

Abstract
Central and peripheral tolerance both contribute to protection against autoimmunity. The pathogenesis of autoimmunity, however, can result from critical deficits or limitations in peripheral and/or central tolerance mechanisms, presenting an opportunity for therapeutic intervention. Recent advances highlight the substantial impact of inhibitory receptors (IRs), which mediate peripheral tolerance, in autoimmunity. Deletion and blockade studies in mice, IR disruption in humans, and correlation with positive disease outcomes all highlight potential clinical benefits of enhancing IR signaling (agonism) - specifically CTLA4, PD1, LAG3, TIM3 and TIGIT - to treat autoimmune disease. Although critical questions remain, IR agonists represent an unappreciated and untapped opportunity for the treatment of autoimmune and inflammatory diseases.
Introduction
Multiple mechanisms of self-tolerance protect against autoimmunity, including central and peripheral tolerance. While central tolerance is critical to delete cells expressing autoreactive B and T cell receptors, it is often imperfect [1]. Peripheral tolerance mechanisms, including ignorance, anergy or apoptosis, and regulatory T cells (Tregs), are therefore necessary to restrain autoreactivity [1]. Recent reports estimate up to ~30% CD4+Foxp3− cells in healthy mice are able to respond to ‘self’ but are restrained by Tregs, and posits a compensatory role for inhibitory receptor (IR) expression in their model lacking Tregs, implying that >30% of CD4+ effectors could be autoreactive [2]. Critical deficits or limitations in tolerance mechanisms, rather than complete failure, can contribute to autoimmunity (Fig. 1). Evidence suggests that the majority of these deficits affect peripheral tolerance, presenting an opportunity for therapeutic intervention [1].
Figure 1 legend:
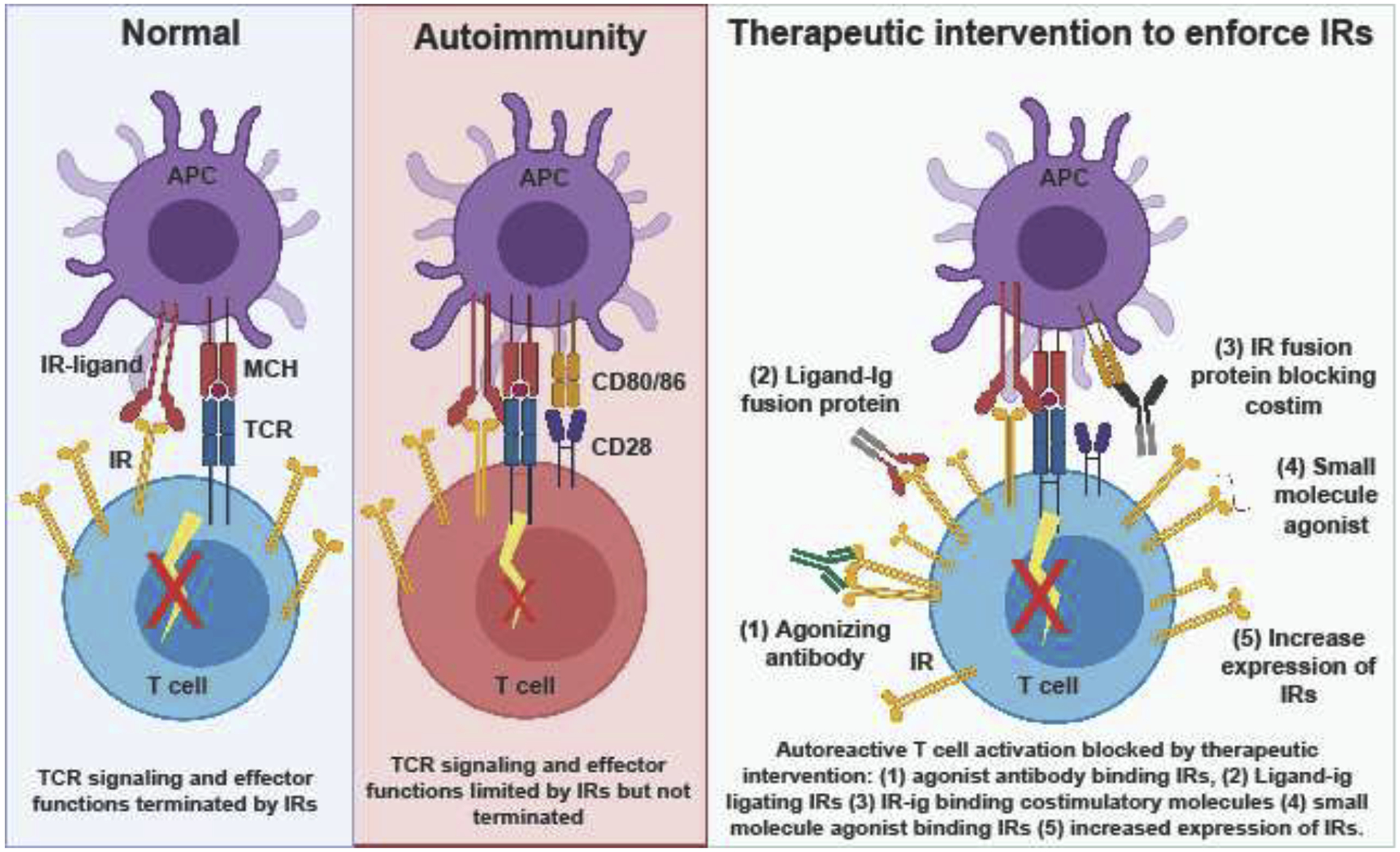
The role of IRs under normal conditions is to limit TCR signaling and terminate an immune response post-infection or to self-antigen. In autoimmune conditions, IRs are present and limit autoimmunity, but are ultimately insufficient to completely prevent autoimmunity, opening up an opportunity for therapeutic intervention. If IR expression or signaling and subsequent downstream effects is enforced therapeutically, autoimmunity may be prevented or managed more easily by patients.
