

Abstract
Dysfunction in mitochondrial dynamics is believed to contribute to a host of neurological disorders and has recently been implicated in cancer metastasis. The outer mitochondrial membrane adapter protein Miro functions in the regulation of mitochondrial mobility and degradation, however, the structural basis for its roles in mitochondrial regulation remain unknown. Here, we report a 1.7Å crystal structure of N-terminal GTPase domain (nGTPase) of human Miro1 bound unexpectedly to GTP, thereby revealing a non-catalytic configuration of the putative GTPase active site. We identify two conserved surfaces of the nGTPase, the “SELFYY” and “ITIP” motifs, that are potentially positioned to mediate dimerization or interaction with binding partners. Additionally, we report small angle X-ray scattering (SAXS) data obtained from the intact soluble HsMiro1 and its paralog HsMiro2. Taken together, the data allow modeling of a crescent-shaped assembly of the soluble domain of HsMiro1/2.
Keywords: Miro, RhoT, Gem1p, GTP-binding protein, mitochondrial dynamics, crystal structure
1. Introduction
Mitochondrial motility [1–3] and other dynamics [4] are critically important in neuronal function, and their dysfunction is a hallmark in neurodegenerative diseases [2,5,6]. Recent evidence suggests an emerging role in cell migration and possibly cancer metastasis as well [1,3,4]. Miro (also called RhoT), a type 2 transmembrane protein of the mitochondrial outer membrane, serves as an adapter of mitochondrial motility via the Kinesin-1/Dynein/TRAK machinery [7,8]. Knockout of Miro causes mis-localization and mitochondrial inheritance phenotypes on a cellular level [9] and knockout of Miro1 is embryonic lethal in mouse models [10,11]. In D.melanogaster Miro has been shown to be required for both antero and retrograde transport along microtubules [12,13]. Miro is involved in actin-based movement through Myosin19 as well [14–16].
Miro comprises two GTPase domains (the nGTPase and cGTPase) which surround a pair of calcium-binding EF-hand domains. A C-terminal transmembrane domain mediates membrane attachment [17] (Fig. 1A). Based on sequence signatures within the nGTPase domain and its apparent localization [18] the protein was initially termed a ‘rho’ GTPase (‘Mitochondrial Rho GTPase’, hence Miro), however, it has since become clear that the Miro proteins, which generally lack the ‘rho insert’ characteristic of the Rho GTPases, likely comprise a distinct subfamily of small GTPases [19,20]. Miro is evolutionarily conserved down to S.cerevisiae [21]. In humans two paralogs of Miro (HsMiro1 and HsMiro2), which overall have ~60% sequence identity, both play a role in mitochondrial dynamics [22]. In yeast, the single Miro homolog is termed Gem1p, with ~30% overall sequence identity to the human Miro1 [21].
Figure 1. Crystal structure of the HsMiro1 nGTPase domain.
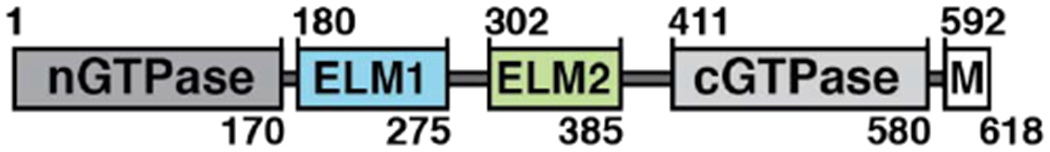
(A) Domain organization of Miro. Numbered residues indicate the limits of structured domains. ELM indicates the EF hand-Ligand mimic pairs. M indicates the transmembrane domain.
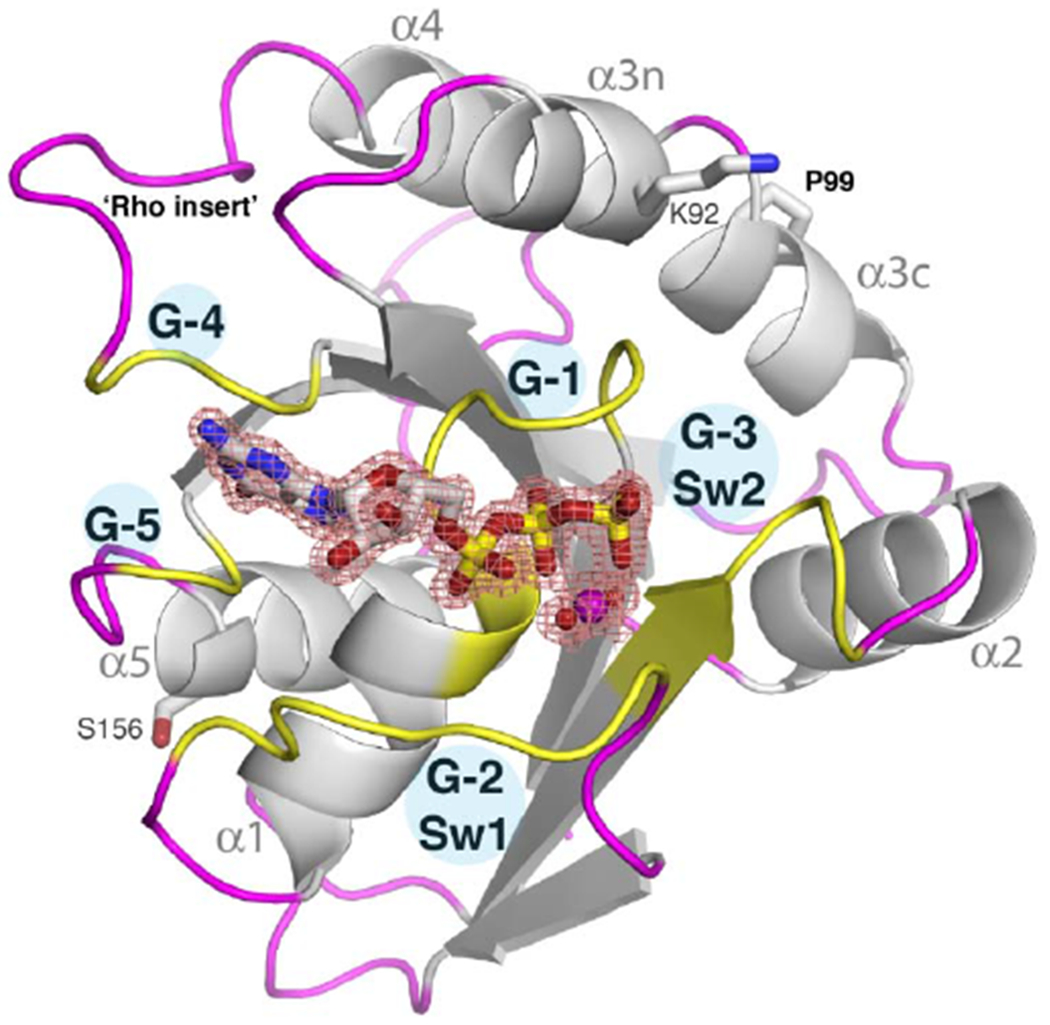
(B) The overall fold of the HsMiro1 nGTPase domain. Guanine nucleotide binding regions (G-1 through G-5) are colored yellow and labeled. The Switch 1 (G-2) and Switch 2 (G-3) peptides are labeled as are helices α1-α5. The bound GTP is shown as sticks while the magnesium ion is shown as a purple sphere. The electron density of the bound Mg2+*GTP is shown contoured at 1.8σ. Pro99, which introduces a kink in helix α3 that is in common with other Rho GTPases, and the location of the ‘rho insert’ absent after G-4, are indicated. The sidechains of Ser156 and Lys92 indicate the positions of two regulatory post-translational modifications of the Miro nGTPase - PINK1 mediated phosphorylation at Ser156 [27,70], and in mouse, acetylation at Lys107 [30].
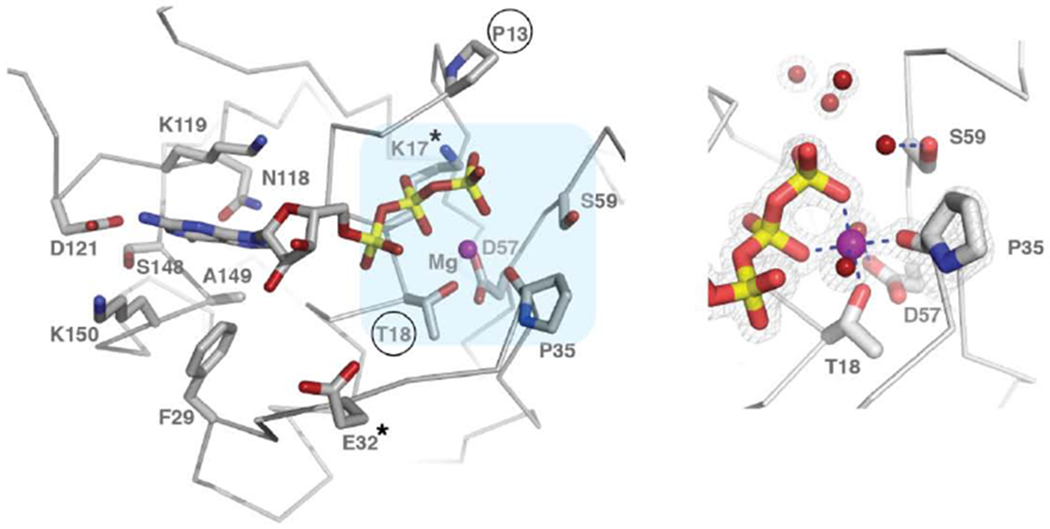
(C) (left) Overview of the active site interactions with bound GTP. Conserved residues that interact directly with GTP are labeled and shown as sticks. The positions of the ‘canonical’ GTPase residues that have been mutated in studies of Miro and Gem1p (Pro13, Lys17, Thr18, Glu32 (Thr33 in Gem1p)) are noted with ‘*’, with Pro13 and Thr18 circled. The light blue area highlights the region shown in closeup. (right) A limited region of the 2Fo-Fc electron density is shown, contoured at 1.8 σ, showing the well-defined coordination interactions with the bound Mg2+ ion and the water structure at the active site. The carbonyl oxygen of Pro35 coordinates directly the active site Mg2+ ion. Water molecules at the putative active center, associated with Ser59, are shown as red spheres. Note that the water molecule interacting with Ser59 side chain hydroxyl is weakly bound (poorly defined in the map). Figures generated using PyMol [49]. A diagrammatic representation of the ligand interactions is shown in Suppl. Fig. 2.
The two EF-hand and C-terminal GTPase domains of Miro form a rigid assembly, termed ‘MiroS’ [17,23]. The EF-hand domains comprise canonical Ca2+ binding motifs paired with ‘hidden’ EF-hands that each interact with a putative ligand-mimic helix, hence are termed ELM1 and ELM2 [17]. While the cGTPase domain is structurally reminiscent of other small GTPases, structures of the human and drosophila cGTPase determined in the apo state or with GDP or GMPPCP bound reveal an open nucleotide binding site that exhibits no significant structural or functional change [17,23]. While mutagenesis studies clearly support a role for calcium binding to the ELM domains and nucleotide binding to the cGTPase domain in the regulation of Miro function [8,22,24–26], the structural mechanisms by which these ligands modulate its activity remain unknown, and their specific roles remain controversial [10,27,28].
The nGTPase domain of Miro is functionally and structurally distinct from the cGTPase, and it is thought that the N-terminal GTPase domain of Miro may be more important for function, as mutations in the N-terminal domain appear to show more robust phenotypes than mutations in the C-terminal domain. For example, it was initially shown that only mutations of the nGTPase domain collapsed the mitochondrial network [22]. Mutation of corresponding residues of the putative GTPase active sites of the N- and C-GTPase domains likewise demonstrate dissimilar behaviors. For example mutation of T18N at the putative nGTPase active center is non-functional in recruiting CENP-F to mitochondria, while the corresponding S432N mutation of the cGTPase behaves like WT [29]. And while both GTPase domains are required for function in yeast Gem1p, only mutations in the nGTPase, and not the cGTPase, alter mammalian mitochondrial transport into axons and dendrites [12]. Furthermore, a recent study demonstrated acetylation of the equivalent residue to Lys92 in humans (mouse Lys105) in the nGTPase domain regulated axonal motility [30]. On this basis it has been suggested that the nGTPase of Miro has the profound role in its function [31] while the cGTPase has a more ‘limited’ role [20]. These observations may additionally explain why the nGTPase shows greater sequence conservation throughout evolution compared to the cGTPase (50% identity between H.sapiens and C.elegans compared to 35% for the cGTPase).
