

Abstract
Ectonucleotide phosphatase/phosphodiesterase 1 (ENPP1) deficiency results in either lethal arterial calcifications (‘Generalized Arterial Calcification of Infancy’ – GACI), phosphate wasting rickets (‘Autosomal Recessive Hypophosphatemic Rickets type 2’ – ARHR2), early onset osteoporosis, or progressive spinal rigidity (‘Ossification of the Posterior Longitudinal Ligament’ – OPLL). As ENPP1 generates a strong endogenous mineralization inhibitor – extracellular pyrophosphate (PPi) – ENPP1 deficiency should not result in reduced bone volume, and therefore the mechanism ENPP1 associated osteoporosis is not apparent given current understanding of the enzyme’s function. To investigate genetic pathways driving the skeletal phenotype of ENPP1 deficiency we compared gene expression in Enpp1asj/asj mice and WT sibling pairs by RNAseq and qPCR in whole bones, and in the liver and kidney by qPCR, directly correlating gene expression with measures of bone microarchitectural and biomechanical phenotypes. Unbiased analysis of the differentially expressed genes compared to relevant human disease phenotypes revealed that Enpp1asj/asj mice exhibit strong signatures of osteoporosis, ARHR2 and OPLL. We found that ENPP1 deficient mice exhibited reduced gene transcription of Wnt ligands in whole bone and increased transcription of soluble Wnt inhibitors in the liver and kidney, suggestive of multiorgan inhibition of Wnt activity. Consistent with Wnt suppression in bone, Collagen gene pathways in bone were significantly decreased and Fgf23 was significantly increased, all of which directly correlated with bone microarchitectural defects and fracture risk in Enpp1asj/asj mice. Moreover, the bone findings in 10-week old mice correlated with Enpp1 transcript counts but not plasma [PPi], suggesting that the skeletal phenotype at 10 weeks is driven by catalytically independent ENPP1 function. In contrast, the bone findings in 23-week Enpp1asj/asj mice strongly correlated with plasma PPi, suggesting that chronically low PPi drives the skeletal phenotype in older mice. Finally, correlation between Enpp1 and Fgf23 transcription suggested ENPP1 regulation of Fgf23, which we confirmed by dosing Enpp1asj/asj mice with soluble ENPP1-Fc and observing suppression of intact plasma FGF23 and ALP. In summary, our findings suggest that osteoporosis associated with ENPP1 deficiency involves the suppression of Wnt via catalytically independent Enpp1 pathways, and validates Enpp1asj/asj mice as tools to better understand OPLL and Paradoxical Mineralization Disorders.
Keywords: ENPP1, FGF23, Osteoporosis, Paradoxical mineralization, RNASeq, Rare bone diseases, Pyrophosphate, Generalized Arterial Calcification of Infancy (GACI), Autosomal Recessive Hypophosphatemic Rickets (ARHR2), Ossification of the Posterior Longitudinal Ligament (OPLL)
1. Introduction
The ecto-nucleotide pyrophosphatase/phosphodiesterase (ENPP) enzymatic family consists of seven human enzymes, all of which generate small molecules that function as extracellular factors that regulate essential physiologic processes. Members of the enzymatic family regulate whole organismal fate in mammals through essential biological pathways that include angiogenesis [1–3], cell motility [4,5], tumor metastasis [6–10], bone mineralization [11–14], vascular calcification [15–17], and hemostasis [18]. ENPP1 was the first enzyme identified and is the only human enzyme capable of generating extracellular pyrophosphate [16].
Homozygous ENPP1 deficiency in humans results in severe disorders of vascular calcification and aberrant bone mineralization. Generalized Arterial Calcification of Infancy (GACI) is a rare and frequently lethal neonatal disorder of arterial calcification in the medium and large arteries beginning as early as the late second trimester and culminating in death in approximately half of the affected infants by 6 months of age. The calcifications result from a generalized deficiency of pyrophosphate (PPi) due to loss of function mutations in ENPP1. We recently demonstrated that enzyme replacement therapy (ERT) with an ENPP1 biologic normalized plasma PPi and eliminated the mortality and sequela of disease in the accepted model of GACI – Enpp1asj/asj mice [19]. Most GACI children who survive to adolescence, as well as some children without a history of infantile vascular calcifications but who are later identified as having homozygous loss of function mutations in ENPP1, develop a phosphate wasting rickets known as Autosomal Recessive Hypophosphatemic Rickets Type 2 (ARHR2). Symptoms associated with ARHR2 include occasional long bone fractures, rachitic skeletal deformities, osteomalacia, and impaired growth and development as children [13,14], and polyarthralgias and rigidity due to tendon calcification as adults [20,21].
In addition to ARHR2, ENPP1 deficiency in humans has also been reported in a disease of progressive spinal rigidity called ‘Ossification of the Posterior Longitude Ligament’ (OPLL) [20,22]. The association of ENPP1 and OPLL is strengthened by murine models of OPLL, which were developed by Japanese researchers based on phenotype but were later discovered to possess loss of function mutations in ENPP1 (a Gly568stop mutation) leading to the OPLL phenotype [11]. OPLL is endemic in the Japanese population with a reported occurrence of 1.5–2.4% in the adult population, and patients typically suffer from nerve root compression resulting in radiculopathy and myelopathy that may severely limit the motility and affect the quality of life in OPLL patients. OPLL is a multifactorial disease where both environmental and genetic factors contribute to the development of the disorder, but signaling pathways of TGF-β and bone morphogenic protein have been identified as playing a key role in the development of the disease, and SNPs in ENPP1 are associated with OPLL, one of which is strongly associated with severe ossification and younger age of onset [23].
Soft tissue calcification resulting from decreased plasma PPi occurs in patients with Pseudoxanthoma Elasticum (PXE), a mineralization disorder unrelated to ENPP1 deficiency which has significant clinical overlap with GACI [24–27]. PXE results from deficiency of ABCC6 which encodes a member of the ATP-binding cassette transporter, subfamily C, member 6 [28–30]. PXE patients typically present in adolescence with characteristic skin calcifications, but many patients progress to ocular calcifications in the Bruch’s membrane, which may result in retinal detachment and blindness in adults. PXE is a monogenic, hereditable connective tissue disease induced by mutations in the ABCC6 gene, and aberrant mineralization may occur in the skin, eye, and vasculature leading to significant morbidity. The local and systemic effects of ABCC6 loss of function mutations result in reduced plasma [PPi] in both humans and mice, an effect currently assumed to result from decreased hepatic ATP secretion [31,32]. ATP is metabolized into PPi and AMP by ENPP1, and it is assumed that reduced extracellular [ATP] in ABCC6 deficiency impairs the generation of PPi by limiting the enzymatic substrate of ENPP1. However, unlike ENPP1 deficiency, patients with ABCC6 deficiency are not prone to fracture risk and osteoporosis despite increased vascular calcifications and decreased plasma PPi [33].
Finally, in collaboration with clinicians and researchers at the Department of Osteology and Biomechanics in Hamburg, we recently reported a cohort of middle age men with hereditary early onset osteoporosis (EOOP) manifested by vertebral and/or radial fractures and low bone mass with heterozygous ENPP1 deficiency. Specifically, these patients exhibit mildly elevated FGF23, decreased serum phosphorous, elevated urinary phosphate clearance, and radiographic evidence (DXA and HR-pQCT scans) of osteoporosis [34]. Evaluation of plasma PPi, performed in a single family to reduce PPi variability, revealed a gene dose effect of ENPP1 deficiency in humans, and the skeletal phenotype and serum biochemistries present in these patients were remarkably similar to a murine model of ENPP1 deficiency – Enpp1asj/asj mice. The close correspondence of the murine and human phenotype validates the use of Enpp1asj/asj mice to investigate the pathogenesis EOOP associated with ENPP1 deficiency. Accordingly, in order to broaden the understanding of this ENPP1 deficiency-driven pathogenesis, we performed RNAseq in whole bones of Enpp1asj/asj mice and wild type sibling pairs at 10 and 23 weeks of age. The whole bone analysis captures osteocytes, osteoclasts, and endothelial cells in bone vasculature to capture the bone mineralization microenvironment. We then correlated the transcriptome analysis with physical parameters of bone mineralization in the animals, including plasma biochemistries (Ca, Pi, PPi, PTH, FGF23, PTH, and CtX), bone mineralization parameters derived from micro-CT (Tb.BV/TV, Tb.Th, Ct.BV/TV, Ct.Th), dynamic histomorphometry (O.Wi, Mlt, OV/BV, BFR/BS, MAR), and 4-point biomechanical testing (Stiffness, Max. Load until Fracture, Post yield deflection) to identify, in an unbiased manner, the bone specific transcriptional alterations associated with ENPP1 deficiency.
2. Materials and methods
2.1. Mouse models
Animal care and maintenance were provided through Yale University Animal Resource Center at Yale University (New Haven). All procedures were approved by the Animal Care and Use Committee of Yale University and complied with the US National Institutes of Health guide for the care and use of laboratory animals. Heterozygous Enpp1asj/+ (genotype C57BL/6J-Enpp1asj/GrsrJ, Jackson Laboratory stock number 012810) breeding pairs were maintained on normal chow throughout the entire experiment. Food and water were delivered ad libitum. The animal colony was housed in pathogen free conditions. All experimental animals were maintained on normal chow in utero through completion of the study. Litters were genotyped on day 8 and weaned at day 21. Following weaning, sibling pairs were sequentially divided into cohorts as described below and assigned to experimental cohorts. Enrollment of the 10 week and 23 week Enpp1wt and Enpp1asj/asj cohorts spanned 4 months. Once the enrollment of an experimental group began, both sexes of the appropriate genotype were consecutively enrolled in an experimental cohort with the exclusion of severely runted animals weighing < 5.5 g at 14 days of life. Following weaning, all experimental animals were housed with littermates to allow for cooperative grooming and nesting. Experimentalists were not blinded during the study. Mature (> 3 months old) Hyp mice and WT sibling pairs were a gift of Steven Tommasini (Yale University).
2.1.1. Phenotypic measurements
The murine skeletons in 10 and 23 week WT and Enpp1asj/asj mice were characterized by histomorphometry, micro-CT, and biomechanically as previously described [34].
2.2. Quantification of plasma PPi
Blood plasma was measured in a blinded fashion. The PPi assay was performed as previously described [31,32] with minor variations. After plasma isolation and filtration, the plasma samples were diluted in 1:1 with 50 mM Tris-Acetate pH 8.0 buffer followed by filtration through a 30 kDa membrane (Amicon) via ultracentrifugation. The samples were aliquoted for single use and stored frozen at −80 °C. The plasma isolation, removal of platelets via filtration, and dilution and freezing of samples were performed within 1 h of blood collection. The PPi assay was carried out by mixing 5 μl plasma samples with 35 μl mixture composed of 50 mM Hepes pH 7.4, 16 μM adenosine phosphosulfate, 32 mU/ml ATP sulfurylase, and 80 mM MgSO4. Standards were prepared using serial dilutions of a freshly prepared 10 mM PPi stock solution (Acros Organics, A0371139), and run in parallel with the samples. All experimental solutions were prepared in a single 96-well PCR plate that was subjected to the following cycle to activate and deactivate ATP sulfurylase: 37 °C 10 min, 90 °C 10 min, 4 °C for hold. A Bactiter-Glo microbial cell viability assay (Promega, G8231) was used by mixing 20 μl product from the reaction and 20 μl of the Bactiter-Glo reagent. Luminescence signal was then read by Bio-Teck synergy HT multi-detection microplate reader at room temperature. Data analysis was performed via GraphPad Prism 7. Background PPi in the samples and water was subtracted and normalized from the samples, and the values were interpolated using Prism (Graphpad 7).
2.3. Measurement of PTH1–84, FGF-23, and ALP
Mouse PTH 1–84 and Mouse/Rat FGF-23 (intact) ELISA kits were purchased from Quidel Corporation (catalog number as 60-2305 and 60-6800, respectively). Mouse Alkaline Phosphatase (ALPL) ELISA kits were purchased from Lifespan BioSciences, Inc. (catalog number as LS-F5741-1). 45 μl plasma samples were used for all the above ELISA experiments. Data analysis was performed via GraphPad Prism 7. All other blood chemistry metrics were measured at the George M. O’Brien Kidney Center at Yale. Data analysis was performed via GraphPad Prism 7.
