

Abstract
Overexpression of the MYC oncoprotein is an initiating step in the formation of several cancers. MYC frequently recruits chromatin-modifying complexes to DNA to amplify the expression of cancer-promoting genes including those regulating cell cycle, proliferation, and metabolism, yet the roles of specific modifiers in different cancer types are not well defined. Here we show that GCN5 is an essential coactivator of cell cycle gene expression driven by MYC overexpression and that deletion of Gcn5 delays or abrogates tumorigenesis in the Eμ-Myc mouse model of B cell lymphoma. Our results demonstrate that Gcn5 loss impacts both expression and downstream functions of MYC.
Keywords: Gcn5, Myc, lymphoma, epigenetics, acetylation
Introduction
The balance between the activities of lysine deacetylases (KDACs) and lysine acetyltransferases (KATs) during development is important in the establishment of gene transcription programs required for proper formation of an organism. This balance is also important in disease states that reflect failures in development, including cancers. Histone lysine deacetylase (HDAC) inhibitors have been used in clinical trials to treat certain types of cancer by modifying the expression or stability of tumor suppressors and oncogenes as well as regulating cell cycle and cell death, albeit with very little success (1), indicating that this therapy may not be appropriate for all cancer types (2). Recent studies indicate that lysine acetyltransferases may provide alternative targets for cancer therapies (3).
The lysine acetyltransferase Gcn5 is conserved from yeast to humans. Gcn5 participates in two major chromatin modifying complexes, the Spt-Ada-Gcn5 acetyltransferase (SAGA) complex and the ATAC complex. Both complexes are recruited to chromatin by transcriptional activators, and Gcn5 acts as a co-activator (4) of gene transcription by acetylating histones and other proteins (5–7). Gcn5 is essential for normal embryonic development in mice (8–10) and it is important as a co-activator for c-Myc in regulating c-Myc target genes in embryonic stem cells and during reprogramming of somatic cells to pluripotency (11). MYC is also a substrate of GCN5 acetyltransferase activity (12); its acetylation of lysine (K) 323 increases MYC stability.
We recently demonstrated that inhibition of GCN5 activity in human Burkitt lymphoma cells in vitro reduces viability and induces apoptosis by reducing MYC expression (13). MYC is the primary driver of the formation of Burkitt lymphoma as well as other B cell malignancies such as diffuse large B cell lymphoma (DLBCL). The primary role for MYC in these cancers is to amplify expression of genes that promote proliferation and growth (14,15). MYC has been reported to work with multiple histone acetyltransferases, including Gcn5, Tip60 (16,17), and CBP (18) to activate its gene targets in cancers as it does in developmental settings. Whether particular HATs are important for specific Myc-driven functions is not yet clear.
In this study we sought to elucidate whether Gcn5 cooperates with Myc to induce the formation of lymphoma using an in vivo model, the Eμ-Myc mouse. We report that deletion of Gcn5 (Kat2A) prolongs the latency and reduces the incidence of lymphoma formation in Myc-overexpressing B cells. Loss of Gcn5 not only ameliorates dysregulation of the cell cycle caused by Myc overexpression, but also affects Myc-induced pathways related to cancer cell growth. Our results demonstrate that Gcn5 is a critical partner for Myc in oncogenesis, suggesting this KAT may be a viable therapeutic target for MYC-overexpressing cancers.
Materials and Methods
Experimental Mouse Models
Eμ-Myc mice were rederived from embryos purchased from the International Mouse Strain Resource (IMSR). CD19-Cre mice were purchased from The Jackson Laboratory (RRID:MGI:4415129). Gcn5 Flox mice were developed by the Dent lab (19). Male CD19-CreTg/0; Eμ-Myc Tg/0; Gcn5Fx/+ mice were crossed with female CD19-CreTg/0; Gcn5Fx/+ mice to generate all experimental mice for mouse lymphoma experiments. Animals were maintained on a C57BL6 background.
Maintenance of Mice
Animals were kept in a 10-hour dark and 14-hour light cycle. Animals were cared for in accordance with guidelines from the Association of Laboratory Animal Care and with the approval of the Institutional Animal Care and Use Committee (IACUC) protocols at the University of Texas MD Anderson Cancer Center Science Park Research Division. Both male and female mice were allocated to experiments following genotyping at 3-4 weeks of age.
Isolation and Preparation of Mouse B cells
Spleen and femur were removed from mouse. The spleen was crushed through 70-micron cell strainer in cell culture dish in 6 ml 1x PBS + 1% BSA, washed in 4 ml 1x PBS + 1% BSA, and resuspended in PBS + 1% BSA. Femurs were spun in an Eppendorf tube to recover bone marrow; recovered cells were resuspended in 10 ml PBS+ 1% BSA. All cells were centrifuged at 4°C 1500 RPM for 3 min. Pellets were resuspend in 1 ml AcK lysis buffer (0.15 M NH4Cl, 10.0 mM KHCO3, 0.1 mM Na2EDTA in dH2O adjusted to pH to 7.2) and incubated for 5 min at room temperature. Lysate was centrifuged at 4°C 1400RPM for 3 min. Cells were washed in 10 ml cold PBS + 1% BSA and centrifuged at 4°C 1500 RPM for 3 min. Pellets were resuspended in 10 ml cold PBS + 1% BSA, and cells were counted. For compensation measurement, 100ul of WT cells were added to one tube for each fluorophore, one tube for propidium iodide (PI) staining, and one tube for unstained cells. 1ul Fc block (TruStain fcX™ (anti-mouse CD16/32) Antibody, Biolgend Cat.101319) was added to each tube. Cells were incubated on ice for 15 min. Single color compensation samples were prepared using predetermined concentrations of each antibody. PI was added at 6 μg/ml PI. For experimental samples a master mix of antibodies was prepared to make 500 μl/sample in PBS + 1% BSA. Tubes were incubated on ice for ~15 min. 6 μg/ml PI was then added to sample tubes.
Flow Cytometry
Flow cytometry analysis and sorting of mouse B cells was performed on a BD FACSARIA™ Fusion. All data was analyzed with FlowJo (FlowJo 10.6.1; FlowJo, RRID:SCR_008520) software. Viable cells were identified based on forward- and side-scatter characteristics as well as propidium iodide exclusion (Invitrogen P3566). CD3+ and CD11b+ gating was used to exclude non-B cells. From the bone marrow B cell populations were, pro-B cells B220+CD43hiIgM−; pre-B B220+CD43lowIgM−; immature B220+CD43lowIgM+IgD−; mature B220+CD43lowIgM+IgD+; plasmablasts B220+CD93−CD138+; plasma cells: B220−CD19−CD138+. From the spleen B cell populations were; follicular B220+CD19+CD23hiCD21low; marginal zone B220+CD19+CD23lowCD21hi; germinal center CD19+GL7+CD95+. Antibodies are listed in Supplementary Table S1.
Protein Lysates
Lysates were prepared as previously described (13). Briefly, cells were pelleted, and washed in 1x PBS. Pellets were resuspended in Buffer C (20 mM Tris-HCl pH 7.9, 20% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.1% NP-40, 0.2 mM EDTA, 0.5 mM DTT, 0.2 mM PMSF, and Sigma Protease inhibitors), vortexed and rocked at 4°C for 20 minutes. After rocking, an equal amount of Buffer A (10 mM HEPES pH 7.5, 1.5 mM MgCl2, 10 mM KCl) was added. Lysate was centrifuged at 4°C for 10 minutes at 10,000 RPM. Supernatant was collected as the whole cell extract and total protein levels were measured by Bradford assay. Antibodies are listed in Supplementary Table S1.
Immunoblotting
Whole cell lysates were run on 4-12% NuPage Bis-Tris gel (Life Technologies Cat. NPO322BOX). Proteins were then transferred to a nitrocellulose membrane. Membranes were blocked in 5% milk in TBS-T at room temperature for 30 minutes then incubated overnight at 4°C with the primary antibodies listed in Supplementary Table S1 in 0.5% milk in TBS-T. Membranes were incubated in horseradish peroxidase (HRP) conjugated secondary antibodies for 1 hour at room temperature. Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare Cat. 45-002-401) was used for chemiluminescent protein detection.