Absent structural information about the nGTPase, dissection of the function of Miro in vivo has generally exploited mutations that are based on inferred functional relationships with the Ras family of GTPases [8,20,21,25,29,31–34]. Such mutations have largely been interpreted as generating what have been termed ‘GTP-bound’ and ‘GDP-bound’ (or ‘active’ and ‘inactive’) states [25,31]. However, as little is known about the actual structure/function relationships of the Miro proteins, whether or not such mutations selected on the basis of analogy impact Miro GTPase function as anticipated remains essentially unknown. Indeed, it is becoming clear that the GTPase fold can be exploited in different ways by different biological systems, with the canonical ‘GTPase switch’ only one of several mechanisms by which GTPase domains can function (e.g. serving as a module for nucleotide-dependent assembly [35–37]), and, therefore, that the biological response of a particular mutation can not necessarily be predicted. Additionally, the limited scope of the mutational library has restricted focus towards function of the Miro GTPase domain as a canonical GTPase switch, whereas clearly a complex set of additional interactions must be required in order for Miro to serve as a tether for regulation of mitochondrial dynamics.
Although the existence of a nucleotide hydrolysis activity by Miro has been inferred from its relationship to other members of the GTPase superfamily, it has received limited biochemical characterization, with the most extensive work carried out using the yeast homolog Gem1p. Indeed, the purified Gem1p readily hydrolyzes GTP, and mutational studies locate the activities to both its N- and C-GTPase domains [20]. The purified Drosophila DmMiro has been shown to hydrolyze GTP, as well [38]. That observations based on Gem1p can be readily extended to the mammalian Miro remains controversial [39–41].
In order to fully understand the regulatory mechanisms and functional interactions of Miro, it will be essential to structurally characterize each of its domains in the context of the full-length protein and to identify the contacts that mediate its intra- and intermolecular assemblies. To that end, we have carried out the X-ray crystallographic and solution scattering studies of the human Miro1 and Miro2 described here. We seek to understand the functional role of the domain organization of Miro and to dissect the mechanisms by which its interactions contribute to regulation of mitochondrial dynamics. Previously, we determined the crystal structures of the DmMiro and HsMiro1 EF-hands and cGTPase domains [17], and the structure of the HsMiro2 cGTPase domain [23]. Here, we have determined the crystal structure of the HsMiro1 nGTPase domain bound, unexpectedly, to un-hydrolyzed GTP. The structure allows us to locate two highly conserved surfaces of the nGTPase domain distinct from the active site which mediate packing contacts in the crystal and that may be poised for functional interaction. In conjunction with small angle X-ray scattering (SAXS) data obtained from both HsMiro1 and HsMiro2, we propose a model for the structure of the entire extra-membranous domain (residues 1-592) of Miro.
2. Experimental
2.1. Cloning and Molecular Biology
HsMiro2 cDNA were graciously provided by the Shaw lab [22]. Fragments were PCR amplified (CloneAmp HiFi PCR Premix, Clonetech) and subcloned into pET-28b vectors (Novagen) as 6xHis tagged proteins via Nco1 and Xho1 restriction sites using ligation-independent cloning (InFusion, Clonetech). Site-directed mutagenesis (Quikchange, Agilent) was employed to create all protein mutants using primers designed in-house (Integrated DNA Technologies). Residue number for the constructs are as follows: HsMiro1 cytoplasmic domain (1-592) c-terminal 6xhis tag (expected MW ~68kD), HsMiro2 cytoplasmic domain (1-588) c-terminal 6xhis tag (expected MW ~66kD), HsMiro1/2 nGTPase (1-180) n-terminal 6xhis tag. Full HsMiro1 (Q8IXI2), HsMiro2 (Q8IXI1), DmMiro (Q8IMX7), and Gem1p (P39722) amino acid sequences are accessible via Uniprot. All DNA constructs were verified by Sanger sequencing (Northwestern Sequencing Core).
2.2. Protein Expression and Purification
HsMiro1 nGTPase.
All recombinant proteins were expressed using E. coli BL21 RP (DE3) strain (Novagen). After inoculating 2 L of TPM media with 2 mL of overnight starter culture, the cells were grown with shaking at 200 rpm for approximately 5 hours at 37°C to OD600=0.45. The temperature was then lowered to 16°C for 1 hour and then cells were induced at OD600=0.90 with 0.125 mM IPTG for approximately 18 hours overnight. The cells were resuspended in 50 mM HEPES*HCl (pH 8.0), 500 mM NaCl, 1 mM MgCl2, 0.5 mM TCEP, 5% sucrose (w/v), and 0.05% Tween20 (v/v) before flash freezing in liquid nitrogen. Cell pastes were stored at −80°C before lysis and purification. Cells were thawed at 37 °C and all lysis and purification steps were performed at 4°C. The cell pastes were sonicated for 4x45 seconds before clarifying at 35,000 rpm for 45 min. The soluble fraction from 8 L of culture was incubated with 8 mL of 50% Co2+ TALON bead slurry, washed (8mM imidazole), then eluted with IMAC Elution Buffer (500 mM imidazole, 200mM NaCl), then slowly diluted 4-fold in Buffer A (25 mM HEPES*HCl pH 7.4, 1 mM MgCl2, 0.5 mM TCEP, 5% sucrose (w/v)) to be loaded onto a 5mL HiTrap Q ion exchange column. The protein was eluted using a 18%-30% 1M NaCl step profile (Buffer A supplemented with 1M NaCl) and concentrated to approximately 2 mL using 10 kDa MWCO spin concentrators (Amicon) before running over a 16/60 Superdex 200 size exclusion column (GE Healthcare) in purification buffer (25 mM HEPES*HCl pH 7.4, 150 mM NaCl, 0.5mM MgCl2, 0.5 mM TCEP, 5% w/v sucrose). Peak fractions were pooled and stored at −80°C. Approximate final yields were ~1 mg pure protein per 2 L of culture. Protein purity was assayed using SDS-PAGE.
HsMiro1 and HsMiro2 cytoplasmic domain.
Expression, lysis and purification of the His-tagged proteins were carried out as described above, with the exception that 1mM CaCl2 was added to all buffers. Subsequently, samples were diluted into Buffer B (25mM Tris pH 8.5, 1mM MgCl2, 1mM CaCl2, 0.5mM TCEP, 2.5% sucrose) to be loaded onto a 5ml HiTrap Q column, followed by elution using a 8-50% gradient (Buffer B supplemented with 1M NaCl). Peak fractions were pooled and injected over a 16/60 S200 gel filtration column equilibrated with 25mM Tris 8.5, 500mM NaCl, 1mM MgCl2, 1mM CaCl2, 0.5mM TCEP, 2.5% sucrose. Peak fractions were concentrated to ~2.5 mg/ml and flash frozen.
The polypeptide identity of the HsMiro 1-180 was confirmed by crystallography (where each side chain with electron density correctly matched the intended target sequence). The polypeptide identity of HsMiro1/2 was previously confirmed via MS-based peptide mapping and western blot [23].
2.3. Crystallization and X-ray Data Collection
Protein was concentrated to ~10 mg/mL in purification buffer (25 mM HEPES*HCl (pH 7.4), 150mM NaCl, 0.5 mM MgCl2, 0.5 mM TCEP, and 5% sucrose (w/v)). Crystallization trials were set up at 22 °C with the commercial sparse matrix screens JCSG+, PACT, PEGs, and PEGs II Suites (Qiagen) in INTELLI-WELL 96-3 LVR sitting drop vapor diffusion plates using a Crystal Phoenix robot (Art Robbins Instruments). 2:3, 1:1, and 3:2 protein:precipitant ratios were tested in a 1.0 μL total drop volume with a 90 μL reservoir. Rod-shaped crystals initially grew after 12 hours only in condition #38 from the PACT Suite (0.1 M MMT buffer pH 5, 25% (w/v) PEG 1500). Crystal diffraction quality was optimized using INTELLI-WELL 24-4 sitting drop vapor diffusion plates (Art Robbins Instruments) over a range of MMT Buffer pH 4.5-5.5 + PEG 1500 10-30% (w/v) concentrations, and different guanine nucleotide conditions. Crystals grew to about 30 μm with rod and cube-shaped morphologies. Cryo-protection was not required during data collection. Measurement of X-ray diffraction data was performed at 100K at the beamlines of Sector 21 of the Life Sciences Collaborative Access Team (LS-CAT) of the Advanced Photon Source (APS) in the Argonne National Laboratory (ANL). Data were measured on a MarMosaic 225 CCD detector and processed using Mosflm [43]. 1 sec exposures were recorded every 1° of rotation over 120° at 65% beam attenuation.
2.4. Structure Determination and Refinement
An initial 3.2Å resolution dataset was obtained from a crystal grown in the presence of a mixture of 1 mM GDP and (as subsequently discovered) GTP. The structure was determined by molecular replacement using the Phenix add-on Ensembler [44] to create an overlapped model of GTPases based on HsMiro1 cGTPase (PDB ID=5KSP), RRas2 (PDB ID=2ERY), and RhoB (PDB ID=2FV8). The model obtained from Phenix Autobuild [45] accounted for 82% of the polypeptide chain (304 of 372 residues in the asymmetric unit). Electron density corresponding to a bound nucleotide was clearly visible and could be unambiguously interpreted as arising from bound guanosine tri-phosphate, not the anticipated guanosine di-phosphate. Subsequently a 1.7Å dataset was obtained from a crystal grown using protein at 10mg/ml supplemented with 1μM GDP, with a 1:1 2.0 μL drop volume over a reservoir of 0.6M MMT pH 5, 22% PEG 1500. This dataset also revealed the protein to be only GTP-bound (as did additional lower-resolution datasets from crystals obtained in the presence of 1mM, rigorously purified, GDP) and it was used for rebuilding and refinement of the final structure with COOT [46] and phenix.refine [47]. The space group is P212121, with two molecules related by a non-crystallographic two-fold symmetry (NCS) axis in the asymmetric unit. Residues 4-175 (chain A) and 4-172 (chain B), as well as the bound Mg2+GTP ligands, were readily built into the electron density map; residues 1-3 and 176/173-180 were not visible and are presumed to be unstructured. Superposition of the NCS-related monomers yields an RMSD of 0.232Å over 136 Ca atoms. GTP ligand geometry restraints were added with Phenix eLBOW [48]. Once the crystallographic statistics reached Rwork= 0.252 and Rfree = 0.296, water molecules were added in every subsequent round of refinement. Final statistics are reported in Suppl. Table 1. PyMOL was used to generate all figures [49]. Superpositions and RMSD values were calculated using the Super routine in PyMOL.
2.5. Homology Modeling.
Homology models of the Gem1p and DmMiro nGTPase domains were generated using I-TASSER [50,51], using the HsMiro1 nGTPase structure to provide the initial template. For Gem1p nGTPase the Idenl (percentage sequence identity in the threading aligned region in the query sequence) with respect to HsMiro1 is 0.30, the C-score (the confidence score for estimating the quality of predicted models by I-TASSER) is 0.41 and the TM-score (a similarity score insensitive to the local modeling error [52]) is 0.77. For DmMiro nGTPase Idenl is 0.66, the C-score is −0.48, and the TM-score is 0.65. The residue-level quality of the protein structure predictions were calculated using ResQ [53] based on local variations of modeling simulations and the uncertainty of homologous alignments. These values, the ‘estimated local accuracy’, are plotted using the PyMol ‘Putty’ representation in Fig. 2B. Additional sequence alignments were carried out with T-COFFEE and EMBOSS-NEEDLE [54,55].
