently transfected with the indicated plasmids (IP, immunoprecipitation; IB, immunoblot). (G) Co-IP experiment from nuclear extract of BMAL1 null MEFs with anti-BMAL1 antibody. (H) Co-IP experiment from liver nuclear extracts using anti-AHCY antibody at indicated zeitgeber times (ZT8 and ZT20). (I) Co-IP experiment from liver cytoplasmic, soluble nuclear, and chromatin-bound fractions using anti-AHCY antibody at the indicated zeitgeber time (ZT8). (J) AHCY ChIP at the Dbp, Per2, and Per1 loci in liver at indicated zeitgeber times. The 3′UTR of Dbp was used as a negative control (mean ± SEM, n = 4 per time point). (K) AHCY ChIP at the Dbp, Per2, and Per1 loci in liver at ZT8 and ZT20 (mean ± SEM, n = 4 per time point).
Identification of AHCY as a potential nuclear BMAL1-associated protein is of particular interest for a number of reasons. First, AHCY was recently found to be associated at chromatin to transcriptionally active genes, including several clock genes (26). Second, AHCY is a highly conserved, ubiquitously expressed enzyme that is uniquely capable of hydrolyzing SAH. Likewise, AHCY has been shown to interact with and enhance the function of the DNA methyltransferase DNMT1 in mammalian cells (27). Last, our MS analysis shows that interaction of AHCY with BMAL1 is prominent at ZT8, indicating that AHCY might be involved in BMAL1-mediated transcriptional activation (Fig. 1, A and B).
Subcellular fractionation confirmed nuclear localization of AHCY in mouse embryonic fibroblasts (MEFs) and in mouse liver (fig. S1, A and B). In MEF cells, nuclear green fluorescent protein (GFP)–AHCY colocalized with red fluorescent protein (RFP)–BMAL1 (Fig. 1C) and fluorescence lifetime imaging of donor (AHCY-GFP) and donor-acceptor (AHCY-GFP/BMAL1-RFP) revealed a shift in the phasor lifetime distribution, which indicates fluorescence resonance energy transfer (FRET) and thus confirms association at common nuclear subdomains (Fig. 1, D and E).
We next confirmed BMAL1-AHCY interaction by performing coimmunoprecipitation (co-IP) assays after ectopic expression of BMAL1, CLOCK, and AHCY in 293T cells (Fig. 1F). Co-IP of AHCY with various BMAL1 truncations revealed that AHCY interacts with the PER-ARNT-SIM (PAS) B domain of BMAL1, which is involved in CLOCK dimerization (28), as well as a region downstream to the PAS-B domain (fig. S1, C and D). Interaction of endogenous AHCY and BMAL1 was also confirmed in MEFs (Fig. 1G) and liver (Fig. 1H) by co-IP assays on nuclear fractions. In liver, consistent with nuclear proteome studies (29, 30), BMAL1 levels did not oscillate. However, association of BMAL1 with AHCY was highest at ZT8 (Fig. 1H), in keeping with the MS data. These results indicate that AHCY is a constituent of the endogenous BMAL1 complex and interaction is greatest during the circadian transcriptional activation phase.
To identify the subnuclear compartment in which the BMAL1-AHCY interaction occurs, we fractionated livers harvested at ZT8 and immunoprecipitated AHCY from the cytoplasmic, soluble nuclear, and chromatin-bound compartments. While AHCY is present in all three fractions, association with BMAL1 occurred mainly at chromatin (Fig. 1I). We next tested whether AHCY is recruited to circadian gene promoters known to be bound by BMAL1. Chromatin IP (ChIP) of livers at ZT8 and ZT20 showed that AHCY bound to E-box regions of Dbp, Per2, and Per1 in a time-dependent manner but not at the 3′ untranslated region (3′UTR) of Dbp that was used as control for specificity (Fig. 1J). Recruitment of AHCY followed the same circadian timing of BMAL1 (Fig. 1K). These results indicate that AHCY may contribute to BMAL1-driven transcriptional control at clock-regulated genomic sites.
Targeting of AHCY dampens circadian transcription
To investigate the functional significance of the AHCY-BMAL1 interaction, we used RNA interference to acutely knock down AHCY expression in U2OS cells stably transfected with a Bmal1-luciferase reporter construct (Bmal1:luc; fig. S2A). Bioluminescence recordings revealed that depletion of AHCY greatly dampened circadian expression and resulted in significant attenuation of the amplitude of oscillations (Fig. 2, A and B). Analogous results were obtained with two other independent small interfering RNAs (siRNAs) targeting AHCY (fig. S2, B to E).
Fig. 2. AHCY is important for amplitude of circadian oscillation.
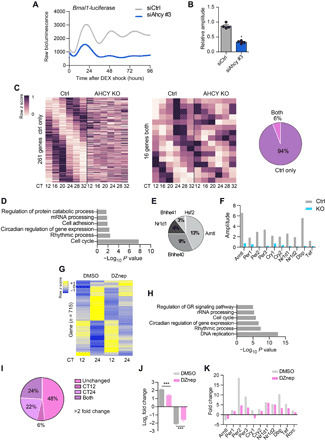
(A) Circadian bioluminescence traces for Bmal1:luc U2OS cells transfected with either siRNA control (siCtrl) or siRNA targeting Ahcy (siAhcy) (mean values shown, n = 4). (B) Bar graph of relative circadian amplitude of U2OS Bmal1:luc siCtrl and siAhcy cells (mean ± SEM, n = 4; *P ≤ 0.05; unpaired Student’s t test). (C) Heatmaps representing genes cyclic in control MEF cells only (Ctrl; 261 genes) and in both conditions (16 genes) (n = 3 per time point, per group; P < 0.01 in each dataset). Pie chart representing the percentage of genes oscillating in both conditions and in control only. (D) Gene Ontology (GO) term enrichment analysis of genes oscillating in the control group only. (E) Transcription factor binding site (TFBS) analysis of rhythmic transcription factors (TFs) on transcripts rhythmic exclusively in control MEFs. Represented as percentage of TFBS. (F) Bar graph of amplitude analysis of clock genes in control (Ctrl) and AHCY null (KO) MEF cells. (G) Heatmap of hierarchical clustering of genes significantly down-regulated or up-regulated in vehicle-treated MEFs (DMSO) between the two analyzed CT points (CT12 and CT24; n = 3 per time point per group; fold change > 2; FDR < 0.05). Heatmap shows gene expression levels of control (DMSO) and DZnep-treated MEFs. (H) GO term enrichment analysis of genes down-regulated or up-regulated in vehicle-treated MEFs (DMSO) between the two analyzed CT points (CT12 and CT24). (I) Pie chart showing the percentage of time-dependent genes identified in (G) (715 genes) differentially expressed in MEFs treated with DZnep (10 μM). (J) Bar graph displaying log2 fold change of gene expression of genes related to (G). (K) Bar graph of fold change analysis of clock genes in vehicle (DMSO)– and DZnep-treated MEF cells. ***P ≤ 0.001; unpaired Student’s t test.
Next, to determine to what extent AHCY affects circadian gene expression, we used CRISPR-Cas9–mediated gene editing, adopting a single-guide RNA targeting exon 4 of Ahcy to generate Ahcy-KO (knockout) MEFs. Two independent clonal lines were generated in which loss of AHCY expression was confirmed (KO-1 and KO-2; fig. S3A). Deletion of AHCY led to the expected shift in the ratio of S-adenosylhomocysteine (SAH) and S-adenosylmethionine (SAM) (fig. S3B). To investigate the effect of AHCY ablation on clock-driven transcription, we performed RNA sequencing (RNA-seq) at different circadian times (CTs) in dexamethasone (DEX)–synchronized control and AHCY-null MEFs (KO-1) (Fig. 2C). The absence of AHCY caused profound changes in circadian gene expression (Fig. 2, C to F). We detected 277 circadian transcripts in control MEFs; of these, only 6% continued to oscillate in AHCY-KO cells (Fig. 2C and data file S2). Gene Ontology (GO) analysis of transcripts rhythmic exclusively in control MEFs revealed enrichment of genes related to circadian rhythms and cell cycle (Fig. 2D). Consistent with the GO results, transcription factor binding site (TFBS) analysis of rhythmic transcription factors targeting the list of genes oscillating only in the control MEFs identified BMAL1 as the top enriched factor (Fig. 2E). We observed a strong effect on the amplitude of expression of clock genes in AHCY-KO MEFs (Fig. 2F), indicating that the clock network was severely compromised.
To confirm the results obtained following AHCY deletion, we turned to a pharmacological approach. We treated confluent DEX-synchronized MEFs with deazaneplanocin A (DZnep), a carbocyclic adenosine analog that specifically inhibits AHCY (31–33). As expected, DZnep-treated cells displayed a shift of SAH/SAM ratio (fig. S3C). Pharmacological targeting of AHCY resulted in marked inhibition of circadian gene expression, confirming our results in the AHCY null cells. To analyze the effect of AHCY inhibition on time-dependent variations of gene expression, we assessed genes whose expression levels were markedly changed [fold change > 2; false discovery rate (FDR) < 0.05] between the two CT points (CT12 and CT24) in vehicle-treated [dimethyl sulfoxide (DMSO)] MEFs and identified 715 differentially expressed genes (Fig. 2G and data file S3). In agreement with the circadian transcriptome from control MEFs (Fig. 2C), GO analysis identified subset of genes enriched for terms related to circadian regulation of gene expression, rhythmic processes, and cell cycle (Fig. 2H). Of the 715 genes displaying time-specific expression, 52% were markedly affected by AHCY inhibition (fold change > 2; FDR < 0.05; Fig. 2I). In addition, cells treated with DZnep displayed a significant attenuation of time-dependent gene expression (Fig. 2, J and K). In DMSO-treated cells, most clock-regulated genes displayed larger than twofold variation in expression levels between CT12 and CT24 (Fig. 2K) and treatment with DZnep greatly reduced oscillation of these genes (Fig. 2K). Acute induction of Per1 after DEX stimulation was unaltered upon deletion or inhibition of AHCY (fig. S3, D and E). Thus, AHCY impairs cyclic gene expression without affecting the glucocorticoid-induced early response that precedes synchronization of cells (34).
AHCY is required for transcriptional activation by BMAL1
To further validate our results, we analyzed the expression of the clock output genes Dbp and Per1 in four experimental conditions: an AHCY-null line (AHCY KO-2; fig. S3A); an inducible Ahcy-knockdown MEF cell line obtained by lentiviral transduction with a tetracycline-inducible short hairpin RNA (shRNA) (fig. S4A); and MEFs treated with DZnep or DZ2002, another specific AHCY inhibitor (35). Supporting our RNA-seq results, oscillation was severely compromised by both deletion and inhibition of AHCY (Fig. 3A and fig. S4B). The inducible knockdown system, as well as the use of pharmacological inhibitors, allows transient targeting of AHCY, thus ruling out any secondary effects due to loss of AHCY and confirming a direct effect of AHCY on circadian genes.
