

Abstract
Light-chain (AL) cardiac amyloidosis (CA) has a worse prognosis than transthyretin (ATTR) CA. In this single-center study, we compared post heart transplant (OHT) survival for AL and ATTR amyloidosis, hypothesizing that these differences would persist post-OHT. Thirty-nine patients with CA (AL, n=18; ATTR, n=21) and 1,023 non-amyloidosis subjects undergoing OHT were included. Cox-proportional hazards modeling was used to evaluate the impact of amyloid subtype and era (early era: from 2001-2007; late era: from 2008-2018) on survival post-OHT. Survival for non-amyloid patients was greater than ATTR (p=0.034) and AL (p<0.001) patients in the early era. One, 3- and 5-year survival rates were higher for ATTR patients than AL patients in the early era (100% vs 75%, 67% vs 50% and 67% vs 33%, respectively for ATTR and AL patients). Survival in the non-amyloid cohort was 87% at 1 year, 81% at 3 years, and 76% at 5 years post-OHT. In the late era, AL and ATTR patients had unadjusted 1-year, 3-year, and 5-year survival rates of 100%, which was comparable to non-amyloid patients (90% vs 84% vs 81%). Overall, these findings demonstrate that in the current era, differences in post-OHT survival for AL compared to ATTR are diminishing; OHT outcomes for selected patients with CA do not differ from non-amyloidosis patients.
Keywords: amyloid, cardiomyopathy, heart transplant, light-chain, transthyretin
Introduction
Cardiac amyloidosis is an infiltrative cardiomyopathy, most commonly caused by either immunoglobulin light chain (AL) or transthyretin (ATTR) protein deposits.1 AL amyloidosis is caused by a plasma cell dyscrasia which results in infiltration of the myocardium by monoclonal immunoglobulin lights chains. ATTR amyloidosis, which is classified as either wild-type (ATTRwt) or mutant (ATTRm) depending on whether a genetic mutation is present on the TTR gene, results from the deposition of insoluble TTR amyloid fibrils. Regardless of subtype, untreated cardiac amyloidosis results in progressive heart failure and is associated with significant rates of morbidity and mortality.2–4 Numerous reports delineate worse outcomes for patients with AL compared to ATTR cardiac amyloidosis.5–8
Recently, emerging therapies for ATTR amyloidosis, specifically tafamidis, have shown to be effective, especially when administered early in the course of disease.9 Despite this, orthotopic heart transplantation (OHT) remains the treatment to most meaningfully extend survival.10 In ATTR amyloidosis, OHT for ATTRwt or combined with orthotopic liver transplantation (OLT) for ATTRm, has been performed11–13, although concerns regarding recurrent amyloid cardiomyopathy and progression of extracardiac manifestations remain.14–16 In AL amyloidosis, the use of OHT in conjunction with either targeted plasma cell therapy or stem cell transplant has been largely restricted to a few centers worldwide.10,17–19 While data from these centers show this therapeutic strategy to have merit, the extent of amyloid deposition in other organs and recurrent disease limit transplantation success and thus the long-term survival of these patients.20–22
Despite these concerns, OHT offers the greatest chance of long-term survival in carefully selected patients with advanced amyloid cardiomyopathy.23 Recent studies, including data from the United Network for Organ Sharing (UNOS) registry, have shown improved post-OHT outcomes for amyloid cardiomyopathy patients over time.10,17,19,23,24 One hypothesis for these improvements may be attributed to differences in outcomes between AL and ATTR patients after transplantation, and a possible shift towards the transplantation of more ATTR patients. However, the UNOS registry is limited by the lack of differentiation in the amyloid subtype; thus, post-OHT outcome comparisons between ATTR and AL cardiac amyloidosis cannot be made. While AL patients have been known to experience worse prognosis than ATTR patients 5–8, a European study including 48 transplant recipients found two year post-transplant survival for cardiac amyloidosis in the current era was comparable to non-amyloid transplants.23 More recently, Barrett et al, described outcomes of 31 OHT recipients and reported longer waitlist times for ATTR compared to AL patients, but no difference in mortality between the amyloid and non-amyloid groups.19 Given the advances in targeted plasma cell therapy for AL amyloidosis25, difference in management strategies by institution and variance of organ availability by region within the United States, we set out to evaluate outcomes at our center.
We compared OHT outcomes in these two subtypes as well as 1,023 non-amyloid patients. The aims of the study were three-fold: 1) To examine whether ATTR patients have better outcomes after OHT than AL patients, 2) To examine whether survival outcomes of patients who underwent OHT from 2008-2018 are better than for those who underwent OHT from 2001-2007, and 3) To examine whether survival after OHT for cardiac amyloidosis patients is comparable to those for patients transplanted for non-amyloid indications.
Methods
Participants
Between July 2001 and June 2018, 39 patients diagnosed with cardiac amyloidosis were followed post-transplant at our center. One patient with AL amyloidosis received a second heart and renal transplant and was censored at the time of second transplant.