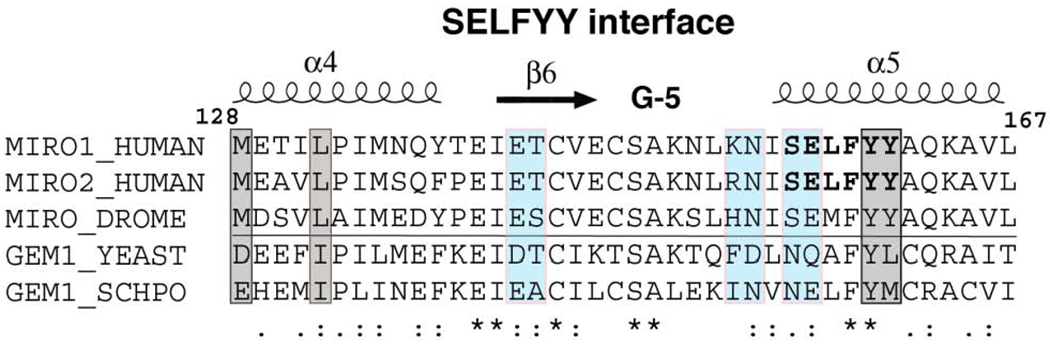
Figure 2. Conserved and divergent regions of the Miro N-terminal GTPases.

(A) Sequence alignment of the nGTPase domains of HsMiro1/2 and Gem1p. The positions of the secondary structural elements are indicated above the alignment, and motifs referenced in the text indicated below the alignment. Residues 4-172 are visible in the crystal structure of the HsMiro1 nGTPase. Sequence alignment generated using T-Coffee and ESPrint [54,93]. (For a more extensive alignment over 36 Miro/Gemp1p nGTPase sequences, see Suppl. Fig. 3)
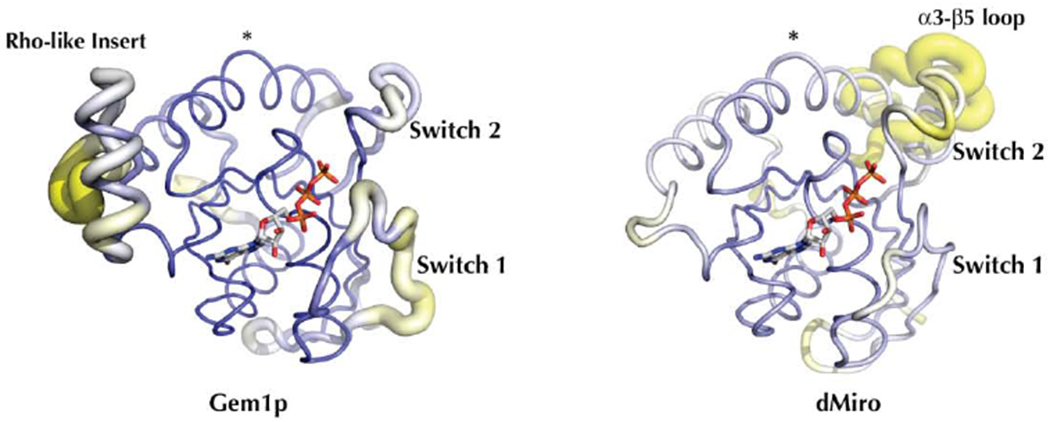
(B) A homology model of Gem1p generated by I-Tasser [50] using the HsMiro1 nGTPase structure as a template. The PyMOL ‘putty’ representation [94] encodes the estimated local accuracy of the model, and highlights that the ‘switch 1’ and ‘rho-like insert’ regions of Gem1p can not be modeled reliably due to divergence between HsMiro and Gem1p. The Miro and Gem1 structures deviate most significantly at Switch 1, and at the Rho-like insert. The position of the α3 break is highlighted with ‘*’. In contrast, the structure of the Drosophila DmMiro nGTPase (at right) can be modeled readily, with the exception of an extended loop inserted between α3 and β5 (labeled). In both images, the poorly ordered N- and C-terminal peptides are omitted – the Gem1p model includes residues 2 to 189 and the DmMiro model includes residues 8 to 198. The position of bound GTP is modeled based on alignment with the HsMiro1 GTP structure.
2.6. Size Exclusion Chromatography - Small Angle X-ray Scattering (SEC-SAXS).
SEC-SAXS Experiments were performed at the BioCAT beamline 18-ID-D at the Advanced Photon Source of Argonne National Laboratory [56]. A Superdex 200 Increase column was preequilibrated with 25 mM Tris, pH 8.5, 500 mM NaCl, 1 mM MgCl2, 0.5 mM TCEP, 2 mM GTP and either 1 mM CaCl2 or 1mM EGTA. Flow rate was 0.75 ml/min. Samples of HsMiro1 (residues 1592 with a C-terminal 6xHis tag) and HsMiro2 (residues 1-588 with a C-terminal 6xHis tag), prepared in 25mM Tris pH 8.5, 500mM NaCl, 1mM MgCl2, 1mM CaCl2, 0.5 mM TCEP-HCl, and 2.5% sucrose, were injected onto the column (HsMiro1: 300 microliters at 2.2 mg/ml and 350 microliters at 5.8 mg/ml; HsMiro2: 400 microliters at 2.7 mg/ml). SAXS was performed in-line with this size exclusion column setup [57]. Photons scattered from the 12 keV X-ray beam were detected with a Dectris Pilatus 1M detector at a distance of 3.5 m from the sample. One second SAXS exposures of fractions of the SEC column, recorded every 3.0s, showed increased x-ray scattering at 3 distinct peaks, as shown in Suppl. Fig. 5. Only the major peak, which represents the monomeric species, was evaluated. Buffer background was obtained by radially binning each exposure, and then averaging over the first 100 fractions. The central portion of the monomer peak was averaged and background subtracted to obtain I vs. q curves (Suppl. Fig. 6, top panels) [58]. The radius of gyration Rg, zero-angle intensity I0, and associated uncertainties for these parameters were obtained by weighted linear regression of log(I) vs. q2 as shown in the Guinier plots in Suppl. Fig. 6. Dmax was determined from the pair distance distribution function P(r) with the program GNOM [59,60]. In determining Dmax, GNOM gives a “total estimate”, which identifies common artifacts encountered by the GNOM method. Dummy atom modeling for SAXS reconstructions shown in Fig. 4A were done with the program DAMMIF [61] (10 runs per SAXS structure) and aligned, averaged, and filtered using SUPCOMB [62], and DAMAVER/DAMFILT [63]. SAXS parameters are reported in Suppl. Table 2. The A260/A280 ratios measured during the runs of 0.67 (HsMiro1 monomer) and 0.81 (HsMiro2 monomer) are consistent with theoretical values calculated from amino acid composition with two nucleotides bound (A260/A280 ratios of 0.70 and 0.75, respectively, when nucleotide-bound; 0.52 and 0.55, respectively, when nucleotide-free), however the nucleotide-bound state was not determined directly.
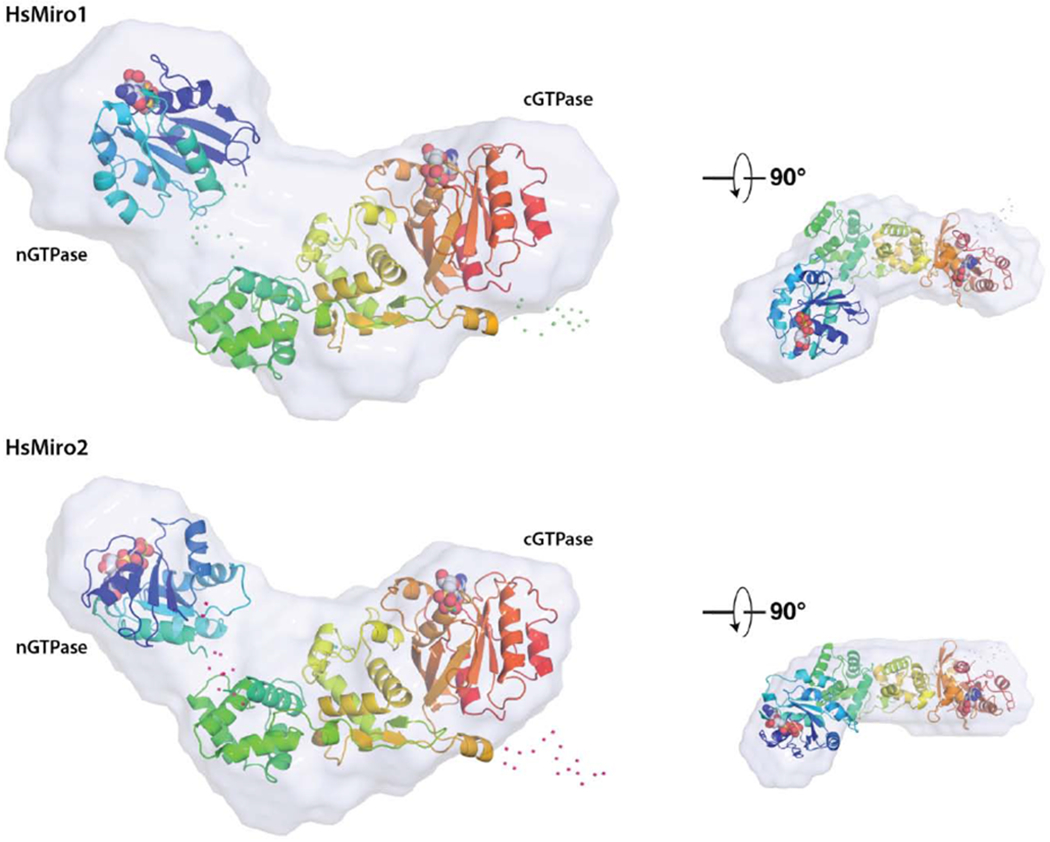
Figure 4. Solution structure of monomeric HsMiro1 and HsMiro2.
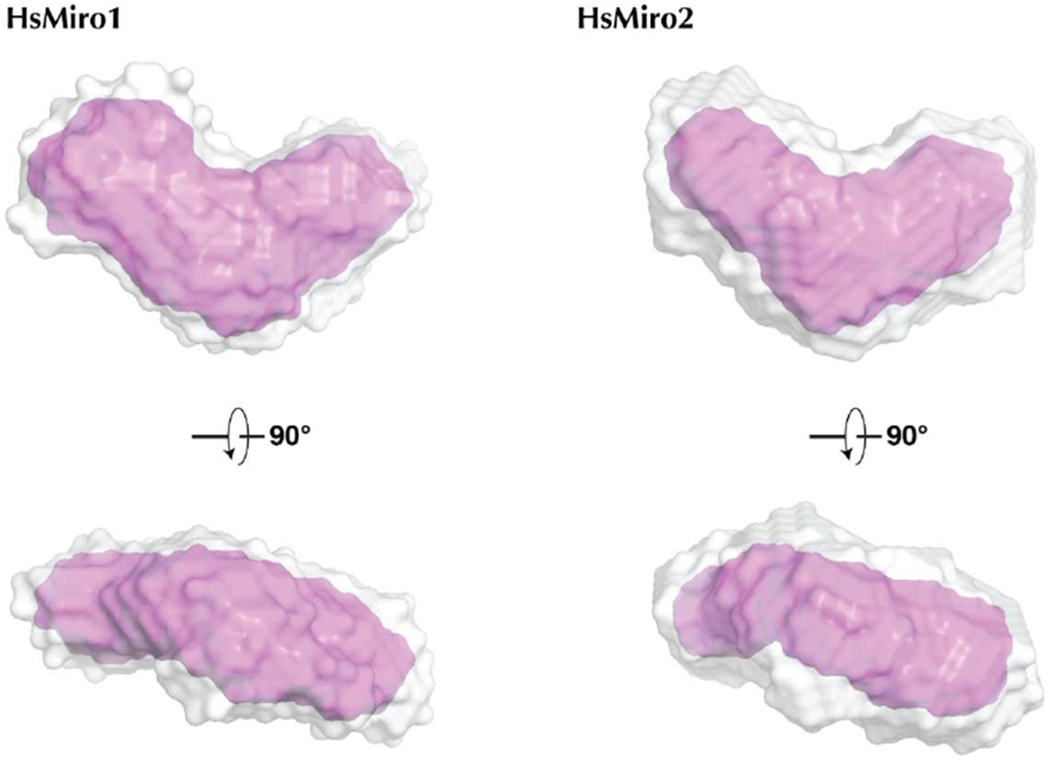
(A) Averaged reconstructions from 10 DAMMIF calculations of the processed SAXS data from HsMiro1 and HsMiro2. The DAMAVER average of the DAMMIF runs is shown in gray and the DAMFILT filtered envelope is shown in pink [61,79]. The dummy atoms used to construct the contours shown were generated with a radius of 4Å.
