

Lysosomes, important centers of metabolic sensing and material recycling, play a key role in the dynamic reshaping of the ER.
Abstract
The endoplasmic reticulum (ER) comprises morphologically and functionally distinct domains: sheets and interconnected tubules. These domains undergo dynamic reshaping in response to changes in the cellular environment. However, the mechanisms behind this rapid remodeling are largely unknown. Here, we report that ER remodeling is actively driven by lysosomes, following lysosome repositioning in response to changes in nutritional status: The anchorage of lysosomes to ER growth tips is critical for ER tubule elongation and connection. We validate this causal link via the chemo- and optogenetically driven repositioning of lysosomes, which leads to both a redistribution of the ER tubules and a change of its global morphology. Therefore, lysosomes sense metabolic change in the cell and regulate ER tubule distribution accordingly. Dysfunction in this mechanism during axonal extension may lead to axonal growth defects. Our results demonstrate a critical role of lysosome-regulated ER dynamics and reshaping in nutrient responses and neuronal development.
INTRODUCTION
The structure of the endoplasmic reticulum (ER) is constantly adapted to the particular needs of the cell: The dynamic transitions between ER sheets and tubules allow it to rapidly respond to changes in the cellular environment. A group of ER-shaping proteins have been identified as maintaining ER morphology (1), mutations in which are linked to diseases such as hereditary spastic paraplegias (HSPs) (2). While these proteins affect global ER topology, they cannot explain the rapid and marked changes in ER morphology observable on time scales of minutes in live-cell imaging experiments (3, 4). Previous work has shown that ER tubule elongation can be driven by three mechanisms: (i) force generation by motors moving along microtubules, which can be classified as sliding (5, 6); (ii) coupling to microtubule growth using a tip assembly complex (7); and (iii) hitchhiking by connecting to other organelles (5, 8). Whether such reshaping in local domains of the ER tubules could lead to the global reorganization and redistribution of ER remains an open question, and if this is the case, how is this process regulated? The ER is known to contact other motile organelles, including endosomes, lysosomes, mitochondria, and peroxisomes (9). Among these, lysosomes are particularly interesting, as they make a great number of contacts with the ER (10) and their positioning is regulated by the cell’s nutritional status (11). Although the ER has been reported to regulate lysosome motions (12), it is not clear whether lysosomes can modulate ER reshaping and distribution, for example, via coupled motion (4). We hypothesized that a causal link exists between lysosome motion and ER redistribution and asked whether this provides a mechanism for the ER’s morphological response to nutritional status, given that lysosomes are known to act as signaling hubs for metabolic sensing (13).
RESULTS
The lysosome-tethered ER network undergoes dynamic morphological changes
We first investigated the correlation between motions of lysosomes and the ER network by rapid live-cell imaging. We visualized ER with enhanced green fluorescent protein (EGFP)–tagged vesicle-associated membrane protein–associated protein A (VAPA) and lysosomes with cathepsin D–specific SiR-Lysosome dye in COS-7 cells (Fig. 1A, i). EGFP-VAPA was found to colocalize strongly with a commonly used ER marker, Sec61β, indicating the high ER specificity of VAPA (fig. S1). We further verified that expression of EGFP-VAPA did not alter the ER tubule percentage when compared with mApple-Sec61β–only expressing control cells (fig. S1). Single-particle tracking (SPT) of the lysosomes revealed a network of motion tracks resembling the network characteristic of the ER (Fig. 1A, ii, and movie S1), featuring path segments, along which lysosomal bidirectional motions take place, and regional nodes, where path segments intersect (Fig. 1A, iii). Compared with lysosomes, early endosomes (labeled with Rab5-GFP) appeared notably more static in COS-7 cells and were substantially less associated with the EGFP-VAPA–labeled ER network (fig. S2 and movie S2). Tracking lysosomes and the ER network simultaneously using dual-color SPT showed that almost all lysosomes (98%) moved synchronously with local ER domains. This was particularly prominent near elongating tubule tips of the ER (Fig. 1B and movie S3). Almost all lysosome-associated tips (99.4%, 174 of 175 in 39 cells) eventually formed new connections with the existing ER network, creating three-way junctions (yellow stars in Fig. 1B). In contrast, ER tips without associated lysosomes failed to sustain their elongation (white arrows) and eventually retracted (yellow arrows) (Fig. 1C and movie S4). These ER tubules normally extended only to about 1 μm, and more than half of them (52.5%, 160 of 305 events in 39 cells) collapsed completely following a retractile phase. Over 90-s periods, ER tubules associated with lysosomes grew significantly longer than their non-associated counterparts (average, 6.72 ± 0.269 μm versus 2.23 ± 0.084 μm; Fig. 1D). In addition, the elongation phase of lysosome-coupled ER tubules persisted longer compared to the lysosome-free cases (9.0 s versus 3.5 s; Fig. 1E). It has previously been reported that 35% of ER tubulation events occur via hitchhiking on late endosomes/lysosomes (5), which is consistent with our observation (31%). However, further quantification scaled by tubule length rather than event number showed that 73% of newly generated tubule length was contributed by lysosome-coupled elongation (Fig. 1F). Together, these findings suggest that lysosomes play an important role in ER growth tip elongation and the reshaping of local domains within the ER network.
Fig. 1. Dynamic ER tubules are coupled to motile lysosomes.
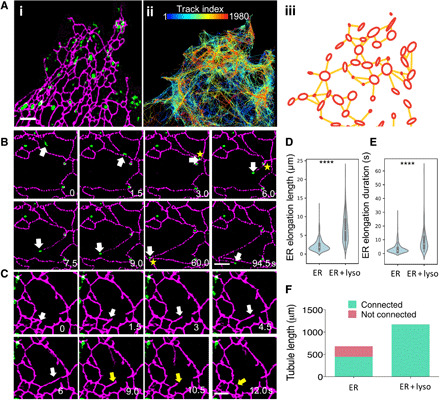
(A) (i) SIM images of a COS-7 cell with EGFP-VAPA–labeled ER (magenta) and SiR dye-labeled lysosomes (green). (ii) SPT of lysosome motion over 12.5 min at 1.5 s per frame, color-coded by track index. (iii) Track density map extracted from SPT data (see movie S1). (B) Coupled motion (white arrows) of the growing tip of a newly formed ER tubule and associated lysosome. The tip forms three-way junctions (yellow stars). See movie S3. (C) An ER tubule, without an associated lysosome, is unstable and retracts (yellow arrows) after a period of elongation (white arrows). See movie S4. (D) Violin plot of ER tubule growth, in a 90-s interval, for growing tips with or without lysosomes attached. (E) Violin plot of maximum duration of the elongation phases of newly formed lysosome-coupled or lysosome-free ER tubules. (F) Sum of the total tubule growth lengths for lysosome-coupled or lysosome-free growing ER tips. Light green, tubules making network connections; red, tubules failing to connect. ****P < 0.0001, Student’s t test in (D) and (E). Scale bars, 2 μm (A to C). For (D) to (F), N = 481 events in 39 cells; see table S1.
ER network distribution and morphology are casually linked to rapid lysosome positioning
VAPA has been identified as the main ER-lysosome anchor protein (14), and we therefore reasoned that abolishing VAPA-mediated anchoring would lead to compromised ER remodeling. To investigate this, we used the K94D/M96D (KD/MD) mutant of VAPA, which is unable to bind the FFAT motif of lysosomal membrane proteins and thus compromises the anchoring of the protein to lysosomes based on a previous study (15). In COS-7 cells overexpressing this protein, we observed frequent detachment of lysosomes from their coupled ER tips during elongation (Fig. 2A and movie S5). While the lysosome was seen to continue along its trajectory, the ER tubule retracted upon detachment. Upon breakage of the link, lysosome speed increased significantly (Fig. 2B), but reduced again once the lysosome reattached to other ER structures, suggesting a resistive force associated with ER growth tip elongation. Furthermore, the depletion of VAPA via small interfering RNA (siRNA) resulted in discontinuity of ER tubules and a reduced tubule percentage (fig. S3), which is consistent with the effects of overexpression of dominant-negative mutant VAPA (KD/MD) (fig. S4). This supports the notion that VAPA-dependent lysosome-ER contacts are important in ER reshaping.
Fig. 2. Remodeling of peripheral ER networks requires contacts with motile lysosomes.
