

Introduction
Except for the filamentous phages, all phages of eubacteria and many phages of the Eurarchaea terminate the infection cycle by lysis of the host and consequent liberation of the progeny virions[1,2]. Besides its fundamental position as the most common cytocidal event in the biosphere, phage lysis is now clinically relevant in its central role in the newly-revived field of phage therapeutics[3]. Here we will present an overview of a type of phage lysis that has not been widely studied and which we have dubbed “Single Gene Lysis” (SGL)[4]. This mode of lysis is used by small single-strand nucleic acid (ssNA) phages of the families Microviridae (ssDNA) and Leviviridae (ssRNA), all of which encode a single lysis protein. Because these genes are so diverse, we have further adopted the convention that the lysis gene and protein in each phage are designated as sgl and Sgl, respectively. This nomenclature is intended for contrast with the Multi-Gene Lysis (MGL) system used by the double-strand nucleic acid phages[2]. In MGL systems, there is always at least one muralytic enzyme, the endolysin, and one cytoplasmic membrane protein, the holin, that exerts precise temporal control on the access of the endolysin to the peptidoglycan (PG). Other proteins in MGL systems include regulators of the holin and endolysin and proteins that target other layers of the envelope.
In overview, the MGL systems can be considered as tightly scheduled, active lysis, in that phage-encoded proteins actively attack the integrity of the envelope at a genetically determined time. In contrast, the known Sgls constitute a passive pathway, in that they encode no degradative activity but instead induce the host cell to undergo autolysis. As a result, lysis is not precisely scheduled but occurs at a time dependent on the cell cycle and/or growth phase of the host. From a distant viewpoint, it should be self-evident that elucidating the molecular basis of these Sgl-induced autolytic events, in the sense that each Sgl is a “magic button” to induce bacterial suicide would lead to the development of new antibiotic strategies. Here we briefly review the molecular basis of five Sgl systems that have been identified in studies that have almost exhausted the known list of plaque-forming lytic ssNA phages. In addition, we will comment on the explosion of ssRNA and ssRNA phage genomes that have suddenly become available from analyses of metagenomics and meta-transcriptomics and focus on the challenges and opportunities presented by this potential bonanza of Sgl information.
Lysis in Microviruses:
ΦX174 E: the founding “protein antibiotic” Sgl
The microviruses are ubiquitous small isometric ssDNA phages that infect a number of bacterial species across different bacterial phyla[5]. The prototypical member of the Microviridae family is the famous coliphage ΦX174, originating in a phage cocktail sold by d’Herelle’s Laboratoire du Bacteriophage and eventually having the honor of being the first sequenced DNA genome of any organism (Figure 1). The sole lysis cistron, gene E, itself occupies a prominent role in the annals of molecular biology. It was both the first gene to be found to be embedded within an alternate reading frame of another gene and the first gene to undergo site-directed mutagenesis[6,7]. However, the mode by which E caused lysis remained controversial for more than two decades after the discovery of the gene, before a genetic approach based on selection for survival after induction of a plasmid-cloned E was implemented[8]. This selection initially generated mutations in the host slyD gene that blocked E lytic function. However, these mutations proved to be recessive, thus identifying SlyD as a host factor required for E function rather than the target of E. Ultimately, similar selections generated dominant E-resistant missense mutations in mraY, which encodes the conserved, membrane-embedded enzyme that forms the first lipid-linked intermediate, Lipid I, in the PG biosynthesis pathway[9]. Furthermore, biochemical assays showed that E and MraY form a complex in vivo and that E is a non-competitive inhibitor of MraY[10]. The primary structure of E is striking, in that its first 34 aa, including a transmembrane domain (TMD), defines the essential lytic domain, whereas the residual 57 residue C-terminal domain (CTD), which is variable in other ΦX174-like phages and is enriched in basic and proline residues, can be replaced by soluble fusion domains like GFP[9,11,12]. SlyD, which is a peptidyl-prolyl cis-trans isomerase of the FKBP type, is absolutely required for E lysis; in the absence of SlyD, E is highly unstable, suggesting that E requires a prolyl-isomerization event for localization and/or binding to MraY[8]. However, SlyD is not required for the lytic function of E-GFP and other fusions where the C-terminal basic domain has been replaced[9]. This suggests that the E-CTD may provide a regulatory handle for regulation of E-mediated lysis; a similar host factor interaction with a dispensable domain has been found for the MS2 Sgl (see below)[13].
Figure 1: Genomes of microviruses of E. coli and intracellular pathogens.

The circular genome maps of members of Bullavirinae (ΦX174) and Gokushovirinae (MH2K, Spv4, and Chp-2) are shown. The genes encoding homologous proteins are numbered and colored the same, with the exception of gene F in ΦX174 which is colored green but labeled as F for historical reasons. The unique genes among Gokushovirinae members are labeled with letters and are colored differently. The only known sgl of Microviridae, E from ΦX174, is colored in blue.
Mutational analyses indicate that the N-terminal residues in the TMD of E are critical for lytic function and likely play a role in interacting with MraY[14]. Although there is a wealth of information from various E mutants, the atomic details of the E-MraY complex are lacking[14,15]. In addition, the role of SlyD in regulating E-mediated lysis is not well understood. It is not clear if SlyD remains associated with E-MraY complex or it dissociates after a successful E-MraY complex formation. In any case, E was the first Sgl to be characterized mechanistically. Clues to its molecular mechanism had been available for decades, going back to electron microscopy (EM) studies by Bradley et al. (1969), who showed that cells infected with alpha3, a close relative of ΦX174, underwent septal catastrophes that resembled the terminal phenotype associated with intoxication by cell-wall antibiotics[16]. E thus qualifies as a “protein antibiotic”[4].
