

Abstract
Multidimensional fractionation-based enrichment methods improve the sensitivity of proteomic analysis for low-abundance proteins. However, a major limitation of conventional multidimensional proteomics is the extensive labor and instrument time required for analyzing many fractions obtained from the first dimension separation. Here, we present a fraction prediction algorithm-assisted two-dimensional LC-based parallel reaction monitoring-mass spectrometry (FRACPRED-2D-PRM) approach for measuring low-abundance proteins in human plasma. Plasma digests were separated by the first dimension high-pH RP-LC with data-dependent acquisition (DDA). We then used the FRACPRED algorithm to predict the retention time of undetectable target peptides according to those of other abundant plasma peptides during the first dimension separation. Fractions predicted to contain target peptides were analyzed by the second dimension low-pH nano RP-LC PRM. We demonstrated the accuracy and robustness of fraction prediction with the FRACPRED algorithm by measuring two low-abundance proteins, aldolase B and carboxylesterase 1, in human plasma. The FRACPRED-2D-PRM proteomics approach demonstrated markedly improved efficiency and sensitivity over conventional 2D-LC proteomics assays. We expect that this approach will be widely used in the study of low-abundance proteins in plasma and other complex biological samples.
Keywords: 2D-LC, fraction prediction algorithm, plasma, low-abundance proteins, PRM
Measuring low-abundance proteins in complex biological samples, such as human plasma, remains a major challenge in the development of mass spectrometry-based proteomics assays.[1] Several sample enrichment methods have been developed to improve the sensitivity of proteomics analysis, including high-abundance protein depletion, immunoaffinity enrichment, and fractionation.[2]
The depletion approach uses immobilized antibodies to remove high-abundance proteins from the matrix, thus increasing the sensitivity of low-abundance protein detection. However, in addition to the associated high cost, these antibodies are often not highly specific, and depletion efficiency varies across different antibody providers, batches, and experiments, introducing poor reproducibility.[3, 4] Notably, detecting low-abundance plasma proteins can still be challenging even after depletion. Immunoaffinity enrichment is commonly used for targeted proteomics analysis, in which specific antibodies capture targeted proteins or peptides in samples. However, this approach suffers from low multiplexing capacity and heavy dependence on high-quality antibodies, which are expensive and not always readily available for the proteins and peptides of interest.[2, 5]
Multidimensional fractionation techniques can increase the sensitivity and dynamic range of proteomic analysis.[2, 6–8] These methods are independent of antibodies, thus eliminating the antibody-associated limitations of the aforementioned enrichment approaches. For the first dimension separation, commonly used techniques include high-pH RP-LC and SCX chromatography. Fractions collected from the first dimension separation are then analyzed by a standard MS-based proteomic method. Conventional multidimensional proteomics is mainly limited by the extensive labor and instrument time required to analyze many first-dimensional fractions. Therefore, being able to predict which fractions contain the peptides of interest would reduce the required analysis, and thus, greatly improve the efficiency of multidimensional fractionation-based proteomics. To this end, Qian and Liu’s groups developed the PRISM-SRM and DD-SRM approaches, which allow precise selection of fractions containing target peptides.[7, 8] In both methods, samples were spiked with high concentrations of stable isotope-labeled internal standard (IS) peptides that can be detected during the first dimension separation, allowing for the tracking of fractions that contain the target peptides. Both PRISM-SRM and DD-SRM employ a low mass resolution triple-quadrupole instrument, and the methods demonstrated ~3–5 orders of magnitude increases in sensitivity relative to conventional proteomics assays. However, some types of stable isotope-labeled IS peptides, such as those generated from SILAC and QconCAT assays, usually contain a small percentage of unlabeled peptides (e.g., ~1%). [9, 10] This small amount of unlabeled peptides has a negligible effect on quantification when the IS is added at a concentration comparable to that of the native peptides in the sample. However, unlabeled peptides could interfere with quantification when IS peptides are added at a concentration much exceeding that of the corresponding native peptides for being detectable during the first dimension separation.