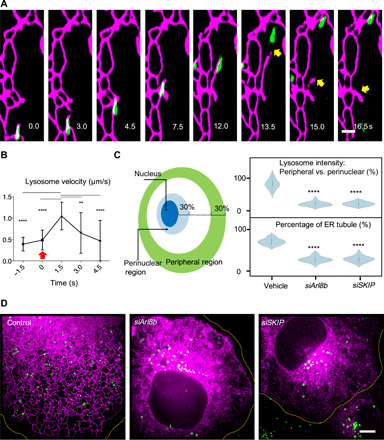
(A) Time-lapse images showing breakage of the connection between the growing tip of a newly formed ER tubule and a lysosome in EGFP-VAPA (KD/MD)–expressing cells. The ER is visualized via EGFP-VAPA (KD/MD), shown in magenta, and lysosomes (green color) are labeled by SiR-Lysosome. See table S2 and movie S5. (B) Average velocities of initially ER-tethered lysosomes that become detached (red arrow) from associated growing ER tips. An example of detachment event is shown in (A). N = 22 events, see table S3. (C) Left: Diagram depicting the regions defined as perinuclear and peripheral regions for the following quantification of lysosome distribution. Right: Violin plots of SiR-Lysosome fluorescent intensity in peripheral versus perinuclear region and percentage of the ER comprising tubules upon knockdown of lysosome motion adaptors. N = 20 cells, see table S4. (D) Representative images showing the distribution of lysosomes and ER tubules in untreated cells and in siRNA-treated cells. The ER is visualized via EGFP-VAPA, shown in magenta, and lysosomes (green color) are labeled by SiR-Lysosome. **P < 0.01 and ****P < 0.0001, Tukey’s one-way analysis of variance (ANOVA) in (B) and (C). Scale bars, 1 μm (A) and 5 μm (D).
Next, we ask whether ER distribution is affected by lysosome positioning. Lysosome movement is highly regulated by machineries such as adenosine diphosphate (ADP) ribosylation factor–like guanosine triphosphatase (GTPase) 8B (Arl8b) and SifA-kinesin interacting protein (SKIP), which are both key proteins that specifically recruit lysosomes to kinesins for anterograde motion toward the cell periphery (16). We have shown that moving lysosomes are tightly coupled with ER growing tips, pulling ER tubules to move. We therefore hypothesized that compromised anterograde motions of lysosomes can result in the reduction of ER tubules in peripheral regions of the cell. Depletion of Arl8b and SKIP (validated in fig. S5), which led to perinuclear clustering of lysosomes (Fig. 2, C and D), strongly reduced the ER tubular domains (Fig. 2, C and D).
To further validate the effect of lysosome positioning on ER distribution, we used a second approach, the iDimerize Inducible Heterodimer System (17), to examine whether the retrograde motion of lysosomes can lead to a reduction of the peripheral distribution of the ER network (Fig. 3A). Administration of the inducer led to recruitment of lysosomes to BicD2, which, in turn, binds to the dynein complex, thus enabling retrograde transport (Fig. 3A). One hour of induction resulted in a significant reduction of lysosome intensity in the peripheral region of the cell, and at the same time, the percentage of ER tubule content within the whole network dropped significantly (Fig. 3, B and C). We proved in a separate experiment that ER stress alone does not lead to the observed ER morphology changes by administering the ER stress inducer thapsigargin (fig. S6).
Fig. 3. Manipulation of lysosome positionings drives the redistribution of ER via reshaping.
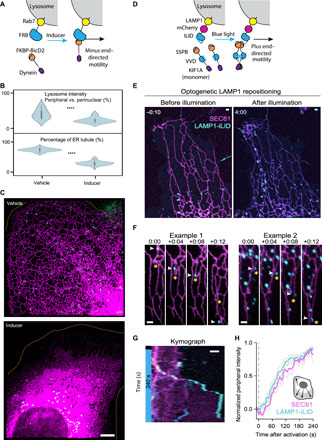
(A) Molecular components of the chemogenetic system. (B) Violin plots of SiR-Lysosome fluorescent intensity and percentage of the ER comprising tubules after 1 hour of vehicle or inducer treatment. N = 10 cells (lysosome intensity) and 20 cells (ER tubule). See table S5. ****P < 0.0001, Student’s t test. (C) Representative images showing the distribution of lysosomes and ER tubules in control and induced cells. The ER is visualized via EGFP-VAPA, shown in magenta, and lysosomes (green color) are labeled by SiR-Lysosome. (D) Molecular components of the optogenetic system for repositioning LAMP1-associated lysosomes. (E to H) Live-cell imaging (E), representative zoom-ins (F), representative kymograph (G), and quantification (H) of LAMP1-mCherry-iLID and YFP-Sec61β in COS-7 cells expressing opto-kinesin before or during activation. White arrows indicate lysosomes pulling ER tubules (yellow asterisks). Quantification shows mean (±SEM) normalized peripheral Sec61β and LAMP1 intensity of eight cells. Blue box indicates illumination with blue light. Scale bars, 5 μm (C) and 1 μm (E to G).
To further validate the causal link between lysosome repositioning and ER redistribution and to investigate the efficiency and speed of this regulatory mechanism, we used light-induced heterodimerization to recruit kinesin to LAMP1-labeled lysosomes in COS-7 cells (Fig. 3D) (18). To this purpose, these cells coexpressed yellow fluorescent protein (YFP)–ER, KIF1A-VVDfast, an anterograde motor protein, and LAMP1-mCherry-iLID, engineered LAMP1 with light-sensitive protein iLID. Upon illumination with blue light, lysosomes were rapidly redistributed by the recruited kinesin from the perinuclear region to the periphery of the cell (Fig. 3E; see movie S6 for raw imaging and reconstructed data). Simultaneously, we observed a burst of ER tubule extension toward the periphery in regions where lysosomes were abundant (Fig. 3E). The rapid elongation of ER tubules driven by lysosomes also led to the formation of new network connections (Fig. 3, E and F). The kymograph and quantification showed a marked and rapid increase in ER fluorescence intensity in the cell periphery following blue light illumination, and the growth of ER intensity strictly followed that of lysosome intensity (Fig. 3, G and H). The observation of ER tubules growing predominantly in lysosome-enriched regions supports the notion of lysosome-controlled redistribution of the ER network.
ER morphology is actively regulated by lysosomes in response to nutritional status
As lysosomes are the main organelles to sense metabolic stimuli, we asked whether this lysosome-guided ER motion is responsive to changing intracellular conditions. We reasoned that intracellular cues that are known to regulate lysosome positioning (Fig. 4A) would result in a redistribution of the ER and a transition between tubular and sheet domain. First, we starved COS-7 cells in serum-free culture for 4 hours to induce autophagy, which resulted in the accumulation of lysosomes in perinuclear regions (Fig. 4B). Lysosome movement toward the perinuclear region reduced ER tubules in the cell periphery and resulted in a concomitant increase of sheet content in central regions of the cell. In contrast, prolonged serum starvation (24 hours) inhibits autophagy and promotes recycling of lysosomes (19), causing their redistribution throughout the whole cytosol. Under this condition, we observed the restoration of the ER tubular network from a previously sheet-dominated state (Fig. 4B). As an example, a marked reshaping of the ER is seen to take place in a protrusion forming at a peripheral region of one cell (Fig. 4B, white box), which is commonly observed under this condition. Motion tracking of lysosomes verified a dominant (>90% among the whole tracks) anterograde motion toward the boxed region (fig. S7 and movie S7). However, in the cells treated with siRNA to deplete Arl8b, which led to compromised anterograde motions of lysosomes, ER was not recovered to its prestimulus state upon prolonged starvation (Fig. 4B). Consistent results were observed in siRNA depletion of SKIP. This provides further evidence that lysosomes’ anterograde motion is essential for the ER tubule extension observed above. Quantitative analysis by measuring lysosome intensity and ER tubule percentage showed that accumulation of lysosomes in the cell center can lead to a significant reduction of ER tubules in peripheral regions (defined as the regions beyond 70% of the cell radius). Furthermore, the extension of ER tubules accompanies the repositioning of lysosomes in the cell periphery (Fig. 4C) and is dependent on Arl8b and SKIP. In addition, metabolic changes can alter ER structure via a selective form of autophagy (known as ER-phagy), which can degrade ER upon nutrient stress (20). To verify whether the reduced ER tubular domain in short-term nutrient starvation might be caused by ER-phagy, we treated the cells with bafilomycin A1 (Baf), which inhibits the fusion of autophagosomes with lysosomes during nutrient starvation. There was no observable difference between Baf-treated samples and the Baf-untreated control (fig. S8), both of which featured a significant reduction in ER tubular content compared with the unstarved control (fig. S8). This confirmed that the decreased ratio between ER tubules and sheet content is not caused by ER-phagy.
Fig. 4. Lysosome-mediated ER reshaping is responsive to nutritional status.
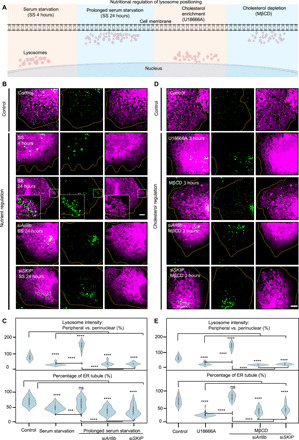
(A) Schematic representation of the nutritional regulation of lysosome positioning. (B) Representative SIM images showing the distribution of lysosomes (green) and ER (magenta) upon disruption of metabolic pathways, same for (D). From top to bottom: no treatment control; 4-hour serum starvation (SS; same in the following text); 24-hour serum starvation (prolonged starvation), see movie S7; siRNA against Arl8b or SKIP in prolonged starvation. (C) Violin plots of SiR-Lysosome fluorescent intensity and percentage of the ER comprising tubules with indicated treatments. (D) From top to bottom: 3-hour U18666A (10 μM) treatment to block lysosome-to-ER cholesterol transfer; 3-hour MβCD (100 μM) treatment to perturb cholesterol sensing; siRNA against Arl8b or SKIP in 3-hour MβCD (100 μM) treatment. (E) Violin plots of SiR-Lysosome fluorescent intensity and percentage of the ER comprising tubules with indicated treatments. ****P < 0.0001, Tukey’s one-way ANOVA. n ≥ 20 cells per condition from three independent experiments, same for (C). See table S6. Scale bars, 5 μm. ns, not significant.