RNA-Sequencing
Total RNA from sorted CD19+ B cells from 5-6-week-old mice was isolated using the RNeasy® Purification Kit (Qiagen). RNA was DNase treated. RNA-Seq sequencing libraries were made using Illumina TruSeq® Stranded Total RNA Library Prep following manufacturer’s protocol. The libraries were sequenced using 2x76 bases paired end protocol on Illumina HiSeq 3000 instrument. Three biological replicates were prepared for each condition. 38-48 million pairs of reads were generated per sample. Each pair of reads represents a cDNA fragment from the library. The reads were mapped to the mouse genome mm10 by TopHat (version 2.0.10; RRID:SCR_013035) (20). By reads, the overall mapping rate is 93-96%. 90-94% fragments have both ends mapped to the mouse genome. For differential expression, the number of fragments in each known gene from GENCODE Release M19 (21) was enumerated using htseq-count from HTSeq package (version 0.6.0; RRID:SCR_005514) (22). Genes with less than 10 fragments in all the samples were removed before differential expression analysis. The differential expression between conditions was statistically assessed by R/Bioconductor package DESeq (23) (version 1.18.0; RRID:SCR_000154). Genes with FDR (false discovery rate) ≤ 0.05 and length > 200bp were called as differentially expressed. Principle Component Analysis (PCA) was performed by R function prcomp using cpm (count of fragments in each gene per million of fragments mapped to all exons) values. The scale option was set as TRUE. The normalized counts from DESeq were used to generate heatmap and dendrogram by Cluster 3.0 (24) (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.html) and Java Treeview (25). The values in each gene were centered by median and rescaled so that the sum of the squares of the values is 1.0. Mapping and differential expression analysis was handled by the MD Anderson Science Park Bioinformatics and Statistics Department.
Quantitative RT-PCR (qRT-PCR)
qRT-PCR was performed using the Power SYBR® RNA-to-Ct kit (Thermo Fisher) Reactions were run in an Applied Biosciences 7500 Fast Block Real Time PCR System. Primers sequences are listed in Supplementary Table S6. ΔΔCT was calculated for all samples relative to Gapdh. Results are expressed as relative mRNA. Primers are listed in Supplementary Table S2.
Statistical Analysis
All data was represented as mean ± standard error of mean. The statistical analysis for qRT-PCR was performed in Microsoft Excel (RRID:SCR_016137). All other statistical analysis was performed in Prism 8 (GraphPad Software 8.0; GraphPad Prism, RRID:SCR_002798) using Students t-test or analysis of variance. Significant P-value was < 0.05. Venn diagram for RNA-seq was produced using BioVenn software (26).
Code Availability
Data in this manuscript are generated using commonly available commercial software and algorithms and are detailed in the corresponding “Methods” section. Specific computer code is not applicable.
Data Availability
RNA-Seq data has been deposited into the Gene Omnibus repository and is available at GSE154108.
Results
Gcn5 loss does not significantly alter normal B cell development in mice
Gcn5 is necessary for normal embryonic development in mice (9,10,27–29). Recent studies have also demonstrated a role for Gcn5 in the development and activation of certain subsets of T cells in vitro (30,31). Therefore, we began our studies by determining whether Gcn5 loss affects the normal development of B cells in mice. We intercrossed Gcn5Fx/Fx mice with mice bearing the B cell specific CD19-Cre transgene. CD19 is expressed in B cells at the Pro B stage and expression continues through the germinal center reaction. Terminally-differentiated plasmablasts and plasma cells lose this marker as they become professional antibody-secreting cells (ASCs). CD19-CreTg/0; Gcn5+/+ and CD19-CreTg/0; Gcn5Fx/Fx mice were born at expected frequencies. Gcn5 deletion was confirmed by immunoblot of proteins from sorted CD19+ splenocytes (Supplementary Fig. S1A).
Comparisons of both bone marrow and splenic cells of 5 to 6-week old CD19-CreTg/0; Gcn5+/+ (Gcn5 WT) and CD19-CreTg/0; Gcn5Fx/Fx mice (Gcn5Fx/Fx) (Fig. 1A–B) revealed no significant differences in the overall number of cells from either tissue. The size of the spleen was also not significantly altered upon loss of Gcn5 (Fig.1C). Next, we profiled B cell populations of the mice by flow cytometry. Percentages of CD19+ cells were comparable between WT and Gcn5Fx/Fx mice (Supplementary Fig. S1B). We assessed numbers of ProB and PreB (Fig. 1D; Supplementary Fig. S1C) cells from bone marrow, and again found no significant changes in the Gcn5 deficient mice. The ratio of immature to mature B cells (Fig. 1E; Supplementary Fig. S1D) from the bone marrow, as well as germinal center cells from the spleen (Supplementary Fig. S1E), were also unaltered upon loss of Gcn5. These results indicate that Gcn5 loss does not impact the development of either early or intermediate stages of B cell maturation, consistent with the lack of changes in cellularity of B cells in both spleen and bone marrow.
Figure 1. Gcn5 loss does not significantly alter normal B cell development in mice.
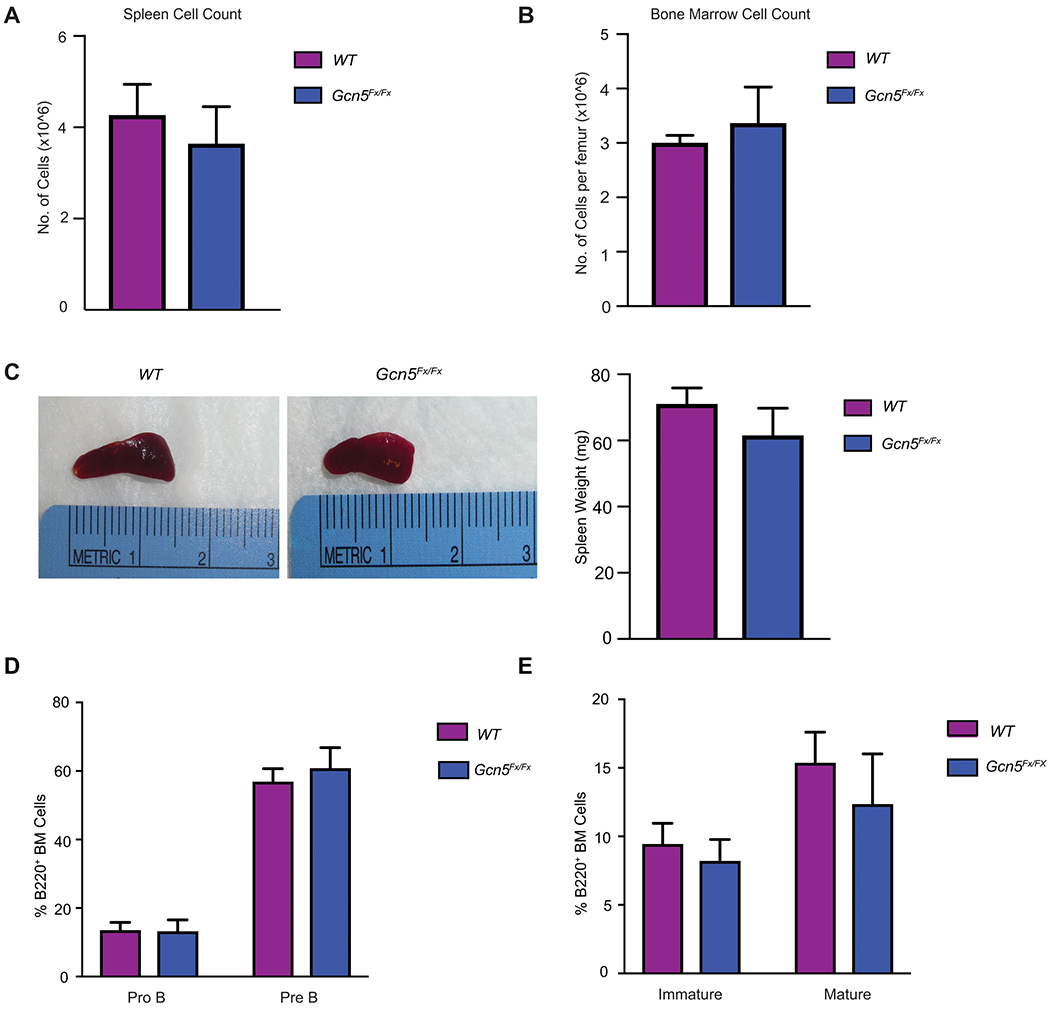
A. Total cellularity of spleen of WT and CD19-Cre; Gcn5Fx/Fx mice 5-6-week-old mice (n=4 WT and n= 4 CD19-Cre; Gcn5Fx/Fx). B. Total femur cellularity of WT and CD19-Cre; Gcn5Fx/Fx 5-6-week-old mice (n=4 WT and n= 4 CD19-Cre; Gcn5Fx/Fx). C. Representative pictures and spleen weights of 5-6-week-old mice (n= 4 WT and n= 4 CD19-Cre Gcn5Fx/Fx). D. Quantification of flow cytometric analysis of B220+ bone marrow ProB (PI−B220+CD43hiIgM−) and PreB (PI−B220+CD43loIgM−) cells. E. Quantification of flow cytometric analysis of Immature (PI−B220+CD43loIgM+IgD−) and Mature (PI−B220+CD43loIgM−IgD+) B cells from bone marrow. Error bars represent mean ± SEM. For flow cytometric analysis n= 7 WT and n=5 CD19-Cre; Gcn5Fx/Fx mice. All p-values determined by unpaired Student’s t-test. *p≤ 0.05; **p≤ 0.01; ***p≤ 0.001; ****p≤ 0.0001.