In the current study, we developed a fraction prediction algorithm-assisted two-dimensional liquid chromatography-based parallel reaction monitoring-mass spectrometry (FRACPRED-2D-PRM) method to measure low-abundance proteins in human plasma using a high-resolution MS instrument. Our approach can precisely predict fractions containing target peptides without using an isotope-labeled IS, thus reducing assay cost and complexity. In addition, we employed data-dependent acquisition (DDA) during the first dimension separation, which enables the selection of high-performance surrogate peptides of proteins of interest for the second dimension PRM proteomics analysis.
The FRACPRED-2D-PRM proteomics workflow is illustrated in Figure 1. The protein samples were prepared using the method we previously reported with minor modifications.[11] Briefly, 1 mg plasma proteins or a mixture of 500 μg human liver microsomes (HLM) and 50 μg plasma proteins were first precipitated by acetone. After reduction and alkylation, the proteins were digested overnight by trypsin. Five hundred μg plasma peptides or 275 μg peptides of the mixture sample (250 μg HLM and 25 μg plasma) were injected for first dimension separation on a ZORBAX Extend 300 C18 column (3.5 μm, 4.6 × 150 mm, Agilent Technologies) using high-pH mobile phases at a flow rate of 1 ml/min. A post-column splitter was used to split the flow at a ratio of 1:200 with 5 μl/min being directed to a mass spectrometer for DDA and 995 ul/min for plasma fraction collection at 30 or 15-second intervals. The proteomic analysis was carried out on an AB Sciex TripleTOF 5600+ mass spectrometer coupled with a Digital PicoView 450 nanospray ion source (New Objective, Inc.) and a Shimadzu HPLC system for the first dimension high-pH RP-LC separation, and an Eksigent 2D plus nano LC system for the second dimension low-pH RP-LC separation.
Figure 1:
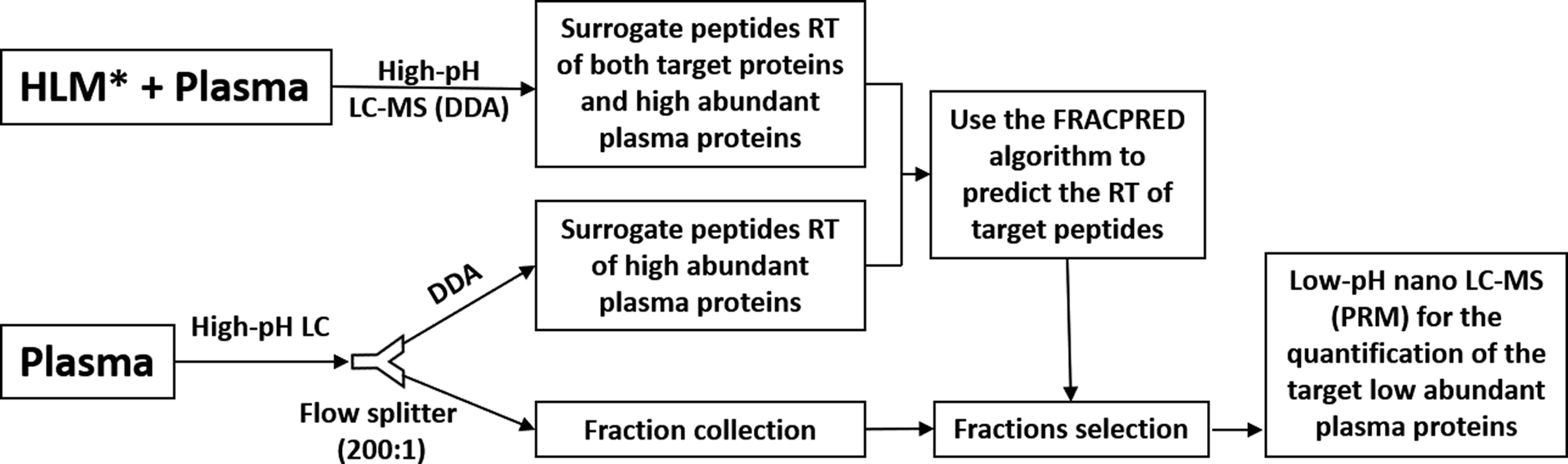
Workflow of the FRACPRED-2D-PRM approach. The digested plasma samples or the mixed plasma and human liver microsomes (HLM) samples are first separated with a high-pH mobile phase on a large diameter C18 column (4.6 mm × 150 mm). A post-column splitter is used to split the flow (1 ml/min) at a 1:200 ratio with 5 μl/min being directed to a mass spectrometer for data-dependent acquisition (DDA) and 995 μl/min for fraction collection at 30 or 15-second intervals. The resulting DDA data are individually searched by the MaxQuant software then imported into the Skyline software to generate a list of peptides and their retention time. The retention time (RT) of target peptides in the plasma samples can be predicted using the FRACPRED algorithm (R package, fracpred), and the corresponding fractions can then be selected for the second dimension low-pH parallel reaction monitoring (PRM) analysis.
