

Abstract
Background
Enteropathogenic Escherichia coli (EPEC) infection causes prolonged, watery diarrhea leading to morbidity and mortality. Although EPEC infection impacts nutrient transporter function and expression in intestinal epithelial cells, the effects of EPEC infection on intestinal absorption of ascorbic acid (AA) have not yet been investigated.
Aims
To investigate the effect of EPEC infection on intestinal AA uptake process and expression of both AA transporters.
Methods
We used two experimental models: Human-derived intestinal epithelial Caco-2 cells and mice. 14C-AA uptake assay, Western blot, RT-qPCR and promoter assay were performed.
Results
EPEC (WT) as well as ΔespF and ΔespG/G2 mutant infected Caco-2 cells showed markedly inhibited AA uptake, while other mutants (ΔescN, ΔespA, ΔespB and ΔespD) did not affect AA uptake. Infection also reduced protein and mRNA expression levels for both hSVCT1 and hSVCT2. EPEC infected mice showed marked inhibitory effect on AA uptake and decreased protein and mRNA expression levels for both mSVCT1 and mSVCT2 in jejunum and colon. MicroRNA regulators of SVCT1 and SVCT2 (miR103a, miR141 and miR200a) were up-regulated significantly upon EPEC infection in both Caco-2 and mouse jejunum and colon. In addition, expression of the accessory protein glyoxalate reductase/hydroxypyruvate reductase (GRHPR), which regulates SVCT1 function, was markedly decreased by EPEC infection in both models.
Conclusions
These findings suggest that EPEC infection causes inhibition in AA uptake through a multifactorial dysregulation of SVCT1 and SVCT2 expression in intestinal epithelial cells.
Keywords: Vitamin C, SVCT1, SVCT2, microRNA, EPEC, GRHPR
Introduction
Vitamin C (Ascorbic acid; AA) is an essential micronutrient which is involved in many biological functions. It is used as a co-factor for many essential enzymatic reactions [1] and is a known antioxidant which counteracts the harmful effect of reactive oxygen species (ROS) [1]. Deficiency of vitamin C is also linked to various elements of immune dysfunction such as impeded response of natural killer (NK) cells to bacterial infection and diminished functionality of cytotoxic T-lymphocytes [2]. Reciprocally, optimizing AA body homeostasis is important to protect against conditions like hepatic and cardiovascular ailments, osteoporosis, cancer, age-related cognitive decline and other neurodegenerative disorders [3–7].
Humans are not capable of producing vitamin C endogenously [8], and therefore they obtain this vitamin through intestinal absorption. Mice, however, can produce vitamin C endogenously, but still rely on vitamin C that exists in the diet to meet their metabolic needs of this micronutrient as demonstrated by studies using knockout mouse models [9, 10]. Uptake of AA by polarized enterocytes occurs via both sodium-dependent vitamin C transporters, SVCT1 (SLC23A1) and SVCT2 (SLC23A2) [11–14]. SVCT1 is predominantly localized at the apical membrane and SVCT2 targeted to the basolateral membrane in enterocytes [15, 16] and are differentially expressed along the gut [17]. Both human and mouse AA transporters exhibit significant sequence homology [14].
Enteropathogenic Escherichia coli (EPEC) infection causes watery diarrhea in humans which can be associated with significant morbidity and mortality in children of developing nations [18, 19]. Malnutrition occurs as a result of EPEC infection, as many infected individuals are already nutritionally compromised; this condition is amplified by severe and/or prolonged infection [20, 21]. EPEC does not produce toxins, rather its pathogenicity involves interaction with intestinal cells to cause attaching and effacing (A/E) lesions that results in formation of actin pedestal, rearrangement of cytoskeleton and damaging microvilli [22]. EPEC injects bacterial products and effectors into host cells using a type III secretion system (T3SS) [22]. These effector molecules (i.e., ΔespF and ΔespG) interact with intracellular targets to affect cell biological (e.g., microtubule stability [23], RNA levels [24]) and physiological aspects of gut epithelia (electrolyte and nutrient transport [25, 26] and cell permeability [27]).
Over the years, considerable strides have been made in understanding the cellular and molecular consequences of EPEC infection on nutrient/electrolyte transport in intestinal epithelia [25, 26, 28–31]. While data on the effects of EPEC on AA uptake is limited, a prolonged and severe EPEC infection could have the potential to drastically affect the AA levels in an infected host, given that the human body has limited storage capacity for AA. Here, we utilized both in vitro (Caco-2 cells) and in vivo (mice) model systems to study the consequences of EPEC infection on intestinal AA uptake.
Experimental Procedures
Reagents
14C-AA (Cat# NEC1460; specific activity of 2.8 mCi/mmol; radiochemical purity > 97%) was from PerkinElmer (Boston, MA). Standard biochemical and molecular biology reagents were from Sigma (St. Louis, MO) or Fisher Scientific (Tustin, CA). Western blot secondary antibodies and blocking buffer were from LI-COR Biosciences (Lincoln, NE). ß-actin antibody (monoclonal) (Cat# sc-47778) was from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell culture
EMEM (Cat# 30–2003; ATCC) with 10% FBS (Cat# 100–106; Gemini Bio Products, West Sacramento, CA), antibiotics (penicillin, streptomycin), were used to culture Caco-2 cells (Cat# HTB-37; ATCC) at 37°C in a 5% CO2 −95% O2 environment. For infection, confluent cells were cultured for 16 h in FBS as well as penicillin and streptomycin free EMEM.
Bacterial culture, in vitro and in vivo infection
Bacterial colonies of EPEC [wild-type (WT) - E2348/69 strain], the non-pathogenic isolate HS4, and EPEC mutants were plated on LB agar, then inoculated overnight in 10 mL of LB broth using a shaking incubator set at 220 rpm and 37°C. Next, bacteria were diluted in 5 mL of serum- and antibiotic-free 0.5% mannose (Cat# 112585; Sigma) containing EMEM and placed in a shaking incubator for 3 h to reach midlog phase (OD ~0.4 at 600 nm). For in vitro studies, Caco-2 cells were infected at a MOI of 100 for 60 min. Post 60 min incubation, bacteria-containing media was replaced with gentamicin (50 μg/mL) (Cat# G1397; Sigma) containing medium for 30 min to completely eliminate all residual bacteria. Further, cells were cultured for 6 h in EMEM at 37°C in a 5% CO2 −95% O2 environment. Six hours later, cells were used for AA uptake and other analyses.
For short-term (‘acute’, 11 days) in vivo infection, WT mice (C57BL/6J male, 6–8 weeks old; Jackson Laboratory) were infected with EPEC (WT, 100 MOI) or vehicle (LB broth) via oral gavage and monitored for 11 days as specified [32, 33]. Every other day, mice weights were measured and their activity evaluated. After 11 days, the mice were sacrificed to harvest jejunum and colon tissues for AA uptake and molecular analyses. For long-lasting effects of EPEC infection in in vivo, mice were bred in-house (UC Davis) and challenged neonatally on post-natal day 7 (P7) with EPEC (50 μl; 105 colony forming units [CFU]) or vehicle via oral gavage [34]. Tissue samples were collected at 6–8 weeks of age. Mice studies were performed in accordance with the Veterans Affairs Medical Center, Long Beach (MIRB# 1378), University of California Irvine, Irvine (AUP-17–227) and University of California Davis, Davis (20072) Institutional Animal Care and Use Committees (IACUC).
14C-AA uptake
Infected [EPEC (WT), HS4, and EPEC mutants] and uninfected (control) Caco-2 cells were incubated with labeled (0.1 μCi) or labeled plus unlabeled (1mM AA) AA containing Krebs-Ringer (KR) buffer (in mM: 1.23 MgSO4:7H2O, 133 NaCl, 10 HEPES, 4.93 KCl, 5 glucose, 0.85 CaCl2 dihydrate, 5 glutamine, 10 MES hydrate; pH 7.4) for 30 min in a 37°C water bath. To determine AA uptake in jejunum and colon tissues, mice were euthanized to extract the jejunum and colonic sheets, which were then opened longitudinally and immediately incubated in KR buffer with either labeled (0.1 μCi) or labeled plus unlabeled (1mM AA) AA at 37°C for 7 min as described [35]. Cells or tissues were lysed with 1N NaOH and heat treated at 80°C for 15 min, followed by neutralization with 10N hydrochloric acid. Subsequently, radioactive content was determined (liquid scintillation counter; Beckman Coulter, Brea, CA). Protein concentrations of cells or intestinal tissues were determined with Protein Assay kit reagents (Bio-Rad, Hercules, CA).
