

Introduction
Paclitaxel is the frontline treatment for many of the most common solid tumors [12; 35]. The major dose-limiting toxicity of paclitaxel is chemotherapy induced peripheral neuropathy (CIPN) [2; 7], the mechanisms of which are complex and yet to be defined in detail. Systemic treatment of rodents with paclitaxel is associated with loss of epidermal nerve fibers (ENFs), and alterations in the expression of dorsal root ganglion (DRG) ion channels, altered intracellular signaling, mitochondrial and oxidative stress, and pathological changes in neurotransmission [4; 27; 36; 40; 45; 48]. Ectopic spontaneous activity and altered excitability in primary afferents [18; 19; 41; 46] that develops appear to underlie the paresthesia, dysesthesia, and pain phenotypically exhibited by CIPN patients [4].
Increased cytokine signaling is another key element of CIPN pathophysiology. Systemic chemotherapy increases release of interleukin (IL) −1β, IL-6, IL-8, tumor necrosis factor (TNF)-α, and C-C chemokine ligand 2 (CCL2) [39]; increases IL-6 and IL-8 are in skin biopsies from CIPN patients [4; 33; 37]; and increases the expression of CCL2 and its receptor, CCR2, in DRG neurons [45]. CCL2/CCR2 inhibitors attenuate the development of mechanical hypersensitivity and prevent the loss of ENFs by paclitaxel [45].
Toll-like receptor 4 (TLR4) also has a key role in paclitaxel-induced CIPN [15; 20; 21]. Exposure of monocytes to paclitaxel in vitro results in the activation of TLR4 downstream mediators including the canonical myeloid differentiation response protein 88 (MyD88) and TIR-domain-containing adapter-inducing interferon-β (TRIF) pathways as well as the non-canonical extracellular signal regulated kinase (ERK) and p38 mitogen-activated protein kinase (MAPK) pathways that both result in activation of nuclear factor kappa B (NF-κB) [11] which induces increased synthesis and release of numerous proinflammatory cytokines [5]. TLR4 downstream pathways are also upregulated in DRG cellular elements after in vivo paclitaxel treatment [20]; and co-treatment with a TLR4 antagonists, including lipopolysaccharide from Rhodobacter sphaeroides (LPS-RS), inhibits paclitaxel-induced intracellular signaling in vivo and prevents the development of the CIPN behavioral phenotype. Similarly, MAPK inhibitors also prevent development of paclitaxel induced CIPN [20; 21].
Clinical studies show pain symptoms often develop within days following the initial paclitaxel infusion [23]; and importantly, higher levels of acute pain is predictive of more severe neuropathy as treatment progresses [31; 34]. This suggests that a more detailed understanding of the acute pathophysiology of paclitaxel on the DRG would be of benefit in identifying potential new therapeutic advances. An in vitro system could be of great benefit in this regard. To this end, we test here whether the biochemical and physiological changes seen in the DRG following in vivo paclitaxel administration (specifically those related to IL-6, CCL2/CCR2, TLR4, and MAPK) are also recruited in the acute responses of DRG neurons to paclitaxel in an in vitro preparation.
Methods
Animals
All experimental protocols were approved by the Institutional Animal Care and Use Committee at The University of Texas MD Anderson Cancer Center and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All procedures were designed to minimize animal discomfort and to use the fewest animals possible for statistical analysis. A total of 48 three-week-old male and female Sprague-Dawley rats (Harlan, Houston, TX) housed in temperature- and light-controlled (12 hour light/dark cycle) conditions with food and water available ad libitum were used.
Primary Culture of DRG Neurons
The rats were decapitated after anesthesia with pentobarbital (390 mg/mL)/phenytoin (50 mg/mL). The spinal column was removed and bisected to allow for DRGs to be dissected free. All ganglia of both sides were removed from cervical, thoracic, and lumbar levels and placed in a small petri dish. Ganglia were placed in type I collagenase (3 mg/mL, Sigma-Aldrich) in a heated (37°C) chamber for 45 min. After the collagenase was removed with PBS washing, 1 mL trypsin (0.25%) was added for 10 min in the same heated chamber. Digestion was inhibited by fetal bovine serum, and the cells were spun at 180 g for 5 min. The supernatant was removed and replaced with fresh DMEM containing fetal bovine serum (FBS). The cells were dissociated by a polished Pasteur pipette and plated on a poly-L-lysine coated 24-well plate, 35-mm plastic dish, or glass sheet for culture [16]. In experiments where paclitaxel was added, it was diluted and added directly into the culture medium. The dose range was chosen based on the level of paclitaxel measured in the DRG following systemic treatment used to establish the initial published model of paclitaxel CIPN in vivo and accounting for the half-life of the agent of 13–52 hours [32; 42]. Paclitaxel was shown in this work to be very persistent in the DRG following systemic treatment.
Enzyme-Linked Immunosorbent Assay
Culture supernatants were collected and assayed at 30 min, 2 h, 4 h, 12 h, 24 h and 48 h and grouped by various paclitaxel doses at 3 nM, 10 nM, 30 nM, 100 nM, 300 nM, 1 μM, 3 μM and 10 μM. The levels of IL-6 and CCL2 were assayed according to protocol with commercial enzyme-linked immunosorbent assay (ELISA) kits provided by R&D Systems Inc.
Western Blot Analysis
After the culture medium was discarded, cultured cells were scratched from the dish bottom and collected into centrifuge tubes. Primary cultured DRG neurons were disrupted in lysis buffer on ice. The supernatant was then transferred and denatured with sample buffer for 10 min at 70°C. Lysates (total protein, 20 μg) were separated on SDS-polyacrylamide gel electrophoresis gels and transferred to polyvinylidene fluoride membranes (Bio-Rad). After the membranes were blocked with 5% fat-free milk in Tris-buffered saline with Tween 20 (137 mM NaCl, 20 mM Tris, 0.1% Tween 20) for 1 h at room temperature, they were then incubated with antisera to anti-IL-6 (mouse anti-rat, 1:5,000; Abcam), anti-CCL2 (rabbit anti-rat, 1:2,000; Abcam), anti-TLR-4 (rabbit anti-rat 1:1000, Proteintech, Chicago, IL), anti-MyD88 (rabbit anti-rat 1:1000, Imgenex), mouse anti-phospho-ERK1/2 and rabbit anti-ERK1/2 antibody (1:2000; Cell Signaling Technology, Beverly, MA), rabbit anti-GAPDH antibody (1:5000; Cell Signaling Technology, Beverly, MA), rabbit anti-phospho-P38 and anti-P38 antibody (1:1000; Cell Signaling Technology, Beverly, MA), rabbit anti-phospho-JNK (1:2000; Abcam, Cambridge, MA) and anti-JNK antibody (1:5000; Cell Signaling Technology, Beverly, MA) and β-actin (mouse anti-rat, 1:10,000; Sigma) in 5% fat-free milk in Tris-buffered saline with Tween 20 overnight at 4°C. After the membranes were washed with Tris-buffered saline with Tween 20, they were incubated with a goat anti-rabbit or goat anti-mouse antibody labeled with horseradish peroxidase (Calbiochem) diluted with 5% fat-free milk in Tris-buffered saline with Tween 20 for 1 h at room temperature and then detected with use of enhanced chemiluminescence reagents (GE Healthcare). The blots were scanned by using Adobe Photoshop (version 8.0; Adobe), and the band densities were detected and compared with use of ImageJ (National Institutes of Health).
Immunohistochemical Analysis
Culture medium for DRG neurons was discarded and replaced with cold 4% paraformaldehyde in 0.1M PB. Cells were washed 10 min later with 0.01M PBS and blocked in 5% normal donkey serum and 0.2% Triton X-100 in PBS for 1 h at room temperature, they were incubated overnight at 4°C in 1% normal donkey serum and 0.2% Triton X-100 in PBS containing primary antibodies against the following targets: IL-6 (mouse anti-rat, 1:2000; Abcam), CCL2 (rabbit anti-rat, 1:500; Abcam), CCR2 (goat anti-rat, 1:200; Santa Cruz), TLR4 (rabbit anti-rat or mouse anti-rat, 1:500; Abcam), IB4 (1:1000 BS-Isolectin B4 FITC Conjugate, Sigma, St. Louis, MO), calcitonin gene-related peptide (CGRP, mouse anti-rat, 1:1000; Abcam, Cambridge, MA), glial fibrillary acid protein (GFAP, mouse anti-rat, 1:1000; Cell Signaling Technology, Beverly, MA), MAP-2 (chicken anti-rat, 1:10000; Abcam), phospho-ERK1/2 (rabbit anti-human, 1:1000; Cell Signaling Technology, Beverly, MA). After the cells were washed, they were then incubated with Cy3-, Cy5- or FITC-conjugated secondary antibodies overnight at 4°C. Culture sheets with cells were viewed under a fluorescent microscope (Eclipse E600; Nikon), and images were collected with use of identical acquisition parameters by experimenters blinded to treatment groups.
MitoSOX Red (Mitochondrial Superoxide Indicator) imaging
A 5 μM working solution of MitoSOX reagent (Thermo Fisher Scientific) was prepared. Cultured DRG neurons were incubated with vehicle or paclitaxel (300 nM) for 2, 4, 24 or 48 h. Coverslips containing adhered DRG neurons were placed in a petri dish and treated with 2 ml of 5 μM MitoSOX working solution for 10 minutes at 37˚C while protected from light exposure. Coverslips were washed three times with warm buffer before images were taken.
Whole Cell Recording
Immunohistochemical and neurophysiological responses of cervical, thoracic and lumbar DRG to systematic treatment with paclitaxel were identical [26; 45], so acute whole cell recordings were done here on L4 and L5 DRG alone. The tissue was excised from rats deeply anesthetized with isoflurane and placed in a culture dish containing trypsin (0.0625 mg/mL; Hyclone) and type IA collagenase (1 mg/mL; Sigma-Aldrich) in DMEM. The dish was shaken for 50 min in a heated (37°C) chamber. The cells were washed, mechanically dispersed with a polished Pasteur pipette and plated on poly-L-lysine coated glass sheets in a culture dish. Whole cell patch recording was performed as described previously [16; 19]. The sheets were transferred to a recording chamber placed on an inverted microscope (Eclipse Ti; Nikon) and perfused with oxygenated ACSF (2 ml/min) at room temperature. Only neurons with a stable resting membrane potential of at least −40 mV and evoked spikes that overshot 0 mV were used. Series resistance was compensated to 70%. Whole cell recordings were completed within 6 h after plating.
Human DRG neuron preparation
Human DRG were collected from patient donors at MD Anderson Cancer Center who had surgical resection of metastatic oncological disease that involved ligation of dorsal roots and had provided legal written consent. The human subjects’ protocol was reviewed and approved by the M.D. Anderson Institutional Review Board. The dissociation method was as in our previous work [17]. Dissociated neurons were placed on coverslips that had been precoated with poly-L-Lysine. Neurons were cultured at 37°C with 5% CO2 in DMEM/F12 supplemented with 10% horse serum, 2 mM glutamine, and 25 ng/ml human nerve growth factor. IHC was performed as described above for rat DRG.