Previous research in DT40 chicken B cells reported a reduction in IgM expressing B cells, correlated with the loss of expression of the IgM heavy chain, upon deletion of GCN5 (32). However, we did not observe a change in the IgM+ population in our mouse model (Supplementary Fig. S1F). Loss of GCN5 in DT40 cells also resulted in reduction in expression of IRF4 and Blimp-1, two genes that are essential for plasma cell differentiation (33). However, we did not observe any difference in expression of IRF4 and Blimp-1 in CD19+ cells from Gcn5Fx/Fx mice (Supplementary Fig. 2). From these analyses, we conclude that loss of Gcn5 does not significantly alter ASC development in mice.
Survival of Eμ-Myc mice is prolonged upon deletion of Gcn5 in CD19+ B cells
Next, we addressed whether loss of Gcn5 affects the formation of Myc-driven B-cell lymphoma in the Eμ-Myc mouse model. Previous studies by others have established that overexpression of Myc in B cells of these mice promotes proliferation of pre B cells, at the expense of differentiation (34). To determine whether Gcn5 is required for Myc functions in this context, we again used the B cell specific CD19-Cre and our Gcn5 floxed allele to delete Gcn5 in B cells in Eμ-Myc mice. We compared timing of lymphoma onset in CD19-Cre0/0; Eμ-MycTg/0; GCN5Fx/+ (hereafter referred to as Eμ-Myc), CD19-CreTg/0; Eμ-Myc Tg/0; GCN5Fx/+ (Eμ-Myc; Gcn5Fx/+) mice and CD19-CreTg/0; Eμ-Myc Tg/0; GCN5Fx/Fx (Eμ-Myc; Gcn5Fx/Fx) mice. Gcn5 protein (Fig. 2A) and RNA levels (Fig. 2B) were increased in the presence of the Eμ-Myc transgenes, consistent with previous findings by our lab and others that Gcn5 is a Myc target gene (11,35).
Figure 2. Gcn5 Loss Delays Formation of B Cell Lymphoma in Mouse Model.
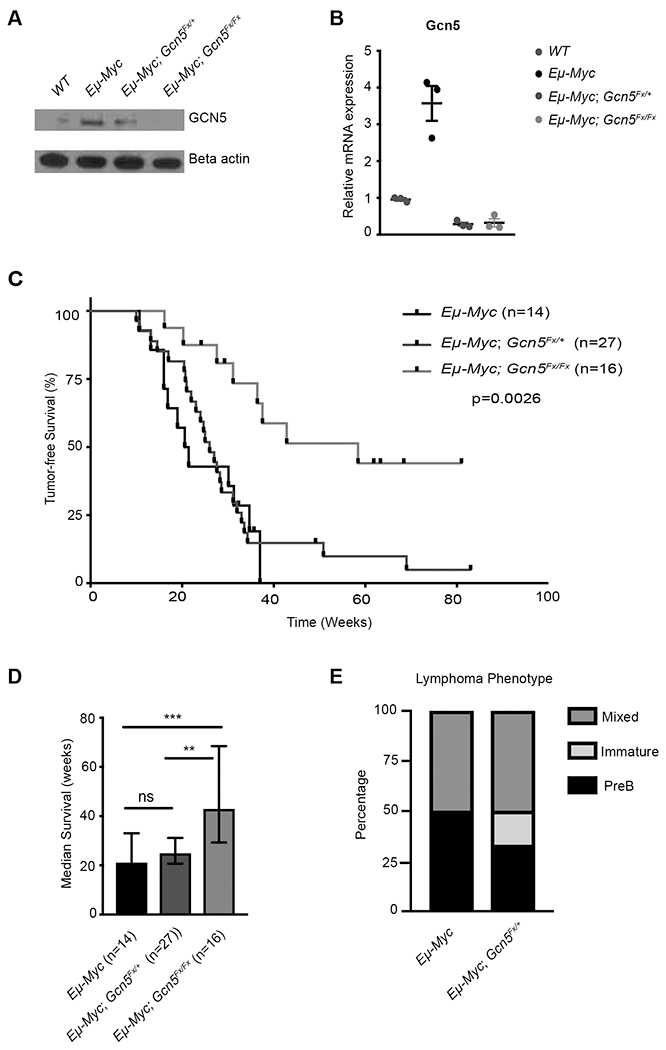
A. Representative immunoblot showing increased expression of Gcn5 in CD19+ B cells of Eμ-Myc transgenic mice. B. Relative Gcn5 mRNA expression in CD19+ B cells of WT, Eμ-Myc, Eμ-Myc; Gcn5Fx/+, and Eμ-Myc; Gcn5Fx/Fx mice. (n=3). All p-values determined by unpaired Student’s t-test. Significant P-value was <0.05 C. Kaplan-Meier curve showing tumor-free survival comparing CD19-Cre0/0; Eμ-Myc; Gcn5Fx/+ (n=14), Eμ-Myc; Gcn5Fx/++ mice (n=27) and Eμ-Myc; Gcn5Fx/Fx (n=16) mice. p=0.0026 determine by Log rank test (χ2= 8.406). Log rank (Mantel-Cox) p= 0.0014 (χ2=11.88), Gehan-Breslow-Wilcoxon test p= 0.0023 (χ2=10.31) D. Median survival time of Eμ-Myc, Eμ-Myc; Gcn5Fx/+ mice and Eμ-Myc; Gcn5Fx/Fx mice. p-values determined by one-way ANOVA *p≤ 0.05;**p≤ 0.01; ***p≤ 0.001; ****p≤ 0.0001. E. Quantification of immune phenotypes of tumors from Eμ-Myc and Eμ-Myc; Gcn5Fx/+ mice.