Lysosomes are known sensors of intracellular cholesterol levels, and their positioning is affected by prevailing cholesterol levels (21). To examine the effects of intracellular cholesterol levels in the context of ER reshaping, we first incubated cells with U18666A, a compound that prevents cholesterol transfer from lysosomes to the ER and leads to lysosome accumulation in perinuclear regions (22), which is what we observed upon 3 hours of treatment (Fig. 4D). In cells with perinuclear lysosome positioning, the ER also retracted toward the cell center and showed reduced tubular content and a prevalence of sheet structure. A recent study demonstrated that long-term cholesterol accumulation in lysosomes via administration of U18666A (18 hours) reduced the number of ER-lysosome contact sites (23). This observation from such a long-term metabolic perturbation could be evidence of a late-onset phenotype of cholesterol accumulation. In our observations, however, contact sites between the ER and lysosomes appear to remain intact for at least 3 hours upon U18666A administration. The observed ER retraction may thus be an early-onset phenotype upon cholesterol accumulation.
In contrast to the above result, depletion of cholesterol in COS-7 cells, via administration of methyl-β-cyclodextrin (MβCD) for 3 hours, prompted a marked redistribution of lysosomes toward the cell periphery and extension of ER tubules into the cell periphery (Fig. 4D). However, cells depleted of Arl8b and SKIP failed to extend their ER tubule network in peripheral regions under 3 hours of treatment with MβCD, which correlated with lysosomes remaining accumulated in the cell centers (Fig. 4D). Quantitative analysis by measuring lysosome intensity and ER tubule percentage showed that the distribution of ER tubular domain is contingent on lysosome positioning, which is, in turn, dependent on Arl8b and SKIP for anterograde motions (Fig. 4E). Further examination by administration of thapsigargin to COS-7 cells showed that the tubule percentage in the global network is relatively consistent with the vehicle control (fig. S6). This suggests that the reshaping of ER tubule in Fig. 4 (B and D) is not due to ER stress that may be induced by the metabolic manipulations. In summary, these findings support the notion that lysosome-driven ER reshaping, which leads to the transition between tubular and sheet domains, is responsive to different metabolic activities.
Lysosome-driven ER elongation promotes axon extension
The observed lysosome-regulated ER redistributions notably affect ER morphology in COS-7 cells, such as the distribution and organization of tubular network and consequently the ratio between tubular and sheet domains. ER-lysosome contacts are also commonly observed in neurons (24), in which the ER displays continuous tubular structure in axons (25). Whether lysosomes can still drive ER tubule to move and thus maintain ER continuity is not clear, and if so, the question arises whether this affects axonal growth. First, to validate the prevalence of lysosome-driven ER motion in a highly polarized system, we studied cultured axons of Xenopus laevis retinal ganglion cell (RGC) neurons, which are a well-established model to study axonal growth (Fig. 5A). We first characterized the ER specificity of fluorescently tagged VAPA and its effects on axonal ER morphology. In EGFP-VAPA–expressing axons incubated with an ER-Tracker dye, all VAPA-labeled structures were ER-Tracker positive (Manders’ coefficient: 0.9296; fig. S9). Live imaging of ER in RGCs with EGFP-VAPA and of lysosomes with LysoTracker showed that 85% (184 of 217) of lysosomes were coupled with the ER. Consistent with our observation in COS-7 cells, lysosomal pulling of the ER was frequently observed along axons. For instance, a lysosome (green) was tethered inside a ring-like structure at an ER tubule free end, pulling it along the axon until it reconnected and fused with another ER segment to generate a continuous ER tubule (Fig. 5B and movie S8). The lysosome was seen to maintain its anterograde motion after successful ER reconnection. In contrast, in RGCs expressing EGFP-VAPA (KD/MD), ER-lysosome connections were disrupted, and the ER failed to stably form a continuous structure (Fig. 5C and movie S9). Quantification shows a significant reduction in the reconnection rate of separate ER tubules in EGFP-VAPA (KD/MD)–expressing axons during 1.5-min imaging sessions (Fig. 5D). ER tubules in the mutant-expressing axons were, on average, 50% shorter than those in the wild-type VAPA-expressing axons (Fig. 5E). Concurrently, the mutant axons were also significantly shorter than the wild-type axons (Fig. 5, F and G) and displayed severe growth defects, with fewer axons exiting from each retinal explant (Fig. 5H). In axons expressing either construct and containing discontinued ER tubules, individual axon lengths were found to be positively correlated with the average ER fragment lengths observed throughout the entire length of the axon (Fig. 5G). Collectively, these observations suggest that the lysosome-driven ER tubule elongation is a frequent event in axons, which enables the efficient reconnection of interrupted ER tubules and supports axonal growth. In contrast, disruption of ER-lysosome contacts leads to ER fragmentation and axon growth defects.
Fig. 5. Disruption of ER-lysosome contacts impairs ER continuity in growing RGC axons.
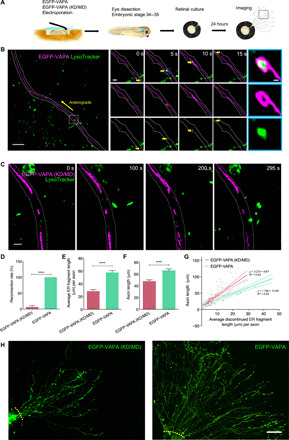
(A) Experimental protocol for Xenopus eye-targeted electroporation, RGC culture, and imaging. (B) Cotransport of a lysosome (green) and an ER tubule tip (magenta) in a representative EGFP-VAPA–expressing axon segment, see movie S8. (C) A representative EGFP-VAPA (KD/MD)–expressing axon fails to join ends of separate ER tubules, see movie S9. (D) Percentage of separate axonal ER tubules successfully connected within 90 s. n = 113 pairs of separate ER tubules. See table S7. (E) Average ER fragment lengths and (F) average axon lengths in EGFP-VAPA (KD/MD)–expressing (n = 152) and EGFP-VAPA–expressing (n = 118) axons. See table S7. (G) Correlation between axon and ER fragment lengths in indicated axons with discontinued ER only. Linear regression analyses are shown by solid lines, and 95% confidence intervals fall between dashed lines. (H) Representative images of cultured EGFP-VAPA (KD/MD)–expressing and EGFP-VAPA–expressing axons from single explants. Yellow dashed lines indicate where axons exit the eye (starting points of length quantification). Mean ± SEM. ****P < 0.0001, Student’s t test. Scale bars, 5 μm (B, left), 1 μm (B, middle), 200 nm (B, right), 1 μm (C), and 20 μm (H).
DISCUSSION
Recent advances in live-cell imaging techniques have revealed new details of the highly dynamic nature of ER structure and its regulation (3–5, 26), posing questions regarding the physiological relevance of this phenomenon. The ER is the main platform for protein and membrane synthesis and quality control of proteins. Lysosomes, on the other hand, are major sensing and recycling centers and are actively relocalized in response to changes in the levels of cholesterol, nutrients, etc. (10, 27). Recent studies have revealed that lysosomes can also be associated with mRNA transport (28) and act as platforms for protein synthesis (29). In this work, we show that movement of ER tubules is actively driven by motile lysosomes, which is facilitated by lysosome-ER anchor protein VAPA. Compromising lysosome-ER contact sites by expressing the VAPA (KD/MD) dominant mutant resulted in discontinuities of ER tubules (Figs. 2A and 5B). VAPA (KD/MD) proteins localized throughout the ER labeled by Sec61β (fig. S4), confirming that the observed ER fragmentation was not due to an altered distribution of VAPA (KD/MD) (Figs. 2A and 5B). Overall, this, in turn, provides a mechanism for the rapid ER network remodeling required to adapt efficiently to local metabolic changes. It has been shown that membrane contact sites (MCSs) enable the ER to promote endosome translocation to the cell periphery (12), but it is not known whether late endosomes or lysosomes can also regulate the global distribution of ER. Our data lead us to propose an independent mechanism of regulation of ER morphology by lysosome positioning, which is supported by previously reported observations. For example, protrudin, a key anchor protein supporting ER-lysosome contacts, is required to maintain ER sheet-to-tubule balance and the distribution of ER tubules (30). Later studies showed that the anterograde motion of late endosomes promotes protrusion and neurite outgrowth (12), which is consistent with our observation of cellular protrusion occurring when lysosomes localized to peripheral regions during prolonged periods of serum starvation (Fig. 4B). This suggests a link between lysosome positioning and ER distribution and neurite outgrowth. We further show that the concomitant reorganization of the ER network can result from chemical-, light-, and metabolic stimulus–induced lysosome relocation. The dynamic rearrangement of ER tubules is strongly dependent on the motile lysosomes in local domains. These contacts modulate the global ER morphology when lysosomes are regulated to move either retro-/anterogradely at the population level, such as upon cholesterol enrichment and nutrient starvation. Therefore, our results imply that lysosomes sense nutritional changes and regulate ER dynamics and morphology in response. How ER morphological changes contribute to the maintenance of cellular homeostasis will be an important question for future study.