2.4. RNA isolation
Tibiae and femora were stripped of soft tissue and an ostectomy was performed at the distal ends of the lower limb long bones followed by centrifugation for 30 s at 2000 RPM to separate the tissues from the bone marrow. The bones were then immersed in 1 ml TRIzol Reagent with two 5 mm steel beads, and homogenized in a Retsch MM400 Mixer Mill at 30 Hz until the tissues were the size of granules (30–60 s). The samples were then snap frozen in liquid nitrogen and finally stored at −80 °C. The TRIzol RNA isolation protocol was used to extract total RNA from the samples. Kidney and liver RNA samples were collected in a similar manner with one kidney or liver lobe being homogenized with one 5 mm steel bead in 1 ml of TRIzol reagent. After storage in −80 °C. The standard TRIzol RNA isolation protocol was used to extract total RNA from all samples.
2.5. RNA sequencing
Whole-bone total RNA, extracted from 10 week and 23 week Enpp1wt and Enpp1asj/asj cohorts of males and females, was submitted to Yale Center for Genomic Analysis (YCGA) for sequencing. Samples passed RIN quality thresholds (> 5) and were then processed using the KAPA Biosystems Hyper Ribo library preparation kit for rRNA depletion Single-end RNA sequencing (75 bp) was performed on an Illlumina HiSeq2500, using four sequencing lanes containing ≤8 samples each, to an average depth of ≈ 31 million reads per sample. The resulting FASTQ files were QC’d using FASTQC [35], and aligned to the Ensembl GRCm38 reference genome [36] using STAR [37], after trimming the adapter sequences using Trimmomatic [38]. Gene expression levels, as read counts, were estimated and filtered by Cook’s distance using DESeq2 [37].
2.6. Differential gene expression analysis
The DESeq2 package [39] was used to identify differential gene expression across development in cohorts of 10-week (Enpp1wt = 8; Enpp1asj/asj = 8) and 23-week (Enpp1wt = 8; Enpp1asj/asj = 8) pooled male and female mice (4 males and 4 females of each genotype at each age) as well as to identify sex specific gene expression differences in each age group. Nominal p-values were FDR corrected for False Discovery Rates (FDR) and a significance threshold was set at FDR < 0.05. To understand their relevance to human disease and to evaluate the predictive power of the Enpp1asj/asj mouse model, we grouped differentially expressed genes into functionally or protein familial categories (Table 2), based on identification to known genetic targets involved in skeletal disorders associated with ENPP1 deficiency – ARHR2, OPLL, and osteoporosis, and evaluated their expression patterns across age groups.
Table 2.
Genes involved in ARHR2, osteoporosis, and OPLL and their associated fold changes in 10 week and 23 week Enpp1asj/asj mice as compared to WT. Statistical significance after FDR correction (< 0.05) are highlighted in blue.
| Cohort | Gene | Fold change | Padj |
|---|---|---|---|
| ARHR2 | |||
| 10-week | Dmp1 | 1.5 | 0.21 |
| Enpp1 | 1.8 | 1.3E–03 | |
| Fgf23 | 6.0 | 5.1E–12 | |
| Gcnt4 | 1.6 | 0.45 | |
| Spp1 | 1.5 | 0.27 | |
| Vdr | 1.4 | 0.02 | |
| 23-week | Alpl | −1.6 | 0.07 |
| Col12a1 | −1.7 | 7.1E–04 | |
| Col6a1 | −1.4 | 0.01 | |
| Col6a2 | −1.5 | 2.2E–03 | |
| Col6a3 | −1.4 | 2.6E–03 | |
| Enpp1 | 1.2 | 0.22 | |
| Fgf23 | 2.6 | 0.03 | |
| Hacd1 | −1.4 | 0.05 | |
| Mmp2 | −1.4 | 2.4E–03 | |
| Spp1 | 1.4 | 0.22 | |
| Osteoporosis | |||
| 10-week | Mepe | −1.5 | 0.37 |
| Sost | −1.6 | 0.19 | |
| Spp1 | 1.5 | 0.27 | |
| Vdr | 1.4 | 0.02 | |
| 23-week | Alpl | −1.6 | 0.07 |
| Esrl | −1.3 | 0.06 | |
| Mepe | −1.4 | 0.38 | |
| Sost | −1.6 | 0.42 | |
| Sp7 | −1.4 | 0.09 | |
| Spp1 | 1.4 | 0.22 | |
| Tgfb3 | −1.4 | 0.08 | |
| Wnt16 | −1.9 | 1.3E-04 | |
| Wnt10b | −1.8 | <0.01 | |
| Lrp1 | −1.2 | <0.05 | |
| OPLL | |||
| 10-week | Ace | −1.2 | 0.33 |
| Enpp1 | 1.8 | 1.3E-03 | |
| Vdr | 1.4 | 0.02 | |
| 23-week | Acvr1 | −1.4 | 0.03 |
| Adamts2 | −1.2 | 0.02 | |
| Antxr1 | −1.4 | 3.8E-03 | |
| C1s1 | −1.3 | 0.03 | |
| Cdon | −1.5 | 4.2E–03 | |
| Col12a1 | −1.7 | 7.1E–04 | |
| Col6a1 | −1.7 | 0.01 | |
| Col3a1 | −1.8 | 0.03 | |
| Col5a1 | −1.4 | 0.02 | |
| Col6a1 | −1.4 | 0.01 | |
| Col6a2 | −1.5 | 2.2E–03 | |
| Col6a3 | −1.4 | 2.6E–03 | |
| Col8a2 | −1.8 | 2.5E–05 | |
| Dchs1 | −1.3 | 0.01 | |
| Ddr2 | −1.4 | 3.8E–04 | |
| Dlx5 | −1.7 | 4.1E–04 | |
| Dnmt3a | −1.2 | 2.6E–03 | |
| Esr1 | −1.3 | 0.06 | |
| Fat4 | −1.4 | 0.01 | |
| Fbn1 | −1.4 | 0.03 | |
| Fgfr1 | −1.3 | 0.08 | |
| Fgfr2 | −1.3 | 0.03 | |
| Gpc3 | −1.5 | 0.02 | |
| Hspb8 | −1.6 | 0.04 | |
| Hspg2 | −1.3 | 0.02 | |
| Itgb6 | −1.7 | 0.02 | |
| Klhl41 | −1.7 | 0.01 | |
| Mylk | −1.3 | 3.3E-05 | |
| Pcolce | −1.3 | 0.01 | |
| Pcolce2 | −1.9 | 0.01 | |
| Prkg1 | −1.6 | 0.04 | |
| Ptch1 | −1.2 | 0.04 | |
| Ror2 | −1.6 | 0.03 | |
| Tgfb3 | −1.4 | 0.08 | |
| Tnxb | −1.8 | 2.9E–03 | |
2.6.1. Functional annotation
The g:GOSt feature on gProfiler software [40] was used under the default parameters to perform functional profiling on significant Differentially Expressed Genes (DEGs). Genes belonging significantly enriched (padj < 0.05) clinical abnormality terms were recorded.
2.6.2. Association of phenotypic traits with gene expression
We calculated Pearson correlation coefficients between phenotypic measurements of Enpp1wt and Enpp1asj/asj mice and expression levels of genes with familial enrichment and/or statistical significance. Unsupervised clustering (hclust R function and method = “average”) on gene expression, biochemical and physical phenotypes (plasma Ca, Pi, PPi, PTH, FGF23, PTH, and CtX), micro-CT derived parameters of Tb.BV/TV, Tb.Th, Ct.BV/TV, Ct.Th, dynamic histomorphometry parameters including O.Wi, Mlt, OV/BV, BFR/BS, MAR, and 4-point biomechanical fracture data including bone stiffness, maximum load until fracture, and post yield deflection was then performed. The resulting analysis was used to generate a correlation matrix in order to identify clusters of strongly correlated genes and co-regulated phenotypic responses. The Student’s two-tailed t-test was used on individual correlation values to determine whether the correlation was significantly non-zero. This was followed by performing a Benjamini-Hochberg correction to all p-values to correct for FDR [41,42].
2.6.3. Genotype-phenotype co-expression networks
Gene/metabolite co-expression networks (gene2source) were inferred by connecting nodes (genes/metabolites) that were strongly correlated (|R| > 0.7). To isolate the interactions related to ENPP1 and plasma [PPi], only nodes that were above this correlation threshold and had a 1st or 2nd degree relationship with ENPP1, plasma [PPi], or a relevant phenotypic measurement in the cohort (i.e., bone micro-architecture as determined by micro-CT, bone histomorphometry, or bone biomechanical properties as determined in 4-point bending experiments) were retained in the network. To relate these gene networks to bone phenotypes, we correlated the same genes observed in the gene2source network with physical parameters in the phys2gene (highly correlated phenotypic and genetic/metabolic measurements within a cohort).
2.6.4. Fgf23 regulation
Relative gene expression levels were correlated to Fgf23 by using genes that were not detected as count outliers using Cook’s distance by DESeq2, creating an extensive set of genes for comparison (17,177 at 10wks and 15,976 at 23wks). Top candidates from this analysis indicate genes with the highest likelihood of having a correlated response to changes in Fgf23. Genes correlated with Fgf23 at |R| > 0.8 within a cohort were plotted to identify relationships and correlations within the genetic hub. Correlations at |R| > 0.8 within this genetic hub were graphed using statnet and network packages [43], where an increasing |R| value results in a thicker edge.
2.7. Validation of genetic pathways using qPCR
To cross-validate our RNAseq results, total RNA was collected from whole bone, kidney, and liver in 23 week old male mice. RNA samples were measured for purity using A260/A280 ratios from a Thermo Scientific NanoDrop Spectrophotometer. RNA samples with A260/A280 ratios > 1.8 were considered pure and proceeded to cDNA synthesis. Superscript IV Reverse Transcriptase by Invitrogen reagents and protocol was used with a few minor modifications to achieve cDNA from the total RNA samples. cDNA was then diluted 1:500 with DEPC-treated RNAse free water. Oligonucleotide primer pairs were designed using benchling primer wizard with all primers used having a penalty score below 1. Oligonucleotide primers were synthesized using Yale Keck Oligonucleotide Synthesis facility. Primers were mixed to a final concentration of 200 nM. Primer sequences used can be found in Supplementary Table 18. The BioRad SYBR green fluorophore and protocol were used for duplicate primer-pair qPCR plate reads on a CFX96 Touch Real-Time PCR Detection System. qPCR runs consisted of forty cycle-thresholds followed by a melt-curve analysis. Primer integrity was assessed via melt-curve. GAPDH was selected as the housekeeping gene after investigating a panel containing GAPDH, ACTIN, B2M, HPRT, and HMBS and discovering GAPDH had the least variance under Enpp1wt and Enpp1asj/asj conditions. Therefore, all primers were normalized to GAPDH. RT-qPCR Ct data was statistically analyzed using student’s two-tailed t-test in GraphPad Prism 7.
2.8. Fgf23 response in ENPP1-Fc dosing
Human ENPP1-Fc was prepared as previously described [19]. Enpp1asj/asj were dosed subcutaneously three times a week for two-weeks with human ENPP1-Fc at 2.5, 5, and 10 mg/kg, or 6 times a week at 10 mg/kg. Plasma was taken before dosing began, and 72 h after the last dose to measure plasma concentrations of PPi, intact FGF23, and Alkaline Phosphatase. Intact FGF23 was measured with an ELISA kit purchased from Quidel Corporation (catalog number as 60-6800).
3. Results
Differential expression analysis between WT and 10-week Enpp1asj/asj mice identified 25 differentially expressed genes (FDR < 0.05); 18 downregulated and 7 upregulated in Enpp1asj/asj (Fig. 1A). In contrast, at 23-weeks, Enpp1asj/asj mice show more pronounced transcriptional alterations with 268 differentially expressed genes, 46 up-regulated and 222 down-regulated in Enpp1asj/asj as compared to wild-type. This included the down-regulation of genes associated with matrisomal and collagen, Wnt, and other pathways implicated in skeletal development.
Fig. 1.