Eμ-Myc mice develop B cell lymphoma as early as 6 weeks of age to as late as 65 weeks (34), consistent with our data showing an average latency period of 21 weeks (p=0.0008) (Fig. 2C, black). However, we observed a dramatic increase in latency of lymphoma development in Eμ-Myc; Gcn5Fx/Fx mice (Fig. 2C, red). Loss of one allele of Gcn5 in the Eμ-Myc; Gcn5Fx/+ mice had minimal effects on survival (Fig. 2C, blue), with lymphoma onset at an average of 25 weeks (p= 0.4959) (Fig. 2D). In contrast, complete loss of Gcn5 extended the median survival of Eμ-Myc; Gcn5Fx/Fx mice to an average of 58.4 weeks (p=0.0026) (Fig. 2D). Survival of the homozygous Eμ-Myc; Gcn5Fx/Fx mice was significantly increased relative to the heterozygous Eμ-Myc; Gcn5Fx/+ mice as well (p=0.0049). Our data suggests Gcn5 haploinsufficiency does not effectively delay lymphomagenesis, however loss of both alleles of Gcn5 inhibits the formation of B cell lymphoma in Eμ-Myc mice.
Loss of Gcn5 alters lymphoma phenotype
Eμ-Myc mice typically present with lymphomas arising from PreB (B220+CD19+IgM−), Immature B (B220+CD19+IgM+), or mixed (both PreB and Immature B) cells (36), reflecting Myc driven expansion of less differentiated B cell states, with impaired differentiation. To determine if reduced expression of Gcn5 affects this expansion, we compared tumor phenotypes by flow cytometry (Supplementary Fig. S3). Because few tumors were available from the Eμ-Myc Gcn5 null mice, we compared tumor phenotypes in Eμ-Myc and Eμ-Myc; Gcn5Fx/+ mice, as these mice did exhibit a slight delay in tumor onset. Eμ-Myc mice presented with an equal number of PreB and mixed lymphomas, as expected (Fig. 2E). Eμ-Myc; Gcn5Fx/+ mice displayed primarily mixed lymphomas (50%) with a fewer number of preB lymphomas than Eμ-Myc mice. These data indicate that loss of Gcn5 retards the expansion of this early population by Myc, allowing further B cell development, and in turn, leading to slower growing lymphomas, or even a block to lymphoma development as seen in some Eμ-Myc; Gcn5Fx/Fx (and even some the Eμ-Myc; Gcn5Fx/+) mice (Fig. 2C).
Pre-lymphomic mice with loss of Gcn5 display reduction in B cell subsets
To define early events that contribute to the increased latency of lymphoma development in the absence of Gcn5, we compared cellular populations in spleens of mice prior to lymphoma development. No discernible difference between spleen size (Fig. 3A) or spleen cell count (Fig. 3B) was observed in pre-lymphomic 5-6-week-old Eμ-Myc; GCN5Fx/Fx and Eμ-Myc mice, and these profiles were similar to those in wild type mice lacking the Eμ-Myc transgene. The percentage of B220+cells in the spleens of Eμ-Myc; GCN5Fx/Fx and Eμ-Myc mice were also similar, but interestingly, the PreB (PI−B220+CD43loIgM−) and Immature B populations (PI−B220+CD43loIgM+IgD−) were significantly lower in the Eμ-Myc; GCN5Fx/Fx mice relative to Eμ-Myc mice (Fig. 3C). The Mature population (PI−B220+CD43loIgM+IgD+) however was not different between the two genotypes. Given that lymphomas in Eμ-Myc mice tend to arise from the PreB and early stage B cell subsets (36), the reductions in these populations upon loss of Gcn5 is consistent with retardation of proliferative capability and increased latency of lymphomagenesis.
Figure 3. Pre-lymphomic Eμ-Myc mice have altered B cell populations with loss of Gcn5.
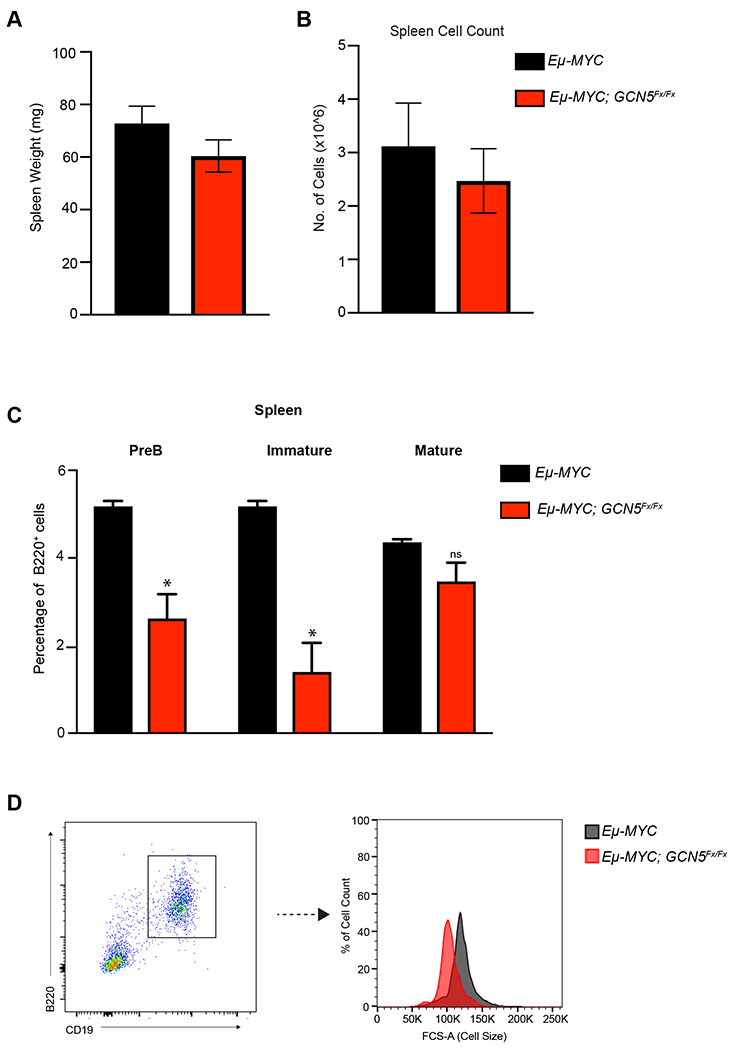
A. Spleen weights of 5-6-week-old Eμ-Myc and Eμ-Myc; Gcn5Fx/Fx mice (n=4). B. Total cellularity of spleen of Eμ-Myc and Eμ-Myc; Gcn5Fx/Fx mice (n=4). C. Quantification of flow cytometric analysis of B220+ spleen PreB (PI−B220+CD43loIgM−) cells, Immature (PI−B220+CD43loIgM+IgD−) and Mature (PI−B220+CD43loIgM−IgD+) B cells. Error bars represent mean ± SEM. For flow cytometric analysis n= 3 for WT and CD19-Cre; Gcn5Fx/Fx mice. All p-values determined by unpaired Student’s t-test. *p≤ 0.05; **p≤ 0.01; ***p≤ 0.001; ****p≤ 0.0001. D. Representative FACS analysis of cells size as measured by forward scatter (FCS-A) of B220+CD19+ B cells from. 5-6-week-old Eμ-Myc and Eμ-Myc; Gcn5Fx/Fx mice (n=3). Black curve is Eμ-Myc. Red curve is Eμ-Myc; Gcn5Fx/Fx.
Since Myc overexpression in Eμ-Myc mice drives increases in B cell size (15,37,38), we also determined whether loss of Gcn5 affects this phenotype. Indeed, Eμ-Myc; Gcn5Fx/Fx B cells were smaller than Eμ-Myc B cells (Fig. 3D), again demonstrating that loss of Gcn5 impacts Myc driven oncogenic phenotypes.
