

Abstract
Triple-negative breast cancer (TNBC) is an aggressive type of cancer characterized by higher metastatic and reoccurrence rates, where approximately one-third of TNBC patients suffer from the metastasis in the brain. At the same time, TNBC shows good responses to chemotherapy, a feature that fuels the search for novel compounds with therapeutic potential in this area. Recently, we have identified novel urea-based compounds with cytotoxicity against selected cell lines and with the ability to cross the blood-brain barrier in vivo. We have synthesized and analyzed a library of more than 40 compounds to elucidate the key features responsible for the observed activity. We have also identified FGFR1 as a molecular target that is affected by the presence of these compounds, confirming our data using in silico model. Overall, we envision that these compounds can be further developed for the potential treatment of metastatic breast cancer.
Graphical Abstract
INTRODUCTION
Breast cancer is the second leading cause of cancer-related death in women. Despite the tremendous advances in cancer treatment in recent decades, over 42,000 women were expected to die of breast cancer in 2020.1 Triple-negative breast cancer (TNBC) is the subset of breast cancer that is characterized by the absence of estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2).2 It accounts for approximately 15–20% of all breast cancer diagnoses,3 and has been categorized as the most aggressive subtype, correlated with poor prognosis.4 Triple-negative tumors possess a higher risk of developing distant metastases, recurrence, and lower five-year survival compared with other subtypes of breast cancer.5, 6 TNBC has a unique propensity for metastasis to the lung and brain that contribute to its lower survival rates.7
In the absence of viable targets for the endocrine and HER-2 targeted therapies, chemotherapy is the mainstream treatment for TNBC.2 Several retrospective studies have concluded that chemotherapy, in combination with whole-brain radiation therapy, gives the most prolonged median survival. At the same time, CALGB trial8 and WSG AM-01 study9 emphasized the benefits of dose-dense and dose-intensive chemotherapeutic regimens in the treatment of TNBC. A retrospective analysis of trastuzumab treatment has identified that this otherwise potent anticancer agent has increased brain metastasis incidents at a rate of 12.6% to 34%.10 The reason for such an increase is seen as “driving” cancer cells to the brain as a sanctuary site, suggesting the treatment of TNBC would be more successful with the use of chemotherapeutic agents with the ability to cross the blood-brain-barrier (BBB). Most available chemotherapies are not able to cross the BBB, not even if the barrier is disrupted by tumor invasion.11, 12 As a result, there is an ongoing drive to develop novel chemotherapeutic agents that are BBB-permeable for the treatment of TNBC patients.
We have targeted the optimization of penfluridol (PFL), an antipsychotic drug, for potential anticancer use. This compound demonstrates antitumor properties in various cancer cell lines such as breast, pancreatic, glioblastoma, and lung cancer cells.13–16 The anticancer effects of PFL were further confirmed in vivo in a variety of models, including the metastatic heart to brain TNBC model.17 Therefore, penfluridol has the potential to be repurposed as a novel antitumor agent.18, 19 Moreover, the ability of PFL to cross the BBB, makes it an attractive candidate to develop anticancer therapeutics to treat brain metastasized cancers. However, this compound has extensive interactions with the majority of G-protein coupled receptors (GPCRs) (Fig. 2A)20 at levels that correspond to the proposed anticancer dosing (estimated as 50 mg of daily dosing in human14, 16, 17, 21). Thus, an off-target CNS activity of penfluridol as an anticancer agent will lead to intensified neurological side effects associated with this agent.22–25
Figure 2.

Inhibitory activity of penfluridol (A) and compound 4a (B) at selected CNS receptors. Primary binding assay was performed at 10 μM concentration and data are expressed as the mean ± SEM of four independent experiments. The red dotted line identifies groups of the receptors being inhibited by >90% and by >50% in the presence of these compounds.
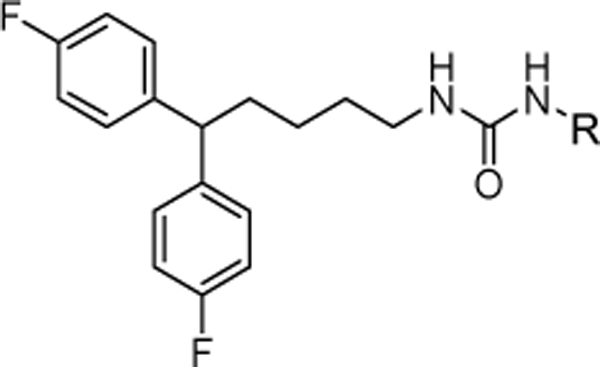
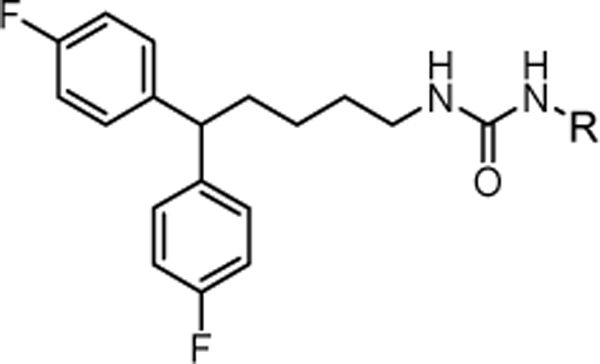
Our group has previously reported the employment of PFL as the hit molecule that can be further optimized for its anticancer properties and the removal of GPCR-related activity.20 We observed that the 4-hydroxypiperidine moiety that was previously shown to be important for its cytotoxicity26, 27 is also essential for the interaction with GPCRs. Here, we report our next step in the optimization of the PFL structure, where a topology-based scaffold hopping approach28–31 was used to identify the second generation of analogs with the retained anticancer activity and alleviated affinity to the GPCRs. To achieve this goal, a set of six diverse chemical fragments was designed to replace the substituted 4-hydroxy-piperidine moiety of PFL (hereafter referred to as the “head” moiety). Selected structural features of the original “head” moiety, such as the presence of a phenyl ring and abundance of heteroatoms, were incorporated in the design of new fragments. The diphenyl-butyl chain (hereafter referred to as the “tail” moiety) was kept unchanged due to its auxophoric character.20, 26 All six molecules were evaluated for their anticancer properties using cytotoxicity assay (see supplemental materials) (Fig. 1), where compound 4a (IC50 = 7.7 μM) was identified as our new “hit” molecule for further evaluation.
Figure 1.

Search for a new hit molecule using scaffold hopping approach. IC50 indicates compound concentration required to inhibit viability of the MDA-MB-231 triple-negative breast cancer cells by 50%. Data are expressed as the mean of three independent experiments, each performed in sextet.
In the next step, compound 4a was assessed for the ability to interact with the GPCRs expressed in the brain (Fig. 2B). We found a significant reduction in the interaction of 4a with the GPCRs, where only six receptors were inhibited at the level above 50%. Under the same test conditions, PFL was shown to inhibit 28 GPCRs at the level above 50%. Previously, it has been demonstrated that neurological side effects of PFL are associated with the inhibition of serotonin and dopamine receptors. 24, 32, 33 Our hit compound (4a) had no significant interaction with the dopamine receptors (subtypes D1-D5) at 10 μM concentration. Similarly, it has no activity at serotonin receptors, except for 5HT-2C, which is a subtype with low expression levels, and its potential role in CNS-induced toxicity can be considered insignificant.34 Thus, for compound 4a, only inhibition of the dopamine transporter (DAT) and histamine 1 receptor (H1R) was noted as sources of possible neurotoxicity.
Further, we evaluated compound 4a for its ability to cross the BBB in vivo, where mice were injected with a dose of 10 mg/kg through intraperitoneal (i.p.) administration35 and sacrificed at selected time points. We observed that 4a reaches the peak values of approximately 1000 nM at 1-hour post-injection (Fig. 3), similarly to the data found for PFL, confirming its retained activity to cross the BBB.36 In summary, our scaffold-hopping design identified compound 4a with cytotoxicity against MDA-MB-231 cells retained the ability to cross the BBB in vivo and limited inhibitory activity on GPCRs. Hence, we envisioned that this compound can be further evaluated as an anticancer agent with the potential therapeutic application in the treatment of the metastatic TNBC. Our current report presents an analysis of the performed structure-activity relationship studies, where a library of 45 analogs was prepared and tested for the anticancer activity. In addition, we investigated potential unwanted overlap in structural requirements for the cytotoxic activity of 4a and its ability to interact with the DAT.
Figure 3.

Distribution of 4a in plasma and brain. Concentration (nM) of compound 4a in plasma and brain at 0, 0.5, 1, 6, 24, and 48 hours after single i.p. administration (10 mg/kg) was measured by liquid chromatography-tandem mass spectrometry. Data are expressed as the mean ± SEM of 2–4 independent experiments.
CHEMISTRY
The synthesis of compounds designed for the scaffold-hopping study is depicted in scheme 1. We have coupled commercially available 4-fluorobenzaldehyde (30), and 4-chlorophenol (31) afforded aryl ether 32 (Scheme 1A) which was subsequently reduced in the presence of lithium aluminum hydride to yield intermediate alcohol 33 and was further coupled with diphenyl alkyl intermediate 27a (scheme 2) to access compound 2.37, 38 Commercially available phenol 34 was protected by benzylation (35), followed by base-mediated hydrolysis of the ester functionality39 to obtain carboxylic acid intermediate 36 (Scheme 1B). A Steglich esterification successfully afforded compound 3.40 Chloro-phenyl isocyanate 48a was coupled with diphenyl alkyl intermediate 27a to afford urea compound 4a (Scheme 1C). A condensation reaction between glycerol (37) and benzaldehyde (38) under acidic conditions afforded 2-phenyl,1,3-dioxane-2-ol 39 (Scheme 1D),41 that was coupled with 27a to form compound 5.42 Synthesis of analog 6 (Scheme 1E) began with the Buchwald-Hartwig43 coupling of aryl iodide 41 using 1,1′-bis(diphenylphosphino)ferrocene (DPPF) as a catalyst to afford diphenylamine intermediate 42.44 Next, coupling with 27a afforded compound 6. Finally, key intermediate 45 was obtained by sulfonylation of piperazine (44) with substituted arylsulfonyl chloride 43,45 and was further coupled with the tail moiety 27a to form compound 7 (Scheme 1F).26
Scheme 1.

Synthesis of compounds selected for a scaffold-hopping study. Reagents and conditions: (a) Cs2CO3, DMF, 85 °C; (b) LiAlH4, THF, rt; (c) 27a, NaH, DMF, 80 °C; (d) Benzyl bromide, K2CO3, DMF, rt; (e) KOH, ethanol, reflux; (f) 25b, DCC, DMAP, DCM, rt to 32 °C; (g) 29a, DCM, 0 °C to 32 °C; (h) conc. H2SO4, 85 °C; (i) 27a, NaH, DMF, rt; (j) dppf (cat.), PdCl2(dppf)• CH2Cl2, KOtBu, anhydrous THF, 100 °C; (k) 27a, NaH, DMF, rt; (l) DIPEA, DCM, rt; (m) 27a, Na2CO3, KI (cat.), acetonitrile, reflux.
Scheme 2.

Synthesis of diphenyl alkyl chain intermediates. Reagents and conditions: (a) Mg, I2, THF, reflux; (b) EtOH, HClconc, reflux; (c) CBr4, Ph3P, DCM, 0 °C to rt; (d) Pd/C (cat.), H2 (80 psi), EtOH, rt; (e) NaN3, acetone: water (2.2:1), rt; (f) Ph3P, anhydrous ether, water; (g) NH4OH, dioxane, 100 °C; (h) conc. H2SO4, conc. HNO3, −15 °C.
The preparation of diphenyl alkyl intermediates (“tail” moieties) that were used to synthesize the final compounds depicted in schemes 1 and 3–6 is outlined in scheme 2. Suitably substituted aryl bromides (21a-e) were converted to the corresponding Grignard reagents, which underwent a double addition reaction with the respective lactones 22a-c to afford dihydroxyl intermediates 23a-g. Acid-catalyzed dehydration of the tertiary alcohol afforded unsaturated aliphatic alcohols 24a-g. Intermediates 26a-g were obtained by subjecting alcohols 24a-g to Appel conditions.46 A palladium-carbon mediated hydrogenation reaction was utilized to obtain saturated intermediates 25a-c, 27a-e, and 27g-h.26 Compound 27f was prepared from 27e using modified nitration conditions to avoid unwanted ortho-substitutions.47, 48 Aliphatic bromides 27a-h were converted to primary amines 29a-h by treatment with ammonia or by Staudinger reduction via the azide intermediates 28a, d-f.49, 50
Scheme 3.

Synthesis of analogues with substitutions at monophenyl “head” moiety. Reagents and conditions: (a) triphosgene, DCM, rt; (b) 29a, DCM, 0 °C to 32 °C; (c) H2N-NH2, Raney-nickel, ethanol, 50 °C; (d) acetic anhydride, DCM. (e) SOCl2, MeOH, reflux; (f) benzyl bromide, K2CO3, DMF, 100 °C; (g) triphosgene, DCM, r.t; (h) 29a, DCM, 0 °C to 32 °C; (i) Pd/C, NaBH4, MeOH.
Scheme 6.

Synthesis of analogs containing methylated urea functionality and analogs with the amide linker. Reagents and conditions: (a) Boc anhydride, TEA, DCM; (b) NaH, CH3I, DMF; (c) TFA, DCM; (d) 48j, DCM. 0 °C to 32 °C; (e) NaH, CH3I, DMF; (f) NaHCO3, isocyanuric chloride, TEMPO (cat.), NaBr (cat.), water: acetone (1:3.5), 0 °C to 25 °C; (g) i) oxalyl chloride, DMF, ii) 47j, TEA, DCM; (h) oxalyl chloride, DMF (cat.), DCM, rt, 1 hr; (i) 29a, TEA, DCM, rt, 12 hrs.
Synthesis of a subgroup of analogs with substitutions at the “head” moiety is depicted in Scheme 3. Diphenyl-alkyl amine 29a (scheme 2) was coupled with freshly prepared isocyanates51 48a-q, to obtain desired urea compounds 4a-d, 4g-m, and 4p-u. Amino substituted derivatives 4e, and 4n were prepared using standard Raney-Ni-catalyzed reduction conditions from the respective nitro-precursors 4d and 4m. They were further acetylated to afford analogs 4f and 4o. Prior to being converted to isocyanates 51a,b via exposure to triphosgene,52 benzoic acid 49 was protected as the methyl,53 or benzyl,54 ester (Scheme 3B). Coupling of 51a,b with 29a afforded analogs 4v-w.55 The final compound in this scheme, acid 4x, was obtained by reductive deprotection of the benzyl ester of 4w.
Analogs with varied substitution at the diphenyl (“tail”) moiety were synthesized according to Scheme 4. The diphenyl-alkyl amine intermediates 29b-f were coupled with the freshly prepared selected isocyanates 48a and 48j-k to produce desired urea compounds 8a-g. Raney-Ni catalyzed reduction was utilized to make compound 8h, which was converted to an acetylated analog 8i. To prepare N-linked diphenyl analogs, 9a-e diphenyl amine 42 was coupled with butyl bromide to obtain 52, which was further converted to the primary alkylamine 53. Final compounds 9a-e were synthesized using 53 and freshly prepared isocyanates 48a, 48b, 48j, 48n, and 48q (Scheme 4B). Synthesis of the last analog (Scheme 4C), compound 10, began with the reduction of 4-fluorophenyl valeric acid (54) with LiAlH4 using a modified literature procedure.56 The resultant primary alcohol 55 was converted to the desired analog 10 in three subsequent steps involving Appel reaction, amination, and urea formation.
Scheme 4.

Synthesis of analogs with substitution at diphenyl (“tail”) moiety. Reagents and conditions: (a) 48a, 48j-k, DCM, 0 °C to 32 °C; (b) H2N-NH2, Raney nickel, ethanol, 50 °C; (c) acetic anhydride, DCM; (d) NaH, 1, 4-dibromobutane, THF, rt; (e) NH4OH, dioxane, 100 °C; (f) 48a, 48b, 48j, 48n, 48q, DCM, 0 °C to 32 °C; (g) LiAlH4, 1N HCl, THF; (h) CBr4, Ph3P, DCM, 0 °C to rt; (i) 48j, DCM, 0 °C to 32 °C.
Several analogs bearing modifications of the linker chain were prepared using conditions outlined in Scheme 5. Alterations in the length of the linker were achieved by reacting intermediates 29g or 29h with isocyanide 48j. The similar synthetic procedure was employed for the preparation of the thiourea analog 12 (Scheme 5B), where thioisocyanate 57 was used as starting material and for the introduction of the carbamate bioisostere 13 (Scheme 5C), where primary alcohol 25a reacted with isocyanate 48j under basic condition.57
Scheme 5.