Recent advances in cancer immunotherapy indicate that IR modulation affects T cell function and disease outcome, which merited the 2018 Nobel Prize in Medicine [3]. IRs mediate peripheral tolerance by antagonizing T cell receptor (TCR) signaling and/or signal propagation in autoreactive T cells, resulting in a dysfunctional state [4]. Evolutionarily, IRs are upregulated in response TCR signaling to dampen the immune response post infection or upon recognition of self-antigen (Fig. 1). Under chronic conditions, IR expression is heightened and T cell effector function is inhibited [5]. Furthermore, evidence suggests that IRs are often upregulated as a module (ie multiple IRs at the same time), implying cooperative and/or synergistic activity [6]. IRs have multiple ligands and different signaling mechanisms (reviewed in [7–9]), but overall result in dampening immune activation. An increasing appreciation for the role of IRs in autoimmunity suggests that IR agonism (increasing inhibitory signaling and its downstream consequences) may help prevent and manage autoimmune disease (Table 1–3). Such evidence is exemplified by deletion or blockade studies in mice, outcomes of IR disruption in humans, and positive correlation of IR expression with favorable disease outcomes. This review briefly summarizes the current state of the field in understanding the role of IRs in autoimmunity, with a focus on the IRs currently in clinical research: cytotoxic T-lymphocyte-associated protein 4 (CTLA4), Programed cell death protein 1 (PD1), Lymphocyte Activating Gene 3 (LAG3), T cell immunoglobulin and mucin domain-containing protein 3 (TIM3), and T cell immunoreceptor with Ig and ITIM domains (TIGIT).
Table 1:
IR agonists showing therapeutic efficacy in mouse models of autoimmunity. MHC – Major histocompatibility complex, MOG - Myelin Oligodendrocyte Glycoprotein, Trail - TNF-related apoptosis-inducing ligand, CD253, ICOS - Inducible T-cell costimulatory, Ig – immunoglobin, Fc – refers to the constant region of an antibody, RA – Rheumatoid Arthritis, DSS – Dextran Sodium Sulfate, CIA – Collagen induced Arthritis
| Enforcing IR engagement in autoimmune mouse models in vivo | |||
|---|---|---|---|
| IR targeted: | Method of reinforcement: | Disease setting: | Ref: |
| PD1 | Dendritic cells transduced to expressed MHC carrying MOG, PDL1, and Trail. | improved EAE outcomes | [53] |
| Adenovirus expressing PDL1 + ICOS inhibitor | suppresses lupus symptoms in disease prone BXSB mice | [54] | |
| Adenovirus expressing PDL1-Fc | ameliorates DSS Colitis | [55] | |
| PDL1-Ig Fusion recombinant proteins | ameliorates T cell mediated Colitis | [55] | |
| PDL1 -Fc fusion protein | PDL1-fc prevented activation in synovial fluid mononuclear cells of RA patients and ameliorated CIA | [56] | |
| PDL1-Ig fusion protein | Ameliorated CIA | [57] | |
| PDL1-Ig fusion protein + anti-CD154 | Improves Islet transplantation tolerance | [58] | |
| CTLA4 | Transgenic mouse expressing membrane bound agonist anti-CTLA4 on B cells | protects NOD mice from autoimmune diabetes onset | [59] |
| TIM3 | Galectin-9 (ligand for TIM3) was administered as a soluble protein | ameliorates CIA | [60] |
| Interferon beta and Galectin-9 fusion proteins | ameliorates EAE | [61] | |
| TIGIT | agonist anti-TIGIT antibody | Improves disease outcomes in EAE | [62] |
Table 3:
Clinical trials/approvals aimed at reinforcing IRs to treat autoimmunity. GVHD – Graph versus host disease
| Current trials or approvals with IR modulators in patients with autoimmune disease | |||
|---|---|---|---|
| Therapeutic targets: |
Method of reinforcement: | Diseases being studied: | Ref: |
| PD1 | PD1 agonist antibody ANB030, phase 1 trials predicted to begin soon | Human alopecia PBMC, humanized model of GVHD | AnaptysBioANB030 |
| PD1 agonist antibody in phase 1 at Eli Lily: Patent US2019/0270818 A1 | Under investigation for autoimmune diseases such as RA and transplant rejection | [67] | |
| https://www.lilly.com/discovery/clinicaldevelopmentpipeline#/ | |||
| CC-90006 Celgene agonist antibody: NCT03337022 | Investigated for psoriatic arthritis | Clinicaltrials.gov | |
| Blocking CD80/86 co-stimulation | Abatacept: CTLA4-ig, blocks CD28 co-stimulation | Summary of selected studies in autoimmune disease settings: | |
| RA (approved) | [68] | ||
| SLE (so far failed to meet clinical trial outcomes) | [69, | ||
| Sojourns Syndrome (promising results in phase 3) NCT02067910 | [70] | ||
| Autoimmune Hepatitis (recruiting for phase 1) NCT04203875 | Clinicaltrials.gov | ||
| Alopecia Areata (phase 2, positive outcomes) NCT02018042 | Clinicaltrials.gov | ||
| Prevention of T1D (Phase 2, delayed, but did not prevent T1 D) NCT01773707 | [71] | ||
| Systemic Sclerosis (Phase 2, well tolerated, not statistically significant outcomes) NCT02161406 | [72] | ||
| Relapsing Remitting MS (Phase 2, well tolerated, not statistically significant outcomes) NCT01116427 | [73] | ||
| Ulcerative Colitis/Crohn’s disease (Failed to show efficacy in Phase 3) NCT00410410 | [74] | ||
| CD80/86 and B cells | Abatacept +Rituxumab: CTLA4-ig blocking CD2P co-stimulation + B cell depletion | T1D (Recruiting for phase 2) NCT03929601 | Clinicaltrials.gov |
| CD80/86 and inflammatory cytokines IL-12/23 | Abatacept + Ustekinumab: anti- IL-12/23 + CTLA4-ig blocking CD28 co-stimulation | Psoriasis Vulgaris (Phase 2, some clinical benefit) CT01999868 | Clinicaltrials.gov |
Genetic deletion and antibody blockade studies in mice
Some of the most compelling evidence for the role of IRs in autoimmunity comes from genetic deletion or antibody blockade studies. Under non-autoimmune prone conditions, genetic ablation of either CTLA4 or PD1 results in development of spontaneous autoimmune symptoms, albeit with slightly different manifestations. Mice deficient in CTLA4 rapidly develop severe lymphoproliferative disease resulting in death by 3–4 weeks of age, whereas PD1-deficient mice develop lupus-like symptoms over time, implying non-redundant functions in maintenance of immune tolerance [10–12]. These data illustrate that simply eliminating PD1 or CTLA4 is sufficient to disrupt baseline immune homeostasis and maintenance of peripheral tolerance. Alternatively, while genetic ablation of either LAG3, TIGIT or TIM3 in mice does not precipitate spontaneous autoimmunity, LAG3/PD1 double-knockout (KO) mice experience multiple organ autoimmunity that manifests more aggressively than PD1 KO alone, suggesting a synergistic effect in limiting autoimmunity [13,14]. This difference in disease progression and manifestation highlights the complexity of targeting each IR in a disease setting, the hierarchy of importance in limiting autoreactivity beginning with CTLA4 and PD1, and possible synergistic effects of ‘tertiary’ IRs such as LAG3, TIGIT and TIM3.