Gcn5 promotes expression of Myc target genes in MYC overexpressing B cells
In order to define the molecular mechanisms for how Gcn5 contributes to lymphoma development upon overexpression of Myc, we performed RNA-seq analyses on total RNA isolated from CD19+ B cells sorted from spleens of 5-6-week-old wild-type, Eμ-Myc, and Eμ-Myc; Gcn5Fx/Fx mice. This time point was chosen to enrich for cells before the onset of lymphoma, in order to define early transcriptional events that require Gcn5 as coactivator. Principal Component Analysis (PCA) showed clear separation between the different genotypes, with the profiles of Eμ-Myc Gcn5Fx/Fx B cells closer to those of wild-type B cells than to B cells from Eμ-Myc mice (Supplementary Fig. S4A). Gene expression heatmap and hierarchical clustering confirmed that patterns of expression of Eμ-Myc; Gcn5Fx/Fx B cells were more similar to those of wild-type B cells than those of Eμ-Myc B cells (Fig. 4A; Supplementary Fig. S4B). We identified 3588 genes as upregulated and 2544 as downregulated in the Eμ-Myc B cells as compared to wild-type (false discovery rate (FDR) <0.05) (Supplementary Fig. S4C). 532 genes were identified to be downregulated and 759 genes upregulated in the Eμ-Myc; Gcn5Fx/Fx B cells as compared to the Eμ-Myc B cells.
Figure 4. Overview of differential gene expression in Gcn5 floxed Eμ-Myc mice.
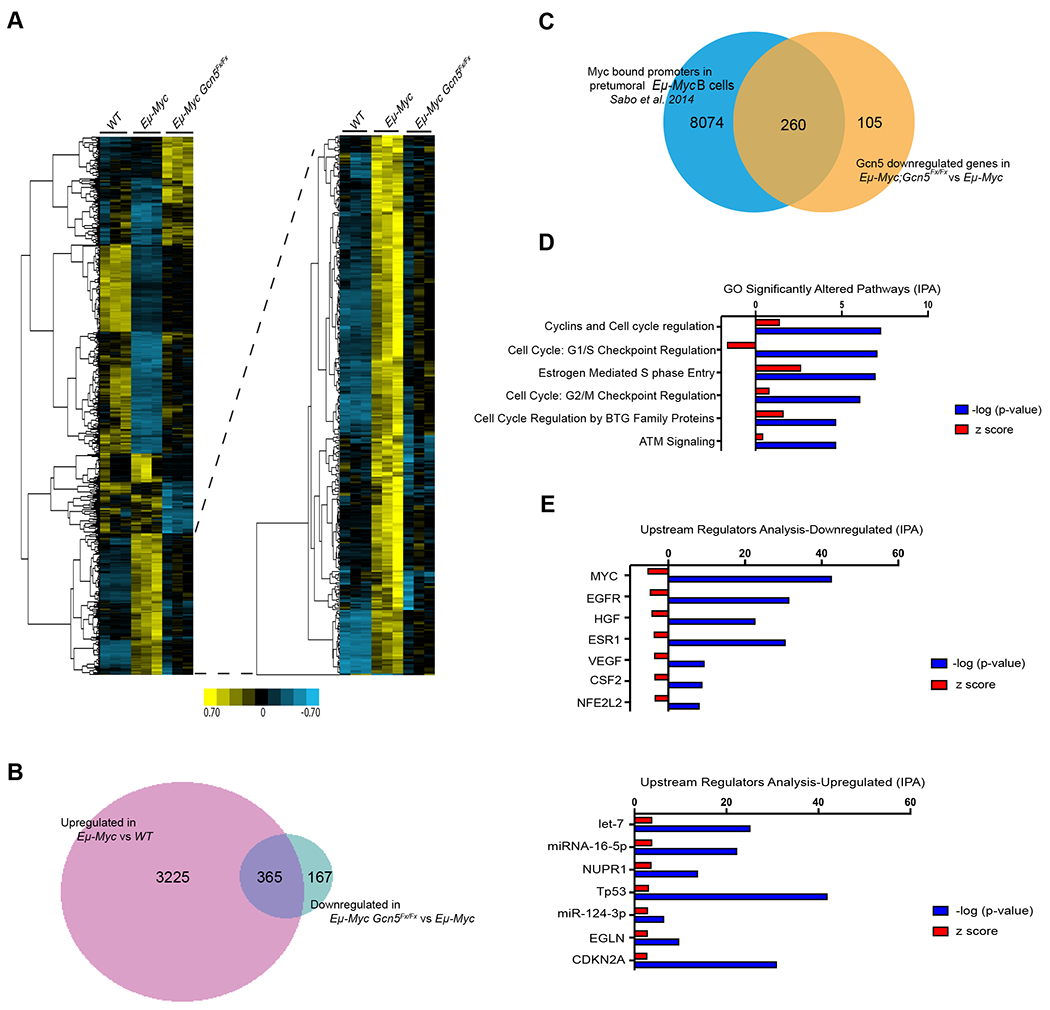
A. Heatmap of gene expression profiles from CD19+ splenic B cells. Expression profiles of CD19+ B cells of 5-6-week-old WT, Eμ-Myc, and Eμ-Myc; Gcn5Fx/Fx mice. (n=3). Yellow and blue indicate up and down regulation relative to the median expression of each gene respectively. B. Venn diagram of genes that were upregulated in Eμ-Myc (3558 genes) compared with downregulated in the Gcn5 floxed B cells (532 genes). C. Comparison of a published dataset of Myc-bound promoters in pretumorous B cells from Eμ-Myc mice to genes down regulated in pretumorous B cells from Eμ-Myc vs Eμ-Myc; Gcn5Fx/Fx mice. D. GO analysis of significantly altered pathways by IPA. E. Predicted upstream regulators whose expression change can lead to the down regulation of genes upon deletion of Gcn5 as compare to Eμ-Myc mice.
Because Gcn5 is a known transcriptional co-activator we focused our analysis on the genes that were both upregulated in the Eμ-Myc B cells relative to wild type and downregulated in Eμ-Myc; Gcn5Fx/Fx B cells relative to Eμ-Myc B cells (FDR≤ 0.05) (Fig. 4A). Of the 3588 genes upregulated in Eμ-Myc B cells, 356 were among the 532 genes downregulated in Eμ-Myc; Gcn5Fx/Fx B cells (Fig. 4B), indicating that Gcn5 is required for activation of these specific Myc target genes. IPA analysis indicated that genes not significantly influenced by loss of Gcn5 in Eμ-Myc B cells are involved in protein synthesis and RNA post-transcriptional modification, damage and repair, whereas Gcn5 influenced genes are linked to cell cycle regulation and gene expression. A previous study defined a number of gene promoters directly bound by Myc in B cells from control, pre-lymphomic Eμ-Myc mice as well as the tumors arising from the mice (15). We compared the genes affected by Gcn5 to the genes shown to be directly regulated by Myc in pre-lymphomic cells. We found a substantial overlap (260 out of the 365 genes) further indicating that Gcn5 is important for activation of these genes by Myc (Fig. 4C).
Gene ontology analysis from IPA (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis) identified cancer as the top related disease (Supplementary Fig. S4D) of these genes, underscoring their importance to Myc driven oncogenesis. Under this category, cell proliferation, cell viability and cell survival of tumor cells were identified as the major biofunctions downregulated in Eμ-Myc; Gcn5Fx/Fx B cells. The pathways altered in Eμ-Myc Gcn5 deficient B cells are predominantly linked with cell cycle regulation (Fig. 4D), including upregulation of cyclins and G2/M checkpoint regulation. In contrast, G1/S checkpoint regulation was downregulated. These results are consistent with previous studies of the transcriptional targets of Gcn5 in non-small cell lung cancer (39). Cell cycle related genes such as Cdc25A, Cdc25B, Btg2 and Foxm1 were significantly downregulated in Eμ-Myc Gcn5 deficient B cells. DAVID software (40,41) confirmed these results (Supplementary Table S3), identifying cell cycle and regulation of transcription as top altered pathways, as well as DNA repair and RNA splicing pathways. These functions have also been previously linked to Gcn5 functions in different cell types (42–44).