Although both early endosomes and late endosomes/lysosomes form MCSs (31), the number of MCSs increases markedly as endosome matures from early to late stages (32). This is consistent with our observations (fig. S2). Furthermore, lysosomes are more motile organelles than early endosomes (fig. S2). These characteristics enable lysosomes to be more efficient than early endosomes in building strong connections with the ER and in regulating ER topology dynamically. Furthermore, lysosomes are central for metabolic sensing, providing for an efficient response for the ER to adapt to cellular changes. There are a number of organelles other than lysosomes forming abundant MCSs with ER; whether and how such organelles, e.g., mitochondria, regulate ER morphology is an interesting topic for future investigation.
Lysosome-driven ER redistributions not only are important in ER reshaping but also facilitate the maintenance and continuity of ER domains in developing axons. A recent study has shown that defects in ER-endosome contacts are associated with HSPs (33). In developing RGCs, expression of KD/MD mutants of VAPA, which are incapable of binding to lysosomes, led to persistent ER fragmentation and axonal growth defects, suggesting an important role of lysosomes in maintaining ER continuity during neuronal development. Therefore, it would be valuable to investigate the role of lysosome-ER interactions in disorders that have in the past been associated with ER network defects (2, 34).
MATERIALS AND METHODS
Cell culture
Key materials in this study are listed in Tables 1 to 6. COS-7 cells were purchased from the American Type Culture Collection (ATCC). COS-7 cells were grown in T75 or T25 flasks or six-well plates by incubation at 37°C in a 5% CO2 atmosphere. Complete medium for normal cell growth consisted of 90% Dulbecco’s modified Eagle’s medium (DMEM), 10% fetal bovine serum (FBS), and 1% streptomycin. Cells were kept in logarithmic phase growth and passaged on reaching 70 to 80% confluence (approximately every 3 to 4 days). Medium was changed every 2 or 3 days. For structured illumination microscopy (SIM) imaging experiments, COS-7 cells were plated onto Nunc Lab-Tek II Chambered Coverglass (Thermo Fisher Scientific, 12-565-335) to achieve ∼70% confluence before transfection.
Table 1. Plasmids.
| pEGFPC1-hVAP-A | Gift from C. Tomasetto |
Addgene plasmid #104447 |
| pEGFPC1-hVAP-A KD/MD | Gift from M. Davidson | Addgene plasmid #104449 |
| mEmerald-Rab5a-7 | Gift from M. Davidson | Addgene plasmid #54243 |
| EGFP-Rab7A | Gift from Q. Zhong | Addgene plasmid #28047 |
| mEmerald-Sec61β-C1 | Gift from J. Lippincott-Schwartz |
Addgene plasmid #90992 |
| mApple-Sec61β-C1 | Gift from J. Lippincott-Schwartz |
Addgene plasmid #90993 |
| ER-GFP | CellLight ER-GFP, BacMam 2.0 |
Cat#C10590 |
| Lysosomes-GFP | CellLight Lysosomes- GFP, BacMam 2.0 |
Cat#C10507 |
| Early endosome-GFP | CellLight Early Endosome-GFP, BacMam 2.0 |
Cat#C10586 |
| pB80-KIF1A(1-365)- VVD fast-FLAG- SSPB(micro) |
Gift from L.K. | (15) |
| pB80-LAMP1-mCherry- iLID |
Gift from L.K. | (15) |
| β-actin YFP-Sec61 | Gift from L.K. | (15) |
| pBa-flag-BicD2 594-FKBP |
Gift from G. Banker | Addgene plasmid #64206 |
Table 6. Software.
| Fiji | Fiji | https://imagej.net/Fiji |
| MATLAB 2016a | MathWorks | https://uk.mathworks.com/products/matlab.html |
| GraphPad Prism | GraphPad Software | https://graphpad.com/scientific-software/prism/ |
Table 2. Antibodies and chemicals.
| SiR-Lysosome | Cytoskeleton Inc. | Cat#CY-SC012 |
| Bafilomycin A1 | Sigma-Aldrich | Cat#19-148 |
| Nocodazole | Sigma-Aldrich | Cat#M1404-10MG |
| Dulbecco’s modified Eagle’s medium | Sigma-Aldrich | Cat#D6546 |
| A/C heterodimerizer | Takara | Cat#635056 |
| Fetal bovine serum | Sigma-Aldrich | Cat#10270-106 |
| Radioimmunoprecipitation assay buffer | Sigma-Aldrich | Cat#R0278 |
| Opti-MEM I Reduced Serum Medium | Invitrogen | Cat#31985062 |
| Anti-VAPA | Abcam | Cat#ab181067 |
| Anti-GAPDH | Sigma-Aldrich | Cat#G8795 |
| Anti-tubulin | Abcam | Cat#ab131205 |
| Anti-PLEKHME (SKIP) | Abcam | Cat#ab91581 |
| Anti-Arl8b | Proteintech | Cat#13049-1-AP |
| Methyl-β-cyclodextrin | Sigma-Aldrich | Cat#332615 |
| U18666A | Sigma-Aldrich | Cat#U3633 |
Table 3. Critical commercial assays.
| QIAprep Spin Miniprep Kit |
QIAGEN | Cat#27104 |
| Lipofectamine 2000 Transfection Reagent |
Invitrogen | Cat#11668030 |
| Lipofectamine RNAiMAX Transfection Reagent |
Invitrogen | Cat#13778030 |
| SuperSignal West Pico PLUS Chemiluminescent Substrate |
Thermo Fisher Scientific |
Cat#34580 |
Table 4. Experimental models.
|
Cercopithecus aethiops: COS-7 |
ATCC | Cat#CRL-1651 |
| Xenopus | Gurdon Institute, Cambridge |
Not available |
Table 5. Oligonucleotides.
| siVAPA | SMARTpool: ON-TARGETplus | Cat#L-021382-00-0005 |
| MISSION siRNA Universal negative control | Sigma-Aldrich | Cat#SIC001 |
| siPLEKHM2 (SKIP) | SMARTpool: ON-TARGETplus | Cat#L-022168-01-0005 |
| siArl8b | SMARTpool: ON-TARGETplus | Cat#L-020294-01-0005 |
COS-7 cells were transfected with plasmid constructs as indicated with Lipofectamine 2000 according to the manufacturer’s protocol 24 to 48 hours before imaging. Cells were stained with SiR-Lysosome at 1 μM 4 hours before imaging. Cells were imaged in a microscope stage top micro-incubator (OKO Lab) with continuous air supply (37°C and 5% CO2). Cells were treated with MβCD at 100 μM for appropriated time window as indicated. Cells were treated with U18666A at 10 μM for appropriated time window as indicated.
siRNA transfection and Western blot
VAPA, SKIP, and Arl8b were depleted using SMARTpool: ON-TARGETplus, Dharmacon. Negative siRNA control (MISSION siRNA Universal negative control) was purchased from Sigma-Aldrich. COS-7 cells were plated in both glass-bottom petri dishes (for imaging) and a six-well plate (for Western blot validation). Cells were transfected with 20 nM siRNA oligonucleotides and 20 nM negative control siRNA using Lipofectamine RNAiMax (Thermo Fisher Scientific) according to the manufacturer’s protocol. After 6 hours of siRNA transfection, the cells were washed and the medium was replaced with complete culture medium. Twenty-four hours after the siRNA transfection, cells were transfected with plasmid DNA indicated in Results using Lipofectamine 2000 (Invitrogen). On the day of imaging, cells were stained with Sir-Lysosome. Cells in glass petri dishes were imaged 24 hours after DNA transfection.
Cells in six-well plates were harvested for Western blot validation 72 hours after siRNA transfection. Protein concentration was measured using a bicinchoninic acid (BCA) protein assay kit. Immunoblotting was performed by standard SDS–polyacrylamide gel electrophoresis/Western protocols. Primary antibody concentrations were as follows: VAPA at 1:20,000; SKIP at 1:5000; Arl8b at 1:5000; GAPDH (glyceraldehyde-3-phosphate dehydrogenase) at 1:30,000; tubulin at 1:5000. Secondary antibodies (Sigma-Aldrich) were used at 1:3000 for all rabbit antibodies and for all mouse antibodies. Signal was detected with SuperSignal West Pico Chemiluminescent Substrate.