Synthesis of analogs with structural modifications at the linker motif. Reagents and conditions: (a) 48j, DCM, 0 °C to 32 °C; (b) DCM, 0 °C to 32 °C; (c) TEA, CCl4, rt.
Scheme 6 depicts reaction conditions for the preparation of mono- and dimethyl substituted urea analogs 14 (Scheme 6A) and 15 (Scheme 6B).58,57 In addition, it describes final modifications of the linker performed in this study, such as the synthesis of N’-phenyl and C’-phenyl amides 16 (Scheme 6C) and 17 (Scheme 6D). To prepare 16, primary alcohol 25c was oxidized under metal-free conditions,59 to afford acid 59, which was subsequently activated as the acid chloride and coupled with substituted aniline 47j. 60 Analog 17, was accessed by the acid chloride of free acid 6060 reacting with diphenyl alkyl amine 29a.
To evaluate the effect of a constrained linker on the anticancer activity of compounds, we prepared analogs 18 and 19a-b following reported literature procedures.52, 61, 62 As depicted in scheme 7, Grignard reaction of substituted Boc-protected piperidine 61a-b with 4-fluorophenylmagnesium bromide afforded tertiary alcohol 62. This compound underwent simultaneous dehydration and Boc-deprotection in the presence of HClconc to obtain unsaturated intermediates 63a-b. Palladium assisted hydrogenation conditions were used to convert 63b to 4-substituted piperidine intermediate 64b. Final compounds 18 and 19b were synthesized by reacting an isocyanate with the respective piperidine intermediates 64a and 64b, whereas the preparation of 19a required treatment of 62a with trifluoroacetic acid and NaBH4 to prepare piperidine 64a prior to the coupling reaction.
Scheme 7.

Synthesis analogues with cyclic alkyl chain. Reagents and conditions: (a) THF, reflux; (b) HClconc, EtOH, reflux; (c) NaBH4, TFA, DCM, 0 °C to rt; (d) H2 (80 psi), Pd/C (cat.), EtOH; (e) 48j, DCM, rt
RESULTS AND DISCUSSION
In vitro cytotoxic activity of synthesized analogs
In our SAR studies, we have used a fragment-based approach to design and synthesize a series of 49 analogs of the hit compound 4a that were further evaluated for cytotoxic potential using the MDA-MB-231 triple-negative breast cancer line (see supplemental materials). The structural modifications were performed in three areas of the hit compound: monophenyl (“head”) moiety, urea linker, and diphenyl (“tail”) moiety (Fig. 4). Thus, we have grouped our in vitro data according to the site of the performed modification.
Figure 4.

Areas of the proposed structural modifications.
Modifications in the mono-phenyl “head” moiety and their effect on the cytotoxicity of analogs.
As shown in table 1, a mono-substitution pattern favors the presence of an electron-withdrawing group located meta- to the urea moiety (compound 4g, 4i-4k) or strong electron-withdrawing functionality at the para-position (4h). Electron-donating groups in both para- (4c, 4e) and meta- (4k) positions cause abrogation of cytotoxic activity. Interestingly, the presence of a hydrogen-bond acceptor is well tolerated in the meta -position (4g vs. 4j), but causes increase in IC50 values in compounds bearing mono para-substitution (4d vs. 4h). We observed that increase in the size of the para-substituents leads to decreased (4b) or complete loss (4f) of cytotoxicity. Another factor that might lead to the observed changes is the loss in the electron-withdrawing properties of substituents when moving from 4d to 4a to 4b, and, finally, to 4f.
Table 1.
In vitro cytotoxic activity of analogs with the mono-substitution at the “head” moiety.
 | |||||
|---|---|---|---|---|---|
| R | MDA-MB-231 aIC50 (μM) | R | MDA-MB-231 aIC50 (μM) | ||
| 4a | 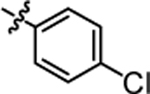 |
7.68 ± 0.21 | 4g | 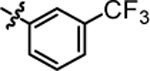 |
3.38 ± 0.08 |
| 4b | 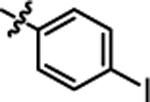 |
11.79 ± 0.40 | 4h | 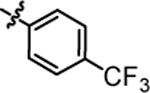 |
2.78 ± 0.09 |
| 4c | 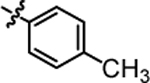 |
>20.00 | 4i | 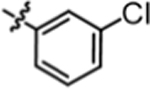 |
5.07 ± 0.33 |
| 4d | 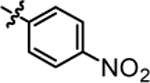 |
6.47 ± 0.84 | 4j | 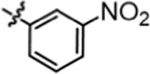 |
3.79 ± 0.22 |
| 4e | 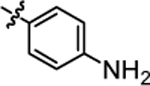 |
>20.0 | 4k | 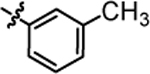 |
>20.0 |
| 4f | >20.0 | ||||
Data are expressed as the mean ± SEM of three independent experiments, each performed in a quartet.
Given that electron-withdrawing groups at para and meta positions were favored, we next prepared a series of di-substituted analogs that possess similar electronic characteristics. As shown in Table 2, a combination of 4g and 4a yielded compound 4l with an activity slightly better than 4g but with a two-fold increase in activity when compared to our hit molecule 4a. Substitution of the trifluoromethyl moiety (4l) with the hydrogen-bond acceptor nitro functionality (4m) led to a slight decrease in overall activity, which was restored in the positional isomer 4l. This data suggests that the meta position prefers non-hydrogen bonding functional groups, a conclusion that was further confirmed by the observed change in the activity of 4o. Although the para position tolerates the presence of H-bond acceptor groups (4l, 4q, 4v), the introduction of a carboxylic acid, an ionizable H-bond donor moiety (4x), led to the loss of cytotoxic activity. We also observed a possible inverse correlation between the size of the meta-substituent and the activity of compounds (4l vs. 4q), suggesting potential size limitations in this part of the binding pocket. Di-substitution with electron-donating groups (4r) abolished the desired activity, whereas a combination of the meta-amino moiety and para-chloro group regained some activity. Finally, we have explored the possibility of ortho-, para- di-substitution pattern, but all of the compounds in this subgroup (4s-4u) were inactive, irrelevant to the electronic properties of substituents. Overall, we have concluded that di-substitution provides compounds with increased cytotoxicity if the meta-, para- pattern is preserved.
Table 2.
In vitro cytotoxic activity of analogs with the di-substitution at the “head” moiety.
 | |||||
|---|---|---|---|---|---|
| R | MDA-MB-231 aIC50 (μM) | R | MDA-MB-231 aIC50 (μM) | ||
| 4l | 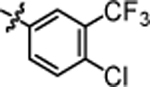 |
3.15 ± 0.11 | 4r |  |
>20.0 |
| 4m |  |
4.06 ± 0.74 | 4s | 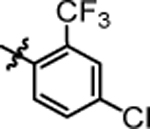 |
>20.0 |
| 4n | 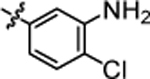 |
9.24 ± 0.29 | 4t | 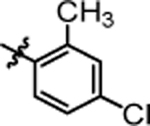 |
>20.0 |
| 4o | 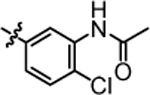 |
9.12 ± 0.43 | 4u | 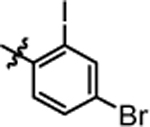 |
>20.0 |
| 4p | 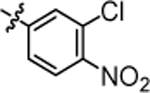 |
3.47 ± 0.14 | 4v | 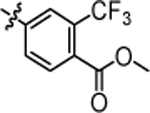 |
4.43 ± 0.23 |
| 4q | 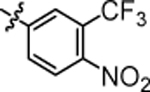 |
4.55 ± 0.12 | 4x | 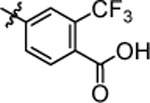 |
>20.0 |
Data are expressed as the mean ± SEM of three independent experiments, each performed in a quartet.
Our structure-activity relationship studies on the “tail” di-phenyl moiety allowed us to draw four main conclusions. First, we observed that the electronic properties of para-substituents do not play a defining role in the activity of the studied analogs, as seen by comparing compounds 4l, 8a, and 8c. However, strong electron-withdrawing groups are favored (8e and 8g). In the subgroup of analogs bearing electron-donating groups of similar size, 8a, and 8h, the ability to form H-bonds was disfavored, although not forbidden. Second, we noted that the size of the para-substitution might be inversely linked to the activity of compounds, as seen in pairs 8a vs. 8c and 8e vs. 8i. Finally, we observed that compounds containing an N-linker have less activity when compared to their carbon-containing bioisosteres (see pairs 9c vs. 4l, 9a vs. 4a, 9b vs. 4b). Finally, compound 10 designed to determine the necessity of the diphenyl moiety showed just a slight decrease in the overall activity, indicating that the diphenyl moiety plays an auxiliary role in binding. However, it might affect the ability of a compound to penetrate the BBB, a hypothesis that is currently under investigation in our laboratory.
The result of structural modifications in the alkyl-urea linker is depicted in Table 4. First, we observed that increase in the length of the linker results in a slight increase in cytotoxicity (4l vs. 11b). Limiting the flexibility of the linker chain by locking it into cyclic urea (19b) and by adding an olefin moiety (18) resulted in a minor decrease of activity, which became more prominent with the simultaneous reduction in the linker length (19a). It is known that substitution at the nitrogen atom of the urea functionality plays an important role in the conformational preferences of these compounds.63 Thus, we have designed and evaluated a subgroup of analogs; the mono-methylated (14, corresponding to the trans,cis configuration) and di-methylated (15, corresponding to the cis,cis configuration) urea, where our results showed a clear preference for the trans,trans configuration of the unsubstituted functionality (4l). These results also show a potential involvement of the phenyl substituted nitrogen in the urea functionality in the hydrogen-bond interaction within the binding pocket. Finally, a substitution of the urea moiety to a thiourea functionality (12) or a carbamate (13) had no significant impact on the activity of the compound. In contrast, amide-containing linkers in 16 and 17 caused a reduction in the cytotoxicity of studied analogs.
Table 4.
In vitro inhibitory activity of analogs with modifications in the linker part.
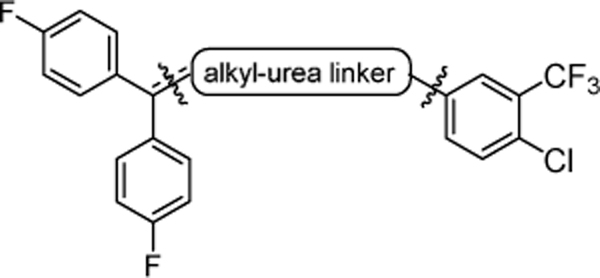 | |||||
|---|---|---|---|---|---|
| MDA-MB-231 aIC50 (μM) | MDA-MB-231 aIC50 (μM) | ||||
| 4l | 3.15 ± 0.11 | 15 | > 20.0 | ||
| 11a | 3.50 ± 0.08 | 16 | 11.44 ± 0.40 | ||
| 11b | 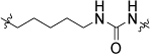 |
2.57 ± 0.07 | 17 | 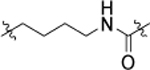 |
11.71 ± 0.38 |
| 12 | 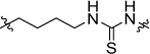 |
3.66 ± 0.12 | 18 | 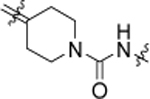 |
5.65 ± 0.43 |
| 13 | 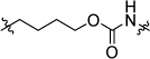 |
4.58 ± 0.25 | 19a | 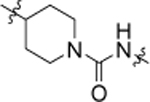 |
11.41 ± 0.32 |
| 14 | 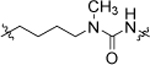 |
6.02 ± 0.09 | 19b | 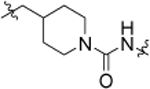 |
4.84 ± 0.17 |
Data are expressed as the mean ± SEM of three independent experiments, each performed in quartet.
Overall, our structure-activity relationship studies highlighted the importance of para-substitution of the “head” moiety with electron-withdrawing functionalities. Similar electronic properties were also favored for the para-substitution of the “tail” moiety. We observed that flexibility of the linker and trans,trans conformation of the urea moiety are preferred, as well as the presence of the three heteroatom-containing moiety. As a result of performed structural modifications, we were able to optimize the cytotoxicity of our “hit” molecule 4a by 6-fold (8g).
In vitro activity of selected analogs against DAT.
To evaluate the effect of performed structural modifications on the ability of compounds to inhibit DAT and estimate the possibility of separating this activity from cytotoxicity, we have selected 13 analogs with different anticancer profile. As shown in Table 5, for monosubstituted analogs 4a and 4b, the ability to inhibit DAT decreases as the size of para-substituent goes up. In a subgroup of disubstituted analogs 4l, 4o, and 4r, there is no direct correlation between the electronic properties of substituents and the observed activity of compounds. We observe that either hydrogen bond acceptor character of meta-group in 4o or slightly larger size may lead to a 100-fold decrease in affinity to DAT when compared to 4l. Similar to the observed trend in anticancer activity, ortho-substitution is not favored here as well (4s and 4t). Unlike in cytotoxicity-related SAR, modifications in the tail moiety (8a-8c) have a notable impact on the interaction with DAT, providing bases for separating pharmacophores responsible for transporter inhibition and anticancer activity. Unlike in other analogs (4a vs. 4l, 9a vs. 9c), a di-substitution pattern in 8a, bearing para-methyl groups at the tail moiety, is less active, than its mono-substituted counterpart 8b. Finally, selected modifications in the linker moiety affect the ability of compounds 9a-19b to interact with the DAT. For example, introduction of a nitrogen atom causes a slight decrease in the inhibitory properties of compounds (4a vs 9a and 4b vs 9c). Similar activity was observed in a series of analogs with the modified linker length (4a vs 11a and 11b). Conformational restrictions introduced by the piperidine moiety result in decreased activity in 19a and abolished DAT activity in 19b. Similarly, an unsubstituted urea moiety is required for DAT activity (4a), whereas the mono-methylated analog 14 loses some of this inhibitory activity, and di-methylated compound 15 is inactive as a DAT inhibitor. Substitution of the urea moiety with carbamate (13) or amide (16, 17) functionalities causes significant loss of DAT activity as well. We have summarized the observed trend in the activity of this set of compounds in Figure 6.
Table 5.
Inhibitory activity of selected analogs at dopamine transporter*.
| Structure | % inhibition at 10 μM | Ki, nM | Structure | % inhibition at 10 μM | Ki, nM | ||
|---|---|---|---|---|---|---|---|
| 4a | 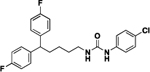 |
82 | 15 | 9a | 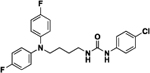 |
68 | 405 |
| 4b | 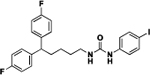 |
24 | ND | 9c | 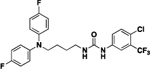 |
85 | 43 |
| 4l | 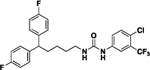 |
90 | 6.5 | 11a | 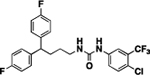 |
86 | 141 |
| 4o | 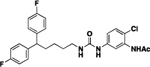 |
76 | 609 | 11b | 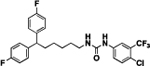 |
77 | 399 |
| 4r |  |
79 | 80 | 13 | 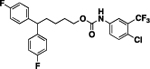 |
49 | ND |
| 4s | 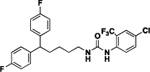 |
20 | ND | 14 | 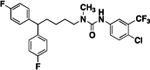 |
73 | 998 |
| 4t | 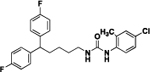 |
58 | 118 | 15 | 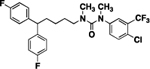 |
26 | ND |
| 8a | 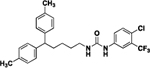 |
38 | ND | 16 | 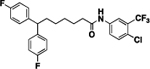 |
33 | ND |
| 8b | 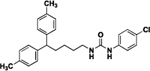 |
59 | 535 | 17 | 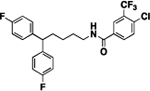 |
51 | 603 |
| 8c | 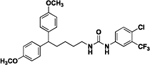 |
75 | 40 | 19a | 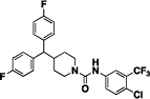 |
52 | 1589 |
| 8d | 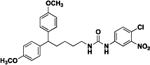 |
64 | 144 | 19b | 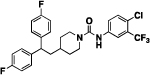 |
24 | ND |
In primary screening assays, compounds were tested in triplicate or quadruplicate at a final concentration of 10 μM. Compounds with a minimum of 50% antagonist activity were subjected to secondary screening (dose-response) assays.
Receptor binding profiles were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271–2013-00017-C (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth MD, Ph.D. at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA. ND− not determined (Ki was not evaluated due to the less than 50% antagonist activity of a compound).
Figure 6.

Heat map of the observed trends in the cytotoxic activity of selected compounds and their ability to inhibit DAT (% inhibition was measured at 10 μM level). Compounds with IC50 >20 μM (tables 1–5) were ascribed IC50value of 30 μM.
Evaluation of microsomal stability in vitro.
As part of the pharmacokinetic assessment of urea analogs in vitro, we have evaluated the metabolic stability of selected analogs (4l and 4q) using standard microsomal stability assay (see supplemental material). The compound’s selection was based on two parameters: the IC50 values and percent of the cell survival 48 hours post-treatment (Fig. S1). If both parameters were similar, we chose compounds with a more diverse substitution pattern. To validate our assay, we have used the clinically approved compound verapamil and PFL as the reference compounds. Both new analogs showed preferred metabolic stability over PFL when analyzed for up to 60 minutes (Fig. 7).
Figure 7.

Stability of verapamil, PFL, 4l, and 4q in liver microsomes following 60-min incubation was measured by liquid chromatography-tandem mass spectrometry. Data are expressed as the mean ± SEM of three independent experiments, each performed in triplicate.
Molecular target analysis.
To elucidate the downstream molecular pathway of 4q-induced antiproliferative effects observed in the cytotoxicity studies, we first explored whether apoptosis is induced. After 48 hours of 4q treatment apoptosis was induced in MDA-MB-231 cells in a dose-dependent manner. Even at the 1 μM concentration, significant induction of apoptosis was observed, whereas 5 μM levels of compound 4q increased the apoptotic index by 10.6-fold (P < 0.01), compared to control cells (Figure 8A). Further, we observed that 4q is capable of caspase-3 cleavage and activation (Supplemental material). At the same time, 4q did not affect the cell cycle in MDA-MB-231 cell line (Figure 8B).
Figure 8.