Fig. 3. AHCY regulates BMAL1 recruitment to DNA.
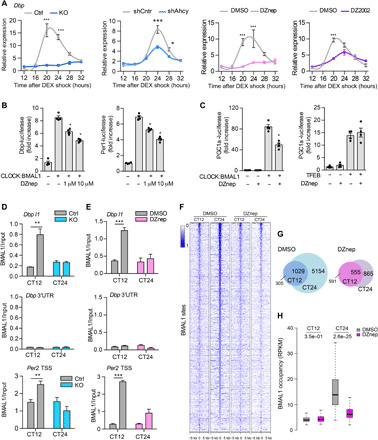
(A) Circadian expression of Dbp in control (Ctrl) or AHCY null (KO-2) MEFs, shCntrl and shAhcy MEFs, and MEFs treated with DMSO, 10 μM DZnep, and 100 μM DZ2002 after DEX shock (mean ± SEM, n = 3 per time point, per group; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ANOVA, Holm-Sidak post hoc). (B) Luciferase assay of Dbp-luciferase (Dbp-luc) and Per2-luciferase (Per2-luc) in HEK293 transfected with CLOCK and BMAL1 plasmids and treated with indicated increasing doses of DZnep for 24 hours (means ± SEM, n = 4; *P ≤ 0.05; unpaired Student’s t test). (C) Luciferase assay of Pgc1a-luciferase (Pgc1a -luc) in HEK293 transfected with CLOCK and BMAL1 (left) or TFEB (right) plasmids and treated with indicated doses of DZnep for 24 hours (means ± SEM, n = 4; *P ≤ 0.05; unpaired Student’s t test). (D) BMAL1 ChIP at the Dbp and Per2 loci in control (Ctrl) or AHCY null MEFs (KO). The 3′UTR of Dbp was used as a negative control (mean ± SEM, n = 4 per time point, per group; **P ≤ 0.01; unpaired Student’s t test). (E) BMAL1 ChIP at the Dbp and Per2 loci in MEFs treated with vehicle (DMSO) or DZnep (10 μM) (mean ± SEM, n = 4 per time point, per group; ***P ≤ 0.001; unpaired Student’s t test). (F) Heatmap of BMAL1 ChIP-seq binding profiles in DMSO- and DZnep-treated MEFs at the two analyzed CT points. (G) Venn diagram of BMAL1 binding sites identified at the two analyzed CT points in vehicle (DMSO) and DZnep-treated MEFs. (H) Boxplots of number of BMAL1 tags per peak at CT12 and CT24 shared peaks in DMSO- and DZnep-treated MEFs (unpaired Student’s t test).
To determine whether catalytic activity of AHCY is required for rhythmic transcription, we expressed hemagglutinin (HA)–tagged wild-type (WT) AHCY or a catalytic dead mutant (K186N) in the tetracycline-inducible shAhcy MEFs (fig. S4, C and D) (36). Cyclic gene expression of Per2 and Dbp was fully restored by reconstitution of WT but not mutant K186N HA-AHCY (fig. S4E).
To further confirm AHCY function on the clock, we analyzed CLOCK-BMAL1–driven transcription in 293 cells transfected with Dbp or Per1 luciferase reporter vectors. DZnep dampened CLOCK-BMAL1–mediated activation of Dbp and Per1 expression in a dose-dependent manner (Fig. 3B), indicating that the enzymatic activity of AHCY contributes to CLOCK-BMAL1–mediated gene transcription. DZnep did not affect the activity of TFEB, another E-box binding transcription factor (Fig. 3C) (37).
We next investigated the mechanism by which AHCY contributes to CLOCK:BMAL1-mediated transcriptional activity. Because BMAL1 and AHCY associate at chromatin (Fig. 1, I and J), we hypothesized that AHCY may influence recruitment of BMAL1 to target promoters. We performed ChIP analyses and assessed BMAL1 binding on Dbp and Per2 E-box regions at CT12 and CT24, comparing control and AHCY KO MEFs. Although recruitment of BMAL1 was strongly time dependent in control MEFs, lack of AHCY completely abolished its rhythmicity (Fig. 3D). The same analyses conducted in MEFs treated with DZnep gave analogous results (Fig. 3E), indicating that enzymatic activity of AHCY is required for proper binding of BMAL1 to promoters. Next, to determine genome-wide binding of BMAL1, we performed ChIP followed by sequencing (ChIP-seq) in MEF cells treated with vehicle (DMSO) or DZnep at two CT points (CT12 and CT24; fig. S5A and data file S4). For the ChIP-seq analyses, inhibition of AHCY was preferred to its deletion to avoid any pleiotropic consequences that could indirectly affect BMAL1 activity. The overall distribution of BMAL1 peaks throughout the genome was similar between time points and conditions (fig. S5B). Genomic binding of BMAL1 was higher at CT24 compared to CT12 in control MEFs (Fig. 3F). Inhibition of AHCY strongly dampened BMAL1 association to chromatin at CT24 (Fig. 3F). In control MEFs, we identified 1334 and 6183 BMAL1 binding sites at CT12 and CT24, respectively (Fig. 3G), which is coherent with reported data (6, 38). In contrast, treatment with DZnep reduced fourfold the number of BMAL1 binding sites at CT24 (6183 CT24 DMSO versus 1420 CT24 DZnep; Fig. 3G), whereas the number of BMAL1 binding sites was virtually unaltered at CT12 (1334 CT12 DMSO versus 1024 CT12 DZnep; Fig. 3G). Of the 1420 binding sites identified in CT24 DZnep MEFs, 77% overlapped with CT24 control-specific sites (fig. S5C), indicating that inhibition of AHCY does not cause divergent genome-wide binding of BMAL1. Because inhibition of AHCY enzymatic activity leads to decreased recruitment of BMAL1 to chromatin, we then focused on genomic sites bound by BMAL1 in all four conditions. While DZnep marginally affected BMAL1 binding strength at shared peaks at CT12, BMAL1 occupancy was strongly decreased at CT24, resulting in dampened rhythmicity (Fig. 3H). Treatment with DZnep did not affect nuclear protein levels of BMAL1 (fig. S6A) or BMAL1 subcellular localization (fig. S6B). Moreover, electrophoretic mobility shift assay (EMSA) using nuclear extracts from MEF cells collected at CT24 showed that neither deletion nor inhibition of AHCY disrupted the ability of BMAL1 to bind to a biotin-labeled promoter E-box motif (fig. S6C). These results suggest that AHCY may mediate time-specific binding of BMAL1 to DNA by modifying the chromatin state.
AHCY is required for rhythmic H3K4 methylation
The activating histone mark H3K4me3 (39) is highly sensitive to changes in the SAM/SAH ratio (40, 41) and has been related to AHCY function (26, 42). Moreover, CLOCK-BMAL1 associate with the histone methyltransferase (HMT) MLL1 and the complex mediates rhythmic H3K4me3 modification, thereby contributing to circadian transcription by generating a chromatin state permissive for CLOCK-BMAL1 recruitment (9, 43). Close proximity of AHCY to chromatin could fuel histone methylation by relieving SAH-mediated feedback inhibition. We thus hypothesized that AHCY may be involved in circadian H3K4me3 modification. We examined genome-wide distribution of H3K4me3 by ChIP-seq (fig. S7A and data file S5). As previously reported, H3K4me3 peaks around transcription start sites (TSS) displayed rhythmicity in MEF cells treated with vehicle (9). Upon treatment with DZnep, rhythmic H3K4me3 levels around TSS of expressed genes were strongly attenuated (Fig. 4A). Following inhibition of AHCY, H3K4me3 accumulation at promoters of expressed genes was significantly reduced at CT24, thereby resulting in arrhythmic H3K4me3 levels (Fig. 4, B and C). Upon inhibition of AHCY, we identified 389 changed peaks compared to vehicle-treated MEFs at CT24 (fold change > 2; P < 0.05). Among the H3K4me3 differential peaks, 97% were decreased and were associated to circadian rhythm–related genes (fig. S7, B and C). ChIP analyses conducted at different CTs in control (DMSO) and DZnep-treated MEFs confirmed loss of rhythmicity of H3K4me3 levels on Dbp and Per2 promoter regions upon inhibition of AHCY (Fig. 4D). Conversely, H3K4me3 and expression levels of the MLL1-controlled gene Hoxa9 displayed no significant change upon AHCY inhibition (fig. S7, D and E). Moreover, loss of cyclic H3K4me3 was coupled with decreased oscillation of H3K9 and H3K14 acetylation (Fig. 4E), other activating marks linked to rhythmic H3K4 methylation (9) and circadian function (44, 45). Inhibition of AHCY could potentially influence the activity the polycomb repressor EZH2 that has been implicated in circadian regulation (46). We thus examined trimethylated H3K27 (H3K27me3) levels on Dbp and Per2 promoters as a readout of EZH2 activity (47). Methylation of H3K27 was not altered upon inhibition of AHCY (fig. S7F). Deletion of AHCY using the MEF null cells (KO) confirmed the results obtained with pharmacological inhibition, resulting in impaired time-dependent accumulation of H3K4me3 levels on Dbp and Per2 genes (Fig. 4F). Thus, AHCY appears to regulate BMAL1 association to genomic target sites and to affect rhythmicity of H3K4me3.
Fig. 4. AHCY mediates oscillation of H3K4me3.
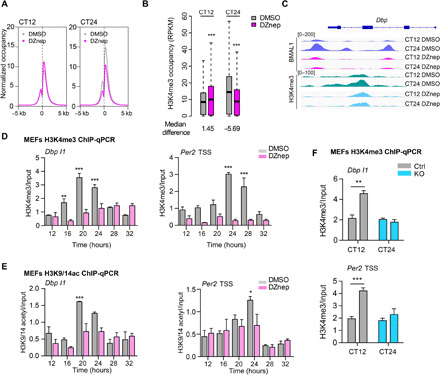
(A) Average H3K4me3 coverage around the TSS of expressed transcripts at the two analyzed CTs. (B) Boxplots of number of H3K4me3 tags per peak within promoter regions of expressed genes (Wilcoxon signed-rank test). (C) IGV (Integrative Genomics Viewer) profile of BMAL1- and H3K4me3-enriched regions over the Dbp locus. (D) H3K4me3 ChIP at the Dbp and Per2 loci in MEFs treated with vehicle (DMSO) or DZnep (10 μM). Samples were collected at the indicated CTs, and immunoprecipitated chromatin was quantified by RT-PCR (mean ± SEM, n = 3 per time point, per group; **P ≤ 0.01; ***P ≤ 0.001; ANOVA, Holm-Sidak post hoc). (E) H3K9 and K14-acetyl ChIP at the Dbp and Per2 loci in MEFs treated with vehicle (DMSO) or DZnep (10 μM) (mean ± SEM, n = 3 per time point, per group; ***P ≤ 0.001; ANOVA, Holm-Sidak post hoc). (F) H3K4me3 ChIP at the Dbp and Per2 loci in control (Ctrl) or AHCY null MEFs (KO) (mean ± SEM, n = 4 per time point, per group; **P ≤ 0.01; unpaired Student’s t test).