Demographic, laboratory, echocardiographic, electrocardiographic, and hemodynamic data among patients with AL and ATTR prior to heart transplantation were analyzed retrospectively. Data were further separated into two eras: early era (2001-2007) and late era (2008-2018) for additional analysis; 18 patients were transplanted in the early era and 21 in the late era. The end of 2007 was chosen to separate the two groups because more stringent patient selection criteria for cardiac amyloidosis was published in 2008 and, importantly, the date also marked the advent of considerable advances in therapy for AL amyloidosis potentially contributing to significant improvement in post-OHT outcomes for AL amyloidosis patients in the past decade. Survival after OHT for AL and ATTR amyloidosis was also compared to 1,023 patients undergoing OHT at our center for non-amyloid cardiomyopathies between July 2001 and June 2018.
All patients underwent our institution’s standard amyloidosis evaluation. Cardiac amyloidosis was determined by typical echocardiographic evidence of infiltrative cardiomyopathy with endomyocardial biopsy-proven congophilic deposits. For AL amyloidosis, evidence of light chain response to pre-OHT chemotherapy was not initially necessary for eligibility, as it proved difficult to achieve in the early era, although over time it became an established criterion when assessing candidacy.
Extracardiac amyloid involvement before OHT was defined according to guidelines from the International Society for Heart and Lung Transplantation.26 Peripheral neuropathy involvement criteria included electromyography confirmation or use of medications taken for neuropathy. Gastrointestinal involvement was defined by alternating constipation, diarrhea, or altered gut motility consistent with amyloid and/or history of gastrointestinal bleeding with biopsy verification for amyloid deposition. Hepatic involvement criteria included an alkaline phosphatase > 1.5 above upper institutional laboratory limit and liver biopsy confirmation. Pleural involvement criteria included chest x-ray and computed tomography to assess for interstitial disease and thoracentesis for a persistent pleural effusion to differentiate amyloidosis from heart failure. Renal involvement criteria required a urinary protein excretion > 3 grams/24 hours or renal biopsy confirmation.26
Statistical analysis
Continuous variables were presented as mean ± standard deviation or median (25th-75th percentile) if highly skewed, and categorical variables were summarized as counts (percentages). The Shapiro-Wilk test was used to test for normality of variables obtained from baseline demographic, echocardiographic, electrocardiographic, and hemodynamic data. Between group differences for continuous variables were tested with the Student’s t-test or Mann-Whitney Test as appropriate, and the χ2 test or the Fisher exact test (when cell counts were small) were used for categorical variables. Kaplan-Meier survival analysis using a log-rank test was performed to compare post-OHT survival between AL and ATTR amyloidosis patients, and eras also stratified by era: early (2001-2007) and late (2008-2018). Univariate Cox proportional hazards models were performed to evaluate whether type of amyloid or era was associated with death after OHT. Analyses were performed using R statistical computing software (R Core Team, Version 3.3.2, Vienna).
Results
Baseline Characteristics
There were 39 patients in this study, 11 women (28%) and 28 men (72%); of the 39, 18 had AL (46%) and 21 had ATTR (54%) amyloidosis. Median age at time of OHT was significantly older in patients with ATTR amyloidosis (63 years vs. 54 years; p = 0.01). More Caucasians were in the AL versus ATTR cohort (83% vs. 48%; p = 0.047), with 10 ATTR patients having Val122Ile mutation, a mutation that almost exclusively affects individuals of Afro-Caribbean descent.27 The remaining breakdown of ATTR patients included 5 ATTRwt, 3 Thr60Ala, 1 Asn38Ala, 1 Ser23Asn, and 1 Thr59Lys. Seven ATTR patients received simultaneous heart and liver transplantation: 4 patients in the early era and 3 in the late era. Other baseline characteristics including demographic, laboratory, clinical and cardiac biomarker data were not statistically different between AL and ATTR cohorts (Table 1).
Table 1:
Baseline characteristics of patients who received heart transplant for light chain (AL) or transthyretin (ATTR) cardiac amyloidosis.