RT-qPCR analysis
Trizol reagent (Cat# 15596018; Life Technologies, Carlsbad, CA) was used to isolate total RNA from cells and mouse intestinal tissues in order to perform cDNA synthesis utilizing DNaseI (Cat# 18068015; Invitrogen, Carlsbad, CA), and iScript kit reagents (Cat# 1708891; Bio-Rad). This cDNA was used to determine mRNA expression levels utilizing appropriate primer combinations. RT-qPCR was performed using iQ SYBR Green reagent (Cat# 1708884; Bio-Rad), human (h) and mouse (m) SVCT1, SVCT2, GRHPR and ß-actin primers (hSVCT1: F, 5′-TCATCCTCTTCTCCCAGTACCT-3′, R, 5′-AGAGCAGCCACACGGTCAT-3′; hSVCT2: F, 5′-TCTTTGTGCTTGGATTTTCGAT-3′, R, 5′-ACGTTCAACACTTGATCGATTC-3′; hGRHPR: F, 5′- GACTCGGATGAGCCCATCC-3′, R, 5′-TCAGGACATCTGGGGTGTAG-3′; hß-actin: F, 5′-CATCCTGCGTCTGGACCT-3′, R, 5′-TAATGTCACGCACGATTTCC-3′; mSVCT1: F, 5′-CAGCAGGGACTTCCACCA-3′, R, 5′-CCACACAGGTGAAGATGGTA-3′; mSVCT2: F, 5′-AACGGCAGAGCTGTTGGA-3′, R, 5′-GAAAATCGTCAGCATGGCAA-3′; mGRHPR: F, 5′-AATTCGGATGACCCCATCC-3′, R, 5′-TCAGGACACCTGGCGTGTAG-3′; mß-actin: F, 5′-ATCCTCTTCCTCCCTGGA-3′, R, 5′-TTCATGGATGCCACAGGA-3′) and a Bio-Rad CFX96 Touch Real-time PCR detection system. Relative SVCT1, SVCT2 and GRHPR mRNA levels were calculated by normalizing threshold cycle values to relative ß-actin as described [36].
Western blot analysis
The soluble protein fractions from Caco-2 cells and mouse small and large intestinal samples were isolated using radioimmunoprecipitation (RIPA) buffer (Cat# R0278; Sigma) comprising protease inhibitor (Cat# PI78410; Roche, NJ) after homogenization, sonication and centrifugation at 14000 rpm for 20 min. Bio-Rad Dc Protein Assay reagents were used to quantify protein concentrations, of which 60 μg was used for Western blot to load an Invitrogen NuPAGE 4–12% Bis-Tris Mini Gel (Cat# NP0321; Invitrogen). After separation, the protein was electroblotted from the gel onto an Immobilon polyvinylidene diflouride (PVDF) membrane (Millipore, Burlington, MA) [37]. LI-COR Odyssey blocking buffer (Cat# 924–40010; LI-COR Biosciences) was used to block the membrane for 10 min, then probed (4°C for overnight on a rotating shaker) with custom generated polyclonal antibodies (Thermo Fisher Scientific, Rockford, IL) against hSVCT1 (amino acids:576–590 SLDQIAIPEDTPENTETAS), or mSVCT1 (amino acids:576–594 RGFSKKTQNQPPVLEDTPD), or human/mouse SVCT2 (amino acids:627–645 GYTWKGLRKSDNSRSSDED) (1:500 dilution for all), and ß-actin (1:2000) antibodies (Santa Cruz Biotechnology). Afterwards the blot was washed 3 times with PBS-0.1% Tween 20, then probed with anti-rabbit IRDye 800 (Cat# NC9401842) and/or anti-mouse IRDye 680 (Cat# NC0046410)(1:30000 dilution) as secondary antibodies and blots were then incubated at room temperature for 45 min. A LI-COR Odyssey infrared imaging system was then used to capture the immunoreactive fluorescent bands for specific band intensity analysis with LI-COR software. The specificity of antibodies was determined using antigenic peptides synthesized against human and mouse SVCT1 and SVCT2 polypeptides.
Transfection and firefly luciferase activity
Ninety percent confluent Caco-2 cells were transiently co-expressed with both Renilla luciferase-thymidine kinase (pRL-TK) (100ng/well) and 3μg (per well) of pGL3-SLC23A1 or pGL3-SLC23A2 constructs [38], or pGL3 basic vector alone utilizing Lipofectamine 2000 (Cat# 11668019; Invitrogen). Twenty four hours later, Caco-2 cells were infected with bacteria as described earlier. The activity of promoter was determined by GloMax 20/20 Luminometer (Promega). The activity of firefly luciferase was normalized against Renilla control reporter (Cat# PRE1960; Promega) activity in the same samples.
Quantification of mature microRNA expression in Caco-2 cells and mouse intestinal samples
To determine the mature microRNA (miRNA) expression levels, miRNA was isolated from different passages of Caco-2 cells, multiple mouse jejunum, and colon tissue samples using miRNeasy Mini Kit (Cat# 217004; Qiagen, Germantown, MD). The miRNA (10 ng) was reverse transcribed using the reverse-transcription primers from TaqMan MicroRNA Reverse Transcription Kit (Cat# A25576; Applied Biosystems, Foster City, CA) to synthesize cDNA. Levels of mature miRNA [miR103a (# 000439), miR141 (# 000463), miR200a (# 000502), RNU6B (# 001093), and U6 (# 001973)] in Caco-2 cells, mouse intestinal segments were determined using TaqMan probes and PCR Master Mix (Cat# 4440039). The RNU6B (human) and U6 (mouse) miRNA were used as representative house-keeping genes (supplied by Applied Biosystems).
Statistical methods
Uptake values are represented as mean ± SE of different experimental determinations using multiple passages of Caco-2 cells, or samples from at least 3 mice, and are presented as a percentage. Carrier-mediated processing was determined as described before [39]. The significance level was determined utilizing Student’s t-test and P < 0.05 or less was set as significant. Western blot analysis, RT-qPCR and promoter activity assay data presented are the results from minimum of triplicate experiments.
Results
EPEC infection inhibits AA uptake
We assessed the effect of EPEC (WT) or non-pathogenic E. coli HS4 (100 MOI) [40] infection on AA uptake (carrier-mediated) in Caco-2 cells. The uptake of AA was determined in confluent Caco-2 cells after 60 min of infection with either EPEC or HS4, followed by 6 h of post infection. EPEC infection caused a marked (P < 0.01) inhibition of AA uptake when compared to control (uninfected) cells (Fig. 1A). Conversely, uptake of AA was not significantly changed in Caco-2 cells incubated with the HS4 (Fig. 1A).
Figure 1. Effect of EPEC (WT) and mutant infection on 14C-AA uptake.