Sgls in Microviruses of intracellular parasites
In the decades following the first characterization of ΦX174 and its related coliphages, plaque-forming Microviruses specific to the exotic intracellular pathogens Bdellovibrio, Spiroplasma, and Chlamydia were isolated[17–19]. The genomes had significant similarities with each other but shared only general genomic architecture with ΦX174; as a result, the ΦX174-like phages were designated as the subfamily Bullavirinae of the family Microviridae, and all the other plaque-formers were designated as Gokushovirinae [5,20]. Certain key genes, such as D, encoding the external scaffolding protein and within which E is embedded in ΦX174, are absent in the Gokushovirinae. In particular, Bdellovibrio phage ΦfMH2K and Chlamydia microviruses (3, 4, Chp2, ΦCPAR39, and ΦCPG1) are similar both in genome organization and the encoded proteins (Figure 1). However, the cell envelope structure in Chlamydia lacks PG and instead has an orthologous structure consisting of a glycanless polypeptide and cysteine-rich proteins[21,22]. Nothing is known about the Sgls of the Gokushovirinae; studying them could provide new insights into the novel cell wall chemistries, growth dynamics, and vulnerabilities of bacterial obligate intracellular parasites.
Metagenomics- a non-trivial gateway to new sgls
Microviruses were underrepresented in early metagenomic studies largely due to the inherent bias towards dsDNA in sample preparation techniques used in standard sequencing approaches such as lllumina’s NexteraXT DNA[23,24]. The diversity of microviruses in various ecological niches was only realized after the implementation of rolling circle amplification technology to amplify the genomes of ssDNA viruses[23,25]. A study on the diversity of microviruses in the gut virome of the sea squirt (Ciona robusta) revealed 258 new genomes, with new being defined as <95% pairwise nucleotide identity [26]. A majority (n=188) of the genomes belonged to the subfamily Gokushovirinae, with nearly half of them sharing less than 70% major capsid protein (MCP) sequence identity with each other. This new haul of microviral genomic information expanded the known diversity in genetic architecture of microviruses, with some families having 11 different patterns of gene organization. There are now thousands of new genomes of microviruses in the database, but most lack rigorous annotations[27].
Based on the experience with the Bullavirinae (above) and the Leviviridae (below), sgl genes are often embedded out of frame in other genes, making them difficult to annotate and essentially invisible to most gene-calling software. A simple protein-protein BLAST of ΦX174 E against the unclassified microviridae (taxid:117574) returned no results, indicating that E-like proteins do not exist in the vast majority of microviruses found in the environment. It is likely that lysis is an essential step in the life cycle of these phages, although, since they were obtained by metagenomic analysis, it is possible that some of these phages are lysis-defective. One recent development is that Gokushovirinae have been found as prophages in some enteric bacteria[28]. Kirchberger and Ochman (2019) were able to resurrect four of these prophages and found one that could form plaques on E. coli K-12[29]. Analysis of the genome reveals 7 unannotated ORFs (>25 codons) with at least one TMD, with three ORFs completely embedded in the gene for major capsid protein. However, none of the putative Sgl candidates were found to have detectable similarity to ΦX174’s E protein or to any known proteins.
In any case, the explosion in microvirus genomics and the absence of E homologs suggest that a systematic approach to identifying the Sgls in these genomes is warranted. One approach is to annotate these genomes and then test the candidate sgl in E. coli or other hosts that have well-established genetic tools for tightly regulated gene expression[30]. This approach is cumbersome and might involve high capital expenditure to synthesize and clone individual genes. Another approach is to make gene-sized libraries with environmental samples enriched for microviruses and then screen/select for plasmids that get released into the media after induction of the pooled transformants. This approach, which we call “plasmid release”, is not only economical but also captures candidate genes sgl genes missed in the gene annotations.
Lysis by ssRNA phages:
With only three to four genes, the ssRNA phages Leviviridae have genomes even simpler than the Microviridae[31]. The three core genes mat, coat, and rep are found in all ssRNA phages and the fourth gene is usually the sgl[4]. The levivrus particle has a T=3 capsid with 178 copies of the Coat protein and one copy of the Mat protein in the shell[32,33]. The Mat protein serves as the “single molecule tail”, in that it recognizes and adsorbs to the pilin subunit of a retractile pilus[34]. The most intensively studied leviviruses are MS2 and Qβ, both specific for the F pilus of enteric bacteria[35]. Surprisingly, despite a history of more than 50 years, there are only 10 distinct ssRNA plaque-forming phages with sequenced genomes (Table 1)[4,36]. This includes the Qβ and MS2, but does not count their >30 close kin that all use the F pilus [37]. The leviviruses are also characterized by their high mutation rate, >10−4 per nt per replication[38]. Of the 10 plaque-forming leviviruses, the sgl genes have evolved in 6 different locations, including 4 cases where they are embedded in an essential gene in a different reading frame (Figure 2); thus sgl genes evolve late and independently, presumably after each speciation to a different retractable pilus[4].
Table 1.