Drug Administration
Pharmaceutical-grade Taxol (TEVA Pharmaceuticals) was diluted with culture medium from the original stock concentration of 6 mg/ml to desired concentration (3, 10, 30, 100, 300 nM and 1, 3, 10 μM). CCL2 (R&D Systems, Inc. Minneapolis, MN, USA) was dissolved in sterile PBS and diluted in sterile ACSF solution to the desired concentration. The TLR4 inhibitor LPS derived from R. sphaeroides (LPS-RS) was from InvivoGen, San Diego, CA, the concentration of LPS-RS is 2 μg/ml in culture medium.
Data Analysis
Data are expressed throughout as the mean ± standard error of mean. Statistical analysis was performed using Prism 8 (GraphPad Software). Western blot, ELISA, rheobase and MitoSOX data were compared using two-way ANOVA. The resting membrane potential (RMP) and current threshold results were compared using the Mann-Whitney U test. CCL2 induced the change of RMP was assessed using paired t-test. P < 0.05 was considered statistically significant.
Results
Upregulation of CCL2 and IL-6 and Co-localization with TLR4 in Primary DRG Culture after Paclitaxel Treatment
Paclitaxel- and vehicle-treated primary DRG cultures were analyzed for CCL2 and IL-6 expression. Western blot data demonstrated elevated CCL2 expression in paclitaxel-treated culture compared to the vehicle-treated culture at 2 h after treatment; this upregulation could not be prevented by LPS-RS (Fig. 1A). CCL2 expression was transiently elevated in culture as Western blots showed that CCL2 expression returned to baseline by 48h of paclitaxel treatment (Fig. 1A, n=3/group/time points). In contrast to CCL2, Western blots revealed no changes in expression of IL-6 after 2h of paclitaxel treatment, but IL-6 was significantly increased after 48h of paclitaxel exposure relative to vehicle treatment (Fig. 1B, n=3/group/time points). Similar to the Western studies, ELISA revealed a significant increase in the release of CCL2 to the culture medium (~1.5–2 fold) at 2 h after incubation with paclitaxel compared with vehicle treatment (Fig. 1D). But, in contrast to the western studies, increased release of CCL2 was blocked by LPS-RS (Fig. 1E). The ELISA experiments paralleled the western blot findings for IL-6, as paclitaxel induced no change in release of IL-6 to the medium at 2h of treatment, but there was a significant increase was observed at 48 h (>2-fold) compared to vehicle treatment (Fig. 1F). LPS-RS prevented the upregulation of IL-6 as measured by both Western blots and ELISA (Fig. 1G). ELISA was repeated 6 times (n=6/concentration/time point).
Figure 1.
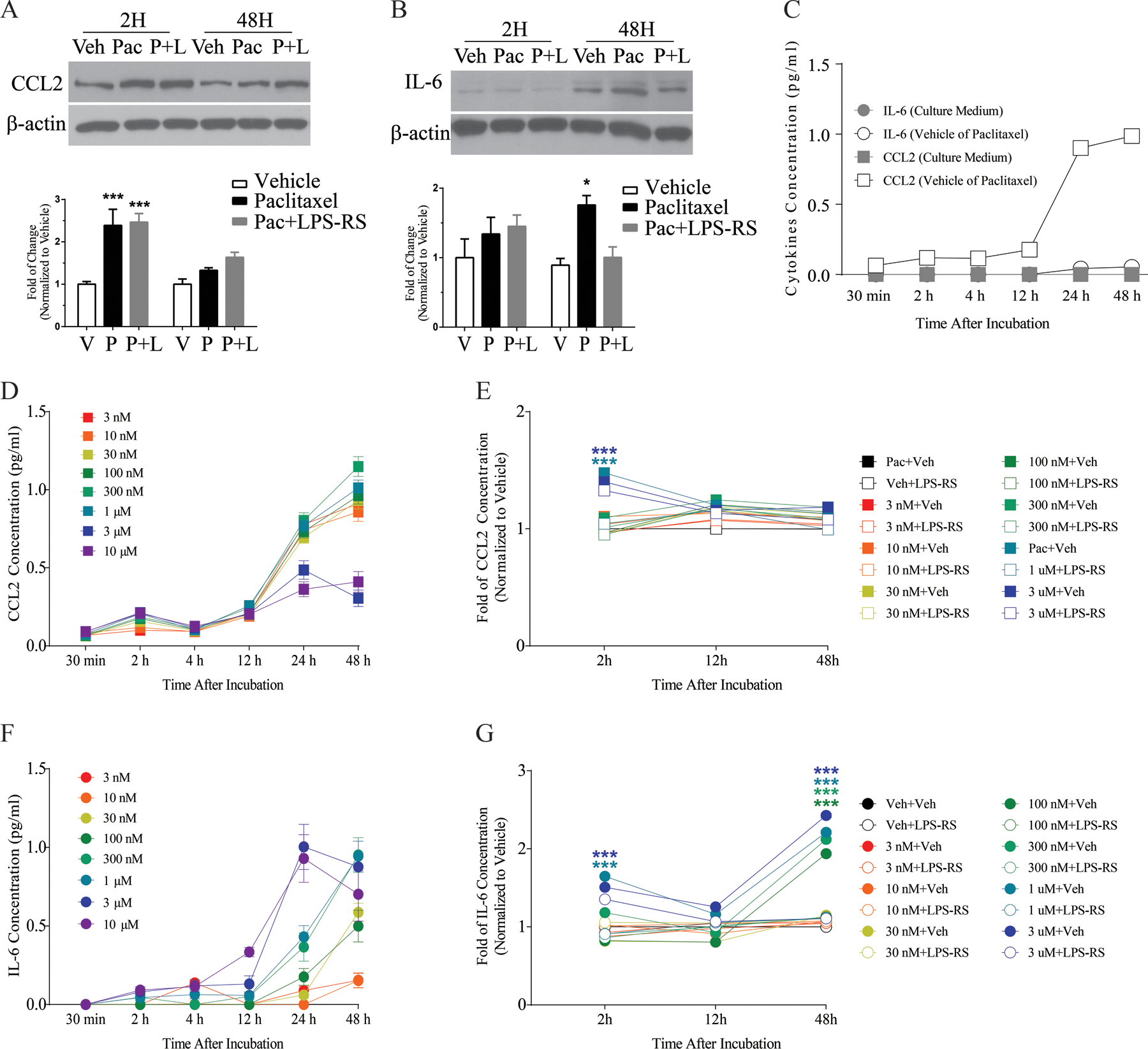
The representative Western blot images and corresponding bar graph summaries illustrate changes in CCL2 (A) and IL-6 (B) expression in primary DRG culture at 2 h and 48 h after paclitaxel (300 nM) incubation. The bar graph in (A) shows that CCL2 was significantly increased in the paclitaxel-treated culture (black bar) compared with the vehicle-treated culture (white bar) at 2 h and that co-treatment of LPS-RS (2μg/ml) with paclitaxel (grey bar) did not eliminate this paclitaxel-induced upregulation (N=3/group/time point). No significant change in expression of CCL2 was observed at 48 h in paclitaxel-treated culture compared with vehicle-treated culture (A). The representative Western blot image and bar graph in (B) illustrate changes in IL-6 expression in primary DRG culture at 2 h and 48 h after paclitaxel (300nM) treatment (N=3/group/time point). The bar graph shows that the change of IL-6 was significantly increased in the paclitaxel-treated culture (black bar) compared with the vehicle-treated culture (white bar) at 48 h and that co-treatment of LPS-RS with paclitaxel (grey bar) blocked paclitaxel-induced upregulation of IL-6. No significant change in expression of IL-6 was observed in the 2 h paclitaxel-treated culture compared with vehicle-treated culture (B). The concentration of CCL2 and IL-6 from ELISA experiments in culture media treated with different paclitaxel doses at different time points is represented in the line and scatter plots (C-G, N=6/concentration/time point). C shows the concentration of CCL2 and IL-6 release from vehicle only or culture medium, where CCL2 was significantly elevated after vehicle treatment at 24 and 48 h. D shows that a significant amount of CCL2 (pg/ml) was released in DRG culture after paclitaxel treatment (1, 3 and 10 μM) compared with vehicle treatment at 2 h and there was no significant difference in the concentration of CCL2 released at the 48 h time point for any dose of paclitaxel compared with vehicle treatment. Cultures co-treated with paclitaxel and LPS-RS demonstrated a significantly lower fold change of CCL2 release compared with paclitaxel alone at the 2 h time point (E). Data from ELISA experiments is represented in F and G, and F shows a significant increase in the concentration of IL-6 released in primary DRG cultures treated with paclitaxel (30, 100, 300 nM and 1, 3, 10 μM) for 48 h compared with cultures treated with vehicle for 48 h. Cultures co-treated with paclitaxel and LPS-RS demonstrated a significantly lower fold change in IL-6 concentration compared with paclitaxel alone at 48 h (G). There was no significant change in the concentration of IL-6 released at 2 h compared with vehicle treatment (F). *p<0.05, **p<0.01, ***p<0.001.
We next examined whether CCL2 or IL-6 were produced by TLR4-expressing neurons using double immunofluorescence. As shown in Figure 2, TLR4 (green, Fig. 2A) was expressed on cultured neurons exposed to vehicle alone as was CCL2 (red, Fig. 2B) and these labels were often co-localized (yellow, Fig. 2C). After 2 h of paclitaxel incubation (300 nM), more neurons were both TLR4 positive (Fig. 2D) and CCL2 positive (Fig. 2E) and these were also typically co-localized (Fig. 2F) in the majority of cells observed. There was also a small population of cells in both sets of cultures that observed to express only TLR4 (CCL2 negative) or CCL2 (TLR4 negative). IL-6 was markedly increased in cultured DRG neurons 48 h after paclitaxel treatment when compared to the low levels observed in vehicle-treated cultures (Fig. 3), consistent with Western blot results (Fig. 1B.) IL-6, like CCL2, was observed in many TLR4-positive cells at 48 h after paclitaxel treatment of primary DRG neuron cultures (Fig. 3).
Figure 2.