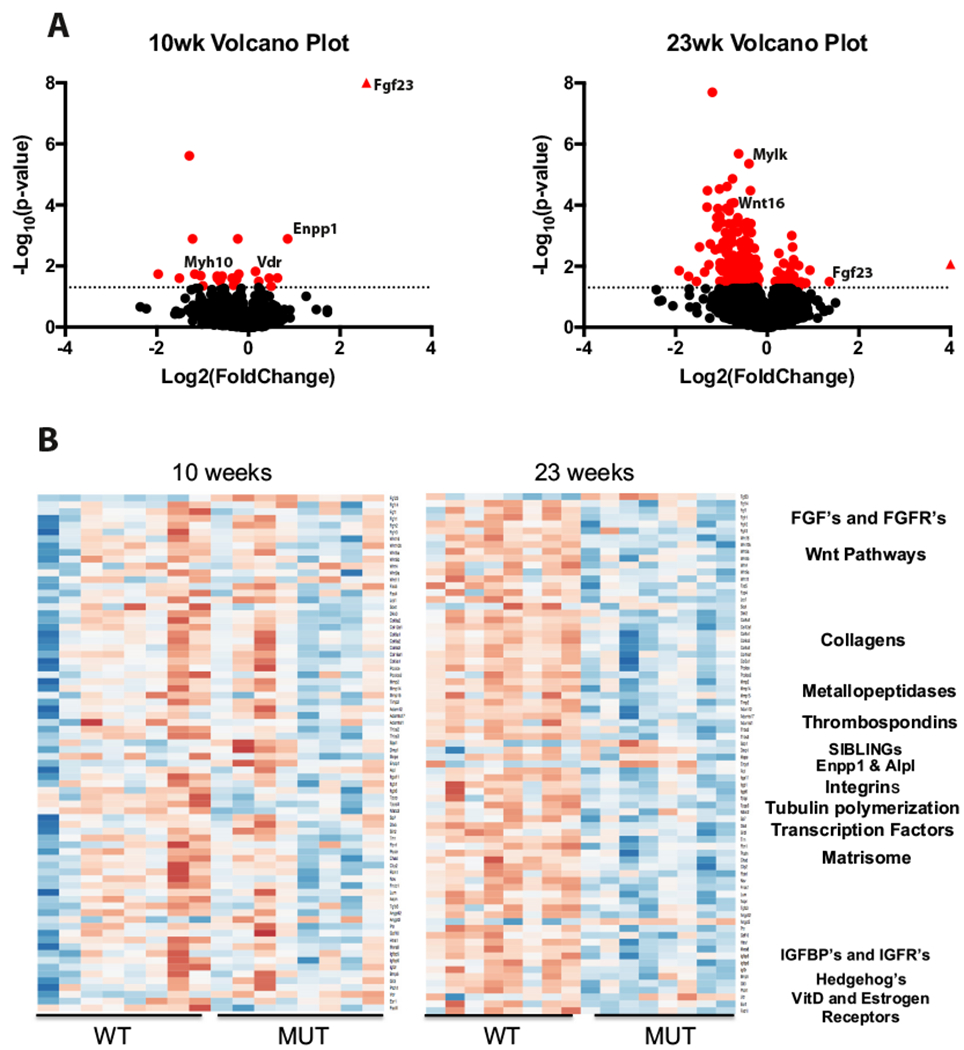
Differential gene expression in Enpp1asj/asj mice as compared to wild-types show integral bone pathways are uniquely affected at 10 and 23 weeks. (A). Volcano plots show genes crossing the significance threshold (red) are more numerous at 23-weeks with the majority displaying decreased expression. In contrast, the expression profile at 10 weeks shows a select few genes as significant with similar proportions displaying increased and decreased expression. Heatmaps (B) of genetic pathways in Enpp1 deficient mice exhibit differential expression in key pathways of Fgf signaling, Wnt activity, Collagens, SIBLING’s, and extensive matrisomal components. Expression differences in these pathways are not identical at 10 and 23 weeks of age, suggesting temporal differences are relevant in Enpp1 deficiency. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Among genes associated with phosphate regulation and osteomalacia, Fgf23 was strongly upregulated at 10 and 23 weeks (Fold Change of 6.0 and 2.6, in Enpp1asj/asj to Enpp1wt comparisons respectively) and Enpp1 was up-regulated at 10-weeks (Fold Change = 1.8) but not significantly changed at 23-weeks. Alpl was not altered at 10-weeks but trended downward at 23-weeks (Fold Change = −1.6, padj = 0.066), while Phex was not differentially expressed at either 10-weeks or 23-weeks. Spp1 (Osteopontin) and Dmp1 trended higher at 10 and 23 weeks without reaching statistical significance. Ank, Nt5e, Clcn5 expression was unaffected, while Abcc6, Slc34a1, and Slc34a3 expression levels were considered as non-expressed and filtered out before analysis.
Examination of functional group expression patterns in genes at 10 and 23 weeks (Fig. 1B) further support the increased progressive misregulation of pathways associated with skeletal morphology (Fgf/Fgfr’s, Wnt pathways, Collagens, Metallopeptidase activity, SIBLINGs), as well as pathways associated with purinergic metabolism, enthesopathy and rickets (matrisomal genes, Fig. 1B; Table 1; Supplementary Tables 1–7). Other notable pathways affected included insulin growth factor pathways, hedgehog transcription factor pathways, hormone metabolism, and others (Fig. 1B; Table 1; Supplementary Tables 8–13).
Table 1.
Targeted genes governing the skeletal phenotype differentially expressed in 10 or 23 week Enpp1asj/asj mice.
| Gene family | Members affected* | Regulation | |
|---|---|---|---|
| Fgf/Fgfr’s | Fgf ligands | Fgf23****, Fgf14* |
↑Fgf23 ↓Fgf14 |
| Fgf receptors | Fgfr2* | ↓ | |
| Wnt pathways | Wnt ligands | Wnt16****, Wnt10b**, | ↓ |
| Wnt co-receptors | Lrp1*, Lrp8* |
↑Lrp8 ↓Lrp1 |
|
| Wnt inhibitors | Dkk3*, Sfrp2* | ↓ | |
| Collagens | Collagens | Col3a1*, Col5a1*, Col6a1*, Col6a2**, Col6a3**, Col8a2****, Col12a1****, Col16a1**, | ↓ |
| Pro-collagens | Pcolce**, Pcolce2** | ↓ | |
| Metallopeptidase activity | Metallopeptidases | Mmp2**, Mmp14****, Timp2**** | ↓ |
| A disintegrin and metallopeptidase | Adam 12**, Adamts2*, Adamts17**** | ↓ | |
| Thrombospondins | Thbs2****, Thbs3**** | ↓ | |
| SIBLING | Dentin matrix protein | Dmp1**(male only) | ↑ |
| Matrix extracellular phosphoglycoprotein | Mepe**(female only) | ↓ | |
| Purinergic metabolism | Enpp1 | Enpp1** | ↑ |
| alkaline phosphatase | Alpl* (male only) | ↓ | |
| Integrins | Integrin alpha | Itga11**** | ↓ |
| Integrin beta | Itgb6* | ↓ | |
| Tubulin polymerization | Tubulin polymerization promoting protein | Tppp*, Tppp3**** | ↓ |
| Transcription factors | Nuclear factor of activated T cells | Nfatc2* | ↓ |
| Distal-less Homeobox | Dlx5**** | ↓ | |
| Matrisome | Matrisomal pathway | Slit2*, Tnn****, Fbn1*, Postn**, Chad**, Cilp2**, Fbln1**, Nov****, Fndc1****, Lum*, Aspn**, Angptl2**, Angpt2**, Rn****, Gdf10*, Htra1****, Mxra8**** |
↑Angpt2 ↓Slit2, Tnn, Fbn1, Postn, Chad, Cilp2, Fbln1, Nov, Fndcl, Lum, Aspn, Angptl2, Ptn, Gdf10, Htra1, Mxra8 |
| Insulin growth factor related | Insulin growth factor related | Igfbp5**, Igfbp6*, Igf2r* | ↓ |
| Hedgehog | Hedgehog signaling | Hhatl****, Ptch1* | ↓ |
| Hormone metabolism | Hormone receptors | Vdr* | ↑Vdr |
Affected members are included based on significant differential gene expression in 10 or 23 week animals.
Padj < 0.05
padj < 0.01
padj < 0.005
padj < 0.001.
Unbiased analysis of the differentially expressed genes to relevant diseased phenotypes using g:Profiler returned significant associations in 10-week samples with Human Phenotype Ontologies (HPO) genes associated with osteomalacia, rickets, and hypophosphatemia, which are all phenotypes characterizing children with ARHR2 (Fig. 2). At 23 weeks, the same analysis returned a more complex pattern with HPO associated genes including significant enrichment for many other disease phenotypes such as scoliosis, abnormality of the hip joint and hand joint mobility, and abnormality of the curvature of the vertebral column, which are phenotypes present in adults with ARHR2 and OPLL.
Fig. 2.
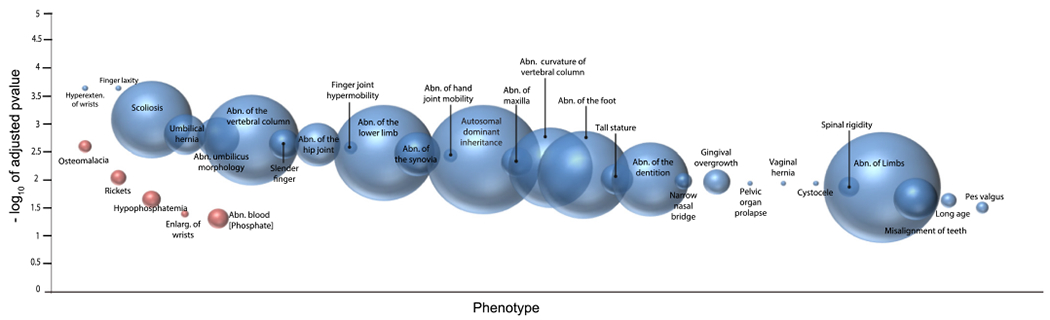
Enriched human phenotype ontological terms associated with differentially expressed genes suggest critical phenotypes in osteomalacia, rickets, enthesopathy, osteoporosis, and OPLL are implicated in Enpp1 deficiency. Phenotypes at 10 (red) and 23 (blue) weeks were plotted as bubbles where increasing bubble size indicates an increasing number of terms contributing to the phenotype and increasing y-axis value indicates increasing statistical significance (−log10(padj)). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.1. Osteoporosis
Established pathways of bone mass disrupted in osteoporosis were affected by ENPP1 deficiency in Enpp1asj/asj mice (summarized in Supplementary Tables 1–13), including Fgf23 [44], Wnt (Supplementary Table 2) [45], Collagens [46] (Supplementary Table 3), Fgf/Fgfr (Supplementary Table 1) [47], genes associated with matrisomal activity (Supplementary Table 7) [48], Metallopeptidases (Supplementary Table 4) [49], and the Hedgehog signaling pathway (Supplementary Table 12) [50]. Additionally, genes associated with osteoporotic fracture in a large GWAS meta-analysis [51], including Mepe, Spp1, Wnt16, and Sost were also differentially expressed Enpp1asj/asj mice. Other genetic pathways associated with human osteoporosis which were differentially expressed in Enpp1 deficiency including Vdr, Esr1, and Tgfb3 (Table 2) [51]. Finally, Alpl and Sp7 expression trended downward at 23 weeks without reaching statistical significance. The list of differentially expressed genes in Enpp1asj/asj mice that are known to play a role in human osteoporosis are cataloged in Table 2.
3.1.1. qPCR validation
A subset of the gene pathways driving skeletal development were probed by RT-qPCR, demonstrating congruence in the directional fold changes of the RNAseq analysis with a greater statistical power to identify differential gene expression. Specifically, the studies confirmed an increase in Fgf23, a decrease in Wnt activity (Wnt5a, Lrp6, and Ctnnb1), and a decrease in the expression of genes involved in bone formation (Bglap, Ibsp, Sp7) and bone resorption/remodeling (Tnfrsf11b and Mmp2) (Table 3). Additionally, the studies identified a previously unobserved increase in Abcc6. Overall, the data demonstrates that the magnitude of fold-change in gene expression is greater in the qPCR data than the RNAseq data. We next investigated systemic effects of Enpp1 deficiency on Wnt signaling in liver and kidney, demonstrating significant increases in the Wnt inhibitor Sfrp1 and significant decreases in the Wnt co-receptor Fzd8 in the liver, and significant increases in the Wnt inhibitor, Wif1 in the kidney. The data confirmed the RNAseq findings demonstrating the disruption of pathways of bone mass by ENPP1 deficiency.
Table 3.