(B) Representative monomer models generated using BUNCH [64] are overlaid with the corresponding SAXS scattering envelope reconstructions. The two models are aligned with respect to the MiroS domain, with the nGTPase at left in each case. The positions of the nucleotides bound to each of the GTPase domains are shown as spheres. Shown smaller at right are the overlays viewed after a ~90° rotation around the horizontal axis. The scattering envelopes reasonably enclose both the rod-like MiroS and nGTPase domains. Note that while the orientations of MiroS in the two structures are similar, the orientations of the nGTPase domains are different, reflecting the fact that its position cannot be fixed based on this data; their relative dispositions suggest that the nGTPase domains are not tightly associated with the ELM1 domain of MiroS. Dots represent amino acids not observed in the crystal structures and modeled as flexible scattering elements.
2.7. SAXS Rigid-Body Modeling
A model of the cytoplasmic domain of HsMiro was obtained using BUNCH [64]. Rigid body optimization of theoretical scattering data to observed SAXS scattering data was based on partial scattering amplitudes obtained from the structures of the nGTPase of HsMiro1 and the EF-hand/cGTPase domains of HsMiro1 (PDB ID: 5KTY) subject to the constraints of the known linker peptide length between the two fragments. The program CRYSOL was used to generate theoretical scattering data from the nGTPase (residues 3-170, with the poorly ordered residues 171-175 deleted) and MiroS (residues 181-582) structures. The Pre-BUNCH input specified the full sequence of the soluble domain (1-600), such that contributions of three peptides (1-2, 171-180, 583-600) were modeled as unknown. Default parameters were used throughout. The modeling runs were carried out against the observed SAXS scattering data obtained from both HsMiro1 and HsMiro2, yielding χ2 values of 1.137 and 4.137 for the two HsMiro1 samples, respectively, and a χ2 of 1.007 for HsMiro2. The generated models of HsMiro1 and HsMiro2 obtained by rigid body optimization against the scattering data were then superimposed on the DAMMIF dummy atom reconstruction obtained from each dataset using SUPCOMB [62]. The normalized spatial discrepancies (NSD) obtained following overlap of the protein model with each dummy atom reconstruction were: HsMiro1 2.0055 and 2.1277; and HsMiro2 1.7645. These superpositions for HsMiro1 and HsMiro2 are shown in Fig. 4B.
3. Results and Discussion
The crystal structure of the Human Miro1 N-terminal GTPase was determined at 1.72 Å resolution with magnesium and GTP bound (Suppl. Table 1), with two molecules in the asymmetric unit. Both polypeptide chains are bound to Mg2+GTP and are very well defined in the electron density map. The overall fold of the HsMiro1 nGTPase is similar to that of other small GTPases (Fig. 1B). The structure can be superimposed on the structure of Ras-GTP with an RMS deviation of 0.902 Å (over 99 Cα atoms, PDB: 5P21), and on the structure of Rho-GTP with an RMSD of 0.700 Å (over 107 Cα atoms, PDB: 3TVD). Portions of the C-terminal peptides of the two protomers, residues 169-172/175, interact across the dimer interface but are poorly ordered, consistent with their likely role as a flexible ~10 residue linker between the N-terminal GTPase and the remainder of the protein, MiroS [17]. The overall structure is shown in Figure 1B.
3.1. Binding interactions include unusual coordination of Mg2+ ion
Small GTPase binding sites are generally characterized by five conserved sequence motifs (G-1 to G-5) [65]. In the HsMiro1 nGTPase, the mainchain atom hydrogen-bonding interactions between the ‘P-loop’ (G-1) motif and the phosphate groups of the bound GTP are typical of members of the small GTPase family. The P-loop also contributes two sidechains, Pro13 and Arg14, which may serve to sequester access to the active site. Both the ‘Switch 1’ (G-2) and ‘Switch 2’ (G-3) motifs, which in other GTPases mediate catalytic activity, appear to be well-ordered and show extensive stabilizing interactions (Fig. 1C and see Suppl. Fig. 1B). However, as the structure contains bound GTP, the configuration of the active site residues and the accompanying water structure seen in the crystal structure are apparently non-catalytic. Interestingly, the magnesium ion coordination mediated by the ‘Switch 1’ (G-2) motif is contributed not by a sidechain atom (e.g. a Thr side chain hydroxyl as is seen in Ras [65]), but rather by the carbonyl oxygen of Pro35 (Fig. 1C and see Suppl. Fig. 2), which is one of a proline-proline (PP) sequence pair the first residue of which is almost universally conserved among the Miro GTPases (Fig. 2A and see Suppl. Fig. 3). There is no threonine sidechain in the vicinity, obviating canonical Switch 1 Thr sidechain-oxygen coordination that would be inferred by analogy to Ras [21,34]. In the yeast Gem1p, to the extent that the Gem1p Switch 1 region can be modeled (see below), a threonine residue (Thr33) occurs six residues N-terminal to the position of a similarly conserved PP motif (Suppl. Fig. 3); its mutation to alanine was found to have no effect on subcellular localization [21,34], presumably indicating that it does not play a role in magnesium coordination. The direct coordination of magnesium by a mainchain carbonyl oxygen is unusual in small GTPases but not unprecedented, as similar carbonyl coordination is found in structures of the RhoA GTPase GDP complex [66,67]. In the nGTPase, the ‘Switch 2’ (G-3) motif contributes three sidechains that are directed towards the active site - Asp57, which canonically stabilizes Mg2+ coordination, Ser59, which appears to form a labile hydrogen bonding interaction with an active site water, and Ala61, which forms a ‘cap’ at one side of the active water structure (Fig. 1C). The water structure is well-defined, with minor differences between the two protomers within the asymmetric unit. In both sites, a water molecule bridges an oxygen of the GTP γ-phosphate with the backbone amide of Gln60, with two additional water molecules completing a tetragonal hydrogen bonding arrangement. This arrangement is conserved, with the addition in one site of the asymmetric unit of an adjacent water molecule bridging and possibly stabilized by both the sidechain hydroxyl of Ser59 and γ-phosphate oxygens of GTP (Fig. 1C). While a water at this position might play a functional role at the active center of the GTPase, how these interactions might have significance here remains unknown. Finally, the interactions of the G-4 motif with the guanine base are mediated canonically by the sidechains of Asp121 (hydrogen bonding to the purine) and Lys 119 (packing against it). The exocyclic oxygen of GTP buried within the active site is stabilized via a hydrogen bond to the mainchain nitrogen of Ala149, which is part of the conserved G-5 motif that occurs just prior to the C-terminal helix of the domain.
3.2. GTP appears to bind tightly in the crystallized N-terminal GTPase domain
The HsMiro1 nGTPase was initially crystallized in the presence of a mixture of 1 mM GDP and GTP (see Methods), however, the bound ligand evident in the resulting 3.2Å electron density map could be readily modeled as a molecule of GTP. Despite the presence of GDP during crystallization, binding heterogeneity could be excluded both by the quality of the density and by the consistency of the B-factors across the phosphate chain with GTP modeled into the site. Subsequently a 1.7Å resolution dataset was obtained from a crystallization carried out in the presence of only a low concentration (1mM) of added nucleotide, and this structure, reported here, also revealed well-defined electron density consistent with binding of only Mg2+-GTP within the active site (Fig. 1B). Additionally, we were unable to exchange GTP for other nucleotides [95]. We conclude, therefore, that GTP is very tightly bound to our HsMiro1 nGTPase domain construct and believe that the bound species originated from nucleotide endogenous to the expression system that co-purified with the protein.
However, the co-purification and evident stability of the GTP observed bound in our crystal structure is surprising, as previous work reported GTP hydrolysis by the purified Drosophila Miro [38], provided qualitative evidence for hydrolysis by a human nGTPase fusion [68], and found robust enzymatic activity with the purified yeast homolog Gem1p [20]. More recently, it has been shown that the HsMiro nGTPase itself can exhibit readily measurable GTP hydrolysis activity [42]. The expression constructs used in the latter study comprised residues 2-169 of HsMiro1/2, terminating just C-terminal to helix α5, and are quite similar to the construct that was crystallized, residues 1-180 of HsMiro1 We cannot yet reconcile our observations with these data, although we note that the additional C-terminal 11 residues are poorly ordered in our structure, and are adjacent to a conserved interface that mediates the non-crystallographic dimer (discussed below). We speculate that the difference in hydrolysis behavior might arise from differences in the protein constructs and purification methods used, as our proteins were subjected to an additional purification step and showed homogeneity by SEC-MALS [23], but further study will be required to resolve this issue.
3.3. Structural relationships to the Rho and C-terminal Miro GTPases
In order to try to relate our crystal structure to functional significance, we compared it to Rho GTPase family members. Based on a number of sequence divergences Miro is no longer considered a bonafide member of the Rho GTPase family [19,20]. Most notable is the absence of the so-called ‘rho-helix’, a conserved ~13 amino acid sequence inserted between the G-4 motif and helix α4 of the Ras-like GTPase fold [19] (Suppl. Fig. 4). Instead, in HsMiro seventeen residues of the rho-helix loop are replaced by a much shorter 4-residue turn. Nevertheless, several elements characteristic of the Rho GTPases are conserved between Miro and Rho and are structurally superimposable: First, a highly conserved WxP motif in helix α3 (see Suppl. Fig. 3) serves to maintain the structural relationship between helices α1, α2, and α3 by introducing a kink in helix α3 at the same position in almost all rho GTPases (Fig. 1B). And, the G-5 motif sequence CSAK characteristic of rho GTPases is structurally conserved, as the serine and alanine residues both play a role in positioning the guanine base. Interestingly, while Pro35 of the PP motif (above) of ‘Switch 1’ appears to be highly conserved in Rho GTPases, the structure of this region, termed the ‘core effector domain’ [19], is generally quite different in Rho GTPases from that in HsMiro1, and the position and orientation of the ‘conserved’ proline is distinct. The absence of several other sequence elements conserved in the Rho family, particularly large hydrophobics that appear to function to position the core effector domain in Rho GTPases, suggests that the apparent conservation of the proline here does not reflect similarity of structure or function.
The recent identification of the Miro cGTPase as an NTP hydrolase [42] suggests that the two GTPase domains of Miro are mechanistically different, and, indeed, they are structurally quite distinct [23] with an overall RMSD for their superposition of 2.09 Å over 104 Cα atoms. The nGTPase domain of Miro exhibits only 22% sequence identity with its cGTPase domain, and notably, sequence and structural alignments show almost no similarity in the switch regions between the nGTPase and cGTPase (Suppl. Fig. 1). Curiously, the sequences of the Miro cGTPase Switch regions themselves are poorly conserved within the Miro protein family (Suppl. Fig. 1A). In contrast to the nGTPase, the Miro cGTPase domain readily exchanges nucleotide, and its structures in GDP-, GDP*Pi-, and GMPPCP-bound states have been determined [17,23]. Particularly notable is the location of its Switch 1 region (Suppl. Fig. 1B), which adopts an open conformation distant from bound nucleotide. The latter is consistent with the possibility that its catalytic and regulatory mechanisms may be unrelated to that of the broader family of GTPases.
3.4. A conserved motif mediates a potential binding interface
The HsMiro1 nGTPase construct crystallized here appears to be monomeric in solution [23]. However, in the crystal structure, the nGTPase domain associates as a two-fold symmetric non-crystallographic dimer (Fig. 3A). The interface between the two protomers is extensive, with a buried surface area of 1225 A2, and a computed ΔG for dimer formation [69] of −9.1 kcal/mol. The interface involves extensive interactions between helices α4, the bridging β-strand, and helix α5, and remarkably, it is mediated by previously unrecognized but highly conserved sequence motif of the Miro nGTPases (Fig. 3B), which we term ‘SELFYY’. The tyrosines of the motif occur along the C-terminal helix, α5, of the nGTPase fold, and they contribute to formation of a complex surface that involves numerous hydrophobic and polar interactions. The penultimate tyrosine residue is almost universally conserved in the Miro/Gem1p family (Suppl. Fig. 4), while the position of the terminal tyrosine, highly conserved in Miro, retains its large hydrophobic character in Gem1p. The tyrosines of the motif are absent in other Rho GTPases, and indeed the fingerprint of the interface rationalizes much of the Miro family sequence conservation observed throughout this region of the molecule (Fig. 3C). Consequently, we infer that the dimer interface observed in the crystal reflects a functionally important interaction of the Miro nGTPase domain. Of note is that Ser156 (‘S’ of the SELFYY motif) is phosphorylated by PINK1 kinase thereby promoting recruitment of the ubiquitin ligase Parkin and triggering Miro ubiquitination and degradation [27,70].