Upstream regulator analysis from IPA identified Myc as the protein whose downregulation is predicted to primarily bring about the most gene expression changes in Eμ-Myc Gcn5 deficient B cells, as expected (p=1.42E-13, z-score: −5.497) (Fig. 4E). Other downregulated pathways include Egfr (p= 3.05E-10, z-score: −4.868), Hgf (p= 0.000000144, z-score: −4.456), and Vegf (p= 0.00137, z-score: −3.786), all of which are related to growth and proliferation of both normal cells and cancers (Supplementary Tables S4–6) (45,46). IPA upstream regulator analysis also identified pathways that were upregulated in Eμ-Myc Gcn5 deficient B cells relative to Eμ-Myc B cells, including let-7, Tpr53, and Cdkn2A (Fig. 4E; Supplementary Table S3). The microRNA let-7a has been shown previously to downregulate Myc, and its proliferation effects in Burkitt lymphoma (47). The Cdkn2A locus encodes the p14ARF and p16ARF tumor suppressors, which protect p53 from degradation (48,49), a crucial initiation step in the development of Burkitt lymphoma (50).
At least 51 direct Myc targets identified as downregulated upon loss of Gcn5 (Fig. 5A, B) are involved in cell cycle regulation, including Ccne2, Chek1, E2f1, Cks2 and Akt1. Interestingly, Csk2 is highly expressed in malignant tumors (51), and our previous work in cell lines connected inhibition of GCN5 with decreased AKT phosphorylation as well as decreased AKT protein levels (13). Our current data indicate that transcription of Akt is also affected by loss of Gcn5. These transcriptional changes indicate that Gcn5 is required for Myc-driven up regulation of genes important for proliferation of pre B cells, leading to decreased expansion of these populations in the Eμ-Myc; Gcn5Fx/Fx mice relative to Eμ-Myc mice.
Figure 5. Most genes downregulated upon loss of Gcn5 are up regulated upon Myc overexpression.
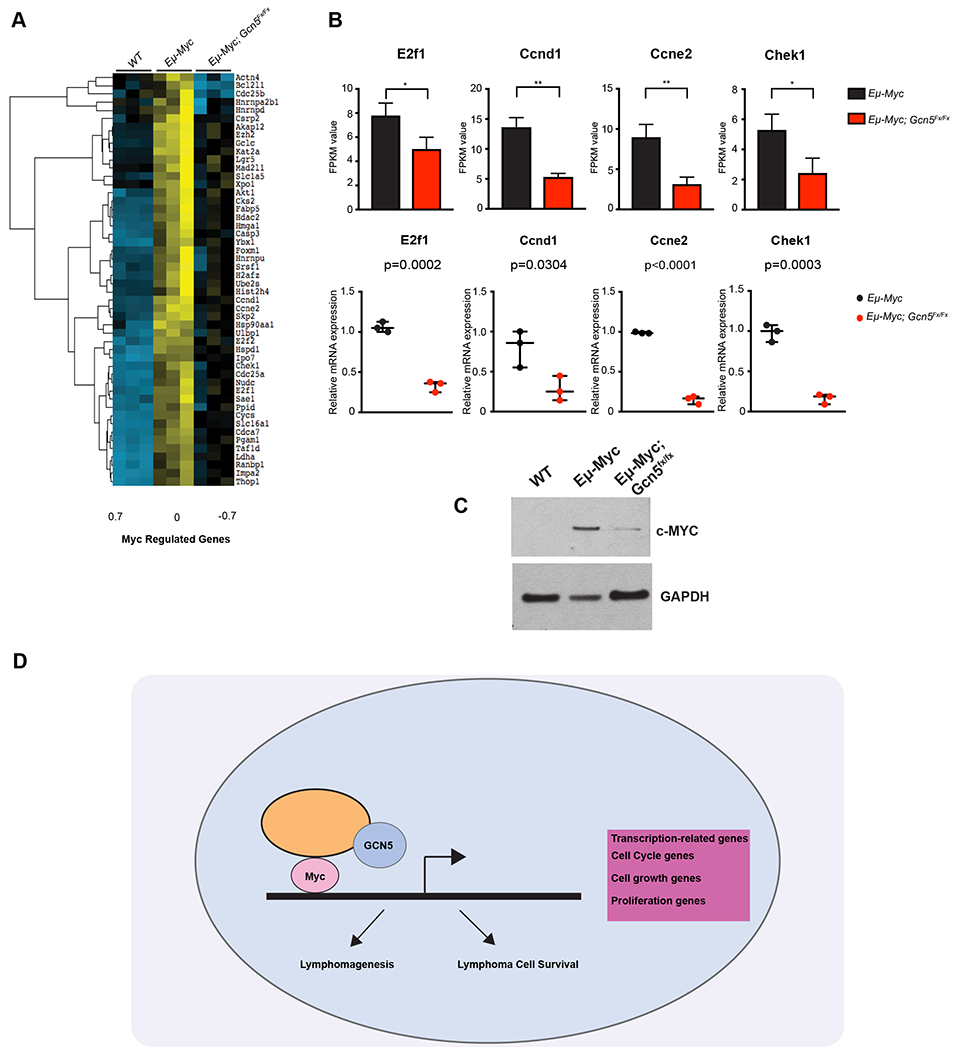
A. Heat Map of differentially expressed genes regulated by Myc. B. RNA-seq values and qRT-PCR validation of genes regulated by Myc. (n=3) All p-values determined by unpaired Student’s t-test. Significant P-value was <0.05. C. Immunoblot of Myc expression relative to Gapdh D. Model of Gcn5 partnership with the Myc oncoprotein in B cell lymphoma. Gcn5 acts as a transcriptional coactivator in the transcription of Myc target genes that promote both lymphomagenesis and lymphoma cell survival.
To determine whether Gcn5 loss affects Myc expression, we examined both Myc RNA and protein levels. Our RNA-seq data indicated that Myc RNA levels were not significantly altered by Gcn5 loss in Eμ-Myc; Gcn5Fx/Fx B cells relative to Eμ-Myc B cells (Supplementary Fig. S4). However, immunoblot analyses revealed that Myc protein levels were substantially decreased in the Eμ-Myc; Gcn5Fx/Fx B cells, although not to levels observed in wild type B cells (Fig. 5C). These findings are consistent with data from our lab and others that Myc is stabilized upon acetylation by Gcn5 and other KATs (12; 13).
Taken together, our data confirm that Gcn5 is required for oncogenic transcription programs related to cell cycle progression and cell proliferation driven by Myc overexpression (Fig. 5D).
Discussion
Genes whose loss delays the onset of lymphomagenesis are also often required for normal B cell development, raising concerns about therapeutic use of specific inhibitors targeting those factors. Our finding that Gcn5 loss does not affect normal B cell development or cellularity is consistent with previous reports that Gcn5 is not required for hematopoietic development or myeloid lineage commitment (52). The lack of effect in these settings might reflect redundancies in functions of Gcn5 and the highly related KAT Pcaf (Kat2B), but such compensation does not appear to occur in Myc overexpressing cells. The difference in effects of Gcn5 on B cell proliferation vs differentiation in wildtype and Eμ-Myc B mice is consistent with oncogene addiction. Eμ-Myc driven lymphomas required sustained high level expression and function of Myc (53) which we show here in turn requires Gcn5.
Myc often acts in concert with another transcription factor important in the regulation of cell growth, E2f1. Interestingly, Gcn5 also functions as a transcriptional co-activator of E2f1 target genes. In small cell lung cancer, E2F1 recruits GCN5 to acetylate H3K9, facilitating transcription of E2F1, CYCLIN E, and CYCLIN D1 (39), all of which promote cellular proliferation and tumor growth. Intriguingly, the effects of Gcn5 deletion in Eμ-Myc mice are similar to those caused by deletion of E2f family members. E2f1 deletion delayed formation of B cell lymphoma in Eμ-Myc mice, affecting Myc induced proliferation in lymphoma without affecting normal B cell development (54). A subsequent study did not find increased survival upon deletion of E2f1, which might reflect either differences in the genetic background or the specific null allele used, but an increased latency in lymphoma onset and alterations in cell cycle were observed upon deletion of E2f2 and E2f4 (55). Our data indicate that Gcn5 loss decreases expression of both E2f1 and E2f2 in Eμ-Myc mice. Skp2, an E3 ligase whose transcription is promoted by E2f1, was also downregulated. Skp2 mediated degradation of p27 is a known contributor in the development of Burkitt lymphoma(56–58). All of these changes in gene expression upon loss of Gcn5 may contribute to delay of lymphoma onset.