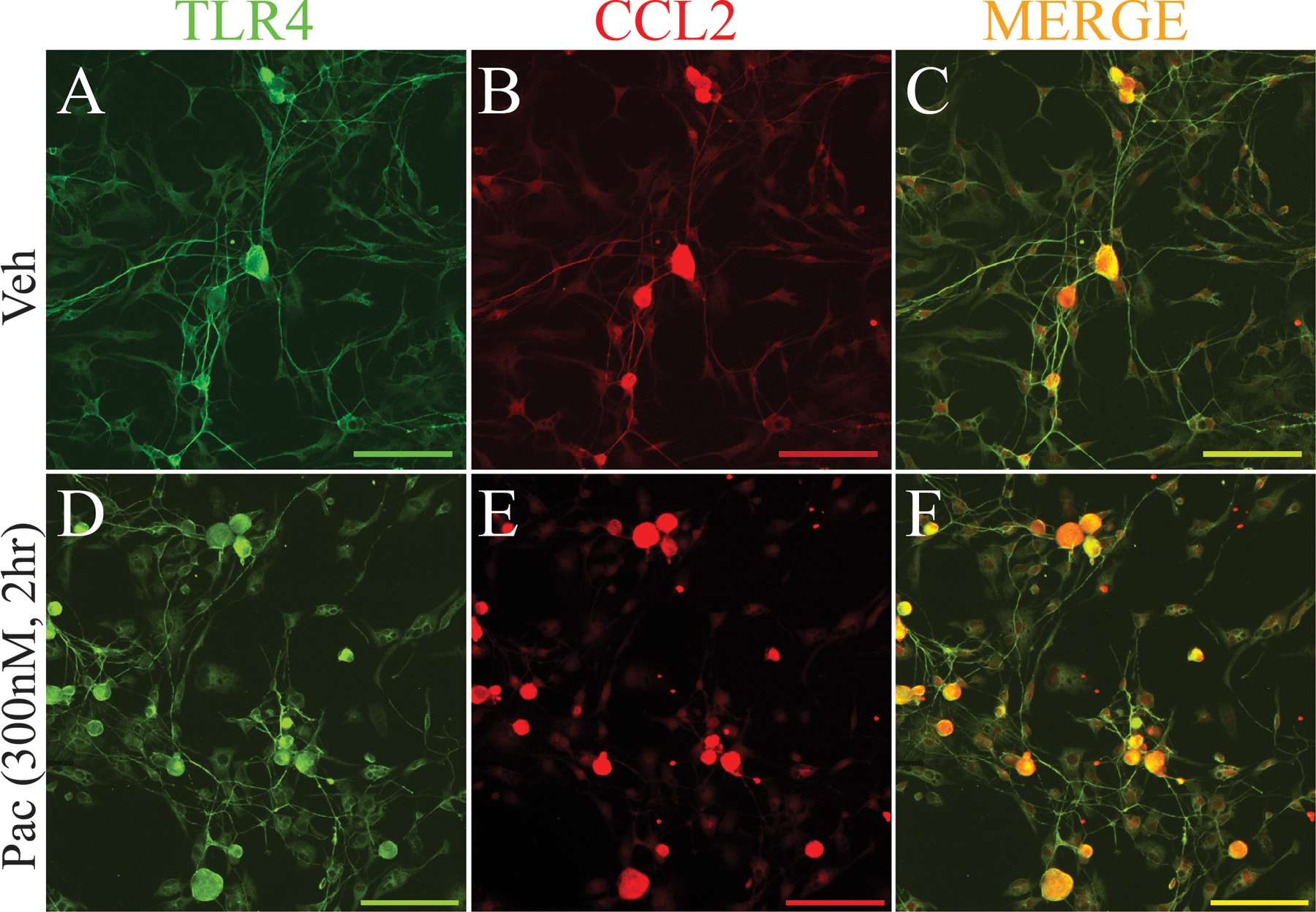
Immunostaining demonstrates increased expression of CCL2 (red) in neurons in paclitaxel-treated culture (300 nM) (E) compared with neurons in vehicle-treated culture (B) at 2 h after paclitaxel incubation. Double labeling (C and F, yellow) indicates that TLR4 (A and D, green) and CCL2 were strongly co-localized in neurons from both paclitaxel and vehicle conditions. Scale bar=100 μm.
Figure 3.
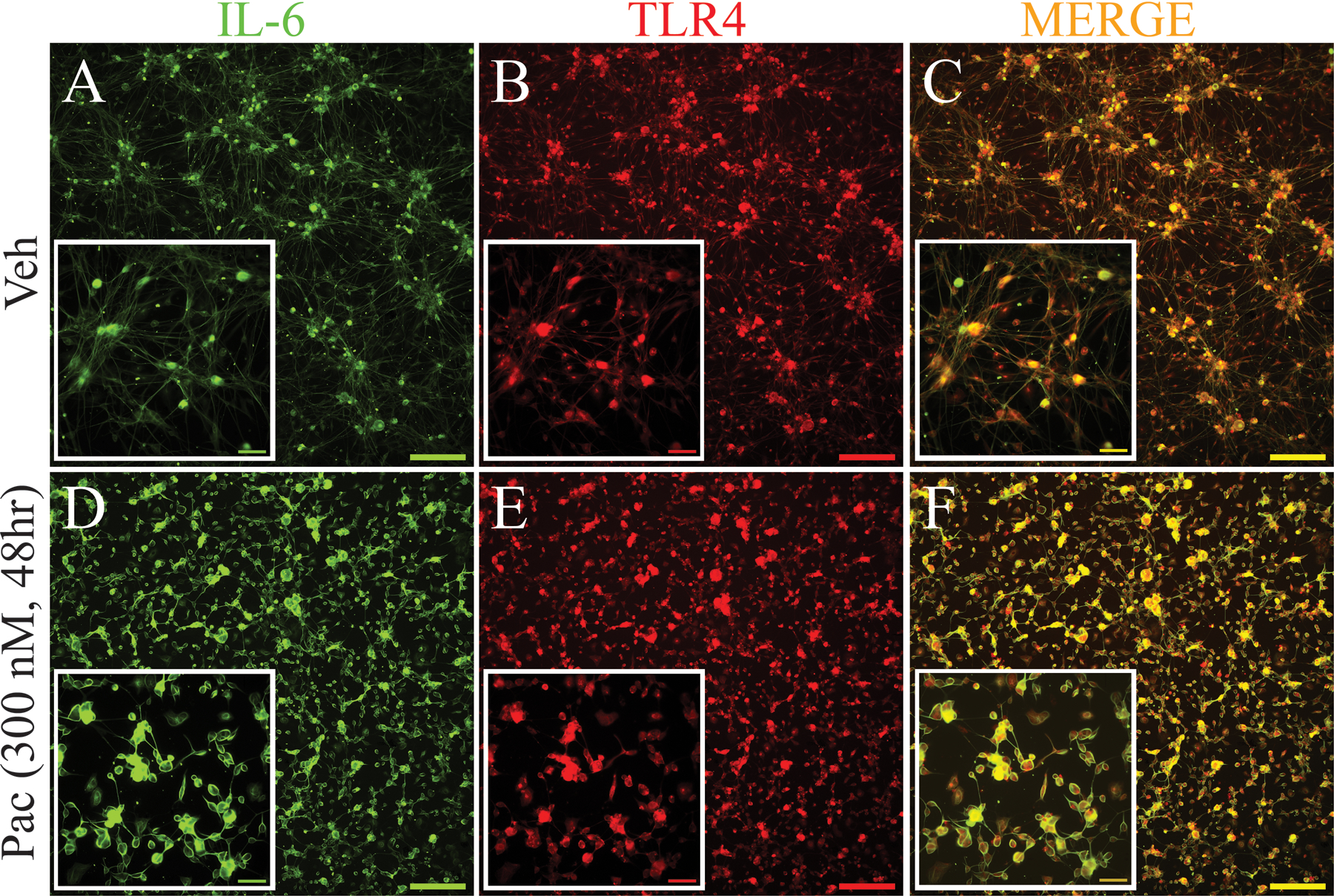
Immunohistochemical stains in (A) and (D) show significant upregulation of IL-6 (green) in primary DRG culture at 48 h after incubation with paclitaxel compared with vehicle-treated culture. The immunohistochemical stains in B and E show significant upregulation of TLR4 (red) in DRG neurons in primary DRG culture at 48 h after incubation with paclitaxel compared with vehicle-treated culture. Double immunohistochemical images in (C) and (F) show that IL-6 (green) colocalized with TLR4 (red) in neurons in both vehicle and paclitaxel conditions. (C and F, colocalization indicated by yellow). Scale bar=200 μm and 100 μm in inserted pictures.
Paclitaxel Upregulates TLR4, MyD88 and pERK Expression in Primary DRG Culture after 48 h of Incubation and pERK and pp38 after 2 h of paclitaxel incubation
Western blots indicated no significant change in the expression of TLR4 or MyD88 at 2 h after paclitaxel treatment (300 nM) compared to that observed in vehicle-treated cultures. However, the expression of both TLR4 and MyD88 was significantly increased after 48 h of paclitaxel treatment (300 nM). This was prevented by co-treatment with LPS-RS (Fig. 4A, B, n=3/group/time point). The expression of pERK was increased at both 2 and 48 h after paclitaxel treatment compared to that observed in vehicle-treated cultures and this also was prevented by co-treatment with LPS-RS (Fig. 4C, n=3/group/time point). The expression of pp38 was also significantly increased at 2 h of paclitaxel treatment, and this was prevented by co-treatment with LPS-RS (Fig. 4D, n=3/group/time point). In contrast to pERK, the expression of pp38 was not different from that in vehicle-treated cell cultures at 48h. There was no significant change in expression of pJNK at either time point after paclitaxel treatment compared to that observed in vehicle-treated cultures (Fig. 4E, n=3/group/time point).
Figure 4.