Structured illumination microscopy
SIM imaging was performed using a custom three-color system built around an Olympus IX71 microscope stage, which we have previously described (35). Laser wavelengths of 488 nm (iBEAM-SMART-488, Toptica), 561 nm (OBIS 561, Coherent), and 640 nm (MLD 640, Cobolt) were used to excite fluorescence in the samples. The laser beam was expanded to fill the display of a ferroelectric binary Spatial Light Modulator (SLM) (SXGA-3DM, Forth Dimension Displays) to pattern the light with a grating structure. The polarization of the light was controlled with a Pockels cell (M350-80-01, Conoptics). A 60×/1.2 numerical aperture (NA) water immersion lens (UPLSAPO 60XW, Olympus) focused the structured illumination pattern onto the sample. This lens also captured the samples’ fluorescent emission light before imaging onto an sCMOS camera (C11440, Hamamatsu). The maximum laser intensity at the sample was 20 W/cm2. Raw images were acquired with the HCImage software (Hamamatsu) to record image data to disk and a custom LabView program (freely available upon request) to synchronize the acquisition hardware. Multicolor images were registered by characterizing channel displacement using a matrix generated with TetraSpeck beads (Life Technologies) imaged in the same experiment as the cells. COS-7 cells expressing EGFP-VAPA (ER marker) and stained with SiR-Lysosome (lysosome marker) were imaged by SIM every 1.5 s (including imaging exposure time of both channels) for 1.5 min.
Reconstruction of the SIM images with LAG SIM
Resolution-enhanced images were reconstructed from the raw SIM data with LAG SIM, a custom plugin for Fiji/ImageJ available in the Fiji Updater. LAG SIM provides an interface to the Java functions provided by fairSIM (36). LAG SIM allows users of our custom microscope to quickly iterate through various algorithm input parameters to reproduce SIM images with minimal artifacts; integration with Squirrel (37) provides numerical assessment of such reconstruction artifacts. Furthermore, once appropriate reconstruction parameters have been calculated, LAG SIM provides batch reconstruction of data so that a folder of multicolor, multiframe SIM data can be reconstructed overnight with no user input.
Image analysis algorithms
Further analysis was performed using custom MATLAB algorithms. The displacement of ER network endpoints, endosomes, and lysosomes was assessed over time with a tracking algorithm. All analysis tools are available from the authors upon request.
Skeletonization of ER tubules
Skeletonization of images was performed using Fiji [National Institutes of Health (NIH)]. Images were manually thresholded to reduce the background noise and increase the contrast of ER network. The preprocessed images were imported to Trainable Weka Segmentation (38), Fiji, to identify tubules. The trained images were then made binary and skeletonized by AnalyzeSkeleton (39) plugin in Fiji.
Growing tip and three-way junction analysis
Growing tips and three-way junctions were determined from customer-built algorism. The input to the program is a skeletonized binary image of ER. The skeleton was created in ImageJ by applying a threshold to create a binary image, followed by the built-in “skeletonize” function to reduce the width of ER tubules to 1 pixel. This results in a network graph of the ER, with connections, nodes, and endpoints. These three features were labeled using MATLAB’s connected component functions. The length of connections was also calculated by fitting splines to the binary tubules and recording the length of the spline. The analysis occurs across a time series of the ER network, allowing results to be drawn about the distribution of tubule length over time, for example. The program ends by saving the labeled image stack as a series of PNG files, as well as several csv files containing statistics describing the network over time.
ER tubule elongation analysis
ER tubule elongation was counted if the de novo ER tubule grew out from the existing ER network. Efficient elongation events are defined as the elongation of tubules over 1 μm and connected with other tubules for over 1 s. Tubular ER generation events were counted if the de novo ER tubules branched from the existing ER and extended more than 1 μm. The tubule elongation analysis (length, duration, efficiency, and proportions) were quantified from 175 events of lysosome-coupled ER motions and 306 events of ER-only motions in 39 cells from four independent experiments.
ER sheet versus tubule ratio
The reconstructed images were imported to Trainable Weka Segmentation (38), Fiji, for segmentation of tubule and sheet domains. Channels containing tubules and sheets were then converted to RGB color format. Channels were then split and converted to Mask and measured for the pixel area. For metabolite assay of ER morphology change, 20 images from three independent experiments were analyzed for each condition. For siRNA assay of ER morphology change, 20 images from three independent experiments were analyzed for each condition.
Lysosome velocity
To examine the driving force in ER-lysosome coupled motions, we transfected COS-7 cells with pEGFPC1-hVAP-A KD/MD and measured the velocity change of lysosomes before and after detachment from the coupled ER tips of 23 events from 11 cells from three independent experiments. We measured the displacement of lysosomes between two frames and divided this with time to obtain the lysosome velocity. Twenty-three events from three independent experiments were analyzed.
Single-particle tracking
COS-7 cells expressing EGFP-VAPA (ER marker) and stained with SiR-Lysosome (lysosome marker) were imaged by SIM every 1.5 s (including imaging exposure time of SiR-Lysosome channel) for 12.5 min (500 frames). EGFP-VAPA and SiR-Lysosome channels were imaged before and after the recording of lysosome motions, which recorded the morphology of ER.
Tracking of lysosome motions was performed by TrackMate (40) in Fiji, which plots the trajectories (2094 tracks) of lysosome motions in 500 frames. Data from SPT allowed us to reconstruct the lysosome motion density. The high-density regions were extracted from the trajectories using a procedure derived from (37): The number of points falling into square bins of width ∆x = 480 nm was counted to construct the density map. From this map, high-density regions, called seeds, were defined as the fraction with 5% higher-density bins. If multiple seeds appeared within a distance of two squares of each other, then only the one with the highest value was kept. For each seed bin, we computed a new density map of size 22 squares, centered on the seed bin center, and with bin size ∆x = 200 nm. From this local map, we kept only the ensemble that has a density of >80% of the central bin value. We collected the trajectory points falling into these bins and from which we computed the corresponding ellipse through their covariance matrix (41). Last, when ellipses overlap, we applied an iterative procedure that merges two overlapping ellipses by computing the ellipse based on the ensemble of points falling in each ellipse. The procedure is iterated until there are no more overlaps. From these high-density regions, by considering the displacements connecting different regions, we reconstructed a network explored by the lysosomes, in which a link (yellow) was added when at least one trajectory started in one and entered to the other. Lysosomes appear to traffic between nodes indicated by ellipses.
Chemogenetics
COS-7 cells expressing mEmerald-Sec61β -C1 (ER marker), pBa-flag-BicD2 594-FKBP, and pBa-FRB-3myc-Rab7 (17) were imaged by SIM. Lysosomes were stained with SiR-Lysosome as described above. To induce the binding between lysosomes and dyneins, inducer A/C Heterodimerizer was added to the cells at 100 nM for an hour before the imaging.
Optogenetics
Plasmids and cloning
β-actin YFP-Sec61β encodes full-length human ER marker Sec61β, fused to EYFP and was a gift from C. Hoogenraad (Utrecht University, Utrecht, The Netherlands). pB80-KIF1A(1-365)-VVDfast-FLAG-SSPB(micro), encoding opto-kinesin, was derived from pB80-KIF1A(1-365)-VVDfast-GFP-SSPB(micro) (18) by replacing GFP with a 2xFLAG-tag (DYKDHDGDYKDHD). pB80-LAMP1-mCherry-iLID was cloned in the mammalian expression vector pβactin-16-pl (chicken β-actin promoter, referred to hereafter as pB80) (42), in which the multiple cloning site was replaced by (Asc I–Xba I–Hind III–Nhe I–Sna BI–Mlu I–Age I). The full-length human lysosome marker LAMP1, without stop codon, was tagged C-terminally with the red fluorescent protein mCherry and the photosensitive heterodimerization module iLID, interspaced by two synthetic 29–amino acid GGGS linkers. The iLID module was derived from pLL7.0-Venus-iLID-Mito (43), a gift from B. Kuhlman (University of North Carolina at Chapel Hill, NC; Addgene plasmid #60413). Full-length wild-type human LAMP1 was derived from LAMP1-mGFP (44), a gift from E. Dell’Angelica (University of California, Los Angeles, CA; Addgene plasmid #34831). All constructs were validated by sequencing of the full open reading frame.
Optogenetic LAMP repositioning experiments
For live optogenetic LAMP1 repositioning experiments, cells were seeded on 24-mm glass coverslips and transfected with 2 μg of plasmid DNA (1:1.3:3 ratio LAMP1:ER:opto-kinesin) and Fugene6 transfection reagent (Promega 1:3) for 20 to 30 hours before imaging. Coverslips were mounted in metal rings; immersed in DMEM without phenol red supplemented with 10% FBS, 2 mM l-glutamine, and penicillin/streptomycin (50 μg/ml); sealed; and maintained at 37°C. Cells were imaged every 2 s for 252 s.