Figure 3. Dimerization and conserved surfaces of the HsMiro nGTPase.
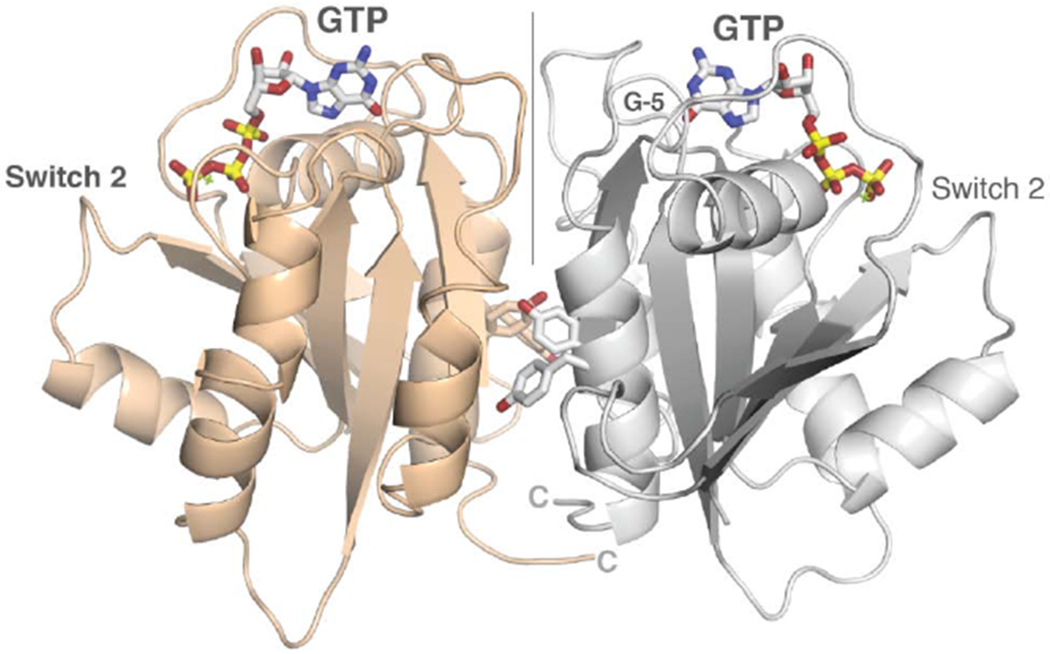
(A) Dimerization of the nGTPase domain in the crystal. The protomers associate across a non-crystallographic two-fold axis. At the center of the interface is a cluster of paired tyrosine sidechains of the ‘SELFYY’ motif. The positions of the GTP molecules are indicated. Note that the interface is distal to the Switch 1 and Switch 2 regions, and therefore is unlikely to be responsive to a specific nucleotide binding state. The G-5 motif in the foreground at right is indicated.
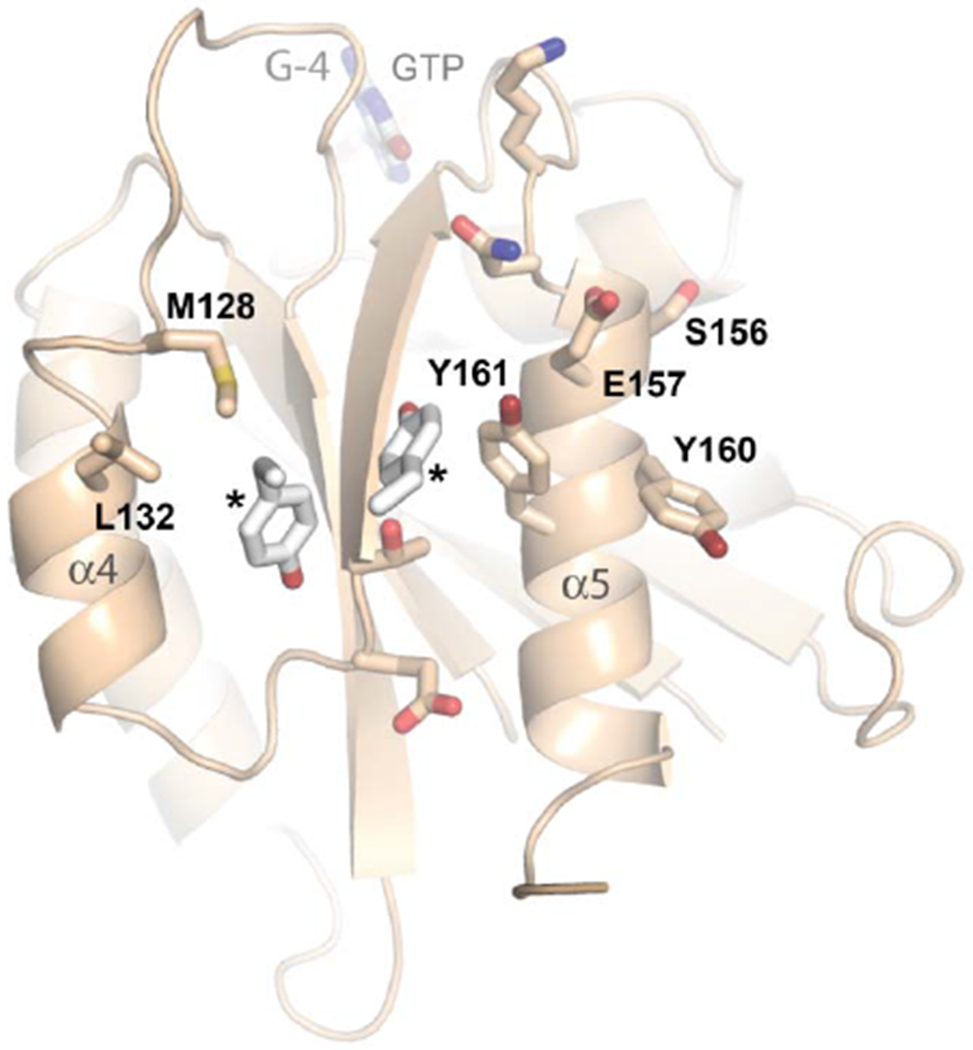
(B) Details of the SELFYY motif interface. One monomer of the dimer is shown rotated 90° relative to (A), with residues substantially buried at the interface shown as sticks. The hydrophobic residues of the interface (M128, L130, Y160, Y161) are labeled, with the positions of the sidechains of the tyrosines extending across the dimer interface (‘*’) shown to highlight their intercalation into a pocket formed by sidechains extending from the α4 and α5 helices. The G-4 loop and GTP are indicated. S156 has been reported to be phosphorylated by PINK1 kinase [70].
(C) Sequence conservation in the SELFYY interface of the Miro nGTPases. The surface residues that are substantially buried upon formation of the interface are highlighted – hydrophobics in grey, polar residues in teal. The associated secondary structure is indicated above the alignment, sequence conservation indicated below it. (For a more extensive alignment over 36 Miro/Gem1p sequences, see Suppl. Fig. 3.)
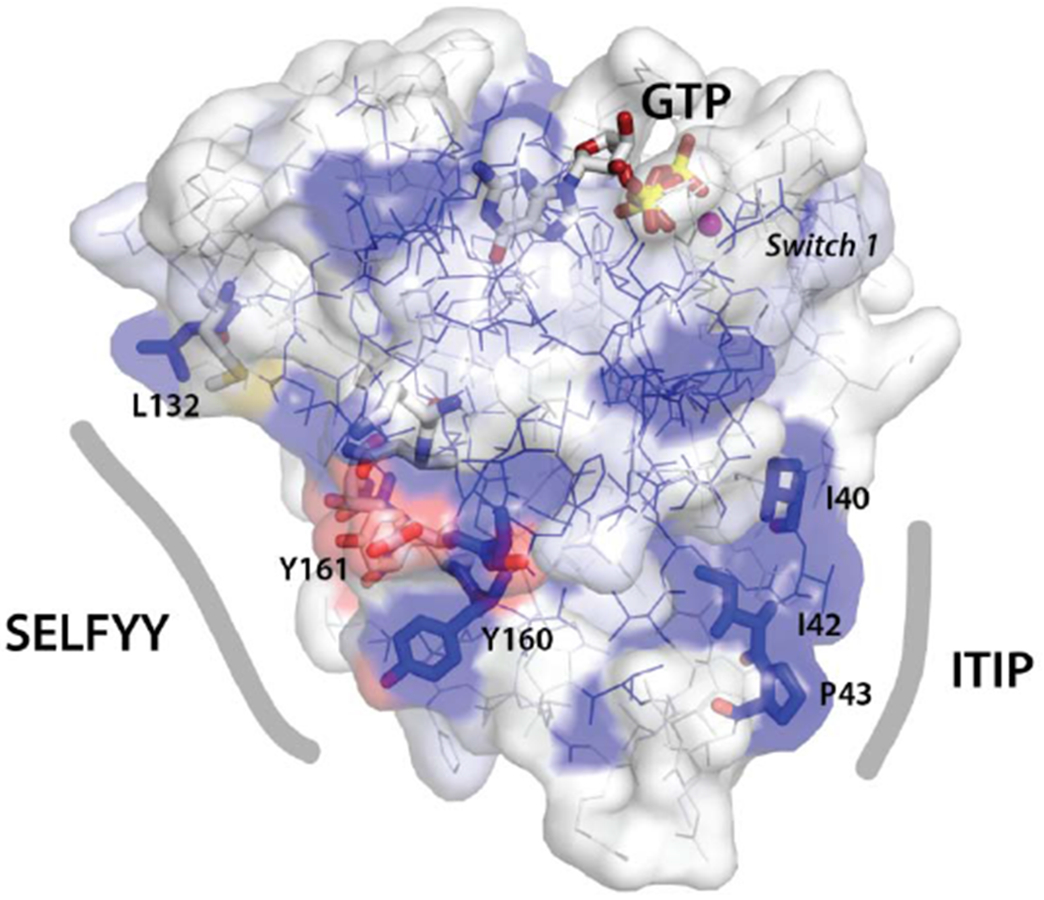
(D) Surface representation of the nGTPase domain. Sequence conservation is mapped as surface color from white (not conserved) to blue (highly conserved). The sidechains of the SELFYY interface and the conserved ‘ITIP’ hydrophobic surface are shown as sticks, and the SW1 region is indicated. The GTP binding site is at top, with GTP shown as sticks. At left, the ‘SELFYY’ surface extends across a broad face of the domain (L132 – Y160); at right the ‘ITIP’ hydrophobic surface (I40 – P43) follows switch 1 (‘SW1’).
The location of the SELFYY interface along a face of the protein fold distal to the switch regions of the nucleotide-binding site suggests that the interface is not responsive to nucleotide-binding state. However, interestingly, a survey of crystal structures of H-Ras has shown that a dimeric form mediated by the corresponding interface, termed the α4/α5 dimer, is preferentially observed when Ras is in its ‘active’ or GTP-bound state [71,72]. This region of the Ras protein has been termed the ‘allosteric lobe’, and although the functional significance of dimerization in Ras is somewhat controversial, there is evidence that it may be functionally coupled to its activity [71,73]. These results raise the interesting, albeit speculative, possibility that the unexpected stabilization of GTP observed in our crystal structure might be coupled to formation of the SELFYY-mediated dimer, and suggest that perturbation of the interface may provide a worthwhile avenue for investigation of Miro function.
We also identify a second conserved hydrophobic surface of the nGTPase, which we term ‘ITIP’ (Fig. 2A and see Suppl. Fig. 3). The motif contributes to a distinct crystal contact and buries a surface area of 599 A2 with a calculated ΔG of −7.8 kcal/mol, as evaluated using PISA [69]. This motif is adjacent to the ‘Switch 1’ region of the nGTPase (Fig. 3D), and thereby configured potentially to be responsive to nucleotide-state. However, although the conservation of the ‘ITIP’ and ‘SELFYY’ motifs on disparate surfaces of the nGTPase domain suggests that they have functional significance, to understand their role it will be essential to establish their interactions within the context of the full-length Miro.