*We chose HLM because the targeted proteins are highly expressed in the liver. Other types of tissues could be used depending on the expression levels of the targeted proteins in the tissues.
In this proof-of-concept study, we targeted two low-abundance plasma proteins, aldolase B (ALDOB) and carboxylesterase 1 (CES1). Both proteins are highly expressed in the liver; thus, they can be readily detected by DDA in the mixed HLM and plasma sample. For each protein, we chose two surrogate peptides for the second dimension PRM analysis based on the responses of their unique peptides in the DDA analysis. To predict which fractions contained the targeted ALDOB and CES1 peptides, we developed a fraction prediction algorithm as follows.
Analyze DDA data obtained from plasma and the mixed HLM and plasma samples;
Generate data sets that contain peptide sequences and retention times;
For the mixed sample data set, keep the four targeted ALDOB and CES1 peptides and those peptides also found in the plasma sample; for the plasma data set, keep the peptides shared with the mixed sample;
-
Calculate retention time of the targeted peptides using the following equation:
where M is the retention time of the targeted peptide measured in the mixed HLM and plasma sample, M− is the retention time of the peptide eluted immediately before the targeted peptide, M+ is the retention time of the peptide eluted immediately after the targeted peptide, N− is the retention time of peptide M− in the plasma sample, and N+ is the retention time of peptide M+ in the plasma sample.(Equation 1) The prediction algorithm is based on the simple assumption that the ratios of the retention time intervals between the three peptides (i.e., one target and two adjacent peptides) are the same in different samples.
Choose fractions for the second dimension PRM analysis based on the predicted retention time of the targeted peptides.
The predicted fractions were dried, reconstituted, and injected for the second dimension low-pH nano RP-LC-PRM analysis. The analysis was performed via a trap-elute configuration, which included a trapping column (Acclaim PepMap C18, 100 Å, 5 μm, 10 × 0.1 mm, Thermo Scientific) and a C18 analytical column (150 × 0.1 mm, 1.7 μm ethylene bridged hybrid particles from Waters Corporation, in-house packed). The mobile phases consisted of water containing 0.1% formic acid as phase A and acetonitrile containing 0.1% formic acid as phase B. Peptides were trapped and cleaned on the trapping column with mobile phase A delivered at a flow rate of 10 μl/min for 5 min before being separated on the analytical column with gradient elution at a flow rate of 300 nl/min. The PRM acquisition consisted of one 250 ms TOF-MS scan from 400 to 1250 Da, followed by product ion scans from 100 to 1500 Da for the target peptides/precursors (Table S1).