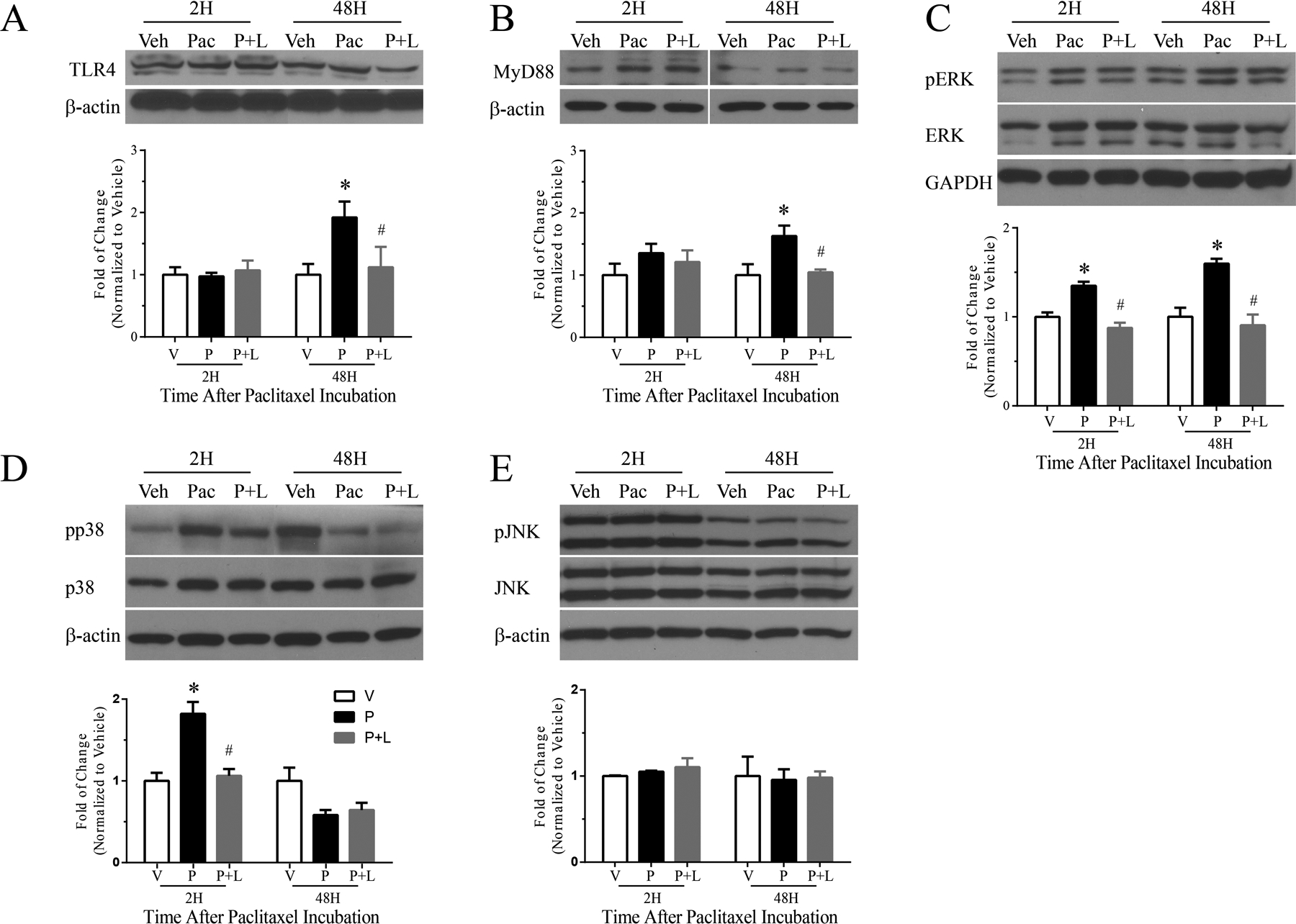
The representative Western blot image and bar graph summary shown in (A) illustrate that the expression of TLR4 in primary DRG culture significantly increased 48 h after paclitaxel treatment (black bar) compared with vehicle-treated culture (white bar), there was no significant change in expression 2 h. LPS-RS co-treatment with paclitaxel (grey bars) prevented the increase in expression of TLR4 in culture incubated at 48 h. The representative Western blot image and bar graph summary shown in (B) similarly show that expression of MyD88 in primary DRG culture significantly increased 48 h after paclitaxel treatment (black bar) compared to vehicle-treated culture (white bar), but no significant increase in MyD88 expression was observed at 2 h. MyD88 expression in cultures co-treated with LPS-RS and paclitaxel (grey bar) was significantly lower than in cultures treated with paclitaxel alone (black bar) 48 h after treatment. The representative Western blot image and bar graph summary in (C) shows the expression pERK is upregulated at both 2 and 48 h after paclitaxel treatment compared to vehicle-treated cultures and the upregulations were prevented by co-treatment with LPS-RS. The expression of pp38 was significantly increased only at 2 h of paclitaxel treatment and this was prevented by co-treatment with LPS-RS (D). There was no significant change in the expression of pJNK at either 2 or 48 h after paclitaxel treatment compared to that observed in vehicle-treated cultures (E). All bar graphs show fold change normalized to vehicle. *p<0.05 versus vehicle; #p<0.05 versus paclitaxel (N=4/group/time point for all experiments).
Distribution of CCL2, CCR2 and IL-6 in Primary DRG Culture
Immunohistochemical analysis was used to determine the distribution of CCL2 and its cognate receptor, CCR2, in primary DRG culture. Double immunostaining revealed that CCL2 was strongly colocalized to TLR4+ cells in both paclitaxel- and vehicle-treated cultures (Fig. 2). In addition, CCL2 was colocalized to IB4+ and CGRP+ neurons (Fig. 5 A–H) in both vehicle- and paclitaxel-treated cultures. CCR2 was primarily expressed in IB4+ small-sized and medium-sized neurons (Fig 5. I–L). Immunohistochemical analysis was used to define the cellular localization of IL-6 in primary DRG culture at increased expression 48 h after paclitaxel treatment (300 nM). As shown in Figure 6, IL-6 co-localized with MAP-2 (neurons) (Fig 6. A–D) and GFAP (glial cells) (Fig 6. E, F) 48 h after both vehicle and paclitaxel treatment. DRG neurons exposed to paclitaxel assumed a rounded morphology with reduced arborization.
Figure 5.
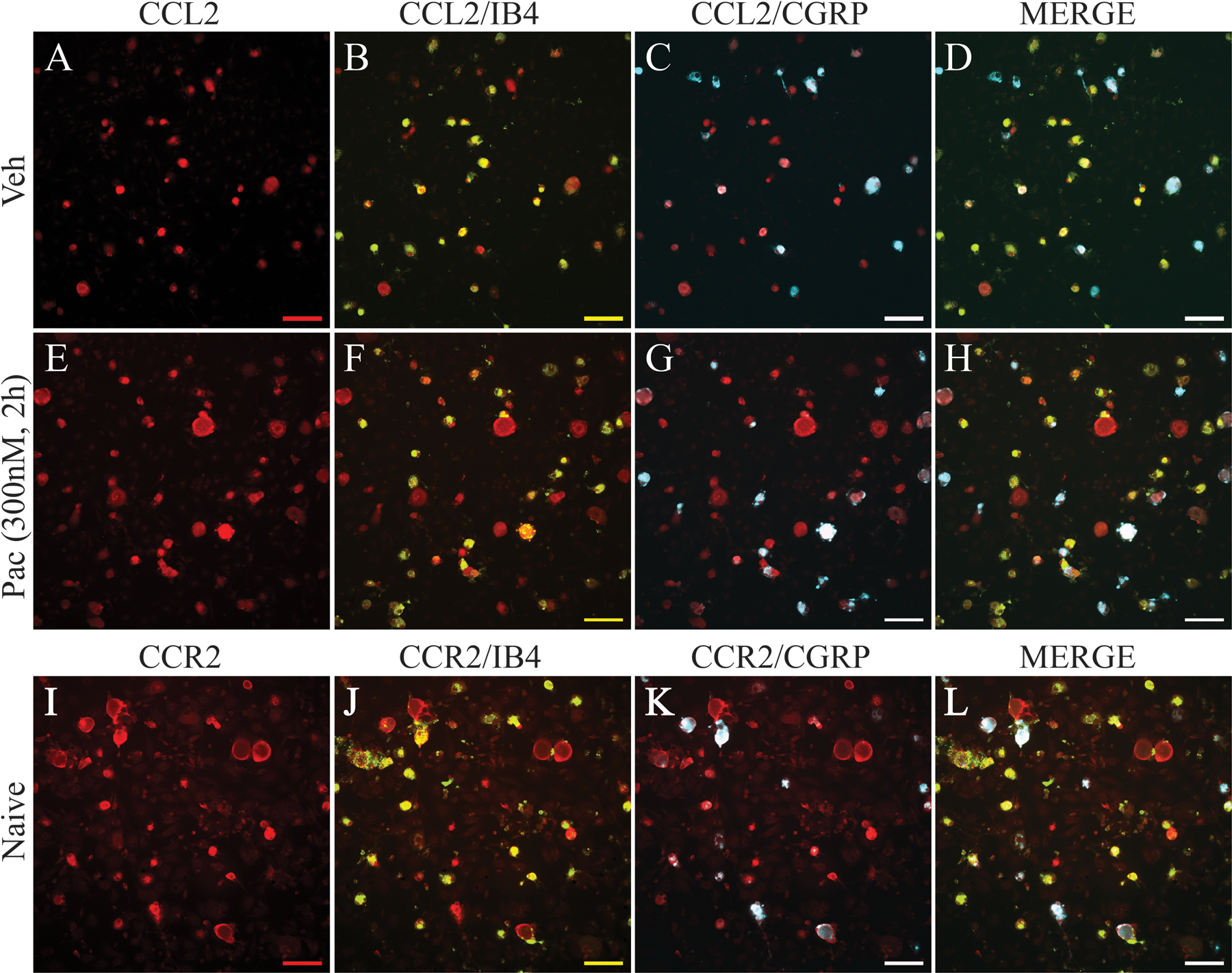
Immunostaining of primary DRG culture demonstrates that expression of CCL2 (red) is upregulated in neurons in paclitaxel-treated culture (E-H) compared with neurons in vehicle-treated culture (A-D) at 2 h after paclitaxel incubation. Images (B) and (F) show that CCL2 is co-localized to IB4-positive neurons (green, double label in yellow), and images (C) and (G) show that CCL2 is co-localized to CGRP-positive neurons (blue, double label in white) in both vehicle and paclitaxel conditions. Images (D) and (H) show CCL2, IB4, and CGRP labels merged. Immunostaining of naïve primary DRG culture (I-L) shows that CCR2 (red) strongly co-localized with IB4-positive neurons (J, green, double label in yellow) and to a lesser extent with CGRP-positive neurons (K, blue, double label in white). Images (L) shows CCR2, IB4, and CGRP labels merged. Scale bar=100 μm.
Figure 6.