Effect of 4q on the induction of apoptosis (A) and cell cycle (B) in MDA-MB-231 triple-negative breast cancer cells. Data are expressed as the mean ± SEM of three independent experiments, each performed in triplicate. **P<0.01
Fibroblast growth factor receptor (FGFR) pathways are significant driver pathways for breast cancer,64 and FGFR overexpression has been proposed as a biomarker for TNBC.65 TNBC presents with gene amplification and protein overexpression of FGFR1, FGFR2 and bFGF, with the latter being shown to have an autocrine effect on tumor growth. TNBCs, along with basal-like breast cancers, show sensitivity to FGFR inhibitors, such as PD173074, while upregulated FGFR signaling is a known resistance mechanism to CDK inhibitors, highlighting the FGFR signaling as a potential therapeutic target for breast cancer, among others.66–68 We, therefore, explored the effect of compound 4q on FGFR1 activation in the presence and absence of bFGF. As shown in Figure 9, compound 4q completely abrogated phosphorylation of FGFR1 in the presence of its natural ligand bFGF at a concentration of 3 μM and 6 μM, indicating that inhibition of the FGFR1 signaling pathway is at least one of the molecular mechanisms responsible for the cytotoxic effects of compound 4q.
Figure 9.

The expression of FGFR1 was significantly reduced by compound 4q. Representative images (left panel) and quantification (right panel) of pFGFR1 expression in MDA-MB-231 cells after 48 hours of treatment with compound 4q at the concentrations depicted in the presence and absence of natural ligand bFGF (n = 3). NS: no significance; *P < 0.05; **P<0.01.
Molecular Modelling.
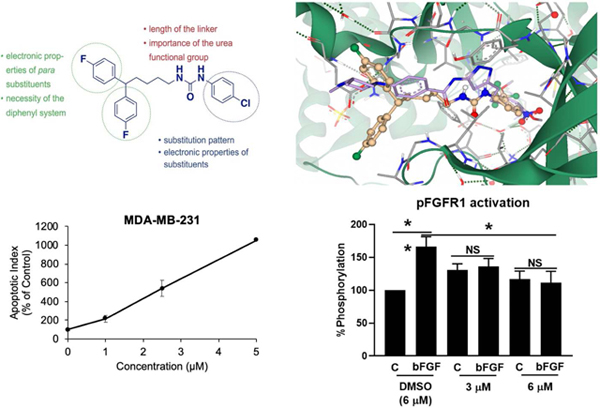
Compound 4q was selected for docking into the FGFR1 protein to gain insights into binding interactions. The crystal structure of FGFR1 in complex with the potent inhibitor AZD454769 (PDB code 4V05) was prepared for docking by removing the ligand and defining the binding site as the 35 amino acid residues surrounding the ligand and an additional 23 residues that account for unoccupied space around the ligand-binding site. Molecular docking simulations afforded binding poses that were scored by HYDE, as described in our previous publication.70 Predicted protein-ligand interactions (Fig. 10) correlate well with experimental data obtained from SAR studies. The compound is predicted to occupy the ATP-binding cleft of FGFR1 in a similar position to the known inhibitor AZD4547.69 The p-NO2 moiety of 4q is predicted to participate in hydrogen bonding interactions with an active site water molecule forming a bridging network that incorporates the LYS514, GLU531, and ASP641 residues. This is facilitated by the length of the linker allowing the bulky diphenyl tail moiety to remain outside the binding pocket and solvent-exposed. The urea moiety of 4q occupies a similar position within the ATP-binding cleft to the pyrazole moiety of AZD4547 (Fig. 11). Calculation of hydrogen bonding distance results in potentially favorable proximity and orientation of a urea NH to the GLU562 residue (3.77 Å), whereas the second urea NH is positioned 4.78 Å away from the ALA564 residue precluding potential binding. This is supported by SAR data that shows a critical role for the hydrogen of the urea nitrogen alpha to the head group. The binding affinity of 4q was predicted to be in the high nanomolar to a low micromolar range, which compares well to its experimentally derived value (IC50 = 4.55 μM).
Figure 10.

Compound 4q (Gold) docked into the ATP-binding cleft of FGFR1 (Grey/Green). The green dashed lines depict putative binding interactions indicating strong hydrogen bonding. Pink lines represent predicted distance between hydrogen bond acceptors and donors..
Figure 11.