DDA data generated from plasma and the mixed samples were individually searched against a human proteome reference using the software MaxQuant (version 1.6.12.0, Max Planck Institute of Biochemistry, Germany) with default settings. The search results of DDA data were imported into Skyline (version 20.1.0.76, University of Washington, Seattle, WA) to generate lists of peptide sequences and retention time. The retention time of the targeted peptides in the plasma sample was predicted using the aforementioned FRACPRED algorithm implemented in the R package fracpred (https://github.com/zhuhaojie/fracpred). PRM data from the second dimension low-pH RP-LC-MS analysis were analyzed by Skyline via automatic matching of MS/MS chromatographic peaks against the spectral library generated from the DDA searches. Detailed methods of the sample preparation, LC-MS settings, and data analysis procedures can be found in Supporting Information.
For the 2D LC-MS/MS-based quantitative proteomics studies, an accurate selection of the first dimension fractions containing target peptides is crucial to improve assay sensitivity and reduce instrument time. Distinct from the PRISM-SRM and DD-SRM methods, we utilized the retention times of high-abundance plasma peptides to predict the retention times of low-abundance plasma peptides that could not be detected during the first dimension RP-LC separation. To accurately locate the fractions containing target peptides, the FRACPRED algorithm uses peptides with retention time adjacent to the targeted peptides (Equation 1). We found that the retention time intervals of two adjacent peptides that bracket target peptides were usually within 30 seconds, allowing for precise retention time prediction. For the second dimension analysis, the selected fractions were analyzed with a low-pH nano RP-LC-PRM method to achieve the highest selectivity and sensitivity. We demonstrated the feasibility of the FRACPRED-2D-PRM proteomics approach by measuring two low-abundance proteins, ALDOB and CES1, in undepleted and depleted human plasma samples, respectively.
ALDOB protein, also known as fructose-bisphosphate aldolase B, is responsible for breaking down fructose and is also involved in glucose metabolism. Defects in ALDOB cause hereditary fructose intolerance.[12] ALDOB is primarily expressed in the liver, but is present at a trace level in plasma.[13] As shown in Figure 2, we accurately predicted the retention time of two unique surrogate peptides of ALDOB, LDQGGAPLAGTNK and ETTIQGLDGLSER, and successfully detected those peptides using the FRACPRED-2D-PRM proteomics approach. We also measured the target peptides in the fractions immediately before and after the predicted fractions. In each fraction, the peak areas of the target peptides were in agreement with their predicted retention time (Figure 2A and 2B). Of note, a relatively small amount of target peptides could appear in the fractions adjacent to the predicted fractions when the target peptide peak spans over two fractions. However, the high retention time prediction accuracy of the FRACPRED algorithm makes it possible to estimate to what extent that the target peptides may reside in the adjacent fractions based on the size of the fraction collection window and the predicted retention time and peak width of the target peptides during the first dimension separation. Thus, for a quantitative proteomics study, it might be desirable to combine the predicted fraction with its adjacent fractions for the second dimension analysis when the prediction indicates that the adjacent fractions also contain the peptides of interest.