Epifluorescence images were acquired using a 40× (Plan Fluor, NA 1.3, Nikon; live-cell imaging) oil-immersion objective on a Nikon Ti inverted microscope equipped with a sample incubator (Tokai-Hit), mercury lamp (Osram), ET-mCherry (49008), and ET 514-nm laser bandpass (49905) filter cubes (all Chroma) and a Coolsnap HQ2 charge-coupled device camera (Photometrics), controlled with μManager 1.4 software (45). To illuminate the cells during live-cell imaging, we used a Polygon 2000 digital mirror device equipped with 405-nm light-emitting diodes (Mightex) that exposed the full field of view between imaging frames with an intensity of ~5 mW cm−2. Light exposure was synchronized with camera frames using camera-evoked Transistor-Transistor Logic (TTL) triggers.
Segmentation using deep convolutional neural networks
Segmentation of the tubular networks of the ER is done with a convolutional neural network (fig. S10). The network architecture of choice is a deep residual network inspired by EDSR (enhanced deep super resolution) and RCAN (residual channel attention network) (46, 47). These models are among a class of residual learning networks (44–46) designed for image restoration, specifically single-image super-resolution (SR), i.e., image upsampling. The state-of-the-art SR architectures generally do not use downsampling between layers (42, 44, 45) but instead make training of deep networks feasible by following the structure of residual networks as first introduced with ResNets (48) intended for image classification and later extended to image restoration with VDSR and SRResNet (49, 50). The design idea of residual networks was taken one step further in the EDSR model with the proposal of a modified residual building block called ResBlock, which was found superior to the previously proposed and more directly adapted ResNet model called SRResNet (51).
Another improvement in this class of residual networks was made with RCAN, which augments the ResBlock with two more convolution layers and a global pooling operation. These extra layers are combined with the layers in the standard ResBlock by a modified skip connection that performs multiplication rather addition, which is believed to allow the model to learn to adaptively rescale channel-wise features by considering the interdependencies among channels. RCAN also adds more regular skip connections in a design that is coined Residual in Residual, where a preset number of ResBlocks are considered a group, and a long skip connection is made from the beginning of the group to the end, whereas the skip connection of each ResBlock only passes by a few convolution layers. This allows abundant low-frequency information to be easily passed on, thus enabling the main network to focus on learning to restore high-frequency information.
Choosing the first part of our segmentation model to have an architecture built for restoration ensures that it is capable of handling images with low signal-to-noise ratio as it can learn to perform denoising in these early layers of its network. A neural network model intended for image restoration will, by default, perform regression to output pixel value predictions in the same color space as the input image. This is achieved during model training by minimizing an appropriate loss function, typically the mean squared error defined over the dataset as
where Θ represents the trainable parameters of the network referred to as F( · ), while D is the training dataset of size N written as consisting of low-quality input images and high-quality target images with pixel size H × W.
Rather than having the model perform restoration via regression followed by thresholding by intensity values to produce binary segmentation maps, the model is directly optimized to output segmentation maps by modifying it to perform classification. A common choice of loss function for classification models is the cross-entropy loss given by
where k and j are iterators over a total of K unique classes, and is a function equal to 1 if the target class for the pixel at (x, y) of the ith image is k, and otherwise, it is equal to 0. With the model set up for classification, the network F( · ) now returns scores for each class for a given input image, from which class probabilities are estimated by applying the softmax function, i.e., the normalized exponential inside the log function. The use of the cross-entropy loss over the mean squared error loss during training greatly improves the performance of models with softmax outputs, because the mean squared error tends to lead to saturation and slow learning (46), which is why the approach of directly optimizing the model to output segmentation maps is preferred. Note that K = 2 for the purposes of the binary segmentation used in this work, in which case the innermost summation in LCE(Θ; D) over the variable k reduces to the addition of two simple terms known as the binary cross-entropy loss.
A proposed network that performs this pixel-wise classification to output segmentation maps is shown in fig. S7. The architecture with a sequence of blocks surrounded by convolutional layers and a long skip connection is the same for both EDSR and RCAN. The definition of the block is different between the two, and for simplicity, the EDSR block is shown, although both blocks were used in testing. In general, the RCAN-based block was preferred. The main difference from the EDSR and RCAN architectures to that of Fig. 1 is the replacement of the SR block, an upsampling module, with a module that decodes all the feature channels from previous convolutional layers into class scores. This is done using feature pooling, in which a convolutional layer with kernel size 1 × 1 reduces the number of feature channels to the number of unique classes in the segmentation map.
Given appropriate training pairs, this network can learn to map low signal-to-noise ratio images into clean segmentation maps. Because the network is capable of restoration, it is not necessary to explicitly remove noise (denoise) images before inputting them to the model. However, to alleviate the complexity of training the network, raw images were first denoised using the denoising method ND-SAFIR (52), which includes a noise parameter estimation of Poisson-Gaussian noise that is typical for optical microscopy.
As for preparing the training data, a very crude segmentation could be performed using grayscale pixel intensity thresholding after applying this denoising method to raw images. A few of these segmentation maps were then manually cleaned and finalized by drawing in a raster graphics editor. These partially hand-drawn segmentation maps then served as targets, i.e., ground truths, in the supervised training of the segmentation network.
To make it feasible for the network to learn to segment the ER images from the relatively small training dataset, a few means of data augmentation were used. First, each segmentation training pair was randomly cropped many times, which shifts the structures from frame to frame and brings the image size down to a manageable size for a graphics card (256 × 256 pixels). This provides a few hundred subimages of different regions of the segmentation examples. Those subimages were then randomly flipped (horizontally and vertically) and rotated (by 90° or 180°) to obtain more training data. Other ways to augment data could have been to randomly change brightness or synthetically add noise to images, but this was not found to be necessary.
Training was done in batches of five images (each being 256 × 256 pixels) with a learning rate of 0.001 using the Adam optimization method for a total of 40 epochs. The learning rate was halved after every 10 epochs. The network was conFigd to have four blocks of the RCAB, which amounts to 40 convolution layers each having 64 filters with a 3 × 3 kernel size, constituting a total of about 1.3 million trainable parameters. The trained network outputs binary segmentation maps that can then be easily skeletonized by a standard thinning algorithm (53).
The implementation has been made with the machine learning library PyTorch. The code as well as training and test data for segmenting ER images is freely available at https://github.com/charlesnchr/ERNet.
Targeted eye electroporation of Xenopus
Targeted eye electroporation can be performed as previously described (54, 55). To anesthetize embryos during electroporation, tricaine methanesulfonate (MS-222) (0.4 mg/ml; Sigma-Aldrich) in 1× Modified Barth’s Saline (MBS) [LM00715 (male), LM00535 (female), Nasco] (pH 7.5) is used. An anesthetized embryo is positioned along the longitudinal channel of a “†”-shaped electroporation Sylgard chamber, with its head positioned at the cross of the longitudinal and transverse channels. A pair of flat-ended platinum electrodes (Sigma-Aldrich) is held in place by a manual micromanipulator (World Precision Instruments) at the ends of the transverse channel. A glass capillary with a fine tip containing plasmid solution is inserted into the eye primordium of stage 26–30 embryos to inject 8 × 5- to 8-nl doses of pEGFPC1-hVAP-A (1 μg/μl; Addgene #104447) or pEGFPC1-hVAP-A KD/MD (Addgene #104449) plasmid driven by an air-pressured injector, such as a Picospritzer (Parker Hannifin). Immediately following the plasmid injection, eight electric pulses of 50-ms duration at 1000-ms intervals are delivered at 18 V by a square wave generator, such as the TSS20 Ovodyne electroporator (Intracel). The embryos are recovered and raised in 0.1× MBS until they reach stage 32–35 as required for retinal cultures.
Xenopus retinal culture and imaging
Glass-bottom dishes (MatTek) are pretreated with 5 M KOH (Sigma-Aldrich) for 1 hour at room temperature to minimize background fluorescence, followed by five to eight rinses with deionized water (Sigma-Aldrich), and are then left to dry in a hood (all culturing procedures should be performed in a laminar flow hood or a microbiological safety cabinet). They are next coated with poly-l-lysine (10 μg/ml; Sigma-Aldrich) overnight at room temperature. Excess poly-l-lysine is discarded from the dishes, which are then rinsed three times with double-distilled water and left in the hood until dry. Subsequently, the dishes are coated with laminin (10 μg/ml; Sigma-Aldrich) in L15 (Gibco) for 1 to 3 hours. Last, the laminin solution is replaced with culture medium [60% (v/v) of L15 and 1% (v/v) antibiotic-antimycotic (100×), in double-distilled water (pH 7.6 to 7.8), sterilized with 0.22-μm pore-size filters], in which dissected eyes can be placed.
Electroporated embryos at stage 32–35 are first screened for EGFP fluorescence to check for successful electroporation. Embryos with fluorescent eyes are then rinsed three times in the embryo wash solution [0.1× MBS with 1% (v/v) antibiotic-antimycotic (100×) (Thermo Fisher Scientific) (pH 7.5), sterilized with 0.22-μm pore-size filters] and anesthetized in MS-222 solution [0.04% (w/v) MS-222 and 1% (v/v) antibiotic-antimycotic (100×) in 1× MBS (pH 7.5), filtered with 0.22-μm filters]. After transfer of the embryos to the Sylgard dish, the electroporated eye primordia are dissected out and washed three times in culture medium. Last, the dissected eye primordia are placed in the center of the dish, and the cultures are incubated at room temperature overnight to allow axon extension.