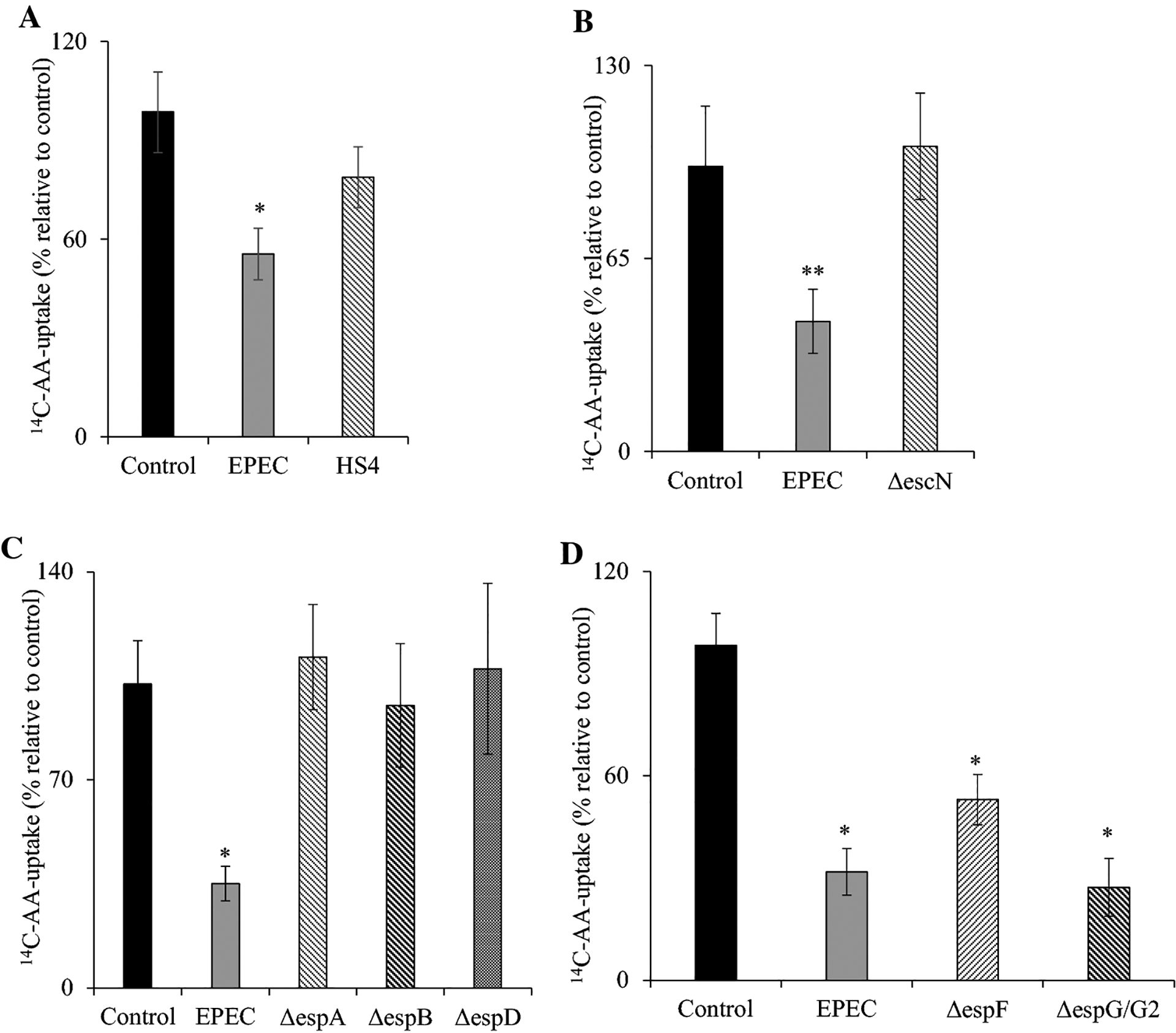
Infecting Caco-2 cells monolayers with EPEC (WT) and non-pathogenic E. coli HS4 (A) and mutants [type III secretory system protein (B), structural proteins (C), and effector proteins (D)] at 100 MOI for 60 min with additional 6 h post incubation after EPEC removal. 14C-AA uptake was subsequently performed as defined (see “Methods”). Values are mean ± SE of at least three individual uptake determinations with different passages of cells. ** P < 0.05; * P < 0.01.
Effect of EPEC infection on AA uptake is functional T3SS-dependent in Caco-2 cells
EPEC has a T3SS and several structural proteins that make up the injectisome to deliver virulence proteins into host cells [41]. This investigation surveyed essential structural components of the T3SS and effectors in contributing to the inhibition of AA uptake seen with WT EPEC. The T3SS has the escN protein, which functions as an ATPase accountable for driving energy for the active transport of effector molecules from bacteria into intestinal cells. Infection of Caco-2 cells with the escN mutant strain (ΔescN) did not show inhibition in AA uptake (Fig. 1B). This indicates that this response is likely dependent on the T3SS system (Fig. 1B). Next, we investigated the role of various structural elements of T3SS: ΔespA (which forms part of the needle tip unit of T3SS), ΔespB, and ΔespD (which form the translocation channel). Infections with each of these mutants failed to inhibit AA uptake in Caco-2 cells (Fig. 1C). Finally, we investigated the role of effector molecules: ΔespF (involved in barrier disruption) and ΔespG/G2 (implicated in microtubule network disruption). Data revealed that Caco-2 cells infected with either of these mutant strains caused a marked (P < 0.01 for both) inhibitory effect on AA uptake (Fig. 1D).
EPEC infection decreases the AA transporters expression levels in Caco-2 cells
Prior research has shown that both hSVCT1 and hSVCT2 mediate intestinal AA uptake [16, 35]. Therefore, we investigated the effect of EPEC infection on vitamin C transporters protein and mRNA levels. Results showed lower (P < 0.05 for hSVCT1; P < 0.01 for hSVCT2) levels of vitamin C transporters protein expression in EPEC infected cells compared to uninfected controls (Fig. 2A, B). Infection with the HS4 failed to induce any significant change in human vitamin C transporters protein expression levels. The specificity of detection of hSVCT1 and hSVCT2 protein was assessed by competition of hSVCT1 and hSVCT2 immunostaining with synthetic antigenic peptides [Fig. 2C (i&ii)]. Additionally, a marked (P < 0.01 for both) reduction in hSVCT1 and hSVCT2 mRNA expression was observed in EPEC infected cells, with no reduction determined after infection with the commensal HS4 strain (Fig. 2D, E).
Figure 2. Effect of EPEC on hSVCT1 and hSVCT2 protein and mRNA levels.
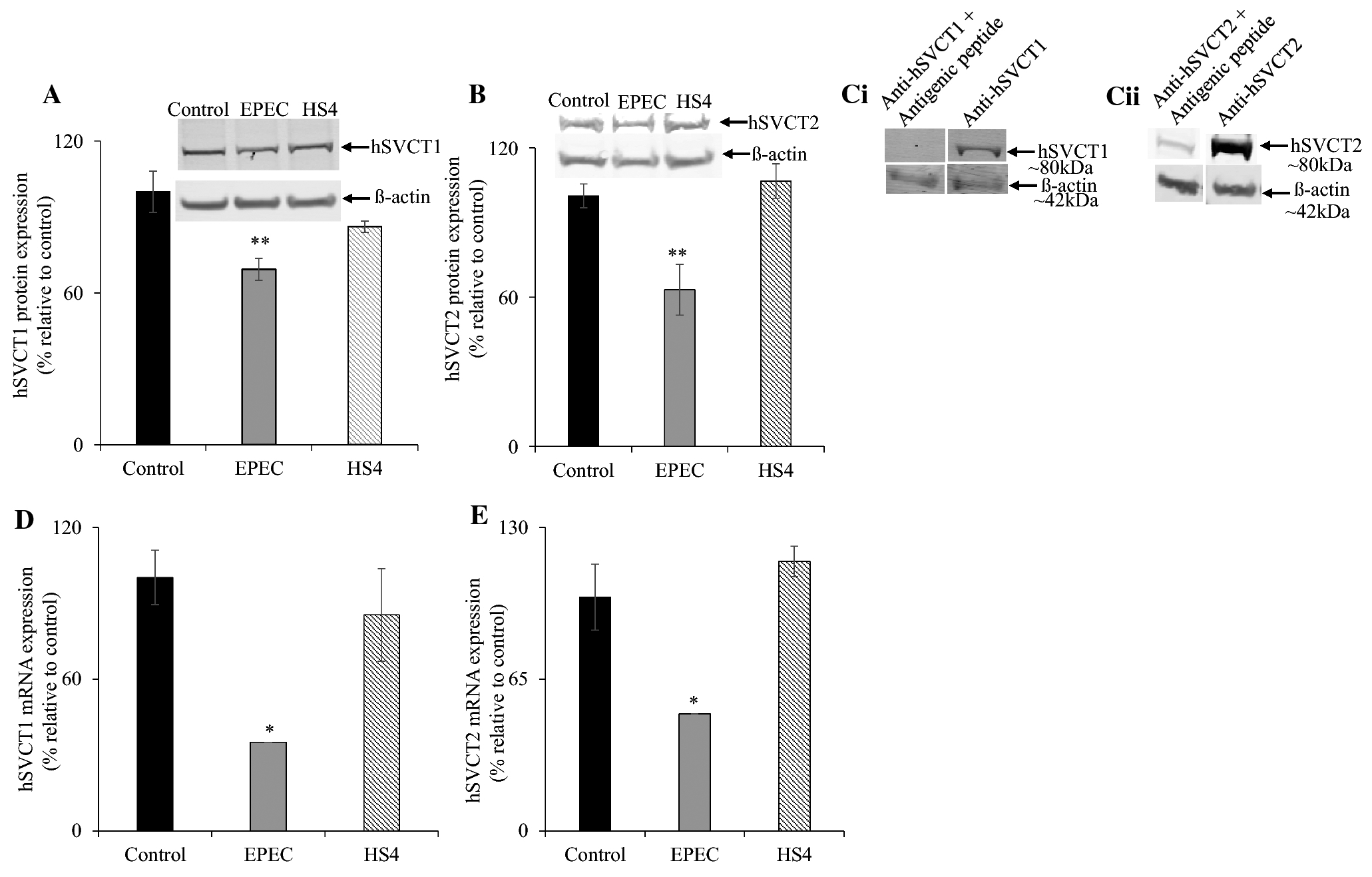
Infecting Caco-2 cells monolayers with EPEC (WT) and HS4 (100 MOI) for 60 min with additional 6 h post incubation after EPEC removal and subjected to Western blot and RT-qPCR analyses as described in “Methods”. The hSVCT1 (A) and hSVCT2 (B) levels of expression were measured by Western blotting using protein isolated from EPEC (WT), HS4, and uninfected control Caco-2 cells. The specificity of hSVCT1 and hSVCT2 specific bands were determined by competing hSVCT1 and hSVCT2 polyclonal antibodies with synthetic antigenic peptides generated against hSVCT1 and hSVCT2 polypeptides (Ci, ii). The mRNA levels of hSVCT1 (D) and hSVCT2 (E) were determined in infected and uninfected control Caco-2 cells by RT-qPCR. Values are mean ± SE of at least triplicate determinations from different passages of cells. ** P < 0.05; * P < 0.01.