3.5. HsMiro1/2 is an elongated ‘crescent’ in solution
To that end, with structures of both the HsMiro1 nGTPase and the HsMiro1 ‘S’ domain comprising the EF-hand and cGTPase domains [23] available, we sought to study the assembly of the cytoplasmic domain of HsMiro with the goal of obtaining a model of the structure of the complete extramembranous Miro polypeptide. Our previous SAXS studies of the DmMiroS [17] indicated good agreement with the structure determined crystallographically. However, in those studies of the Drosophila Miro we noted some heterogeneity in the SAXS data from the cytoplasmic domain of the protein likely arising from aggregation and/or formation of multimers. To obviate this previously noted heterogeneity, we took advantage of a SEC-SAXS facility available at the BioCAT beamline that allowed us to readily resolve scattering from dimers and aggregates from that of the monomeric full length protein [57] (Suppl. Fig. 5). Experiments were carried out with two independently purified samples of the cytoplasmic domain of HsMiro1, and from a sample of its paralog HsMiro2. Measurements carried out in the presence and absence of 1 mM calcium chloride revealed a negligible effect on the SAXS scattering parameters for HsMiro1/2, consistent with our previous studies showing that changes in calcium occupancy of the EF hands of MiroS altered neither the solution structure nor the crystal structure of MiroS [17]. Significantly, the scattering parameters were very consistent among the three samples (Suppl. Fig. 6). We generated ab initio scattering models from each dataset. The reconstructions revealed an overall crescent-shaped structure which was similar amongst the HsMiro1 and HsMiro2 samples (Fig. 4A), and which was consistent with the rod-like shape of the fragment of HsMiro determined previously [17,23]. The reproducibility of the reconstructions obtained was indicated by DAMMIF normalized spatial discrepancy (NSD) values of 0.62 and 0.63 for HsMiro1 and HsMiro2, respectively (Suppl. Table 2). From this we conclude that each of the reconstructions generated for HsMiro1/2 are both similar to each other and, as expected from our crystal structures, do not exhibit substantial inter-domain disorder.
We generated structural models of the cytoplasmic domain of Miro1/2 using the programs BUNCH [64] and CORAL [79], combining our SAXS data with the previously reported crystal structures of the HsMiro1 EF-hand and cGTPase domains (‘MiroS’; PDB ID: 5KTY) and the nGTPase domain. We included the unmodeled N- and C-termini, and modeled the short linker peptide between the nGTPase and MiroS (170-180) as a flexible restraint between two rigid-body domains. The overall fit of the resulting structural models yielded in the best case χ2 values of 1.137 for HsMiro1 and 1.007 for HsMiro2. In each of the models we found the position of the nGTPase domain to be consistent, located in one lobe of the ‘crescent’ but extended from the remainder of the MiroS (Fig. 4B). While the position of the nGTPase relative to MiroS must be constrained by the relatively short linker between the two, its positions in our models suggests that its relative orientation is less so, and that the nGTPase does not form a stable interface with the rest of the molecule. However, we cannot say this with certainty given the inherent low-resolution of SAXS scattering data. Thus, the deviation of the χ2 values may arise both from the poorly defined polypeptide ‘linker regions’ (at the N- and C-termini, and the nGTPase boundary, and internal to the otherwise rodlike assembly of the HsMiro S domain), as well as from the distribution of orientations available to the nGTPase relative to the rest of the protein. Nevertheless, the consistent observation of a similar overall shape derived from the scattering data obtained from the three samples is striking. We conclude that the nGTPase domain has limited mobility relative to the remainder of the molecule, but, in contrast to the extensive interface of the cGTPase domain the second ELM2 domain [17], the nGTPase domain of HsMiro does not appear to be tightly coupled to its EF-hand domains.
We speculate that such conformational freedom could facilitate interactions with distinct binding partners across multiple interfaces of the nGTPase domain. The sterically accessible orientations of the nGTPase modeled within the scattering envelope suggest that one or both of the conserved surface motifs we identify above might mediate association of the nGTPase with the rest of the intact Miro. However, in the BUNCH models, the SELFYY motif in particular is potentially exposed at the surface of the HsMiro1/2 extra-membranous domain (Fig. 4B). HsMiro1 has been shown to occur in a protein complex with HsMiro2 [29,74,75], and its binding partners Kinesin-1, TRAK1/2, DISC1, Myosin19, OGT, Mfn1/2, p150glued, and PINK1 self-associate in cells [75–78]. We speculate, therefore, that one or both of these putative binding surfaces of the HsMiro1 nGTPase domain could mediate dimerization of Miro and/or functionally interact with its binding partners.
4. Conclusions
Here, we report the structure of the nGTPase of HsMiro1 in complex with GTP. We found no evidence for hydrolysis of GTP and conclude that the nucleotide is tightly bound and that the crystallized N-terminal GTPase construct is catalytically inactive. Extensive interactions with the ligand, including a novel backbone carbonyl coordination of magnesium, appear to stabilize the bound Mg2+GTP. The absence of hydrolysis is unexpected, as recent studies using a similar nGTPase construct have shown GTP hydrolysis activity similar to that of the small GTPase Ras [42]. Additionally, GTP hydrolysis has been reported for both the yeast Miro ortholog Gem1p, and the Drosophila DmMiro [20,38]. Protein crystal structures necessarily image limited structural states, and the intriguing possibility that the snapshot captured in the crystal form reported here, while well-ordered and fully consistent with what is understood of GTPase structure and function, may represent one of a myriad of functional states, perhaps some of which are catalytically active, has yet to be explored. Nevertheless, the structure should provide an improved framework for modeling studies [42] and mutational analysis [32]. Mutational studies have established that disruption of the putative active centers of both the N- and C-terminal Miro GTPases have phenotypic effect [8,25,80], however whether these mutations stabilize conformationally distinct ‘GTP-bound’ and ‘GDP-bound’-like states, as has been claimed [32], or whether they destabilize the GTPase domain structure and/or its functional interactions remains unknown. The structure confirms that the T18N mutation almost certainly disrupts magnesium coordination and hence nucleotide binding, however its effect at this point is impossible to predict. Does the resultant nucleotide-free protein promote interaction with a regulatory guanine nucleotide exchange factor (GEF), as appears to be the case with Ras [81,82], or does it more simply disrupt the structure and hence function of the protein? Indeed, some members of the GTPase family, such as Rnd1 [83] and RhoE/Rnd3 [84] are thought to remain GTP-bound and regulated by expression rather than by GTP/GDP switching [85]. The effect of the P13V mutation at the surface of the nucleotide binding site is also impossible to predict. In Ras, the corresponding mutation disrupts the catalytic interactions of the cognate GTPase activating protein (RasGAP) [86,87] thereby limiting stimulation of GTP hydrolysis and, as a second order effect, stabilizing the ‘active’ GTP-bound state. As yet there is no evidence that a corresponding interaction occurs with Miro. Further studies addressing the regulation of the GTPase activity of the nGTPase domain, whether by intrinsic and/or extrinsic interactions, will be needed to resolve these issues. We suggest Miro “GTPases” may be more accurately described as “GTP-binding proteins”.
The structure of the N-terminal domain of the HsMiro1 also potentially provides a structural model for the yeast homolog Gem1p. However, while the overall sequence identity between human and yeast is 30% over the nGTPase of Miro, there are several interesting features that suggest that structural and functional inferences may be problematic - which may be particularly important given the apparently distinct functional roles played by the homologs in yeast and metazoans. It has previously been noted that Gem1p nGTPase lacks both a rho-like ‘Switch 2’ (G-3) sequence consensus and rho-like helix sequence [21]. In contrast to HsMiro, however, a ~17 residue insertion occurs in Gem1p at the position of the ‘rho helix’ (Fig. 2B), which though divergent in sequence, retains the polar character [66] of the rho-specific insert (9 of 17 residues are Asn/Glu/Asp). This sequence is absent in HsMiro1 and hence we cannot model it. There is limited conservation of the sequence of the G-3 (‘Switch 2’) motif amongst Miro/Gem1p nGTPases (Fig. 2A and see Suppl. Fig. 3), most notably at Ser59 which is directed towards the water structure at the putative active center in HsMiro1. A serine at the corresponding position in the ‘constitutively activated’ (i.e. non-hydrolyzing) GTPase RhoE/Rnd3 has, interestingly, been argued to play a role in limiting its hydrolysis [84]. With respect to the G-2 (‘Switch 1’) motif, however, establishing a relationship between Miro and Gem1p is problematic, as the sequence of the motif is poorly conserved (Suppl. Fig. 3). Interestingly, a ‘PP’ motif is partially conserved within the Gem1p family, but its position relative to elements that can be reliably modeled based on our structure (i.e. which correspond in this region to the hydrophobic beta strands at the core of the nGTPase fold) is shifted four residues C-terminal; consequently it is impossible to model reliably the structure of the G-2 (Switch 1) region in Gem1p, as shown, graphically, in Fig. 2B. Since both the G-2 and G-3 GTPase motifs play a direct role in GTP hydrolysis and exchange [88], differences in their structural arrangements and nucleotide interactions presumably lead to distinct functional and catalytic behaviors. The extensive interactions of the G-2 motif in the HsMiro nGTPase domain with nucleotide may significantly stabilize the bound species, consistent with our observation of co-purification of GTP. If those interactions were absent or different in the yeast homolog, the biochemical behavior of the two proteins would likely be quite distinct.
With respect to the apparent lack of hydrolytic activity, recently, biochemical characterization of HsMiro1/2 domain constructs corresponding to the nGTPase, cGTPase, and MiroS have suggested that both putative GTPase domains exhibit readily measurable GTP nucleotide hydrolysis activity in vitro [42]. The rate constants for hydrolysis for both domains towards GTP are similar to that of the Ras GTPase. Remarkably, however, the activity of the cGTPase was found to be promiscuous, hydrolyzing ATP and CTP in addition to GTP. The authors propose a structural basis for these differences based on modeling studies, which have yet to be validated [42]. It is worth noting that GTPases that occur only in the GTP bound state are known, where certain structural pecularities in the Switch II region cause intrinsic hydrolytic deficiency [96–98]. And in the case of RhoE, this “GTP-bound” state is the only species found in cells. The intriguing possibility that Miro behaves similarly is raised by our structure, but at this point there is insufficient evidence available to make a conclusion. Our crystal structure and structural models could aid in the design of hydrolysis and binding mutants to test the functionality of Miro’s “GTPase cycle” in vivo.
We also report a low resolution envelope for the structure of the intact HsMiro1 and HsMiro2 based on small angle X-ray scattering (SAXS) data, revealing a crescent-shaped structure consistent with positioning of the nGTPase domain at one end of, but not colinear with, the rodlike MiroS (comprising the two EF-hand and cGTPase domains). The short linker peptide between the nGTPase and the rest of the protein (~10 residues) likely constrains its position. However, the low resolution of the scattering envelope does not allow determination of the extent to which the relative positions of the nGTPase and MiroS domains under these solution conditions are fixed or conformationally dynamic. The structure does seem to exclude direct interaction of the nGTPase with the face of the ELM1 domain [17].
Miro has been found in the context of the cell to occur as a protein complex [7], and many of its reported binding partners are thought to be dimeric or multimeric (e.g. Kinesin-1, TRAK1/2, Mfn1/2) [70,77,89,90]. The exact stoichiometry and oligomeric state of Miro in its complexes in vivo is unknown. Here, we locate two conserved and largely hydrophobic surfaces of the nGTPase domain that potentially function as binding interfaces. The first, which we term ‘SELFYY’ is quite extensive and mediates homodimerization in the crystal, suggesting the intriguing possibility that situated at the constrained surface of the mitochondrial membrane it might mediate dimerization of the intact Miro as well. Based on its relationship to the GTPase protein fold, this surface is less likely to be responsive to nucleotide-binding state than the canonical ‘switch’ regions that are located adjacent to the phosphate chain. We also identify a second interface in the crystal that is mediated by conserved residues adjacent to the nGTPase ‘switch 1’ region. However, even if functional as bonafide interaction motifs, their roles are likely regulated by the presence of ligands and/or macromolecular binding partners. Further studies will be needed to determine whether and how each of these interfaces contributes to Miro function.