Gcn5 may also contribute to oncogenesis through modification of non-histone proteins. In many cases of acute lymphoblastic leukemia (ALL), a translocation event fuses E2A with the pre-B-cell leukemia transcription factor 1 (PBX1) creating the chimeric transcription factor E2A-PBX1 which drives oncogenesis (59). GCN5 acetylates this fusion protein increasing its stability in ALL cells (60). Other studies indicate that the overall cellular acetylation state can affect tumorigenesis and cancer cell survival. Reduction of Acetyl-CoA, and thereby total acetylation levels, reduces tumor formation in mammalian hepatocellular carcinoma and triple negative breast cancer cells (61). Also, knockdown of HDAC1 and HDAC2 accelerated leukemogenesis in a mouse model of acute promyelocytic leukemia (APL) as well as lymphomagenesis in the Eμ-Myc mouse model, however both knockdown and pharmacologic inhibition of HDAC1 was efficacious in already formed APL cells (62). Indeed, the balance of HAT and HDAC activities is likely critical in both developmental and disease contexts (3).
A majority of human cancers overexpress Myc (63), but no effective means to inhibit Myc activity in tumors yet exists. Our findings indicate that Gcn5 loss inhibits the ability of the Eμ-Myc transgene to drive B cell lymphomagenesis in mice, likely due to down regulation of cell cycle genes normally induced by Myc overexpression. These data are consistent with functional connections between Gcn5 and Myc in stem cell self-renewal (64), in ES cell differentiation (29), and during reprogramming of somatic cells to induced pluripotent cells (11). We also previously demonstrated that Gcn5 works with Myc to co-transcribe genes that promote oncogenic growth and potential in human Burkitt lymphoma cell lines (13). Therefore, inhibition of Gcn5 provides an attractive avenue for specifically inhibiting Myc-driven oncogenesis and cancer cell survival. Specific inhibitors of Gcn5 might prove effective in Myc driven cancers without affecting normal cells. Combinations of Gcn5 inhibitors, to cripple Myc functions, with inhibitors that lower Myc expression, such as BET domain inhibitors or inhibitors of CBP/P300, might be especially efficacious.
Supplementary Material
Significance:
Our results provide important proof of principle for Gcn5 functions in formation and progression of Myc driven cancers, suggesting that GCN5 may be a viable target for development of new cancer therapies.
Acknowledgments
We would like to thank FCCIC and Pam Whitney for her support on flow cytometry experiments. We thank RASF, Amanda Martin, and Tim Macatee for their help with animal husbandry. We would like to thank the Science Park NGS Core for their work on the RNA sequencing, which was supported by Cancer Prevention and Research Institute of Texas Core Facility Support grant (RP170002 to JS). We would also like to thank all members of the Dent lab for their advice in experimental procedures. This work was funded by the National Institutes of Health to SYRD (RO1 GM0677182; R35 GM131678) and the Sowell-Huggins Professorship/Fellowship in Cancer Research to ATF and SYRD.
Funding Information: This work was funded by the National Institutes of Health to SYRD (RO1 GM0677182; R35 GM131678) and the Sowell-Huggins Professorship/Fellowship in Cancer Research to ATF and SYRD.
Footnotes
Conflict of Interest Statement: The authors declare no potential conflicts of interest.
References
- 1.Newbold A, Falkenberg KJ, Prince MH, Johnstone RW. How do tumor cells respond to HDAC inhibition? Febs J 2016 [DOI] [PubMed] [Google Scholar]
- 2.Li Y, Seto E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med 2016;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farria A, Li W, Dent SY. KATs in cancer: functions and therapies. Oncogene 2015;34:4901–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flinn EM, Wallberg AE, Hermann S, Grant PA, Workman JL, Wright AP. Recruitment of Gcn5-containing complexes during c-Myc-dependent gene activation. Structure and function aspects. J Biol Chem 2002;277:23399–406 [DOI] [PubMed] [Google Scholar]
- 5.Zhang N, Ichikawa W, Faiola F, Lo SY, Liu X, Martinez E. MYC interacts with the human STAGA coactivator complex via multivalent contacts with the GCN5 and TRRAP subunits. Biochim Biophys Acta 2014;1839:395–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu X, Tesfai J, Evrard YA, Dent SY, Martinez E. c-Myc transformation domain recruits the human STAGA complex and requires TRRAP and GCN5 acetylase activity for transcription activation. J Biol Chem 2003;278:20405–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kenneth NS, Ramsbottom BA, Gomez-Roman N, Marshall L, Cole PA, White RJ. TRRAP and GCN5 are used by c-Myc to activate RNA polymerase III transcription. Proc Natl Acad Sci U S A 2007;104:14917–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phan HM, Xu AW, Coco C, Srajer G, Wyszomierski S, Evrard YA, et al. GCN5 and p300 share essential functions during early embryogenesis. Dev Dyn 2005;233:1337–47 [DOI] [PubMed] [Google Scholar]
- 9.Bu P, Evrard YA, Lozano G, Dent SY. Loss of Gcn5 acetyltransferase activity leads to neural tube closure defects and exencephaly in mouse embryos. Mol Cell Biol 2007;27:3405–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin W, Zhang Z, Srajer G, Chen YC, Huang M, Phan HM, et al. Proper expression of the Gcn5 histone acetyltransferase is required for neural tube closure in mouse embryos. Dev Dyn 2008;237:928–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirsch CL, Coban Akdemir Z, Wang L, Jayakumaran G, Trcka D, Weiss A, et al. Myc and SAGA rewire an alternative splicing network during early somatic cell reprogramming. Genes Dev 2015;29:803–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel JH, Du Y, Ard PG, Phillips C, Carella B, Chen CJ, et al. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Molecular and cellular biology 2004;24:10826–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farria AT, Mustachio LM, Akdemir ZHC, Dent SYR. GCN5 HAT inhibition reduces human Burkitt lymphoma cell survival through reduction of MYC target gene expression and impeding BCR signaling pathways. Oncotarget 2019;10:5847–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012;151:56–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sabo A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014;511:488–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan HM, et al. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep 2003;4:575–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ravens S, Yu C, Ye T, Stierle M, Tora L. Tip60 complex binds to active Pol II promoters and a subset of enhancers and co-regulates the c-Myc network in mouse embryonic stem cells. Epigenetics Chromatin 2015;8:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vervoorts J, Luscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, et al. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep 2003;4:484–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin W, Srajer G, Evrard YA, Phan HM, Furuta Y, Dent SY. Developmental potential of Gcn5(−/−) embryonic stem cells in vivo and in vitro. Dev Dyn 2007;236:1547–57 [DOI] [PubMed] [Google Scholar]
- 20.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013;14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mudge JM, Harrow J. Creating reference gene annotation for the mouse C57BL6/J genome assembly. Mamm Genome 2015;26:366–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31:166–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010;11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics 2004;20:1453–4 [DOI] [PubMed] [Google Scholar]
- 25.Saldanha AJ. Java Treeview--extensible visualization of microarray data. Bioinformatics 2004;20:3246–8 [DOI] [PubMed] [Google Scholar]