Wide-field imaging of Xenopus axons and analysis
Twenty-four hours after culturing, fluorescent RGC axons are identified and imaged under an Olympus IX81 inverted microscope with a 20× 0.45 NA air objective at room temperature. As the axons extended beyond the field of view, multiple positions of axons projecting from the same eye are imaged to cover all the axons.
Images of Xenopus RGC axons are processed and analyzed using Fiji (NIH). Images at multipositions are mapped and integrated. EGFP-labeled axons are manually traced by “freehand line” from the distal tips to the proximal segments until reaching the explants. The axon length is quantified by “Measure-Analyze” in Fiji. Three independent experiments were performed, in which 446 axons from 12 EGFP-VAPA–expressing eyes and 396 axons from 11 EGFP-VAPA (KD/MD)–expressing eyes were quantified.
SIM imaging of Xenopus RGC axons
To visualize lysosomes, retinal cultures are incubated with 50 nM LysoTracker Deep Red (Thermo Fisher Scientific) for 30 min at room temperature and rinsed three times with culture medium. Time-lapse recordings of ER and lysosomes are taken using a custom three-color system built around an Olympus IX71 microscope stage, which has been described above. Laser wavelengths of 488 nm (iBEAM-SMART-488, Toptica) and 640 nm (MLD 640, Cobolt) were used to excite fluorescence in the samples. Multicolor images were registered by characterizing channel displacement using a matrix generated with TetraSpeck beads (Life Technologies) imaged in the same experiment as the cells.
Samples were imaged at 3 or 5 s per frame for 60 frames immediately following the LysoTracker staining. Resolution-enhanced images were reconstructed from the raw SIM data with LAG SIM as described above. The axon outlines were drawn by increasing the brightness of fluorescence from LysoTracker staining in the analysis step. Two hundred twenty-one EGFP-VAPA (KD/MD)–expressing axons from 55 time-lapse images and 85 EGFP-VAPA–expressing axons from 32 time-lapse images were acquired from three independent experiments.
Data visualization
Videos of time-lapse imaging and analysis were performed using Fiji (NIH).
Statistical analysis
Statistical significance between two values was determined using a two-tailed, unpaired Student’s t test (GraphPad Prism). Statistical analysis of three or more values was performed by one-way analysis of variance with Tukey’s post hoc test (GraphPad Prism). All data are presented as the mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Statistical parameters including the exact value of n, the mean, median, dispersion and precision measures (mean ± SEM), and statistical significance are reported in the figures and figure legends. Data are judged to be statistically significant when P < 0.05 by two-tailed Student’s t test. In the figures, asterisks denote statistical significance as calculated by Student’s t test (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001). Statistical analysis was performed using both Origin and Excel unpaired t test.
Supplementary Material
Acknowledgments
We thank A. I. Fernández Villegas, L. Wunderlich, and W. Li for helping with the cell culture. We thank E. Ward for helping with the image processing. Funding: This research was funded by Infinitus (China) Company Ltd. (supporting M.L. and C.F.K.); a UKRI Engineering and Physical Sciences Research Council (EPSRC) grant (EP/L015889/1) awarded to the Centre for Doctoral Training in Sensor Technologies and Applications (supporting F.W.v.T.); a Sir Henry Wellcome Postdoctoral Fellowship from the Wellcome Trust (215943/Z/19/Z, to J.Q.L.); the Netherlands Organization for Scientific Research (NWO) (supporting W.N.); the European Research Council (ERC) (supporting L.K.); Wellcome Trust Collaborative Grant (203249/Z/16/Z to C.E.H. and C.F.K.); and the UK Dementia Research Institute, which receives its funding from UK DRI Ltd., funded by the UK Medical Research Council, Alzheimer’s Society, and Alzheimer’s Research UK (supporting E.A. and C.F.K.). D.H.’s research is supported by a PSL-Cambridge grant and an ERC grant, agreement no. 882673. Author contributions: M.L. designed, conducted, and interpreted experiments and wrote the article. J.Q.L. and F.W.v.T. performed experiments with M.L. and edited the article. W.N. performed the optogenetic experiments and analyzed the data with M.L. P.P., M.F., and C.N.C. provided the computational support and analysis methods. E.A., C.E.H., A.T., D.H., L.K., and G.S.K.S. gave advice throughout the project and edited the article. C.F.K. supervised the research, coordinated the study, and wrote the article. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabc7209/DC1
REFERENCES AND NOTES
- 1.Westrate L. M., Lee J. E., Prinz W. A., Voeltz G. K., Form follows function: The importance of endoplasmic reticulum shape. Annu. Rev. Biochem. 84, 791–811 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Blackstone C., Cellular pathways of hereditary spastic paraplegia. Annu. Rev. Neurosci. 35, 25–47 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nixon-Abell J., Obara C. J., Weigel A. V., Li D., Legant W. R., Xu C. S., Pasolli H. A., Harvey K., Hess H. F., Betzig E., Blackstone C., Lippincott-Schwartz J., Increased spatiotemporal resolution reveals highly dynamic dense tubular matrices in the peripheral ER. Science 354, aaf3928 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo Y., Li D., Zhang S., Yang Y., Liu J.-J., Wang X., Liu C., Milkie D. E., Moore R. P., Tulu U. S., Kiehart D. P., Hu J., Lippincott-Schwartz J., Betzig E., Li D., Visualizing intracellular organelle and cytoskeletal interactions at nanoscale resolution on millisecond timescales. Cell 175, 1430–1442.e17 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Lee C., Chen L. B., Dynamic behavior of endoplasmic reticulum in living cells. Cell 54, 37–46 (1988). [DOI] [PubMed] [Google Scholar]
- 6.Friedman J. R., Webster B. M., Mastronarde D. N., Verhey K. J., Voeltz G. K., ER sliding dynamics and ER–mitochondrial contacts occur on acetylated microtubules. J. Cell Biol. 190, 363–375 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waterman-Storer C. M., Gregory J., Parsons S. F., Salmon E. D., Membrane/microtubule tip attachment complexes (TACs) allow the assembly dynamics of plus ends to push and pull membranes into tubulovesicular networks in interphase Xenopus egg extracts. J. Cell Biol. 130, 1161–1169 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zajac A. L., Goldman Y. E., Holzbaur E. L. F., Ostap E. M., Local cytoskeletal and organelle interactions impact molecular-motor-driven early endosomal trafficking. Curr. Biol. 23, 1173–1180 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phillips M. J., Voeltz G. K., Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 17, 69–82 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valm A. M., Cohen S., Legant W. R., Melunis J., Hershberg U., Wait E., Cohen A. R., Davidson M. W., Betzig E., Lippincott-Schwartz J., Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 546, 162–167 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neefjes J., Jongsma M. M. L., Berlin I., Stop or go? Endosome positioning in the establishment of compartment architecture, dynamics, and function. Trends Cell Biol. 8, 580–594 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Raiborg C., Wenzel E. M., Pedersen N. M., Olsvik H., Schink K. O., Schultz S. W., Vietri M., Nisi V., Bucci C., Brech A., Johansen T., Stenmark H., Repeated ER–endosome contacts promote endosome translocation and neurite outgrowth. Nature 520, 234–238 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Lawrence R. E., Zoncu R., The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 21, 133–142 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Zhao Y. G., Liu N., Miao G., Chen Y., Zhao H., Zhang H., The ER contact proteins VAPA/B interact with multiple autophagy proteins to modulate autophagosome biogenesis. Curr. Biol. 28, 1234–1245.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Alpy F., Rousseau A., Schwab Y., Legueux F., Stoll I., Wendling C., Spiegelhalter C., Kessler P., Mathelin C., Rio M.-C., Levine T. P., Tomasetto C., STARD3 or STARD3NL and VAP form a novel molecular tether between late endosomes and the ER. J. Cell Sci. 126 ( pt. 23), 5500–5512 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Rosa-Ferreira C., Munro S., Arl8 and SKIP act together to link lysosomes to kinesin-1. Dev. Cell 21, 1171–1178 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kapitein L. C., Schlager M. A., van der Zwan W. A., Wulf P. S., Keijzer N., Hoogenraad C. C., Probing intracellular motor protein activity using an inducible cargo trafficking assay. Biophys. J. 99, 2143–2152 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nijenhuis W., van Grinsven M. M. P., Kapitein L. C., An optimized toolbox for the optogenetic control of intracellular transport. J. Cell Biol. 219, e201907149 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu L., McPhee C. K., Zheng L., Mardones G. A., Rong Y., Peng J., Mi N., Zhao Y., Liu Z., Wan F., Hailey D. W., Oorschot V., Klumperman J., Baehrecke E. H., Lenardo M. J., Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465, 942–946 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nthiga T. M., Kumar Shrestha B., Sjøttem E., Bruun J.