Summary of known plaque-forming ssRNA phages.
| phage | pilus | host | sgl location | Sgl target | Sgl size (aa) |
|---|---|---|---|---|---|
| MS2 | F | E. coli | coat-rep junction | L-target | 75 |
| Qβ | F | E. coli | = A2 | MurA | 420 |
| M | RIP69 (IncM) | E. coli | distal rep | MurJ | 37 |
| Hgal1 | R27 (IncH) | E. coli | coat-rep junction | L-target | 65 |
| C-1 | RA1(IncC) | E. coli | coat-rep junction | L-target | 65 |
| phiCB5 | polar | C. crescentus | middle rep | ? | 136 |
| PP7 | polar type IV | P. aeruginosa | coat-rep junction | L-target | 55 |
| LeviOr01 | polar type IV | P. aeruginosa | ? | ? | ? |
| PRR1 | R1822 (IncP-1) | P. aeruginosa | coat-rep junction | L-target | 54 |
| AP205 | type IV twitching | Acinetobacter | 5’ of mat | L-target | 35 |
Figure 2. Genome organization of ssRNA phages with unique sgl genetic architecture.
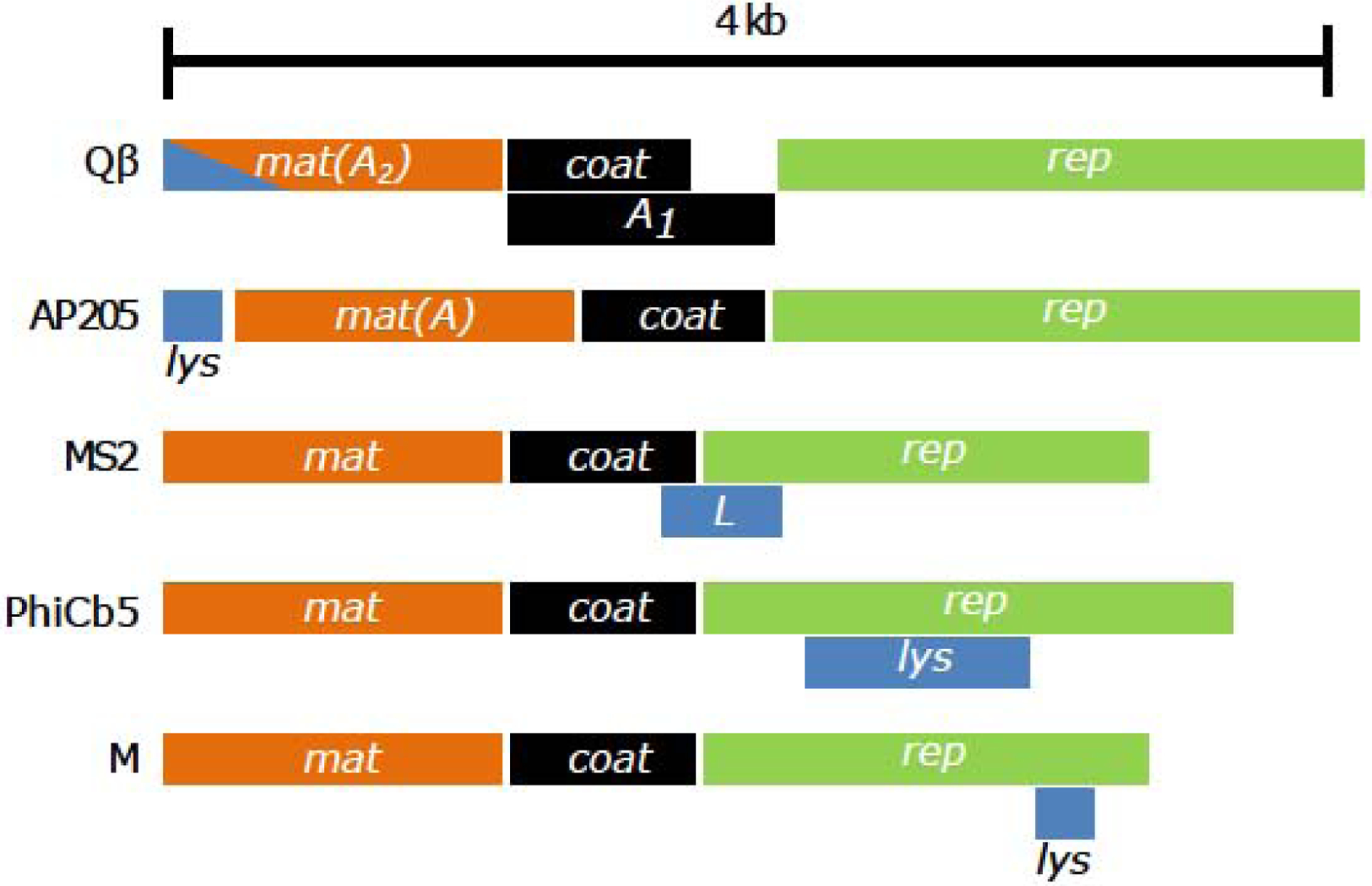
The three core genes mat (orange), coat (black), and rep (green) are found in all ssRNA phages in the same gene order. For historical reasons, the mat genes in MS2 and Qβ are also known as A and A2, respectively. The A1 gene in Qβ encodes the read-through product of the coat gene. With the exception of Qβ, the sgls (blue) in all the other phages exists as a separate gene, often embedded within the reading frame of at least one other gene. The A2 gene product in Qβ serves the dual function of both pili recognition and also host lysis.