Last, we analyzed whether AHCY could influence the binding of other transcription factors. We examined binding of c-MYC because, similarly to BMAL1, it is a basic helix-loop-helix (bHLH) protein that binds E-box sequences in a H3K4me3-dependent manner (48). Inhibition of AHCY enzymatic activity did not change binding efficiency of c-MYC to target regions (fig. S7G). In addition, gene expression levels of c-MYC responsive genes (49) were not affected by DZnep treatment (fig. S7H).
AHCY regulates circadian locomotor activity
The master mammalian clock resides within the suprachiasmatic nucleus (SCN) in the hypothalamus that operates as the primary regulator of circadian behavior and physiology (50). Having explored how AHCY affects circadian gene expression, we set out to study the role of AHCY in circadian physiology in vivo. We delivered DZnep or saline into the hypothalamus of mice by direct infusion and examined circadian rhythms of locomotor activity in free running conditions dark/dark (D/D); Fig. 5, A and B]. As expected, control mice displayed a clear pattern of increased activity during the subjective night (CT12-24). In contrast, diurnal locomotor activity of DZnep-infused mice was greatly dampened, exhibiting equal distribution between subjective day and night (Fig. 5C and fig. S8, A and B). To assess the robustness of circadian activity rhythms, we estimated the amplitude of circadian rhythm by performing spectral analysis using fast Fourier transform (FFT). Mice receiving saline infusion exhibited robust rhythmic behavior, whereas mice infused with DZnep showed significantly lower amplitudes of circadian activity (Fig. 5D and fig. S8C). Moreover, DZnep-treated mice displayed a significantly longer circadian period of activity compared to controls (saline, 23.7 ± 0.03 hours; DZnep, 24.9 ± 0.06 hours; P < 0.001; Fig. 5, B and E, and fig. S8D).
Fig. 5. In vivo inhibition of AHCY modulates circadian locomotor activity and rhythmic transcription in the SCN.
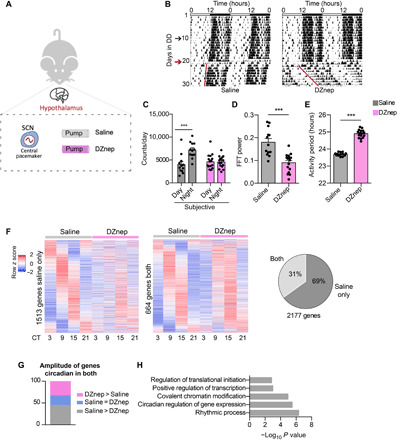
(A) Schematic of the in vivo experimental protocol. (B) Representative double-plot actograms for diurnal locomotor activity of mice infused with saline or DZnep (100 μM). In each actogram, the first days were recorded under 12-hour light/12-hour dark conditions, after which the light was turned off and recording was continued in constant darkness (DD; black arrow indicates start of DD; red arrow indicates day of surgery and starting point for all analyses). (C) Bar graph of locomotor activity during subjective night and day (mean ± SEM, n = 12 saline and n = 16 DZnep; ***P ≤ 0.001; ANOVA, Holm-Sidak post hoc). (D) Bar graph of amplitude of circadian rhythm represented by fast Fourier transform (FFT) in the circadian range (mean ± SEM, n = 12 saline and n = 16 DZnep; ***P ≤ 0.001; unpaired Student’s t test). (E) Bar graph of period lengths (mean ± SEM, n = 12 saline and n = 16 DZnep; ***P ≤ 0.001; unpaired Student’s t test). (F) Heatmaps of genes cyclic in saline-infused mice only (1513 genes) and in both conditions (saline and DZnep, 664 genes) (n = 4 per time point, per group; P < 0.05 in each dataset). Pie chart representing the percentage of genes oscillating in both conditions and in saline only. (G) Amplitude analysis of genes circadian in both conditions in the SCN. In the graph, the percentages of genes with amplitude higher, lower, or equal to saline condition are reported. (H) GO analysis of genes displaying lower amplitude in DZnep-infused mice compared to saline.
AHCY influences circadian transcription in the SCN
To characterize the transcriptional changes induced by DZnep in vivo, we isolated SCNs and profiled gene expression at four CT points by RNA-seq (data file S6). We identified 2177 rhythmic transcripts in control mice (Fig. 5F, left). Of these, 69% displayed impaired oscillation in DZnep-treated mice (Fig. 5F), demonstrating that AHCY contributes to circadian transcription in vivo. Genes displaying rhythmicity exclusively in control mice were enriched for biological processes such as transcription, cell cycle, chromatin modification, and protein phosphorylation (fig. S9A). Among genes cyclic in both groups (Fig. 5F, middle), 45% of transcripts displayed lower amplitude in DZnep-infused mice compared to controls (Fig. 5G). Genes with decreased amplitude included CCGs such as Dbp, Per1, and Nampt, and GO terms included rhythmic process as one of the top terms (Fig. 5H). On the other hand, genes whose amplitude increased in DZnep-treated mice were enriched in other unrelated biological processes (fig. S9B). Together, these results show that the enzymatic activity of AHCY plays a critical role in the function of the circadian clock in vivo.
DISCUSSION
A conceptual advance in the field of gene regulation relates to the finding that some metabolic enzymes may directly participate in transcriptional control by physically interacting with chromatin remodeling systems (22, 51, 52). The underlying implication of these findings is that enzymes at chromatin may use and/or generate localized pockets of metabolites required for functional posttranslational modifications of histones and nonhistone proteins. A paradigmatic example is constituted by the metabolic enzyme acetyl–coenzyme A (CoA) synthetase 2 (ACSS2) that directly regulates histone acetylation in neurons by controlling acetyl-CoA levels (22). How these enzymes get to chromatin and what regulates the dynamics of their recruiting is yet to be established. Here, we report that the metabolic enzyme AHCY physically associates in a time-specific manner with the core clock regulator BMAL1 at chromatin. Thus, in the case of AHCY, it appears that the dynamics of its function at chromatin is directed by an essential transcription factor.
Despite the existence of two AHCY paralogs, AHCY is the only mammalian enzyme capable of hydrolyzing SAH, and as such, it is a key enzyme for many biological functions (53, 54). Consequently, complete loss of AHCY is embryonic lethal (55). In addition, stable knockdown experiments of AHCY have revealed its critical role for cancer (56–58) and, more recently, stem cells (26). Here, by means of transient perturbation of AHCY, we uncover a further function linked to circadian transcriptional regulation. Considering that AHCY is involved in the regulation of numerous cellular functions, we anticipate that it may do so by promoting specific gene expression programs through interaction with selected transcription factors and chromatin regulators. The finding that AHCY interacts with CLOCK and BMAL1 to coordinately regulate circadian transcription can be readily exploited to other biological contexts where AHCY may cooperate with additional regulators.
Accumulating evidence has implicated several chromatin modifiers in circadian control (4). Our study uncovers a functional cross-talk between the molecular clock and a metabolic enzyme. AHCY associates with CLOCK-BMAL1 at chromatin during the circadian activation phase, serving to promote circadian transcription by supporting histone methylation (Fig. 4). AHCY was recently identified as a high-confident chromatin-bound protein (26), and subcellular fractionation confirmed AHCY presence at chromatin. Chromatin binding of BMAL1 peaks at ZT8 and is low at ZT20 (6). Hence, we believe that close proximity of BMAL1 and AHCY at chromatin sites promotes their interaction and functional interplay. AHCY-BMAL1 interaction at chromatin is likely to enable quick and dynamic removal of local SAH accumulation at sites undergoing rapid chromatin and circadian transcriptional changes, thereby facilitating a transcription-permissive environment.
Because AHCY metabolizes SAH generated by methyltransferase reactions, the AHCY-BMAL1 complex likely cooperates with or is in close proximity to chromatin remodeling factors known to drive cyclic H3K4 trimethylation, such as MLL1 and MLL3 (9, 43). It has been shown that MLL1 promotes binding of CLOCK-BMAL1 to chromatin by generating a permissive chromatin state (9). On a genome-wide level, H3K4me3 is highly correlated with H3ac at active promoters (59). MLL1-deficient MEFs display impaired oscillation of H3K4me3 and H3K9/14ac (9). Likewise, inhibition of AHCY generates comparable loss of rhythmicity, supporting a strong interdependence of cyclic histone H3 methylation and acetylation. Our findings favor a scenario where BMAL1 binds to AHCY at chromatin, thereby promoting local hydrolysis of SAH and activity of HMTs at clock-regulated sites, leading to increased methylation and consequently CLOCK-BMAL1 binding to promoters. Previous work has established histone acetyltransferase (HAT) activity of CLOCK to be essential for histone H3ac rhythmicity (45). Thus, the observed changes in H3K9/14ac at clock-regulated sites likely reflect impaired recruitment of CLOCK to DNA. Thus, our results add an additional layer to the known mechanisms of clock regulation whereby the molecular clock relies not only on the activity of chromatin remodelers but also on that of a metabolic enzyme. Further investigations aimed at identifying the cyclic nuclear interactome of AHCY will provide crucial insights into the mechanisms by which metabolic signaling affects circadian biology.
Despite the biological importance of transmethylation reactions (60), their role in circadian biology is not fully explored. One finding relates to inhibition of N6-methyladenosine mRNA methylation that was shown to lengthen the mammalian circadian period in vitro and in vivo (61, 62). On the other hand, AHCY was shown to be localized in the nucleus in Arabidopsis, Xenopus laevis, and mammalian cells (27, 63–66), suggesting that localized activity of AHCY at specific sites may promote activity of HMTs that would otherwise be inhibited by SAH. Nicely paralleling our study, AHCY was shown to be enriched at TSS of transcriptionally active genes marked with H3K4me3, including several circadian genes (26). Here, we identify BMAL1 as a binding partner of AHCY, a finding that can help explain how metabolic enzymes may regulate epigenetic modifications at specific sites. It still remains poorly understood how these enzymes can affect chromatin marks at specific loci and, in turn, regulate expression of distinct genes (67). In our view, while it is likely that AHCY might interact with other TFs, our study provides a notable insight by which specificity is achieved in the context of circadian biology.
There is an intimate molecular link between the circadian clock and the aging process (68–70). Furthermore, mutation in the circadian gene BMAL1 is associated with a premature aging phenotype and reduced life span (71). In our study, inhibition of AHCY activity in the SCN results in a prolonged period of rhythmic activity, recapitulating what is observed in aged mice (68). Depletion of AHCY in Drosophila results in a reduced life span and an aging phenotype characterized by accumulation of SAH (42). An intriguing possibility is that decline in AHCY activity during aging may promote circadian deregulation and decay of circadian function. Additional studies will further delineate the physiological implications of this newly established cross-talk between cellular metabolism, chromatin regulation, and circadian rhythms.