Regardless of their role in baseline immune homeostasis or spontaneous autoimmunity, blocking or genetically deleting IRs exacerbates disease in autoimmune-prone backgrounds or induced disease models (reviewed in [15]). For example, in the non-obese diabetic (NOD) mouse model of autoimmune diabetes, a spontaneous model in which 80% of female mice develop diabetes between 10–30 weeks of age, treatment with anti-PD1 or anti-LAG3 induces autoimmune diabetes with 100% penetrance within only a few weeks post administration, regardless of timing [16]. Similar results are obtained in global PD1, LAG3 or CTLA4 KOs on a NOD background [16–20]. Conceptually, these results are mirrored on the MRL-lpr background, which is susceptible to spontaneous lupus-like autoimmunity. On the MRL-lpr background, PD1, CTLA4 and B- and T-lymphocyte attenuator (BTLA) have been shown to limit disease progression in studies with KO mice and/or antibody blockade [15,21].
Additionally, IR blockade or deletion worsens autoimmunity in inducible settings. Studies using the experimental autoimmune encephalomyelitis model (EAE; a mouse model of multiple sclerosis), demonstrated that treatment with anti-PD1 or genetic deletion of PD1, TIM3, and TIGIT exacerbated disease symptoms [22–24]. Similarly, IRs may control environmentally-induced autoimmunity, as studies with LAG3 KO mice highlight a role for this IR in limiting mercury-induced autoimmune dysfunction [25].
To add further complexity, IRs play different roles depending on the cell type. For example, Tregs can use IRs such as LAG3 and CTLA4 as mechanisms of suppression [26,27]. Indeed, non-autoimmune prone C57Bl/6 mice with Treg-restricted deletion of CTLA4 develop extensive lymphoproliferative disease, similar to Ctla4 global KO or Foxp3−/− mice, implying Treg suppressive capacity is severely impaired in the absence of CTLA4 and that Tregs may be driving the phenotype behind the global CTLA4 KO [28]. Conversely, however, Treg-restricted deletion of LAG3 improves disease outcomes in autoimmune diabetes, as LAG3 was shown to limit Treg proliferation in this disease setting [29].
Finally, the timing of IR blockade administration or induction of IR deletion can give rise to differing disease outcomes, highlighting another layer to consider when targeting IRs in an autoimmune setting. For example, anti-CTLA4 treatment of NOD mice induces autoimmune diabetes with 100% penetrance only when administered prior to insulitis onset [18,19]. Similarly, Treg-restricted temporal deletion of Ctla4 in C57Bl/6 mice in adulthood does not cause the hallmark systemic autoimmunity of global or Treg specific CTLA4 KO [30]. Both observations suggest that the suppressive capacity of CTLA4 may be more important during the T cell priming phase of disease [18,19,31]. Unexpectedly, although quite similar to Treg-restricted LAG3 deletion in the NOD model, adulthood Treg-restricted CTLA4 deletion confers protection from EAE, which is attributed to increased Treg numbers and an upregulation of compensatory IRs on CD4+ effector T cells [30]. The aforementioned dichotomous results imply a need to further understand the role of these IRs temporally, and in a cell type and disease specific manner prior to targeting them therapeutically.
IR disruption in humans
While animal studies are useful to gain a mechanistic understanding of IRs, there is good evidence that IR disruption also contributes to autoimmunity in humans. Notably, many large-scale genome-wide association studies (GWAS) have linked hundreds of SNPs to specific autoimmune diseases, many of which are immune related and are within genes encoding IRs [1]. This implies that IR function is critical to maintain self-tolerance, although these studies only inform correlation and not causation. SNPs have varying levels of association in autoimmune-prone individuals compared to the general population. Databases compiling GWAS studies in conjunction with other published data can now score genes for association with a particular disease [32] (Open Target Platforms URL: Targetvalidation.org). Interestingly, there is an almost hierarchical level of association of IRs with autoimmune disease. CTLA4 is very strongly implicated in almost every autoimmune disease, and thus could be considered a primary IR. The strongest associations linked to CTLA4 SNPs are type 1 diabetes (T1D), Graves’ disease, and systemic lupus erythematosus (SLE) [33]. PDCD1 (gene for PD1) is associated with many diseases but not all, specifically SLE [34], and thus could be considered a secondary IR. Tertiary IRs, such as TIGIT, LAG3 and HAVCR2 (TIM3), have lower association scores but are still linked to certain autoimmune diseases [9].
The use of IR blockade as a cancer therapeutic provides a unique setting to study the role of IRs in humans. Many patients with no prior history of autoimmunity who receive treatment with anti-CTLA4, anti-PD1, or anti-PDL1 experience autoimmune side-effects; as many as 85%, 37%, and 24% of patients, respectively [31]. The majority of IR blockade-induced autoimmunity results in organ specific immune infiltration and damage, and often occurs in a location distal to that of the primary malignancy (reviewed in [31,35]). These autoimmune side effects are difficult to predict and interpret, but they posit the idea that individuals may be poised for or experiencing subclinical autoreactivity, which is restrained by IRs, and only by blocking IR function is clinical autoimmunity revealed.
Signatures of IR expression are predictive of prognosis in human autoimmunity
Signatures of IR expression and inhibition are actively being implicated in autoimmune disease progression and outcomes. IRs are particularly upregulated in chronic diseases, and often result in a state termed “exhaustion” in which T cells lose effector functions and experience genome wide transcriptional and epigenetic changes [5]. Most autoimmune diseases are chronic and thus T cells from these patients show an IR-rich phenotype. The dichotomy of expressing IRs while still remaining pathogenic is somewhat of an enigma in autoimmunity; yet, this phenomenon highlights the fact that normal regulatory processes are present but insufficient to prevent autoimmunity (Fig. 1). This provides a potential window for therapeutic intervention, possibly even supporting the idea of therapeutically induced exhaustion to treat autoimmunity [36].