Two protein antibiotic Sgls found in Leviviridae
In Qβ, the Sgl activity is a second function of the Mat protein (A2). Using the same selection as used for ΦX174 E, rat mutants (resistant to A2) were isolated and mapped to murA, encoding the enzyme that catalyzes the first committed step in PG biosynthesis[39]. Genetic and biochemical analysis suggested that A2 bound across the active site cleft of MurA once it had closed over the MurNac and PEP substrates[40]. Recently, this was confirmed when a cryo-EM structure of the Qβ particle via its single molecule of A2 was determined, making Qβ the “world’s largest enzyme inhibitor”[41]. Thus, A2 induced autolysis in the same general mode as ΦX174 E, by blocking the flow of Lipid II to the PG synthesis apparatus, making A2 the second member of the protein antibiotic class of Sgls. Recently, another member of this group was found using similar genetic selections. LysM, the Sgl of phage M, Lys M, was found to be a specific inhibitor of MurJ, the conserved lipid II flippase, which externalizes the PG precursor to the exterior of the cell[42]. Thus in all three cases, with E, A2 and LysM, the Sgl proteins block the incorporation of new precursors into the PG, leading to a septal catastrophe and lysis when the infected cell attempts to undergo cell division[4].
The mysterious L Sgl
The molecular mechanism of lysis of the fourth ssRNA phage Sgl, L from phage MS2, is not understood but operationally it is significantly different from the protein-antibiotic class in that it does not block net precursor incorporation into PG[43]. L, 75 aa in length, has a mirror-image likeness to E, in that it has a dispensable, highly basic N-terminal domain[44,45] . Selections using a cloned L gene failed to obtain the expected dominant mutations in a host locus encoding the putative L target[13]. Nevertheless, this analysis revealed that DnaJ was required as a chaperone at 30°C for L to cause lysis; moreover, like the case of SlyD and E, removal of the dispensable basic domain eliminated chaperone dependence, again suggesting that the host factor, in this case DnaJ, could be a mechanism for regulating lysis. In any case, the target of L remains mysterious. However, detailed genetic analysis of L itself showed that despite its short length, it had four domains, including a conserved LS dipeptide encoded in all of the MS2-like F-specific Leviviridae L genes [45]. Moreover, despite the nearly total lack of sequence similarity between the 10 unique levivirus genomes (Figure 3), with only LeviOr01 sharing some sequence similarity with PP7, the L domain structure could be recognized in five of the seven remaining Leviviridae. In four of these, the “L-like” genes have evolved partially embedded within both coat and rep genes. In a fifth, the Acinetobacter phage AP205, the L-like gene has evolved as a separate gene at the 5-prime end of the genome[46]. Thus, despite the L mechanism still remaining unsolved, the most parsimonious interpretation is that six of the 10 unique Leviviridae have evolved Sgls with the L-target (Table 1).
Figure 3. Multiple nucleotide dot plots of 10 unique leviviruses.
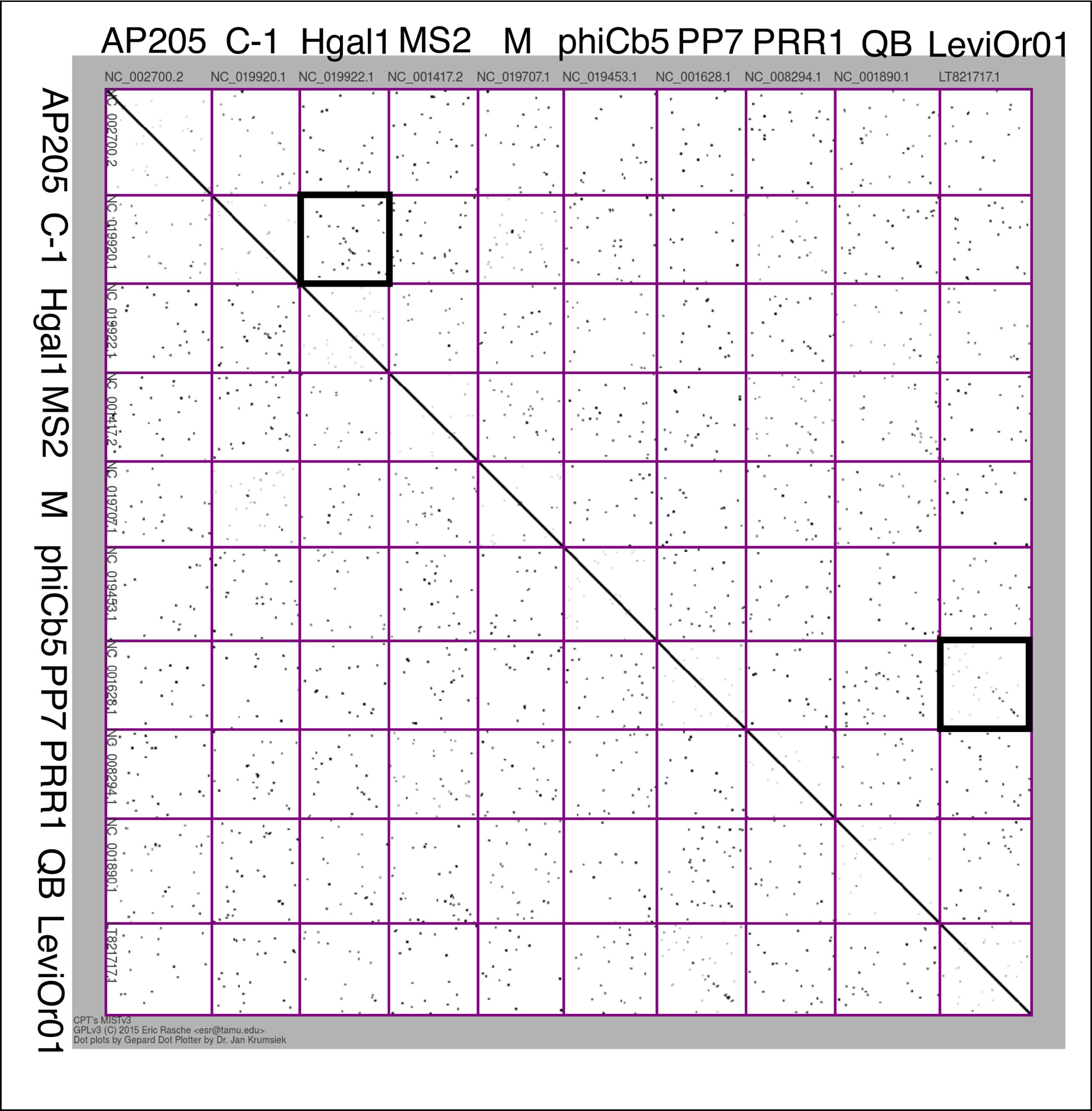
A 10X10 multiple sequence dot plot was constructed with Multiple Interrelated Sequence doTplotter (MIST) v3 (cpt.tamu.edu/galaxy) with each pixel corresponding to 50 bases in the sequence. The ssRNA phage names and the corresponding accession numbers are shown on the two axes. The individual dot plots of the genome pairs that share sequence similarity with each other are highlighted by black squares.