MATERIALS AND METHODS
Animals
Male C57BL/6 mice (2 months old) were housed under 12-hour light/12-hour dark (LD) cycles. Infusion of 100 μM DZnep into the third ventricle was performed with Alzet Osmotic Pump (Alzet, USA) as previously described (72). Locomotor activity of mice was recorded for at least 1 week in a standard LD 12:12, followed by 10 days in constant darkness (DD) to calculate the circadian period before operation. The transfer to DD and subsequent operation was timed so that each group of four replicate animals, whether vehicle- or DZnep-treated, was operated at the same CT (based on 10 days in DD) after exactly 10 days in DD. Only one group of four replicates was operated per day. Ten days after operation, each group of four replicate animals was sacrificed at the same CT (based on the 10 days after operation), one group per day. Locomotor activity was measured using optical beam motion detection (Philips Respironics), and data were collected using the Mini-Mitter VitalView 5.0 data acquisition software. Actograms and activity profiles were calculated using ClockLab software (Actimetrics). Period and amplitude were calculated by obtaining the slope of onset of the free running activity and computed by χ2 periodogram analysis. The Institutional Animal Care and Use Committee of the University of California, Irvine, and the Animal Experimental Committee of Kyoto University approved all experiments.
Plasmids and antibodies
Myc-BMAL1, Myc-CLOCK, mDbp, and mPer2 promoters fused to luciferase have been described previously (9). mPGC1α promoter fused to luciferase and pCMV TFEB were a gift from A. Ballabio. The GFP-AHCY plasmid was obtained by cloning the AHCY complementary DNA (cDNA) obtained by polymerase chain reaction (PCR) amplification from total mouse cDNA into the Eco RI and Bam HI sites of the pEGFP-C3 vector (Evrogen). The BMAL-RFP plasmid was obtained by cloning the BMAL1 cDNA obtained by PCR amplification from the Myc-BMAL1 plasmid into the Eco RI and Bam HI sites of the pTagRFP-N vector (Evrogen). The lentiCRISPRv2 plasmid was purchased from Addgene (#52961). The HA-AHCY plasmid was purchased from VectorBuilder, and the K186N mutation was created using the Q5 Site-Directed Mutagenesis Kit (NEB). Ahcy inducible shRNA vectors and shcontrol vector were purchased from Dharmacon (SMARTvector Inducible Lentiviral shRNA vector).
The following antibodies were used: anti-AHCY (#RN126PW, MBL, IP, ChIP; #ab134966, Abcam, Western blot; #ab151734, Abcam, Western blot), anti-BMAL1 (#ab93806, Abcam, IP, ChIP, Western blot; #NB-100-2288, Novus, IF), anti-CLOCK (#A302-618A, Bethyl, IP, Western blot), anti-GFP (#ab290, Abcam, IP, Western blot), anti-HA (#05-904 Millipore), anti-myc (#05419, Millipore, IP, Western blot), anti-p84 (#GTX70220, GeneTex, Western blot), anti-tubulin (#T5168, Sigma-Aldrich, Western blot), anti-actin (#A5441, Sigma-Aldrich, Western blot), anti-H3 (#ab1791, Abcam, Western blot), anti-H3K4me3 (#39915, Active Motif, ChIP), anti-H3K9/14ac (#C15410200, Diagenode, ChIP), anti-H3K27me3 (#07-449, Millipore), c-Myc (#AF3696, R&D Systems, ChIP), and normal rabbit immunoglobulin G (IgG) (#10500C, Invitrogen).
GST constructs, expression, and purification
cDNAs encoding amino acid residues 1–134 for F1, 135–265 for F2, 266–399 for F3, 400–506 for F4, and 507–626 for F5 from mouse BMAL1 [National Center for Biotechnology Information (NCBI) reference sequence, NM_007489.4] were cloned into the vector pGEX4T1 (GE Healthcare) with a TEV-cleavable N-terminal glutathione S-transferase (GST) tag and a C-terminal HA tag using Eco RI/Not I restriction sites. All constructs were confirmed by DNA sequencing. Expression and purification of GST-tagged constructs were done as described previously (73). Briefly, fusion proteins were expressed in Escherichia coli BL21 (DE3) and purified using glutathione Sepharose 4 Fast Flow resin according to the manufacturer’s protocol (GE Healthcare).
Cell culture
MEF human embryonic kidney (HEK) 293T cells and U2OS cells were cultured in Dulbecco’s modified Eagle’s medium (4.5 g liter−1 glucose, HyClone) supplemented with 10% fetal bovine serum (Gibco) and antibiotics (Gibco). MEFs from BMAL1-deficient mice were obtained as previously described (74). Confluent cells were synchronized with DEX (Sigma-Aldrich) dissolved in ethanol. MEFs were synchronized with 100 nM DEX for 1 hour. U2OS cells were synchronized with 200 nM DEX for 2 hours. DEX was washed out, and cells were placed in fresh medium for the indicated time. In addition, 3-DZnep (Cayman) was dissolved in DMSO and used at the indicated doses.
siRNA transfection of U2OS cells
U2OS cells stably transfected with Arntl-luc reporter were plated in standard medium. The next day, medium was changed to an antibiotic-free medium and cells were transfected with the following Stealth siRNA (Invitrogen) against human Ahcy (siRNA #1, 5′-gaggccugucaggagggcaacaucu-3′; siRNA #2, 5′-cccuggacauugcugagaacgagau-3′; and siRNA #3; 5′-acaggcuguauugacaucauccuug-3′) or with negative controls (Stealth RNAiT siRNA Negative Control, Med GC #1, #2, and #3) using Lipofectamine 2000 (Invitrogen). Medium was changed 6 to 8 hours after transfection. The following day, cells were synchronized with DEX, and after 2 hours, medium was replaced with medium containing 1 mM luciferin. Luminescence was recorded in successive 20-min bins at 37°C and analyzed as previously described (61).
Generation of Ahcy-KO MEF
The CRISPR-Cas9 system was used to generate Ahcy mutant MEF cells. A lentiCRISPRv2 vector with a guide RNA targeting EGFP was used as control. To delete Ahcy, a guide RNA targeting exon 4 (5′-tgcgcacctgacagaagctg-3′) was cloned into the lentiCRISPRv2 vector. The resulting plasmid was verified by sequencing and transfected into HEK293T cells together with psPAX2 and VSVG second-generation lentiviral packaging systems using BioT reagent (Bioland Scientific LLC), according to the manufacturer’s instructions. After 48 hours, lentiviral particles in the medium were collected, filtered, and used to infect MEFs. Forty-eight hours after transduction, the cells were subjected to puromycin (Sigma-Aldrich) selection. Monoclonal cell lines were generated and tested for loss of AHCY by Western blot analysis. Sanger sequence analysis of PCR amplicons generated using primers 5′-CACCTCCTCACCAATGTCCT-3′ (forward) and 5′-CTGTGGTGCATTGAGCAGAC-3′ (reverse) confirmed AHCY mutation in clones #1 and #2.
Generation of inducible Ahcy-shRNA MEF
To generate inducible shRNA-knockdown MEF, lentiviral transduction with a tetracycline-inducible shRNA (SMARTvector Inducible Lentiviral shRNA, Dharmacon) targeting 3′UTR of Ahcy (5′-TGACCAGACGGCCTTCAGC-3′) or a nontargeting vector (#VSC11651) was conducted as above. Following puromycin selection, cells were plated and GFP induction was confirmed with fluorescence microscopy upon doxycycline induction (1 μg ml−1). Cells were synchronized with DEX 24 hours after doxycycline induction.
Generation of stable AHCY MEF
Inducible Ahcy-knockdown MEFs were transfected with a linearized plasmid encoding WT or K186N mutant mouse AHCY using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s protocol. Two days after transfection, G418 (InvivoGen) was added at a final concentration of 1 mg/ml for 3 weeks to allow selection. Cells were synchronized with DEX 24 hours after doxycycline induction.
Mass spectrometry
IP and on-bead digestion
Whole mouse livers were divided into three equal pieces, and each was minced in 5 ml of buffer A [10 mM Hepes (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol (DTT), supplemented with protease inhibitor cocktail (Roche), 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 20 mM NaF, 1 μM trichostatin A (TSA), and 10 mM nicotinamide (NAM)]. Tissue was homogenized and spun at 1600 rpm (600g) for 10 min at 4°C. The pellet was resuspended in 3 ml of buffer S1 [250 mM sucrose, 10 mM MgCl2, supplemented with protease inhibitor cocktail (Roche), 0.5 mM PMSF, 20 mM NaF, 1 μM TSA, and 10 mM NAM], layered over 3 ml of buffer S2 [350 mM sucrose, 0.5 mM MgCl2, supplemented with protease inhibitor cocktail (Roche), 0.5 mM PMSF, 20 mM NaF, 1 μM TSA, and 10 mM NAM], and spun at 2500 rpm (1430g) for 5 min at 4°C. All three nuclear pellets were combined using buffer S2, sonicated, layered over 3 ml of buffer S3 [880 mM sucrose, 0.5 mM MgCl2, supplemented with protease inhibitor cocktail (Roche), 0.5 mM PMSF, 20 mM NaF, 1 μM TSA, and 10 mM NAM], and spun at 3500 rpm (2800g) for 10 min at 4°C. Fractions were collected and resuspended in 1× radioimmunoprecipitation assay (RIPA) [50 mM tris (pH 7.5), 150 mM NaCl,1% NP-40] or mixed with 5× RIPA, briefly sonicated, rocked for 30 min at 4°C, and spun at 12,500 rpm (18,000g) for 10 min at 4°C, and enriched lysates were collected. Supernatant was retained as the enriched nucleoplasmic fraction and diluted with 5× RIPA [250 mM tris (pH 7.5), 750 mM NaCl, 5% NP-40, supplemented with protease inhibitor cocktail (Roche), 0.5 mM PMSF, 20 mM NaF, 1 μM TSA, and 10 mM NAM]. The nucleolar pellet was resuspended in 500 μl of buffer S2 and spun at 3500 rpm (2800g) for 5 min at 4°C. The cleaned nucleolar pellet was resuspended in 1× RIPA [50 mM tris (pH 7.5), 150 mM NaCl, 1% NP-40, supplemented with protease inhibitor cocktail (Roche), 0.5 mM PMSF, 20 mM NaF, 1 μM TSA, and 10 mM NAM]. Fractions were briefly sonicated, rocked for 30 min at 4°C, and spun at 12,500 rpm (18,000g) for 10 min at 4°C, and enriched lysates were collected.