Given the recent advances in systems immunology and in tracking patients over time, we can now better investigate the relationship between IR expression and disease outcomes in humans. In 2015, the first implication of IR induced T cell exhaustion was linked to better prognosis in a long-term study of patients with antibody-associated vasculitis (AAV) [37]. Indeed, gene set enrichment for an exhaustion signature predicted better flare-free survival in this AAV cohort, as well as in inflammatory bowel disease (IBD) and SLE cohorts [37]. Patients were followed for up to 2,000 days post initial analysis, providing valuable information on flare-free survival as opposed to disease activity at time of blood draw, for which IR signature is not predictive [37]. Similarly, recently diagnosed T1D patients with an enriched exhausted phenotype on CD8+ T cells are reported to have slower disease progression and longer maintenance of c-peptide levels, a biomarker of insulin secretion [38]. Both of these studies identified an “exhaustion” signature based on gene or protein expression from peripheral blood, and relied mainly on IR expression, though it should be noted that this is just one aspect of an exhausted T cell. A current research limitation is obtaining adequate numbers of IR-expressing cells from human peripheral blood for analysis that would prove that these cells are functionally exhausted or for epigenetic analysis. Furthermore, an even more distinct exhausted signature may be evident in the target organ of an autoimmune disease, which presents another limitation of human studies.
Finally, there are novel immunotherapeutic strategies that show efficacy by indirectly modulating IR expression. T1D studies make an interesting case for targeting IR expression and function therapeutically, as many novel therapeutics directly target T cell function. Novel immunotherapies for T1D are limited in that they must be more safe and efficacious than daily insulin injections. Therefore, novel T1D therapies must specifically target T cell function and several therapeutics in trial modulate IRs to meet this goal.
The first example is teplizumab, an anti-CD3 blocking antibody, which is in clinical trial to prevent T1D in at-risk individuals (NCT03875729) [39]. Antagonizing CD3 blocks TCR signal propagation and subsequent activation. In both mice and humans, anti-CD3 induces remission and delays the onset of diabetes [39]. The mechanism of action of anti-CD3 therapy has been attributed not only to preserving Tregs, but also to inducing a “partially exhausted” or “potentially anergic” state in effector CD4+ and CD8+ T cells [40,41]. In fact, patients treated with anti-CD3 in trial showed multiple IR modulating effects, including transiently increased PD1+ Tregs, increased potentially anergic CD4+ effectors (CD57−KLRG1−PD1+) and increased partially exhausted CD8+ T cell populations (CD57−KLRG1+PD1+ and TIGIT+KLRG1+), characterized by increased exhaustion related transcription factor, EOMES, and downregulation of memory marker CD127 [40]. Furthermore, those TIGIT+KLRG1+ CD8+ T cells from responding patients showed transcriptional enrichment for markers of exhaustion including EOMES and IRs Tigit, Lag3, Cd160, and Tim3 [41]. Although anti-CD3 does not directly target IRs, anti-CD3 modulates IR expression and thus T cell function for therapeutic benefit, revealing the potential untapped power that reinforcing IRs may have on disease outcome. In summary, signatures of IR expression, either naturally or therapeutically induced, have been correlated to positive disease outcomes in T1D, AAV, SLE, and IBD in humans.
Similar to anti-CD3, though yet to be translated to humans, nanoparticles containing an insulin–ChgA hybrid peptide show therapeutic induction of anergy in the NOD model [42]. Nanoparticles can induce an antigen-specific response by delivering disease-specific antigens to Tregs. Interestingly, insulin–ChgA hybrid peptides not only increase the Treg:CD4+Foxp3− T effector (Teff) ratio, but also alter the Teff phenotype. RNAseq shows this modulated phenotype is characterized by high expression of IRs Lag3 and Pdcd1, as well as several markers of anergy, thus contributing to disease protection [42].
Finally, signatures of IR upregulation and T cell dysfunction have been shown in other autoimmune settings. For example, features of T cell exhaustion, including IR upregulation, have also been observed in the kidney of MRL-lpr mice with nephritis [43]. Similarly, anergic CD4+ T cells can differentiate into Tregs and prevent autoimmune arthritis and IBD in an adoptive transfer settings, in which IR upregulation is observed [44]. However, an expanded PD1+CD8+ metabolically active population was recently found in IBD, juvenile idiopathic arthritis and atopic dermatitis, suggesting that single IR expression may simply mark an activated state, as opposed to a truly dysfunctional state [45]. Identifying these IR-expressing populations is the first step to determine clinical impact and provides evidence to warrant deeper investigation into their function in autoimmunity. This poses the question - can IR agonism, in monotherapy or in combination with other therapeutics, delay or prevent the onset of autoimmunity and/or limit symptoms?
Therapeutically targeting IRs
The success of cancer immunotherapy highlighted IRs as a meaningful and significant target to impact disease outcome. In autoimmunity, novel therapies that induce IR upregulation prevent or delay disease. However, therapeutics that directly agonize or enhance IR signaling are very limited and are, at most, in early phases of clinical trial (reviewed in [46]) (Fig. 1, Table 3). However, successful mouse studies and in vitro human studies have highlighted the potential therapeutic benefits that IR agonists may have in the treatment of autoimmunity (Table 1 and Table 2).
Table 2:
IR reinforcement studies that will be translated to treat autoimmune patients. FGL1 – Fibrinogen-like protein 1, PBMC – Peripheral blood mononuclear cells, MS – multiple sclerosis.
| Observations that may be translated to autoimmune disease | |||
|---|---|---|---|
| IR targeted: | Method of reinforcement: | Setting studied: | Ref: |
| LAG3 | FGL1 -Ig suppresses T cell proliferation in vitro | Treatment with FGL1-ig prevents Lag3+ T cell activation | [63] |
| Crosslinking LAG3 with CD3 prevented T cell activation | Cross-linking prevents activation of human PBMC | [64] | |
| LAG3 agonist antibody IMP761 | prevents activation of human PBMC, and delayed type hypersensitivity in cynomolgus macaque | [65] | |
| TIGIT | Agonist anti-TIGIT | decreases proliferation and cytokine production in vitro from CD4+ T cells collected from MS patients | [66] |
Despite some success, key questions remain that will inform the development of IR-agonist therapeutics. (1) Which cell types are targeted by IR agonism, and what is the impact on disease outcomes? While such therapeutics aim to target self-reactive CD4+ and CD8+ effector T cells, inevitably Tregs will also be influenced by IR agonists, the results of which are largely unknown. Additionally, agonizing certain IRs over others may preferentially affect effector T cell activation over Tregs, for example anti-CTLA4, as evidenced by temporal Treg-specific deletion studies [30].