Two other sgl genes from ΦCb5 (lysPhiCb5) and coliphage M (lysM) are embedded completely within the rep gene[47,48]. Although these two Sgls do not share any significant sequence identity, they do have similar predicted membrane topologies (N-in and C-out). The topology of LysM was experimentally verified using a functional eGFP-LysM fusion construct [42]. LysΦCb5 has a large N-terminal cytoplasmic domain with several proline residues, reminiscent of the cytoplasmic domain of E from ΦX174. The similarity in membrane topology and the presence of a proline-rich cytoplasmic domain suggests that LysPhiCb5 and E convergently evolved to target MraY, in different species of bacteria. Although this will require experimental testing, the simplest perspective is that of 10 unique ssRNA plaque-forming phages it appears that there are four different Sgl-host target pairs, and more than half share the L-target.
ssRNA phages with no lysis gene
The newest Pseudomonas levivirus, LeviOr01, forms clear plaques of different sizes only on the clinical P. aeruginosa strain PcyII-10 but forms turbid plaques on other Pseudomonas strains (C9–11, PcyII-11, PcyII-36 and PcyII-57) [36]. LeviOr01 shares homology with another Pseudomonas phage PP7 at the protein level, with the core proteins sharing ~50% sequence identity. Although a sgl candidate was annotated in the same genomic location as PP7 and PRR1, the authors chose an arbitrary leucine codon (TTA) as the start codon (Figure 4). Moreover, the putative Sgl lacks a predicted TMD. Taken together, it is likely that LeviOr01 does not have the typical sgl found in other Pseudomonas leviviruses, which suggests the evolution of a new sgl at a new location within the genome or no sgl at all. The later explanation is supported by the observation that LeviOr01 exists in a carrier state and releases progeny virions sporadically every few rounds of cell division. This strategy of having the infected cell undergoing lysis only every few generations might be more prevalent in the environment. Phages with this characteristic might not be able to form plaques in traditional phage hunts. This perspective might explain the lack of diversity among the leviviruses isolated as plaque-formers and sequenced thus far.
Figure 4. Leviviruses of Pseudomonas.

The genetic architecture of the three leviviruses known to infect and plaque on different Pseudomonas strains are shown. The gene color scheme is the same as Figure 2, the putative sgl in LeviOr01 is shown as a grey rectangle. The full-length genome size is shown as a black bar above the respective genes with 1 kb size intervals shown as short orthogonal bars.
Explosion in available ssRNA genomes
The rapid expansion in the available ssRNA phage genomes began with a metagenomic study of sewage from the San Francisco bay area, from which two new ssRNA phage genomes (EC and MB) of unknown host specificity were found [49]. A subsequent study in 2016 increased the available genomes by ~10-fold by mining publicly-available transcriptomic and RNA-inclusive metagenomic datasets for contigs that contained the signatures of ssRNA phage core proteins (RdRp or Coat or Maturation)[50]. This approach yielded 158 partial genomes of which 122 were novel ssRNA phage sequences. From these new genomes it was clear that even with limited coding space ssRNA phages could exhibit a high degree of genetic novelty. Furthermore, the study reported 91 new ORFs that had no known homologs, an unusually large Maturation protein (>95 kDa). Notably, out of these 158 genomes, only one Sgl homolog was detected, a protein with weak homology to MS2 L (38% sequence identity). During the same period, another 66 new ssRNA phage genomes were reported in one of the largest studies of RNA virome of invertebrate species [51]. In still another very recent study, nearly 1300 ssRNA phage genomes were reconstructed from soil that was sampled multiple times from four distinct soil habitats over a 22-day period[52]. A majority of the new genomes are phylogenetically more distant to previously discovered genomes, with the genomes found in bulk soil clustering into separate clades. The genomes were reported to have new genetic architectures with one genome having five genes including two putative lysis genes. Several putative sgl candidates were annotated in these genomes, mainly based on the presence of a predicted export signal. A closer inspection revealed that most of the sgl candidates lack a start codon and are likely not true sgls. The validity of any sgl annotations have to be experimentally confirmed in the native hosts or a model organism such as E. coli. To find the possible hosts of these phages, the authors also identified a pool of 355 Proteobacterial species, based on the phylogeny of ribosomal protein S3, as possible hosts for the leviviruses in the soil. A more recent study identified 15,611 partial and near-complete genomes from sewage and lake water ecosystems[53]. With this study, the available ssRNA phage genomic space increased by another 60-fold. Therefore, in the past four years the number of available ssRNA phage genomes have rapidly expanded from tens to tens of thousands. This represents a vastly under-explored genomic resource for mining new Sgls with new modes of action and ultimately unravelling new factors in bacterial envelope homeostasis. In the next few years, the available ssRNA phage genomes are expected to proportionally increase with the ever-expanding metatranscriptomic studies of various ecological systems.