Expression-fold changes in targeted genes driving skeletal phenotype in 23-week old males (n = 8).
| Gene family | Gene | qPCR expression | RNAseq expression |
|---|---|---|---|
| Bone | |||
| †Fgf activity | Fgf23 | +6.0** | +2.8** |
| †Wnt activity | Lrp6 | −2.5* | −1.2 |
| Wnt5a | −2.8* | −2.1 | |
| Ctnnb1 | −1.8* | −1.1 | |
| †Collagens | Col1a1 | −3.3 | −1.5 |
| †Metallopeptidases | Mmp2 | −2.5* | −1.5 |
| †Purinergic | Alpl | −2.8* | −1.4 |
| Abcc6 | +6.9** | Below detection limit | |
| Cd73 | −2.4** | Below detection limit | |
| †Bone formation | Bglap (OCN) | −2.6** | −1.3 |
| Sp7 | −1.8** | −1.4 | |
| Ibsp (BSP) | −2.1* | −1.3 | |
| †Bone resorption | Tnfrsf11b | −4.6* | −1.1 |
| Liver | |||
| ‡Wnt activity | Sfrp1 | +2.1* | N/A |
| Fzd8 | −3.4** | N/A | |
| Kidney | |||
| ‡Wnt activity | Wif1 | +11.7* | N/A |
Comparison of fold-changes by RT-qPCR and RNAseq.
RT-qPCR evaluation of systemic Wnt activity in select organs.
p < 0.05,
p < 0.01,
Student’s unpaired t-test. The following genes were tested in 23-week old males, and demonstrated no statistically significant differences: Sost, Wisp2, Lrp5, Csnk1a1, Ctnnb1, Apc, Axin1, Dvl1, and Gsk3b.
Blue cells denote organs from which RNA was extracted and gene expression measured.
3.1.2. Relationship of bone mass and Enpp1 catalytic activity
ENPP1 deficient mice exhibit severe osteoporosis with mild osteomalacia that is only detected on histology [34]. ENPP1 deficiency results in decreased PPi which should allow for (at least) appropriate apposition of mineral in the available bone matrix, rather than the overall reduction in bone mass observed in an osteoporotic animal. The mechanism by which ENPP1 deficiency results in osteoporosis is therefore not apparent given our understanding of the protein’s catalytic activity. To address this paradox we evaluated transcriptional networks associated with either ENPP1 catalytic activity (plasma PPi) or ENPP1 transcription counts in order to detect catalytically independent signaling networks. We found that 10-weeks Enpp1 gene counts correlated with Fgf23, Spp1, Dmp1, Bmp3, Bmp8a, Lrp5, and metabolic blood measurements such as non-fasted blood glucose and intact FGF23, while plasma [PPi] correlated with Nfatc2, Clec4e, and plasma parathyroid hormone (PTH) (Fig 3A). Moreover, gene2source and phys2gene networks illustrated that Enpp1 transcription counts strongly correlated with bone mass through physical parameters that included tibial fracture load, tibial maximal Load, tibial stiffness, and bone micro-architecture in parameters that included trabecular thickness (Tb.Th), trabecular bone surface/bone volume (BS/BV), and the structure model index (SMI) (Fig. 4A). In contrast, plasma [PPi] at 10 weeks strongly correlated with osteomalacia in physical measures such as bone compliance (tibial yield load and work-to-yield), osteoid volume (OV/BV), and osteoid thickness (O.Wi). Our analysis suggests that at 10 weeks ENPP1 catalytic activity is linked to osteomalacia, but surprisingly, catalytically independent Enpp1 signaling may regulate bone mass at 10-weeks.
Fig. 3.
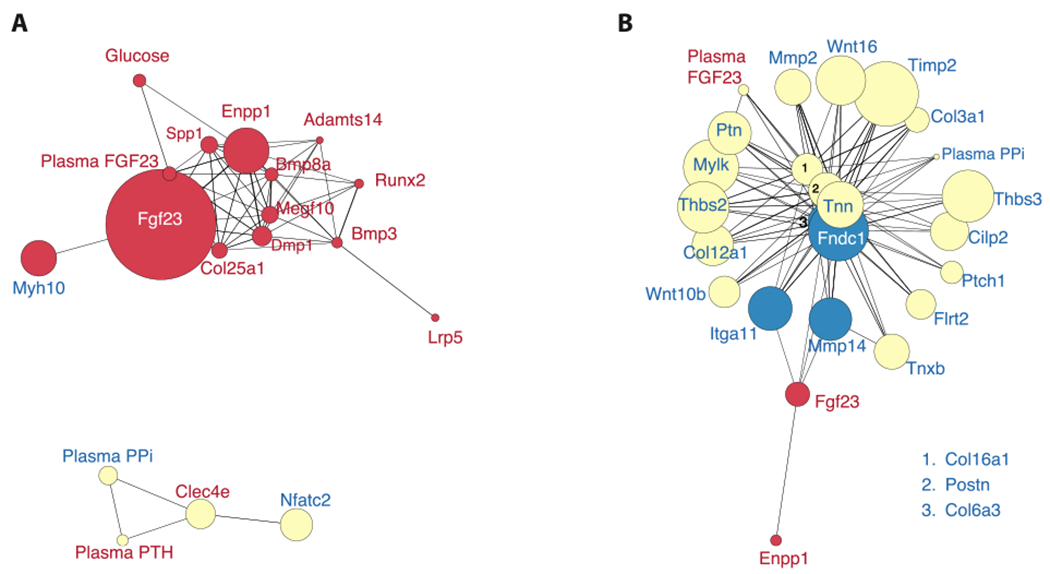
Age dependent bone mineralization pathways in Enpp1 deficiency show a temporal shift of Enpp1 catalytic and non-catalytic activity on gene co-expression networks. Red nodes correlate with Enpp1 transcription, yellow nodes correlate with Plasma PPi measurements, and blue nodes correlate with both. Increasing node size represents increasing significance of differential gene expression, where −log10(p-value) was used to calculate the node’s size. (A) At 10wks, non-catalytic Enpp1 signaling triggers a network containing Fgf23, Opn, Dmp1, and other elements regulating bone formation. Plasma PPi appears less determinative at this stage of development with the exception of Parathyroid hormone and Nfatc2, both of which regulate bone resorption. (B) Co-expression data at 23wks demonstrates the importance of PPi on disease progression with time. At 23 weeks, all the major pathways regulating bone mineralization including the Wnt pathway and matrisomal genes now correlate with plasma PPi. Enpp1 transcription remains correlated to Fgf23 transcription, but intact plasma Fgf23 levels also correlate with plasma PPi at 23 weeks. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 4.
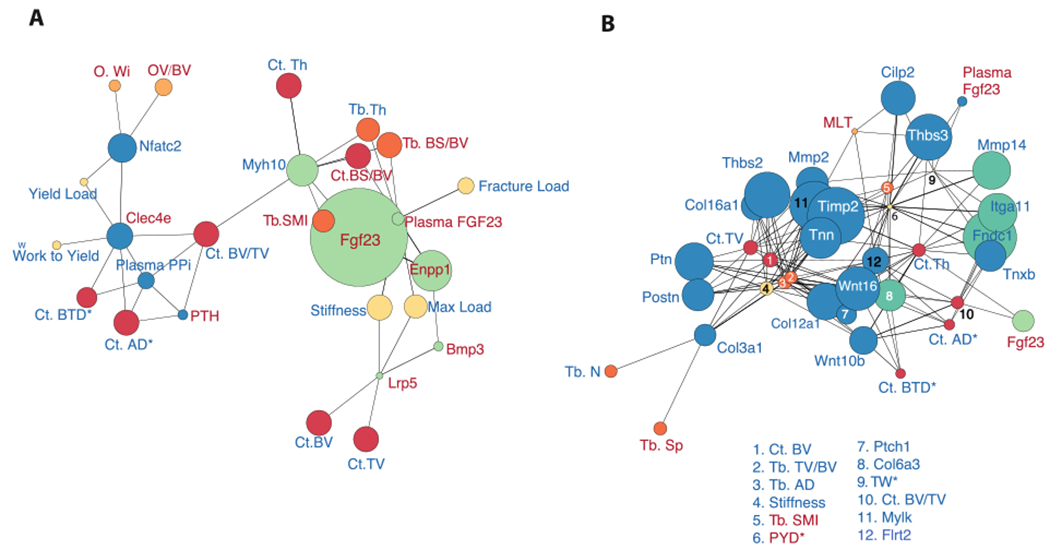
Networks of correlated bone phenotypes with plasma PPi, Enpp1 gene expression levels, and their downstream targets indicate Enpp1 catalytic and non-catalytic activity temporally influence distinct aspects of Enpp1 deficient phenotypes. Green nodes correspond to elements correlated with Enpp1 gene expression levels and blue nodes represent elements correlated with plasma PPi concentrations. Biomechanic, trabecular microarchitecture, cortical microarchitecture, and Osteoid phenotypes are displayed as yellow, dark orange, red, and light orange, respectively. (A) Combined genetic/phenotypic networks show Enpp1 gene expression levels at 10-weeks to be correlated with biomechanic phenotypes such as bone strength and microarchitectural measures of trabecular and cortical bone mass (dark orange and red nodes). Genes associated with the catalytic effects of ENPP1 (plasma PPi) correlate with cortical measurements of tissue density and bone volume, as well as strong correlation with measurements of osteoid volume and thickness. Fgf23 appears associated with microarchitectural deficits and osteopenia, consistent with recent associations of Fgf23 in humans [65], while plasma PPi is associated with osteomalacia. (B) At 23 weeks, the associations are reversed, with plasma PPi now associated with osteoporotic phenotypes, including multiple metrics of bone strength and mass. Enpp1 transcription counts are now solely correlated with Fgf23, which is associated with cortical bone volume and thickness. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
At 23 weeks the skeletal phenotype of Enpp1asj/asj mice transforms into a low bone turnover osteoporosis with decreased PTH, dramatically reduced bone formation rates, and a severe osteoporosis [34], and the genotype correlations at 23 weeks now shows plasma [PPi] as the dominant signaling network responsible for this phenotype, with the effects of Enpp1 signaling limited to Fgf23 (Fig. 3B). At 23 weeks plasma [PPi] now becomes associated with Wnt pathways, Collagens, Metallopeptidases, and various other Matrisomal genes, suggesting that the low bone turnover osteoporosis may be driven by the accumulated effects of low plasma PPi. An increase in network complexity as evidenced by an increase in high degree nodes indicates the more influential role of PPi on skeletal phenotype at later stages of development (Fig. 4B).
3.2. Ossification of the Posterior Longitudinal Ligament (OPLL)
Evidence of OPLL in murine ENPP1 deficiency was observed by multiple analyses. Results from using gProfiler to identify annotation enrichment of the human phenotype disease ontological terms revealed the significant overrepresentation of genes associated with Scoliosis (padj < 0.01) and Spinal Rigidity (padj < 0.01) in differentially expressed genes from Enpp1asj/asj 23-week mice (Fig. 2; Supplementary Table 17). The Scoliosis ontology is defined to represent “The presence of an abnormal lateral curvature of the spine” and consists of differential expression in Mylk, Ddr2, Dlx5, Col12a1, Col6a1,Col6a2, Col6a3, Dnmt3a, Tnxb, Antxr1, Cdon,Dchs1, Fat4, Klhl41, Hspg2, Adamts2, Col5a1,Gpc3, Itgb6, Acvr1, Cls1, Fgfr2, Col3a1,Ror2, Fbn1, Prkg1, Hspb8, and Ptch1 genes, and spinal rigidity is defined to predict “Reduced ability to move the vertebral column with a resulting limitation of neck and trunk flexion” and consists of differential expression of Col6a1, Col6a2, Col6a3,Col12a1, Klhl41, Hspg2, and Acvr1. The differentially expressed genes associated with the OPLL phenotype observed in this experiment are cataloged in Table 2.
OPLL specific genes altered in Enpp1asj/asj mice include Vdr, and Enpp1 (differentially expressed at 10wks) and Col6a1 and Ptch1 (Table 2). Wnt activity, which is suppressed in human OPLL [52], was discussed above (Supplementary Table 2), and collagen expression (also decreased in human OPLL) is also significantly decreased in 23-week Enpp1asj/asj mice (e.g. Col3a1, Col5a1, Col6a1, Col6a2, Col6a3, Col8a2, Col12a1, Col16al, Pcolce, Pcolce2, Table 1).
3.3. Autosomal Recessive Hypophosphatemic Rickets type 2 (ARHR2)
At 10-weeks, g:Profiler returned human phenotype clinical abnormality terms of osteomalacia (padj < 0.01), rickets (padj < 0.01), hypophosphatemia (padj < 0.05), enlargement of the Wrists (Padj < 0.05), and abnormal blood phosphate concentration (Padj < 0.05), all phenotypes observed in ARHR2 [20,22], (Fig. 2, Supplementary Table 17). Fgf23 is up-regulated at 10-weeks (log fold change = 6.0, padj < 0.0001) in a correlative pattern with Enpp1, Spp1, and Dmp1, demonstrating that the regulation of FGF23 by ENPP1 occurs at a genetic (as opposed to posttranscriptional) level (Fig. 5A and B, Supplementary Tables 14 and 16, Supplementary Fig. 1). Dmp1 and Spp1, which have been associated with Fgf23 activity [53], correlate with elements of the Fgf23 network but with less overlap than ENPP1, suggesting that ENPP1 may regulate Fgf23 transcription or that the two may be co-regulated by the same upstream element (Fig. 5A and B).