-A., Bowitz Larsen K., Bhujabal Z., Lamark T., Johansen T., CALCOCO1 acts with VAMP-associated proteins to mediate ER-phagy. EMBO J. 39, e103649 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pu J., Guardia C. M., Keren-Kaplan T., Bonifacino J. S., Mechanisms and functions of lysosome positioning. J. Cell Sci. 129, 4329–4339 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Willett R., Martina J. A., Zewe J. P., Wills R., Hammond G. R. V., Puertollano R., TFEB regulates lysosomal positioning by modulating TMEM55B expression and JIP4 recruitment to lysosomes. Nat. Commun. 8, 1580 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Höglinger D., Burgoyne T., Sanchez-Heras E., Hartwig P., Colaco A., Newton J., Futter C. E., Spiegel S., Platt F. M., Eden E. R., NPC1 regulates ER contacts with endocytic organelles to mediate cholesterol egress. Nat. Commun. 10, 4276 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu Y., Whiteus C., Xu C. S., Hayworth K. J., Weinberg R. J., Hess H. F., De Camilli P., Contacts between the endoplasmic reticulum and other membranes in neurons. Proc. Natl. Acad. Sci. U.S.A. 114, E4859–E4867 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Terasaki M., Axonal endoplasmic reticulum is very narrow. J. Cell Sci. 131, jcs210450 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Holcman D., Parutto P., Chambers J. E., Fantham M., Young L. J., Marciniak S. J., Kaminski C. F., Ron D., Avezov E., Single particle trajectories reveal active endoplasmic reticulum luminal flow. Nat. Cell Biol. 20, 1118–1125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lim C.-Y., Davis O. B., Shin H. R., Zhang J., Berdan C. A., Jiang X., Counihan J. L., Ory D. S., Nomura D. K., Zoncu R., ER–lysosome contacts enable cholesterol sensing by mTORC1 and drive aberrant growth signalling in Niemann–Pick type C. Nat. Cell Biol. 21, 1206–1218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liao Y.-C., Fernandopulle M. S., Wang G., Choi H., Hao L., Drerup C. M., Qamar S., Nixon-Abell J., Shen Y., Meadows W., Vendruscolo M., Knowles T. P. J., Nelson M., Czekalska M. A., Musteikyte G., Gachechiladze M. A., Stephens C. A., Pasolli H. A., Forrest L. R., St. George-Hyslop P., Lippincott-Schwartz J., Ward M. E., RNA granules hitchhike on lysosomes for long-distance transport, using annexin A11 as a molecular tether. Cell 179, 147–164.e20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cioni J.-M., Lin J. Q., Holtermann A. V., Koppers M., Jakobs M. A. H., Azizi A., Turner-Bridger B., Shigeoka T., Franze K., Harris W. A., Holt C. E., Late endosomes act as mRNA translation platforms and sustain mitochondria in axons. Cell 176, 56–72.e15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang J., Lee S., Blackstone C., Protrudin binds atlastins and endoplasmic reticulum-shaping proteins and regulates network formation. Proc. Natl. Acad. Sci. U.S.A. 110, 14954–14959 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rowland A. A., Chitwood P. J., Phillips M. J., Voeltz G. K., ER contact sites define the position and timing of endosome fission. Cell 159, 1027–1041 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Friedman J. R., DiBenedetto J. R., West M., Rowland A. A., Voeltz G. K., Endoplasmic reticulum–endosome contact increases as endosomes traffic and mature. Mol. Biol. Cell 24, 1030–1040 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allison R., Edgar J. R., Pearson G., Rizo T., Newton T., Günther S., Berner F., Hague J., Connell J. W., Winkler J., Lippincott-Schwartz J., Beetz C., Winnder B., Reid E., Defects in ER–endosome contacts impact lysosome function in hereditary spastic paraplegia. J. Cell Biol. 216, 1337–1355 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blackstone C., O’Kane C. J., Reid E., Hereditary spastic paraplegias: Membrane traffic and the motor pathway. Nat. Rev. Neurosci. 12, 31–42 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Young L. J., Ströhl F., Kaminski C. F., A guide to structured illumination TIRF microscopy at high speed with multiple colors. J. Vis. Exp. 111, e53988 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Müller M., Mönkemöller V., Hennig S., Hübner W., Huser T., Open-source image reconstruction of super-resolution structured illumination microscopy data in ImageJ. Nat. Commun. 7, 10980 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Culley S., Albrecht D., Jacobs C., Pereira P. M., Leterrier C., Mercer J., Henriques R., Quantitative mapping and minimization of super-resolution optical imaging artifacts. Nat. Methods 15, 263–266 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arganda-Carreras I., Kaynig V., Rueden C., Eliceiri K. W., Schindelin J., Cardona A., Seung H. S., Trainable Weka Segmentation: A machine learning tool for microscopy pixel classification. Bioinformatics 33, 2424–2426 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Arganda-Carreras I., Fernández-González R., Muñoz-Barrutia A., Ortiz-De-Solorzano C., 3D reconstruction of histological sections: Application to mammary gland tissue. Microsc. Res. Tech. 73, 1019–1029 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Tinevez J.-Y., Perry N., Schindelin J., Hoopes G. M., Reynolds G. D., Laplantine E., Bednarek S. Y., Shorte S. L., Eliceiri K. W., TrackMate: An open and extensible platform for single-particle tracking. Methods 115, 80–90 (2017). [DOI] [PubMed] [Google Scholar]
- 41.P. Parutto, J. Heck, M. Heine, D. Holcman, Reconstructing wells from high density regions extracted from super-resolution single particle trajectories. bioRxiv 642744 [Preprint]. 20 May 2019. https://doi.org/10.1101/642744.
- 42.Kaech S., Ludin B., Matus A., Cytoskeletal plasticity in cells expressing neuronal microtubule-associated proteins. Neuron 17, 1189–1199 (1996). [DOI] [PubMed] [Google Scholar]
- 43.Guntas G., Hallett R. A., Zimmerman S. P., Williams T., Yumerefendi H., Bear J. E., Kuhlman B., Engineering an improved light-induced dimer (iLID) for controlling the localization and activity of signaling proteins. Proc. Natl. Acad. Sci. U.S.A. 112, 112–117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Falcón-Pérez J. M., Nazarian R., Sabatti C., Dell’Angelica E. C., Distribution and dynamics of Lamp1-containing endocytic organelles in fibroblasts deficient in BLOC-3. J. Cell. Sci. 118 ( Pt. 22), 5243–5255 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Edelstein A., Amodaj N., Hoover K., Vale R., Stuurman N., Computer control of microscopes using μManager. Curr. Protoc. Mol. Biol. 92, 14.20.1–14.20.17 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.B. Lim, S. Son, H. Kim, S. Nah, K. M. Lee, Enhanced deep residual networks for single image super-resolution, in The IEEE Conference on Computer Vision and Pattern Recognition (CVPR Workshops, Hawaii Convention Center, 2017). [Google Scholar]
- 47.Y. Zhang, K. Li, K. Li, L. Wang, B. Zhong, Y. Fu, Image super-resolution using very deep residual channel attention networks, in Computer Vision – ECCV 2018. ECCV 2018. Lecture Notes in Computer Science (Springer, 2018), pp. 294–310.
- 48.K. He, X. Zhang, S. Ren, J. Sun, Delving deep into rectifiers: Surpassing human-level performance on imagenet classification, in 2015 IEEE International Conference on Computer Vision (ICCV) (IEEE, 2015), pp. 1026–1034. [Google Scholar]
- 49.J. Kim, J. K. Lee, K. M. Lee, Accurate image super-resolution using very deep convolutional networks, in 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR) (IEEE, 2016), pp. 1646–1654. [Google Scholar]
- 50.C. Ledig, L. Theis, F. Huszár, J. Caballero, A. Cunningham, A. Acosta, A. Aitken, A. Tejani, J. Totz, Z. Wang, W. Shi, Photo-realistic single image super-resolution using a generative adversarial network, in 2017 IEEE Conference on Computer Vision and Pattern Recognition (CVPR) (IEEE, 2017), pp. 105–114. [Google Scholar]
- 51.I. Goodfellow, Y. Bengio, A. Courville, Deep Learning (MIT Press, 2016). [Google Scholar]
- 52.J. Boulanger, “Non-parametric estimation and contributions to image sequence analysis: Modeling, simulation and estimation of the intracellular traffic in video-microscopy image sequences,” thesis, Université de Rennes 1, Mention Traitement du Signal et des Télécommunications (2007). [Google Scholar]
- 53.Lee T. C., Kashyap R. L., Chu C. N., Building skeleton models via 3-D medial surface axis thinning algorithms. CVGIP: Graph. Models Image Process. 56, 462–478 (1994). [Google Scholar]
- 54.Falk J., Drinjakovic J., Leung K. M., Dwivedy A., Regan A. G., Piper M., Holt C. E., Electroporation of cDNA/Morpholinos to targeted areas of embryonic CNS in Xenopus. BMC Dev. Biol. 7, 107 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wong H.-W., Holt C. E., Targeted electroporation in the CNS in Xenopus embryos. Methods Mol. Biol. 1865, 119–131 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabc7209/DC1