Overlay of compound 4q (Gold) and the known inhibitor AZD4547 (Magenta) in the ATP-binding site of FGFR1
CONCLUSION
We have described a novel series of cytotoxic compounds that possess cytotoxicity towards the MDA-MB-231 cell line. The first compound in this series, our “hit” molecule 4a, was shown to cross the BBB in vivo and to have minimum interaction with CNS receptors when compared to the original penfluridol molecule. The first round of structure-activity relationship studies was designed to understand the structural requirements for the anticancer activity of these compounds. Observed results highlighted the importance of electron-withdrawing substitution with the hydrogen-bonding properties on both tail and head moieties of studied analogs, as well as the preferred length of a linker and its flexibility. Selected derivatives were subjected to a primary binding assay to investigate their affinity to the DAT. Based on the current data, we have concluded that the toluene-like structure of the tail-moiety will allow preserving the anticancer activity of urea analogs while eliminating unwanted interactions with the DAT. These results are in line with the reported SAR studies for DAT inhibitors,71 and we will continue exploring the effect of electron-withdrawing groups in the tail moiety to clarify DAT-related pharmacophore of the urea compounds. Similarly, certain modification of the linker motif will allow preservation of the anticancer activity of compounds, while eliminating unwanted interactions with the DAT. Using a representative molecule, we have shown that this class of agents can induce apoptosis in MDA-MB-231 cell lines in a concentration-dependent manner while having no effect on cell cycle. Further, we observe that the tested analog was able to cleave caspase-3. Our initial mechanistic studies have identified FGFR1 as one of the potential targets modulated by this class of agents. Our molecular modeling studies confirmed the emphasized findings from the SAR studies. We envision that these compounds can be further developed for the potential treatment of metastatic breast cancer. In vivo validation of the observed in vitro data, as well as further validation of FGFR1 as a potential target will be reported shortly.
EXPERIMENTAL SECTION
General chemistry procedures.
All reactions were carried out in oven- or flame-dried glassware under positive nitrogen/ argon pressure unless otherwise noted. All solvents and chemicals were reagent grade. Unless otherwise noted, all reagents and solvents were purchased from commercial vendors and used as received. The purity and characterization of compounds were established by a combination of methods, including TLC, HPLC, mass spectrometry, NMR analysis. 1H and 13C NMR spectra were recorded on a Bruker 400 MHz Advance III HD spectrometer using chloroform-d, methanol-d, or DMSO-d6 with tetramethyl (TMS) (0.00 ppm) or solvent peaks as the internal standard. Chemical shits (δ) are recorded in ppm relative to the reference signal, and coupling constant (J) values are recorded in hertz (Hz). Multiplicates are indicated by s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet), br (broad). Thin-layer chromatography (TLC) was performed on EMD precoated silica gel 6-F254 plates, and spots were visualized with UV light or iodine staining. Flash column chromatography was performed with silica gel (40–63 μm, 60 Å) using the mobile phase indicated or on a Teledyne Isco (CombiFlash Rf UV/Vis). High-resolution mass spectra were obtained using TripleTOF 5600 mass spectrometer. The purity of all final compounds was greater than 95%. The purity was determined by Waters Acquity UPLC using C18 column (Cortecs, 1.6μm, 2.1×50 mm): eluent A, 0.1 % aqueous CF3COOH and eluent B. CH3CN containing 0.1 % CF3COOH, gradient elution (0 min: 95% A, 5% B; 2 min: 50% A, 50% B; 4 min: 50% A, 50% B; 6 min: 10% A, 90% B; 9 min: 10% A, 90% B; 10 min: 95% A, 5%), with a flow rate of 0.2 ml/min.
General procedure A.
A substituted phenyl isocyanate (0.50 mmol) dissolved in dichloromethane (2 mL) was added to a solution of selected alkyl amine (0.50 mmol) in dichloromethane (3 mL) at 0 °C under inert environment. After stirring for 6 hours at room temperature, the solvent was evaporated under reduced pressure, and the formed residue was purified by flash chromatography with ethyl acetate /hexane (1:9 to 1:1) to obtain the desired compound.
General procedure B.
Hydrazine hydrate (0.50 mL) was added to a solution of a nitroaromatic compound (1.00 eq, 0.40 mmol) in methanol (10 mL). After stirring for 15 min at 50 °C, an excess of Raney®-Nickel was added (approx. 2.00 eq.). A reaction mixture was kept stirring at 50 °C for an hour then filtered using Celite® bed. The organic solvent was removed under reduced pressure, and formed residue was purified by flash chromatography with methanol/ dichloromethane (1:99 to 1:19) to obtain the reduced product.
General procedure C.
An excess of acetic acid (0.03 mL) was added to a solution of an aniline derivative (1.00 equiv., 0.21 mmol) in dichloromethane (10 mL). After overnight stirring at room temperature, the reaction solvent was removed under reduced pressure and formed residue was purified by flash chromatography using methanol/ dichloromethane (1:99 to 1:19) to obtain desired N-phenylacetamide derivative.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(4-chlorophenyl)urea (4a).
Compound 4a was prepared in 56% yield from compound 29a and 48a using the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.12–1.27 (m, 2H), 1.46 (quin, 2H, J = 7.46 Hz), 1.87–2.01 (m, 2H), 3.11 (q, 2H, J = 6.85 Hz), 3.79 (t, 1H, J = 7.83 Hz), 5.00 (t, 1H, J = 5.62 Hz), 6.83 (s, 1H), 6.89–7.04 (m, 4H) 7.05–7.14 (m, 4H), 7.14–7.23 (m, 4H). 13C NMR (100 MHz, CDCl3) δC 25.18, 30.00, 35.47, 40.11, 49.67, 115.19, 115.40, 121.62, 128.99, 129.07, 129.14, 137.28, 140.36, 140.40, 160.12, 162.55. HRMS-ESI: (m/z) calculated for C24H23ClF2N2O, 429.1545 [M+H]+; found 429.1590. Purity: 98.67%
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(4-iodophenyl)urea (4b).
Compound 4b was prepared in 31% yield from compounds 29a and 48b using the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.17–1.32 (m, 2H), 1.47–1.56 (m, 2H), 1.90–2.05 (m, 2H), 3.11–3.23 (m, 2H), 3.83 (t, 1H, J = 7.95 Hz), 4.64 (t, 1H, J = 5.62 Hz), 6.31 (s, 1H), 6.89–7.00 (m, 4H), 7.00–7.09 (m, 2H), 7.09–7.21 (m, 4H), 7.51–7.63 (m, 2H). 13C NMR (100 MHz, CDCl3) δC 25.19, 29.99, 35.48, 40.18, 49.68, 86.48, 115.21, 115.42, 122.27, 129.03, 129.11, 138.08, 138.47, 140.42, 160.13, 162.57. HRMS-ESI: (m/z) calculated for C24H23F2IN2O, 521.0901 [M+H]+; found 521.0897. Purity: 97.41%.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(p-tolyl)urea (4c).
Compound 4c was prepared in 84% yield from compounds 29a and 48c using the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.13–1.24 (m, 2H), 1.48 (quin, 2H, J = 7.40 Hz), 1.88–2.01 (m, 2H), 2.29 (s, 3 H), 3.06–3.20 (m, 2H), 3.80 (t, 1H, J = 7.83 Hz), 4.94 (t, 1H, J = 5.62 Hz), 6.59 (s, 1H), 6.88–7.01 (m, 4H), 7.03–7.19 (m, 8 H). 13C NMR (100 MHz, CDCl3) δC 20.80, 25.16, 30.02, 35.48, 40.05, 49.65, 115.16, 115.36, 122.01, 129.02, 129.10, 129.88, 133.96, 135.71, 140.45, 140.49, 156.31, 160.10, 162.53. HRMS-ESI: (m/z) calculated for C25H26F2N2O, 409.2091 [M+H]+; found 409.2127. Purity: >99%.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(4-nitrophenyl)urea (4d).
Compound 4d was prepared in 81% yield from compounds 29a and 48d using the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.21–1.31 (m, 2H), 1.55 (quin, 2H, J = 7.46 Hz), 1.93–2.03 (m, 2H), 3.22 (q, 2H, J = 6.85 Hz), 3.82 (t, 1H, J = 7.83 Hz), 5.13 (t, 1H, J = 5.50 Hz), 6.87–7.00 (m, 4H), 7.04–7.17 (m, 4H), 7.22 (s, 1H), 7.45–7.53 (m, 2H), 8.07–8.19 (m, 2H). 13C NMR (100 MHz, CDCl3) δC 25.19, 29.90, 35.47, 39.99, 49.69, 115.18, 115.39, 117.47, 117.52, 125.31, 128.99, 129.07, 140.35, 140.39, 141.73, 146.10, 160.10, 162.54. HRMS-ESI: (m/z) calculated for C24H23F2N3O3, 440.1785 [M+H]+; found 440.1830. Purity: 99.70%.
1-(4-aminophenyl)-3-(5,5-bis(4-fluorophenyl)pentyl)urea (4e).
Compound 4e was prepared in 81% yield from compound 4d using the general procedure B. 1H NMR (400 MHz, CDCl3) δH 1.12–1.27 (m, 2H), 1.48 (quin, 2H, J = 7.30 Hz), 1.90–2.04 (m, 2H), 3.16 (q, 2H, J = 6.85 Hz) 3.69, (br. s., 2H), 3.83 (t, 1H, J = 7.83 Hz), 4.52 (t, 1H, J = 5.50 Hz), 5.98 (s, 1H), 6.56–6.70 (m, 2H), 6.89–7.05 (m, 6H), 7.08–7.22 (m, 4H). 13C NMR (100 MHz, CDCl3) δC 25.13, 30.03, 35.52, 40.05, 49.65, 115.16, 115.37, 115.76, 126.69, 128.23, 129.05, 129.13, 140.51, 140.54, 144.74, 157.13, 160.12, 162.55. HRMS-ESI: (m/z) calculated for C24H25F2N3O, 410.2044 [M+H]+; found 410.2080. Purity: >99%.
N-(4-(3-(5,5-bis(4-fluorophenyl)pentyl)ureido)phenyl)acetamide (4f).
Compound 4f was prepared in 81% yield from compound 4e using the general procedure C. 1H NMR (400 MHz, DMSO-d6) δH 1.11–1.25 (m, 2H), 1.43–1.48 (m, 2H), 1.92–2.07 (m, 5H), 3.02 (q, 2H, J = 6.77 Hz), 3.97 (t, 1H, J = 7.83 Hz), 6.03 (t, 1H, J = 5.75 Hz), 7.08 (dd, 4H, J = 8.90, 8.90 Hz), 7.23–7.37 (m, 6H), 7.41 (d, 2H, J = 8.80 Hz), 8.25–8.40 (m, 1H), 9.77 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC 24.29, 25.29, 30.13, 35.15, 49.30, 115.43, 115.64, 118.35, 120.08, 129.70, 129.77, 133.44, 136.43, 141.72, 141.75, 155.73, 159.85, 162.26, 168.16. HRMS-ESI: (m/z) calculated for C26H27F2N3O2, 452.2149 [M+H]+; found 452.2186. Purity: >99%.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(3-(trifluoromethyl)phenyl)urea (4g).
Compound 4g was prepared in 88% yield from compounds 29a and 48e using the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.08–1.21 (m, 2H), 1.42 (quin, 2H, J = 7.40 Hz), 1.82–1.95 (m, 2H), 3.02–3.16 (m, 2H), 3.74 (t, 1H, J = 7.70 Hz), 5.53 (t, 1H, J = 5.26 Hz), 6.84–6.97 (m, 4H), 7.00–7.12 (m, 4 H), 7.13–7.20 (m, 1H), 7.20–7.28 (m, 1H), 7.37 (d, 1H, J = 8.31 Hz), 7.50 (s, 1H), 7.60 (s, 1H). 13C NMR (100 MHz, CDCl3) δC 25.17, 29.89, 35.43, 40.06, 49.64, 115.16, 115.37, 116.16, 116.20, 119.41, 122.60, 125.25, 128.95, 129.03, 129.49, 139.49, 140.35, 140.39, 156.00, 160.11, 162.54. HRMS-ESI: (m/z) calculated for C25H23F5N2O, 463.1809 [M+H]+; found 463.1845. Purity: >99%.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(4-(trifluoromethyl)phenyl)urea (4h).
Compound 4h was prepared in 73% yield from compounds 29a and 48f using the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.15–1.24 (m, 2H), 1.47 (quin, 2H, J = 7.43 Hz), 1.87–1.99 (m, 2H), 3.13 (q, 2H, J = 6.85 Hz), 3.78 (t, 1H, J = 7.76 Hz), 5.18 (t, 1H, J = 5.50 Hz), 6.80–7.00 (m, 4H), 7.02–7.13 (m, 4H), 7.15 (s, 1H), 7.35 (m, 2H, J = 8.56 Hz), 7.46 (m, 2H, J = 8.56 Hz). 13C NMR (100 MHz, CDCl3) δC 25.18, 29.93, 35.45, 40.14, 49.68, 115.19, 115.40, 118.87, 126.26, 126.30, 126.33, 128.97, 129.05, 140.32, 140.35, 142.05, 155.33, 160.14, 162.56. HRMS-ESI: (m/z) calculated for C25H23F5N2O, 463.1809 [M+H]+; found 463.1764. Purity: 99.67%.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(3-chlorophenyl)urea (4i).
Compound 4i was prepared in 66% yield from compounds 29a and 48g using the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.14–1.27 (m, 2H) 1.49, (t, 2H, J = 7.09 Hz), 1.95 (q, 2H, J = 7.62 Hz), 3.07–3.22 (m, 2H), 3.80 (t, 1H, J = 7.76 Hz), 4.99 (br. s., 1H), 6.78 (br. s., 1H), 6.89–7.02 (m, 5H), 7.02–7.21 (m, 6H), 7.33 (s, 1H). 13C NMR (100 MHz, CDCl3) δC 25.17, 29.96, 35.47, 40.16, 49.67, 115.18, 115.39, 118.07, 120.13, 123.36, 129.01, 129.09, 130.11, 134.75, 139.99, 140.40, 140.43, 155.42, 160.12, 162.55. HRMS-ESI: (m/z) calculated for C24H23ClF2N2O, 429.1545 [M+H]+; found 429.1505. Purity: 99.91%.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(3-nitrophenyl)urea (4j).
Compound 4j was prepared in 44% yield from compounds 29a and 48h using the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.22–1.30 (m, 2H), 1.53 (quin, 2H, J = 7.40 Hz), 1.87–2.00 (m, 2H), 3.20 (q, 2H, J = 6.85 Hz), 3.81 (t, 1H, J = 7.76 Hz), 5.20 (t, 1H, J = 5.44 Hz), 6.86–6.98 (m, 4H), 7.05–7.16 (m, 4H), 7.35 (t, 1H, J = 8.13 Hz), 7.70 (dd, 1H, J = 8.19, 1.34 Hz), 7.78 (dt, 1H, J = 8.19, 1.10 Hz), 8.10 (t, 1H, J = 2.14 Hz). 13C NMR (100 MHz, CDCl3) δC 25.19, 29.91, 35.47, 40.19, 49.67, 76.71, 113.65, 115.18, 115.39, 117.29, 124.90, 128.99, 129.07, 129.77, 140.32, 140.37, 140.40, 148.52, 155.28, 160.11, 162.55. HRMS-ESI: (m/z) calculated for C24H23F2N3O3, 440.1785 [M+H]+; found 440.1750. Purity: 99.65%.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(m-tolyl)urea (4k).
Compound 4k was prepared in 76% yield from compounds 29a and 48i using the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.18–1.25 (m, 2H), 1.52 (quin, 2H, J = 7.40 Hz), 1.91–2.03 (m, 2H), 2.31 (s, 3H), 3.08–3.25 (m, 2H), 3.83 (t, 1H, J = 7.83 Hz), 4.75 (t, 1H, J = 5.62 Hz), 6.29 (s, 1H), 6.85–7.03 (m, 6H), 7.04–7.22 (m, 6H). 13C NMR (100 MHz, CDCl3) δC 21.43, 25.17, 30.01, 35.50, 40.14, 49.68, 115.18, 115.39, 118.68, 122.38, 125.07, 129.03, 129.11, 129.18, 138.27, 139.41, 140.46, 155.83, 160.13. HRMS-ESI: (m/z) calculated for C25H26F2N2O, 409.2091 [M+H]+; found 409.2051. Purity: 98.34%.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(4-chloro-3-(trifluoromethyl)phenyl)urea (4l).
Compound 4l was prepared in 60% yield from compounds 29a and 48j using the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.11–1.34 (m, 2H), 1.38–1.56 (m, 2H), 1.84–2.03 (m, 2H), 3.14 (q, 2H, J = 6.60 Hz), 3.79 (t, 1H, J = 7.70 Hz), 5.09 (br. s., 1H), 6.93 (t, 4H, J = 8.56 Hz), 7.10 (dd, 4H, J = 8.44, 5.26 Hz), 7.31 (d, 1H, J = 8.56 Hz), 7.42 (dd, 1H, J = 8.68, 2.32 Hz), 7.50–7.59 (m, 1H). 13C NMR (100 MHz, CDCl3) δC 25.18, 29.91, 35.45, 40.21, 49.69, 115.21, 115.42, 123.33, 128.99, 129.06, 131.96, 137.74, 140.35, 155.13, 160.13, 162.56. HRMS-ESI: (m/z) calculated for C25H22ClF5N2O, 497.1419 [M+H]+; found 497.1455. Purity: 98.08%.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(4-chloro-3-nitrophenyl)urea (4m).
Compound 4m was prepared in 59% yield from compounds 29a and 48k using the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.17–1.29 (m, 2 H), 1.52 (quin, 2H, J = 7.46 Hz), 1.90–2.03 (m, 2H), 3.17 (q, 2H, J = 6.85 Hz), 3.81, (t, 1H, J = 7.83 Hz), 5.17 (t, 1H, J = 5.50 Hz), 6.85–6.99 (m, 4H), 7.05–7.19 (m, 4H), 7.28 (s, 1H), 7.34 (d, 1H, J = 8.80 Hz), 7.43 (dd, 1H, J = 8.93, 2.57 Hz), 7.90 (d, 1H, J = 2.69 Hz). 13C NMR (100 MHz, CDCl3) δC 25.20, 29.86, 35.46, 40.25, 49.66, 115.20, 115.41, 115.53, 119.82, 123.33, 129.00, 129.08, 132.07, 138.72, 140.33, 140.36, 147.73, 154.92, 160.11, 162.55. HRMS-ESI: (m/z) calculated for C24H22ClF2N3O3, 474.1396 [M+H]+; found 474.1444. Purity: 100.00%.
1-(3-amino-4-chlorophenyl)-3-(5,5-bis(4-fluorophenyl)pentyl)urea (4n).