| AL (n = 18) | ATTR (n = 21) | p-value | |
|---|---|---|---|
| Demographics | |||
| Sex, female | 7 (38.9) | 4 (19.1) | 0.31 |
| Age at OHT, years | 54 (48.5-62.0) | 63 (58-65) | 0.01 |
| Time from diagnosis to OHT, days | 133 (91.2-589) | 376 (207-561) | 0.26 |
| Time from listing to OHT, days | 62 (24.8-139) | 53 (37-149) | 0.92 |
| Race, Caucasian | 15 (83.3) | 10 (47.6) | 0.05 |
| Laboratory | |||
| BMI, kg/ m2 | 25.8 (24.6-27.9) | 25.2 (24.4-28.2) | 0.98 |
| mBMI, kg/m2 | 1030 (876-1190) | 1090 (988-1160) | 0.27 |
| Creatinine, mg/dL | 1.1 (1.0-1.5) | 1.3 (1.0-1. 5) | 0.50 |
| eGFR, ml/min/1.73 m2 | 55.8 (46.4-70.9) | 59.1 (46.4-74.6) | 0.90 |
| Proteinuria, mg/dL | 442 (116-796) | 288 (109-318) | 0.09 |
| Serum albumin, g/liter | 3.9 (3.5-4.5) | 4.2 (4.1-4.3) | 0.29 |
| Total Bilirubin, mg/dL | 0.8 (0.6-0.98) | 0.9 (0.7-1.4) | 0.14 |
| Alkaline Phosphatase, U/L | 96 (86-112) | 80 (74-127) | 0.46 |
| INR | 1.2 (1.1-1.5) | 1.3 (1.2-1.7) | 0.44 |
| Clinical Data | |||
| Average NYHA class | 3.6 ± 0.5 | 3.8 ± 0.4 | 0.18 |
| Hypertension, n (%) | 6 (33.3) | 9 (42.9) | 0.68 |
| Diabetes, n (%) | 4 (22.2) | 3 (14.3) | 0.68 |
| Coronary artery disease, n (%) | 6 (33.3) | 6 (33.3) | 1.00 |
| Peripheral neuropathy, n (%) | 1 (5.6) | 3 (14.3) | 0.61 |
| Nephrotic syndrome/renal involvementa | 1 (7.1) | 0 (0) | 1.00 |
| Organs affected by amyloidosis | 0.39 | ||
| Isolated cardiac | 14 (77.8) | 18 (85.7) | |
| 2 organs affected | 4 (22.2) | 3 (14.3) | |
| Cardiac Biomarkers | |||
| Troponin I, ng/mL | 0.2 (0.1-1.6) | 0.1 (0.06-0.22) | 0.24 |
| BNP, ng/mL | 986 (542-1550) | 685 (391-961) | 0.29 |
AL= light chain amyloidosis; ATTR = transthyretin amyloidosis; BMI = body mass index; BNP = brain natriuretic peptide; eGFR, by MDRD = estimated glomerular filtration rate; INR = international normalized ratio; mBMI = modified body mass index; NYHA = New York Heart Association Class; OHT = orthotopic heart transplantation.
Values are presented as median (25th-75th percentile), mean ± standard deviation, or counts (%).
Nephrotic range proteinuria defined as > 3 g of protein in the urine
The presence of electrophysiologic devices before OHT and data from invasive hemodynamics at diagnosis were not significantly different between AL and ATTR groups (Table 2). The median cardiac indices were comparable for AL and ATTR patients (1.6 vs. 1.7 liters/min/m2, p=0.88). Echocardiographic measurements between groups showed a greater left ventricular mass index (175 vs. 132 g/m2; p < 0.01), septal wall thickness (1.8 vs. 1.6 cm; p = 0.03), and posterior wall thickness (1.8 vs. 1.4 cm; p = 0.01) in the ATTR cohort (Table 2).
Table 2:
Baseline echocardiographic, electrocardiographic and invasive hemodynamic data for patients who received heart transplant for light chain (AL) or transthyretin (ATTR) cardiac amyloidosis.
| AL (n = 18) | ATTR (n = 21) | p-value | |
|---|---|---|---|
| Echocardiographic | |||
| Left ventricular | |||
| EDD, cm | 4.2 (4.0-4.4) | 4.2 (4.1-4.6) | 0.89 |
| EF, % | 50 (35.0-55.0) | 37.8 (21.8-52.5) | 0.18 |
| Mass Index, g/m2 | 132 (90.2-148.0) | 175 (134.0-218.0) | 0.01 |
| LA diameter, cm | 4.6 (4.2-5.3) | 4.7 (4.4-4.8) | 0.80 |
| Septal wall, cm | 1.6 (1.2-1.8) | 1.8 (1.7-2.1) | 0.03 |
| Posterior wall, cm | 1.4 (1.1-1.8) | 1.8 (1.6-2.0) | 0.01 |
| Electrocardiographic | |||
| Non-sinus rhythm | 4 (22.2) | 11 (52.4) | 0.11 |
| Atrial fibrillation/flutter | 2 (11.1) | 5 (23.8) | 0.42 |
| PR interval, msec | 176 (152-199) | 180 (152-196) | 0.86 |
| QRS, msec | 94 (87-104) | 106 (88-116) | 0.29 |
| QTc, msec | 439 (404-483) | 480 (456-487) | 0.24 |
| Low voltage | 8 (44.4) | 11 (52.4) | 0.86 |
| Pseudoinfarct | 12 (85.7) | 5 (26.3)a | 0.003 |
| Electrophysiologic Devices | |||
| Pacemaker | 5 (27.8) | 13 (61.9) | 0.07 |
| Biventricular Pacemaker | 1 (5.6) | 0 (0.0) | 0.46 |
| ICD | 1 (5.6) | 9 (42.9) | 0.01 |
| Invasive Hemodynamics | |||
| Cardiac Index, liters/min/m2 | 1.6 (1.4-2.1) | 1.7 (1.3-2.1) | 0.88 |
| PCWP, mmHg | 20.5 (17.5-22.8) | 18 (10.5-24.5) | 0.83 |
| Mean RAP, mmHg | 8.5 (5.3-11.5) | 9 (4.3-15.0) | 0.92 |
| PAs, mmHg | 41 (34.5-43.8) | 41.5 (33.0-48.2) | 0.87 |
| PAd, mmHg | 19.5 (17.5-21.8) | 19 (14.0-20.8) | 0.63 |
| PA saturation, % | 58 (52-66) | 61 (57.0-65.1) | 0.53 |
| PVR, WU | 2.9 (1.9-4.9) | 3.9 (2.8-5.6) | 0.35 |
AL = light chain amyloidosis; ATTR = transthyretin amyloidosis, EDD = end-diastolic diameter, EF = ejection fraction, ESD = end-systolic diameter; LA = left atrium; ICD = implantable cardioverter defibrillator; PAd = diastolic pulmonary arterial pressure; PAs = systolic pulmonary arterial pressure; PCWP = pulmonary capillary wedge pressure; PVR = pulmonary vascular resistance; RAP = right atrial pressure; WU = wood units. Values are presented as median (25th-75th percentile), mean ± standard deviation, or counts (%).