Fig. 5.
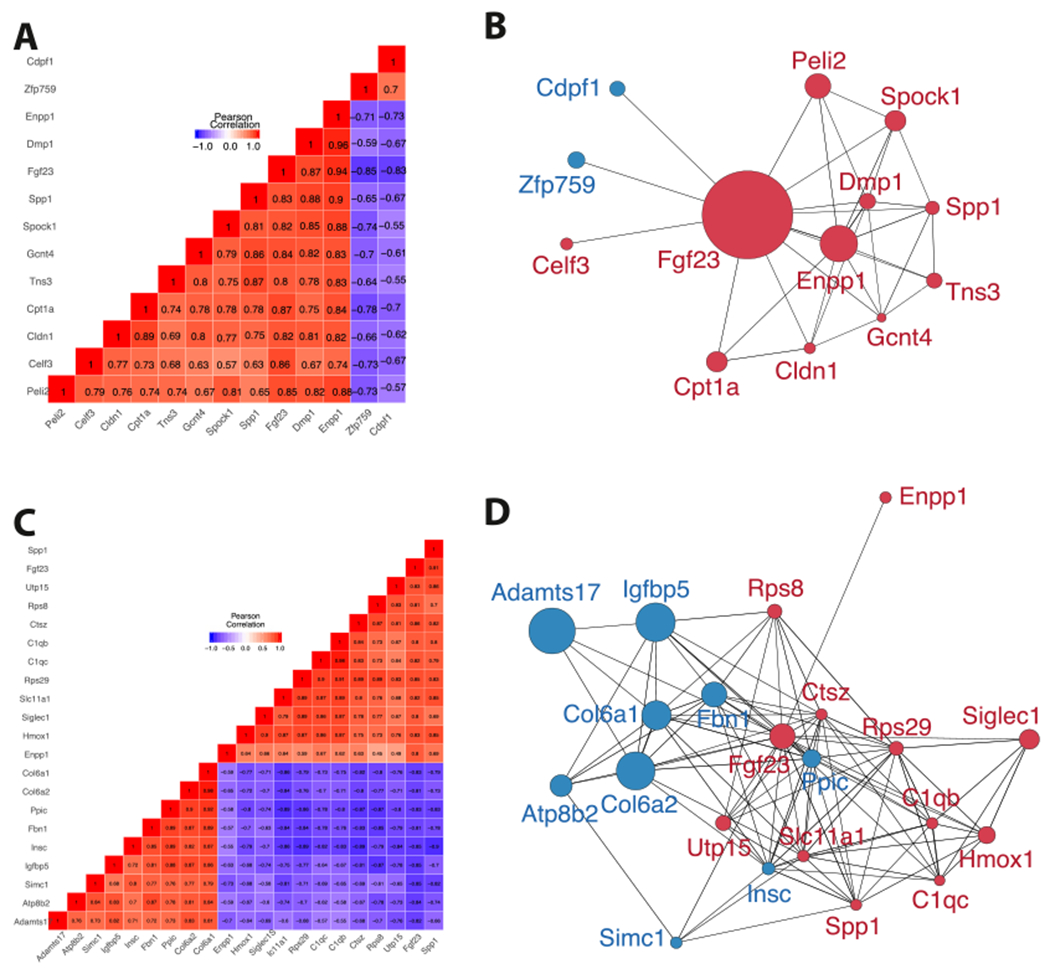
Unsupervised clustering and correlation networks suggest that Enpp1 transcription contributes to the regulation of Fgf23 expression. Red and blue represent positive and negative correlation, respectively, with increasing color intensity denoting increased correlation in co-expression plots. In the networks, red nodes represent genes that were up-regulated and blue nodes represent genes that were down-regulated. (A) The 10 week correlation plot reveals a gene cluster linking Fgf23 with Enpp1, Spp1, and Dmp1. (B) Genes correlated with Fgf23 at 10 weeks were graphed to highlight correlations with other network members. Genes with higher connectivity to network members are more likely to be up-stream elements triggering a response within the co-expressed module, suggesting that Enpp1, Dmp1, Spp1, and Gcnt4 are most likely triggering the network associated with Fgf23 activity. (C) The co-expression plot at 23 weeks displays clustered genes correlating with Fgf23 transcription, revealing connectivity of Fgf23 and Spp1, and a less strongly correlated module containing Fgf23, Spp1, Enpp1, Hmox1, Siglec1, Slc11a1, Rps29, C1qc, C1qb, Ctsz, Rsp8, and Utp15. (D) Genes correlated with Fgf23 at 23 weeks are graphed to include correlations among other network members. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
At 23 weeks, ontological findings of enthesopathy are present in Enpp1asj/asj mice, including decreases in “Ankle flexion contracture” (padj < 0.05) and “Increased endomysial connective tissue” (padj < 0.05) HPO’s (Supplementary Table 17). The Ankle flexion contracture ontology, reported in tendon calcification and enthesopathy, was detected in Enpp1asj/as mice through the down-regulation of Col6a1, Col6a2, Col6a3, Col12a1, Hacd1, and Mmp2, and increased endomysial connective tissue, which combines with perimysium and epimysium to form the collagen fibers of tendons, to which Col6a1, Col6a2, Col6a3, and Col12a1 contribute. Finally, at 23-weeks Alpl is down-regulated in males (Table 1). Fgf23 correlations at 23-weeks are more numerous than at 10-weeks, and connectivity is increased (Fgf23 node degrees increase from 12 to 20, Fig. 5C and D), and Spp1 remains heavily embedded in the Fgf23 network.
Patterns observed in the networks of correlated responses identify additional coexpression targets for Fgf23 regulation. Elements that correlate with Fgf23 at both 10 and 23-weeks are Enpp1 and Spp1 (Fig 6A). Enpp1 correlates with Fgf23 in both WT and Enpp1asj/asj mice while Spp1 only correlates with Fgf23 in Enpp1asj/asj mice, suggesting that the Spp1 may be an adaptive response to the increased FGF23 transcription induced by ENPP1 deficiency (Fig. 6B).
Fig. 6.
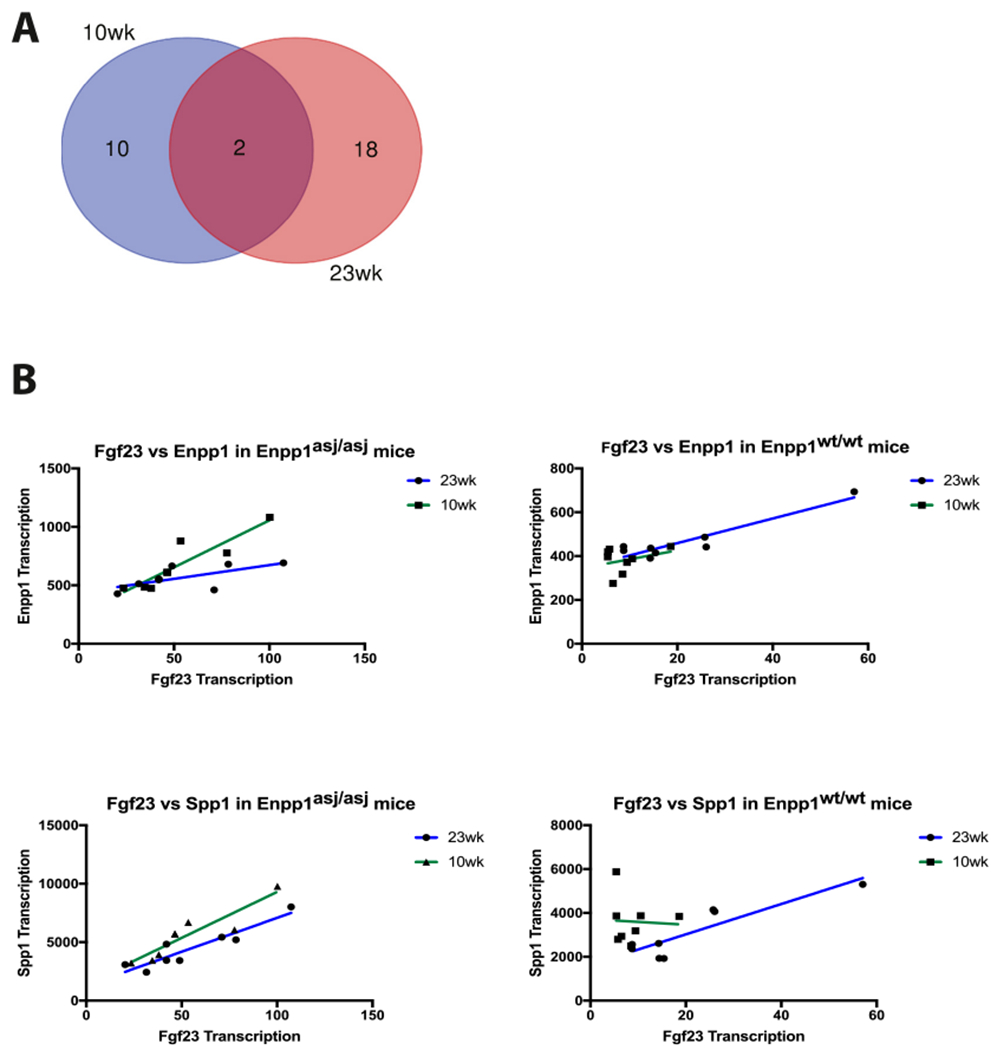
FGF23 correlations (A) only two genes are correlated with Fgf23 transcription at both 10 and 23-week – Enpp1 and Spp1. (B) Correlation of Fgf23 with Enpp1 and Spp1 at 10 and 23 weeks in Enpp1asj/asj and Enpp1wt mice. Enpp1 transcription correlations with Fgf23 in both Enpp1asj/asj and Enpp1wt mice, but Spp1 correlates with Fgf23 in only Enpp1asj/asj mice, suggesting that Spp1 transcription may be responding to ENPP1 deficiency rather than regulating Fgf23.
3.4. Correlation of ENPP1 and FGF23
The relationship observed in Enpp1 and Fgf23 transcription counts (Fig. 6B) suggested an inverse relationship between their gene products – ENPP1 and FGF23. To determine whether ENPP1 and/or plasma [PPi] could directly suppress FGF23 we administered soluble ENPP1 protein in the form of human ENPP1-Fc to Enpp1asj/asj mice and measured concentrations of intact plasma FGF23, ALP, PPi, and Pi in the animals before and after two weeks of dosing to quantify changes to steady state levels of these analytes. Plasma PPi in Enpp1asj/asj mice normalized to WT levels (2.0 μM) at the lowest subcutaneous dose tested – 7.5 mg/Kg ENPP1-Fc per week – and continued to increase at doses up to 30 mg/Kg per week (Fig. 7B). Intact FGF23 trended downwards with increasing ENPP1-Fc doses, becoming significantly suppressed at doses of 30 mg/kg per week (Fig. 6A), confirming our suspicion that ENPP1 or plasma PPi is a direct inhibitor of FGF23. However, when the ENPP1-Fc dose was further increased to 60 mg/kg per week, plasma PPi elevated well above WT levels to 7 μM, and FGF23 now increased and ALP decreased (Fig. 7C), but plasma [Pi] remained unchanged (Fig 7D). Conversely, to assess whether FGF23 is capable of suppressing ENPP1, we measured plasma PPi in Hyp mice, an established murine model of X-Linked Hypophosphatemia with elevated plasma FGF23 (due to large deletion in Phex). We found that compared to WT sibling pairs, plasma PPi in Hyp mice is significantly reduced by about half (Fig. 7E) and intact FGF23 is significantly elevated (Fig. 7E). The combined data demonstrates that ENPP1 and FGF23 counterregulate, and that human exogenous ENPP1 directly suppresses FGF23 when plasma PPi levels are within WT range, but at ENPP1-Fc doses where plasma PPi increases above WT levels, ALP and FGF23 will adjust to maintain physiologically levels of [Pi].
Fig. 7.