Compound 4n was prepared in 97% yield from compound 4m using the general procedure B. 1H NMR (400 MHz, CDCl3) δH 1.15 (quin, 2H, J = 7.70 Hz), 1.42 (quin, 2H, J = 7.40 Hz), 1.82–1.97 (m, 2H), 3.06 (q, 2H, J = 6.85 Hz), 3.76 (t, 1H, J = 7.70 Hz), 3.95 (br. s., 1H), 5.26–5.38 (m, 1H), 6.44 (dd, 1H, J = 8.56, 2.20 Hz), 6.79 (d, 1H, J = 2.20 Hz), 6.85–7.00 (m, 4H), 7.00–7.18 (m, 6H). 13C NMR (100 MHz, CDCl3) δC 25.19, 30.00, 35.46, 40.05, 49.65, 107.49, 110.99, 113.72, 115.17, 115.38, 129.01, 129.09, 129.64, 138.36, 140.43, 140.45, 143.51, 156.09, 160.09, 162.52. HRMS-ESI: (m/z) calculated for C24H24ClF2N3O, 444.1654 [M+H]+; found 444.1699. Purity: 99.74%.
N-(5-(3-(5,5-bis(4-fluorophenyl)pentyl)ureido)-2-chlorophenyl)acetamide (4o).
Compound 4o was prepared in 78% yield from compound 4n following the general procedure C. 1H NMR (400 MHz, DMSO-d6) δH 1.08–1.25 (m, 2H), 1.45 (quin, 2H, J = 7.27 Hz), 2.00 (q, 2H, J = 7.83 Hz), 2.08 (s, 3H), 3.03 (q, 2H, J = 6.77 Hz) 3.97, (t, 1H, J = 7.70 Hz), 6.06 (t, 1H, J = 5.50 Hz), 7.09 (t, 4H, J = 8.80 Hz), 7.21–7.42 (m, 6H), 7.74 (s, 1H), 8.59 (s, 1H), 9.38 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC 25.27, 30.02, 35.14, 49.29, 115.43, 115.64, 129.59, 129.69, 129.77, 135.43, 140.23, 141.74, 155.40, 159.86, 162.26. HRMS-ESI: (m/z) calculated for C26H26ClF2N3O2, 486.1760 [M+H]+; found 486.1811. Purity: 99.86%.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(3-chloro-4-nitrophenyl)urea (4p).
Compound 4p was prepared in 72% yield from compound 29a and 48l following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.19–1.30 (m, 2H), 1.54 (quin, 2H, J = 7.40 Hz), 1.88–2.02 (m, 2H), 3.20 (q, 2H, J = 6.85 Hz), 3.81 (t, 1H, J = 7.83 Hz), 5.40 (t, 1H, J = 5.50 Hz), 6.84–6.99 (m, 4H), 7.01–7.17 (m, 4H), 7.30 (dd, 1H, J = 9.05, 2.20 Hz), 7.52–7.67 (m, 2H), 7.90 (d, 1H, J = 9.05 Hz). 13C NMR (100 MHz, CDCl3) δC 25.21, 29.81, 35.46, 40.23, 49.68, 115.20, 115.41, 116.25, 120.37, 127.65, 128.98, 129.06, 129.34, 140.30, 140.33, 140.76, 144.58, 154.46, 160.12, 162.55. HRMS-ESI: (m/z) calculated for C24H22ClF2N3O3, 474.1396 [M+H]+; found 474.1446. Purity: 99.03%.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(4-nitro-3-(trifluoromethyl)phenyl)urea (4q).
Compound 4q was prepared in 71% yield from compound 29a and 48m following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.28 (quin, 2H, J = 7.70 Hz), 1.57 (quin, 2H, J = 7.46 Hz), 2.01 (q, 2H, J = 7.82 Hz), 3.24 (q, 2H, J = 6.77 Hz), 3.84 (t, 1H, J = 7.83 Hz), 4.90 (br. s., 1H), 6.87–7.05 (m, 5H), 7.14 (dd, 4H, J = 8.44, 5.50 Hz), 7.70–7.85 (m, 2H), 7.95 (d, 1H, J = 8.80 Hz). 13C NMR (100 MHz, CDCl3) δC 25.15, 29.76, 35.44, 40.32, 49.69, 115.23, 115.43, 116.90, 120.45, 127.51, 129.01, 129.08, 140.30, 140.34, 160.14, 162.57. HRMS-ESI: (m/z) calculated for C25H22F5N3O, 508.1659 [M+H]+; found 508.1715. Purity: 97.34%.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(3,4-dimethylphenyl)urea (4r).
Compound 4r was prepared in 73% yield from compound 29a and 48n following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.18–1.29 (m, 2H), 1.52 (dt, 2H, J = 14.67, 7.34 Hz), 1.93–2.02 (m, 2H), 2.22 (s, 6H), 3.13–3.25 (m, 2H), 3.83 (t, 1H, J = 7.83 Hz), 4.69 (t, 1H, J = 5.38 Hz), 6.14 (s, 1H), 6.88–7.02 (m, 6H), 7.05 (d, 1H, J = 7.82 Hz), 7.09–7.22 (m, 4H). 13C NMR (100 MHz, CDCl3) δC 19.16, 19.85, 25.15, 30.01, 35.50, 40.11, 49.66, 115.16, 115.37, 124.09, 129.03, 129.11, 130.45, 135.72, 137.87, 156.14, 160.11. HRMS-ESI: (m/z) calculated for C26H28F2N2O, 423.2248 [M+H]+; found 423.2292. Purity: 96.99%.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(4-chloro-2-(trifluoromethyl)phenyl)urea (4s).
Compound 4s was prepared in 83% yield from compound 29a and 48o following the general procedure A.1H NMR (400 MHz, CDCl3) δH 1.18–1.28 (m, 2H), 1.50 (quin, 2H, J = 7.40 Hz), 1.91–2.03 (m, 2H), 3.08–3.20 (m, 2H), 3.82 (t, 1H, J = 1.00 Hz), 5.11 (t, 1H, J = 5.14 Hz), 6.62 (s, 1H), 6.88–7.05 (m, 4H), 7.05–7.20 (m, 4H), 7.41 (dd, 1H, J = 8.93, 2.32 Hz), 7.51 (d, 1H, J = 2.20 Hz), 7.89 (d, 1H, J = 9.05 Hz). 13C NMR (100 MHz, CDCl3) δC 25.13, 29.83, 35.48, 40.31, 49.67, 115.20, 115.41, 125.91, 126.12, 126.18, 128.79, 129.00, 129.08, 132.72, 135.14, 140.38, 140.40, 154.80, 160.14, 162.57. HRMS-ESI: (m/z) calculated for C25H22ClF5N2O, 497.1419 [M+H]+; found 497.1480. Purity: 99.04%.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(4-chloro-2-methylphenyl)urea (4t).
Compound 4t was prepared in 94% yield from compound 29a and 48p following the general procedure A. 1H NMR (400 MHz, CDCl3) δH ppm 1.13–1.29 (m, 2H), 1.50 (quin, 2H, J = 7.34 Hz), 1.90– 2.05 (m, 2H), 3.17 (d, 2H, J = 5.26 Hz), 3.83 (t, 1H, J = 7.76 Hz), 4.49 (br. s., 1 H), 5.94 (s, 1H), 6.95 (t, 4H, J = 8.68 Hz), 7.13 (dd, 5 H, J = 8.38, 5.56 Hz), 7.18–7.22 (m, 1H), 7.28 (s, 1H). 13C NMR (100 MHz, CDCl3) δC 17.76, 25.14, 30.04, 35.50, 40.18, 49.66, 115.18, 115.39, 126.68, 127.12, 129.01, 129.09, 130.83, 131.22, 134.46, 134.56, 140.41, 140.44, 160.12, 1 162.55. HRMS-ESI: (m/z) calculated for C25H25ClF2N2O, 443.1701 [M+H]+; found 443.1705. Purity: >99%.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(4-bromo-2-iodophenyl)urea (4u).
Compound 4u was prepared in 64% yield from compound 29a and 48q following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.19–1.33 (m, 2H), 1.55 (quin, 2H, J = 7.40 Hz), 1.93–2.04 (m, 2H), 3.12–3.26 (m, 2H), 3.83 (t, 1H, J = 7.70 Hz), 4.93 (t, 1H, J = 5.38 Hz), 6.51 (s, 1H), 6.84–7.03 (m, 4H), 7.03–7.21 (m, 4H), 7.26 (s, 1H), 7.38 (dd, 1H, J = 8.68, 2.32 Hz), 7.74–7.90 (m, 2H). 13C NMR (100 MHz, CDCl3) δC 25.20, 29.92, 35.50, 40.43, 49.67, 90.75, 115.22, 115.42, 116.30, 122.83, 129.01, 129.09, 132.16, 138.57, 140.37, 140.41, 140.50, 154.66, 160.12, 162.55. HRMS-ESI: (m/z) calculated for C24H22BrF2IN2O, 599.9959 [M+H]+; found 599.9942. Purity: 99.07%.
methyl 4-(3-(5,5-bis(4-fluorophenyl)pentyl)ureido)-2-(trifluoromethyl)benzoate (4v).
Compound 4v was prepared in 63% yield from compound 51a and 29a following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.09–1.25 (m, 2H), 1.46 (quin, 2H, J = 7.40 Hz), 1.90 (q, 2H, J = 7.83 Hz), 3.13 (q, 2H, J = 6.60 Hz), 3.75 (t, 1H, J = 7.83 Hz), 3.83 (s, 3H), 5.78 (d, 1H, J = 5.38 Hz), 6.82–6.99 (m, 4H), 6.99–7.17 (m, 4 H), 7.50 (d, 1H, J = 8.56 Hz), 7.63 (s, 1H), 7.71 (d, 1H, J = 8.56 Hz), 8.00–8.15 (m, 1H). 13C NMR (100 MHz, CDCl3) δC 25.20, 29.85, 35.42, 40.08, 49.65, 52.63, 115.15, 115.36, 120.40, 128.95, 129.03, 132.41, 140.33, 140.38, 142.64, 155.61, 160.10, 162.53, 166.64. HRMS-ESI: (m/z) calculated for C27H25F5N2O3, 521.1863 [M+H]+; found 521.1867. Purity: 98.84%.
4-(3-(5,5-bis(4-fluorophenyl)pentyl)ureido)-2-(trifluoromethyl)benzoic acid (4x).
NaBH4 (0.02 g, 0.52 mmol) was added portion wise to a suspension of compound 4w (0.15 g, 0.39 mmol) and Pd/C (0.02g, cat.) in methanol (15 mL). The reaction mixture was stirred for 20 min, filtered using Celite® bed, and washed with methanol. Organic solvent was removed under reduced pressure and formed residue was re-dissolved in methanol (5 mL). Precipitation by dropwise addition of dichloromethane afforded compound 4x (0.08 g, 61%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δH 1.10–1.26 (m, 2H), 1.47 (quin, 2H, J = 7.06 Hz), 2.00 (q, 2H, J = 7.78 Hz), 3.03 (q, 2H, J = 6.44 Hz), 3.97 (t, 1H, J = 7.89 Hz), 4.13 (d, 1H, J = 4.89 Hz), 7.00 (br. s., 1H), 7.07 (t, 4H, J = 8.86 Hz), 7.21–7.50 (m, 6H), 7.81 (d, 1H, J = 1.83 Hz), 9.47 (br. s., 1H). 13C NMR (100 MHz, DMSO-d6) δC 25.29, 30.02, 35.16, 49.05, 49.30, 115.39, 115.60, 120.14, 125.96, 129.69, 129.77, 129.92, 140.12, 141.73, 155.92, 159.84, 162.24, 171.05. HRMS-ESI: (m/z) calculated for C26H23F5N2O3, 507.1707 [M+H]+; found 507.1715. Purity: 98.34%.
1-(4-chloro-3-(trifluoromethyl)phenyl)-3-(5,5-di-p-tolylpentyl)urea (8a).
Compound 8a was prepared in 41% yield from compounds 29b and 48j following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.17–1.29 (m, 2H), 1.47 (quin, 2H, J = 7.37 Hz), 1.97 (q, 2 H, J = 7.78 Hz), 2.26 (s, 6H), 3.11 (q, 2H, J = 6.85 Hz), 3.75 (t, 1H, J = 7.76 Hz), 5.11 (t, 1H, J = 5.44 Hz), 6.95–7.10 (m, 8H), 7.15 (s, 1H), 7.20–7.32 (m, 1H), 7.37 (dd, 1H, J = 8.74, 2.51 Hz). 7.54 (d, 1H, J = 2.57 Hz). 13C NMR (100 MHz, CDCl3) δC 20.93, 25.30, 29.88, 35.27, 40.19, 50.43, 118.25, 118.31, 123.26, 125.36, 127.55, 129.14,131.87, 135.56, 137.79, 142.12, 155.39. HRMS-ESI: (m/z) calculated for C27H28ClF3N2O, 489.1920 [M+H]+; found 489.1923. Purity: 99.80%.
1-(4-chlorophenyl)-3-(5,5-di-p-tolylpentyl)urea (8b).
Compound 8b was prepared in 39% yield from compounds 29b and 48a following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.23–1.32 (m, 2H), 1.48 (quin, 2H, J = 7.40 Hz), 1.92–2.02 (m, 2H), 2.27 (s, 6H), 3.12 (q, 2H, J = 6.85 Hz), 3.77 (t, 1H, J = 7.76 Hz), 4.85 (t, 1H, J = 5.56 Hz), 6.64 (s, 1H), 7.01–7.12 (m, 8H), 7.12–7.22 (m, 4H). 13C NMR (100 MHz, CDCl3) δC 20.96, 25.33 29.97, 35.33, 40.20, 50.43, 121.67, 127.58, 128.51, 129.14, 135.52, 137.30, 142.17, 155.55. HRMS-ESI: (m/z) calculated for C26H29ClN2O, 420.1968 [M+H]+; found 421.2037. Purity: 99.08%.
1-(5,5-bis(4-methoxyphenyl)pentyl)-3-(4-chloro-3-(trifluoromethyl)phenyl)urea (8c).
Compound 8c was prepared in 65% yield from compounds 29c and 48j following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.16–1.27 (m, 2H) 1.40–1.51 (m, 2H), 1.93 (q, 2H, J = 7.78 Hz), 3.11 (q, 2H, J = 6.72 Hz), 3.65–3.79 (m, 7H), 5.24 (t, 1H, J = 5.32 Hz), 6.72–6.84 (m, 4H), 7.07 (d, 4H, J = 8.68 Hz), 7.26 (d, 1H, J = 8.68 Hz), 7.31–7.41 (m, 2H), 7.54 (d, 1H, J = 2.45 Hz). 13C NMR (100 MHz, CDCl3) δC 25.23, 29.79, 35.49, 40.12, 49.48, 55.21, 113.83, 118.14, 118.19, 123.15, 123.96, 125.21, 128.41, 128.57, 128.71, 131.86, 137.44, 137.89, 155.49, 157.81. HRMS-ESI: (m/z) calculated for C27H28ClF3N2O3, 521.1819 [M+H]+; found 521.1909. Purity: >99%.
1-(5,5-bis(4-methoxyphenyl)pentyl)-3-(4-chloro-3-nitrophenyl)urea (8d).
Compound 8d was prepared in 25% yield from compounds 29c and 48k following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.23–1.30 (m, 2H), 1.51 (quin, 2H, J = 7.31 Hz), 1.96 (q, 2H, J = 7.83 Hz), 3.16 (q, 2H, J = 6.72 Hz), 3.68–3.80 (m, 7H), 5.03 (t, 1H, J = 5.44 Hz), 6.79 (d, 4H, J = 8.68 Hz), 7.00–7.19 (m, 5H), 7.32 (d, 1H, J = 8.80 Hz), 7.46 (dd, 1H, J = 8.80, 2.57 Hz), 7.85 (d, 1H, J = 2.57 Hz). 13C NMR (100 MHz, CDCl3) δC 25.19, 29.70, 35.45, 40.19, 49.46, 55.25, 113.85, 115.43, 119.64, 123.24, 128.61, 132.01, 137.46, 138.82, 147.74, 154.84, 157.82. Purity: HRMS-ESI: (m/z) calculated for C26H28ClN3O5, 498.1795 [M+H]+; found 498.1782. Purity: 99.93%.
1-(5,5-bis(4-(trifluoromethoxy)phenyl)pentyl)-3-(4-chloro-3-(trifluoromethyl)phenyl)urea (8e).
Compound 8e was prepared in 62% yield from compounds 29d and 48j following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.17–1.25 (m, 2H), 1.49 (quin, 2H, J = 7.40 Hz), 1.97 (q, 2H, J = 7.91 Hz), 3.15 (q, 2H, J = 6.72 Hz), 3.85 (t, 1H, J = 7.76 Hz), 5.04 (t, 1H, J = 5.50 Hz), 7.05 (s, 1H), 7.10 (d, 4H, J = 8.31 Hz), 7.17 (d, 4H, J = 8.68 Hz), 7.32 (d, 1H, J = 8.80 Hz), 7.44 (dd, 1H, J = 8.68, 2.45 Hz), 7.56 (d, 1H, J = 2.45 Hz). 13C NMR (100 MHz, CDCl3) δC 25.12, 29.72, 29.93, 35.25, 40.13, 49.97, 121.08, 123.33, 128.93, 131.97, 137.73, 142.87, 147.70, 147.72, 155.19. HRMS-ESI: (m/z) calculated for C27H22ClF9N2O3, 629.1253 [M+H]+; found 629.1231. Purity: 98.40%.
1-(4-chloro-3-(trifluoromethyl)phenyl)-3-(5,5-diphenylpentyl)urea (8f).
Compound 8f was prepared in 25% yield from compounds 29e and 48j following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.25–1.32 (m, 2H), 1.46–1.56 (m, 2H), 1.99–2.10 (m, 2H), 3.09–3.23 (m, 2H), 3.86 (t, 1H, J = 7.76 Hz), 4.73 (t, 1H, J = 5.56 Hz), 6.63 (s, 1H), 7.16–7.29 (m, 10H), 7.34 (d, 1H, J = 8.68 Hz), 7.46 (dd, 1H, J = 8.68, 2.57 Hz), 7.58 (d, 1H, J = 2.45 Hz). 13C NMR (100 MHz, CDCl3) δC 25.22 29.81, 35.17, 40.21, 51.26, 118.25, 118.30, 123.31, 123.95, 126.19, 127.80, 128.48, 131.95, 137.77, 144.87, 154.83. HRMS-ESI: (m/z) calculated for C25H24ClF3N2O, 461.1607 [M+H]+; found 461.1594. Purity: 99.35%.
1-(5,5-bis(4-nitrophenyl)pentyl)-3-(4-chloro-3-(trifluoromethyl)phenyl)urea (8g).
Compound 8g was prepared in 29% yield from compounds 29f and 48j following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.27–1.33 (m, 2H), 1.56 (d, 2H, J = 7.21 Hz), 2.12 (q, 2H, J = 7.74 Hz), 3.21 (q, 2H, J = 6.81 Hz), 4.11 (t, 1H, J = 7.70 Hz), 4.79 (t, 1H, J = 5.56 Hz), 6.65 (s, 1H), 7.31–7.42 (m, 5H), 7.50 (dd, 1H, J = 8.74, 2.38 Hz), 7.62 (d, 1H, J = 2.45 Hz), 8.14 (d, 4H, J = 8.80 Hz). 13C NMR (100 MHz, CDCl3) δC 24.95, 29.71, 29.93, 34.59, 39.98, 50.96, 118.19, 118.24, 123.27, 124.10, 128.65, 132.03, 137.73, 146.84, 150.56, 154.78. HRMS-ESI: (m/z) calculated for C25H22ClF3N4O5, 551.1309 [M+H]+; found 551.1303. Purity: >99%.
1-(5,5-bis(4-aminophenyl)pentyl)-3-(4-chloro-3-(trifluoromethyl)phenyl)urea (8h).
Compound 8h was prepared in 85% yield from compound 8g following the general procedure B. 1H NMR (400 MHz, CDCl3) δH 1.13–1.23 (m, 2H), 1.34–1.45 (m, 2H), 1.81–1.94 (m, 2H), 3.05 (q, 2H, J = 6.56 Hz), 3.53 (br. s., 4H), 3.61 (t, 1H, J = 7.76 Hz), 5.16 (t, 1H, J = 5.38 Hz), 6.55 (d, 4H, J = 8.44 Hz), 6.95 (d, 4H, J = 8.44 Hz), 7.19–7.29 (m, 1H), 7.31–7.41 (m, 2H), 7.55 (d, 1H, J = 2.32 Hz). 13C NMR (100 MHz, CDCl3) δC 25.30, 28.55, 35.38, 45.31, 49.37, 115.23, 123.86, 123.91, 128.52, 128.98, 130.06, 132.98, 135.65, 144.34, 180.69. HRMS-ESI: (m/z) calculated for C25H26ClF3N4O, 491.1825 [M+H]+; found 491.1842. Purity: 96.38%.
N,N’-((5-(3-(4-chloro-3-(trifluoromethyl)phenyl)ureido)pentane-1,1-diyl)bis(4,1 phenylene)) diacetamide (8i).
Compound 8i was prepared in 51% yield from compound 8h following the general procedure C. 1H NMR (400 MHz, DMSO-d6) δH 1.11–1.25 (m, 2H), 1.40–1.52 (m, 2H), 1.89–2.05 (m, 8H) 3.03, (q, 2H, J = 6.60 Hz), 3.79 (t, 1H, J = 7.83 Hz), 6.29 (t, 1H, J = 5.62 Hz), 7.18 (d, 4H, J = 8.56 Hz), 7.45 (d, 4H, J = 8.56 Hz), 7.49–7.61 (m, 2H), 8.05 (d, 1H, J = 1.96 Hz), 8.92 (s, 1H), 9.84 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δC 20.91, 24.35, 25.43, 30.01, 35.21, 49.94, 119.58, 121.65, 122.69, 128.08, 132.26, 137.66, 140.47, 140.67, 155.29, 168.49. HRMS-ESI: (m/z) calculated for C29H30ClF3N4O3, 575.2037 [M+H]+; found 575.2048. Purity: 99.79%.
1-(4-(bis(4-fluorophenyl)amino)butyl)-3-(4-chlorophenyl)urea (9a).
Compound 9a was prepared in 51% yield from compounds 53 and 48a following the general procedure A. 1H NMR (400 MHz, DMSO-d6) δH 1.40–1.50 (m, 2H) 1.50–1.60 (m, 2H), 3.08 (q, 2H, J = 6.36 Hz), 3.63 (t, 2H, J = 7.21 Hz), 6.21 (t, 1H, J = 5.50 Hz), 6.89–7.02 (m, 4H), 7.02–7.15 (m, 4H), 7.21–7.29 (m, 2H), 7.36–7.48 (m, 2H), 8.60 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC 24.71, 27.61, 52.17, 116.27, 116.48,119.52, 122.59, 122.67, 124.82, 128.89, 140.04, 144.80, 144.83, 155.54,156.35, 158.72. HRMS-ESI: (m/z) calculated for C23H22ClF2N3O, 430.1497 [M+H]+; found 430.1508. Purity: >99%.
1-(4-(bis(4-fluorophenyl)amino)butyl)-3-(4-iodophenyl)urea (9b).