Of those with an interpretable rhythm.
Survival rates after heart transplantation
In the early era (2001-2007), for AL patients, unadjusted 1-year, 3-year, and 5-year survival rates were 75%, 50%, and 33%, respectively. For ATTR patients, unadjusted 1-year, 3-year, and 5-year survival rates were 100%, 67%, and 67%, respectively. In comparison, 1-year, 3-year, and 5-year survival rates for non-amyloid cardiomyopathies were 87%, 81% and 76%. In the late era (2008-2018), for AL patients and ATTR patients, unadjusted 1-year, 3-year, and 5-year survival rates were all 100%. The 1-year, 3-year, and 5-year survival rates for non-amyloid cardiomyopathies were 90%, 84% and 81%. Kaplan-Meier curves comparing survival for cardiac amyloid subtypes with those of non-amyloid cardiomyopathies, separated by era, are shown in Figure 1 and Figure 2. From 2001-2007, survival post-OHT for non-amyloid cardiomyopathy was found to be significantly better than those of both ATTR (p=0.03) and AL (p<0.001) amyloidosis (Figure 1). However, in the late era (2008-2018), there was no significant difference found between post-OHT outcomes in the non-amyloid cardiomyopathy vs ATTR or AL cohorts (p=0.12 and p=0.91, respectively) (Figure 2).
Figure 1:
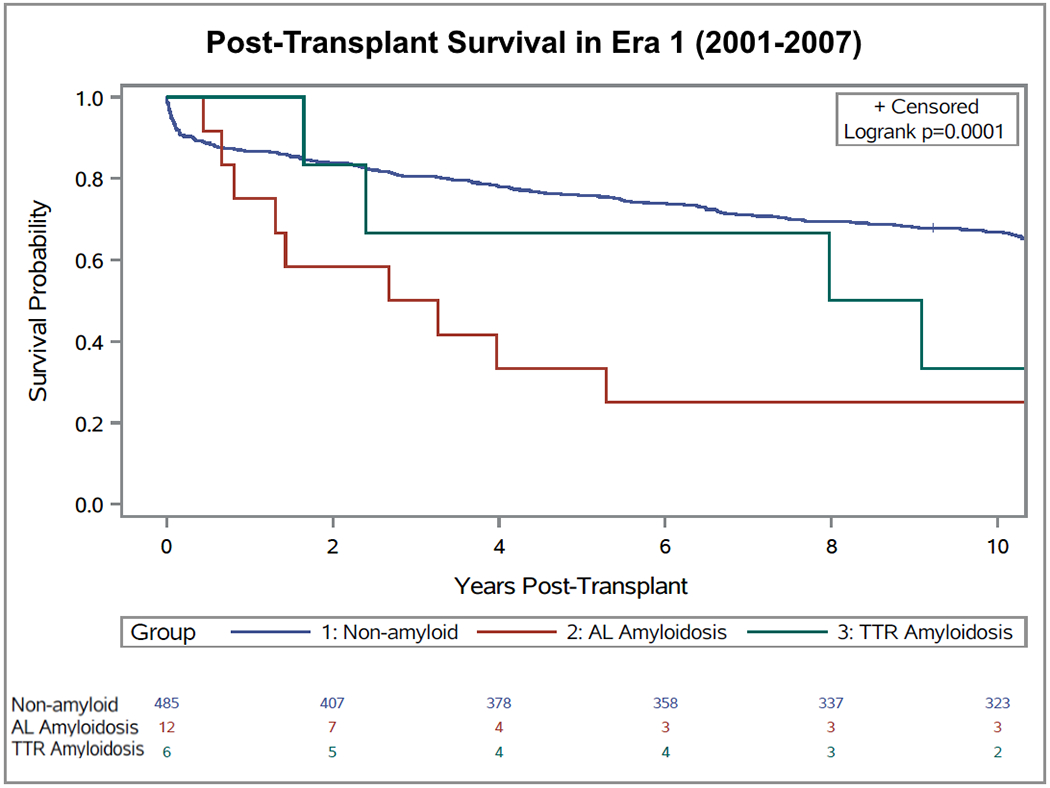
Survival of patients with transthyretin cardiac amyloidosis (n = 6), light chain amyloidosis (n = 12) and non-amyloid cardiomyopathy (control group; n = 485) after heart transplantation in the early era (2001-2007).