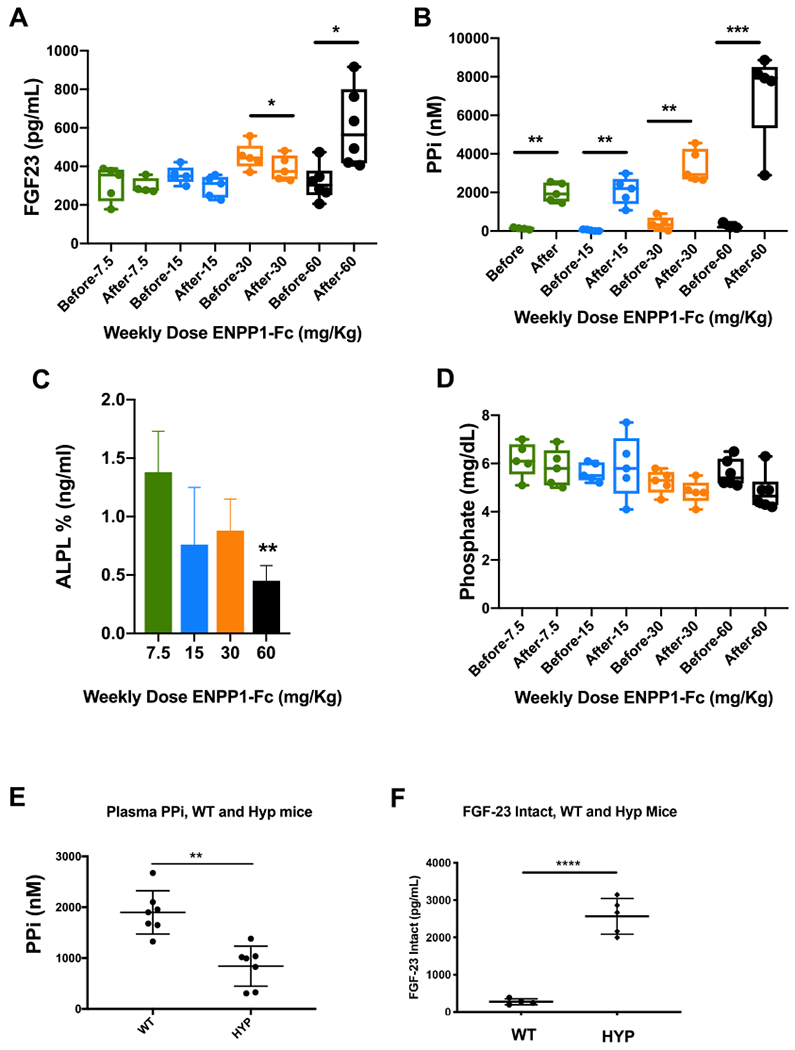
Response of intact FGF23, alkaline phosphatase, and plasma PPi and Pi to ENPP1 (A–D). Enpp1asj/asj mice were dosed with increasing concentrations of soluble ENPP1, and the listed analytes were quantified before and after 2 weeks of dosing. ALP levels are expressed as fractional changes compared to predosed animals. (E) Plasma PPi and (F) intact FGF23 in sibling pairs of WT and Hyp mice. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 Student’s paired (A–D) and unpaired (E–F) t-test.
4. Discussion
ENPP1 deficient mice first came to the attention of researchers in Japan [54], and were named ‘tip toe walking’ mice (ttw) because they developed a severe myelopathy simulating aspects of the human disease ‘Ossification of the Posterior Longitudinal Ligament (OPLL)’ [55]. The phenotype was due to extensive calcifications of cartilage and ligaments resulting from a spontaneous mutation in Enpp1 (Gly568stop) [11]. Only later were ttw mice found to exhibit decreased bone mass [56,57], which was soon followed by several additional murine models of ENPP1 deficiency, all of which demonstrated diminished skeletal bone mass [58,59]. The murine findings were discordant with the only known skeletal phenotype present in human ENPP1 deficiency at the time, which was a rickets associated with homozygous ENPP1 deficiency. Our understanding of ENPP1 on bone mass in humans increased when we, in collaboration with colleagues at the Univ. of Hamburg, reported early onset osteoporosis (EOOP) in humans with heterozygous ENPP1 deficiency. The skeletal and biochemical phenotypes of the homozygous deficient mice and the heterozygous deficient humans are nearly identical [34], inviting the use of these animals to further investigate the relationship between ENPP1 and bone mass in mammals.
Accordingly, we employed RNAseq in tibial bone of Enpp1wt and Enpp1asj/asj mice to understand the genetic pathways disrupted by ENPP1 deficiency. ENPP1 generates PPi, which is a strong inhibitor of accrual of bone mineral in the extant bone matrix, and one would anticipate that low PPi would not lead to an overall reduction in bone volume. The RNAseq analysis revealed genetic signatures in Enpp1asj/asj mice also disrupted in human osteoporosis, most notable in the Wnt pathway in bone through the reduction of Wnt ligands Wnt10b and Wnt16. The phenotype of Wnt10b and Wnt16 knockout mice is known, and we note that the skeletal phenotype of Wnt10b knockout mice closely corresponds to Enpp1asj/asj mice. Specifically, Wnt10b knockout mice exhibit a sex-independent age-related osteopenia that is not driven by increased bone resorption, as demonstrated by normal serum CTX [60]. The osteoporosis in Wnt10b knockout mice mainly effects trabeculae, while the osteoporosis in Wnt16 knockout mice effects primarily the bone cortex [61]. Enpp1asj/asj mice also exhibit a sex-independent osteopenia that effects both trabeculae and cortex, progresses with age, and is independent of osteoclast resorption as demonstrated by normal serum CTX similar to Wnt10b knockout mice [34]. To investigate the mechanism further we performed qPCR on select Wnt pathways in other tissues in 23-week male mice, detecting increased expression of soluble Wnt inhibitors – Sfrp1 in liver and Wif1 in kidney. Although they must be confirmed by additional experiments, the findings support the notion that ENPP1 deficiency induces systemic inhibition of the Wnt pathway through the Sfrp family of soluble Wnt inhibitors, providing a genetic mechanism for the low bone mass observed in heterozygous ENPP1 deficiency (Fig. 8A).
Fig. 8.
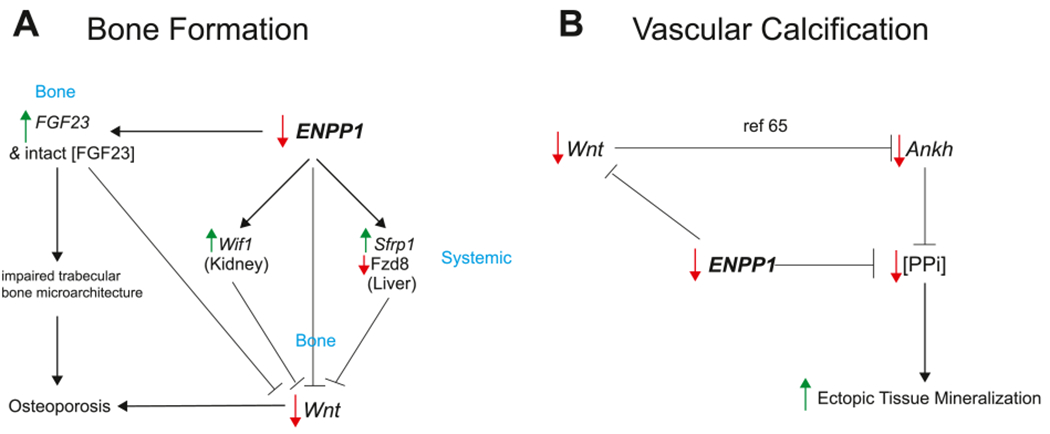
ENPP1 deficiency induces pathways of paradoxical mineralization. (A) ENPP1 deficiency increases FGF23 gene transcription in bone and intact [FGF23] in plasma. Increased [FGF23] is associated with impaired trabecular bone microarchitecture in osteoporosis [65]. ENPP1 deficiency decreases Wnt transcription in bone, increases transcription of the Wnt inhibitors Wif1 in kidney and Sfrp1 in liver, and decreases the transcription of Wnt ligand Fzd8 in liver, all leading to decreased Wnt signaling, contributing to decreased bone mass. Increased FGF23 has also been shown to suppress Wnt in animal models of CKD [68], further exacerbating low bone mass. (B) ENPP1 deficiency directly leads to decreased plasma [PPi] through inefficient hydrolysis of ATP, inducing ectopic mineralization. A second pathway of ectopic mineralization from Wnt inhibition has been described in animal models of CKD via suppression of Ankh, which reduces extracellular PPi [69].
To investigate the mechanism by which Enpp1 regulates bone mass we looked at catalytically dependent and independent networks, correlating plasma [PPi] or Enpp1 transcription counts with skeletal phenotypes in each mouse. We found that at 10 weeks Enpp1 transcription counts strongly correlated with bone mass, being tied to skeletal phenotypes including biomechanical measurements of tibial bone stiffness, maximal load, and fracture load, as well as cortical bone volume and thickness and trabecular thickness. At 23-weeks the contribution of plasma [PPi] to the bone phenotype dramatically shifts and now becomes strongly associated with bone mass. The findings suggest that at 10 weeks the bone phenotype may be independent of plasma [PPi], but at 23 weeks the osteoporotic phenotype is now strongly associated with plasma PPi. Although suggested at 10 weeks, additional experiments are required to discern whether a catalytical independent mechanism driving low bone mass in ENPP1 deficiency is present.
The effects of the ENPP1 deficiency on bone mass compound with age, as indicated by the more complex genetic signature at 23 weeks demonstrated by the volcano and network plots. The 10-week cohort revealed 25 differentially expressed genes while the 23-week animals differentially expressed 268 genes. The strongest genetic signals observed at 23 weeks were associated with Fgf, Wnt, Collagen, or matrisomal activity, signatures which were less apparent or absent at 10-weeks. Generated network estimates also indicated a shift from smaller-scale networks in earlier life stages to more scale-free network with more node of high degree density.
Although only long bones were examined, unbiased ontogeny analysis of the RNAseq data returned phenotypes strongly associated with OPLL such as scoliosis, abnormality of the vertebral column, abnormality of the curvature of the vertebral column, and spinal rigidity (Fig. 2, Supplementary Table 17). Disruptions in several genes previously identified in OPLL patients were also present in ENPP1 deficient mice, such as Vdr, Col6a1, and Ptch1 [52,62], providing validation for the use of axial bones in the model for further study of the genetic signatures in OPLL. Recently, OPLL disease progression and severity has been linked to elevations of plasma FGF23 [63] and reductions in plasma [Pi] [64], plasma biochemistries that we recently identified in heterozygous ENPP1 deficiency [21,34], findings which additionally link ENPP1 deficiency with the pathophysiology of OPLL.
The relationship between ENPP1 and FGF23 has been unclear since reports of hypophosphatemic rickets in ENPP1 deficient children first appeared, and it is therefore notable that Fgf23 is among the most significantly increased genes in Enpp1asj/asj mice, a finding consistent with previous observations of increased circulating FGF23 protein in Enpp1−/− mice [59]. We found that Fgf23 and intact Fgf23 inversely correlated with phenotypic markers of bone biomechanical integrity and microarchitecture in the mice, a finding closely paralleling observations in humans [65], demonstrating the close correspondence of genetic mechanisms of skeletal mass in mice and humans. Correlations observed between Fgf23, osteoid width, and mineralization lag time are consistent with our understanding of Fgf23 in hypophosphatemic rickets, and correlations between Fgf23 and matrisomal and collagen genes suggest additional connections with bone organization and remodeling.
The only genes co-regulated with Fgf23 at 10 and 23-weeks are Enpp1 and Spp1, and only Fgf23 and Enpp1 correlate in both ENPP1 deficient and WT mice, suggesting that Spp1 is responding to ENPP1 deficiency whereas Fgf23 may be regulated by ENPP1. To determine whether ENPP1 protein regulates intact Fgf23, we dosed Enpp1asj/asj mice with a soluble form of ENPP1 (ENPP1-Fc [19]), and monitored plasma PPi, Pi, ALP, and intact Fgf23 before and after dosing to observe responsive changes in enzymes and analytes in metabolic pathways regulating plasma [Pi]. We observed that intact Fgf23 decreases with increasing doses of ENPP1-Fc, becoming significant when plasma [PPi] reaches the upper limits of normal (3–4 μM). When plasma [PPi] is elevated well above normal (6–8 μM), FGF23 now elevates. Similarly, ALP trends downward with increased ENPP1-Fc and PPi concentrations, becoming significantly suppressed at the highest ENPP1-Fc doses and PPi concentrations. Decreases in [PPi] and increases in [ALP] are thought to drive disease morbidity in PXE, a disease closely linked with GACI [32,66]. Our studies demonstrate suppression of murine Fgf23 and Alp by human ENPP1-Fc, albeit with limited potency, and require further studies to eliminate potentially confounding effects of a human soluble protein on a murine physiologic response and investigate the requirements of therapeutic localization. Using an in vivo physiologic system to remove the confounding effects of bone targeting and human/murine sequence variations, we demonstrated suppression of plasma PPi in Hyp mice which manifest elevated circulating FGF23 levels, consistent with the notion that ENPP1 and FGF23 counterregulate one another in vivo.