Figure 2:
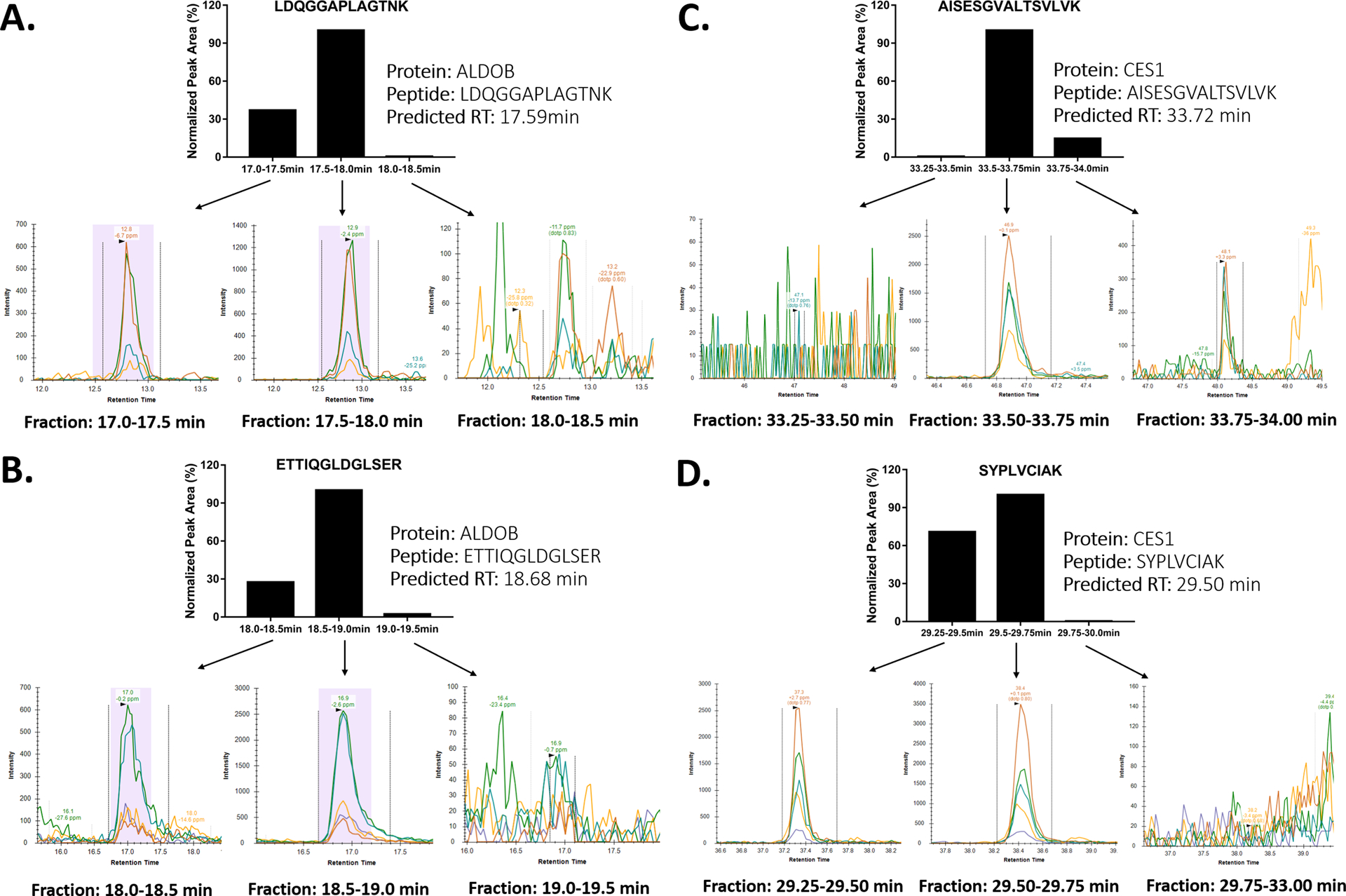
Chromatograms of the ALDOB surrogate peptides LDQGGAPLAGTNK (A), ETTIQGLDGLSER (B), and CES1 surrogate peptides AISESGVALTSVLVK (C), SYPLVCIAK (D) from the fractions predicted by the FRACPRED algorithm and their adjacent fractions from human plasma. The peak areas of the target peptides measured in each fraction were in agreement with the prediction.