The paralogs Miro1 and Miro2 are differentially expressed in cells [32] and it is becoming clear that their roles are functionally distinct [15]. Studies of Miro1 in mouse models have shown that Miro2 protein levels are not sufficient to replace Miro1 knockout dysfunction at a subcellular, cellular or organismal level [10,11]. Differential binding localization studies suggest that Miro1 and Miro2 interact differently with the motor adapters TRAK1 and TRAK2 [74]. The nGTPase domains of HsMiro1 and HsMiro2, which are believed to play the key role in the interactions with TRAK1/2 [14], are structurally quite similar, as they exhibit 73% sequence identity and 88% sequence similarity. Consequently, the set of 21 nonhomologous amino acid substitutions between them presumably differentiate the functions of the two proteins. Mapping these differences on the structure of HsMiro1 we find, interestingly, that they locate to one face of the domain, primarily along loops adjacent to the GTP binding site and to the C-terminal end of the switch 2 helix (Fig. 5). Notably, the ‘SELFYY’ surface, opposite, is highly conserved between HsMiro1 and HsMiro2, perhaps consistent with a common function as an interaction interface.
Figure 5. Differences between the nGTPases of HsMiro1 and HsMiro2 are localized to one face of the domain.
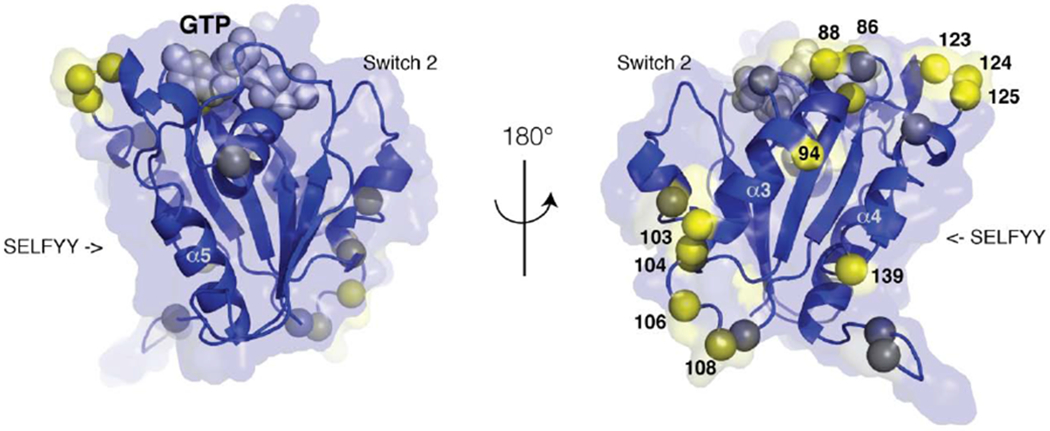
The sequences of the nGTPases of HsMiro1/2 are 73% identical and 88% similar, with no gaps. The location of the 21 non-homologous substitutions are mapped on the structure of HsMiro1, using a color scale from blue (conservative) to yellow (non-conservative substitution). The two orientations related by 180° highlight the sidedness of the distribution of sequence differences. At right, towards the helices α3 and α4 of the GTPase fold, clusters of sequence divergence occur; select amino acid positions are labeled. Both views are perpendicular to the SELFFY interface, which is highly conserved. The positions of the Switch 1 and Switch 2 loops are indicated, and a CPK model of the GTP nucleotide is shown in grey.
Miro presents an interesting puzzle in that it is a calcium-regulated protein that contains two distinct GTPase domains of unknown function. Previous studies of HsMiro GTPase function primarily focused on observation of mitochondrial dynamics phenotypes in cells [8,25,80], and studies of the GTPase domains have been largely framed in terms of canonical models for GTPase structure and function [65,91]. Whether the Miro GTPases even act, like Ras, as ‘on/off switches’, or whether they serve assembly-activated ‘docking’ modules [36,92], remains unknown. By providing a structural model of the complete extramembranous HsMiro polypeptide and identifying surfaces that are likely to be functionally important, our structures should provide a framework for more extensive interrogation of the structure/function relationships of this important mitochondrial protein.
Supplementary Material
Acknowledgements
We thank Samuel Light and the Anderson lab for crystallization supplies and thoughtful feedback. We thank Melissa Gonzalez for assistance, and thank Theint Aung, Chi-Hao Luan, and David Haselbach for assisting with data collection and interpretation. We thank Priscilla Yeung and Jodi Nunnari for thoughtful discussions. K.P.S. and J.L.K. were supported by T32GM008382 and the Walter S. and Lucienne Driskill Graduate Training Program in Life Sciences at Northwestern University. S.E.R. was supported by R01GM107209. This work used resources of the Northwestern University Structural Biology Facility and the Keck Biophysics Facility, supported by NCI-CCSG-P30-CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the Pilatus 3 1M detector was provided by grant 1S10OD018090-01 from NIGMS. Use of the LS-CAT was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (085P1000817). This project was supported by grant 9 P41 GM103622 from the National Institute of General Medical Sciences of the National Institutes of Health. The authors declare no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Additional Information
Accession Codes: Coordinates and structure factors have been deposited at the wwPDB (PDB) under the accession code 6D71.
The authors declare no conflicts of interest.
References
- 1.Schuler M-H, Lewandowska A, Caprio GD, Skillern W, Upadhyayula S, Kirchhausen T, Shaw JM and Cunniff B (2017) Miro1-mediated mitochondrial positioning shapes intracellular energy gradients required for cell migration. Mol Biol Cell (Schuler M-H, Lewandowska A, Caprio GD, Skillern W, Upadhyayula S, Kirchhausen T, Shaw JM, and Cunniff B, eds.) 28, 2159–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sheng Z-H and Cai Q (2012) Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci 13, 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caino MC, Seo JH, Aguinaldo A, Wait E, Bryant KG, Kossenkov AV, Hayden JE, Vaira V, Morotti A, Ferrero S, et al. (2016) A neuronal network of mitochondrial dynamics regulates metastasis. Nat Commun, Nature Publishing Group 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murley A and Nunnari J (2016) The Emerging Network of Mitochondria-Organelle Contacts. Mol Cell 61,648–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Z, Okamoto K-I, Hayashi Y and Sheng M (2004) The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. [DOI] [PubMed] [Google Scholar]
- 6.Burté F, Carelli V, Chinnery PF and Yu-Wai-Man P (2015) Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol 11, 11–24. [DOI] [PubMed] [Google Scholar]
- 7.Glater EE, Megeath LJ, Stowers RS and Schwarz TL (2006) Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol 173, 545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.MacAskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D and Kittler JT (2009) Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61,541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung JY-M, Steen JA and Schwarz TL (2016) Phosphorylation-Induced Motor Shedding Is Required at Mitosis for Proper Distribution and Passive Inheritance of Mitochondria. CellReports 16, 2142–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen TT, Oh SS, Weaver D, Lewandowska A, Maxfield D, Schuler M-H, Smith NK, MacFarlane J, Saunders G, Palmer CA, et al. (2014) Loss of Miro1-directed mitochondrial movement results in a novel murine model for neuron disease. Proceedings of the National Academy of Sciences 111, E3631–E3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.López-Doménech G, Higgs NF, Vaccaro V, Roš H, Arancibia-Carcamo IL, MacAskill AF and Kittler JT (2016) Loss of Dendritic Complexity Precedes Neurodegeneration in a Mouse Model with Disrupted Mitochondrial Distribution in Mature Dendrites. CellReports 17, 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL and Zinsmaier KE (2005) The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47, 379–393. [DOI] [PubMed] [Google Scholar]
- 13.Russo GJ, Louie K, Wellington A, Macleod GT, Hu F, Panchumarthi S and Zinsmaier KE (2009) Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. Journal of Neuroscience 29, 5443–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oeding SJ, Majstrowicz K, Hu X-P, Schwarz V, Freitag A, Honnert U, Nikolaus P and Bähler M (2018) Identification of Miro1 and Miro2 as mitochondrial receptors for myosin XIX. Journal of Cell Science 131. [DOI] [PubMed] [Google Scholar]
- 15.López-Doménech G, Covill Cooke C, Ivankovic D, Halff EF, Sheehan DF, Norkett R, Birsa N and Kittler JT (2018) Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J 37, 321–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bocanegra JL, Fujita BM, Melton NR, Cowan JM, schinski EL, Tamir TY, Major MB and Quintero OA (2019) MYO19 interacts weakly with Miro2 GTPases on the mitochondrial outer membrane. BioRxiv. [Google Scholar]
- 17.Klosowiak JL, Focia PJ, Chakravarthy S, Landahl EC, Freymann DM and Rice SE (2013) Structural coupling of the EF hand and C-terminal GTPase domains in the mitochondrial protein Miro. EMBO Rep 14, 968–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okumoto K, Ono T, Toyama R, Shimomura A, Nagata A and Fujiki Y (2017) New splicing variants of mitochondrial Rho GTPase-1 (Miro1) transport peroxisomes. J Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wennerberg K and Der CJ (2004) Rho-family GTPases: it’s not only Rac and Rho (and I like it). J Cell Sci 117, 1301–1312. [DOI] [PubMed] [Google Scholar]
- 20.Koshiba T, Holman HA, Kubara K, Yasukawa K, Kawabata S-I, Okamoto K, MacFarlane J and Shaw JM (2011) Structure-function analysis of the yeast mitochondrial Rho GTPase, Gem1p: implications for mitochondrial inheritance. J Biol Chem 286, 354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frederick RL, McCaffery JM, Cunningham KW, Okamoto K and Shaw JM (2004) Yeast Miro GTPase, Gem1p, regulates mitochondrial morphology via a novel pathway. J Cell Biol 167, 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fransson A, Ruusala A and Aspenström P (2006) The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem. Biophys. Res. Commun. 344, 500–510. [DOI] [PubMed] [Google Scholar]
- 23.Klosowiak JL, Park S, Smith KP, French ME, Focia PJ, Freymann DM and Rice SE (2016) Structural insights into Parkin substrate lysine targeting from minimal Miro substrates. Sci. Rep. 6, 33019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacAskill AF, Brickley K, Stephenson FA and Kittler JT (2009) GTPase dependent recruitment of Grif-1 by Miro1 regulates mitochondrial trafficking in hippocampal neurons. Mol. Cell. Neurosci. 40, 301–312. [DOI] [PubMed] [Google Scholar]
- 25.Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R and Hajnoczky G (2008) Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proceedings of the National Academy of Sciences 105, 20728–20733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu X and Hajnóczky G (2009) Ca2+-dependent regulation of mitochondrial dynamics by the Miro-Milton complex. Int. J. Biochem. Cell Biol. 41, 1972–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ and Schwarz TL (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang KT, Niescier RF and Min K-T (2011) Mitochondrial matrix Ca2+ as an intrinsic signal regulating mitochondrial motility in axons. Proceedings of the National Academy of Sciences 108, 15456–15461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanfer G, oux TCE, Peterka M, Meier S, Soste M, Melnik A, Reis K, m, P AO., Peter M, Picotti P, et al. (2015) Mitotic redistribution of the mitochondrial network by Miro and Cenp-F. Nat Commun 6, 8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalinski AL, Kar AN, Craver J, Tosolini AP, Sleigh JN, Lee SJ, Hawthorne A, Brito-Vargas P, Miller-Randolph S, Passino R, et al. (2019) Deacetylation of Miro1 by HDAC6 blocks mitochondrial transport and mediates axon growth inhibition. J Cell Biol 218, 1871–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Babic M, Russo GJ, Wellington AJ, Sangston RM, Gonzalez M and Zinsmaier KE (2015) Miro’s N-Terminal GTPase Domain Is Required for Transport of Mitochondria into Axons and Dendrites. Journal of Neuroscience 35, 5754–5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fransson A, Ruusala A and Aspenstrom P (2003) Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem 278, 6495–6502. [DOI] [PubMed] [Google Scholar]
- 33.Wang X and Schwarz TL (2009) The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell 136, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kornmann B, Osman C and Walter P (2011) The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections. Proceedings of the National Academy of Sciences 108, 14151–14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chappie JS, Acharya S, Leonard M, Schmid SL and Dyda F (2010) G domain dimerization controls dynamin’s assembly-stimulated GTPase activity. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Focia PJ, Shepotinovskaya IV, Seidler JA and Freymann DM (2004) Heterodimeric GTPase core of the SRP targeting complex. Science 303, 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan L, Qi Y, Huang X, Yu C, Lan L, Guo X, Rao Z, Hu J and Lou Z (2018) Structural basis for GTP hydrolysis and conformational change of MFN1 in mediating membrane fusion. Nat Struct Mol Biol 25, 233–243. [DOI] [PubMed] [Google Scholar]
- 38.Lee S, Lee K-S, Huh S, Liu S, Lee D-Y, Hong SH, Yu K and Lu B (2016) Polo Kinase Phosphorylates Miro to Control ER-Mitochondria Contact Sites and Mitochondrial Ca. Dev Cell, Elsevier Inc. 37, 174–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS and Walter P (2009) An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lewis SC, Uchiyama LF and Nunnari J (2016) ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 353, aaf5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen TT, Lewandowska A, Choi J-Y, Markgraf DF, Junker M, Bilgin M, Ejsing CS, Voelker DR, Rapoport TA and Shaw JM (2012) Gem1 and ERMES do not directly affect phosphatidylserine transport from ER to mitochondria or mitochondrial inheritance. Traffic 13, 880–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peters DT, Kay L, Eswaran J, Lakey JH and Soundararajan M (2018) Human Miro Proteins Act as NTP Hydrolases through a Novel, Non-Canonical Catalytic Mechanism. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Battye TGG, Kontogiannis L, Johnson O, Powell HR and Leslie AGW (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X and Snoeyink J (2008) Defining and computing optimum RMSD for gapped and weighted multiple-structure alignment. IEEE/ACM Trans Comput Biol Bioinform 5, 525–533. [DOI] [PubMed] [Google Scholar]
- 45.Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung L-W, Read RJ and Adams PD (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D 64, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emsley P, Lohkamp B, Scott WG and Cowtan K (2010) Features and development of Coot. Acta Crystallogr D 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH and Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moriarty NW, Grosse-Kunstleve RW and Adams PD (2009) electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr D 65, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeLano WL (2002) The PyMOL Molecular Graphics System. The PyMOL Molecular Graphics System.