The genetic pathways associated with Enpp1 deficiency has direct relevance to rare and common diseases of mineralization imbalance, including GACI, PXE, ARHR2, OPLL, early onset osteoporosis, and the poorly understood mineralization imbalances in Chronic Kidney Disease Bone Mineralization Disorder (CKD-MBD). CKD-MBD patients exhibit ‘Paradoxical Mineralization’ in which increased vascular calcification, concurrent loss of bone mass, and increased fracture risk occur, although a satisfying physiologic mechanism for these mineralization imbalances has never been identified. The now established association of decreased plasma [PPi] in renal failure patients [67] together with the suppression of bone formation pathways we have identified in murine Enpp1 deficiency suggests that ENPP1 deficiency may serve as a genetic mechanism underlying the paradoxical mineralization defects and altered bone mass accrual that are exhibited in CKD-MBD (Fig. 8), a hypothesis we will further investigate in future work.
Supplementary Material
Acknowledgments
Funding
Funding from Inozyme Pharma and the National Institutes of Health through R01 DK121326 to DTB. Plasma analytes in mice were performed by the George M O’Brien Kidney Center at Yale, and funded by NIH grant P30 DK079310.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bone.2020.115656.
Declaration of competing interest
D.T.B. is an inventor of patents owned by Yale University which describe therapeutics for ENPP1 deficiency. D.T.B is an equity holder and receives research and consulting support from Inozyme Pharma, Inc.
References
- [1].van Meeteren LA, Ruurs P, Christodoulou E, Goding JW, Takakusa H, Kikuchi K, Perrakis A, Nagano T, Moolenaar WH, Inhibition of autotaxin by lysophosphatidic acid and sphingosine 1-phosphate, J. Biol. Chem. 280 (22) (2005) 21155–21161. [DOI] [PubMed] [Google Scholar]
- [2].Tanaka M, Okudaira S, Kishi Y, Ohkawa R, Iseki S, Ota M, Noji S, Yatomi Y, Aoki J, Arai H, Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid, J. Biol. Chem 281 (35) (2006) 25822–25830. [DOI] [PubMed] [Google Scholar]
- [3].Nam SW, Clair T, Kim YS, McMarlin A, Schiffmann E, Liotta LA, Stracke ML, Autotaxin (NPP-2), a metastasis-enhancing motogen, is an angiogenic factor, Cancer Res. 61 (18) (2001) 6938–6944. [PubMed] [Google Scholar]
- [4].Manning TJ Jr., Rosenfeld SS, Sontheimer H, Lysophosphatidic acid stimulates actomyosin contraction in astrocytes, J. Neurosci. Res 53 (3) (1998) 343–352. [DOI] [PubMed] [Google Scholar]
- [5].Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K, Yamori T, Mills GB, Inoue K, Aoki J, Arai H, Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production, J. Cell Biol 158 (2) (2002) 227–233 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nam SW, Clair T, Campo CK, Lee HY, Liotta LA, Stracke ML, Autotaxin (ATX), a potent tumor motogen, augments invasive and metastatic potential of rastransformed cells, Oncogene. 19 (2) (2000) 241–247. [DOI] [PubMed] [Google Scholar]
- [7].Hoelzinger DB, Nakada M, Demuth T, Rosensteel T, Reavie LB, Berens ME, Autotaxin: a secreted autocrine/paracrine factor that promotes glioma invasion, J. Neuro-Oncol 86 (3) (2008) 297–309. [DOI] [PubMed] [Google Scholar]
- [8].Saunders LP, Ouellette A, Bandle R, Chang WC, Zhou H, Misra RN, De La Cruz EM, Braddock DT, Identification of small-molecule inhibitors of autotaxin that inhibit melanoma cell migration and invasion, Mol. Cancer Ther 7 (10) (2008) 3352–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liu S, Umezu-Goto M, Murph M, Lu Y, Liu W, Zhang F, Yu S, Stephens LC, Cui X, Murrow G, Coombes K, Muller W, Hung MC, Perou CM, Lee AV, Fang X, Mills GB, Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases, Cancer Cell 15 (6) (2009) 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kishi Y, Okudaira S, Tanaka M, Hama K, Shida D, Kitayama J, Yamori T, Aoki J, Fujimaki T, Arai H, Autotaxin is overexpressed in glioblastoma multiforme and contributes to cell motility of glioblastoma by converting lysophosphatidylcholine to lysophosphatidic acid, J. Biol. Chem 281 (25) (2006) 17492–17500. [DOI] [PubMed] [Google Scholar]
- [11].Okawa A, Nakamura I, Goto S, Moriya H, Nakamura Y, Ikegawa S, Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine, Nat. Genet 19 (3) (1998) 271–273 Epub 1998/07/14 10.1038/956. [DOI] [PubMed] [Google Scholar]
- [12].Nakamura I, Ikegawa S, Okawa A, Okuda S, Koshizuka Y, Kawaguchi H, Nakamura K, Koyama T, Goto S, Toguchida J, Matsushita M, Ochi T, Takaoka K, Nakamura Y, Association of the human NPPS gene with ossification of the posterior longitudinal ligament of the spine (OPLL), Hum. Genet 104 (6) (1999) 492–497 (Epub 1999/08/24.). [DOI] [PubMed] [Google Scholar]
- [13].Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N, Bercovich D, Chalifa-Caspi V, Manor E, Buriakovsky S, Hadad Y, Goding J, Parvari R, Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene, Am. J. Hum. Genet 86 (2) (2010) 273–278 Epub 2010/02/09 10.1016/j.ajhg.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lorenz-Depiereux B, Schnabel D, Tiosano D, Hausler G, Strom TM, Loss-offunction ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets, Am. J. Hum. Genet 86 (2) (2010) 267–272, 10.1016/j.ajhg.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Hohne W, Schauer G, Lehmann M, Roscioli T, Schnabel D, Epplen JT, Knisely A, Superti-Furga A, McGill J, Filippone M, Sinaiko AR, Vallance H, Hinrichs B, Smith W, Ferre M, Terkeltaub R, Nurnberg P, Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification, Nat. Genet 34 (4) (2003) 379–381, 10.1038/ng1221. [DOI] [PubMed] [Google Scholar]
- [16].Rutsch F, Vaingankar S, Johnson K, Goldfine I, Maddux B, Schauerte P, Kalhoff H, Sano K, Boisvert WA, Superti-Furga A, Terkeltaub R, PC-1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification, Am. J. Pathol. 158 (2) (2001) 543–554, 10.1016/S0002-9440(10)63996-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li Q, Guo H, Chou DW, Berndt A, Sundberg JP, Uitto J, Mutant Enpp1asj mice as a model for generalized arterial calcification of infancy, Dis. Model. Mech 6 (5) (2013) 1227–1235, 10.1242/dmm.012765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Albright RA, Chang WC, Robert D, Ornstein DL, Cao W, Liu L, Redick ME, Young JI, De La Cruz EM, Braddock DT, NPP4 is a procoagulant enzyme on the surface of vascular endothelium, Blood 120 (22) (2012) 4432–4440 Epub 2012/09/22 10.1182/blood-2012-04-425215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Albright RA, Stabach P, Cao W, Kavanagh D, Mullen I, Braddock AA, Covo MS, Tehan M, Yang G, Cheng Z, Bouchard K, Yu ZX, Thorn S, Wang X, Folta-Stogniew EJ, Negrete A, Sinusas AJ, Shiloach J, Zubal G, Madri JA, De La Cruz EM, Braddock DT, ENPP1-Fc prevents mortality and vascular calcifications in rodent model of generalized arterial calcification of infancy, Nat. Commun. 6 (2015) 10006 Epub 2015/12/02 10.1038/ncomms10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mehta P, Mitchell A, Tysoe C, Caswell R, Owens M, Vincent T, Novel compound heterozygous mutations in ENPP1 cause hypophosphataemic rickets with anterior spinal ligament ossification, Rheumatology (Oxford) 51 (10) (2012) 1919–1921 Epub 2012/04/28 10.1093/rheumatology/kes089. [DOI] [PubMed] [Google Scholar]
- [21].Kotwal A, Ferrer A, Kumar R, Singh RJ, Murthy V, Schultz-Rogers L, Zimmermann M, Lanpher B, Zimmerman K, Stabach PR, Klee E, Braddock DT, Wermers RA, Clinical and biochemical phenotypes in a family with ENPP1 mutations, J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res (2019), 10.1002/jbmr.3938 Epub 2019/12/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Saito T, Shimizu Y, Hori M, Taguchi M, Igarashi T, Fukumoto S, Fujitab T, A patient with hypophosphatemic rickets and ossification of posterior longitudinal ligament caused by a novel homozygous mutation in ENPP1 gene, Bone. 49 (4) (2011) 913–916, 10.1016/j.bone.2011.06.029. [DOI] [PubMed] [Google Scholar]
- [23].Koshizuka Y, Kawaguchi H, Ogata N, Ikeda T, Mabuchi A, Seichi A, Nakamura Y, Nakamura K, Ikegawa S, Nucleotide pyrophosphatase gene polymorphism associated with ossification of the posterior longitudinal ligament of the spine, J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res 17 (1) (2002) 138–144 Epub 2002/01/05 10.1359/jbmr.2002.17.1.138. [DOI] [PubMed] [Google Scholar]
- [24].Nitschke Y, Baujat G, Botschen U, Wittkampf T, du Moulin M, Stella J, Le Merrer M, Guest G, Lambot K, Tazarourte-Pinturier MF, Chassaing N, Roche O, Feenstra I, Loechner K, Deshpande C, Garber SJ, Chikarmane R, Steinmann B, Shahinyan T, Martorell L, Davies J, Smith WE, Kahler SG, McCulloch M, Wraige E, Loidi L, Hohne W, Martin L, Hadj-Rabia S, Terkeltaub R, Rutsch F, Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6, Am. J. Hum. Genet 90 (1) (2012) 25–39, 10.1016/j.ajhg.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nitschke Y, Rutsch F, Generalized arterial calcification of infancy and pseudoxanthoma elasticum: two sides of the same coin, Front. Genet 3 (2012) 302, 10.3389/fgene.2012.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Le Boulanger G, Labreze C, Croue A, Schurgers LJ, Chassaing N, Wittkampf T, Rutsch F, Martin L, An unusual severe vascular case of pseudoxanthoma elasticum presenting as generalized arterial calcification of infancy, Am. J. Med. Genet. A 152A (1) (2010) 118–123, 10.1002/ajmg.a.33162. [DOI] [PubMed] [Google Scholar]
- [27].Li Q, Brodsky JL, Conlin LK, Pawel B, Glatz AC, Gafni RI, Schurgers L, Uitto J, Hakonarson H, Deardorff MA, Levine MA, Mutations in the ABCC6 gene as a cause of generalized arterial calcification of infancy: genotypic overlap with pseudoxanthoma elasticum, The Journal of investigative dermatology 134 (3) (2014) 658–665, 10.1038/jid.2013.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bergen AA, Plomp AS, Schuurman EJ, Terry S, Breuning M, Dauwerse H, Swart J, Kool M, van Soest S, Baas F, ten Brink JB, de Jong PT, Mutations in ABCC6 cause pseudoxanthoma elasticum, Nat. Genet 25 (2) (2000) 228–231, 10.1038/76109. [DOI] [PubMed] [Google Scholar]
- [29].Le Saux O, Urban Z, Tschuch C, Csiszar K, Bacchelli B, Quaglino D, Pasquali-Ronchetti I, Pope FM, Richards A, Terry S, Bercovitch L, de Paepe A, Boyd CD, Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum, Nat. Genet 25 (2) (2000) 223–227, 10.1038/76102. [DOI] [PubMed] [Google Scholar]