CES1 is the most abundant hepatic serine hydrolase and plays important roles in the metabolism of various ester, thioester, amide, and carbamate compounds, including many ester prodrugs such as oseltamivir, dabigatran etexilate, sacubitril, and angiotensin-converting-enzyme inhibitors.[14–18] CES1 was also proposed as a plasma protein biomarker for hepatocellular carcinoma.[19] However, quantifying CES1 in human plasma is difficult due to its low concentration. A previous study used a magnetic bead-based immunoprecipitation method to enrich CES1 protein prior to LC-MS analysis; however, this approach is expensive and heavily depends on high-quality antibodies. [19] With the FRACPRED-2D-PRM approach, we were able to accurately locate the fractions containing the CES1 surrogate peptides AISESGVALTSVLVK and SYPLVCIAK and detected both peptides in the predicted fractions. In addition, the measured peak areas of the target peptides in the predicted fractions and the adjacent ones further support the accuracy of retention time prediction of the FRACPRED algorithm (Figure 2C and 2D).
To further demonstrate the accuracy and robustness of the FRACPRED-2D-PRM approach, we prepared two data sets from the mixed HLM and human plasma samples. From one data set, we removed 272 randomly selected plasma peptides across different retention time, such that this mixed sample could be used as a sham plasma sample for retention time prediction in conjunction with the second data set. As shown in Figure 3, the fitted linear curve between the observed and predicted retention time of these plasma peptides is almost identical to the line of identity with R2 equal to 1 (Figure 3A). Notably, most of the prediction bias was within 15 seconds, and none exceeded 30 seconds (Figure 3B), indicating the excellent accuracy and robustness of this retention time prediction algorithm. The FRACPRED algorithm demonstrated superior retention time prediction accuracy over other approaches that do not require stable isotope-labeled IS. For example, the iRT (indexed retention time) approach developed by Bruderer et al. reported a median delta iRT value of 0.53 min (0.27% of the gradient) for a large set of peptides.[20] In comparison, the median delta retention time was 0.040 min (0.033% of the gradient) for the randomly selected 272 plasma peptides in our study. Moreover, Ma et al. established a deep learning method (DeepRT) for peptide retention time prediction.