Conclusions:
Considerable progress has been made in understanding the molecular mechanisms of lysis in small lytic phages. To recap, the Sgls of prototypical phages of Microviridae (E from ΦX174) and Leviviridae (L from MS2 and A2 from Qβ) were studied in detail over the past few decades. The molecular mechanisms of E and A2 are solved, while the mechanism of L has been an enduring mystery since the discovery of the L gene 41 years ago. Genetic and comparative analyses of L and other Sgls from ssRNA phages has revealed a common domain architecture of L-like proteins, despite lacking overall sequence similarity. Moreover, the L-like proteins appear to be the most prevalent Sgls (6 out of 10) among the unique plaque forming ssRNA phages. Recently, LysM the Sgl from coli phage M was shown to be a specific inhibitor of MurJ, the lipid II flippase. The Sgl of Caulobacter phage PΦCb5 (LysΦCb5) is proposed to be an inhibitor of MraY. Among microviruses, E-like proteins appear to be only found in the members of Bullavirinae and no E-homologs have yet been identified in Gokushovirinae. In fact, the microviruses of the Gokushovirinae sub-family lack annotated lysis genes and there is no evidence to-date that suggest that these phages use an SGL system. It would be interesting to see if these viruses might have evolved a MGL system.
In the last five years, these “meta-omics” approaches have rapidly expanded the total known genomic space of both Microviridae and Leviviridae. This massive influx of new genetic information has enhanced our understanding of their abundance and diversity in different ecological niches and the pivotal role these phages play in carbon and other nutrient cycling in the environment[52]. In addition, it has also provided hints at the hidden biology waiting to be discovered. For example, some new proteins have no known homologs, which suggests that these are fairly new additions to the molecular repertoire of these phages. However, challenges remain in finding the native hosts of these phages and in annotating sgls. Among all the new ssRNA phage genomes, there was only one annotated lysis gene (sglAVE017) that was related to a known sgl (MS2 L), with 38% protein sequence identity[50]. Although SglAVE017 remains to be experimentally verified as a L-like protein, this finding highlights the incredible sequence diversity that exists in Leviviridae Sgls. In the recent study, the presence of a predicted export signal was used to narrow down putative sgl candidates[52]. Although this was a relatively systematic approach to finding sgls, the workflow failed to capture candidate reading frames with a proper start site, as most of the candidates did not start with a start codon. A more systematic search for new sgls may illuminate further the regulatory processes of host cell wall regulation and how they are exploited by phage to effect host lysis. The subsequent characterization of their molecular mechanisms are likely to yield new host targets for antibiotic development. In any case, it seems clear that phage biology will continue to be enriched by the study of the intricate evolutionary interplay between the Sgl lysis systems and the genes involved in host envelope synthesis and homeostasis.
Highlights:
Small lytic phages (ssNA) use a single protein (Sgl) to cause host lysis.
The 3 well-characterized Sgls target different steps in peptidoglycan biosynthesis.
Hyperexpansion of the sequenced ssNA genomes in the recent years.
Potential to discover new protein antibiotics.
Acknowledgements:
We thank past and current members of the Young laboratory and the Center for Phage Technology for advice. We also thank Daisy Wilbert for clerical assistance.
Funding:
This work was supported by National Institutes of Health Grant GM27099 and by the Center for Phage Technology at Texas A&M University, jointly sponsored by Texas A&M AgriLife.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References:
- 1.Quemin ER, Quax TE: Archaeal viruses at the cell envelope: entry and egress. Front Microbiol 2015, 6:552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cahill J, Young R: Phage Lysis: Multiple Genes for Multiple Barriers. Adv Virus Res 2019, 103:33–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schooley RT, Biswas B, Gill JJ, Hernandez-Morales A, Lancaster J, Lessor L, Barr JJ, Reed SL, Rohwer F, Benler S, et al. : Development and Use of Personalized Bacteriophage-Based Therapeutic Cocktails To Treat a Patient with a Disseminated Resistant Acinetobacter baumannii Infection. Antimicrob Agents Chemother 2017, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chamakura K, Young R: Phage single-gene lysis: Finding the weak spot in the bacterial cell wall. J Biol Chem 2019, 294:3350–3358.* Most recent review on Single-gene lysis.