EPEC infection inhibits AA uptake and decreases its transporters expression in mouse intestine
Next, we assessed the effect of EPEC on small (jejunum; where the majority of vitamin C absorption occurs) and large (colon; where EPEC colonize during post-infection) intestinal segments in WT mice. AA uptake was performed within these intestinal segments after acute infection. Results revealed a marked (P < 0.01) inhibitory effect in jejunal uptake in EPEC infected animals relative to uninfected (age- and sex-matched) controls (Fig. 3A). This inhibition was correlated with decreased expression of both mSVCT1 and mSVCT2 (P < 0.05 for both) proteins in jejunum mucosa (Fig. 3B, C). The specificity of detection of mSVCT1 and mSVCT2 protein was again demonstrable by co-incubating mSVCT1 and mSVCT2 polyclonal antibodies with synthetic antigenic peptides [Fig. 3D (i&ii)]. In parallel, mRNA (Fig. 3E, F) expression of both mSVCT1 and mSVCT2 in jejunum mucosa were also (P < 0.01 for mSVCT1; P < 0.05 for mSVCT2) reduced in EPEC infected animals when compared to their appropriate controls. Similar results in jejunum were observed when mice were exposed neonatally with EPEC, but assessed in adulthood (6–8 weeks later; Fig. 3G–J).
Figure 3. Effect of acute and neonatal exposure of mice to EPEC infection on 14C-AA uptake, mSVCT1 and mSVCT2 expression levels in mouse jejunum.
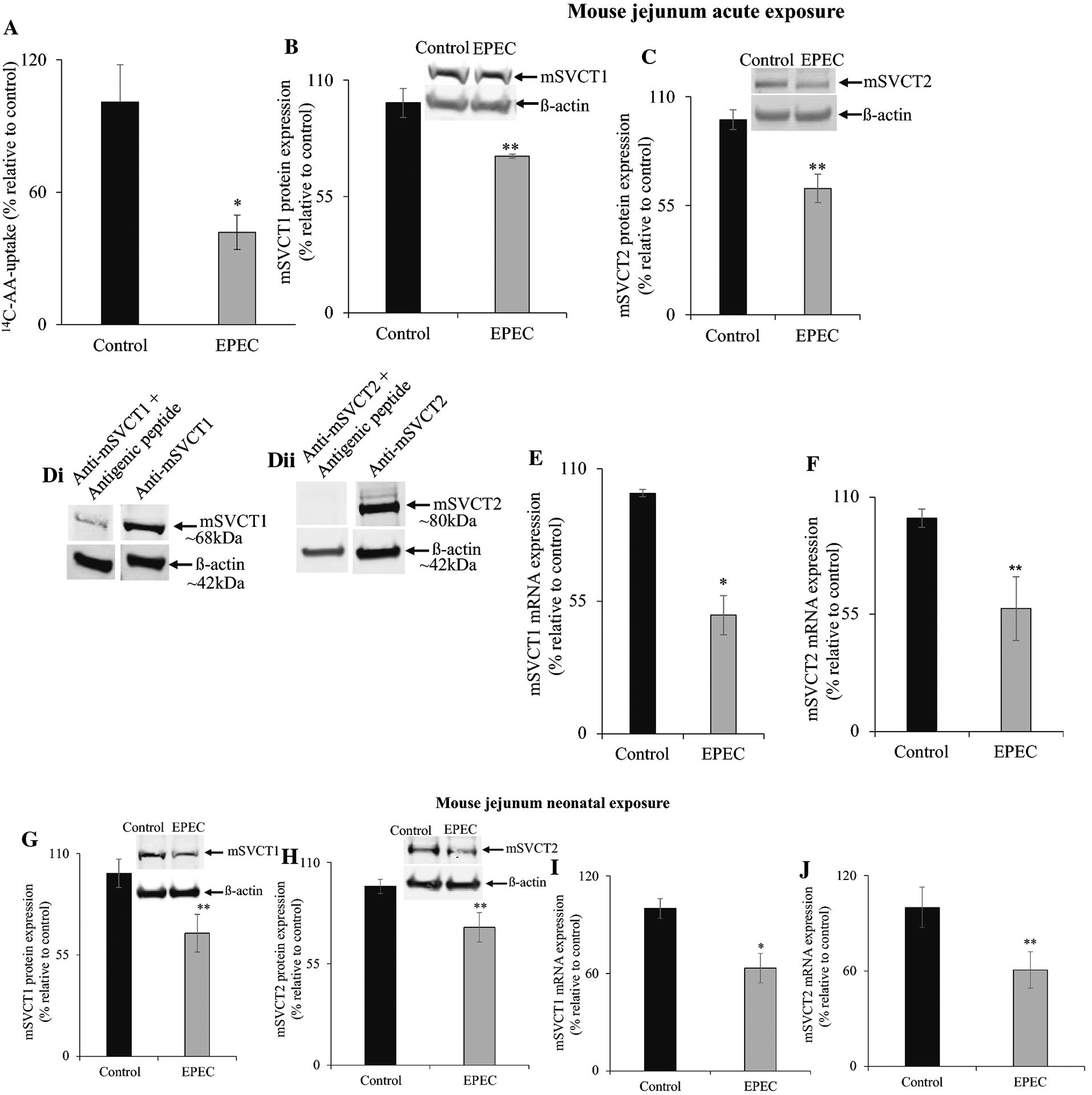
14C-AA uptake was measured in mouse jejunum after acute exposure of mice with EPEC (WT) infection (see “Methods”) (A). Western blot was done using 60 μg of protein obtained from both acute and neonatal exposed jejunum mucosa in order to measure the mSVCT1 (B, G) and mSVCT2 (C, H) protein levels. The specificity of mSVCT1 and mSVCT2 specific bands were determined by competing with mSVCT1 and mSVCT2 polyclonal antibodies against synthetic antigenic peptides (Di, ii). The mSVCT1 (E, I) and mSVCT2 (F, J) mRNA expression levels in mouse jejunum were determined by RT-qPCR. Values are mean ± SE of at least triplicate determinations from different mice. ** P < 0.05; * P < 0.01.
In colon, we also determined the consequence of EPEC infection on AA uptake. Results revealed a marked (P < 0.05) inhibitory effect in colonic uptake of AA in acute EPEC infected mice in comparison with uninfected mice (Fig. 4A). Again, inhibitory effect coincided with a marked decrease in both mSVCT1 and mSVCT2 protein (P < 0.05 for both) and mRNA (P < 0.05 for both) expression (Fig. 4B–E). Similar results in colon were obtained when mice were subjected to longer EPEC infection (Fig. 4F–I).
Figure 4. Effect of acute and neonatal exposure of mice to EPEC infection on 14C-AA uptake, mSVCT1 and mSVCT2 expression levels in mouse colon.