[21] The reported Δt95% (the minimal time window containing the deviations between observed and predicted retention times for 95% of the peptides) ranged from 1.42 min (2.8% of the gradient) to 25.88 min (10.8% of the gradient) for the eight tested datasets, whereas our FRACPRED method exhibited a Δt95% value of 0.13 min (0.11% of the gradient) for the 272 plasma peptides. Given that the fraction collection intervals were set to be 0.25 min or 0.5 min in our study, the two aforementioned methods may not provide adequate accuracy to predict the fractions containing the target peptides.
Figure 3:
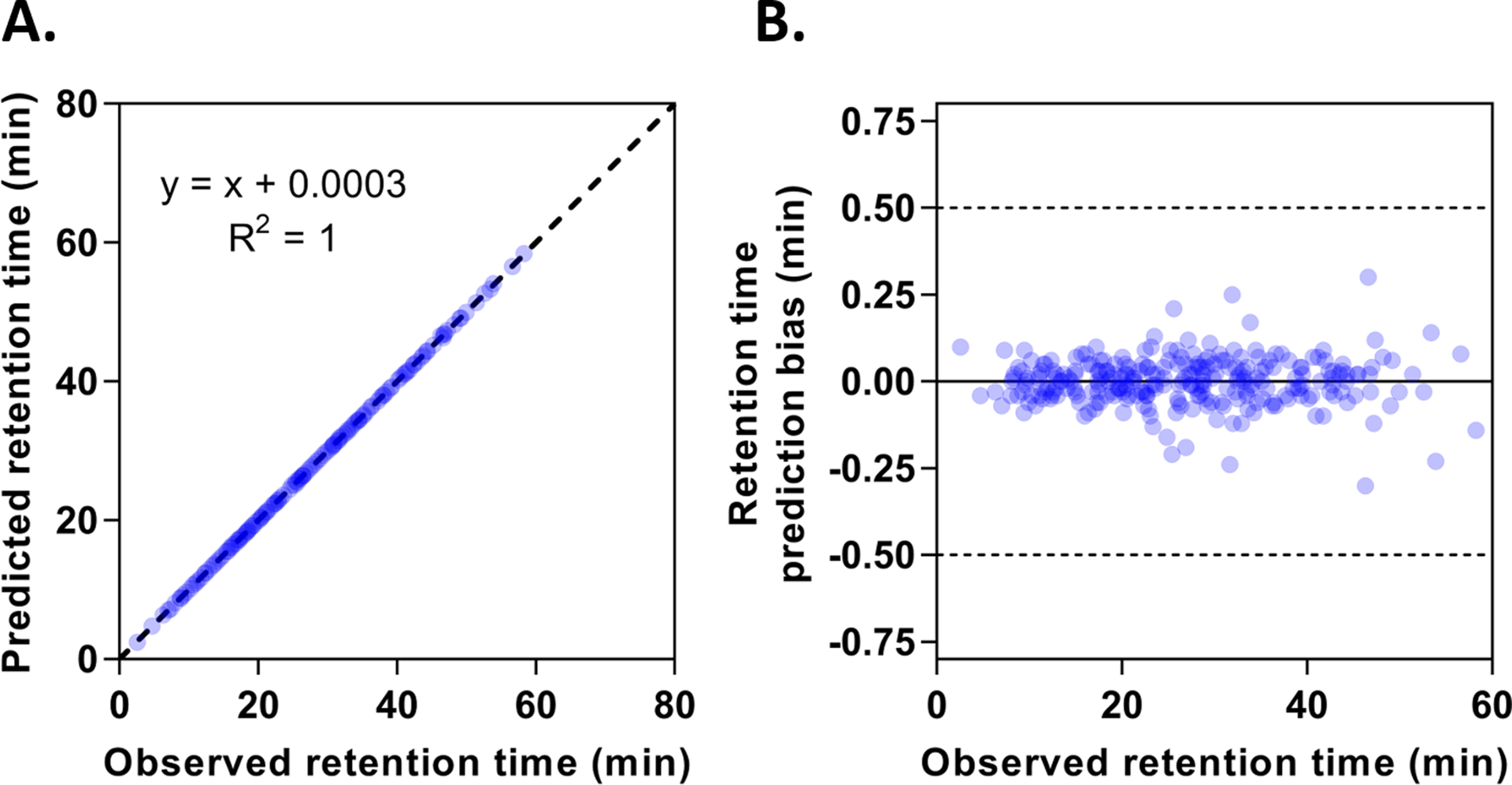
Retention time prediction of target peptides by the FRACPRED algorithm. (A) shows the correlation between the observed and predicted retention time of randomly selected 272 human plasma peptides. The dotted line represents the line of identity. (B) shows the retention time prediction bias for the 272 peptides across different retention time.
In summary, we report here an antibody-free, two-dimensional fractionation-based proteomics approach, FRACPRED-2D-PRM, for the measurement of low-abundance proteins in human plasma. This approach allows for the accurate prediction of fractions containing peptides of interest without requiring isotope-labeled IS, thus markedly reducing the cost and instrument time needed for the second dimension analysis. Furthermore, DDA analysis during the first dimension separation facilitates the selection of high-performance surrogate peptides of proteins of interest for use in the second dimension PRM analysis. This approach holds great potential to be widely applied to the study of low-abundance proteins in plasma and other complex biological samples.
Supplementary Material
Acknowledgements
This work was partially supported by the University of Michigan MCubed program, the National Institutes of Health National Heart, Lung, and Blood Institute [Grant R01HL126969, Hao-Jie Zhu], the Eunice Kennedy Shriver National Institute of Child Health and Human Development [Grant R01HD093612, John S. Markowitz and Hao-Jie Zhu], and the National Science Foundation [NSF 1904146, Robert Kennedy].
Abbreviations:
- ALDOB
aldolase B
- CES1
carboxylesterase 1
- FRACPRED-2D-PRM
A fraction prediction algorithm-assisted two-dimensional liquid chromatography-based parallel reaction monitoring-mass spectrometry
- DDA
data-dependent acquisition
- HLM
human liver microsomes
- IS
internal standard
Footnotes
Conflict of Interest
The authors have declared no conflict of interest.