- [30].Ringpfeil F, Lebwohl MG, Christiano AM, Uitto J, Pseudoxanthoma elasticum: mutations in the MRP6 gene encoding a transmembrane ATP-binding cassette (ABC) transporter, Proc. Natl. Acad. Sci. U. S. A 97 (11) (2000) 6001–6006, 10.1073/pnas.100041297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jansen RS, Kucukosmanoglu A, de Haas M, Sapthu S, Otero JA, Hegman IE, Bergen AA, Gorgels TG, Borst P, van de Wetering K, ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release, Proc. Natl. Acad. Sci. U. S. A 110 (50) (2013) 20206–20211, 10.1073/pnas.1319582110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jansen RS, Duijst S, Mahakena S, Sommer D, Szeri F, Varadi A, Plomp A, Bergen AA, Oude Elferink RP, Borst P, van de Wetering K, ABCC6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation-brief report, Arterioscler. Thromb. Vasc. Biol 34 (9) (2014) 1985–1989, 10.1161/ATVBAHA.114.304017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Martin L, Hoppe E, Kauffenstein G, Omarjee L, Navasiolava N, Henni S, Willoteaux S, Leftheriotis G, Early arterial calcification does not correlate with bone loss in pseudoxanthoma elasticum, Bone. 103 (2017) 88–92 Epub 2017/06/29 10.1016/j.bone.2017.06.017. [DOI] [PubMed] [Google Scholar]
- [34].Oheim R, Zimmerman K, Maulding ND, Stürznickel J, Kroge S. v., Kavanagh D, Stabach P, Kornak U, Tommasini SM, Horowitz M, Amling M, Thompson D, Schinke T, Busse B, Carpenter TO, Braddock DT, Human heterozygous ENPP1 deficiency is associated with early onset osteoporosis, a phenotype recapitulated in a mouse model of Enpp1 deficiency, J. Bone Miner. Res (2019), 10.1002/jbmr.3911 Epub Nov. 5th. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Andrews S, FastQC: a quality control tool for high throughput sequence data, Available onlline at http://www.bioinformatics.babraham.ac.uk/projects/fastqc2010. [Google Scholar]
- [36].Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Giron CG, Gil L, Gordon L, Haggerty L, Haskell E, Hourlier T, Izuogu OG, Janacek SH, Juettemann T, To JK, Laird MR, Lavidas I, Liu Z, Loveland JE, Maurel T, McLaren W, Moore B, Mudge J, Murphy DN, Newman V, Nuhn M, Ogeh D, Ong CK, Parker A, Patricio M, Riat HS, Schuilenburg H, Sheppard D, Sparrow H, Taylor K, Thormann A, Vullo A, Walts B, Zadissa A, Frankish A, Hunt SE, Kostadima M, Langridge N, Martin FJ, Muffato M, Perry E, Ruffier M, Staines DM, Trevanion SJ, Aken BL, Cunningham F, Yates A, Flicek P, Ensembl 2018, Nucleic Acids Res. 46 (D1) (2018) D754–D761 Epub 2017/11/21 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner, Bioinformatics 29 (1) (2013) 15–21 Epub 2012/10/30 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bolger AM, Lohse M, Usadel B, Trimmomatic: a flexible trimmer for Illumina sequence data, Bioinformatics 30 (15) (2014) 2114–2120 Epub 2014/04/04 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome Biol. 15 (12) (2014) 550 Epub 2014/12/18 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, Vilo J, g:Profiler-a web server for functional interpretation of gene lists (2016 update), Nucleic Acids Res. 44 (W1) (2016) W83–W89 Epub 2016/04/22 10.1093/nar/gkw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Benjamini Y, Hochberg Y, Controlling the false discovery rate: a practical and powerful approach to multiple testing, J. R. Stat. Soc. Ser. B Methodol 57 (1) (1995) 289–300. [Google Scholar]
- [42].Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I, Controlling the false discovery rate in behavior genetics research, Behav. Brain Res 125 (1–2) (2001) 279–284 Epub 2001/10/30 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- [43].Butts CT, Network: classes for relational data, https://CRAN.R-project.org/package=network2015. [Google Scholar]
- [44].Xu L, Zhang L, Zhang H, Yang Z, Qi L, Wang Y, Ren S, The participation of fibroblast growth factor 23 (FGF23) in the progression of osteoporosis via JAK/STAT pathway, J. Cell. Biochem 119 (5) (2018) 3819–3828 Epub 2017/08/08 10.1002/jcb.26332 ). [DOI] [PubMed] [Google Scholar]
- [45].Manolagas SC, Wnt signaling and osteoporosis, Maturitas 78 (3) (2014) 233–237 Epub 2014/05/13 10.1016/j.maturitas.2014.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ralston SH, Genetic control of susceptibility to osteoporosis, J. Clin. Endocrinol. Metab 87 (6) (2002) 2460–2466 Epub 2002/06/07 10.1210/jcem.87.6.8621. [DOI] [PubMed] [Google Scholar]
- [47].Fromigue O, Modrowski D, Marie PJ, Growth factors and bone formation in osteoporosis: roles for fibroblast growth factor and transforming growth factor beta, Curr. Pharm. Des 10 (21) (2004) 2593–2603 Epub 2004/08/24. [DOI] [PubMed] [Google Scholar]
- [48].Sroga GE, Vashishth D, Effects of bone matrix proteins on fracture and fragility in osteoporosis, Curr Osteoporos Rep. 10 (2) (2012) 141–150 Epub 2012/04/27 10.1007/s11914-012-0103-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Krane SM, Inada M, Matrix metalloproteinases and bone, Bone. 43 (1) (2008) 7–18 Epub 2008/05/20 10.1016/j.bone.2008.03.020. [DOI] [PubMed] [Google Scholar]
- [50].Hu Z, Chen B, Zhao Q, Hedgehog signaling regulates osteoblast differentiation in zebrafish larvae through modulation of autophagy, Biol Open 8 (5) (2019), 10.1242/bio.040840 Epub 2019/04/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rivadeneira F, Makitie O, Osteoporosis and bone mass disorders: from gene pathways to treatments, Trends Endocrinol. Metab 27 (5) (2016) 262–281 Epub 2016/04/16 10.1016/j.tem.2016.03.006. [DOI] [PubMed] [Google Scholar]
- [52].Choi BW, Song KJ, Chang H, Ossification of the posterior longitudinal ligament: a review of literature, Asian Spine J. 5 (4) (2011) 267–276 Epub 2011/12/14 10.4184/asj.2011.5.4.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Martin A, Liu S, David V, Li H, Karydis A, Feng JQ, Quarles LD, Bone proteins PHEX and DMP1 regulate fibroblastic growth factor Fgf23 expression in osteocytes through a common pathway involving FGF receptor (FGFR) signaling, FASEB J. 25 (8) (2011) 2551–2562 Epub 2011/04/22 10.1096/fj.10-177816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hosoda Y, Yoshimura Y, Higaki S, A new breed of mouse showing multiple osteochondral lesions—twy mouse, Ryumachi. 21 (Suppl) (1981) 157–164. [PubMed] [Google Scholar]
- [55].Stapleton CJ, Pham MH, Attenello FJ, Hsieh PC, Ossification of the posterior longitudinal ligament: genetics and pathophysiology, Neurosurg. Focus 30 (3) (2011) E6, , 10.3171/2010.12.FOCUS10271. [DOI] [PubMed] [Google Scholar]
- [56].Kobayashi Y, Goto S, Tanno T, Yamazaki M, Moriya H, Regional variations in the progression of bone loss in two different mouse osteopenia models, Calcif. Tissue Int. 62 (5) (1998) 426–436. [DOI] [PubMed] [Google Scholar]
- [57].Okawa A, Goto S, Moriya H, Calcitonin simultaneously regulates both periosteal hyperostosis and trabecular osteopenia in the spinal hyperostotic mouse (twy/twy) in vivo, Calcif. Tissue Int 64 (3) (1999) 239–247. [DOI] [PubMed] [Google Scholar]
- [58].Babij P, Roudier M, Graves T, Han CY, Chhoa M, Li CM, Juan T, Morony S, Grisanti M, Li X, Yu L, Dwyer D, Lloyd DJ, Bass MB, Richards WG, Ebeling C, Amato J, Carlson G, New variants in the Enpp1 and Ptpn6 genes cause low BMD, crystal-related arthropathy, and vascular calcification, J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res 24 (9) (2009) 1552–1564, 10.1359/jbmr.090417. [DOI] [PubMed] [Google Scholar]
- [59].Mackenzie NC, Zhu D, Milne EM, van’t Hof R, Martin A, Darryl Quarles L, Millan JL, Farquharson C, MacRae VE, Altered bone development and an increase in FGF-23 expression in Enpp1(−/−) mice, PLoS One 7 (2) (2012) e32177.Epub 2012/02/24 10.1371/journal.pone.0032177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Stevens JR, Miranda-Carboni GA, Singer MA, Brugger SM, Lyons KM, Lane TF, Wnt10b deficiency results in age-dependent loss of bone mass and progressive reduction of mesenchymal progenitor cells, J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res 25 (10) (2010) 2138–2147 Epub 2010/05/26 10.1002/jbmr.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ohlsson C, Henning P, Nilsson KH, Wu J, Gustafsson KL, Sjogren K, Tornqvist A, Koskela A, Zhang FP, Lagerquist MK, Poutanen M, Tuukkanen J, Lerner UH, Moverare-Skrtic S, Inducible Wnt16 inactivation: WNT16 regulates cortical bone thickness in adult mice, J. Endocrinol 237 (2) (2018) 113–122 Epub 2018/03/14 10.1530/JOE-18-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Nam DC, Lee HJ, Lee CJ, Hwang SC, Molecular pathophysiology of Ossification of the Posterior Longitudinal Ligament (OPLL), Biomol Ther (Seoul) 27 (4) (2019) 342–348 Epub 2019/05/03 10.4062/biomolther.2019.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kawaguchi Y, Kitajima I, Nakano M, Yasuda T, Seki S, Suzuki K, Yahara Y, Makino H, Ujihara Y, Ueno T, Kimura T, Increase of the serum FGF-23 in ossification of the posterior longitudinal ligament, Global Spine J 9 (5) (2019) 492–498 Epub 2019/08/23 10.1177/2192568218801015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kawaguchi Y, Nakano M, Yasuda T, Seki S, Suzuki K, Yahara Y, Makino H, Kitajima I, Kimura T, Serum biomarkers in patients with ossification of the posterior longitudinal ligament (OPLL): inflammation in OPLL, PLoS One 12 (5) (2017) e0174881.Epub 2017/05/04 10.1371/journal.pone.0174881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Rupp T, Butscheidt S, Vettorazzi E, Oheim R, Barvencik F, Amling M, Rolvien T, High FGF23 levels are associated with impaired trabecular bone microarchitecture in patients with osteoporosis, Osteoporos. Int 30 (8) (2019) 1655–1662 Epub 2019/05/03 10.1007/s00198-019-04996-7. [DOI] [PubMed] [Google Scholar]
- [66].Sanchez-Tevar AM, Garcia-Fernandez M, Murcia-Casas B, Rioja-Villodres J, Carrillo JL, Camacho M, Van Gils M, Sanchez-Chaparro MA, Vanakker O, Valdivielso P, Plasma inorganic pyrophosphate and alkaline phosphatase in patients with pseudoxanthoma elasticum, Ann Transl Med. 7 (24) (2019) 798 Epub 2020/02/12 10.21037/atm.2019.12.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].O’Neill WC, Sigrist MK, McIntyre CW, Plasma pyrophosphate and vascular calcification in chronic kidney disease, Nephrol. Dial. Transplant. 25 (1) (2010) 187–191, 10.1093/ndt/gfp362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Carrillo-Lopez N, Panizo S, Alonso-Montes C, Roman-Garcia P, Rodriguez I, Martinez-Salgado C, Dusso AS, Naves M, Cannata-Andia JB, Direct inhibition of osteoblastic Wnt pathway by fibroblast growth factor 23 contributes to bone loss in chronic kidney disease, Kidney Int. 90 (1) (2016) 77–89 Epub 2016/05/12 10.1016/j.kint.2016.01.024. [DOI] [PubMed] [Google Scholar]
- [69].Chen B, Zhao Y, Han D, Zhao B, Mao Y, Cui ZK, Chu YC, Feng L, Yin S, Wang CY, Wang X, Xu MJ, Zhao G, Wnt1 inhibits vascular smooth muscle cell calcification by promoting ANKH expression, J. Mol. Cell. Cardiol 135 (2019) 10–21 Epub 2019/07/30 10.1016/j.yjmcc.2019.07.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.