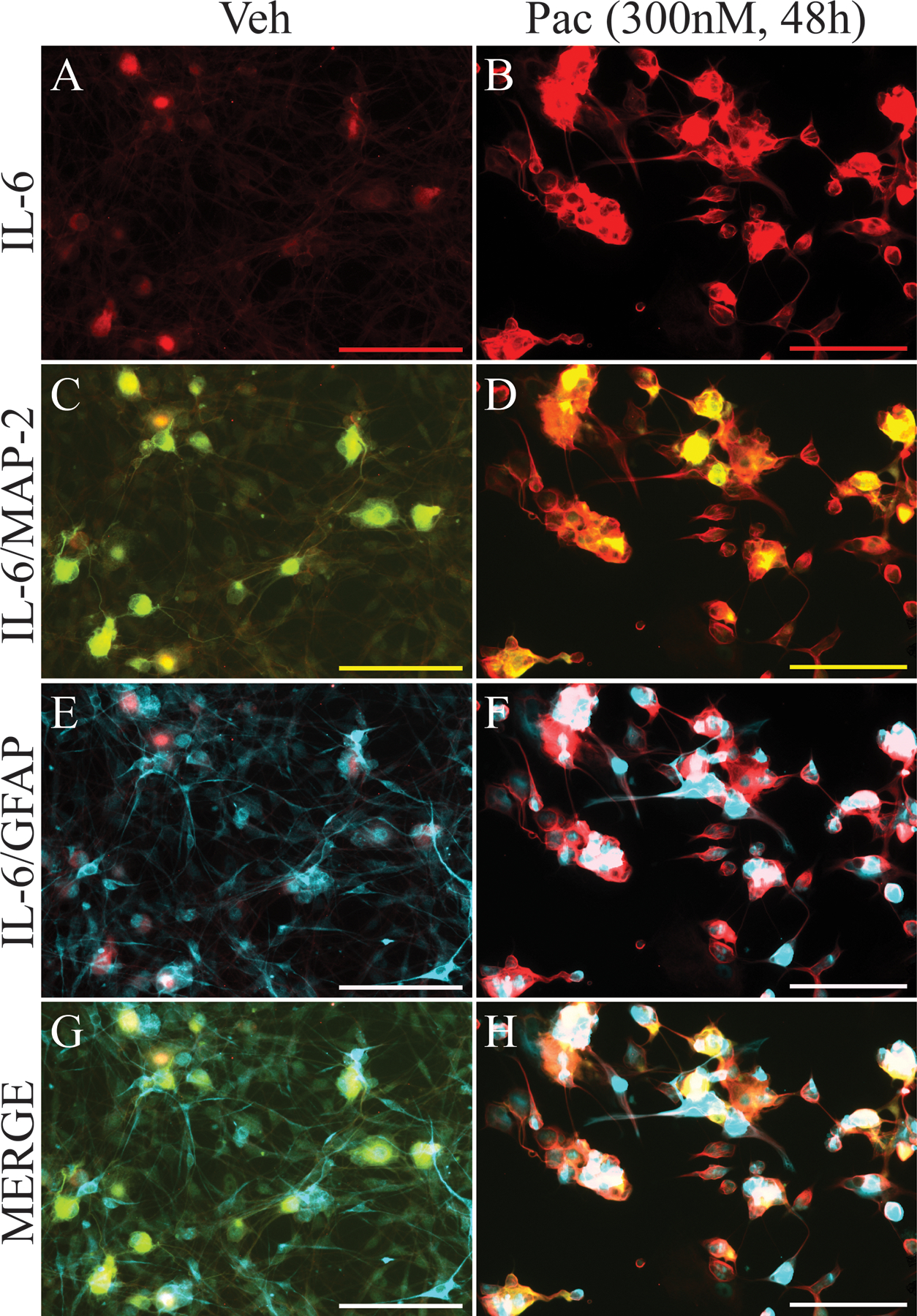
IL-6 is increased and co-localized to subsets of cultured DRG neurons after paclitaxel incubation. The representative immunohistochemical images in (A) and (B) show that the expression of IL-6 (red) in the DRG is low in vehicle-treated culture incubated for 48 h but that there is a marked increase in expression in culture treated with paclitaxel and incubated for 48 h. Double immunohistochemical images shown in (C-H) indicate that IL-6 co-localized to MAP2 (green)-positive cells (C and D, co-localization indicated by yellow) and GFAP (blue)-positive cells (E and F, co-localization indicated by purple). Images G and H show IL-6, MAP-2, and GFAP labels merged. Scale bar=100 μm.
Paclitaxel-Induced Changes in Neurophysiological Properties of Primary DRG Neurons in Culture.
Whole-cell patch clamp recordings were performed to assess the impact of acute paclitaxel exposure on the physiological properties of DRG neurons in vitro. Cultured naïve DRG neurons were incubated with paclitaxel (300 nM) for up to 48 h. Depolarizing spontaneous fluctuations (DSFs) in resting membrane potential (RMP) were observed at 2h and 48h, but no spontaneous activity was present (Fig. 7). Mean RMP was decreased after 48 h incubation but no changes were seen in current thresholds (Fig. 7 A, B, n=15~25/group). The results of recordings testing intrinsic excitability of DRG neurons from neurons following incubation with vehicle or paclitaxel are shown in Fig.7C (n=10/group/current strength). Evoked action potential frequencies (spikes/500ms) from 3X to 5X rheobase were higher for neurons incubated with paclitaxel compared to those neurons incubated with vehicle.
Figure 7.
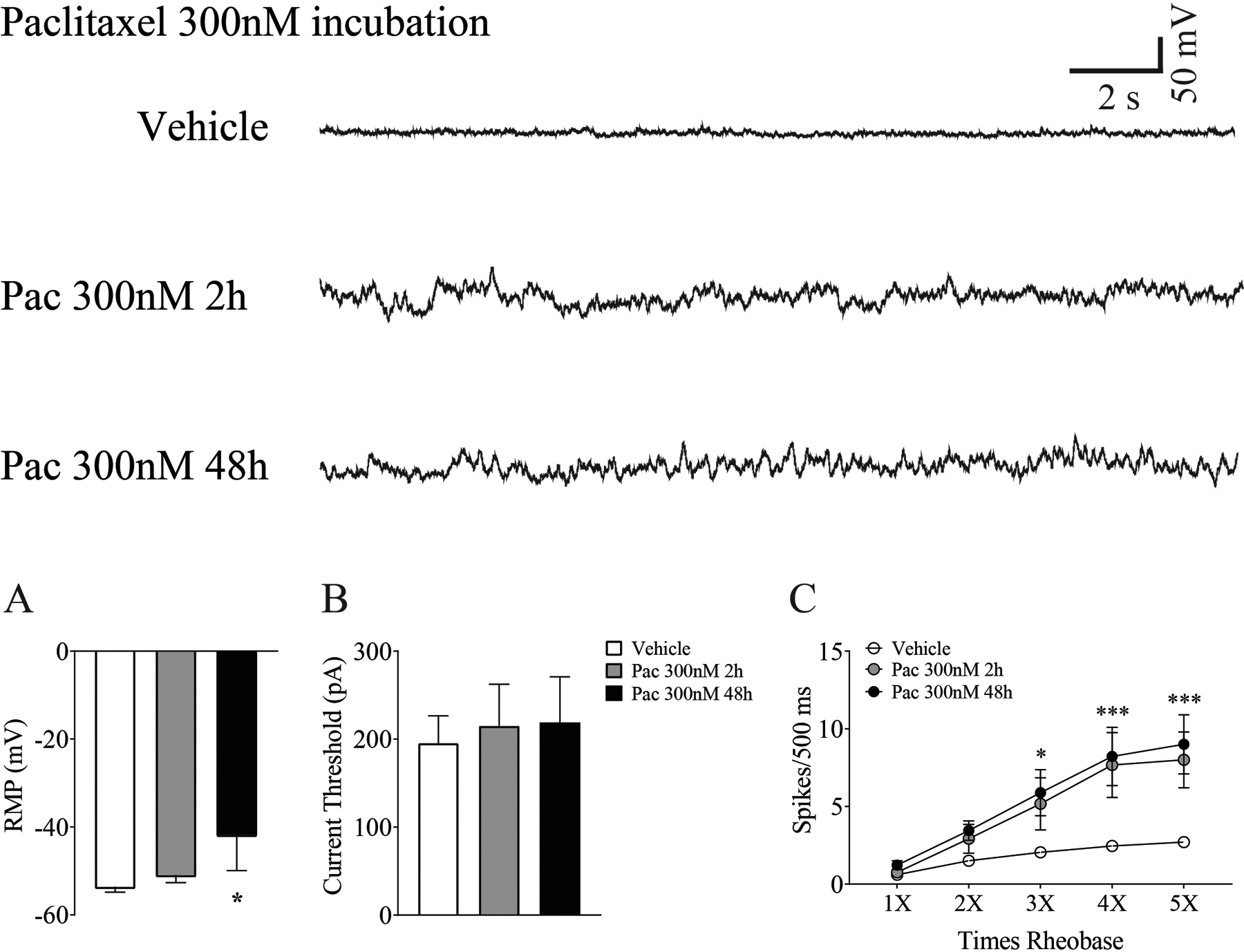
Whole cell patch clamp recordings reveal paclitaxel-induced changes in DRG neurons in vitro. Depolarizing spontaneous fluctuations (DSFs) in membrane potential were seen after 2h and 48 h of paclitaxel incubation as presented in the traces at the top of the figure. DSFs were accompanied by a significant decrease in mean membrane potential (A), but no change in current threshold was observed at either time point (B) (n=15–25 cells/group). As summarized in C, neurons from cultures incubated with paclitaxel showed more evoked action potentials at 3 −5X rheobase compared to those incubated with vehicle (n=10/group/current strength). *p <0.05, ***p < 0.001.
CL2-Induced Spontaneous Activity in Primary DRG Culture
Whole-cell patch clamp recordings were taken to investigate the role of CCL2 in inducing spontaneous neuronal activity. Naïve DRG neurons were recorded before and after exposure to CCL2. Neurons tested with a low dose of CCL2 (5 or 10 pg/ml) showed no evoked action potentials (not shown). In contrast, as shown in the representative recordings in Fig. 8A this was commonly seen following application of a higher concentration of CCL2 (50pg/ml) and this was accompanied by a change from a well regulated resting membrane potential (Fig. 8B) to less regulated with DSFs being generated (Fig. 8C). The bar graphs in Fig. 8D and E show detailed grouped results. Application of CCL2 at the lower concentrations (5 or 10 pg/ml) resulted in membrane depolarization in 4 of 7 neurons (Fig.8 D, black line), while 3 of 7 neurons showed no change in membrane potential (Fig. 8D, gray line). Yet, the overall mean membrane potential was not significantly changed by CCL2 applied at this concentration (Fig. 8E, left). When tested at the higher concentration (50 pg/ml), 6 of 8 neurons were depolarized during CCL2 administration at this concentration (Fig. 8D, solid and dashed black line) and 5 of 8 neurons showed evoked action potentials (Fig. 8D, dashed line). The group mean membrane potential was significantly depolarized at the higher concentration of CCL2 compared to pre-treatment (Fig. 8E). Interestingly, CCL2 treatment did not induce any action potentials in small-sized neurons at any of the concentrations tested but only produced action potentials in medium-sized neurons. This result was consistent with a previous report on the distribution of CCR2 in DRG neurons [45].
Figure 8.

Whole cell patch clamp recordings indicate that CCL2 treatment in naïve neurons rapidly induced spontaneous activity in vitro. A representative trace (A) shows CCL2-induced changes in membrane potential and spontaneous discharge. The area indicated by the dotted lines in (A) is presented in (B), and shows normal, low-level fluctuations at baseline, while the area indicated by the solid lines is presented in (C) and shows enhanced fluctuations in membrane potential and action potentials after CCL2 administration. Spontaneous activity ceased after a brief washout period. Application of CCL2 at the lower concentrations (5 or 10 pg/ml) resulted in membrane depolarization in 4 of 7 neurons (D, black line), while 3 of 7 neurons showed no change in membrane potential (D, gray line). Yet, the overall mean membrane potential was not significantly changed by CCL2 applied at this concentration (E, left). When tested at the higher concentration (50 pg/ml), 6 of 8 neurons were depolarized during CCL2 administration at this concentration (D, solid and dashed black line) and 5 of 8 neurons showed evoked action potentials (dashed line). Relative to baseline, mean resting membrane potential was significantly lower after administration of the higher concentration (50 pg/ml) of CCL2 (E, right, paired t-test, p<0.05).