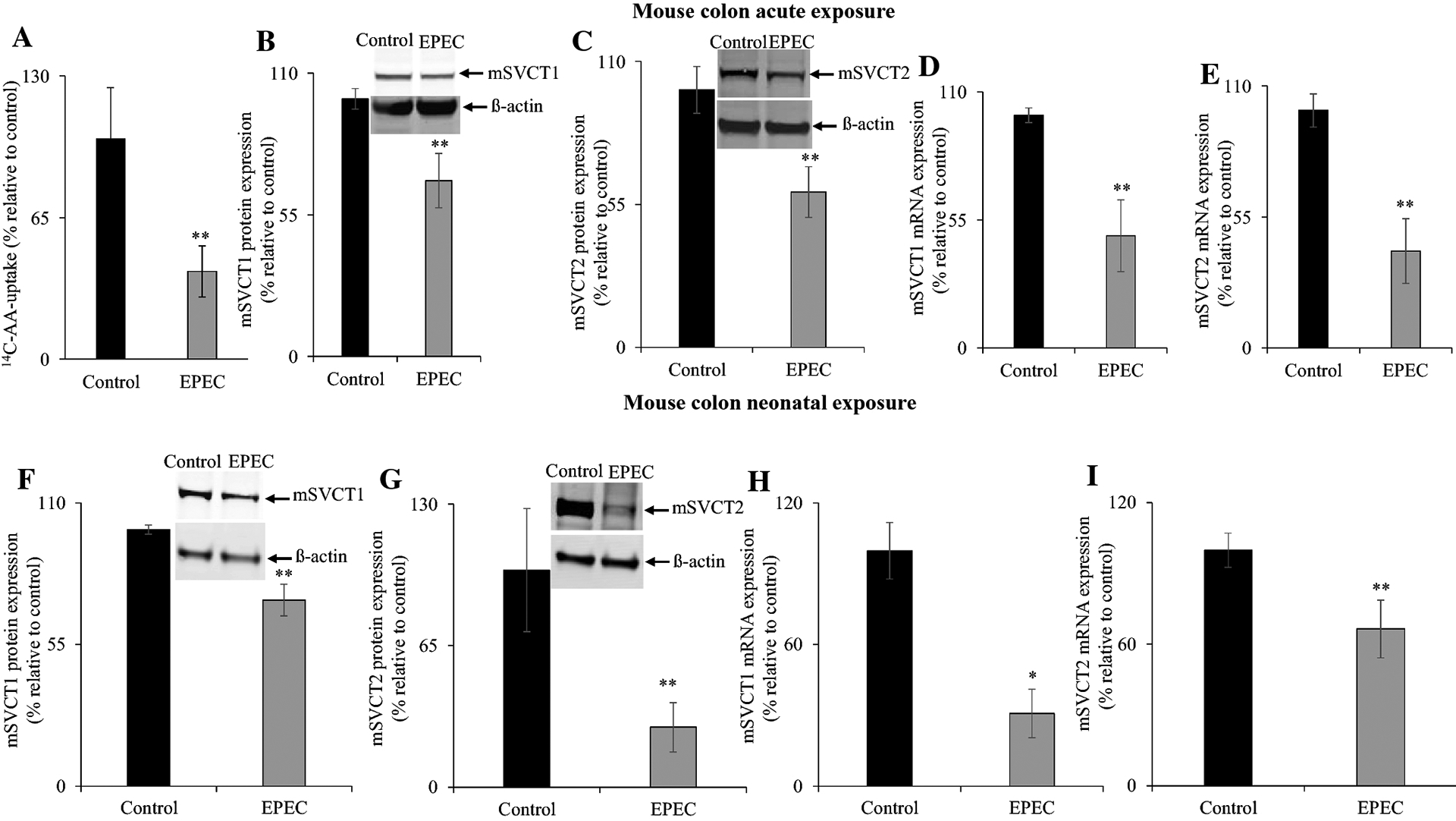
14C-AA uptake was determined in mouse colon after acute exposure of EPEC (WT) infection (see “Methods”) (A). Western blot was conducted using 60 μg of protein obtained from both acute and neonatal exposed colon mucosa in order to assess the mSVCT1 (B, F) and mSVCT2 (C, G) protein levels. The mSVCT1 (D, H) and mSVCT2 (E, I) mRNA expression levels in mouse colon were measured using RT-qPCR. Data are mean ± SE of at least triplicate determinations from different mice. ** P < 0.05.
EPEC infection does not affect hSVCT1 (SLC23A1) and hSVCT2 (SLC23A2) promoter activity and up-regulates microRNA expression
Changes observed in mRNA levels upon EPEC infection could occur via changes in either transcriptional and/or post-transcriptional mechanisms. To determine if a transcriptional mechanism(s) were involved in decreasing vitamin C transporters mRNA levels, we assessed the effect of EPEC on both pGL3-SLC23A1 and pGL3-SLC23A2 promoter activity after transfection into Caco-2 cells. EPEC infection had no effect on luciferase activity of either of the promoter constructs compared to controls (uninfected) cells (Fig. 5A, B). These data suggest that the observed inhibition upon EPEC infection in intestinal AA uptake may be mediated via post-transcriptional mechanism(s).
Figure 5. Effect of EPEC on SLC23A1 and SLC23A2 promoter activity.
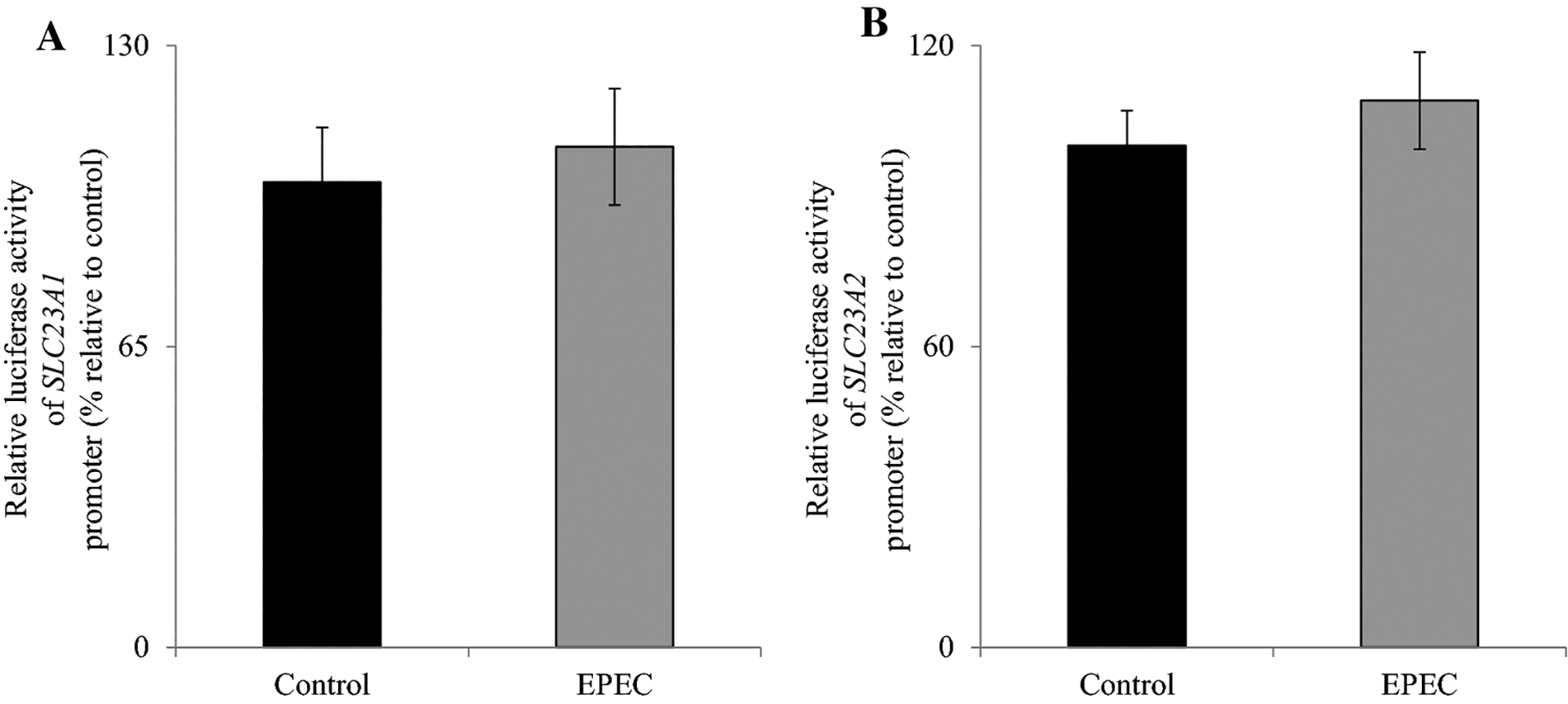
Caco-2 cells were transiently transfected with pGL3-SLC23A1 (A) and pGL3-SLC23A2 (B) promoter constructs followed by infection with EPEC as described in “Methods”. Values are mean ± SE of at least triplicate determinations with different passages of cells.
Next, we investigated the role of post-transcriptional mechanisms. Recent studies have determined that epigenetic mechanisms are involved in mediating effects caused by bacterial infection in the intestine [42, 43]. Here, we investigated the role of microRNAs miR103a, miR141, and miR200a that have been identified as regulators of SVCT1 and SVCT2 [44, 45]. Results indicated the expression levels of mature miR103a, miR141, and miR200a were all significantly increased (P < 0.05 for all) after EPEC infection of Caco-2 cells (Fig. 6A–C). Similarly, acute infection of adult mice with EPEC also resulted in a significant increase in all three mature miRNAs expression levels in both mouse jejunum (P < 0.01 for both miR103a and miR141; P < 0.05 for miR200a) (Fig. 6D–F) and in mouse colon (P < 0.01 for all) (Fig. 6G–I). These data implicate a role for post-transcriptional regulation in the observed inhibition caused by EPEC infection on vitamin C transport in intestinal epithelia.