Figure 2:
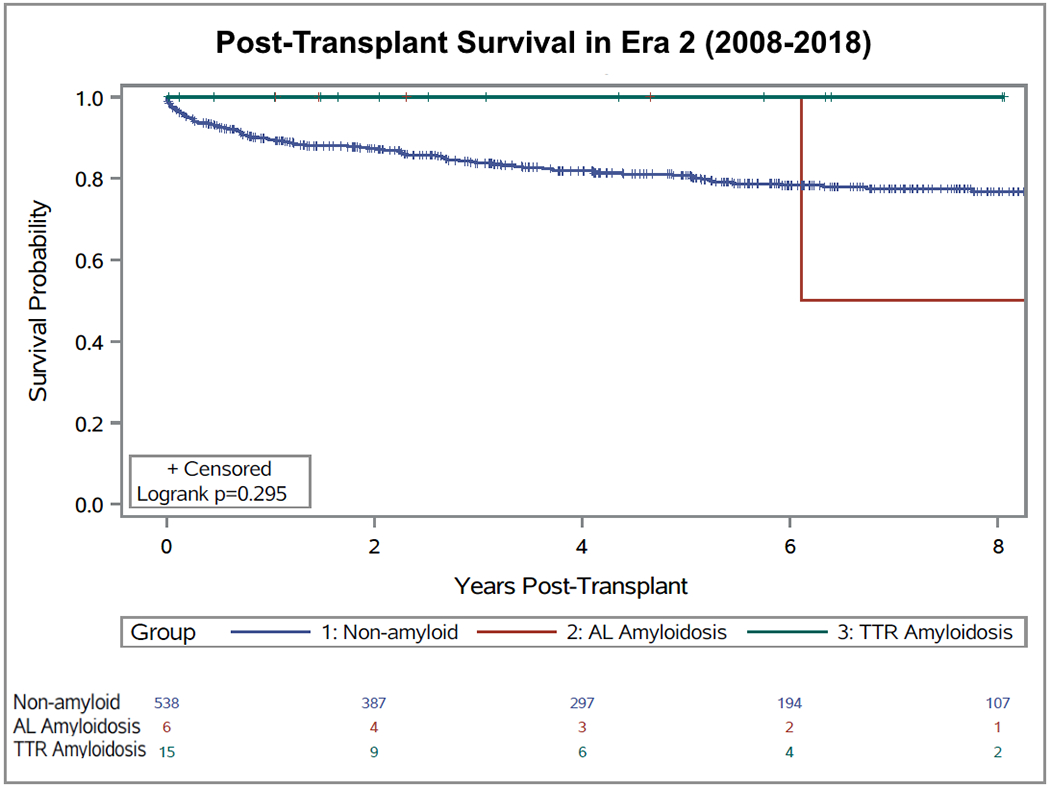
Survival of patients with transthyretin cardiac amyloidosis (n = 15), light chain amyloidosis (n = 6) and non-amyloid cardiomyopathy (control group; n = 539) after heart transplantation in the late era (2008-2018).
Predictors of Death Post-OHT for Cardiac Amyloidosis
In Cox proportional hazards modelling, heart transplantation in the late era was associated with a decreased risk of mortality (hazard ratio (HR), 0.09; 95% confidence interval (CI), 0.01-0.67; p=0.02) compared to the early era, while the AL amyloidosis subtype was found to have a higher but non-significant risk of mortality after OHT (HR, 2.35; 95% CI, 0.80-6.92; p = 0.12) (data not shown).
Given that transplantation in the late era was found to have the most significant association with improved survival after OHT, baseline characteristics of patients before transplantation were separated temporally and compared in detail (Table S1). Moving from the early era (2001-2007) to the late era (2008-2018), there was a shift to transplanting more ATTR patients (from 6 to 15 ATTR patients) and fewer AL patients (from 12 to 6 AL patients). For both subtypes, there was a trend towards less extra-cardiac involvement over time, though this trend did not reach statistical significance as transplantation in both eras was largely limited to patients with isolated cardiac amyloidosis. Between 2008-2018, only one ATTR and one AL patient were transplanted with known extra-cardiac involvement. The AL patient exhibited peripheral neuropathic involvement and the ATTR patient experienced gastrointestinal manifestations of amyloidosis.22
Discussion
This study analyzed the post-heart transplant outcomes of cardiac amyloidosis at a single center. In the early era, survival in ATTR and AL patients was decreased compared to the non-amyloid transplant patients, most particularly in the AL amyloid cohort. However, both AL and ATTR patients in the late era had survival comparable to that of non-amyloid patients.