(2) At what timepoint do IR agonists best influence disease outcome? This question is complicated because the answer may depend on the disease state as well as the IR in question. For example, CTLA4 functions in a time-restricted window in T1D; this window may be shifted or protracted in other immune settings. In many autoimmune cases, we may need to administer immunotherapeutic treatment prior to disease onset to reveal the true potential in disease prevention, which raises further questions regarding identification of patients at high risk for disease [39]. We must begin to assess timepoints for efficacy as well as how best to incorporate IR agonism into current treatments for autoimmunity, for example, as a monotherapy or in combination with other therapeutics, either dual IR-agonism or standard of care treatments.
(3) Are there potentially detrimental off-target effects of IR agonism through general immune suppression? IR blockade-induced autoimmunity occurs in a significant proportion of cancer patients [31]. One can envision opposing effects in autoimmune patients treated with IR agonists, such as poor response to vaccinations, impaired viral clearance, or even increased risk of cancer. Thus, the goal must be to minimize these possible adverse events for clinical treatment.
(4) What is the best way to target IRs in an agonistic manner (Fig. 1, Tables 1–3)? Agonist antibodies can be challenging to generate, but potent examples exist, such as the CD28 agonist TGN1412, which caused detrimental cytokine storms in phase 1 trials [47]. Further, there is great potential and value in producing novel IR agonists (Tables 1–3), some of which are showing promising therapeutic efficacy. Can similarly potent agonistic effects be achieved using small-molecule agonists or ligand (e.g., PDL1-Ig fusion to induce PD1 signaling)? Can IR fusion proteins (e.g., CTLA4-Ig, abatacept, Table 3) be used to adequately block co-stimulation? Can we achieve similar results by simply increasing expression of IRs? These issues are complicated further by an incomplete understanding of IR transcriptional regulation, ligands, and signaling pathways [15]. Furthermore, certain ligands, for example CD80/86 or CD155, may be shared between IRs and co-stimulatory molecules, which complicates the use of ligands to enhance IR signaling. Lastly, in choosing an approach to enhance IR signaling and function, one must consider that many IRs can also be produced and/or shed as soluble receptors in serum, such as LAG3, TIM3, CTLA4 and PD1, and may regulate T cell activity and/or interfere with any therapeutic approach [48–51]. Of note, a soluble CTLA4 isoform has been found to be elevated in many autoimmune settings and influences T cell proliferation and cytokine production [50]. In contrast, soluble LAG3, which is generated by ADAM metalloprotease-mediated cell surface shedding, does not appear to impact T cell function [51,52]. Thus care must be taken when using IR enforcement strategies to evaluate any potential impact of soluble IRs on efficacy and/or dosing. Levels of soluble IRs may vary between patients, which also may complicate evaluating the appropriate therapeutic window for IR agonists, and may require analysis on a per-patient basis. In summary, we need a better understanding of whether there are specific IRs expressed in unique disease settings and/or on unique cell types, the temporal usage of IRs, IR signaling and function, and patient prognosis information.
The role of IRs in autoimmunity holds promise for the development of novel therapeutic approaches to treat autoimmunity. There is strong rationale and supportive evidence for targeting IRs in autoimmunity through deletion or blockade studies, information gathered from human IR disruptions, and attempts to identify IR correlations with disease progression and outcomes. Although many questions remain, the future of immunotherapy for autoimmune disease may involve the use of IR agonists.
Acknowledgements
The authors thank Angela Gocher and Creg J. Workman for helpful comments. This work was supported by the National Institutes of Health [F31 AI147638 and T32 AI089443 to S.G.; R01 DK089125, R01 AI144422 and P01 AI108545 to D.A.A.V.].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Declaration of interest: D.A.A.V. has submitted patents on LAG3 and NRP1 that are issued or pending and is entitled to a share in net income generated from licensing of these patent rights for commercial development.
Bibliography
Papers of particular interest, published within the period of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Theofilopoulos AN, Kono DH, Baccala R: The multiple pathways to autoimmunity. Nat. Immunol 2017, 18:716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2**.Cebula A, Kuczma M, Szurek E, Pietrzak M, Savage N, Elhefnawy WR, Rempala G, Kraj P, Ignatowicz L: Dormant pathogenic CD4+ T cells are prevalent in the peripheral repertoire of healthy mice. Nat. Commun 2019, 10:4882. [DOI] [PMC free article] [PubMed] [Google Scholar]; The prevalence of auto-antigen specific CD4+ effector T cells is re-examined, finding that in the absence of Tregs, ~30% of the peripheral CD4+ effector T cell repitoire is capable of recognizing ubiquituous self peptides presented on MCHII by conventional dendritic cells. This paper highlights the importance of peripheral tolerance in mediating immune homeostasis.
- 3.Zang X: 2018 Nobel Prize in medicine awarded to cancer immunotherapy: Immune checkpoint blockade - A personal account. Genes Dis. 2018, 5:302–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walker LSK, Abbas AK: The enemy within: keeping self-reactive T cells at bay in the periphery. Nat. Rev. Immunol 2002, 2:11–19. [DOI] [PubMed] [Google Scholar]
- 5.Wherry EJ, Kurachi M: Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol 2015, 15:486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6*.Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, Nyman J, Marjanovic ND, Kowalczyk MS, Wang C, et al. : Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 2018, 558:454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]; A co-inhibitory receptor module is identified and includes expression of PD1, TIM3, LAG3 and TIGIT and is controlled by transcription factors PRDM1 and c-MAF. Given that IRs are disregulated in autoimmunity, this provides a potential module that may be of interest when desigining novel therapeutics for autoimmune disease.