Figure 6. Effect of EPEC on mature microRNA levels in Caco-2 cells and mouse intestinal mucosa.
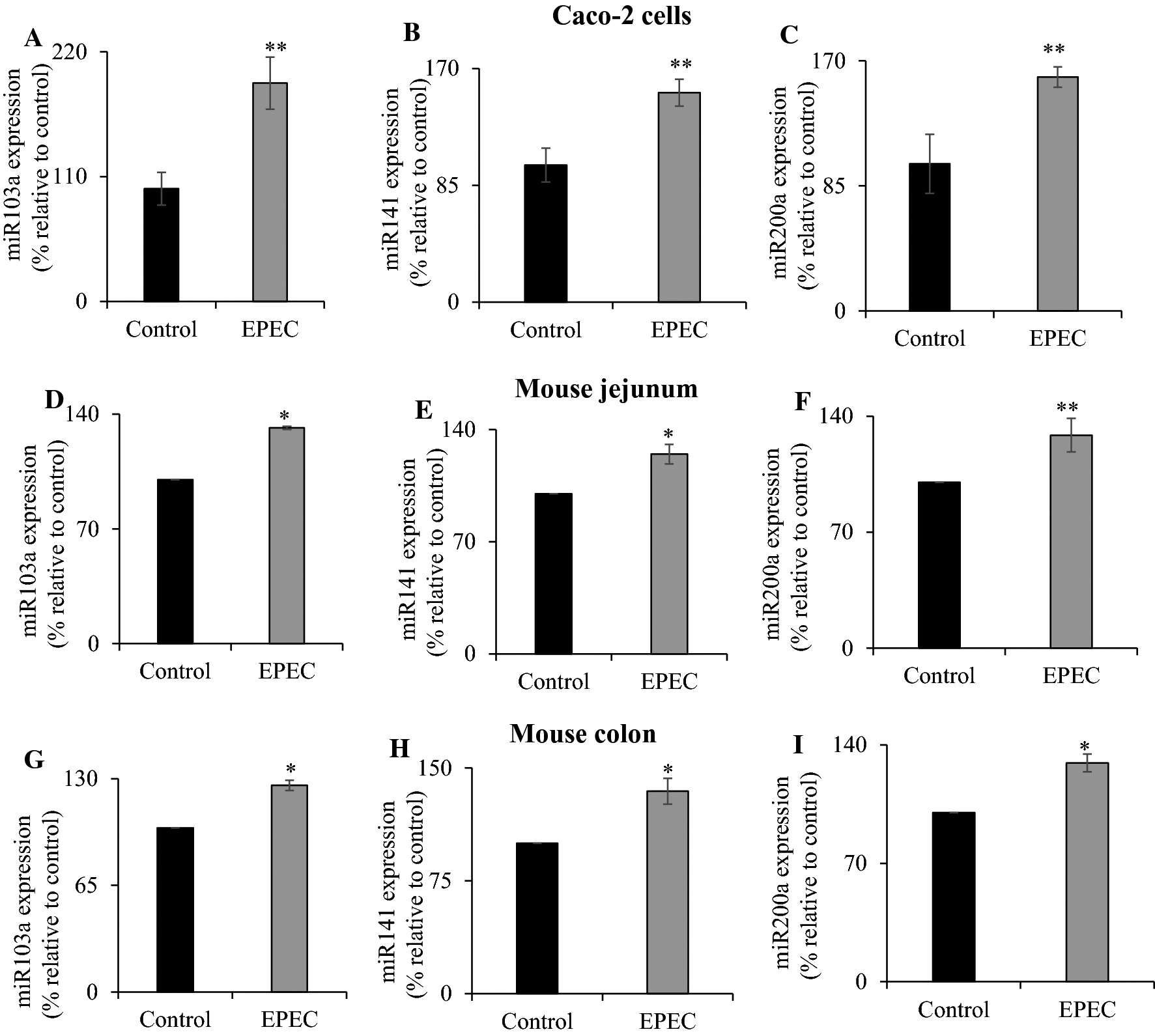
Levels of mature miR103a (A, D, G), miR141 (B, E, H), and miR200a (C, F, I) expression were determined in EPEC infected Caco-2 cells, mouse jejunum and colon as detailed in “Methods”. Data are mean ± SE of at least triplicate determinations using different passages of Caco-2 cells or tissue from multiple mice. ** P < 0.05; * P < 0.01.
EPEC infection decrease the GRHPR expression in in vitro and in vivo models
Our laboratory has identified human glyoxalate reductase/hydroxypyruvate reductase (hGRHPR) to be an important protein interactor of hSVCT1 and this interaction induces AA uptake [46]. Here, we assessed the role of GRHPR (both human and mouse share > 85% of identity at the amino acid level) in modulating the observed effects of EPEC on AA transport. hGRHPR mRNA levels were markedly (P < 0.05) lower upon EPEC infection in Caco-2 cells (Fig. 7A). Similarly, acute infection of mice with EPEC caused a marked reduction in mGRHPR mRNA levels in mouse jejunum and colon (P < 0.01 for both) (Fig. 7B, C). These results demonstrates that GRHPR may also be involved in the observed inhibition of vitamin C uptake in both systems.
Figure 7. Effect of EPEC infection on GRHPR mRNA expression in in vitro and in vivo models.
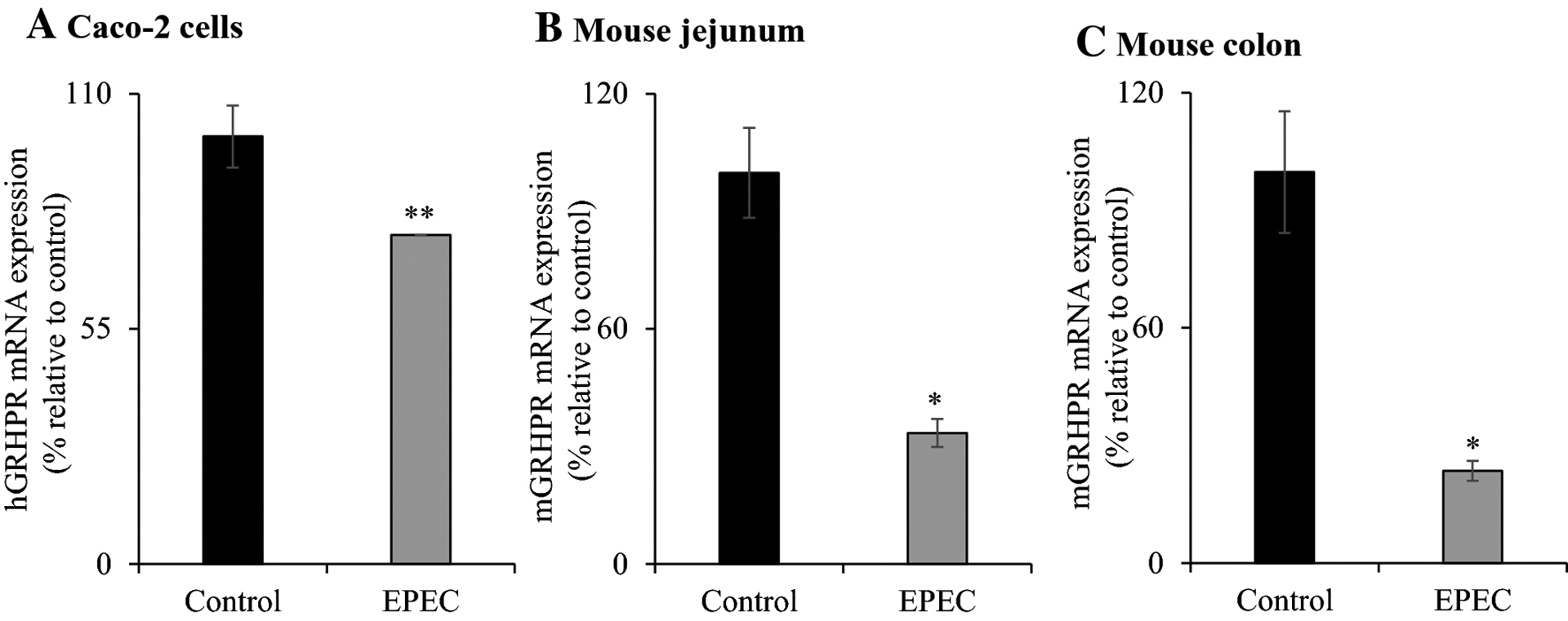
The level of GRHPR mRNA expression in EPEC infected Caco-2 cells (A), mouse jejunum (B) and colon (C) were measured by PCR amplification. Data are mean ± SE of at least 3 individual observations from multiple samples. ** P < 0.05; * P < 0.01.