Compound 9b was prepared in 29% yield from compounds 53 and 48b following the general procedure A. 1H NMR (400 MHz, DMSO-d6) δH 1.39–1.50 (m, 2H) 1.50–1.59 (m, 2H), 3.07 (q, 2H, J = 6.32 Hz), 3.63 (t, 2H, J = 7.27 Hz) 6.18 (t, 1H, J = 5.62 Hz), 6.87–7.00 (m, 4H), 7.02–7.14 (m, 4H), 7.24 (m, 2H, J = 8.80 Hz), 7.52 (m, 2H, J = 8.80 Hz), 8.54 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC 24.72, 27.61, 52.17, 83.87, 116.27, 116.49, 120.36, 121.01, 122.59, 122.67, 137.59, 137.78, 140.97, 144.83, 155.45 HRMS-ESI: (m/z) calculated for C23H22F2IN3O, 522.0854 [M+H]+; found 522.0864.
1-(4-(bis(4-fluorophenyl)amino)butyl)-3-(4-chloro-3-(trifluoromethyl)phenyl)urea (9c).
Compound 9c was prepared in 65% yield from compounds 53 and 48j following the general procedure A. 1H NMR (400 MHz, DMSO-d6) δH 1H NMR (400 MHz, CDCl3) δH 1.50–1.58 (m, 2H), 1.59–1.65 (m, 2H), 3.22 (q, J = 6.56 Hz, 2H), 3.59, (t, J = 7.09 Hz, 2H), 4.93 (t, J = 5.56 Hz, 1H), 6.77 (s, 1H), 6.80–6.88 (m, 4H), 6.88–6.99 (m, 4H), 7.34 (d, J = 8.68 Hz, 1H), 7.44 (dd, J = 8.68, 2.57 Hz, 1H), 7.58 (d, J = 2.45 Hz, 1H). 13C NMR (100 MHz, CDCl3) δC 24.79, 27.56, 40.19, 52.36, 115.89, 116.11, 118.36, 118.41, 122.28, 122.35, 123.40, 132.00, 137.59, 144.46, 144.48, 155.02,156.77, 159.17. HRMS-ESI: (m/z) calculated for C24H21ClF5N3O, 498.1371 [M+H]+; found 498.1373. Purity: 97.52%.
1-(4-(bis(4-fluorophenyl)amino)butyl)-3-(3,4-dimethylphenyl)urea (9d).
Compound 9d was prepared in 57% yield from compound 53 and 48n following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.47–1.57 (m, 2H), 1.58–1.66 (m, 2H), 2.12–2.27 (m, 6H), 3.21 (q, 2H, J = 6.68 Hz), 3.58 (t, 2H, J = 7.21 Hz), 4.82 (t, 1H, J = 5.62 Hz), 6.27 (s, 1H), 6.79–6.89 (m, 4H), 6.89–6.98 (m, 5H), 6.98–7.02 (m, 1H), 7.04 (d, 1H, J = 8.07 Hz). 13C NMR (100 MHz, CDCl3) δC 19.14, 19.84, 24.74, 27.75, 40.02, 52.46, 115.83, 116.05, 119.98, 122.23, 122.31, 123.96, 130.44, 133.18, 135.72, 137.84, 144.49, 144.52, 156.25, 156.72, 159.11. HRMS-ESI: (m/z) calculated for C25H27F2N3O, 424.2200 [M+H]+; found 424.2189. Purity: >99%.
1-(4-(bis(4-fluorophenyl)amino)butyl)-3-(4-bromo-2-iodophenyl)urea (9e).
Compound 9e was prepared in 51% yield from compounds 53 and 48q following the general procedure A. 1H NMR (400 MHz, DMSO-d6) δH 1.43–1.53 (m, 2H) 1.53–1.62 (m, 2H), 3.09 (q, 2H, J = 6.32 Hz), 3.64 (t, 2H, J = 7.21 Hz), 6.90–7.02 (m, 4H), 7.02–7.18 (m, 5H), 7.47 (dd, 1H, J = 8.93, 2.32 Hz), 7.80, (d, 1H, J = 8.93 Hz), 7.94 (d, 1H, J = 2.32 Hz). 13C NMR (100 MHz, DMSO-d6) δC 24.81, 27.45, 52.16, 114.62, 116.28, 116.51, 122.60, 122.68, 123.46, 131.62, 140.47, 140.76, 144.81, 144.83, 155.18, 156.38, 158.74. HRMS-ESI: (m/z) calculated for C23H21BrF2IN3O, 599.9959 [M+H]+; found 599.9942. Purity: 99.07%.
1-(4-chloro-3-(trifluoromethyl)phenyl)-3-(5-(4-fluorophenyl)pentyl)urea (10).
Compound 10 was prepared in 58% yield from compounds 56 and 48j following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.24–1.30 (m, 2H), 1.45 (quin, 2H, J = 7.46 Hz), 1.53 (quin, 2H, J = 7.64 Hz), 2.50 (t, 2H, J = 7.64 Hz), 3.07–3.22 (m, 2H), 5.56 (t, 1H, J = 5.01 Hz), 6.79–6.98 (m, 2H), 7.04 (dd, 2H, J = 8.38, 5.56 Hz), 7.20–7.31 (m, 1H), 7.31–7.38 (m, 1H), 7.59 (d, 1H, J = 2.45 Hz), 7.70 (s, 1H). 13C NMR (100 MHz, CDCl3) δC 26.35, 29.85, 31.11, 34.88, 40.28, 114.89, 115.10, 118.37, 118.43,123.34, 125.46, 129.54, 129.61, 131.87, 137.75, 137.79, 137.81, 155.90, 159.99, 162.42. HRMS-ESI: (m/z) calculated for C19H19ClF4N2O, 403.1200 [M+H]+; found 403.1203. Purity: 99.35 %.
1-(4,4-bis(4-fluorophenyl)butyl)-3-(4-chloro-3-(trifluoromethyl)phenyl)urea (11a).
Compound 11a was prepared in 83% yield from compounds 29g and 48j following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.34 (dt, 2H, J = 14.92, 7.46 Hz), 1.82–1.96 (m, 2H), 3.14 (q, 2H, J = 6.85 Hz), 3.74 (t, 1H, J = 7.70 Hz), 5.52 (t, 1H, J = 5.38 Hz), 6.90 (t, 4H, J = 8.56 Hz), 7.05 (dd, 4H, J = 8.56, 5.38 Hz), 7.23 (d, 1H, J = 8.80 Hz), 7.29 (dd, 1H, J = 8.68, 2.32 Hz), 7.55 (d, 1H, J = 2.20 Hz), 7.66 (s, 1H). 13C NMR (100 MHz, CDCl3) δC 28.44, 33.00, 40.11, 49.40, 115.26, 115.47, 118.30, 118.35, 123.29, 125.52, 128.77, 128.90, 128.98, 131.90, 137.70, 140.03, 155.83, 160.17, 162.61. HRMS-ESI: (m/z) calculated for C24H20ClF5N2O, 483.1262 [M+H]+; found 483.1294. HRMS-ESI: (m/z) calculated for C24H20ClF5N2O, 483.1262 [M+H]+; found 483.1294. Purity: >99%.
1-(6,6-bis(4-fluorophenyl)hexyl)-3-(4-chloro-3-(trifluoromethyl)phenyl)urea (11b).
Compound 11b was prepared in 42.86% yield from compounds 29h and 48j following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.20–1.29 (m, 2H), 1.29–1.41 (m, 2H) 1.48 (quin, 2H, J = 7.24 Hz), 1.96 (q, 2H, J = 7.74 Hz), 3.20 (q, 2H, J = 6.85 Hz), 3.83 (t, 1H, J = 7.83 Hz), 4.67 (t, 1H, J = 5.50 Hz), 6.49 (s, 1H), 6.96 (t, 4H, J = 8.62 Hz), 7.13 (dd, 4H, J = 8.44, 5.50 Hz), 7.33–7.42 (m, 1H), 7.51 (dd, 1H, J = 8.74, 2.26 Hz), 7.63 (d, 1H, J = 2.32 Hz). 13C NMR (100 MHz, CDCl3) δC 26.79, 27.62, 29.92, 35.79, 40.41, 49.70, 115.16, 115.36, 118.36, 118.42, 123.41, 125.62, 128.99, 129.07, 131.97, 137.69, 140.52, 140.55, 155.08, 160.11, 162.54. HRMS-ESI: (m/z) calculated for C26H24ClF5N2O, 511.1575 [M+H]+; found 511.1566. Purity: 96.81%.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(4-chloro-3-(trifluoromethyl)phenyl)thiourea (12).
Compound 12 was prepared in 51% yield from compounds 29a and 57 following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.20–1.32 (m, 2H), 1.64 (quin, 2H, J = 7.52 Hz), 1.95–2.08 (m, 2H), 3.59 (d, 2H, J = 5.38 Hz), 3.86 (t, 1H, J = 7.83 Hz), 5.86 (br. s., 1H), 6.87–7.02 (m, 4H), 7.07–7.21 (m, 4H), 7.30 (d, 1H, J = 7.34 Hz), 7.47–7.56 (m, 2H), 7.74 (br. s., 1H). 13C NMR (100 MHz, CDCl3) δC 25.16, 28.64, 35.35 45.25, 49.57, 115.24, 115.45, 123.88, 123.93, 129.02, 129.10, 133.10, 140.25, 140.30, 160.16, 162.59, 180.62. HRMS-ESI: (m/z) calculated for C25H22ClF5N2S, 513.1190 [M+H]+; found 513.1199. Purity: >99%.
5,5-bis(4-fluorophenyl)pentyl (4-chloro-3-(trifluoromethyl)phenyl)carbamate (13).
A substituted isocyanate 48j (0.12 g, 0.54 mmol) was added drop wise to a solution of intermediate 25a (0.15 g, 0.54 mmol) in chloroform (5 mL), followed by addition of TEA (113 μL, 0.81 mmol). The reaction mixture was stirred for 9 hours. Upon completion, organic solvent was removed under reduced pressure and obtained residue was purified using column chromatography with ethyl acetate/ hexanes (1:19 to 1:3) to obtain desired compound 13 (0.11 g, 40%) as colorless liquid. 1H NMR (400 MHz, CDCl3) δH 1.26–1.38 (m, 2H), 1.70 (quin, 2H, J = 7.15 Hz), 1.95–2.07 (m, 2H), 3.86 (t, 1H, J = 7.83 Hz), 4.14, (t, 2H, J = 6.60 Hz), 6.62 (s, 1H), 6.88–7.03 (m, 4H), 7.06–7.21 (m, 4H), 7.41 (d, 1H, J = 8.80 Hz), 7.52 (d, 1H, J = 8.07 Hz), 7.71 (d, 1H, J = 2.20 Hz). 13C NMR (100 MHz, CDCl3) δC 24.20, 28.69, 35.45, 49.69, 65.49, 115.22, 115.43, 129.02, 129.10, 132.05, 136.80, 140.34, 140.38, 153.16, 160.16, 162.59. HRMS-ESI: (m/z) calculated for C25H21ClF5NO2, 498.1259 [M+H]+; found 498.1247. Purity: 97.51%.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(4-chloro-3-(trifluoromethyl)phenyl)-1-methylurea (14).
Compound 14 was prepared in 36% yield from compounds 58 and 48j following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.20–1.30 (m, 2H), 1.60 (quin, 2H, J = 7.55 Hz), 1.94–2.09 (m, 2H), 2.95 (s, 3H), 3.30 (t, 2H, J = 7.46 Hz), 3.84 (t, 1H, J = 7.76 Hz), 6.42 (s, 1H), 6.86–6.99 (m, 4H), 7.07–7.19 (m, 4H), 7.37 (d, 1H, J = 8.68 Hz), 7.54 (dd, 1H, J = 8.80, 2.57 Hz), 7.68 (d, 1H, J = 2.57 Hz). 13C NMR (100 MHz, CDCl3) δC25.17, 27.84, 34.57, 35.59, 48.86, 49.75, 115.19, 115.40, 118.56, 118.62, 123.65, 129.02, 129.10, 131.72, 138.13, 140.36, 140.39, 154.64, 160.14, 162.58. HRMS-ESI: (m/z) calculated for C26H24ClF5N2O, 511.1575 [M+H]+; found 511.1571. Purity: 100 %.
1-(5,5-bis(4-fluorophenyl)pentyl)-3-(4-chloro-3-(trifluoromethyl)phenyl)-1,3-dimethylurea (15).
Sodium hydride (0.07 g, 1.70 mmol) was added portion wise to a solution of compound 4l (0.20 g, 0.42 mmol) in dimethylformamide (5 mL) at 0 °C under inert environment and reaction mixture was stirred for 10 min. Methyl iodide (0.20 g, 1.70 mmol) dissolved in dimethylformamide (1 mL) was added to the reaction mixture dropwise and stirring continued for 3 hours at room temperature. Upon reaction completion, dimethylformamide was removed by air flow, formed residue was re-suspended in dichloromethane (50 mL) and filtered. Filtrate was washed with aqueous solution of LiCl (5%) (5 × 50 mL), dried over Na2SO4 and concentrated in vacuum. The obtained residue was purified by column chromatography with ethyl acetate/ hexanes (1:19 to 1:2) to afford 15 (0.12 g, 56%) as colorless semi-solid. 1H NMR (400 MHz, CDCl3) δH 1.17 (quin, 2H, J = 7.76 Hz), 1.51 (quin, 2H, J = 7.64 Hz), 1.90–2.04 (m, 2H), 2.59 (s, 3H), 3.07–3.22 (m, 5H), 3.83 (t, 1H, J = 7.70 Hz), 6.91–7.01 (m, 4H), 7.07 (dd, 1H, J = 8.68, 2.57 Hz), 7.10–7.23 (m, 4H), 7.31 (d, 1H, J = 2.69 Hz), 7.38 (d, 1H, J = 8.56 Hz). 13C NMR (100 MHz, CDCl3) δC 25.23, 27.06, 35.59, 36.14, 38.93, 49.75, 60.40, 115.21, 115.42, 121.17, 121.22, 123.90, 126.25, 126.39, 129.02, 129.11, 132.38, 140.40, 140.44, 145.53, 160.17, 160.87, 162.60. HRMS-ESI: (m/z) calculated for C27H26ClF5N2O, 525.1732 [M+H]+; found 525.1739. Purity: 95.31%.
N-(4-chloro-3-(trifluoromethyl)phenyl)-6,6-bis(4-fluorophenyl)hexanamide (16).
Carboxylic acid 59 (0.20 g, 0.66 mmol) and aromatic amine 81f (0.15 g, 0.77 mmol) were dissolved in dimethylformamide (9 ml), followed by addition of DIEA (0.51 g, 3.94 mmol) and BOP (0.87 g, 1.97 mmol). The reaction mixture was stirred overnight at room temperature, diluted with ethyl acetate (40 mL), washed with brine (5 × 25 mL), and dried over MgSO4. Organic solvent was removed under reduced pressure and formed residue was purified with column chromatography using ethyl acetate/ hexanes (1:19 to 1:1) to afford compound 16 (0.14 g, 43.08%.) as yellow semi solid. 1H NMR (400 MHz, CDCl3) δH 1.27–1.35 (m, 2H), 1.75 (quin, 2H, J = 7.61 Hz), 1.96–2.08 (m, 2H), 2.31 (t, 2H, J = 7.46 Hz), 3.86 (t, 1H, J = 7.83 Hz), 6.87–7.03 (m, 4H), 7.06–7.19 (m, 4H), 7.20–7.26 (m, 1H), 7.42 (d, 1H, J = 8.68 Hz), 7.67 (dd, 1H, J = 8.68, 2.20 Hz), 7.79 (d, 1H, J = 2.32 Hz). 13C NMR (100 MHz, CDCl3) δC 25.16, 27.48, 35.63, 37.38, 49.52, 115.21, 115.42, 118.62, 118.67, 123.65, 129.02, 129.09, 132.00, 136.61, 140.37, 140.40, 160.15, 162.58, 171.21. HRMS-ESI: (m/z) calculated for C25H21ClF5NO, 482.1310 [M+H]+; found 482.1306. Purity: 96.14%.
N-(5,5-bis(4-fluorophenyl)pentyl)-4-chloro-3-(trifluoromethyl)benzamide (17).
To a solution of substituted benzoic acid 60 (0.10 g, 0.45 mmol) in dichloromethane (2 mL), oxalyl chloride (77.00 μL, 0.95 mmol) was added drop wise. A catalytic amount of dimethyformamide was added and reaction mixture was stirred at room temperature for an hour. Upon reaction completion, the reaction mixture was concentrated under reduced pressure and formed residue was re-dissolved in dichloromethane (2 mL). Compound 29a (0.12 g, 0.45 mmol) was added to the reaction mixture and solution was stirred overnight. Upon completion of the reaction, reaction mixture was washed with water (5 × 10 mL), and dried over MgSO4. Organic solvent was removed under reduced pressure and formed residue was purified with column chromatography using ethyl acetate/ hexanes (1:19 to 1:3) to obtain compound 17 (0.05 g, 22.35%.) as white semi solid. 1H NMR (400 MHz, CDCl3) δH 1.28–1.37 (m, 2H), 1.66 (quin, 2H, J = 7.46 Hz), 1.99–2.09 (m, 2 H), 3.43 (q, 2H, J = 6.93 Hz), 3.86 (t, 1H, J = 7.83 Hz), 6.22 (br. s., 1H), 6.90–7.03 (m, 4H), 7.10–7.23 (m, 4H), 7.58 (d, 1H, J = 8.31 Hz), 7.79 (dd, 1H, J = 8.31, 1.96 Hz), 8.00 (d, 1H, J = 1.71 Hz). 13C NMR (100 MHz, CDCl3) δC 25.15, 29.25, 35.35, 40.28, 49.61, 115.23, 115.44, 126.10, 126.16, 129.01, 129.09, 131.20, 131.97, 132.96, 135.98, 140.28, 140.32, 160.16, 162.59, 165.91. HRMS-ESI: (m/z) calculated for C25H21ClF5NO, 482.1310 [M+H]+; found 482.1318. Purity: 99.98%.
4-(bis(4-fluorophenyl)methylene)-N-(4-chloro-3-(trifluoromethyl)phenyl) piperidine-1-carboxamide (18).
Compound 18 was prepared in 46% yield from compounds 63a and 48j following the general procedure A. 1H NMR (400 MHz, DMSO-d6) δ ppm 2.30 (t, 4H, J = 5.50 Hz), 3.54 (t, 4H, J = 5.50 Hz), 7.10–7.24 (m, 8H) 7.57 (d, 1H, J = 1.00 Hz), 7.79 (d, 1H, J = 1.00 Hz), 8.06 (s, 1H), 8.97 (s, 1H). 13C NMR (100 MHz, CDCl3) δC 31.19, 45.10, 115.14, 115.35, 118.63, 118.68, 123.71, 131.09, 131.17, 131.81, 133.46, 136.26, 137.50, 137.53, 138.00, 154.12, 160.52, 162.97. HRMS-ESI: (m/z) calculated for C26H20ClF5N2O, 507.1262 [M+H]+; found 507.1260. Purity: 97.98%.
4-(bis(4-fluorophenyl)methyl)-N-(4-chloro-3-(trifluoromethyl)phenyl)piperidine-1-carboxamide (19a).
Compound 19a was prepared 17.97% yield from compounds 64a and 48j following the general procedure A in. 1 H NMR (400 MHz, CDCl3) δH 1.15 (q, 2H, J = 11.00 Hz), 1.62 (q, 2H, J = 12.00 Hz), 2.23 (m, 1H, J = 9.50 Hz), 2.86 (t, 2H, J = 12.23 Hz), 3.49 (d, 1H, J = 9.80 Hz), 4.02 (t, 2H, J = 12.00 Hz), 6.50 (d, 1H, J = 1.00 Hz), 6.87–7.10 (m, 4H), 7.11–7.24 (m, 4H), 7.37 (d, 1H, J = 7.34 Hz), 7.52 (s, 1H), 7.64 (s, 1H). 13C NMR (100 MHz, CDCl3) δC 30.97, 39.96, 44.54, 57.03, 115.52, 115.74, 118.59, 118.64, 123.68, 129.15, 129.23, 131.76, 138.07, 138.57, 138.60, 154.12, 160.29, 162.73. HRMS-ESI: (m/z) calculated for C26H22ClF5N2O, 509.1419 [M+H]+; found 509.1418. Purity: 98.87%.
4-(2,2-bis(4-fluorophenyl)ethyl)-N-(4-chloro-3-(trifluoromethyl)phenyl) piperidine-1 carboxamide (19b).
Compound 19b was prepared in 35% yield from compounds 64b and 48j following the general procedure A. 1H NMR (400 MHz, CDCl3) δH 1.25 (td, 2H, J = 12.26, 3.73 Hz), 1.36 (m, 1H), 1.76 (d, 2H, J = 10.76 Hz), 1.95 (dd, 2H, J = 7.58, 6.97 Hz), 2.68–2.84 (m, 2H), 3.92–4.08 (m, 3H), 6.55 (s, 1H), 6.89–7.06 (m, 4H), 7.09–7.22 (m, 4H), 7.36 (d, 1H, J = 8.80 Hz), 7.53 (dd, 1H, J = 8.68, 2.57 Hz), 7.65 (d, 1H, J = 2.57 Hz). 13C NMR (100 MHz, CDCl3) δC 32.00, 33.37, 42.63, 44.45, 46.36, 115.36, 115.58, 118.57, 118.63, 123.68, 125.30, 128.31, 128.96, 129.04, 131.73, 138.17, 140.10, 140.14, 154.10, 160.21, 162.64. HRMS-ESI: (m/z) calculated for C27H24ClF5N2O, 523.9518 [M+H]+; found 523.1563. Purity: 98.86%.
Biological evaluation. Cell lines and culture condition.
The human breast cancer cells MDA-MB-231 were used for cytotoxicity studies. Cells were cultured at 37 °C, 95% humidity, and 5% (v/v) CO2 in a Napco series 8000 WJ CO2 incubator (ThermoScientific). MDA-MB-231 cells were maintained in Dulbecco’s Modified Eagle Media (DMEM) supplemented with 10% heat-inactivated FBS and 1% antibiotic-antimycotic. Adherent cells were passaged via the addition of trypsin/0.05% EDTA solution (GIBCO), followed by centrifugation and resuspension in fresh media. All cell lines were routinely tested for the presence of mycoplasma infection.