Paclitaxel Induced Oxidative Stress in Mitochondria of Cultured DRG Neurons
Oxidative stress induced by incubation with paclitaxel (300 nM) was evaluated using mitochondrial superoxide imaging of cultured DRG neurons with the mitochondrial-specific ROS detector, MitoSOX Red. DRG neurons treated with vehicle or paclitaxel for 2, 4, 24, and 48 h were incubated with MitoSOX Red and imaged (Fig. 9 A–H, n=5/group/time point). The intensity of fluorescence produced after MitoSOX Red exposure was quantified for analysis (Fig. 8 I). Relative to vehicle, mitochondrial superoxide was lower after 2 h paclitaxel incubation and unchanged after 4 h. In contrast, mitochondrial superoxide was substantially higher in DRG neurons incubated for 24 h or 48 h.
Figure 9.
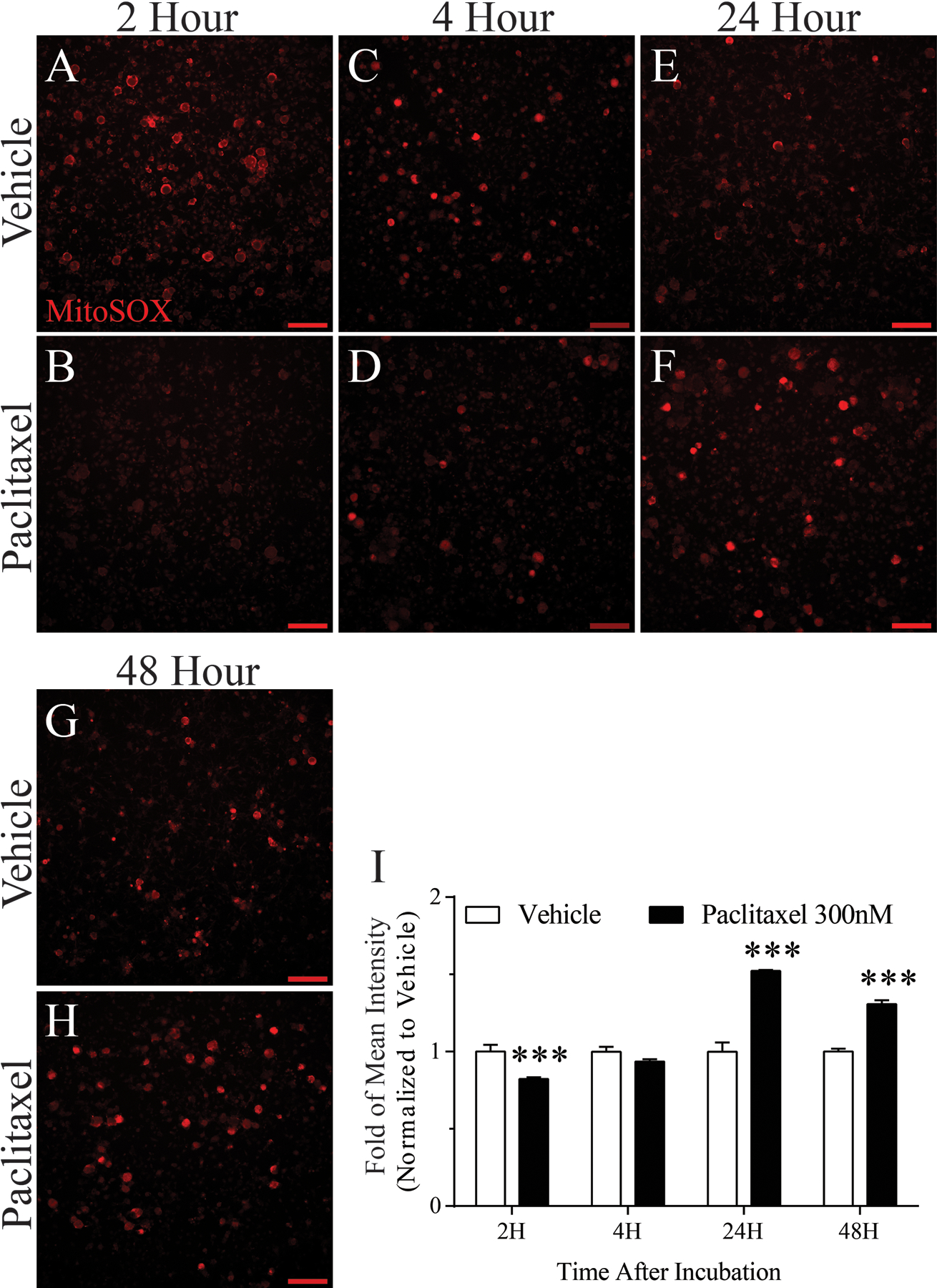
Effects of paclitaxel-induced mitochondrial superoxide. Mitochondrial superoxide production in live DRG neurons was measured by fluorescence microscopy using MitoSOX Red dye. Representative fluorescence images show MitoSOX Red fluorescence at 2, 4, 24, and 48 h after vehicle or paclitaxel incubation (n=5/group/time point). Fluorescence intensity summaries in bar graph I, show that relative to vehicle, mitochondrial superoxide was lower after 2 h paclitaxel incubation and unchanged after 4 h. In contrast, mitochondrial superoxide was substantially higher in DRG neurons incubated with paclitaxel for 24 h or 48 h. ***p < 0.001, Scale bar 100 μm.
Paclitaxel Upregulates pERK Expression in Primary Human DRG Culture at 2 h and 48 h of Incubation
We performed an immunohistochemistry experiment to test the translatability of our findings to human DRG. The results show that like in rats, human DRG shows an up-regulation of p-ERK at both 2 and 48 h after paclitaxel incubation. Immunohistochemistry indicated the fluorescence intensity of pERK was increased at both 2 h (IHC picture not shown, n=10 neurons/group) and 48 h after paclitaxel treatment (D, n=30 neurons/group) compared to that observed in vehicle-treated cultures (A) and both were prevented by co-treatment with LPS-RS (G). Co-localization images shown in C, F and I indicate that pERK co-localized to MAP2 (B, E, and H, green)-positive cells.
Discussion
A key finding in this work is that exposure of cellular elements found in naïve DRG to paclitaxel in vitro reproduces many of the biochemical and neurophysiological alterations that are observed in the DRG with systemic chemotherapy-induced CIPN. In the systemic rat model, paclitaxel induces a TLR4-triggered, MyD88-NFκB-MAP kinase cascade that results in the synthesis and release of multiple cytokines and chemokines, including the pro-inflammatory agents IL-6 and CCL2[5; 22; 29; 44]. CCL2, in particular was shown to play a key role in the development of paclitaxel-induced CIPN in vivo [21; 45], as it draws macrophages into the DRG that make a key contribution to the induction of CIPN. Blockade of TLR4, any of its downstream signals, CCL2 or elimination of macrophages results in failure of the behavioral and anatomical phenotypic signs of CIPN to develop [45]. Finally, the development of ectopic spontaneous activity (SA) in DRG neurons appears to be a key final consequence of these TLR4-related signal pathways in that agents blocking this SA also relieve CIPN-related behavioral hypersensitivity. Here, the expression and release of CCL2 and the expression of the MAP kinases pp38 and pERK are increased as soon as at 2 h after paclitaxel, while the expression of TLR4 and its immediate downstream signal MyD88 are increased after 48h of incubation. IL-6 expression was also increased 48 h after incubation, and ELISA data showed that paclitaxel induced the release of IL-6 into the culture media at this same time point. The TLR4 antagonist LPS-RS prevented the paclitaxel-induced upregulation of pERK, TLR4, MyD88, and IL-6, and immunostaining revealed that TLR4 was co-localized with both IL-6 and CCL2. IL-6 was co-localized with both microtubule associated protein 2 (MAP-2) and glial fibrillary acid protein (GFAP). An inhibition of neurite outgrowth in DRG neurons and arborization in DRG glial GFAP+ cells was also observed in cultures incubated with paclitaxel for 48 h, an effect that has been previously observed in vitro using cultured rat and mouse neurons [6; 38; 43]. This result is best illustrated in Figure 6G and H. Pathological changes in neuron morphology such as these were partially blocked in TLR4 knockout mice, providing evidence that TLR4 signaling is also involved in processes leading to axonal degeneration during paclitaxel treatment [38]. Finally, neurophysiological data showed that neurons treated with paclitaxel in vitro develop spontaneous depolarizing fluctuations (DSFs) of membrane potential and hyper-responsiveness to depolarizing stimuli. Hence, the findings here confirm what had long been suspected: the cellular elements in the DRG itself, both neurons and glial cells, are a direct key target of chemotherapy drugs to initiate CIPN. The finding that CCL2 was strongly co-localized to both calcitonin gene-related peptide+ (CGRP) and isolectin-B4+ (IB4) neurons in this study, which has also been observed in the in vivo model, further indicates a selectivity of paclitaxel for subsets of neurons in vitro that parallels that seen in vivo.
However, there were also some notable differences in the results here compared to our previous studies in vivo. This could reflect the fact that young rats were used here while adult rats were used previously. But, the differences in results could also further confirm the importance of recruitment to the DRG of immune cells to produce the full CIPN phenotype [14; 47]. Increased expression of pp38, a key signal pathway engaged by activated macrophages, was persistent over time in the DRG following systemic treatment with paclitaxel, but was only transiently increased here in cultured DRG with in vitro treatment of paclitaxel. Another key difference between systemic versus in vitro paclitaxel treatment was the impact on the physiological properties of DRG neurons. When given systemically, DRG neurons develop spontaneous activity (SA) and hyper-excitability to injection of depolarizing current injection as evidenced by a leftward shift in current action potential (AP) function. SA following systemic treatment is supported by the development of DSFs in membrane potential, depolarization in resting membrane potential (RMP), and a hyperpolarization of AP threshold [30]. Here, hyper-excitability to current injection, increased DSFs and depolarization of RMP were observed, but hyperpolarization of AP threshold and SA did not occur. This finding would suggest that infiltrating macrophages contribute a final key element to support SA in DRG neurons that presumably drive the on-going spontaneous pain that characterizes CIPN in patients.
Macrophages infiltrating the DRG could contribute to the development of CIPN through direct cell-cell contact with neurons or other cellular elements in the ganglia, or by the release of inter-cellular signals. CCR2 was co-localized to IB4+ small- and medium-sized neurons. CCL2/CCR2 signaling has been previously shown to depolarize neurons and induce action potentials [45], and a similar finding was shown here in medium-sized neurons from naïve animals in vitro. Naive neurons treated with CCL2 displayed on-going AP discharges, which continued after washout of CCL2 and thus may contribute to both the initiation and maintenance of this feature of CIPN, especially given the significant increase in the expression of CCR2 on DRG neurons following in vivo paclitaxel treatment [45]. This is of particular significance, considering the role of SA in the experience of allodynia and hyperalgesia observed in CIPN and other neuropathies [3; 13]. CCL2 has previously been shown to alter the function of TTX-resistant sodium channel Nav1.8 in DRG neurons of naïve rats through a CCR2-dependent mechanism [1]. A study in CFA-treated rats demonstrated that the depolarization of DRG neurons induced by CCL2 treatment occurred as a result of changes in the activity of tetrodotoxin (TTX)-sensitive sodium channels that led to increased sodium currents [24]. However, the changes in neuronal function elicited by CCL2 in this study were limited to neurons isolated from CFA-treated rats. Further study is needed to determine how paclitaxel treatment itself impacts the effect of CCL2/CCR2 on sodium channel function.