Four milligrams of nucleoplasmic and nucleolar fractions was prepared in 1× RIPA [50 mM tris (pH 7.5), 150 mM NaCl,1% NP-40, supplemented with protease inhibitor cocktail (Roche), 0.5 mM PMSF, 20 mM NaF, 1 μM TSA, and 10 mM NAM]. Lysates were precleared with 20 μl of protein G Sepharose beads (Sigma-Aldrich) while rocking at 4°C for 1 hour. After removal of preclearing beads, lysates were incubated with 4 ug of BMAL1 antibody (Abcam) and an additional pooled sample was prepared for incubation with 4 μg of rabbit IgG (Santa Cruz Biotechnology) and rocked overnight at 4°C. The next day, 40 μl of fresh protein G Sepharose beads was added to the lysates and rocked at 4°C for 2 hours. Beads were washed three times with 1× RIPA. Beads were washed three times with 50 mM NH4HCO3 and incubated with trypsin (10 ng/μl) in 1 M urea–50 mM NH4HCO3 for 30 min and washed with 50 mM NH4HCO3, and the supernatant was digested overnight in the presence of 1 mM DTT. Digested peptides were alkylated and desalted before liquid chromatography (LC)–MS analysis.
Mass spectrometry
For LC-MS/MS purposes, desalted peptides were injected in an UltiMate 3000 RSLCnano system (Thermo Fisher Scientific) and separated in a 15-cm analytical column (75 μm inside diameter home-packed with ReproSil-Pur C18-AQ 2.4 μm from Dr. Maisch) with a 50-min gradient from 5 to 60% acetonitrile in 0.1% formic acid. The effluent from the high-performance liquid chromatography (HPLC) was directly electrosprayed into an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific) operated in data-dependent mode to automatically switch between full-scan MS and MS/MS acquisition. MS and MS/MS acquisition. Survey full-scan MS spectra [from mass/charge ratio (m/z), 300 to 2000] were acquired in the Orbitrap with resolution R = 60,000 at m/z 400 (after accumulation to a “target value” of 500,000 in the linear ion trap). The six most intense peptide ions with charge states between 2 and 4 were sequentially isolated to a target value of 10,000 and fragmented in the linear ion trap by collision-induced dissociation (CID). All fragment ion spectra were recorded in the LTQ part of the instrument. For all measurements with the Orbitrap detector, three lock-mass ions from ambient air were used for internal calibration as described before (75). Typical MS conditions were as follows: spray voltage, 1.5 kV; no sheath and auxiliary gas flow; heated capillary temperature, 200°C; normalized CID energy, 35%; activation q = 0.25; activation time, 30 ms. MaxQuant 1.5.2.8 was used to identify proteins and quantify by iBAQ with the following parameters: database, UP000000589_10090_Mmusculus_151030; MS tol, 10 ppm; MS/MS tol, 0.5 Da; peptide FDR, 0.1; protein FDR, 0.01; minimum peptide length, 5; variable modifications, oxidation (M); fixed modifications, carbamidomethyl (C); peptides for protein quantitation, razor and unique; minimum peptides, 1; and minimum ratio count, 2. Identified proteins were considered nucleoplasmic enriched interaction partners if their MaxQuant iBAQ values were greater than log2 twofold enrichment and P = 0.05 [analysis of variance (ANOVA)] when compared to the control.
Protein extraction and IP
For total extracts, cells were washed with phosphate-buffered saline (PBS) and lysed for 15 min at 4°C in RIPA buffer [50 mM tris-HCl (pH 8.0), 150 mM NaCl, 5 mM EDTA, 15 mM MgCl2, 1% NP-40, 0.5% deoxycholate (DOC), 1× protease inhibitor cocktail (Roche), and 1 mM PMSF]. Preparation of fractionated protein extracts from cells was as follows. After PBS washes, cells were resuspended and washed in low-salt buffer [10 mM Hepes-NaOH (pH 8.0), 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, and 1× protease inhibitor cocktail (Roche)]. The pellets were resuspended in low-salt buffer supplemented with 1% NP-40 and incubated on ice for 15 min. Following a centrifugation step, the supernatant was recovered and stored at −80°C as the cytoplasmic fraction. The pellet containing the nuclear fraction was washed twice with low-salt buffer, resuspended in high-salt buffer [10 mM Hepes-NaOH, 500 mM NaCl, 1.5 mM MgCl2, 0.5 mM DTT, 1% NP-40, 10% glycerol, 1× protease inhibitor cocktail (Roche), and 1 mM PMSF], and incubated on ice for 30 min. After a 10-min centrifugation at 14,000 rpm at 4°C, the supernatant was stored at −80°C as the soluble nuclear fraction. The nucleo-insoluble pellet was washed twice with high-salt buffer, resuspended in RIPA buffer, and sonicated (Qsonica) on ice. After centrifugation, the pellet was stored at −80°C as the soluble chromatin-bound fraction.
Subcellular extracts from liver were obtained as follows: 200 mg of liver was homogenized with a motorized tissue grinder in TMS buffer [50 mM tris-HCl (pH 8), 5 mM MgCl2, 250 mM 20% sucrose, 1× protease inhibitor cocktail (Roche), and 1 mM PMSF], passed through a 100-μm filter, and centrifuged at 800g at 4°C. Samples were washed with Cyto buffer [10 mM Hepes-NaOH, 25 mM KCl, 0.65 mM spermidine, 1 mM EDTA, 1 mM EGTA, 0.34 M sucrose, 1% NP-40, 1 mM DTT, 1× protease inhibitor cocktail (Roche), and 0.5 mM PMSF], incubated on ice for 10 min, and centrifuged at 3000 rpm for 10 min at 4°C. The supernatant was recovered and stored at −80°C as the cytoplasmic fraction, and the pellet was washed twice in low-salt buffer [10 mM Hepes-NaOH, 25 mM KCl, 0.65 mM spermidine, 1 mM EDTA, 1 mM EGTA, 20% glycerol, 1 mM DTT, 1× protease inhibitor cocktail (Roche), and 0.5 mM PMSF], with centrifugation at 3000 rpm for 10 min at 4°C. The pellets were then resuspended in high-salt buffer [10 mM Hepes-NaOH, 25 mM KCl, 0.65 mM spermidine, 1 mM EDTA, 1 mM EGTA, 400 mM NaCl, 20% glycerol, 1% NP-40, 1 mM DTT, 1× protease inhibitor cocktail (Roche), and 0.5 mM PMSF], rocked at 4°C for 30 min, and centrifuged at 14,000 rpm for 10 min. The supernatant was recovered and stored at −80°C as the soluble nuclear fraction, and the pellet was resuspended in RIPA buffer and sonicated (Qsonica) on ice. After centrifugation, the pellet was stored at −80°C as the soluble chromatin-bound fraction.
For co-IP experiments, protein extracts were incubated with the indicated antibodies overnight at 4°C, followed by incubation with Dynabeads (protein G, Thermo Fisher Scientific) for 2 hours at 4°C. Beads were washed four times with RIPA buffer [50 mM tris-HCl (pH 8.0), 150 mM NaCl, 5 mM EDTA, 15 mM MgCl2, 1% NP-40, 0.5% DOC, 1× protease inhibitor cocktail (Roche), and 1 mM PMSF], and samples were eluted with SDS loading buffer.
SAM and SAH quantification
Levels of SAM and SAH in cells were determined with the SAM and SAH ELISA Combo Kit (STA-671-C, Cell Biolabs).
AHCY activity assay
To measure the activity of AHCY, samples were incubated in homogenization buffer provided by the Adenosylhomocysteinase (AHCY) Activity Fluorometric Assay Kit (BioVision, K807-100) for 15 min at 4°C, followed by centrifugation at 14,000 rpm for 10 min. HA-tagged AHCY was immunoprecipitated overnight at 4°C, followed by incubation with Dynabeads (protein G, Thermo Fisher Scientific) for 2 hours at 4°C. Beads were washed four times with homogenization buffer (BioVision) and resuspended in AHCY assay buffer (BioVision), and the assay was prepared according to the manufacturer’s instructions. The assay was performed in 96-well white plates, and the fluorescence was measured in kinetic mode for 30 min at 37°C using a Varioskan LUX multimode microplate reader (Thermo Fisher Scientific).
Immunofluorescence
For the detection of AHCY and BMAL1 colocalization, MEF cells were grown on Nunc Lab-Tek chamber slides (Thermo Fisher Scientific) and cotransfected with GFP-AHCY and BMAL1-RFP plasmids using Lipofectamine 3000 (Invitrogen). After 36 hours, cells were fixed in 4% Paraformaldehyde for 10 min at room temperature. Cells were washed four times in PBS and permeabilized with 0.1% Triton X-100 in PBS for 10 min. DRAQ7 (Thermo Fisher Scientific) was used to visualize nuclei. The slides were imaged using a Leica confocal microscope, and colocalization was quantified using the JaCoP plugin of Fiji.
Live cell imaging was carried out as follows: Fluorescence lifetime images of the donor only (GFP) and donor-acceptor (GFP-RFP) were measured using a modified Olympus FluoView 1000 microscope equipped with a Spectra-Physics Mai Tai HP laser and FLIMBox acquisition card. The samples were excited with the 900-nm laser line using a 63× water immersion objective (1.2 numerical aperture, Olympus Plan Apo) applying a two-photon excitation scheme. The fluorescence was split using a dichroic mirror (FF495-Di03-25x36, Semrock) and then passed through two separate filters (520/35 nm to 641/75 nm, Semrock), collected using two separate photomultiplier tube (Hamamatsu), and recorded using FLIMBox. The pixel dwell time for the acquisitions was 32 μs, and the images were taken with sizes of 256 × 256 pixels. To have high signal-to-noise ratio, 30 to 50 frames were collected. The data from each pixel were recorded and analyzed using the SimFCS software (developed by E. Gratton in Laboratory for Fluorescence Dynamics, University of California, Irvine, CA). The intensity decays collected at each pixel of the image were transformed to the Fourier space, and the phasor coordinates were calculated using the following relations
where gi,j(ω) and si,j(ω) are the x and y coordinates of the phasor plot, respectively, and n and ω are the harmonic number and the angular frequency of excitation, respectively. The transformed data were then plotted in the phasor plot in a way that the data from each individual pixel are transformed to a point in the phasor plot. The phasor histogram was analyzed using the fractional intensity distribution between two phasor components: phasor positions of non-FRET donor-only component and that of the decreased lifetime of the donor in donor-acceptor sample. According to the linear combination of the phasor, the shift in the phasor at image pixels, where both donor only and donor-acceptor samples are present, will be along the linear combination of the individual phasor positions of the donor lifetime in donor only and donor-acceptor samples. The relative fractions of the two components—donor only and donor-acceptor—will determine the phasor position of that image pixel according to their relative fractional intensity contribution. In nucleus, if the positions of the donor and donor-acceptor samples are not segmented—as is the situation here—this will represent a shift of the fractional intensity distribution to the shorter lifetime component of the donor lifetime in donor-acceptor species (76, 77). An increase in the fraction of phasor points in the fractional intensity component of the shorter lifetime is indicative of the presence of FRET. This method of fractional intensity contribution distribution (76) has been used here to show that an increasing fraction of pixels has higher contribution to the short lifetime (and corresponding phasor) of the donor-acceptor FRET samples.