Cytotoxicity assay of analogs on MDA-MB-231 cells.
Cytotoxicity of synthesized compounds was determined by MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) assay. MDA-MB-231 (8000 cells per well) were seeded into 96-well plates in 100 μL of 10% FBS containing DMEM media and incubated for 24 hours. The percentage of cell viability at 20 μM concentration of synthesized analogs was assessed initially. The compounds showed more than 50% growth inhibition at 20 μM concentration were subjected to IC50 determination. DMSO stocks synthesized analogs were diluted in culture media (2% FBS containing DMEM) to obtain a series of concentrations (25, 20, 15, 10, 7.5, 5, 2.5, and 0 μM). The final concentration of DMSO in the treatment medium was 0.1% for all wells. After 48 hours of treatment, MTT solution was added to each well (0.5 mg/mL) and incubated at 37 °C for 2 hours. The media was removed from each well, and the formed blue formazan crystals were dissolved in 100 μL of DMSO. The optical density was determined at the wavelength of 570 nm. For each time point, we have performed triplicate data, and mean cell viability was calculated. IC50 values (± SEM) were calculated using GraphPad Prism 8 Software.
Evaluation of microsomal stability in vitro.
Liver microsomal stability of selected compounds was determined by following modified literature procedures72, 73 and a protocol provided by Waters74. Instead of 96-well plates, microcentrifuge tubes were used as reaction pot/vessel. In brief, 0.5 mM stock solutions of the compounds in DMSO were prepared and then diluted to 5 μM with dilution buffer. Microsomes (The Pooled Male Rat Liver Microsomes, Sprague-Dawley, Cat. # 452501, Corning®,) were rapidly thawed, then diluted with warm potassium-phosphate buffer to prepare a 6.2% microsomal mixture. This mixture was maintained at 37°C (for <30 minutes) while NADPH solution (NADPH Regenerating System - Solution A, Cat. # 451220, Corning® and Solution B, Cat. # 451200, Corning®) in potassium-phosphate buffer was prepared and kept in 4°C. 100 μL of NADPH solution was transferred to each microcentrifuge tube arranged in a tray. 50 μL of each of the 5 μM samples were added to the respective microcentrifuge tube and warmed to 37 °C in a shaker (100 rpm) for approximately 10 minutes. After warming to 37 °C, 100 μL of liver microsome solution was added to each tube and incubated at 37 °C on the shaker (100 rpm) for an hour. 20 μL samples from each tube were collected at 5, 15, 30, and 60 min time points, and 200 μL ice-cold acetonitrile was added to quench the reaction. For the 0 min time point (baseline), 12 μL of the compound-NADPH solution, and 8 μL of liver microsome solution was added to 200 μL ice-cold acetonitrile. Samples were centrifuged for 15 minutes at 4°C, and collected supernatant solutions were analyzed using the LC-MS/MS according to in-house developed LC-MS/MS method. The percent remaining of a compound was calculated relative to the amount of compound found at time point T= 0 using peak area ratios.
In vivo BBB permeability.
All animal studies were carried out according to the Texas Tech University Health Sciences Center Institutional Animal Care and Use Committee (IACUC)-approved protocols. Eight weeks old C57 mice, male and female mice weighing ~20 grams, were used for the experiments. Compound 4a was administered intraperitoneally at a dose 10 mg/kg (vehicle: ethanol 2.5%, DMSO 2.5%, Tween 80 5.0%, PEG 400 25% and PBS 65%). At different time points (0, 0.5, 1, 6, 24, and 48 hours), mice were anesthetized, and blood samples were collected. Brains were collected after performing cardiac perfusion. Plasma and brain were stored at −80 °C until the following LC-MS/MS analysis.
LC-MS/MS method development.
Briefly, an AB SCIEX QTRAP® 5500 tandem mass spectrometer (Framingham, MA), attached to the UHPLC system and electrospray ionization (ESI) interface, maintained in positive ionization mode, was used. All chemicals and solvents (liquid chromatography-mass spectrometry grade) were obtained from Fisher Scientific. The analytes were quantified by multiple-reaction monitoring (MRM) method with the transitions of the parent ions to the product ions of m/z 524.1→149.1 for penfluridol, 455.0→ 165.3 for verapamil, 429.1→ 180.3 for 4a, 497.2→180.3 for 4l, 508.0→412.1 for 4q and m/z 395.1→213.0 for rotenone (internal standard) respectively. Standard calibration curves were constructed by analyzing a series of the blank matrix of corresponding organs from untreated mice spiked with a known amount of tested compounds and internal standard.
The organs and plasma samples collected in animal studies were stored at −80°C prior to protein precipitation and LC-MS/MS analysis. The samples were homogenized in phosphate buffer saline (1:3, w/v). The standards and study samples (30 μl) were extracted using 200 μl acetonitrile containing a mixture of 100 ng/ml of the internal standard. Upon protein precipitation by centrifugation at 14,000 rpm for 10 min, 150 μl of clear supernatant from the top was subjected to analysis. Compounds were eluted using a Kinetex C18 column (50 × 2.1 mm, 1.3 μm; Phenomenex) with a mobile phase consisting of 0.1% formic acid in water-acetonitrile (gradient flow, 5 min) at a flow rate of 0.5 ml/min. The concentration of test compounds in each unknown sample was determined using the linear calibration curve equation for each corresponding drug/internal standard ratio. The peaks of analytes were integrated and quantified using Analyst 1.6.2 software (AB Sciex).
Immunoblot analysis.
The immunoblot analysis was performed according to reported literature.75 The cells were treated for 48 hours and then lysed on ice in RIPA buffer (10 mmol/L Tris-HCl, 1 mmol/L EDTA, 0.5 mmol/L EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS and 140 mmol/L NaCl), supplemented with protease and phosphatase inhibitors (Halt Protease and Phosphatase Inhibitor Cocktail; Thermo Scientific). Cell lysates were centrifuged at 13,000 rpm for 10 min at 4 °C, and each supernatant was mixed with the appropriate volume of 5x SDS loading buffer, heated to 95–100 °C for 5 min and briefly centrifuged. Equal amounts of proteins were subjected to SDS-PAGE and transferred onto an Immobilon P, polyvinylidene difluoride membrane (Millipore, Billerica, MA). The membranes were then incubated with the appropriate primary antibodies: The membranes were then incubated with the appropriate primary antibodies: pFGFR1 (Cat# 3471; 1:1000), Cleaved Caspase-3 (Cat# 9664; 1:1000), Caspase-3 (Cat# 9668; 1:1000) and β-tubulin (1:2000) (all from Cell Signaling Technology, Beverly, MA). As a secondary antibody, goat anti-rabbit was used (1:50000). The antigens were visualized using the Immobilon Western Chemiluminescent HRP substrate (Millipore), according to the manufacturer’s instructions. The protein levels that corresponded to immunoreactive bands were quantified using the Image PC image analysis software (Scion Corp., Frederick, MD) and ImageJ image analysis software (National Institutes of Health).
Docking study.
The docking study was performed with SeeSAR version 10.0; BioSolveIT GmbH, Sankt Augustin, Germany, 2020, www.biosolveit.de/SeeSAR, using the FGFR1 protein structure from the RCSB protein data bank (PDB code: 4V05).
Supplementary Material
Figure 5.
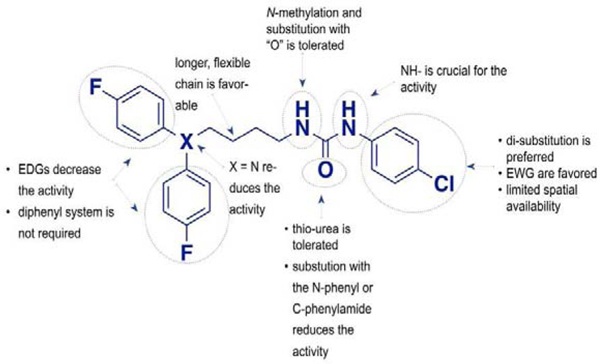
Summary of the SAR data for the anticancer activity of studied urea analogs
Table 3.
In vitro cytotoxic activity of analogs with the modifications at the “tail” di-phenyl moiety.
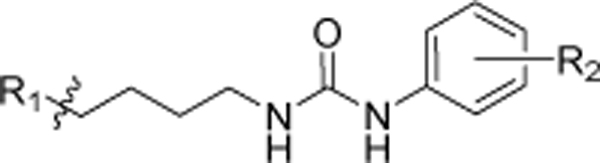 | |||
|---|---|---|---|
| R1 | R2 | MDA-MB-231 aIC50 (μM) | |
| 4l | 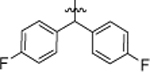 |
4-Cl, 3-CF3 | 3.15 ± 0.11 |
| 8a | 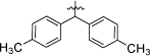 |
4-Cl, 3-CF3 | 2.51 ± 0.09 |
| 8b | 4-Cl | 17.93 ± 0.37 | |
| 8c | 4-Cl, 3-CF3 | 3.30 ± 0.32 | |
| 8d | 4-Cl, 3-NO2 | 3.99 ± 0.44 | |
| 8e | 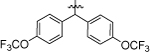 |
4-Cl, 3-CF3 | 1.69 ± 0.11 |
| 8f | 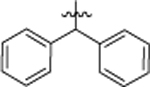 |
4-Cl, 3-CF3 | 5.72 ± 0.26 |
| 8g |  |
4-Cl, 3-CF3 | 1.23 ± 0.20 |
| 8h |  |
4-Cl, 3-CF3 | 12.20 ± 0.59 |
| 8i |  |
4-Cl, 3-CF3 | >20.0 |
| 9a |  |
4-Cl | >20.0 |
| 9b | 4-I | > 20.0 | |
| 9c | 4-Cl, 3-CF3 | 5.04 ± 0.14 | |
| 9d | 4-CH3, 3-CH3 | >20.0 | |
| 9e | 4-Br, 2-I | > 20.0 | |
| 10 | 4-Cl, 3-CF3 | 5.44 ± 0.59 | |
Data are expressed as the mean ± SEM of three independent experiments, each performed in a quartet.
HIGHLIGHTS.
TNBC is a highly metastatic type of breast cancer with poor prognosis
We have identified novel urea-based compounds with the activity against TNBC and ability to cross the BBB in vivo
We identified potential off-target activity at DAT and structural requirements associated with this activity
Our compounds induce apoptosis and modulate FGFR1 expression in MDA-MB-231 triple-negative breast cancer cell lines
ACKNOWLEDGMENTS
Ki determinations and receptor binding profiles were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271–2013-00017-C (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth MD, Ph.D. at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA.
This work is supported by the NIH 1R15CA231339–0 to N.G. and C.M., CPRIT RP170003 to T.P. and N.G, NIH R01 CA186662 and R01CA214019 to R.Z., and American Cancer Society (ACS) grant RSG-15–009-01-CDD to W.W.
ABBREVIATIONS USED
- BBB
blood brain barrier
- bFGF
basic fibroblast growth factor
- Boc
tertbutoxycarbonyl
- CALGB
Cancer and Leukemia Group B
- CNS
central nervous system
- D1–5
dopamine receptor subtypes
- DAT
dopamine transporter
- DCM
dichloromethane
- DMEM
Dulbecco’s Modified Eagle Medium
- DMSO
dimethyl sulfoxide
- DPPH
1,1′-bis(diphenylphosphino)ferrocene
- EMEM
Eagle’s Minimum Essential Medium
- ER
estrogen receptor
- EtOH
ethanol
- FGFR
fibroblast growth factor receptor
- GPCR
G-protein coupled receptor
- HER2
human epidermal growth factor receptor 2
- H1R
histamine 1 receptor
- 5HT
serotonin receptors
- HYDE
hydrogen bond and dehydration energies
- IC50
half-maximum inhibitory concentration
- i.p.
intraperitoneal injection
- Ki
inhibition constant
- LC
liquid chromatography
- LC-MS
liquid chromatography-mass spectrometry
- MDA-MB-231
human triple-negative breast cancer cell line
- MTT
3-[4,5-dimethylthiazol-2-yl]-2,5-dimethyltetrazolium bromide
- NADPH
Nicotinamide adenine dinucleotide phosphate-H
- NIMH
National Institute of Mental Health
- NMR
nuclear magnetic resonance
- PDB
protein data bank
- PDSP
Psychoactive Drug Screening Program
- PFL
penfluridol
- PR
progesterone receptor
- rt
room temperature
- SAR
structure-activity relationship
- SEM
standard error of the mean
- THF
tetrahydrofuran
- TLC
thin-layer chromatography
- TNBC
triple-negative breast cancer
- WSG
West German Study Group
Footnotes
Declaration of interests
☒ The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Siegel RL; Miller KD; Jemal A, Cancer statistics, 2020. CA: A Cancer Journal for Clinicians 2020, 70 (1), 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Oualla K; El-Zawahry HM; Arun B; Reuben JM; Woodward WA; Gamal El-Din H; Lim B; Mellas N; Ueno NT; Fouad TM, Novel therapeutic strategies in the treatment of triple-negative breast cancer. Therapeutic Advances in Medical Oncology 2017, 9 (7), 493–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lehmann BD; Bauer JA; Chen X; Sanders ME; Chakravarthy AB; Shyr Y; Pietenpol JA, Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. Journal of Clinical Investigation 2011, 121 (7), 2750–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dent R; Trudeau M; Pritchard KI; Hanna WM; Kahn HK; Sawka CA; Lickley LA; Rawlinson E; Sun P; Narod SA, Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clinical Cancer Research 2007, 13 (15), 4429–4434. [DOI] [PubMed] [Google Scholar]
- 5.Freedman GM; Anderson PR; Li T; Nicolaou N, Locoregional recurrence of triple-negative breast cancer after breast-conserving surgery and radiation. Cancer 2009, 115 (5), 946–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Voduc KD; Cheang MCU; Tyldesley S; Gelmon K; Nielsen TO; Kennecke H, Breast Cancer Subtypes and the Risk of Local and Regional Relapse. Journal of Clinical Oncology 2010, 28 (10), 1684–1691. [DOI] [PubMed] [Google Scholar]
- 7.Kennecke H; Yerushalmi R; Woods R; Cheang MCU; Voduc D; Speers CH; Nielsen TO; Gelmon K, Metastatic Behavior of Breast Cancer Subtypes. Journal of Clinical Oncology 2010, 28 (20), 3271–3277. [DOI] [PubMed] [Google Scholar]
- 8.Liedtke C; Mazouni C; Hess KR; Andre F; Tordai A; Mejia JA; Symmans WF; Gonzalez-Angulo AM; Hennessy B; Green M; Cristofanilli M; Hortobagyi GN; Pusztai L, Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol 2008, 26 (8), 1275–81. [DOI] [PubMed] [Google Scholar]
- 9.Gluz O; Nitz UA; Harbeck N; Ting E; Kates R; Herr A; Lindemann W; Jackisch C; Berdel WE; Kirchner H; Metzner B; Werner F; Schutt G; Frick M; Poremba C; Diallo-Danebrock R; Mohrmann S; West German Study G, Triple-negative high-risk breast cancer derives particular benefit from dose intensification of adjuvant chemotherapy: results of WSG AM-01 trial. Ann Oncol 2008, 19 (5), 861–70. [DOI] [PubMed] [Google Scholar]
- 10.Bendell JC; Domchek SM; Burstein HJ; Harris L; Younger J; Kuter I; Bunnell C; Rue M; Gelman R; Winer E, Central nervous system metastases in women who receive trastuzumab-based therapy for metastatic breast carcinoma. Cancer 2003, 97 (12), 2972–7. [DOI] [PubMed] [Google Scholar]
- 11.Leone JP; Leone BA, Breast cancer brain metastases: the last frontier. Experimental Hematology & Oncology 2015, 4 (1), 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adkins CE; Mohammad AS; Terrell-Hall TB; Dolan EL; Shah N; Sechrest E; Griffith J; Lockman PR, Characterization of passive permeability at the blood-tumor barrier in five preclinical models of brain metastases of breast cancer. Clin Exp Metastasis 2016, 33 (4), 373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ranjan A; German N; Mikelis C; Srivenugopal K; Srivastava SK, Penfluridol induces endoplasmic reticulum stress leading to autophagy in pancreatic cancer. Tumour Biol 2017, 39 (6). [DOI] [PubMed] [Google Scholar]
- 14.Ranjan A; Srivastava SK, Penfluridol suppresses glioblastoma tumor growth by Akt-mediated inhibition of GLI1. Oncotarget 2017, 8 (20), 32960–32976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu L; Liu YY; Li ZX; Zhao Q; Wang X; Yu Y; Wang YY; Wang YQ; Luo F, Anti-tumor effects of penfluridol through dysregulation of cholesterol homeostasis. Asian Pac J Cancer Prev 2014, 15 (1), 489–94. [DOI] [PubMed] [Google Scholar]