- 26.Hulsen T, de Vlieg J, Alkema W. BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 2008;9:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu W, Edmondson DG, Evrard YA, Wakamiya M, Behringer RR, Roth SY. Loss of Gcn5l2 leads to increased apoptosis and mesodermal defects during mouse development. Nat Genet 2000;26:229–32 [DOI] [PubMed] [Google Scholar]
- 28.Lin W, Zhang Z, Chen CH, Behringer RR, Dent SY. Proper Gcn5 histone acetyltransferase expression is required for normal anteroposterior patterning of the mouse skeleton. Dev Growth Differ 2008;50:321–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L, Koutelou E, Hirsch C, McCarthy R, Schibler A, Lin K, et al. GCN5 Regulates FGF Signaling and Activates Selective MYC Target Genes during Early Embryoid Body Differentiation. Stem Cell Reports 2018;10:287–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Yun C, Gao B, Xu Y, Zhang Y, Wang Y, et al. The Lysine Acetyltransferase GCN5 Is Required for iNKT Cell Development through EGR2 Acetylation. Cell Rep 2017;20:600–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao B, Kong Q, Zhang Y, Yun C, Dent SYR, Song J, et al. The Histone Acetyltransferase Gcn5 Positively Regulates T Cell Activation. J Immunol 2017;198:3927–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kikuchi H, Nakayama M, Kuribayashi F, Imajoh-Ohmi S, Nishitoh H, Takami Y, et al. GCN5 is involved in regulation of immunoglobulin heavy chain gene expression in immature B cells. Gene 2014;544:19–24 [DOI] [PubMed] [Google Scholar]
- 33.Kikuchi H, Nakayama M, Kuribayashi F, Imajoh-Ohmi S, Nishitoh H, Takami Y, et al. GCN5 is essential for IRF-4 gene expression followed by transcriptional activation of Blimp-1 in immature B cells. J Leukoc Biol 2014;95:399–404 [DOI] [PubMed] [Google Scholar]
- 34.Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. The E mu-myc transgenic mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J Exp Med 1988;167:353–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, McMahon SB, Eisenman RN. Myc influences global chromatin structure. EMBO J 2006;25:2723–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langdon WY, Harris AW, Cory S, Adams JM. The c-myc oncogene perturbs B lymphocyte development in E-mu-myc transgenic mice. Cell 1986;47:11–8 [DOI] [PubMed] [Google Scholar]
- 37.Iritani BM, Eisenman RN. c-Myc enhances protein synthesis and cell size during B lymphocyte development. Proc Natl Acad Sci U S A 1999;96:13180–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barna M, Pusic A, Zollo O, Costa M, Kondrashov N, Rego E, et al. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 2008;456:971–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen L, Wei T, Si X, Wang Q, Li Y, Leng Y, et al. Lysine acetyltransferase GCN5 potentiates the growth of non-small cell lung cancer via promotion of E2F1, cyclin D1, and cyclin E1 expression. J Biol Chem 2013;288:14510–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4:44–57 [DOI] [PubMed] [Google Scholar]
- 41.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009;37:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo R, Chen J, Mitchell DL, Johnson DG. GCN5 and E2F1 stimulate nucleotide excision repair by promoting H3K9 acetylation at sites of damage. Nucleic Acids Res 2011;39:1390–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gunderson FQ, Johnson TL. Acetylation by the transcriptional coactivator Gcn5 plays a novel role in co-transcriptional spliceosome assembly. PLoS Genet 2009;5:e1000682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hirsch CL, Coban Akdemir Z, Wang L, Jayakumaran G, Trcka D, Weiss A, et al. Myc and SAGA rewire an alternative splicing network during early somatic cell reprogramming. Genes Dev 2015;29:803–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saryeddine L, Zibara K, Kassem N, Badran B, El-Zein N. EGF-Induced VEGF Exerts a PI3K-Dependent Positive Feedback on ERK and AKT through VEGFR2 in Hematological In Vitro Models. PLoS One 2016;11:e0165876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jucker M, Gunther A, Gradl G, Fonatsch C, Krueger G, Diehl V, et al. The Met/hepatocyte growth factor receptor (HGFR) gene is overexpressed in some cases of human leukemia and lymphoma. Leuk Res 1994;18:7–16 [DOI] [PubMed] [Google Scholar]
- 47.Sampson VB, Rong NH, Han J, Yang Q, Aris V, Soteropoulos P, et al. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res 2007;67:9762–70 [DOI] [PubMed] [Google Scholar]
- 48.Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J 1998;17:5001–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, et al. p14ARF links the tumour suppressors RB and p53. Nature 1998;395:124–5 [DOI] [PubMed] [Google Scholar]
- 50.Schmitz R, Ceribelli M, Pittaluga S, Wright G, Staudt LM. Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harb Perspect Med 2014;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.You H, Lin H, Zhang Z. CKS2 in human cancers: Clinical roles and current perspectives (Review). Mol Clin Oncol 2015;3:459–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bararia D, Kwok HS, Welner RS, Numata A, Sarosi MB, Yang H, et al. Acetylation of C/EBPalpha inhibits its granulopoietic function. Nat Commun 2016;7:10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choi PS, van Riggelen J, Gentles AJ, Bachireddy P, Rakhra K, Adam SJ, et al. Lymphomas that recur after MYC suppression continue to exhibit oncogene addiction. Proc Natl Acad Sci U S A 2011;108:17432–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baudino TA, Maclean KH, Brennan J, Parganas E, Yang C, Aslanian A, et al. Myc-mediated proliferation and lymphomagenesis, but not apoptosis, are compromised by E2f1 loss. Mol Cell 2003;11:905–14 [DOI] [PubMed] [Google Scholar]
- 55.Rempel RE, Mori S, Gasparetto M, Glozak MA, Andrechek ER, Adler SB, et al. A role for E2F activities in determining the fate of Myc-induced lymphomagenesis. PLoS Genet 2009;5:e1000640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lim MS, Adamson A, Lin Z, Perez-Ordonez B, Jordan RC, Tripp S, et al. Expression of Skp2, a p27(Kip1) ubiquitin ligase, in malignant lymphoma: correlation with p27(Kip1) and proliferation index. Blood 2002;100:2950–6 [DOI] [PubMed] [Google Scholar]
- 57.Martins CP, Berns A. Loss of p27(Kip1) but not p21(Cip1) decreases survival and synergizes with MYC in murine lymphomagenesis. EMBO J 2002;21:3739–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Old JB, Kratzat S, Hoellein A, Graf S, Nilsson JA, Nilsson L, et al. Skp2 directs Myc-mediated suppression of p27Kip1 yet has modest effects on Myc-driven lymphomagenesis. Mol Cancer Res 2010;8:353–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kamps MP, Look AT, Baltimore D. The human t(1;19) translocation in pre-B ALL produces multiple nuclear E2A-Pbx1 fusion proteins with differing transforming potentials. Genes Dev 1991;5:358–68 [DOI] [PubMed] [Google Scholar]
- 60.Holmlund T, Lindberg MJ, Grander D, Wallberg AE. GCN5 acetylates and regulates the stability of the oncoprotein E2A-PBX1 in acute lymphoblastic leukemia. Leukemia 2013;27:578–85 [DOI] [PubMed] [Google Scholar]
- 61.Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, et al. Acetate dependence of tumors. Cell 2014;159:1591–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Santoro F, Botrugno OA, Dal Zuffo R, Pallavicini I, Matthews GM, Cluse L, et al. A dual role for Hdac1: oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood 2013;121:3459–68 [DOI] [PubMed] [Google Scholar]
- 63.Bouvard C, Lim SM, Ludka J, Yazdani N, Woods AK, Chatterjee AK, et al. Small molecule selectively suppresses MYC transcription in cancer cells. Proc Natl Acad Sci U S A 2017;114:3497–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seruggia D, Oti M, Tripathi P, Canver MC, LeBlanc L, Di Giammartino DC, et al. TAF5L and TAF6L Maintain Self-Renewal of Embryonic Stem Cells via the MYC Regulatory Network. Mol Cell 2019;74:1148–63 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq data has been deposited into the Gene Omnibus repository and is available at GSE154108.