Luciferase reporter assay
HEK293T cells were transfected with 25 ng of luciferase reporter and LacZ plasmids, with or without 50 ng of Myc-CLOCK and Myc-BMAL1 or TFEB with BioT transfection reagent (Bioland). At 16 hours after transfection, culture medium was replaced and luciferase activity was measured 36 hours after transfection as described previously (9). Luciferase activity was normalized to transfection efficiency by a colorimetric β-galactosidase assay.
Electrophoretic mobility shift assay
EMSA was carried out using a Light-Shift Chemiluminescent EMSA kit (Thermo Fisher Scientific, 20148) according to the manufacturer’s instructions. Briefly, 6 μg of nuclear extracts was mixed with EMSA binding buffer in the presence of a previously described biotin-labeled probe (38). The mixture was incubated at room temperature for 30 min, and the protein-DNA complexes were loaded onto a 4.5% nondenaturing polyacrylamide gel.
RNA extraction and quantification of gene expression
Confluent ctrl or AHCY KO MEFs were synchronized with DEX, and total RNA was isolated at the indicated time points using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. For the inhibitor experiment, confluent MEF cells were treated with DMSO or 10 μM DZnep for 8 hours and synchronized with DEX. At the indicated time points, total RNA was isolated using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. cDNA was obtained by retrotranscription of 1 μg of RNA with the Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Quantitative real-time (qRT)–PCR analysis was performed using QuantStudio 3 (Applied Biosystems) with PowerUp SYBR Green Master Mix (Applied Biosystems). 18S was used as an endogenous control. Primer sequences are listed in table S1.
Chromatin immunoprecipitation
MEF cells were cross-linked at the indicated time points with 1% formaldehyde at room temperature for 10 min, and the reaction was quenched by addition of glycine. Cells were rinsed with PBS, flash-frozen in liquid nitrogen, and stored at −80°C until all the time points were processed. For tissue, 250 mg of frozen tissue was homogenized in PBS containing 1× protease inhibitor cocktail (Roche) and 1 mM PMSF. Liver homogenates were crossed-linked with 1% formaldehyde at room temperature for 10 min, and the reaction was quenched by addition of glycine. Samples were centrifuged at 1000 rpm for 10 min at 4°C.
Pellets were then lysed in lysis buffer [5 mM Pipes (pH 8.0), 85 mM KCl, 0.5% NP-40, and 1× protease inhibitor cocktail (Roche)], and nuclei were collected by centrifugation (3000 rpm, 10 min, 4°C). Nuclei were resuspended in sonication buffer [1% SDS, 10 mM EDTA, 50 mM tris-HCl (pH 8), and 1× protease inhibitor cocktail (Roche)] and incubated on ice for 30 min. Samples were then sonicated using a Bioruptor Pico sonication device (Diagenode) until chromatin was sheared to an average DNA fragment length of 0.2 to 0.5 kb. After centrifugation (14,000 rpm, 10 min, 4°C), supernatants were diluted with tris-EDTA buffer (TE) and IP reactions were set up as follows: 1% Triton X-100, 0.1% DOC, 1× protease inhibitor cocktail (Roche), and magnetic beads (#11203D, Dynal Invitrogen) coupled with the indicated antibody (#RN126PW, MBL; #ab93806, Abcam; #39915, Active Motif; #C15410200, Diagenode; #07-449, Millipore) and incubated overnight at 4°C. Immunocomplexes were washed eight times with RIPA buffer [50 mM Hepes (pH 8.0), 1 mM EDTA, 1% NP-40, 0.7% DOC, and 0.5 M LiCl] and once with TE. To reverse the cross-linking, washed beads were resuspended in elution buffer [10 mM tris (pH 8.0), 1 mM EDTA, and 1% SDS] and incubated overnight at 65°C. The following day, samples were incubated for 2 hours at 37°C in proteinase K mix (TE, glycogen, and proteinase K). Ribonuclease A treatment was carried out for 30 min at 37°C, and then DNA was purified by phenol-chloroform extraction using phase lock tubes (#2302820, Quantabio). The DNA pellet was resuspended in deoxyribonuclease-free water (Gibco) and stored at −20°C.
BMAL1 ChIP for sequencing was performed using the ChIP-IT high Sensitivity Kit (#53040, Active Motif) according to the manufacturer’s instructions using 2 μg of BMAL1 (#ab93806, Abcam). Purified DNA was used for qRT-PCR analysis using QuantStudio 3 (Applied Biosystems) with PowerUp SYBR Green Master Mix (Applied Biosystems). Primer sequences are listed in table S2.
Next-generation sequencing sample preparation and analysis
RNA sequencing
To generate libraries, total RNA was isolated from Ctrl MEF, AHCY KO MEF, DMSO, and 10 μM DZnep–treated MEF using TRIzol (three biological replicates). Libraries were prepared from three biological replicates according to TruSeq RNA Library Prep Kit v2.
Libraries were assessed for quality and quantity using BioAnalyzer (Agilent) and qPCR (Kapa Biosystems). The multiplexed libraries were sequenced on eight lanes using single-end 100-cycle chemistry for HiSeq 4000 [100 base pairs (bp), single-end reads]. The version of HiSeq control software was HCS 3.3.76 with real-time analysis software, RTA 2.7.6. Demultiplexed FASTQ files were aligned to the reference genome assembly mm10 using TopHat-2.1.1 with default parameters. Aligned reads were mapped to genomic features and quantified using Cufflinks 2.2.1. Reads per kilobase of transcript per million mapped reads (RPKM) were used to quantify the relative abundance of each transcript in a sample and to perform gene expression analyses. Genes systematically observed with no expression in all replicates were not considered in the study.
To generate libraries from SCN samples, RNA was extracted from SCN punches using the RNeasy Micro Kit (QIAGEN) according to the manufacturer’s instructions (four biological replicates for all CT points with the exception of CT21 saline that has three biological replicates). Libraries were prepared using SMARTer Stranded Total RNA-Seq Kit v2, Pico Input Mammalian (Clontech) according to the manufacturer’s instructions. Libraries were assessed for quality and quantity using BioAnalyzer (Agilent) and qPCR (Kapa Biosystems). The multiplexed libraries were sequenced on eight lanes using single-end 100-cycle chemistry for HiSeq 4000 (100 bp, paired-end reads). After trimming the first three nucleotides from the 5′ end of the paired end read, FASTQ files were aligned to the reference genome assembly mm10 as described above. For time series datasets, rhythmic transcripts were detected with the BIO_CYCLE algorithm (78) and results were confirmed with JTK algorithm (79). Output measures including amplitude and phase of rhythms were also determined. Genes were considered rhythmic over the circadian cycle if their permutation-based, adjusted P value was <0.01 for MEF samples and <0.05 for SCN samples. Differential analysis of gene expression at individual CTs was defined using Cyber-T, a differential analysis program using a Bayesian-regularized t test (80). GO biological process enrichment analysis was conducted using The Database for Annotation, Visualization and Integrated Discovery (DAVID) version 6.8 (81). TFBS enrichment was performed using MotifMap (82), and ChIP-seq experiments were obtained from ENCODE. RNA-seq data are deposited in, and are available through, the CircadiOmics web portal at http://circadiomics.igb.uci.edu/ (83).
ChIP sequencing
Samples were prepared as described above in the ChIP section. Library construction was performed from two biological replicates using a PrepX Illumina Chip-seq kit and the PrepXComplete ILMN DNA barcodes on the Apollo324 System. DNA quantity was measured by Qubit DNA High Sensitivity, and the fragment size was measured by Agilent Bioanalyzer 2100 High Sensitivity DNA chip assay. The input amount of DNA was 4 ng for H3K4me3 and 0.5 ng for BMAL1. The ends of the DNA were repaired and adenylated. The reaction mixture was then cleaned up using AMPure XP magnetic beads, and Illumina barcoded adapters were ligated onto the blunt-end/adenylated product. The adapter ligated product was cleaned using AMPure XP beads and then amplified for adapter ligated products using 15 cycles of PCR. The resulting library was cleaned with AMPure XP beads and quantified by qPCR with Kapa Sybr Fast universal for Illumina Genome Analyzer kit. The library size was determined by analysis using the Bioanalyzer 2100 DNA High Sensitivity Chip, and libraries were sequenced on the HiSeq 4000 Sequencer (100 bp, single-end reads). Sequencing was performed using single read flowcell chemistry and 100 cycles with additional cycles for the index read. The HiSeq real-time analysis software (RTA 2.7.7) converted the images into intensities and base calls. Postprocessing of the run to generate the FASTQ files was performed at the Institute for Genomics and Bioinformatics (UCI IGB).
The raw reads were mapped to mouse reference genome NCBI build 37 (mm9) by bowtie v1.1.0, allowing up to one mismatch. To adjust the difference of library size, uniquely mapped reads of each sample were down-sampled to the lowest read count among all samples including input. Then, the ChIP-seq peaks were called by MACS v1.4.2 (84) with a cutoff of P ≤ 1 × 10−8, and clonal reads were automatically removed by MACS. The peaks that overlapped with any “negative peaks” (peaks are called by swapping the ChIP and input) were discarded. As the ChIP-seq profile of two replicates is highly correlated (H3K4me3 Pearson’s R ≥ 0.96, BMAL1 Pearson’s R ≥ 0.76), data from replicate 1 were applied in downstream analysis. For peak overlap analysis, peaks from all four samples were merged together as consensus regions. Then, peaks of each sample, which have at least 500 bp or 50% overlap with the consensus regions, were defined as overlapped peaks. To reduce false-positive calls, BMAL1 peaks with lower than twofold enrichment over input in all four samples were also discarded. The ChIP-seq read density was determined by deepTools v2.3.4 (85), and then the average binding profile was visualized using R v3.2.3. Gene promoter regions were defined as 1 kb upstream to 1 kb downstream of gene TSS. H3K4me3 occupancy at gene promoters was quantified as RPKM, which was normalized by both promoter length and total read number in each sample. The differential analysis of H3K4me3 occupancy was conducted between DMSO and DZnep samples using two biological replicates. The differential analysis was visualized by volcano plots using R v3.6.0. The genes with at least twofold difference and P < 0.05 were defined as significant differentially bound genes.
Statistical analysis
For each experiment, the number of biological replicates, statistical test, significance threshold, and visual representation information can be found in the figure legends or main text. Statistics were performed with GraphPad Prism 6.0 software or R software.