- 7.Schildberg FA, Klein SR, Freeman GJ, Sharpe AH: Coinhibitory Pathways in the B7-CD28Ligand-Receptor Family. Immunity 2016, 44:955–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andrews LP, Yano H, Vignali DAA: Inhibitory receptors and ligands beyond PD-1, PDL1 and CTLA-4: breakthroughs or backups. Nat. Immunol 2019, 20:1425–1434. [DOI] [PubMed] [Google Scholar]
- 9.Anderson AC, Joller N, Kuchroo VK: Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44:989–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishimura H, Nose M, Hiai H, Minato N, Honjo T: Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11:141–151. [DOI] [PubMed] [Google Scholar]
- 11.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH: Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3:541–547. [DOI] [PubMed] [Google Scholar]
- 12.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW: Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270:985–988. [DOI] [PubMed] [Google Scholar]
- 13.Woo S-R, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, et al. : Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72:917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okazaki T, Okazaki I, Wang J, Sugiura D, Nakaki F, Yoshida T, Kato Y, Fagarasan S, Muramatsu M, Eto T, et al. : PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J. Exp. Med 2011, 208:395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Q, Vignali DAA: Co-stimulatory and Co-inhibitory Pathways in Autoimmunity. Immunity 2016, 44:1034–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bettini M, Szymczak-Workman AL, Forbes K, Castellaw AH, Selby M, Pan X, Drake CG, Korman AJ, Vignali DAA: Cutting edge: accelerated autoimmune diabetes in the absence of LAG-3. J. Immunol 2011, 187:3493–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T: Establishment of NOD-Pdcd1−/− mice as an efficient animal model of type I diabetes. Proc. Natl. Acad. Sci. USA 2005, 102:11823–11828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lühder F, Höglund P, Allison JP, Benoist C, Mathis D: Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) regulates the unfolding of autoimmune diabetes. J. Exp. Med 1998, 187:427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luhder F, Chambers C, Allison JP, Benoist C, Mathis D: Pinpointing when T cell costimulatory receptor CTLA-4 must be engaged to dampen diabetogenic T cells. Proc. Natl. Acad. Sci. USA 2000, 97:12204–12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khattri R, Auger JA, Griffin MD, Sharpe AH, Bluestone JA: Lymphoproliferative disorder in CTLA-4 knockout mice is characterized by CD28-regulated activation of Th2 responses. J. Immunol 1999, 162:5784–5791. [PubMed] [Google Scholar]
- 21.Oya Y, Watanabe N, Kobayashi Y, Owada T, Oki M, Ikeda K, Suto A, Kagami S, Hirose K, Kishimoto T, et al. : Lack of B and T lymphocyte attenuator exacerbates autoimmune disorders and induces Fas-independent liver injury in MRL-lpr/lpr mice. Int. Immunol 2011, 23:335–344. [DOI] [PubMed] [Google Scholar]
- 22.Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, Sharpe AH, Kuchroo VK: Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J. Immunol 2011, 186:1338–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, Manning S, Greenfield EA, Coyle AJ, Sobel RA, et al. : Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415:536–541. [DOI] [PubMed] [Google Scholar]
- 24.Carter LL, Leach MW, Azoitei ML, Cui J, Pelker JW, Jussif J, Benoit S, Ireland G, Luxenberg D, Askew GR, et al. : PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. J. Neuroimmunol 2007, 182:124–134. [DOI] [PubMed] [Google Scholar]
- 25.Jha V, Workman CJ, McGaha TL, Li L, Vas J, Vignali DAA, Monestier M: Lymphocyte Activation Gene-3 (LAG-3) negatively regulates environmentally-induced autoimmunity. PLoS One 2014, 9:e104484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vignali DAA, Collison LW, Workman CJ: How regulatory T cells work. Nat. Rev. Immunol 2008, 8:523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang C-T, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, Hipkiss EL, Ravi S, Kowalski J, Levitsky HI, et al. : Role of LAG-3 in regulatory T cells. Immunity 2004, 21:503–513. [DOI] [PubMed] [Google Scholar]
- 28.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S: CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322:271–275. [DOI] [PubMed] [Google Scholar]
- 29**.Zhang Q, Chikina M, Szymczak-Workman AL, Horne W, Kolls JK, Vignali KM, Normolle D, Bettini M, Workman CJ, Vignali DAA: LAG3 limits regulatory T cell proliferation and function in autoimmune diabetes. Sci. Immunol 2017, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]; LAG3+ Tregs are found to have impaired proliferation and function in autoimmune diabetes, highlighting the unexpected result that Lag3L/LFoxp3Cre-GFP.NOD mice are protected from autoimmune diabetes. Despite it’s known role as a mechanism of Treg suppression, LAG3 in autoimmune diabetes is found to limit Treg function, highlighting a need to further understand IR function in a cell type and disease specific manner.
- 30.Paterson AM, Lovitch SB, Sage PT, Juneja VR, Lee Y, Trombley JD, Arancibia-Cárcamo CV, Sobel RA, Rudensky AY, Kuchroo VK, et al. : Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J. Exp. Med 2015, 212:1603–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pauken KE, Dougan M, Rose NR, Lichtman AH, Sharpe AH: Adverse events following cancer immunotherapy: obstacles and opportunities. Trends Immunol. 2019, 40:511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32*.Carvalho-Silva D, Pierleoni A, Pignatelli M, Ong C, Fumis L, Karamanis N, Carmona M, Faulconbridge A, Hercules A, McAuley E, et al. : Open Targets Platform: new developments and updates two years on. Nucleic Acids Res. 2019, 47:D1056–D1065. [DOI] [PMC free article] [PubMed] [Google Scholar]; Open Targets Platform provides a database compiling evidence from GWAS studies in conjunction with published data from animal models, somatic mutations, therapeutics, and systems biology to provide association scores to mapped gene regions and diseases susceptibility. The information provided on this platform can inform drug target identification and the importance of certain SNPs in autoimmunity.
- 33.Romo-Tena J, Gómez-Martín D, Alcocer-Varela J: CTLA-4 and autoimmunity: new insights into the dual regulator of tolerance. Autoimmun. Rev 2013, 12:1171–1176. [DOI] [PubMed] [Google Scholar]
- 34.Zamani MR, Aslani S, Salmaninejad A, Javan MR, Rezaei N: PD-1/PD-L and autoimmunity: A growing relationship. Cell Immunol. 2016, 310:27–41. [DOI] [PubMed] [Google Scholar]
- 35.June CH, Warshauer JT, Bluestone JA: Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat. Med 2017, 23:540–547. [DOI] [PubMed] [Google Scholar]
- 36.McKinney EF, Smith KG: T cell exhaustion and immune-mediated disease-the potential for therapeutic exhaustion. Curr. Opin. Immunol 2016, 43:74–80. [DOI] [PubMed] [Google Scholar]
- 37**.McKinney EF, Lee JC, Jayne DRW, Lyons PA, Smith KGC: T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 2015, 523:612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study is the first to link T cell exhaustion to clinical outcomes of autoimmunity in humans. Here, a transcriptional signature reflecting CD8 T-cell exhaustion, primarily by IR expression, is positively correlated to flare-free survival in AAV, SLE, and IBD. Furthermore, surrogate markers of co-stimulation are negatively correlated with disease outcomes on independent datasets of T1D, AAV, SLE, idiopathic pulmonary fibrosis and dengue haemorrhagic fever.