- 5.Doore SM, Fane BA: The microviridae: Diversity, assembly, and experimental evolution. Virology 2016, 491:45–55. [DOI] [PubMed] [Google Scholar]
- 6.Sanger F, Air GM, Barrell BG, Brown NL, Coulson AR, Fiddes JC, Hutchison CA III, Slocombe PM, Smith M: Nucleotide sequence of bacteriophage ϕX174 DNA. Nature 1977, 265:687–695. [DOI] [PubMed] [Google Scholar]
- 7.Smith HO, Hutchison CA 3rd, Pfannkoch C, Venter JC: Generating a synthetic genome by whole genome assembly:ϕX174 bacteriophage from synthetic oligonucleotides. Proc Natl Acad Sci U S A 2003, 100:15440–15445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roof WD, Horne SM, Young KD, Young R: slyD, a host gene required for ϕX174 lysis, is related to the FK506-binding protein family of peptidyl-prolyl cis-trans-isomerases. J Biol Chem 1994, 269:2902–2910. [PubMed] [Google Scholar]
- 9.Bernhardt TG, Roof WD, Young R: Genetic evidence that the bacteriophage ϕX174 lysis protein inhibits cell wall synthesis. Proc Natl Acad Sci U S A 2000, 97:4297–4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng Y, Struck DK, Young R: Purification and functional characterization of ϕX174 lysis protein E. Biochemistry 2009, 48:4999–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maratea D, Young K, Young R: Deletion and fusion analysis of theϕX174 lysis gene E. Gene 1985, 40:39–46. [DOI] [PubMed] [Google Scholar]
- 12.Struck DK, Maratea D, Young R: Purification of hybrid á-galactosidase proteins encoded by f X174 E f l acZ and E. coli prlA f lacZ : a general method for the isolation of lacZ fusion polypeptides produced in low amounts. J.Mol.Appl.Genet 1985, 3:18–25. [PubMed] [Google Scholar]
- 13.Chamakura KR, Tran JS, Young R: MS2 lysis of Escherichia coli depends on host chaperone DnaJ. J Bacteriol 2017, 199. [DOI] [PMC free article] [PubMed]
- 14.Tanaka S, Clemons WM Jr.: Minimal requirements for inhibition of MraY by lysis protein E from bacteriophage ϕX174. Mol Microbiol 2012, 85:975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng Y, Struck DK, Bernhardt TG, Young R: Genetic analysis of MraY inhibition by the ϕX174 protein E. Genetics 2008, 180:1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bradley DE, Dewar CA, Robertson D: Structural changes in Escherichia coli infected with a ϕX174 type bacteriophage. J Gen Virol 1969, 5:113–121. [DOI] [PubMed] [Google Scholar]
- 17.Liu BL, Everson JS, Fane B, Giannikopoulou P, Vretou E, Lambden PR, Clarke IN: Molecular characterization of a bacteriophage (Chp2) from Chlamydia psittaci 2. Journal of Virology 2000, 74:3464–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brentlinger KL, Hafenstein S, Novak CR, Fane BA, Borgon R, McKenna R, Agbandje-McKenna M: Microviridae, a family divided: Isolation, characterization, and genome sequence of f MH2K, a bacteriophage of the obligate intracellular parasitic bacterium Bdellovibrio bacteriovorus J. Bacteriol 2002, 184:1089–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Renaudin J, Pascarel MC, Bove JM: Spiroplasma virus 4: nucleotide sequence of the viral DNA, regulatory signals, and proposed genome organization 3. J.Bacteriology 1987, 169:4950–4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Young R, Wang IN: Phage lysis In The Bacteriophages, edn 2nd Edited by Calendar R: Oxford University Press; 2006:104–126. [Google Scholar]
- 21.Ghuysen J-M, Goffin C: Lack of Cell Wall Peptidoglycan versus Penicillin Sensitivity: New Insights into the Chlamydial Anomaly. Antimicrob. Agents Chemother 1999, 43:2339–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kosma P: Chlamydial lipopolysaccharide. Biochim. Biophys. Acta 1999, 1455:387–402. [DOI] [PubMed] [Google Scholar]
- 23.Szekely AJ, Breitbart M: Single-stranded DNA phages: from early molecular biology tools to recent revolutions in environmental microbiology. FEMS Microbiol Lett 2016, 363. [DOI] [PubMed] [Google Scholar]
- 24.Roux S, Solonenko NE, Dang VT, Poulos BT, Schwenck SM, Goldsmith DB, Coleman ML, Breitbart M, Sullivan MB: Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ 2016, 4:e2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim MS, Park EJ, Roh SW, Bae JW: Diversity and abundance of single-stranded DNA viruses in human feces. Appl Environ Microbiol 2011, 77:8062–8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Creasy A, Rosario K, Leigh BA, Dishaw LJ, Breitbart M: Unprecedented Diversity of ssDNA Phages from the Family Microviridae Detected within the Gut of a Protochordate Model Organism (Ciona robusta). Viruses 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, Ling Y, Shan T, Yang S, Xu H, Deng X, Delwart E, Zhang W: Gut virome of mammals and birds reveals high genetic diversity of the family Microviridae. Virus Evol 2019, 5:vez013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krupovic M, Forterre P: Microviridae goes temperate: microvirus-related proviruses reside in the genomes of Bacteroidetes. PLoS One 2011, 6:e19893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirchberger PC, Ochman H: Resurrection of a global, metagenomically defined Gokushovirus. Elife 2020, 9.**The authors discovered several Gokushoviruses as integrated prophages in the genomes of E. coli and other Enterobacteriaceae. The authors for the first time reconstitute viable gokusho-phage particles from a synthetic circular dsDNA construct and show that the reconstructed virions can plaque on E. coli lawns.