AL amyloidosis is more often associated with extra-cardiac involvement, is associated with a poorer prognosis than ATTR and cannot be cured, thus with risk of recurrence. As such, it is not surprising that over the 17-year duration of our study AL amyloidosis was associated with worse post-OHT outcomes than ATTR in our cohort of patients (data not shown). However, contrary to our hypothesis, when separated temporally the current study found that post-OHT survival in the late era (2008-2018) was comparable to non-amyloid transplants in both AL and ATTR cohorts, and better than the early era (2001-2007). In univariable analysis of survival post-OHT, we report that the era of OHT was the only variable significantly associated with improved survival.
The end of 2007 was chosen to separate the two groups for analysis for two main reasons. First, more stringent patient selection criteria regarding extra-cardiac organ involvement of amyloidosis was published in 2008 by the Mayo Clinic; this, in turn, led to further studies that helped refine the selection criteria for OHT in amyloid cardiomyopathy.10,17,28–30 This is numerically reflected in our ATTR cohort with 93.33% of OHT confined to isolated cardiac patients since 2008 (Table S1). Second, the introduction of considerable advances in therapy for AL amyloidosis in the past decade has contributed to significant improvements in post-OHT outcomes for AL amyloidosis patients.31,32 Specifically, the development of multiple novel therapies targeted at the monoclonal protein production, including bortezomib, lenalidomide and more recently daratumumab25 has made long-term control of plasma cell dyscrasia possible. In our study, most AL patients received stem cell transplants; however, with the advent of newer plasma cell therapies, induction and consolidation therapies have become more common over time (Table S2). While there are many possible mechanisms as to why the post-OHT outcomes have improved, the strongest reasons from our data include the use of more targeted novel chemotherapy and the transplantation of relatively more ATTR patients than AL.
The improvement in survival by era found in our study is consistent with findings from recent studies, including data from the UNOS registry, that have shown improved survival for amyloid cardiomyopathy of either subtype in the past decade.10,23,24 Studies have attributed these improvements to more refined patient selection resulting in reduced extracardiac involvement and the introduction of new chemotherapeutic agents.23 However, given that the UNOS registry does not delineate the type of amyloid, assessment of the interaction of amyloid type on improved outcomes after OHT is indeterminable from the UNOS database. Our study expands on the UNOS data and is in agreement with the findings of Kristen et al23 and Barrett et al19 who found survival in the modern era to be comparable to non-amyloid transplants, in both AL and ATTR cohorts. Kristen et al. attributed improvements in their cohort to a more refined patient selection resulting in reduced extracardiac involvement and the introduction of new chemotherapeutic agents. Furthermore, our finding of a shift towards transplanting relatively more ATTR patients than AL, may add to the explanation for improved outcomes seen in the UNOS data. Additionally, our findings that the early era was associated with a significantly higher risk of mortality than the late era, irrespective of amyloid subtype, suggest that advances in plasma cell therapy administered after OHT for AL amyloidosis are contributing significantly to improved outcomes in AL amyloidosis.
Another consideration is the lengthy wait time associated with transplantation for cardiac amyloidosis patients. Interestingly, Barrett et al19 noted a significantly longer waitlist time for ATTR patients than AL, however, there was no appreciable difference in our cohort. In some instances, as was the case for four of our subjects, patients have opted to undergo transplantation in other regions with shorter wait times. Importantly, the 2018 Adult Heart Allocation Policy and companion guidance document specifically providing a pathway to transplant in cardiac amyloidosis may decrease wait times in this population, though this has yet to be determined.33 Novel mechanisms to transplant amyloid patients may be needed as they are still at a disadvantage with the new allocation system. Given the scarcity of organ availability, the continued increase in the demand for transplants, and the increased supply of HCV-positive organs in recent years 34, the use of HCV-positive donor organs presents an opportunity to reduce wait time. In our study, two HCV-negative patients were transplanted with HCV-positive donors. This may be particularly relevant for patients with amyloid cardiomyopathy who have non-dilated left ventricles and are often not eligible for durable mechanical support to bridge them successfully to transplantation.