- 50.Zhang Y (2009) I-TASSER: fully automated protein structure prediction in CASP8. Proteins: Structure, Function, and Bioinformatics 77 Suppl 9, 100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang J and Zhang Y (2015) I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res 43, W174–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y and Skolnick J (2004) Scoring function for automated assessment of protein structure template quality. Proteins: Structure, Function, and Bioinformatics 57, 702–710. [DOI] [PubMed] [Google Scholar]
- 53.Yang J, Wang Y and Zhang Y (2016) ResQ: An Approach to Unified Estimation of B-Factor and Residue-Specific Error in Protein Structure Prediction. J Mol Biol, Elsevier Ltd 428, 693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Di Tommaso P, Orobitg M, Guirado F, Cores F, Espinosa T and Notredame C (2010) Cloud-Coffee: implementation of a parallel consistency-based multiple alignment algorithm in the T-Coffee package and its benchmarking on the Amazon Elastic-Cloud. Bioinformatics 26, 1903–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rice P, Longden I and Bleasby A (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16, 276–277. [DOI] [PubMed] [Google Scholar]
- 56.Mathew E, Mirza A and Menhart N (2004) Liquid-chromatography-coupled SAXS for accurate sizing of aggregating proteins. Journal of synchrotron radiation 11, 314–318. [DOI] [PubMed] [Google Scholar]
- 57.Malaby AW, Chakravarthy S, Irving TC, Kathuria SV, Bilsel O and Lambright DG (2015) Methods for analysis of size-exclusion chromatography-small-angle X-ray scattering and reconstruction of protein scattering. J. Appl. Cryst 48, 1102–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ and Svergun DI (2003) sw: a Windows PC-based system for smallangle scattering data analysis. J. Appl. Cryst 36, 1277–1282. [Google Scholar]
- 59.Bussler H, Linder M, Linder D and Reinwald E (1998) Determination of the disulfide bonds within a B domain variant surface glycoprotein from Trypanosoma congolense. J Biol Chem 273, 32582–32586. [DOI] [PubMed] [Google Scholar]
- 60.Svergun DI (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst 25, 495–503. [Google Scholar]
- 61.Franke D and Svergun DI (2009) DAMMIF, a program for rapid ab-initioshape determination in small-angle scattering. J. Appl. Cryst, International Union of Crystallography 42, 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kozin MB and Svergun DI (2001) Automated matching of high-and low-resolution structural models. J. Appl. Cryst, International Union of Crystallography 34, 33–41. [Google Scholar]
- 63.Volkov VV and Svergun DI (2003) Uniqueness of ab initioshape determination in smallangle scattering. J. Appl. Cryst, International Union of Crystallography 36, 860–864. [Google Scholar]
- 64.Petoukhov MV and Svergun DI (2005) Global Rigid Body Modeling of Macromolecular Complexes against Small-Angle Scattering Data. Biophys J 89, 1237–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bourne H, Sanders D and McCormick F (1991) The GTPase superfamily: conserved structure and molecular mechanism. Nature 349, 117–127. [DOI] [PubMed] [Google Scholar]
- 66.Wei Y, Zhang Y, Derewenda U, Liu X, Minor W, Nakamoto RK, Somlyo AV, Somlyo AP and Derewenda ZS (1997) Crystal structure of RhoA-GDP and its functional implications. Nat Struct Biol 4, 699–703. [DOI] [PubMed] [Google Scholar]
- 67.Pellegrini E and Bowler MW (2016, June 29) RhoA GDP with novel switch II conformation.
- 68.Suzuki M, Danilchanka O and Mekalanos JJ (2014) Vibrio cholerae T3SS Effector VopE Modulates Mitochondrial Dynamics and Innate Immune Signaling by Targeting Miro GTPases. Cell Host and Microbe, Elsevier Inc. 16, 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Krissinel E and Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- 70.Shlevkov E, Kramer T, Schapansky J, LaVoie MJ and Schwarz TL (2016) Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proceedings of the National Academy of Sciences 113, E6097–E6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Spencer-Smith R, Koide A, Zhou Y, Eguchi RR, Sha F, Gajwani P, Santana D, Gupta A, Jacobs M, Herrero-Garcia E, et al. (2017) Inhibition of RAS function through targeting an allosteric regulatory site. Nat Chem Biol 13, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Güldenhaupt J, Rudack T, Bachler P, Mann D, Triola G, Waldmann H, Kötting C and Gerwert K (2012) N-Ras forms dimers at POPC membranes. Biophys J 103, 1585–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fetics SK, Guterres H, Kearney BM, Buhrman G, Ma B, Nussinov R and Mattos C (2015) Allosteric Effects of the Oncogenic RasQ61L Mutant on Raf-RBD. Structure, Elsevier Ltd 23, 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van Spronsen M, Mikhaylova M, Lipka J, Schlager MA, van den Heuvel DJ, Kuijpers M, Wulf PS, Keijzer N, Demmers J, Kapitein LC, et al. (2013) TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron 77, 485–502. [DOI] [PubMed] [Google Scholar]
- 75.Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR and Selkoe DJ (2009) Pink1 Forms a Multiprotein Complex with Miro and Milton, Linking Pink1 Function to Mitochondrial Trafficking. Biochemistry, American Chemical Society 48, 2045–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koutsopoulos OS, Laine D, Osellame L, Chudakov DM, Parton RG, Frazier AE and Ryan MT (2010) Human Miltons associate with mitochondria and induce microtubule-dependent remodeling of mitochondrial networks. Biochim Biophys Acta 1803, 564–574. [DOI] [PubMed] [Google Scholar]
- 77.Cao Y-L, Meng S, Chen Y, Feng J-X, Gu D-D, Yu B, Li Y-J, Yang J-Y, Liao S, Chan DC, et al. (2017) MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 542, 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leliveld SR, Hendriks P, Michel M, Sajnani G, Bader V, Trossbach S, Prikulis I, Hartmann R, Jonas E, Willbold D, et al. (2009) Oligomer assembly of the C-terminal DISC1 domain (640-854) is controlled by self-association motifs and disease-associated polymorphism S704C. Biochemistry 48, 7746–7755. [DOI] [PubMed] [Google Scholar]
- 79.Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HDT, Konarev PV and Svergun DI (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Cryst 45, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Murley A, Lackner LL, Osman C, West M, Voeltz GK, Walter P, Nunnari J and Youle RJ (2013) ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. eLife, eLife Sciences Publications Limited 2, e00422. [Google Scholar]
- 81.Feig LA (1999) Tools of the trade: use of dominant-inhibitory mutants of Ras-family GTPases. Nature Cell Biology 1, E25–7. [DOI] [PubMed] [Google Scholar]
- 82.Fiordalisi JJ, Holly SP, Johnson RL, Parise LV and Cox AD (2002) A distinct class of dominant negative Ras mutants: cytosolic GTP-bound Ras effector domain mutants that inhibit Ras signaling and transformation and enhance cell adhesion. J Biol Chem 277, 10813–10823. [DOI] [PubMed] [Google Scholar]
- 83.Foster R, Hu KQ, Lu Y, Nolan KM, Thissen J and Settleman J (1996) Identification of a novel human Rho protein with unusual properties: GTPase deficiency and in vivo farnesylation. Mol Cell Biol 16, 2689–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Garavini H, Riento K, Phelan JP, McAlister MSB, Ridley AJ and Keep NH (2002) Crystal structure of the core domain of RhoE/Rnd3: a constitutively activated small G protein. Biochemistry 41,6303–6310. [DOI] [PubMed] [Google Scholar]
- 85.Nobes CD, Lauritzen I, Mattei MG, Paris S, Hall A and Chardin P (1998) A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J Cell Biol 141, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lu S, Jang H, Nussinov R and Zhang J (2016) The Structural Basis of Oncogenic Mutations G12, G13 and Q61 in Small GTPase K-Ras4B. Sci. Rep. 6, 21949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Khrenova MG, Mironov VA, Grigorenko BL and Nemukhin AV (2014) Modeling the role of G12V and G13V Ras mutations in the Ras-GAP-catalyzed hydrolysis reaction of guanosine triphosphate. Biochemistry 53, 7093–7099. [DOI] [PubMed] [Google Scholar]
- 88.Cherfils J and Zeghouf M (2013) Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiological Reviews 93, 269–309. [DOI] [PubMed] [Google Scholar]
- 89.Lovas JR and Wang X (2013) The meaning of mitochondrial movement to a neuron’s life. Biochim Biophys Acta 1833, 184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Daumke O and Roux A (2017) Mitochondrial Homeostasis: How Do Dimers of Mitofusins Mediate Mitochondrial Fusion? Curr Biol 27, R353–R356. [DOI] [PubMed] [Google Scholar]
- 91.Bourne HR, Sanders DA and McCormick F (1990) The GTPase superfamily: a conserved switch for diverse cell functions. Nature 348, 125–132. [DOI] [PubMed] [Google Scholar]
- 92.Qi Y, Yan L, Yu C, Guo X, Zhou X, Hu X and Huang X (2016) Structures of human mitofusin 1 provide insight into mitochondrial tethering. J Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Robert X and Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42, W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mura C (2004) Development & implementation of a PyMOL “putty” representation. Biomolecules (q-bio.BM) 1–3. [Google Scholar]
- 95.Kanie T and Jackson P (2018) Guanine nucleotide exchange assay using fluorescent MANT-GDP. bio-protocol 8(7): e2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nobes C, Lauritzen I, Mattei M-G, Paris S, Hall A, and Chardin P (1998) A new member of the rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J Cell Biol. 141(1):187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bergbrede T, Pylypenko O, Rak A, Alexandrov K (2005) Structure of the extremely glow GTPase Rab6A in the GTP bound form at 1.8Å resolution. J Struct Biol. 152, 235–238. [DOI] [PubMed] [Google Scholar]
- 98.Gao Z, Xing K, Zhang C, Qi J, Wang L, Gao S, Lai R (2019) Crystal structure and function of Rbj: A constitutively GTP-bound small G protein with an extra DnaJ domain. 10, 760–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.