- 30.Guzman LM, Belin D, Carson MJ, Beckwith J: Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol 1995, 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bollback JP, Huelsenbeck JP: Phylogeny, genome evolution, and host specificity of single-stranded RNA bacteriophage (family Leviviridae). J.Mol.Evol 2001, 52:117–128. [DOI] [PubMed] [Google Scholar]
- 32.Dai X, Li Z, Lai M, Shu S, Du Y, Zhou ZH, Sun R: In situ structures of the genome and genome-delivery apparatus in a single-stranded RNA virus. Nature 2017, 541:112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gorzelnik KV, Cui Z, Reed CA, Jakana J, Young R, Zhang J: Asymmetric cryo-EM structure of the canonical Allolevivirus Qβ reveals a single maturation protein and the genomic ssRNA in situ. Proc Natl Acad Sci U S A 2016, 113:11519–11524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meng R, Jiang M, Cui Z, Chang JY, Yang K, Jakana J, Yu X, Wang Z, Hu B, Zhang J: Structural basis for the adsorption of a single-stranded RNA bacteriophage. Nat Commun 2019, 10:3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zinder ND: RNA phages. Annu Rev Microbiol 1965, 19:455–472. [DOI] [PubMed] [Google Scholar]
- 36.Pourcel C, Midoux C, Vergnaud G, Latino L: A carrier state is established in Pseudomonas aeruginosa by phage LeviOr01, a newly isolated ssRNA levivirus. J Gen Virol 2017, 98:2181–2189. [DOI] [PubMed] [Google Scholar]
- 37.Kannoly S, Shao Y, Wang IN: Rethinking the evolution of single-stranded RNA (ssRNA) bacteriophages based on genomic sequences and characterizations of two R-plasmid-dependent ssRNA phages, C-1 and Hgal1. J Bacteriol 2012, 194:5073–5079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Domingo E, Holland JJ: RNA virus mutations and fitness for survival. Annu Rev Microbiol 1997, 51:151–178. [DOI] [PubMed] [Google Scholar]
- 39.Bernhardt TG, Wang IN, Struck DK, Young R: A protein antibiotic in the phage Qβ virion: diversity in lysis targets. Science 2001, 292:2326–2329. [DOI] [PubMed] [Google Scholar]
- 40.Reed CA, Langlais C, Kuznetsov V, Young R: Inhibitory mechanism of the Qβ lysis protein A2. Mol Microbiol 2012, 86:836–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cui Z, Gorzelnik KV, Chang JY, Langlais C, Jakana J, Young R, Zhang J: Structures of Qβ virions, virus-like particles, and the Qβ-MurA complex reveal internal coat proteins and the mechanism of host lysis. Proc Natl Acad Sci U S A 2017, 114:11697–11702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chamakura KR, Sham LT, Davis RM, Min L, Cho H, Ruiz N, Bernhardt TG, Young R: A viral protein antibiotic inhibits lipid II flippase activity. Nat Microbiol 2017, 2:1480–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holtje JV, van Duin J: MS2 phage induced lysis of E. coli depends upon the activity of the bacterial autolysins In Microbial Cell Wall Synthesis and Autolysis Edited by Nombela C: Elsevier Science Publishers; 1984:195–199. [Google Scholar]
- 44.Berkhout B, de Smit MH, Spanjaard RA, Blom T, van Duin J: The amino terminal half of the MS2-coded lysis protein is dispensable for function: implications for our understanding of coding region overlaps. EMBO J 1985, 4:3315–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chamakura KR, Edwards GB, Young R: Mutational analysis of the MS2 lysis protein L. Microbiology 2017, 163:961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klovins J, Overbeek GP, van den Worm SH, Ackermann HW, van Duin J: Nucleotide sequence of a ssRNA phage from Acinetobacter: kinship to coliphages. J Gen Virol 2002, 83:1523–1533. [DOI] [PubMed] [Google Scholar]
- 47.Kazaks A, Voronkova T, Rumnieks J, Dishlers A, Tars K: Genome structure of Caulobacter phage phiCb5. J Virol 2011, 85:4628–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rumnieks J, Tars K: Diversity of pili-specific bacteriophages: genome sequence of IncM plasmid-dependent RNA phage M. BMC Microbiol 2012, 12:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greninger AL, DeRisi JL: Draft Genome Sequences of Leviviridae RNA Phages EC and MB Recovered from San Francisco Wastewater. Genome Announc 2015, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krishnamurthy SR, Janowski AB, Zhao G, Barouch D, Wang D: Hyperexpansion of RNA Bacteriophage Diversity. PLoS Biol 2016, 14:e1002409.* First major expansion of ssRNA phage genomes. The authors mined publicly available metatranscriptomes and RNA-inclusive metagenomes for signatures of ssRNA phage core proteins. A total of 122 novel ssRNA phage genomes were discovered in this one study with some genomes having new ORFs in unusual genetic architectures.
- 51.Shi M, Lin XD, Tian JH, Chen LJ, Chen X, Li CX, Qin XC, Li J, Cao JP, Eden JS, et al. : Redefining the invertebrate RNA virosphere. Nature 2016, 540:539–543. [DOI] [PubMed] [Google Scholar]
- 52.Starr EP, Nuccio EE, Pett-Ridge J, Banfield JF, Firestone MK: Metatranscriptomic reconstruction reveals RNA viruses with the potential to shape carbon cycling in soil. Proc Natl Acad Sci U S A 2019, 116:25900–25908.**Temporal dynamics in the diversity and abundance of RNA viruses in soil metatranscriptomes and this study added another 1300 new ssRNA phage genomes to the NCBI database.
- 53.Callanan J, Stockdale SR, Shkoporov A, Draper LA, Ross RP, Hill C: Expansion of known ssRNA phage genomes: From tens to over a thousand. Sci Adv 2020, 6:eaay5981.** The authors use Hidden Markov Model to mine ssRNA phage genomes in the publicly available metranscriptomes sourced from sewage and freshwater ecosystems. Approximately 14,000 new partial and ~1000 near-complete ssRNA phage genomes were discovered.