Supplementary Material
Acknowledgments
In memory of Paolo Sassone-Corsi, an exceptional mentor and scientist. We thank C. Partch and all members of the P.S.-C. laboratory for discussion and support. We thank A. Ballabio (TIGEM) for the pCMV TFEB plasmid. We also thank M. Oakes, S.-A. Chung, and V. Ciobanu of the Genomics High Throughput Facility. Funding: C.M.G. was supported by the National Cancer Institute of the NIH under award number T32CA009054 and by the European Union’s Horizon 2020 research and innovation programme under Marie Sklodowska-Curie grant agreement 749869. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. M.C. was supported by the NIH F31 Kirschstein-NRSA Predoctoral Fellowship (F31GM117942) and the American Heart Association Predoctoral Fellowship (17PRE33410952). The work of N.C., M.S., and P.B. was, in part, supported by NIH grant GM123558 to P.B. Work in the H.O. laboratory was supported by the CREST Japan Science and Technology Agency (JPMJCR14W3). Work in the E.G. laboratory was supported by NIH grants P50 GM076516 and NIH P41GM103540. Work in the P.S.-C. laboratory is supported by NIH grants R21DK114652 and R21AG053592 and a Challenge Grant from the Novo Nordisk Foundation (NNF-202585). K.K. was supported by a fellowship from the Japan Society for the Promotion of Science (JSPS). Work in the A.I. laboratory is partly funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project ID 325871075—SFB 1309. Author contributions: C.M.G. and P.S.-C. conceived and designed the study and wrote the manuscript. M.C. performed the biochemistry experiment to identify BMAL1 nuclear interactors. K.B.K. performed molecular biology analyses J.-M.F., K.I., and R.K. performed U2OS cell studies and in vivo studies. N.C., M.S., J.S., and P.B. performed bioinformatics analyses. I.F. and A.I. performed MS analyses. S.R. performed FRET analyses. J.G. and K.K. provided technical support for project design and data analysis. E.G., P.B., A.I., W.L., and H.O. provided technical support. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The data that support the findings of this study are available upon request from the corresponding author. The RNA-seq ChIP-seq data have been deposited in the Gene Expression Omnibus (GEO) with accession numbers GSE126851 (RNA-seq) and GSE125720 (ChIP-seq).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabc5629/DC1
REFERENCES AND NOTES
- 1.Greco C. M., Sassone-Corsi P., Circadian blueprint of metabolic pathways in the brain. Nat. Rev. Neurosci. 20, 71–82 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bass J., Circadian topology of metabolism. Nature 491, 348–356 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Partch C. L., Green C. B., Takahashi J. S., Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 24, 90–99 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papazyan R., Zhang Y., Lazar M. A., Genetic and epigenomic mechanisms of mammalian circadian transcription. Nat. Struct. Mol. Biol. 23, 1045–1052 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aguilar-Arnal L., Sassone-Corsi P., Chromatin landscape and circadian dynamics: Spatial and temporal organization of clock transcription. Proc. Natl. Acad. Sci. U.S.A. 112, 6863–6870 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koike N., Yoo S.-H., Huang H.-C., Kumar V., Lee C., Kim T.-K., Takahashi J. S., Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 338, 349–354 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vollmers C., Schmitz R. J., Nathanson J., Yeo G., Ecker J. R., Panda S., Circadian oscillations of protein-coding and regulatory RNAs in a highly dynamic mammalian liver epigenome. Cell Metab. 16, 833–845 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Etchegaray J.-P., Lee C., Wade P. A., Reppert S. M., Rhythmic histone acetylation underlies transcription in the mammalian circadian clock. Nature 421, 177–182 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Katada S., Sassone-Corsi P., The histone methyltransferase MLL1 permits the oscillation of circadian gene expression. Nat. Struct. Mol. Biol. 17, 1414–1421 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aguilar-Arnal L., Katada S., Orozco-Solis R., Sassone-Corsi P., NAD+-SIRT1 control of H3K4 trimethylation through circadian deacetylation of MLL1. Nat. Struct. Mol. Biol. 22, 312–318 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DiTacchio L., Le H. D., Vollmers C., Hatori M., Witcher M., Secombe J., Panda S., Histone lysine demethylase JARID1a activates CLOCK-BMAL1 and influences the circadian clock. Science 333, 1881–1885 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J. Y., Kwak P. B., Weitz C. J., Specificity in circadian clock feedback from targeted reconstitution of the NuRD corepressor. Mol. Cell 56, 738–748 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Katada S., Imhof A., Sassone-Corsi P., Connecting threads: Epigenetics and metabolism. Cell 148, 24–28 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Lu C., Thompson C. B., Metabolic regulation of epigenetics. Cell Metab. 16, 9–17 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X., Egervari G., Wang Y., Berger S. L., Lu Z., Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat. Rev. Mol. Cell Biol. 19, 563–578 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boukouris A. E., Zervopoulos S. D., Michelakis E. D., Metabolic enzymes moonlighting in the nucleus: Metabolic regulation of gene transcription. Trends Biochem. Sci. 41, 712–730 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Reid M. A., Dai Z., Locasale J. W., The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol. 19, 1298–1306 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Locasale J. W., Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 13, 572–583 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiang P. K., Gordon R. K., Tal J., Zeng G. C., Doctor B. P., Pardhasaradhi K., McCann P. P., S-Adenosylmethionine and methylation. FASEB J. 10, 471–480 (1996). [PubMed] [Google Scholar]
- 20.Richon V. M., Johnston D., Sneeringer C. J., Jin L., Majer C. R., Elliston K., Jerva L. F., Scott M. P., Copeland R. A., Chemogenetic analysis of human protein methyltransferases. Chem. Biol. Drug Des. 78, 199–210 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Campbell S. L., Wellen K. E., Metabolic signaling to the nucleus in cancer. Mol. Cell 71, 398–408 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Mews P., Donahue G., Drake A. M., Luczak V., Abel T., Berger S. L., Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 546, 381–386 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gekakis N., Staknis D., Nguyen H. B., Davis F. C., Wilsbacher L. D., King D. P., Takahashi J. S., Weitz C. J., Role of the CLOCK protein in the mammalian circadian mechanism. Science 280, 1564–1569 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Lipton J. O., Yuan E. D., Boyle L. M., Ebrahimi-Fakhari D., Kwiatkowski E., Nathan A., Güttler T., Davis F., Asara J. M., Sahin M., The circadian protein BMAL1 regulates translation in response to S6K1-mediated phosphorylation. Cell 161, 1138–1151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cervantes M., Forné I., Ranjit S., Gratton E., Imhof A., Sassone-Corsi P., BMAL1 associates with NOP58 in the nucleolus and contributes to pre-rRNA processing. iScience 23, 101151 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aranda S., Alcaine-Colet A., Blanco E., Borràs E., Caillot C., Sabidó E., Croce L. D., Chromatin capture links the metabolic enzyme AHCY to stem cell proliferation. Sci. Adv. 5, eaav2448 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ponnaluri V. K. C., Estève P.-O., Ruse C. I., Pradhan S., S-adenosylhomocysteine hydrolase participates in DNA methylation inheritance. J. Mol. Biol. 430, 2051–2065 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Huang N., Chelliah Y., Shan Y., Taylor C. A., Yoo S.-H., Partch C., Green C. B., Zhang H., Takahashi J. S., Crystal structure of the heterodimeric CLOCK:BMAL1 transcriptional activator complex. Science 337, 189–194 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robles M. S., Humphrey S. J., Mann M., Phosphorylation is a central mechanism for circadian control of metabolism and physiology. Cell Metab. 25, 118–127 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Wang Y., Song L., Liu M., Ge R., Zhou Q., Liu W., Li R., Qie J., Zhen B., Wang Y., He F., Qin J., Ding C., A proteomics landscape of circadian clock in mouse liver. Nat. Commun. 9, 1553 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glazer R. I., Hartman K. D., Knode M. C., Richard M. M., Chiang P. K., Tseng C. K., Marquez V. W., 3-Deazaneplanocin: A new and potent inhibitor of S-adenosylhomocysteine hydrolase and its effects on human promyelocytic leukemia cell line HL-60. Biochem. Biophys. Res. Commun. 135, 688–694 (1986). [DOI] [PubMed] [Google Scholar]
- 32.Tseng C. K., Marquez V. E., Fuller R. W., Goldstein B. M., Haines D. R., McPherson H., Parsons J. L., Shannon W. M., Arnett G., Hollingshead M., Driscoll J. S., Synthesis of 3-deazaneplanocin A, a powerful inhibitor of S-adenosylhomocysteine hydrolase with potent and selective in vitro and in vivo antiviral activities. J. Med. Chem. 32, 1442–1446 (1989). [DOI] [PubMed] [Google Scholar]
- 33.Chiang P. K., Biological effects of inhibitors of S-adenosylhomocysteine hydrolase. Pharmacol. Ther. 77, 115–134 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Balsalobre A., Brown S. A., Marcacci L., Tronche F., Kellendonk C., Reichardt H. M., Schütz G., Schibler U., Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science 289, 2344–2347 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Wu Q.-L., Fu Y.-F., Zhou W.-L., Wang J.-X., Feng Y.-H., Liu J., Xu J.-Y., He P.-L., Zhou R., Tang W., Wang G.-F., Zhou Y., Yang Y.-F., Ding J., Li X.-Y., Chen X.-R., Yuan C., Lawson B. R., Zuo J.-P., Inhibition of S-adenosyl-l-homocysteine hydrolase induces immunosuppression. J. Pharmacol. Exp. Ther. 313, 705–711 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Takata Y., Yamada T., Huang Y., Komoto J., Gomi T., Ogawa H., Fujioka M., Takusagawa F., Catalytic mechanism of S-adenosylhomocysteine hydrolase. Site-directed mutagenesis of Asp-130, Lys-185, Asp-189, and Asn-190. J. Biol. Chem. 277, 22670–22676 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Napolitano G., Ballabio A., TFEB at a glance. J. Cell Sci. 129, 2475–2481 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ripperger J. A., Schibler U., Rhythmic CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp transcription and chromatin transitions. Nat. Genet. 38, 369–374 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Berger S. L., The complex language of chromatin regulation during transcription. Nature 447, 407–412 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Mentch S. J., Mehrmohamadi M., Huang L., Liu X., Gupta D., Mattocks D., Padilla P. G., Ables G., Bamman M. M., Thalacker-Mercer A. E., Nichenametla S. N., Locasale J. W., Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab. 22, 861–873 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai Z., Mentch S. J., Gao X., Nichenametla S. N., Locasale J. W., Methionine metabolism influences genomic architecture and gene expression through H3K4me3 peak width. Nat. Commun. 