Finally, with the approval of the TTR stabilizer tafamidis in May, 2019 for treatment of ATTR cardiac amyloidosis and more agents undergoing clinical trials, the landscape for transplant of ATTR cardiac amyloidosis is ever-changing. With greater awareness of disease and improvements in noninvasive diagnostic techniques, earlier diagnosis and treatment of these patients may reduce the number who develop end stage heart failure necessitating heart transplant. For those who do proceed to OHT without concurrent liver transplantation, deposition of TTR continues in the extracardiac organs and possibly the transplanted heart. Addition of TTR stabilizer or silencer agents to the standard post-transplant regimen, may improve post OHT outcomes even further, though this is purely speculative at this time.
We acknowledge several limitations to this study. First, the nonrandomized design precludes our ability to exclude the possibility that confounding variables such as selection bias may have influenced the survival of transplanted patients within subgroups. Second, although this is one of the largest cohorts of patients undergoing OHT for cardiac amyloidosis, this study is limited by a small sample size and by the number of patients with extended follow-up data. Third, the decision to select 2007 as the cut-off point between transplant eras, while justified, may introduce inherent biases to the study.
In conclusion, our data for mid-term follow-up post-OHT for AL and ATTR amyloidosis show encouraging results, particularly in the past decade. Taking into consideration that these patients are typically not candidates for durable mechanical circulatory support, these data suggest that amyloidosis should not be considered a contraindication to transplant. However, longer-term follow-up and larger multicenter studies are warranted to confirm and expand on these preliminary findings and compare to those of non-amyloid cardiomyopathies, particularly in the context of new and advancing therapies.
Supplementary Material
Acknowledgements:
Dr. Maurer is supported by a K24 grant from NIA (AG AG036778) entitled Midcareer Mentoring Award in Geriatric Cardiology.
Disclosure: Dr. Maurer reports grant support from National Institutes of Health [R01HL139671-01], [R21AG058348] and [K24AG036778], consulting income from Pfizer, GSK, EIdos, Prothena, Akcea and Alnylam, and institution received clinical trial funding from Pfizer, Prothena, Eidos and Alnylam. Dr. Clerkin is supported by the National Heart, Lung, and Blood Institute (Grant K23HL148528).
Abbreviations:
- AL
light-chain
- ATTR
transthyretin
- ATTRm
mutated transthyretin
- ATTRwt
wild type transthyretin
- HCV
hepatitis C virus
- OHT
orthotopic heart transplantation
- OLT
orthotopic liver transplantation
- UNOS
United Network for Organ Sharing
References
- 1.Maurer MS, Raina A, Hesdorffer C, et al. Cardiac transplantation using extended-donor criteria organs for systemic amyloidosis complicated by heart failure. Transplantation. 2007;83(5):539–545. [DOI] [PubMed] [Google Scholar]
- 2.Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation. 2005;112(13):2047–2060. [DOI] [PubMed] [Google Scholar]
- 3.Ruberg FL, Maurer MS, Judge DP, et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the Transthyretin Amyloidosis Cardiac Study (TRACS). Am Heart J. 2012;164(2):222–228 e221. [DOI] [PubMed] [Google Scholar]
- 4.Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016;387(10038):2641–2654. [DOI] [PubMed] [Google Scholar]
- 5.Holmgren G, Ericzon BG, Groth CG, et al. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet. 1993;341(8853):1113–1116. [DOI] [PubMed] [Google Scholar]
- 6.Ng B, Connors LH, Davidoff R, Skinner M, Falk RH. Senile systemic amyloidosis presenting with heart failure: a comparison with light chain-associated amyloidosis. Arch Intern Med. 2005;165(12):1425–1429. [DOI] [PubMed] [Google Scholar]
- 7.Rapezzi C, Merlini G, Quarta CC, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120(13):1203–1212. [DOI] [PubMed] [Google Scholar]
- 8.Connors LH, Prokaeva T, Lim A, et al. Cardiac amyloidosis in African Americans: comparison of clinical and laboratory features of transthyretin V122I amyloidosis and immunoglobulin light chain amyloidosis. Am Heart J. 2009;158(4):607–614. [DOI] [PubMed] [Google Scholar]
- 9.Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med. 2018. [DOI] [PubMed] [Google Scholar]
- 10.Davis MK, Lee PH, Witteles RM. Changing outcomes after heart transplantation in patients with amyloid cardiomyopathy. J Heart Lung Transplant. 2015;34(5):658–666. [DOI] [PubMed] [Google Scholar]
- 11.Pilato E, Dell’Amore A, Botta L, Arpesella G. Combined heart and liver transplantation for familial amyloidotic neuropathy. Eur J Cardiothorac Surg. 2007;32(1):180–182. [DOI] [PubMed] [Google Scholar]