- 38*.Wiedeman AE, Muir VS, Rosasco MG, DeBerg HA, Presnell S, Haas B, Dufort MJ, Speake C, Greenbaum CJ, Serti E, et al. : Autoreactive CD8+ T cell exhaustion distinguishes subjects with slow type 1 diabetes progression. J. Clin. Invest 2020, 130:480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]; The rate of progression of patients recently diagnosed with type 1 diabetes, measured by c-peptide level, was found to be slower in patients with a CD8+ T cell profile indicative of exhaustion. This was characterized by the expression of multiple IRs, limited cytokine production, and impaired proliferation by mass cytometry and analyzed using the new analytical method DISCOV-R to characterize rare populations.
- 39.Warshauer JT, Bluestone JA, Anderson MS: New frontiers in the treatment of type 1 diabetes. Cell Metab. 2020, 31:46–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40**.Long SA, Thorpe J, Herold KC, Ehlers M, Sanda S, Lim N, Linsley PS, Nepom GT, Harris KM: Remodeling T cell compartments during anti-CD3 immunotherapy of type 1 diabetes. Cell Immunol. 2017, 319:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]; Clinical outcome of anti-CD3 immunotherapy is attributed to the remodeling of T cell compartments to reflect a transiently increased PD1+ Tregs, increased potentially anergic CD4+ effectors and increased partially exhausted CD8+ T cell populations. The mechanism of action of anti-CD3 therapy is attributed to a modulation T cell states to reflect increased IR expression by flow cytometry. This supports the idea that IR modulation is an effective treatment for autoimmune disease.
- 41**.Long SA, Thorpe J, DeBerg HA, Gersuk V, Eddy J, Harris KM, Ehlers M, Herold KC, Nepom GT, Linsley PS: Partial exhaustion of CD8 T cells and clinical response to teplizumab in new-onset type 1 diabetes. Sci. Immunol 2016, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]; Anti-CD3 therapy induces a partial exhausted transcriptional signature in the CD8+ T cells of responding patients. This partial exhaustion is characterized by increased IR expression, supporting the hypothesis that IR enforcement limits autoimmune disease.
- 42*.Jamison BL, Neef T, Goodspeed A, Bradley B, Baker RL, Miller SD, Haskins K: Nanoparticles Containing an Insulin-ChgA Hybrid Peptide Protect from Transfer of Autoimmune Diabetes by Shifting the Balance between Effector T Cells and Regulatory T Cells. J. Immunol 2019, 203:48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]; Insulin-ChgA Hybrid Peptide nanoparticles induce an anergic state in Effector T cells in the NOD mouse model of autoimmune diabetes. This anergic state is characterized by markers of anergy Egr2, Egr3, Rnf128 (genes encoding GRAIL), Spry1, and Izumo1r (gene encoding FR-4), as well as IRs Pdcd1 and Lag3. This provides another example of how mechanistically, therapeutics confering protection from autoimmunity result in the upregulation of IRs.
- 43*.Tilstra JS, Avery L, Menk AV, Gordon RA, Smita S, Kane LP, Chikina M, Delgoffe GM, Shlomchik MJ: Kidney-infiltrating T cells in murine lupus nephritis are metabolically and functionally exhausted. J. Clin. Invest 2018, 128(11):4884–489. [DOI] [PMC free article] [PubMed] [Google Scholar]; CD4 and CD8+ T cell exhaustion is shown to be present in the kidneys of MRL-lpr mice experinecing lupus nephritis. These cells express multiple IRs, have impaired cytokine production and proliferation, and experience metabolic defects characteristic of T cell exhaustion. Importantly, this highlights the presence of IR expressing cells in the MRL-lpr mouse model and suggests their function in limiting disease in SLE.
- 44.Kalekar LA, Schmiel SE, Nandiwada SL, Lam WY, Barsness LO, Zhang N, Stritesky GL, Malhotra D, Pauken KE, Linehan JL, et al. : CD4(+) T cell anergy prevents autoimmunity and generates regulatory T cell precursors. Nat. Immunol 2016, 17:304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45*.Petrelli A, Mijnheer G, van Konijnenburg DPH, van der Wal MM, Giovannone B, Mocholi E, Vazirpanah N, Broen JC, Hijnen D, Oldenburg B, et al. : PD-1+CD8+ T cells are clonally expanding effectors in human chronic inflammation. J. Clin. Invest 2018, 128(10):4669–4681. [DOI] [PMC free article] [PubMed] [Google Scholar]; Importantly, IR expression is not sufficient to imply T cell exhaustion or functional inhibition. Here, a metabolically active PD1+CD8+ T cell subset is found to contribute to auto-inflammatory disease, juvenile idiopathic arthritis. This population has no indications of being exhausted, highlighting the care with which research must be done to ensure IR expression is limiting self-reactivity, as opposed to simply marking an activated effector cell type.