9, 1955 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parkhitko A. A., Binari R., Zhang N., Asara J. M., Demontis F., Perrimon N., Tissue-specific down-regulation of S-adenosyl-homocysteine via suppression of dAhcyL1/dAhcyL2 extends health span and life span in Drosophila. Genes Dev. 30, 1409–1422 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valekunja U. K., Edgar R. S., Oklejewicz M., van der Horst G. T. J., O’Neill J. S., Tamanini F., Turner D. J., Reddy A. B., Histone methyltransferase MLL3 contributes to genome-scale circadian transcription. Proc. Natl. Acad. Sci. U.S.A. 110, 1554–1559 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakahata Y., Kaluzova M., Grimaldi B., Sahar S., Hirayama J., Chen D., Guarente L. P., Sassone-Corsi P., The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 134, 329–340 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Doi M., Hirayama J., Sassone-Corsi P., Circadian regulator CLOCK is a histone acetyltransferase. Cell 125, 497–508 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Etchegaray J.-P., Yang X., DeBruyne J. P., Peters A. H. F. M., Weaver D. R., Jenuwein T., Reppert S. M., The polycomb group protein EZH2 is required for mammalian circadian clock function. J. Biol. Chem. 281, 21209–21215 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Cao R., Zhang Y., The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Genet. Dev. 14, 155–164 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Guccione E., Martinato F., Finocchiaro G., Luzi L., Tizzoni L., Dall’ Olio V., Zardo G., Nervi C., Bernard L., Amati B., Myc-binding-site recognition in the human genome is determined by chromatin context. Nat. Cell Biol. 8, 764–770 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Zeller K. I., Jegga A. G., Aronow B. J., O’Donnell K. A., Dang C. V., An integrated database of genes responsive to the Myc oncogenic transcription factor: Identification of direct genomic targets. Genome Biol. 4, R69 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takahashi J. S., Molecular neurobiology and genetics of circadian rhythms in mammals. Annu. Rev. Neurosci. 18, 531–553 (1995). [DOI] [PubMed] [Google Scholar]
- 51.Katoh Y., Ikura T., Hoshikawa Y., Tashiro S., Ito T., Ohta M., Kera Y., Noda T., Igarashi K., Methionine adenosyltransferase II serves as a transcriptional corepressor of Maf oncoprotein. Mol. Cell 41, 554–566 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Wang Y., Guo Y. R., Liu K., Yin Z., Liu R., Xia Y., Tan L., Yang P., Lee J.-H., Li X.-J., Hawke D., Zheng Y., Qian X., Lyu J., He J., Xing D., Tao Y. J., Lu Z., KAT2A coupled with the α-KGDH complex acts as a histone H3 succinyltransferase. Nature 552, 273–277 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De La Haba G., Cantoni G. L., The enzymatic synthesis of S-adenosyl-L-homocysteine from adenosine and homocysteine. J. Biol. Chem. 234, 603–608 (1959). [PubMed] [Google Scholar]
- 54.Devogelaere B., Sammels E., De Smedt H., The IRBIT domain adds new functions to the AHCY family. Bioessays 30, 642–652 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Dickinson M. E., Flenniken A. M., Ji X., Teboul L., Wong M. D., White J. K., Meehan T. F., Weninger W. J., Westerberg H., Adissu H., Baker C. N., Bower L., Brown J. M., Caddle L. B., Chiani F., Clary D., Cleak J., Daly M. J., Denegre J. M., Doe B., Dolan M. E., Edie S. M., Fuchs H., Gailus-Durner V., Galli A., Gambadoro A., Gallegos J., Guo S., Horner N. R., Hsu C.-W., Johnson S. J., Kalaga S., Keith L. C., Lanoue L., Lawson T. N., Lek M., Mark M., Marschall S., Mason J., Mc Elwee M. L., Newbigging S., Nutter L. M. J., Peterson K. A., Ramirez-Solis R., Rowland D. J., Ryder E., Samocha K. E., Seavitt J. R., Selloum M., Szoke-Kovacs Z., Tamura M., Trainor A. G., Tudose I., Wakana S., Warren J., Wendling O., West D. B., Wong L., Yoshiki A.; The International Mouse Phenotyping Consortium, Wurst W., MacArthur D. G., Tocchini-Valentini G. P., Gao X., Flicek P., Bradley A., Skarnes W. C., Justice M. J., Parkinson H. E., Moore M., Wells S., Braun R. E., Svenson K. L., de Angelis M. H., Herault Y., Mohun T., Mallon A.-M., Henkelman R. M., Brown S. D. M., Adams D. J., Lloyd K. C. K., Kerlie C. M., Beaudet A. L., Bućan M., Murray S. A., High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beluzic L., Grbeša I., Belužić R., Park J. H., Kong H. K., Kopjar N., Espadas G., Sabidó E., Lepur A., Rokić F., Jerić I., Brkljačić L., Vugrek O., Knock-down of AHCY and depletion of adenosine induces DNA damage and cell cycle arrest. Sci. Rep. 8, 14012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park S. J., Kong H. K., Kim Y. S., Lee Y. S., Park J. H., Inhibition of S-adenosylhomocysteine hydrolase decreases cell mobility and cell proliferation through cell cycle arrest. Am. J. Cancer Res. 5, 2127–2138 (2015). [PMC free article] [PubMed] [Google Scholar]
- 58.Li Q., Mao L., Wang R., Zhu L., Xue L., Overexpression of S-adenosylhomocysteine hydrolase (SAHH) in esophageal squamous cell carcinoma (ESCC) cell lines: Effects on apoptosis, migration and adhesion of cells. Mol. Biol. Rep. 41, 2409–2417 (2014). [DOI] [PubMed] [Google Scholar]
- 59.The ENCODE Project Consortium , Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ducker G. S., Rabinowitz J. D., One-carbon metabolism in health and disease. Cell Metab. 25, 27–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fustin J.-M., Doi M., Yamaguchi Y., Hida H., Nishimura S., Yoshida M., Isagawa T., Morioka M. S., Kakeya H., Manabe I., Okamura H., RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 155, 793–806 (2013). [DOI] [PubMed] [Google Scholar]
- 62.Fustin J. M., Kojima R., Itoh K., Chang H.-Y., Ye S., Zhuang B., Oji A., Gibo S., Narasimamurthy R., Virshup D., Kurosawa G., Doi M., Manabe I., Ishihama Y., Ikawa M., Okamura H., Two Ck1δ transcripts regulated by m6A methylation code for two antagonistic kinases in the control of the circadian clock. Proc. Natl. Acad. Sci. U.S.A. 115, 5980–5985 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Radomski N., Kaufmann C., Dreyer C., Nuclear accumulation of S-adenosylhomocysteine hydrolase in transcriptionally active cells during development of Xenopus laevis. Mol. Biol. Cell 10, 4283–4298 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee S., Doxey A. C., McConkey B. J., Moffatt B. A., Nuclear targeting of methyl-recycling enzymes in Arabidopsis thaliana is mediated by specific protein interactions. Mol. Plant 5, 231–248 (2012). [DOI] [PubMed] [Google Scholar]
- 65.Grbeša I., Kalo A., Belužić R., Kovačević L., Lepur A., Rokić F., Hochberg H., Kanter I., Simunović V., Muńoz-Torres P. M., Shav-Tal Y., Vugrek O., Mutations in S-adenosylhomocysteine hydrolase (AHCY) affect its nucleocytoplasmic distribution and capability to interact with S-adenosylhomocysteine hydrolase-like 1 protein. Eur. J. Cell Biol. 96, 579–590 (2017). [DOI] [PubMed] [Google Scholar]
- 66.Kloor D., Hermes M., Fink K., Schmid H., Klingel K., Mack A., Grenz A., Osswald H., Expression and localization of S-adenosylhomocysteine-hydrolase in the rat kidney following carbon monoxide induced hypoxia. Cell. Physiol. Biochem. 19, 57–66 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Schvartzman J. M., Thompson C. B., Finley L. W. S., Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol. 217, 2247–2259 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chang H.-C., Guarente L., SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 153, 1448–1460 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Solanas G., Peixoto F. O., Perdiguero E., Jardí M., Ruiz-Bonilla V., Datta D., Symeonidi A., Castellanos A., Welz P.-S., Caballero J. M., Sassone-Corsi P., Muñoz-Cánoves P., Benitah S. A., Aged stem cells reprogram their daily rhythmic functions to adapt to stress. Cell 170, 678–692 e20 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Sato S., Solanas G., Peixoto F. O., Bee L., Symeonidi A., Schmidt M. S., Brenner C., Masri S., Benitah S. A., Sassone-Corsi P., Circadian reprogramming in the liver identifies metabolic pathways of aging. Cell 170, 664–677.e11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kondratov R. V., Kondratova A. A., Gorbacheva V. Y., Vykhovanets O. V., Antoch M. P., Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 20, 1868–1873 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yamaguchi Y., Suzuki T., Mizoro Y., Kori H., Okada K., Chen Y., Fustin J.-M., Yamazaki F., Mizuguchi N., Zhang J., Dong X., Tsujimoto G., Okuno Y., Doi M., Okamura H., Mice genetically deficient in vasopressin V1a and V1b receptors are resistant to jet lag. Science 342, 85–90 (2013). [DOI] [PubMed] [Google Scholar]
- 73.Delvecchio M., Gaucher J., Aguilar-Gurrieri C., Ortega E., Panne D., Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nat. Struct. Mol. Biol. 20, 1040–1046 (2013). [DOI] [PubMed] [Google Scholar]
- 74.Hirayama J., Sahar S., Grimaldi B., Tamaru T., Takamatsu K., Nakahata Y., Sassone-Corsi P., CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature 450, 1086–1090 (2007). [DOI] [PubMed] [Google Scholar]
- 75.Olsen J. V., de Godoy L. M. F., Li G., Macek B., Mortensen P., Pesch R., Makarov A., Lange O., Horning S., Mann M., Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell Proteomics 4, 2010–2021 (2005). [DOI] [PubMed] [Google Scholar]
- 76.Ranjit S., Datta R., Dvornikov A., Gratton E., Multicomponent analysis of phasor plot in a single pixel to calculate changes of metabolic trajectory in biological systems. J. Phys. Chem. A 123, 9865–9873 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Ranjit S., Malacrida L., Jameson D. M., Gratton E., Fit-free analysis of fluorescence lifetime imaging data using the phasor approach. Nat. Protoc. 13, 1979–2004 (2018). [DOI] [PubMed] [Google Scholar]
- 78.Agostinelli F., Ceglia N., Shahbaba B., Sassone-Corsi P., Baldi P., What time is it? Deep learning approaches for circadian rhythms. Bioinformatics 32, 3051 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hughes M. E., Hogenesch J. B., Kornacker K., JTK_CYCLE: An efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. J. Biol. Rhythms 25, 372–380 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kayala M. A., Baldi P., Cyber-T web server: Differential analysis of high-throughput data. Nucleic Acids Res. 40, W553–W559 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huang D. W., Sherman B. T., Lempicki R. A., Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 82.Daily K., Patel V. R., Rigor P., Xie X., Baldi P., MotifMap: Integrative genome-wide maps of regulatory motif sites for model species. BMC Bioinformatics 12, 495 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ceglia N., Liu Y., Chen S., Agostinelli F., Eckel-Mahan K., Sassone-Corsi P., Baldi P., CircadiOmics: Circadian omic web portal. Nucleic Acids Res. 46, W157–W162 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ramirez F., Ryan D. P., Grüning B., Bhardwaj V., Kilpert F., Richter A. S., Heyne S., Dündar F., Manke T., deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabc5629/DC1