Discussion
EPEC is a major diarrheagenic pathogen which significantly impacts developing nations and its infections are associated with thousands of deaths every year [47]. EPEC does not produce toxins, rather its pathogenicity is initiated by attaching to host intestinal epithelial cells and injecting virulence factors using T3SS. As a result, host cells are subject to significant alterations in cytoskeletal networks, ion transport, inflammatory signaling and tight junction function [23, 27, 30, 48]. Although a significant effort has been made to address the underlying cellular and molecular mechanisms through which EPEC infection effects on nutrient/electrolyte transport [25, 26], no studies to date have evaluated the consequence of EPEC infection on intestinal absorption of AA. Therefore, we utilized both in vivo and in vitro models to address this issue.
Our findings demonstrated that EPEC (WT) markedly inhibits AA uptake in intestinal epithelial cells compared to uninfected or commensal HS4 infected cells. Similar effects have been reported previously for other transporters [26, 30, 31]. EPEC possesses an intact T3SS, which contains a number of structural components to deliver bacterial virulence proteins into host cells [49]. The T3SS comprises ΔescN, the putative ATPase that drives T3SS [41]. Lack of AA inhibition upon infecting with ATPase defective EPEC mutant (ΔescN) suggests that this response is T3SS-dependent. In addition, expression of ΔespA, ΔespB and ΔespD mutants (lack of T3SS ability) also failed to inhibit the AA uptake. On the other hand, the EPEC-secreted effector proteins (i. e., ΔespF and ΔespG/G2) showed a marked inhibitory effect on AA uptake. Collectively, these observations envisage that the effect of EPEC on AA uptake in intestinal epithelia requires functional T3SS.
The observed inhibitory effect of EPEC on AA uptake was accompanied by dysregulated expression of SVCT1 and SVCT2 protein and mRNA in both in vitro and in vivo. Change in a given mRNA expression levels could be an indication of the involvement of transcriptional and/or post-transcriptional mechanism(s) [50]. To investigate the role of transcriptional mechanism(s), we assessed the effect of EPEC on activity of SLC23A1 and SLC23A2 promoters and found no effect on the activity of these promoters. In contrast, the unrelated vitamin thiamine transporter gene promoter activity was markedly inhibited upon EPEC infection [26]. This information envisage that the EPEC mechanism of action is specific for a given nutrient transport. Next, we examined the other possibility that post-transcriptional mechanism(s) (epigenetic mechanisms; microRNA) mediate the observed effect of EPEC infection on AA uptake and SVCT1 and SVCT2 expression. It is interesting to mention here that recent studies have implicated that microRNAs mediate pathogenic bacterial infection in intestine [42, 43]. Therefore, we determined the involvement of microRNAs miR103a (regulator of SLC23A1 [45]) and both miR141 and miR200a (regulators of SLC23A2 [44]) in Caco-2 cells and mice small and large intestinal segments. Our findings showed that all three mature microRNAs (miR103a, miR141, and miR200a) expression levels were markedly up-regulated upon EPEC infection in Caco-2 cells and mouse intestinal segments. These findings demonstrate that post-transcriptional mechanisms play a role, at least in part, in the observed AA inhibitory effect in both model systems. Subsequent studies are needed to explore the possible involvement of other epigenetic mechanisms such as DNA methylation and histone acetylation in mediating the observed AA inhibitory effect of EPEC infection.
In another study, we have recently established the protein-protein interaction between hGRHPR and hSVCT1 [46], the rate-limiting intestinal vitamin C transporter which expresses apically in polarized intestinal epithelia. In the current study, we have investigated the role of this novel interacting partner in facilitating the observed effect on uptake of vitamin C in in vitro and in vivo intestinal models. Interestingly, the expression of GRHPR in both model systems was markedly decreased upon EPEC infection. These findings further demonstrate that the interacting protein associated with hSVCT1 may also be partly responsible for mediating the observed AA inhibition in both in vivo and in vitro model systems.
In summary, our findings demonstrate that EPEC infection inhibits intestinal vitamin C absorption via dysregulation of SVCT1 and SVCT2 expression and is, to some extent, partially mediated through post-transcriptional mechanisms.
Acknowledgments
The current study was supported by the National Institutes of Health grants DK107474 (VSS), DK56061 (HMS), AA018071 (HMS) and GM088790 (JSM), MH108154 (MGG), as well as a grant from the Department of Veterans Affairs (HMS). We thank Dr. Gail Hecht (Loyola University, Chicago) for providing EPEC mutant strains.
Footnotes
Conflict of interests The authors declare that there are no conflicts of interest.
References
- 1.Packer L, Fuchs J. Vitamin C in health and disease. Marcel Dekker Inc, New York, USA. 1997. [Google Scholar]
- 2.Carr AC, Maggini S. Vitamin C and immune function. Nutrients. 2017; 9:1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carr AC, Frei B. Toward a new recommended dietary allowance for Vitamin C based on antioxidant and health effects in humans. Am J Clin Nutr. 1999; 69:1086–1107. [DOI] [PubMed] [Google Scholar]
- 4.Simon JA, Hudes ES. Serum ascorbic acid and gallbladder disease prevalence among US adults. Arch Intern Med. 2000; 160:931–936. [DOI] [PubMed] [Google Scholar]
- 5.Harrison SA, Torgerson S, Hayashi P, et al. Vitamin E and vitamin C treatment improves fibrosis in patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2003; 98:2485–2490. [DOI] [PubMed] [Google Scholar]
- 6.Li Y, Schellhorn HE. New developments and novel therapeutic perspectives for vitamin. J Nutr. 2007; 137:2171–2184. [DOI] [PubMed] [Google Scholar]
- 7.Harrison FE. A critical review of vitamin C for the prevention of age-related cognitive decline and Alzheimer’s disease. J Alzheimers Dis. 2012; 29:711–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishikimi M, Fukuyama R, Minoshima S, et al. Cloning and chromosomal mapping of the human nonfunctional gene for L-gulono-γ-lactone oxidase, the enzyme for L-ascorbic acid biosynthesis missing in man. J Biol Chem. 1994; 269:13685–13688. [PubMed] [Google Scholar]
- 9.Sotiriou S, Gispert S, Cheng J, Wang Y, Chen A, Hoogstraten-Miller S, Miller GF, Knon O, Levine M, Guttentag SH, Nussbaum RL. Ascorbic-acid transporter Slc23a2 is essential for vitamin C transport into the brain and for perinatal survival. Nat Med. 2002; 8: 514–517. [DOI] [PubMed] [Google Scholar]
- 10.Corpe CP, Tu H, Eck P, Wang J, Faulhaber-Walter R, Schnermann J, Margolis S, Padayatty S, Sun H, Wang Y, Nussbaum RL, Espey MG, Levine M. Vitamin C transporter Slc23a1 links renal reabsorption, vitamin C tissue accumulation, and perinatal survival in mice. J Clin Invest. 2010; 120:1069–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daruwala R, Song J, Koh WS, et al. Cloning and functional characterization of the human sodium-dependent vitamin C transporters hSVCT1 and hSVCT2. FEBS Lett. 1999; 460:480–484. [DOI] [PubMed] [Google Scholar]
- 12.Rajan PD, Huang W, Dutta B, et al. Human placental sodium dependent vitamin C transporter (SVCT2): molecular cloning and transport function. Biochem Biophys Res Commun. 1999; 262:762–768. [DOI] [PubMed] [Google Scholar]
- 13.Liang WJ, Johnson D, Jarvis SM. Vitamin C transport systems of mammalian cells. Molec Membr Biol. 2001; 18:87–95. [DOI] [PubMed] [Google Scholar]
- 14.Savini I, Rossi A, Pierro C, et al. SVCT1 and SVCT2: key proteins for vitamin C uptake. Amino Acids. 2008; 34:347–355. [DOI] [PubMed] [Google Scholar]
- 15.Subramanian VS, Marchant JS, Boulware MJ, et al. A C-terminal region dictates the apical plasma membrane targeting of the human sodium-dependent vitamin C transporter-1 in polarized epithelia. J Biol Chem. 2004; 279:27719–27728. [DOI] [PubMed] [Google Scholar]
- 16.Boyer JC, Campbell CE, Sigurdson WJ, et al. Polarized localization of vitamin C transporters, SVCT1 and SVCT2, in epithelial cells. Biochem Biophys Res Commun. 2005; 334:150–156. [DOI] [PubMed] [Google Scholar]