Increased oxidative stress and mitochondrial toxicity may be another key mechanism by which that infiltrating macrophages produce the full CIPN phenotype. Vacuolated mitochondria were the standout feature of the early initial ultrastructural studies on peripheral axons and DRG neurons in the rat paclitaxel CIPN model [9]; and this observation was complemented by studies showing that anti-oxidants were effective in protecting and reversing signs of paclitaxel-induced CIPN [8; 10]. Similar observations have been extended to include oxaliplatin-, bortezomib- [49; 50] and cisplatin-induced CIPN [25]. We observed here evidence that paclitaxel does indeed produce increased oxidative stress in DRG neurons beginning at 24 hours after paclitaxel exposure. Interestingly, the signs of oxidative stress coincided with the production of IL-6 in the culture medium which itself promotes the production of superoxides, while inhibition of TLR4 signaling prevented the other biochemical sequelae of paclitaxel incubation, suggesting that oxidative stress may be linked to TLR4 signaling. A key mechanism by which pro-inflammatory macrophages exert their bactericidal function is through the production of superoxides, and thus infiltration of these into the DRG following paclitaxel exposure in vivo would likely exacerbate the oxidative stress produced by paclitaxel itself. Superoxides are also activators of TLR4 signaling, so production of these by infiltrating macrophages would provide a positive feedback mechanism to amplify the direct effects of paclitaxel in the DRG. This amplification step may be the key that finally leads to the generation of ectopic SA, leading to spontaneous pain. Future follow-up co-culture experiments should help better define this possibility.
A final key, albeit preliminary, finding is that cultures of human DRG appear to reveal many shared acute biochemical responses with rodent DRG to paclitaxel exposure. TLR4 has proven to have many shared effects on human and rodent DRG [15; 18; 19]; and here is shown to engage p-ERK signaling on exposure to paclitaxel in vitro. This suggests that future studies focusing on the responses of human DRG to chemotherapy agents could be a promising pathway for therapeutic discovery. Indeed, transcriptomic profiling of human DRG before and then following chemotherapy exposure could be a particularly exciting approach in this regard [28].
In summary, this study provides new data regarding TLR4 signaling during acute paclitaxel treatment and confirms the involvement of downstream pro-inflammatory cytokines that have previously been implicated in the development of CIPN. The TLR4 signaling pathway involving IL-6 and CCL2 may provide novel targets for the prevention and treatment of the acute pain syndrome and peripheral neuropathy that limit the tolerated dose of paclitaxel in cancer treatment.
Figure 10.
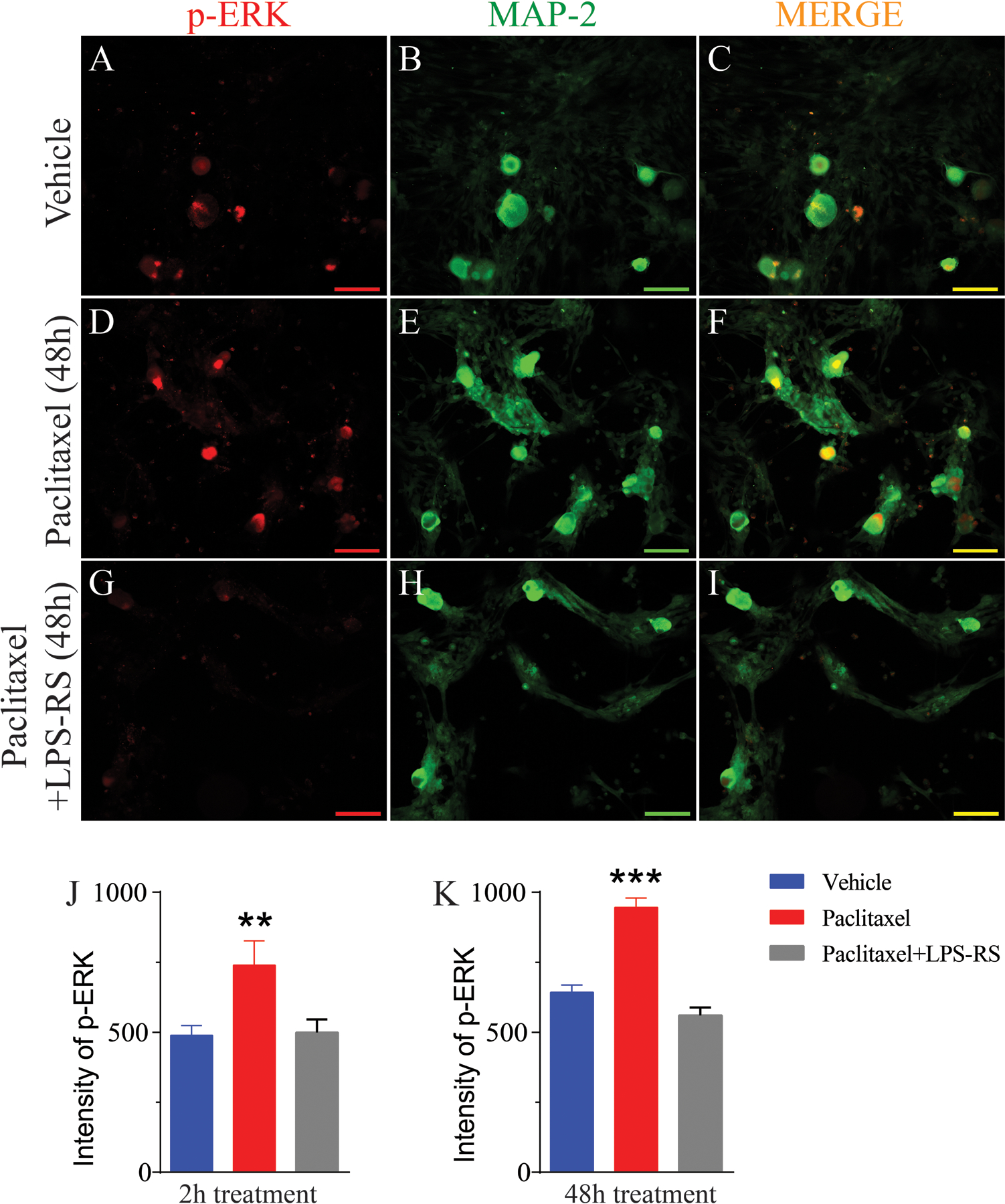
Double IHC revealed that pERK is increased in cultured human DRG neurons after incubation with paclitaxel. The representative immunohistochemical images in (A) and (D) show that the expression of pERK (red) in the DRG is low in vehicle-treated culture incubated for 48 h but that there is a marked increase in expression in culture treated with paclitaxel and incubated for 48 h. Upregulation of pERK were prevented by co-treatment with LPS-RS (G). Double immunohistochemical images shown in (C, F and I) indicate that pERK co-localized to MAP2 (B, E, and H, green)-positive cells (co-localization indicated by yellow). The bar graph in (J and K) shows that a significant increased fluorescence intensity of pERK at both 2 (n=10 neurons/group) and 48 h after paclitaxel treatment compared to vehicle-treated cultures and the upregulations were prevented by co-treatment with LPS-RS. Scale bar= 100 μm. ** = p <0.01, *** = P < 0.001.
Acknowlegements:
This work was supported by grants from the National Institutes of Health (CA200263, NS), the Thompson Family Foundation Initiative and the H.E.B. Professorship in Cancer Research.
Footnotes
The authors declare no conflict of interest.