- 46.Paluch C, Santos AM, Anzilotti C, Cornall RJ, Davis SJ: Immune checkpoints as therapeutic targets in autoimmunity. Front. Immunol 2018, 9:2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N: Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med 2006, 355:1018–1028. [DOI] [PubMed] [Google Scholar]
- 48.Zhu X, Lang J: Soluble PD-1 and PD-L1: predictive and prognostic significance in cancer. Oncotarget 2017, 8:97671–97682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gonçalves Silva I, Yasinska IM, Sakhnevych SS, Fiedler W, Wellbrock J, Bardelli M, Varani L, Hussain R, Siligardi G, Ceccone G, et al. : The Tim-3-galectin-9 Secretory Pathway is Involved in the Immune Escape of Human Acute Myeloid Leukemia Cells. EBioMedicine 2017, 22:44–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simone R, Pesce G, Antola P, Rumbullaku M, Bagnasco M, Bizzaro N, Saverino D: The soluble form of CTLA-4 from serum of patients with autoimmune diseases regulates T-cell responses. Biomed Res. Int 2014, 2014:215763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li N, Wang Y, Forbes K, Vignali KM, Heale BS, Saftig P, Hartmann D, Black RA, Rossi JJ, Blobel CP, et al. : Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J. 2007, 26:494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li N, Workman CJ, Martin SM, Vignali DAA: Biochemical analysis of the regulatory T cell protein lymphocyte activation gene-3 (LAG-3; CD223). J. Immunol 2004, 173:6806–6812. [DOI] [PubMed] [Google Scholar]
- 53.Hirata S, Matsuyoshi H, Fukuma D, Kurisaki A, Uemura Y, Nishimura Y, Senju S: Involvement of regulatory T cells in the experimental autoimmune encephalomyelitis-preventive effect of dendritic cells expressing myelin oligodendrocyte glycoprotein plus TRAIL. J. Immunol 2007, 178:918–925. [DOI] [PubMed] [Google Scholar]
- 54.Ding H, Wu X, Wu J, Yagita H, He Y, Zhang J, Ren J, Gao W: Delivering PD-1 inhibitory signal concomitant with blocking ICOS co-stimulation suppresses lupus-like syndrome in autoimmune BXSB mice. Clin. Immunol 2006, 118:258–267. [DOI] [PubMed] [Google Scholar]
- 55.Song M-Y, Hong C-P, Park SJ, Kim J-H, Yang B-G, Park Y, Kim SW, Kim KS, Lee JY, Lee S-W, et al. : Protective effects of Fc-fused PD-L1 on two different animal models of colitis. Gut 2015, 64:260–271. [DOI] [PubMed] [Google Scholar]
- 56.Raptopoulou AP, Bertsias G, Makrygiannakis D, Verginis P, Kritikos I, Tzardi M, Klareskog L, Catrina AI, Sidiropoulos P, Boumpas DT: The programmed death 1/programmed death ligand 1 inhibitory pathway is up-regulated in rheumatoid synovium and regulates peripheral T cell responses in human and murine arthritis. Arthritis Rheum. 2010, 62:1870–1880. [DOI] [PubMed] [Google Scholar]
- 57.Wang G, Hu P, Yang J, Shen G, Wu X: The effects of PDL-Ig on collagen-induced arthritis. Rheumatol. Int 2011, 31:513–519. [DOI] [PubMed] [Google Scholar]
- 58.Gao W, Demirci G, Strom TB, Li XC: Stimulating PD-1-negative signals concurrent with blocking CD154 co-stimulation induces long-term islet allograft survival. Transplantation 2003, 76:994–999. [DOI] [PubMed] [Google Scholar]
- 59.Fife BT, Griffin MD, Abbas AK, Locksley RM, Bluestone JA: Inhibition of T cell activation and autoimmune diabetes using a B cell surface-linked CTLA-4 agonist. J. Clin. Invest 2006, 116:2252–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seki M, Oomizu S, Sakata K-M, Sakata A, Arikawa T, Watanabe K, Ito K, Takeshita K, Niki T, Saita N, et al. : Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin. Immunol 2008, 127:78–88. [DOI] [PubMed] [Google Scholar]
- 61.Hamana A, Takahashi Y, Tanioka A, Nishikawa M, Takakura Y: Safe and effective interferon-beta gene therapy for the treatment of multiple sclerosis by regulating biological activity through the design of interferon-beta-galectin-9 fusion proteins. Int. J. Pharm 2018, 536:310–317. [DOI] [PubMed] [Google Scholar]
- 62.Dixon KO, Schorer M, Nevin J, Etminan Y, Amoozgar Z, Kondo T, Kurtulus S, Kassam N, Sobel RA, Fukumura D, et al. : Functional Anti-TIGIT Antibodies Regulate Development of Autoimmunity and Antitumor Immunity. J. Immunol 2018, 200:3000–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, Chen L, Chen Y, Zhu G, Yin W, et al. : Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176:334–347.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hannier S, Tournier M, Bismuth G, Triebel F: CD3/TCR complex-associated lymphocyte activation gene-3 molecules inhibit CD3/TCR signaling. J. Immunol 1998, 161:4058–4065. [PubMed] [Google Scholar]
- 65.Angin M, Brignone C, Triebel F: A LAG-3-Specific Agonist Antibody for the Treatment of T Cell-Induced Autoimmune Diseases. J. Immunol 2020, 204:810–818. [DOI] [PubMed] [Google Scholar]
- 66.Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA: The TIGIT/CD226 axis regulates human T cell function. J. Immunol 2012, 188:3869–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yachi PP, Chai Q, Feng Y, Newburn KP, Truhlar SM, Petra V: PD-1 AGONIST ANTIBODIES AND USES THEREOF [Internet]. 2019, [no volume]. [Google Scholar]
- 68.Blair HA, Deeks ED: Abatacept: A review in rheumatoid arthritis. Drugs 2017, 77:1221–1233. [DOI] [PubMed] [Google Scholar]
- 69.Pimentel-Quiroz VR, Ugarte-Gil MF, Alarcón GS: Abatacept for the treatment of systemic lupus erythematosus. Expert Opin. Investig. Drugs 2016, 25:493–499. [DOI] [PubMed] [Google Scholar]
- 70.Meiners PM, Vissink A, Kroese FGM, Spijkervet FKL, Smitt-Kamminga NS, Abdulahad WH, Bulthuis-Kuiper J, Brouwer E, Arends S, Bootsma H: Abatacept treatment reduces disease activity in early primary Sjögren’s syndrome (open-label proof of concept ASAP study). Ann. Rheum. Dis 2014, 73:1393–1396. [DOI] [PubMed] [Google Scholar]
- 71.Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, Gottlieb PA, Greenbaum CJ, Marks JB, Monzavi R, et al. : Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet 2011, 378:412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khanna D, Spino C, Johnson S, Chung L, Whitfield ML, Denton CP, Berrocal V, Franks J, Mehta B, Molitor J, et al. : Abatacept in Early Diffuse Cutaneous Systemic Sclerosis: Results of a Phase II Investigator-Initiated, Multicenter, Double-Blind, Randomized, Placebo-Controlled Trial. Arthritis Rheumatol. 2020, 72:125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khoury SJ, Rochon J, Ding L, Byron M, Ryker K, Tosta P, Gao W, Freedman MS, Arnold DL, Sayre PH, et al. : ACCLAIM: A randomized trial of abatacept (CTLA4-Ig) for relapsing-remitting multiple sclerosis. Mult. Scler 2017, 23:686–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sandborn WJ, Colombel J-F, Sands BE, Rutgeerts P, Targan SR, Panaccione R, Bressler B, Geboes K, Schreiber S, Aranda R, et al. : Abatacept for Crohn’s disease and ulcerative colitis. Gastroenterology 2012, 143:62–69.e4. [DOI] [PubMed] [Google Scholar]