- 17.Subramanian VS, Srinivasan P, Wildman AJ, et al. Molecular mechanism(s) involved in differential expression of vitamin C transporters along the intestinal tract. Am J Physiol Gastrointest Liver Physiol. 2017; 312:G340–G347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adhikari M, Coovadi Y, Hewitt J. Enteropathogenic Escherichia coli (EPEC) and enterotoxigenic (ETEC) related diarrhoeal disease in a neonatal unit. Ann Trop Paediatr. 1985; 5: 19–22. [DOI] [PubMed] [Google Scholar]
- 19.Croxen MA, Finaly BB. Molecular mechanisms of Escherichia coli pathogenicity. Nat Rev Microbiol. 2010; 8:26–38. [DOI] [PubMed] [Google Scholar]
- 20.Fagundes-Neto U, Scaletsky IC. The gut at war: the consequences of enteropathogenic Escherichia coli infection as a factor of diarrhea and malnutrition. São Paulo Med J. 2000; 118: 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guerrant RL, Oriá RB, Moore SR, et al. Malnutrition as an enteric infectious disease with long-term effects on child development. Nutr Rev. 2008; 66:487–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Viswanathan VK, Hodges K, Hecht G. Enteric infection meets intestinal function: how bacterial pathogens cause diarrhoea. Nat Rev Microbiol. 2009; 7:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomson FL, Viswanathan VK, Kanack KJ, et al. Enteropathogenic Escherichia coli EspG disrupts microtubules and in conjunction with Orf3 enhances perturbation of the tight junction barrier. Mol Microbiol 2005; 56:447–464. [DOI] [PubMed] [Google Scholar]
- 24.Zyrek AA, Cichon C, Helms S, et al. Molecular mechanisms underlying the probiotic effects of Escherichia coli Nissle 1917 involve ZO-2 and PKCzeta redistribution resulting in tight junction and epithelial barrier repair. Cell Microbiol. 2007; 9:804–816. [DOI] [PubMed] [Google Scholar]
- 25.Collington GK, Booth IW, Knutton S. Rapid modulation of electrolyte transport in Caco-2 cell monolayers by enteropathogenic Escherichia coli (EPEC) infection. Gut. 1998; 42:200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashokkumar B, Kumar JS, Hecht GA, et al. Enteropathogenic Escherichia coli inhibits intestinal vitamin B1 (thiamin) uptake: studies with human-derived intestinal epithelial Caco-2 cells. Am J Physiol Gastrointest Liver Physiol. 2009; 297:G825–G833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spitz J, Yuhan R, Koutsouris A, et al. Enteropathogenic Escherichia coli adherence to intestinal epithelial monolayers diminishes barrier function. Am J Physiol. 1995; 268:G374–G379. [DOI] [PubMed] [Google Scholar]
- 28.Hecht G, Hodges K, Gill RK, et al. Differential regulation of Na+/H+ exchange isoform activities by enteropathogenic E. coli in human intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2004; 287:G370–G378. [DOI] [PubMed] [Google Scholar]
- 29.Borthakur A, Gill RK, Hodges HK, et al. Enteropathogenic Escherichia coli inhibits butyrate uptake in Caco-2 cells by altering the apical membrane MCT1 level. Am J Physiol Gastrointest Liver Physiol. 2006; 290:G30–G35. [DOI] [PubMed] [Google Scholar]
- 30.Gill RK, Borthakur A, Hodges K, et al. Mechanism underlying inhibition of intestinal apical Cl-/OH- exchange following infection with enteropathogenic E. coli. J Clin Invest. 2007; 117: 428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Esmaili A, Nazir SF, Borthakur A, et al. Enteropathogenic Escherichia coli infection inhibits intestinal serotonin transporter function and expression. Gastroenterology. 2009; 137:2074–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi HJ, Kim J, Do KH, et al. Prolonged NF-κB activation by a macrophage inhibitory cytokine 1-linked signal in enteropathogenic Escherichia coli-infected epithelial cells. Infect Immun. 2013; 81:1860–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith AD, Yan X, Chen C, et al. Understanding the host-adapted state of Citrobacter rodentium by transcriptomic analysis. Arch Microbiol. 2016; 198:353–362. [DOI] [PubMed] [Google Scholar]
- 34.Dupont A, Sommer F, Zhang K, et al. Age-Dependent Susceptibility to Enteropathogenic Escherichia coli (EPEC) Infection in Mice. PLOS Pathog. 2016; 12:e1005616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subramanian VS, Sabui S, Moradi H, et al. Inhibition of intestinal ascorbic acid uptake by lipopolysaccharide is mediated via transcriptional mechanisms. Biochim Biophys Acta Biomembr. 2018; 1860:556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta DeltaC (T)) method. Methods. 2001; 25:402–408. [DOI] [PubMed] [Google Scholar]
- 37.Anandam KY, Alwan OA, Subramanian VS, et al. Effect of the proinflammatory cytokine TNF-α on intestinal riboflavin uptake: inhibition mediated via transcriptional mechanism(s). Am J Physiol Cell Physiol. 2018; 315:C653–C663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reidling JC, Subramanian VS, Dahhan T, et al. Mechanism and regulation of vitamin C uptake: Studies of the hSVCT systems in human liver epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2008; 295:G1217–G1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Subramenium GA, Sabui S, Marchant JS, et al. Enterotoxigenic Escherichia coli heat liable enterotoxin inhibits intestinal ascorbic acid uptake via a cAMP-dependent NF-κB-mediated pathway. Am J Physiol Gastrointest Liver Physiol. 2019; 316:G55–G63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roxas JL, Monasky RS, Roxas BAP, et al. Enteropathogenic Escherichia coli EspH-Mediated Rho GTPase Inhibition Results in Desmosomal Perturbations. Cell Mol Gastroenterol Hepatol. 2018; 6: 163–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gauthier A, Puente JL, Finlay BB. Secretin of the enteropathogenic Escherichia coli type III secretion system requires components of the type III apparatus for assembly and localization. Infect Immun. 2003; 71:3310–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen HT, Dalmasso G, Müller S, et al. Crohn’s Disease-Associated Adherent Invasive Escherichia coli Modulate Levels of microRNAs in Intestinal Epithelial Cells to Reduce Autophagy. Gastroenterology. 2014; 146:508–519. [DOI] [PubMed] [Google Scholar]
- 43.Sabharwal H, Cichon C, Ölschläger TA, et al. Interleukin-8, CXCL1, and MicroRNA miR-146a Responses to Probiotic Escherichia coli Nissle 1917 and Enteropathogenic E. coli in Human Intestinal Epithelial T84 and Monocytic THP-1 Cells after Apical or Basolateral Infection. Infect Immun. 2016; 84:2482–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sangani R, Periyasamy-Thandavan S, Kolhe R, et al. MicroRNAs-141 and 200a regulate the SVCT2 transporter in bone marrow stromal cells. Mol Cell Endocrinol. 2015; 410:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subramanian VS, Sabui S, Marchant JS, et al. MicroRNA-103a regulates sodium-dependent vitamin C transporter-1 expression in intestinal epithelial cells. J Nutr Biochem. 2019; 65:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Subramanian VS, Nabokina SM, Patton JR, et al. Glyoxalate reductase/hydroxypyruvate reductase interacts with the sodium-dependent vitamin C transporter-1 to regulate cellular vitamin C homeostasis. Am J Physiol Gastrointest Liver Physiol. 2013; 304:G1079–G1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu J, Torres AG. Enteropathogenic Escherichia coli: foe or innocent bystander? Clin Microbiol Infect. 2015; 21:729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhuang X, Chen Z, He C, et al. Modulation of host signaling in the inflammatory response by enteropathogenic Escherichia coli virulence proteins. Cell Mol Immunol. 2017; 14:237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glotfelty LG, Zahs A, Hodges K, et al. Enteropathogenic E. coli effectors EspG1/G2 disrupt microtubules, contribute to tight junction perturbation and inhibit restoration. Cell Microbiol. 2014; 16:1767–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Subramanian VS, Sabui S, Heskett CW, et al. Sodium Butyrate Enhances Intestinal Riboflavin Uptake via Induction of Expression of Riboflavin Transporter-3 (RFVT3). Dig Dis Sci. 2019; 64:84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]