References
- [1].Belkouch M, Dansereau MA, Reaux-Le GA, Van SJ, Beaudet N, Chraibi A, Melik-Parsadaniantz S, Sarret P. The chemokine CCL2 increases Nav1.8 sodium channel activity in primary sensory neurons through a Gbetagamma-dependent mechanism. J Neurosci 2011;31(50):18381–18390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bhatnagar B, Gilmore S, Goloubeva O, Pelser C, Medeiros M, Chumsri S, Tkaczuk K, Edelman M, Bao T. Chemotherapy dose reduction due to chemotherapy induced peripheral neuropathy in breast cancer patients receiving chemotherapy in the neoadjuvant or adjuvant settings: a single-center experience. Springerplus 2014;3:366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Boyette-Davis JA, Cata JP, Driver LC, Novy DM, Bruel BM, Mooring DL, Wendelschafer-Crabb G, Kennedy WR, Dougherty PM. Persistent chemoneuropathy in patients receiving the plant alkaloids paclitaxel and vincristine. Cancer Chemother Pharmacol 2013;71:619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Boyette-Davis JA, Walters ET, Dougherty PM. Mechanisms involved in the development of chemotherapy-induced neuropathy. Pain Manag 2015;5(4):285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Byrd-Leifer CA, Block EF, Takeda K, Akira S, Ding A. The role of MyD88 and TLR4 in the LPS-mimetic activity of taxol. Eur J Immunol 2001;31:2448–2457. [DOI] [PubMed] [Google Scholar]
- [6].Cashman CR, Hoke A. Mechanisms of distal axonal degeneration in peripheral neuropathies. Neurosci Lett 2015;596:33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chaudhry V, Rowinsky EK, Sartorius SE, Donehower RC, Cornblath DR. Peripheral neuropathy from taxol and cisplatin combination chemotherapy: Clinical and electrophysiological studies. Ann Neurol 1994;35:304–311. [DOI] [PubMed] [Google Scholar]
- [8].Flatters SJ, Bennett GJ. Ethosuximide reverses paclitaxel- and vincristine-induced painful peripheral neuropathy. Pain 2004;109(1–2):150–161. [DOI] [PubMed] [Google Scholar]
- [9].Flatters SJ, Bennett GJ. Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain 2006;122(3):245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Flatters SJ, Xiao WH, Bennett GJ. Acetyl-L-carnitine prevents and reduces paclitaxel-induced painful peripheral neuropathy. Neurosci Lett 2006;397(3):219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal 2001;13:85–94. [DOI] [PubMed] [Google Scholar]
- [12].Hagiwara H, Sunada Y. Mechanism of taxane neurotoxicity. Breast Cancer 2004;11(1):82–85. [DOI] [PubMed] [Google Scholar]
- [13].Han Y, Smith MT. Pathobiology of cancer chemotherapy-induced peripheral neuropathy (CIPN). Front Pharmacol 2011;4:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Krukowski K, Eijkelkamp N, Laumet G, Hack CE, Li Y, Dougherty PM, Heijnen CJ, Kavelaars A. CD8+ T Cells and Endogenous IL-10 Are Required for Resolution of Chemotherapy-Induced Neuropathic Pain. J Neurosci 2016;36(43):11074–11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li Y, Adamek P, Zhang H, Tatsui CE, Rhines LD, Mrozkova P, Li Q, Kosturakis AK, Cassidy RM, Harrison DS, Cata JP, Sapire K, Zhang H, Kennamer-Chapman RM, Jawad AB, Ghetti A, Yan J, Palecek J, Dougherty PM. The cancer chemotherapeutic paclitaxel increases human and rodent sensory neuron responses to TRPV1 by activation of TLR4. J Neurosci 2015;35(39):13487–13500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li Y, Cai J, Han Y, Xiao X, Meng XL, Su L, Liu FY, Xing GG, Wan Y. Enhanced function of TRPV1 via up-regulation by insulin-like growth factor-1 in a rat model of bone cancer pain. Eur J Pain 2014;18(6):774–784. [DOI] [PubMed] [Google Scholar]
- [17].Li Y, North RY, Rhines LD, Tatsui CE, Rao G, Edwards DD, Cassidy RM, Harrison DS, Johansson CA, Zhang H, Dougherty PM. DRG Voltage-Gated Sodium Channel 1.7 Is Upregulated in Paclitaxel-Induced Neuropathy in Rats and in Humans with Neuropathic Pain. J Neurosci 2018;38(5):1124–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Li Y, North RY, Rhines LD, Tatsui CE, Rao G, Edwards DD, Cassidy RM, Harrison DS, Johansson CA, Zhang H, Dougherty PM. DRG voltage-gated sodium channel 1.7 is upregulated in paclitaxel-induced neuropathy in rats and in humans with neuropathic pain. JNeurosci 2018;38(5):1124–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Li Y, Tatsui CE, Rhines LD, North RY, Harrison DS, Cassidy RM, Johansson CA, Kosturakis AK, Edwards DD, Zhang H, Dougherty PM. Dorsal root ganglion neurons become hyperexcitable and increase expression of voltage-gated T-type calcium channels (Cav3.2) in paclitaxel-induced peripheral neuropathy. Pain 2017;158:417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li Y, Zhang H, Kosturakis AK, Cassidy RM, Zhang H, Kennamer-Chapman RM, Jawad AB, Colomand CM, Harrison DS, Dougherty PM. MAPK signaling downstream to TLR4 contributes to paclitaxel-induced peripheral neuropathy. Brain Behav Immun 2015;49:255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li Y, Zhang H, Zhang H, Kosturakis AK, Jawad AB, Dougherty PM. Toll-like receptor 4 signaling contributes to paclitaxel-induced peripheral neuropathy. J Pain 2014;15(7):712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liu XJ, Liu T, Chen G, Wang B, Yu XL, Yin C, Ji RR. TLR signaling adaptor protein MyD88 in primary sensory neurons contributes to persistent inflammatory and neuropathic pain and neuroinflammation. Sci Rep-Uk 2016;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Loprinzi CL, Reeves BN, Dakhil SR, Sloan JA, Wolf SL, Burger KN, Kamal A, Le-Lindqwister NA, Soori GS, Jaslowski AJ, Novotny PJ, Lachance DH. Natural history of paclitaxel-associated acute pain syndrome: prospective cohort study NCCTG N08C1. J Clin Oncol 2011;29(11):1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Luo P, Shao J, Jiao Y, Yu W, Rong W. CC chemokine ligand (CCL2) enhances TTX-sensitive sodium channel activity of primary afferent neurons in the complete Freud adjuvant-induced inflammatory pain model. Acta BiochimBiophysSin 2018;50(12):1219–1226. [DOI] [PubMed] [Google Scholar]
- [25].Maj MA, Ma J, Krukowski KN, Kavelaars A, Heijnen CJ. Inhibition of mitochondrial p53 accumulation by PFT-mu prevents cisplatin-induced peripheral neuropathy. FrontMolNeurosci 2017;10:108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Megat S, Ray PR, Moy JK, Lou TF, Barragan-Iglesias P, Li Y, Pradhan G, Wanghzou A, Ahmad A, Burton MD, North RY, Dougherty PM, Khoutorsky A, Sonenberg N, Webster KR, Dussor G, Campbell ZT, Price TJ. Nociceptor Translational Profiling Reveals the Ragulator-Rag GTPase Complex as a Critical Generator of Neuropathic Pain. J Neurosci 2019;39(3):393–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Melli G, Taiana M, Camozzi F, Triolo D, Podini P, Quattrini A, Taroni F, Lauria G. Alpha-lipoic acid prevents mitochondrial damage and neurotoxicity in experimental chemotherapy neuropathy. Exp Neurol 2008;214:276–284. [DOI] [PubMed] [Google Scholar]
- [28].North RY, Li Y, Ray P, Rhines LD, Tatsui CE, Rao G, Johansson CA, Zhang H, Kim YH, Zhang B, Dussor G, Kim TH, Price TJ, Dougherty PM. Electrophysiologic and transcriptomic correlates of neuropathic pain in human dorsal root ganglion neurons. Brain 2019;In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].O’Brien JM Jr., Wewers MD, Moore SA, Allen JN. Taxol and colchicine increase LPS-induced pro-IL-1 beta production, but do not increase IL-1 beta secretion. A role for microtubules in the regulation of IL-1 beta production. J Immunol 1995;154(8):4113–4122. [PubMed] [Google Scholar]
- [30].Odem MA, Bavencoffe AG, Cassidy RM, Lopez ER, Tian J, Dessauer CW, Walters ET. Isolated nociceptors reveal multiple specializations for generating irregular ongoing activity associated with ongoing pain. Pain 2018;159(11):2347–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pachman DR, Qin R, Seisler D, Lavoie Smith EM, Kaggal S, Novotny P, Ruddy KJ, Lafky JM, Ta LE, Beutler AS, Wagner-Johnston ND, Staff NP, Grothey A, Dougherty PM, Cavaletti G, Loprinzi CL. Comparison of oxaliplatin and paclitaxel-induced neuropathy (Alliance A151505). Support Care Cancer 2016;24:5059–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Polomano RC, Mannes AJ, Clark US, Bennett GJ. A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. Pain 2001;94(3):293–304. [DOI] [PubMed] [Google Scholar]
- [33].Pusztai L, Mendoza TR, Reuben JM, Martinez MM, Willey JS, Lara J, Syed A, Fritsche HA, Bruera E, Booser D. Changes in plasma levels of inflammatory cytokines in response to paclitaxel chemotherapy. Cytokine 2004;25(3):94–102. [DOI] [PubMed] [Google Scholar]
- [34].Reeves BN, Dakhil SR, Sloan JA, Wolf SL, Burger KN, Kamal A, Le-Lindqwister NA, Soori GS, Jaslowski AJ, Kelaghan J, Novotny PJ, Lachance DH, Loprinzi CL. Further data supporting that paclitaxel-associated acute pain syndrome is associated with development of peripheral neuropathy: North Central Cancer Treatment Group trial N08C1. Cancer 2012;118(20):5171–5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rowinsky EK, Cazenave LA, Donehower RC. Taxol: a novel investigational antimicrotubule agent. J Natl Cancer Inst 1990;82(15):1247–1259. [DOI] [PubMed] [Google Scholar]
- [36].Siau C, Xiao W, Bennett GJ. Paclitaxel- and vincristine-evoked painful peripheral neuropathies: Loss of epidermal innervation and activation of Langerhans cells. Exp Neurol 2006;201:507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Uceyler N, Kafke W, Riediger N, He L, Necula G, Toyka KV, Sommer C. Elevated proinflammatory cytokine expression in affected skin in small fiber neuropathy. Neurology 2010;74(22):1806–1813. [DOI] [PubMed] [Google Scholar]
- [38].Ustinova EE, Shurin GV, Gutkin DW, Shurin MT. The role of TLR4 in the paclitaxel effects on neuronal growth in vitro. PL0S One 2013;8(2):e56886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wang XM, Lehky TJ, Brell JM, Dorsey SG. Discovering cytokines as targets for chemotherapy-induced painful peripheral neuropathy. Cytokine 2012;59(1):3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Warwick RA, Hanani M. The contribution of satellite glial cells to chemotherapy-induced neuropathic pain. Eur J Pain 2013;17:571–580. [DOI] [PubMed] [Google Scholar]
- [41].Xiao WH, Bennett GJ. Chemotherapy-evoked neuropathic pain: Abnormal spontaneous discharge in A-fiber and C-fiber primary afferent neurons and its suppression by acetyl-L-carnitine. Pain 2008;135(3):262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Xiao WH, Zheng H, Zheng FY, Nuydens R, Meert TF, Bennett GJ. Mitochondrial abnormality in sensory, but not motor, axons in paclitaxel-evoked painful peripheral neuropathy in the rat. Neuroscience 2011;199:461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yang IH, Siddique R, Hosmane S, Thakor N, Hoke A. Compartmentalized microfluidic culture platform to study mechanism of paclitaxel-induced axonal degeneration. ExpNeurol 2009;218(1):124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zaks-Zilberman M, Zaks TZ, Vogel SN. Induction of proinflammatory and chemokine genes by lipopolysaccharide and paclitaxel (Taxol) in murine and human breast cancer cell lines. Cytokine 2001;15(3):156–165. [DOI] [PubMed] [Google Scholar]
- [45].Zhang H, Boyette-Davis JA, Kosturakis AK, Li Y, Yoon SY, Walters ET, Dougherty PM. Induction of monocyte chemoattractant protein-1 (MCP-1) and its receptor CCR2 in primary sensory neurons contributes to paclitaxel-induced peripheral neuropathy. J Pain 2013;14(10):1031–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhang H, Dougherty PM. Enhanced excitability of primary sensory neurons and altered gene expression of neuronal ion channels in dorsal root ganglion in paclitaxel-induced peripheral neuropathy. Anesthesiology 2014;120(6):1463–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhang H, Li Y, de Carvalho-Barbosa M, Kavelaars A, Heijnen CJ, Albrecht PJ, Dougherty PM. Dorsal root ganglion infiltration by macrophages contributes to paclitaxel chemotherapy-induced peripheral neuropathy. J Pain 2016;17(7):775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhang H, Yoon S-Y, Zhang H, Dougherty PM. Evidence that spinal astrocytes but not microglia contribute to the pathogenesis of paclitaxel-induced painful neuropathy. J Pain 2012;13(3):293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zheng H, Xiao WH, Bennett GJ. Functional deficits in peripheral nerve mitochondria in rats with paclitaxel- and oxaliplatin-evoked painful peripheral neuropathy. Exp Neurol 2011;232(2):154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zheng H, Xiao WH, Bennett GJ. Mitotoxicity and bortezomib-induced chronic painful peripheral neuropathy. Exp Neurol 2012;238(2):225–234. [DOI] [PubMed] [Google Scholar]