- 16.Hedrick E; Li X; Safe S, Penfluridol Represses Integrin Expression in Breast Cancer through Induction of Reactive Oxygen Species and Downregulation of Sp Transcription Factors. Mol Cancer Ther 2017, 16 (1), 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ranjan A; Gupta P; Srivastava SK, Penfluridol: An Antipsychotic Agent Suppresses Metastatic Tumor Growth in Triple-Negative Breast Cancer by Inhibiting Integrin Signaling Axis. Cancer Res 2016, 76 (4), 877–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw V; Srivastava S; Srivastava SK, Repurposing antipsychotics of the diphenylbutylpiperidine class for cancer therapy. Semin Cancer Biol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tuan NM; Lee CH, Penfluridol as a Candidate of Drug Repurposing for Anticancer Agent. Molecules 2019, 24 (20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashraf-Uz-Zaman M; Sajib MS; Cucullo L; Mikelis CM; German NA, Analogs of penfluridol as chemotherapeutic agents with reduced central nervous system activity. Bioorganic & Medicinal Chemistry Letters 2018, 28 (23–24), 3652–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nair AB; Jacob S, A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 2016, 7 (2), 27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang RI; Larson C; Treul SJ, Study of penfluridol and chlorpromazine in the treatment of chronic schizophrenia. J Clin Pharmacol 1982, 22 (5–6), 236–42. [DOI] [PubMed] [Google Scholar]
- 23.Chouinard G; Annable L, Penfluridol in the treatment of newly admitted schizophrenic patients in a brief therapy unit. Am J Psychiatry 1976, 133 (7), 850–3. [DOI] [PubMed] [Google Scholar]
- 24.Jackson DM; Anden NE; Engel J; Liljequist S, The effect of long-term penfluridol treatment on the sensitivity of the dopamine receptors in the nucleus accumbens and in the corpus striatum. Psychopharmacologia 1975, 45 (2), 151–5. [DOI] [PubMed] [Google Scholar]
- 25.Ota KY; Kurland AA; Slotnick VB, Safety evaluation of penfluridol, a new long-acting oral antipsychotic agent. J Clin Pharmacol 1974, 14 (4), 202–9. [DOI] [PubMed] [Google Scholar]
- 26.Chen G; Xia H; Cai Y; Ma D; Yuan J; Yuan C, Synthesis and SAR study of diphenylbutylpiperidines as cell autophagy inducers. Bioorg Med Chem Lett 2011, 21 (1), 234–9. [DOI] [PubMed] [Google Scholar]
- 27.Ananda Kumar CS; Benaka Prasad SB; Vinaya K; Chandrappa S; Thimmegowda NR; Kumar YC; Swarup S; Rangappa KS, Synthesis and in vitro antiproliferative activity of novel 1benzhydrylpiperazine derivatives against human cancer cell lines. Eur J Med Chem 2009, 44 (3), 1223–9. [DOI] [PubMed] [Google Scholar]
- 28.Cramer RD; Jilek RJ; Guessregen S; Clark SJ; Wendt B; Clark RD, “Lead hopping”. Validation of topomer similarity as a superior predictor of similar biological activities. J Med Chem 2004, 47 (27), 6777–91. [DOI] [PubMed] [Google Scholar]
- 29.Cramer RD; Poss MA; Hermsmeier MA; Caulfield TJ; Kowala MC; Valentine MT, Prospective identification of biologically active structures by topomer shape similarity searching. J Med Chem 1999, 42 (19), 3919–33. [DOI] [PubMed] [Google Scholar]
- 30.Sun H; Tawa G; Wallqvist A, Classification of scaffold-hopping approaches. Drug Discov Today 2012, 17 (7–8), 310–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mauser H; Guba W, Recent developments in de novo design and scaffold hopping. Curr Opin Drug Discov Devel 2008, 11 (3), 365–74. [PubMed] [Google Scholar]
- 32.Penfluridol in the treatment of newly admitted schizophrenic patients in a brief therapy unit. American Journal of Psychiatry 1976, 133 (7), 850–853. [DOI] [PubMed] [Google Scholar]
- 33.Ota KY; Kurland AA; Slotnick VB, Safety Evaluation of Penfluridol, a New Long-Acting Oral Antipsychotic Agent. The Journal of Clinical Pharmacology 1974, 14 (4), 202–209. [DOI] [PubMed] [Google Scholar]
- 34.Dale E; Pehrson AL; Jeyarajah T; Li Y; Leiser SC; Smagin G; Olsen CK; Sanchez C, Effects of serotonin in the hippocampus: how SSRIs and multimodal antidepressants might regulate pyramidal cell function. CNS Spectr 2016, 21 (2), 143–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Al Shoyaib A; Archie SR; Karamyan VT, Intraperitoneal Route of Drug Administration: Should it Be Used in Experimental Animal Studies? Pharmaceutical Research 2019, 37 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Airoldi L; Marcucci F; Mussini E; Garattini S, Distribution of penfluridol in rats and mice. Eur J Pharmacol 1974, 25 (3), 291–5. [DOI] [PubMed] [Google Scholar]
- 37.Bouillot Anne Marie J; Dodic N; Gellibert Francoise J; Mirguet O. Thiazole Compounds As Activators Of Soluble Guanylate Cyclase. WO 2010/015652 A2, 2009/08/05, 2010. [Google Scholar]
- 38.Wang C; Yang G; Li J; Mele G; Slota R; Broda M; Duan M; Vasapollo G; Zhang X; Zhang F, Novel meso-substituted porphyrins: Synthesis, characterization and photocatalytic activity of their TiO2-based composites. Dyes and Pigments 2009, 80 (3), 321–328. [Google Scholar]
- 39.Chen T-HCPPD Synthetic epigallocatechin gallate (egcg) analogs. 2012. [Google Scholar]
- 40.Hassib ST; Hassan GS; El-Zaher AA; Fouad MA; Abd El-Ghafar OA; Taha EA, Synthesis and biological evaluation of new prodrugs of etodolac and tolfenamic acid with reduced ulcerogenic potential. European Journal of Pharmaceutical Sciences 2019, 140. [DOI] [PubMed] [Google Scholar]
- 41.Carlsen PHJ; Sørbye K; Ulven T; Aasbø K; Klinga M; Romerosa A, Synthesis of Benzylidene-Protected Dihydroxyacetone. Acta Chemica Scandinavica 1996, 50, 185–187. [Google Scholar]
- 42.Jurayi J. F. e. a. Preparation of trans-1,2-disubstituted cycloalkanes from 1,2-cycloalkanols and related compounds. 12/24/2003, 2003. [Google Scholar]
- 43.Dorel R; Grugel CP; Haydl AM, The Buchwald–Hartwig Amination After 25 Years. Angewandte Chemie International Edition 2019, 58 (48), 17118–17129. [DOI] [PubMed] [Google Scholar]
- 44.Cao J; Lever JR; Kopajtic T; Katz JL; Pham AT; Holmes ML; Justice JB; Newman AH, Novel azido and isothiocyanato analogues of [3-(4-phenylalkylpiperazin-1-yl)propyl]bis(4-fluorophenyl)amines as potential irreversible ligands for the dopamine transporter. J Med Chem 2004, 47 (25), 6128–36. [DOI] [PubMed] [Google Scholar]
- 45.Xiang J; Wan ZK; Li HQ; Ipek M; Binnun E; Nunez J; Chen L; McKew JC; Mansour TS; Xu X; Suri V; Tam M; Xing Y; Li X; Hahm S; Tobin J; Saiah E, Piperazine sulfonamides as potent, selective, and orally available 11beta-hydroxysteroid dehydrogenase type 1 inhibitors with efficacy in the rat cortisone-induced hyperinsulinemia model. J Med Chem 2008, 51 (14), 4068–71. [DOI] [PubMed] [Google Scholar]
- 46.Appel R, Tertiary Phosphane/Tetrachloromethane, a Versatile Reagent for Chlorination, Dehydration, and P?N Linkage. Angewandte Chemie International Edition in English 1975, 14 (12), 801–811. [Google Scholar]
- 47.Davis M; Deady LW; Paproth TG, The Nitration of alkylbenzenes: A lecture demonstration. Journal of Chemical Education 1978, 55 (1). [Google Scholar]
- 48.Esteves PM; de M. Carneiro JW; Cardoso SP; Barbosa AGH; Laali KK; Rasul G; Prakash GKS; Olah GA, Unified Mechanistic Concept of Electrophilic Aromatic Nitration: Convergence of Computational Results and Experimental Data. Journal of the American Chemical Society 2003, 125 (16), 4836–4849. [DOI] [PubMed] [Google Scholar]
- 49.Benati L; Bencivenni G; Leardini R; Minozzi M; Nanni D; Scialpi R; Spagnolo P; Zanardi G, Radical reduction of aromatic azides to amines with triethylsilane. J Org Chem 2006, 71 (15), 5822–5. [DOI] [PubMed] [Google Scholar]
- 50.Saito Y; Matsumoto K; Bag SS; Ogasawara S; Fujimoto K; Hanawa K; Saito I, C8-alkynyl- and alkylamino substituted 2′-deoxyguanosines: a universal linker for nucleic acids modification. Tetrahedron 2008, 64 (16), 3578–3588. [Google Scholar]
- 51.Liu D; Tian Z; Yan Z; Wu L; Ma Y; Wang Q; Liu W; Zhou H; Yang C, Design, synthesis and evaluation of 1,2-benzisothiazol-3-one derivatives as potent caspase-3 inhibitors. Bioorg Med Chem 2013, 21 (11), 2960–7. [DOI] [PubMed] [Google Scholar]
- 52.Chen J-N; Wang X-F; Li T; Wu D-W; Fu X-B; Zhang G-J; Shen X-C; Wang H-S, Design, synthesis, and biological evaluation of novel quinazolinyl-diaryl urea derivatives as potential anticancer agents. European Journal of Medicinal Chemistry 2016, 107, 12–25. [DOI] [PubMed] [Google Scholar]
- 53.Yadagiri B; Holagunda UD; Bantu R; Nagarapu L; Guguloth V; Polepally S; Jain N, Rational design, synthesis and anti-proliferative evaluation of novel benzosuberone tethered with hydrazide-hydrazones. Bioorg Med Chem Lett 2014, 24 (21), 5041–4. [DOI] [PubMed] [Google Scholar]
- 54.Brough PA; Baker L; Bedford S; Brown K; Chavda S; Chell V; D’Alessandro J; Davies NG; Davis B; Le Strat L; Macias AT; Maddox D; Mahon PC; Massey AJ; Matassova N; McKenna S; Meissner JW; Moore JD; Murray JB; Northfield CJ; Parry C; Parsons R; Roughley SD; Shaw T; Simmonite H; Stokes S; Surgenor A; Stefaniak E; Robertson A; Wang Y; Webb P; Whitehead N; Wood M, Application of Off-Rate Screening in the Identification of Novel Pan-Isoform Inhibitors of Pyruvate Dehydrogenase Kinase. J Med Chem 2017, 60 (6), 2271–2286. [DOI] [PubMed] [Google Scholar]
- 55.Chayah M; Camacho ME; Carrión MD; Gallo MA; Romero M; Duarte J, N,N′-Disubstituted thiourea and urea derivatives: design, synthesis, docking studies and biological evaluation against nitric oxide synthase. MedChemComm 2016, 7 (4), 667–678. [Google Scholar]
- 56.Ponzano S; Berteotti A; Petracca R; Vitale R; Mengatto L; Bandiera T; Cavalli A; Piomelli D; Bertozzi F; Bottegoni G, Synthesis, biological evaluation, and 3D QSAR study of 2-methyl-4-oxo-3-oxetanylcarbamic acid esters as N-acylethanolamine acid amidase (NAAA) inhibitors. J Med Chem 2014, 57 (23), 10101–11. [DOI] [PubMed] [Google Scholar]
- 57.Sumita A; Kurouchi H; Otani Y; Ohwada T, Acid-Promoted Chemoselective Introduction of Amide Functionality onto Aromatic Compounds Mediated by an Isocyanate Cation Generated from Carbamate. Chemistry - An Asian Journal 2014, 9 (10), 2995–3004. [DOI] [PubMed] [Google Scholar]
- 58.Manickam M; Jalani HB; Pillaiyar T; Boggu PR; Sharma N; Venkateswararao E; Lee YJ; Jeon ES; Son MJ; Woo SH; Jung SH, Design and synthesis of sulfonamidophenylethylureas as novel cardiac myosin activator. Eur J Med Chem 2018, 143, 1869–1887. [DOI] [PubMed] [Google Scholar]
- 59.De Luca L; Giacomelli G; Masala S; Porcheddu A, Trichloroisocyanuric/TEMPO Oxidation of Alcohols under Mild Conditions: A Close Investigation. The Journal of Organic Chemistry 2003, 68 (12), 4999–5001. [DOI] [PubMed] [Google Scholar]
- 60.Winterton SE; Capota E; Wang X; Chen H; Mallipeddi PL; Williams NS; Posner BA; Nijhawan D; Ready JM, Discovery of Cytochrome P450 4F11 Activated Inhibitors of Stearoyl Coenzyme A Desaturase. Journal of Medicinal Chemistry 2018, 61 (12), 5199–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Walsh DA; Franzyshen SK; Yanni JM, Synthesis and antiallergy activity of 4-(diarylhydroxymethyl)-1-[3-(aryloxy)propyl)piperidines and structurally related compounds. Journal of Medicinal Chemistry 1989, 32 (1), 105–118. [DOI] [PubMed] [Google Scholar]
- 62.Sander K; Galante E; Gendron T; Yiannaki E; Patel N; Kalber TL; Badar A; Robson M; Johnson SP; Bauer F; Mairinger S; Stanek J; Wanek T; Kuntner C; Kottke T; Weizel L; Dickens D; Erlandsson K; Hutton BF; Lythgoe MF; Stark H; Langer O; Koepp M; Årstad E, Development of Fluorine-18 Labeled Metabolically Activated Tracers for Imaging of Drug Efflux Transporters with Positron Emission Tomography. Journal of Medicinal Chemistry 2015, 58 (15), 6058–6080. [DOI] [PubMed] [Google Scholar]
- 63.Ghosh AK; Brindisi M, Urea Derivatives in Modern Drug Discovery and Medicinal Chemistry. J Med Chem 2020, 63 (6), 2751–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Perez-Garcia J; Muñoz-Couselo E; Soberino J; Racca F; Cortes J, Targeting FGFR pathway in breast cancer. The Breast 2018, 37, 126–133. [DOI] [PubMed] [Google Scholar]
- 65.Lehmann BD; Pietenpol JA, Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. The Journal of Pathology 2014, 232 (2), 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee HJ; Seo AN; Park SY; Kim JY; Park JY; Yu JH; Ahn J-H; Gong G, Low Prognostic Implication of Fibroblast Growth Factor Family Activation in Triple-negative Breast Cancer Subsets. Annals of Surgical Oncology 2014, 21 (5), 1561–1568. [DOI] [PubMed] [Google Scholar]
- 67.Sharpe R; Pearson A; Herrera-Abreu MT; Johnson D; Mackay A; Welti JC; Natrajan R; Reynolds AR; Reis-Filho JS; Ashworth A; Turner NC, FGFR Signaling Promotes the Growth of Triple-Negative and Basal-Like Breast Cancer Cell Lines Both In Vitro and In Vivo. Clinical Cancer Research 2011, 17 (16), 5275–5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.De Luca A; Frezzetti D; Gallo M; Normanno N, FGFR-targeted therapeutics for the treatment of breast cancer. Expert Opinion on Investigational Drugs 2017, 26 (3), 303–311. [DOI] [PubMed] [Google Scholar]
- 69.Tucker Julie A.; Klein T; Breed J; Breeze Alexander L.; Overman R; Phillips C; Norman Richard A., Structural Insights into FGFR Kinase Isoform Selectivity: Diverse Binding Modes of AZD4547 and Ponatinib in Complex with FGFR1 and FGFR4. Structure 2014, 22 (12), 1764–1774. [DOI] [PubMed] [Google Scholar]
- 70.Wang H; Huwaimel B; Verma K; Miller J; Germain TM; Kinarivala N; Pappas D; Brookes PS; Trippier PC, Synthesis and Antineoplastic Evaluation of Mitochondrial Complex II (Succinate Dehydrogenase) Inhibitors Derived from Atpenin A5. ChemMedChem 2017, 12 (13), 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Slack RD; Ku TC; Cao J; Giancola JB; Bonifazi A; Loland CJ; Gadiano A; Lam J; Rais R; Slusher BS; Coggiano M; Tanda G; Newman AH, Structure-Activity Relationships for a Series of (Bis(4-fluorophenyl)methyl)sulfinyl Alkyl Alicyclic Amines at the Dopamine Transporter: Functionalizing the Terminal Nitrogen Affects Affinity, Selectivity, and Metabolic Stability. J Med Chem 2020, 63 (5), 2343–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Di L; Kerns EH; Hong Y; Kleintop TA; McConnell OJ; Huryn DM, Optimization of a higher throughput microsomal stability screening assay for profiling drug discovery candidates. J Biomol Screen 2003, 8 (4), 453–62. [DOI] [PubMed] [Google Scholar]
- 73.Bera R; Kundu A; Sen T; Adhikari D; Karmakar S, In Vitro Metabolic Stability and Permeability of Gymnemagenin and Its In Vivo Pharmacokinetic Correlation in Rats - A Pilot Study. Planta Med 2016, 82 (6), 544–50. [DOI] [PubMed] [Google Scholar]
- 74.Alden D. S. a. P. Determination of Microsomal Stability by UPLC/MS/MS. https://www.waters.com/waters/library.htm?locale=en_us&lid=10070741 (accessed March 27). [Google Scholar]
- 75.Zahra FT; Sajib MS; Ichiyama Y; Akwii RG; Tullar PE; Cobos C; Minchew SA; Doçi CL; Zheng Y; Kubota Y; Gutkind JS; Mikelis CM, Endothelial RhoA GTPase is essential for in vitro endothelial functions but dispensable for physiological in vivo angiogenesis. Scientific Reports 2019, 9 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.