- 12.Barreiros AP, Post F, Hoppe-Lotichius M, et al. Liver transplantation and combined liver-heart transplantation in patients with familial amyloid polyneuropathy: a single-center experience. Liver Transpl. 2010;16(3):314–323. [DOI] [PubMed] [Google Scholar]
- 13.Nelson LM, Penninga L, Sander K, et al. Long-term outcome in patients treated with combined heart and liver transplantation for familial amyloidotic cardiomyopathy. Clin Transplant. 2013;27(2):203–209. [DOI] [PubMed] [Google Scholar]
- 14.Liepnieks JJ, Zhang LQ, Benson MD. Progression of transthyretin amyloid neuropathy after liver transplantation. Neurology. 2010;75(4):324–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sabatino M, Potena L, Longhi S, et al. Outcomes of Heart Transplantation for Transthyretin-Related Amyloid Cardiomyopathy. The Journal of Heart and Lung Transplantation. 2016;35(4):S63–S64. [Google Scholar]
- 16.Rosenbaum AN, AbouEzzeddine OF, Grogan M, et al. Outcomes After Cardiac Transplant for Wild Type Transthyretin Amyloidosis. Transplantation. 2018;102(11):1909–1913. [DOI] [PubMed] [Google Scholar]
- 17.Gray Gilstrap L, Niehaus E, Malhotra R, et al. Predictors of survival to orthotopic heart transplant in patients with light chain amyloidosis. J Heart Lung Transplant. 2014;33(2):149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kpodonu J, Massad MG, Caines A, Geha AS. Outcome of heart transplantation in patients with amyloid cardiomyopathy. J Heart Lung Transplant. 2005;24(11):1763–1765. [DOI] [PubMed] [Google Scholar]
- 19.Barrett CD, Alexander KM, Zhao H, et al. Outcomes in Patients With Cardiac Amyloidosis Undergoing Heart Transplantation. J Am Coll Cardiol HF. 2020;8:461–468. [DOI] [PubMed] [Google Scholar]
- 20.Hosenpud JD, DeMarco T, Frazier OH, et al. Progression of systemic disease and reduced long-term survival in patients with cardiac amyloidosis undergoing heart transplantation. Follow-up results of a multicenter survey. Circulation. 1991;84(5 Suppl):III338–343. [PubMed] [Google Scholar]
- 21.Gillmore JD, Goodman HJ, Lachmann HJ, et al. Sequential heart and autologous stem cell transplantation for systemic AL amyloidosis. Blood. 2006;107(3):1227–1229. [DOI] [PubMed] [Google Scholar]
- 22.Mehra MR, Canter CE, Hannan MM, et al. The 2016 International Society for Heart Lung Transplantation listing criteria for heart transplantation: A 10-year update. J Heart Lung Transplant. 2016;35(1):1–23. [DOI] [PubMed] [Google Scholar]
- 23.Kristen AV, Kreusser MM, Blum P, et al. Improved outcomes after heart transplantation for cardiac amyloidosis in the modern era. J Heart Lung Transplant. 2018;37(5):611–618. [DOI] [PubMed] [Google Scholar]
- 24.Davis MK, Kale P, Liedtke M, et al. Outcomes after heart transplantation for amyloid cardiomyopathy in the modern era. Am J Transplant. 2015;15(3):650–658. [DOI] [PubMed] [Google Scholar]
- 25.Roussel M, Merlini G, Chevret S, et al. A prospective phase 2 trial of daratumumab in patients with previously treated systemic light-chain amyloidosis. Blood. 2020;135(18):1531–1540. [DOI] [PubMed] [Google Scholar]
- 26.Mehra MR, Canter CE, Hannan MM, et al. The 2016 International Society for Heart Lung Transplantation listing criteria for heart transplantation: A 10-year update. J Heart Lung Transplant. 2016;35(1):1–23. [DOI] [PubMed] [Google Scholar]
- 27.Buxbaum J, Alexander A, Koziol J, Tagoe C, Fox E, Kitzman D. Significance of the amyloidogenic transthyretin Val 122 Ile allele in African Americans in the Arteriosclerosis Risk in Communities (ARIC) and Cardiovascular Health (CHS) Studies. Am Heart J. 2010;159(5):864–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dispenzieri A, Gertz MA, Kyle RA, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol. 2004;22(18):3751–3757. [DOI] [PubMed] [Google Scholar]
- 29.Varr BC, Liedtke M, Arai S, Lafayette RA, Schrier SL, Witteles RM. Heart transplantation and cardiac amyloidosis: approach to screening and novel management strategies. J Heart Lung Transplant. 2012;31(3):325–331. [DOI] [PubMed] [Google Scholar]
- 30.Grogan M, Gertz M, McCurdy A, et al. Long term outcomes of cardiac transplant for immunoglobulin light chain amyloidosis: The Mayo Clinic experience. World J Transplant. 2016;6(2):380–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359(9):906–917. [DOI] [PubMed] [Google Scholar]
- 32.Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation. 2017;135(14):1357–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Committee OUTOT. Review Board Guidance for Hypertrophic and Restrictive Cardiomyopathy Exception Requests. 2018.
- 34.Levitsky J, Formica RN, Bloom RD, et al. The American Society of Transplantation Consensus Conference on the Use of Hepatitis C Viremic Donors in Solid Organ Transplantation. Am J Transplant. 2017;17(11):2790–2802. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.