References
- [1].Geyer PE, Holdt LM, Teupser D, Mann M, Molecular systems biology, 2017, 13, 942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Shi T, Su D, Liu T, Tang K, Camp DG 2nd, Qian WJ, Smith RD, Proteomics, 2012, 12, 1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bellei E, Bergamini S, Monari E, Fantoni LI, Cuoghi A, Ozben T, Tomasi A, Amino Acids, 2011, 40, 145. [DOI] [PubMed] [Google Scholar]
- [4].Tu C, Rudnick PA, Martinez MY, Cheek KL, Stein SE, Slebos RJ, Liebler DC, J Proteome Res, 2010, 9, 4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Acharya P, F1000Res, 2017, 6, 851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dowell JA, Frost DC, Zhang J, Li L, Analytical chemistry, 2008, 80, 6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shi T, Fillmore TL, Sun X, Zhao R, Schepmoes AA, Hossain M, Xie F, Wu S, Kim JS, Jones N, Moore RJ, Pasa-Tolic L, Kagan J, Rodland KD, Liu T, Tang K, Camp DG 2nd, Smith RD, Qian WJ, Proceedings of the National Academy of Sciences of the United States of America, 2012, 109, 15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nie S, Shi T, Fillmore TL, Schepmoes AA, Brewer H, Gao Y, Song E, Wang H, Rodland KD, Qian WJ, Smith RD, Liu T, Analytical chemistry, 2017, 89, 9139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chen X, Wei S, Ji Y, Guo X, Yang F, Proteomics, 2015, 15, 3175. [DOI] [PubMed] [Google Scholar]
- [10].Pratt JM, Simpson DM, Doherty MK, Rivers J, Gaskell SJ, Beynon RJ, Nature protocols, 2006, 1, 1029. [DOI] [PubMed] [Google Scholar]
- [11].Shi J, Wang X, Lyu L, Jiang H, Zhu HJ, Drug metabolism and pharmacokinetics, 2018, 33, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Santer R, Rischewski J, von Weihe M, Niederhaus M, Schneppenheim S, Baerlocher K, Kohlschutter A, Muntau A, Posselt HG, Steinmann B, Schneppenheim R, Human mutation, 2005, 25, 594. [DOI] [PubMed] [Google Scholar]
- [13].Geyer PE, Kulak NA, Pichler G, Holdt LM, Teupser D, Mann M, Cell systems, 2016, 2, 185. [DOI] [PubMed] [Google Scholar]
- [14].Wang X, Liang Y, Liu L, Shi J, Zhu HJ, Rapid Commun Mass Spectrom, 2016, 30, 553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shi J, Wang X, Nguyen JH, Bleske BE, Liang Y, Liu L, Zhu HJ, Biochem Pharmacol, 2016, 119, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shi J, Wang X, Nguyen J, Wu AH, Bleske BE, Zhu HJ, Drug Metab Dispos, 2016, 44, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang X, Wang G, Shi J, Aa J, Comas R, Liang Y, Zhu HJ, The pharmacogenomics journal, 2016, 16, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shi J, Wang X, Eyler RF, Liang Y, Liu L, Mueller BA, Zhu HJ, Basic Clin Pharmacol Toxicol, 2016, 119, 555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Na K, Lee EY, Lee HJ, Kim KY, Lee H, Jeong SK, Jeong AS, Cho SY, Kim SA, Song SY, Kim KS, Cho SW, Kim H, Paik YK, Proteomics, 2009, 9, 3989. [DOI] [PubMed] [Google Scholar]
- [20].Bruderer R, Bernhardt OM, Gandhi T, Reiter L, Proteomics, 2016, 16, 2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ma C, Ren Y, Yang J, Ren Z, Yang H, Liu S, Analytical chemistry, 2018, 90, 10881. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.