

Abstract
Histone deacetylase 6 (HDAC6) is a zinc-dependent HDAC that mainly modulates the acetylation status of non-histone substrates, such as α-tubulin and heat shock protein 90 (HSP90). The activity of HDAC6 plays a critical role in cell proliferation, protein trafficking and degradation, cell shape, migration, as well as regulation of immunomodulatory factors. For this reason, HDAC6 influences the progress of cancers, neurodegenerative disorders, and autoimmune responses. In the last few years, the discovery of selective HDAC6 inhibitors (HDAC6is) has become an attractive research area as five HDAC6is are being investigated in phase I/II clinical trials. However, the hydroxamic acid functional group still represents the predominant zinc-binding group (ZBG), that often suffers from poor pharmacokinetics and mutagenic potential, thus impairing the application of hydroxamate-based HDAC6is for long-term therapies. On the other hand, mercaptoacetamide (MCA)-based HDAC6is comprise a class of compounds that, in some cases, display nanomolar HDAC6 potency and a thousand-fold selectivity over class I HDAC isozymes. Moreover, MCA-based HDAC6is lack the mutagenicity associated with the hydroxamate function and display pharmacological effects, demonstrating the potential of this particular ZBG to improve upon the drug-like properties of HDAC6is. Herein, we summarize for the first time the structure-activity relationships (SARs) of MCA-based HDAC6is, discuss their HDAC6 selectivity at the molecular level using inhibitor-HDAC co-crystal structures, and further provide our perspective regarding their drug metabolism, pharmacokinetics, and pharmacological properties.
Keywords: Histone acetylation, Tubulin acetylation, Epigenetics, Sulfur, Zinc-binding group
1. Introduction
Histone deacetylases (HDACs) are a family of enzymes that balance the acetylation/deacetylation state of lysine residues on the histone tails around which the DNA is wrapped; histone acetylation is essential to promote chromatin unfolding, thus enabling transcriptional activation [1]. At present, the 18 known mammalian HDACs are divided into four classes according to their similarity to yeast deacetylases. Class I (HDAC1-3, and 8), class IIa (HDAC4, 5, 7, and 9), class IIb (HDAC6 and 10), and class IV (HDAC11) isozymes are zinc-dependent amidohydrolases that are capable of deacetylating the ε-amino group of the lysine residues on histones and non-histone proteins. In contrast, class III HDACs, also called sirtuins (SIRT1-7), exert their enzymatic activity through a nicotinamide adenine dinucleotide (NAD+)-dependent pathway, which hydrolyzes not only acetyl moieties but also crotonyl and propionyl groups [2-4]. HDAC inhibition has emerged as a practical therapeutic approach for cancer therapy based on the successful launch of five HDAC inhibitors (HDACis), namely vorinostat (SAHA, 1), panobinostat (LBH-589, 2), belinostat (PXD-101, 3), romidepsin (FK228, 4), and chidamide (CS055/HBI-8000, 5) (Fig. 1) [5]. However, most of them act as pan-HDAC inhibitors [6-8]. Their broad-spectrum HDAC inhibitory activities were found to be associated with various adverse effects (e.g., liver impairment and bone loss) and dose-limited toxicity [9,10]. Moreover, hydroxamate-based HDACis (e.g., vorinostat, panobinostat, and belinostat), the predominant class of HDACis, often exhibit unfavorable pharmacokinetic profiles (rapid clearance rates and short half-lives), which may result in inadequate drug exposure and thus result in a need for repeated administration [11,12]. It is also noted that hydroxamate-based HDACis have the potential to be mutagenic, which may cause the development of secondary cancers, preventing their use for some chronic diseases as well as neurological disorders [5]. Therefore, in the last decade, researchers have turned their interest to both the discovery of subtype- and isoform-selective HDACis and to the identification of alternative zinc-binding groups to replace the genotoxic hydroxamate moiety [13].
Fig. 1.
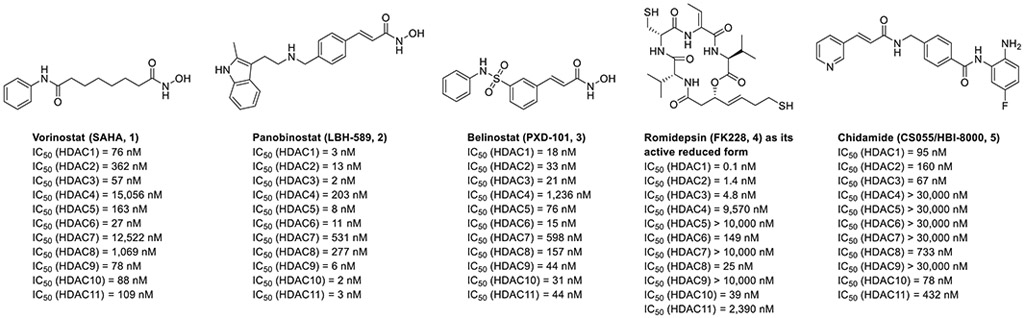
Structures and HDAC inhibitory activities of approved HDACis. The HADC IC50 values for vorinostat, panobinostat, and belinostat were extracted from Ref [6]. The HADC IC50 values for romidepsin were extracted from Ref [8]. The HDAC IC50 values for chidamide were extracted from Ref [7].
In comparison with the other HDAC isozymes, HDAC6 has become an attractive target related to its unique structural features, distinct biological function, and extensive therapeutic potentials. Unlike the other HDAC isoforms, HDAC6 is primarily found in the cytosol rather than in the nucleus, and it is structurally unique because it possesses two homologous catalytic domains (CD1 and CD2) [4]. Both domains are required for deacetylase activity, even though CD2 has been observed as the major catalytic domain involved in the deacetylation of cytosolic peptides [14,15]. CD2 is responsible for deacetylating peptides with an internal acetyllysine residue, such as α-tubulin and tau [16,17]. On the other hand, the function of CD1 is not fully understood, but there is evidence to support that CD1 serves as an E3 ubiquitin ligase [18]. Recent studies have also revealed that CD1 deacetylates the ATP-dependent RNA helicase DDX3X. In addition, CD1 was found to have narrower substrate specificity, compared to CD2, which solely recognizes peptide substrates containing C-terminal acetyllysine residues [19-23]. Furthermore, HDAC6 is the only isoform that has the ability to deacetylate non-histone substrates unrelated to the chromatin environment, including α-tubulin, heat shock protein 90 (HSP90), cortactin, phospho-binding protein 14-3-3ζ, as well as peroxiredoxin [24-27]. α-Tubulin was identified as the first substrate of HDAC6 whose acetylation status at lysine 40 can be regulated by HDAC6. Reduced α-tubulin acetylation has been observed in phenotypic animal models of neurodegenerative disorders, such as Alzheimer’s disease (AD) [28], Charcot-Marie-Tooth disease (CMT) [29-32], amyotrophic lateral sclerosis (ALS) [33,34], and Rett Syndrome (RTT) [35,36], which is associated with defective axonal transport or impaired brain-derived neurotrophic factor (BDNF) trafficking. Accordingly, upregulation of the levels of acetylated α-tubulin (Ac-Tub) by selective HDAC6 inhibition has been considered a potential therapeutic approach for neurodegenerative disorders [37]. Moreover, it has been recently reviewed that HDAC6 could also be a potential target for other rare diseases, such as inherited retinal diseases (IRDS) and idiopathic pulmonary fibrosis (IPF), attributed to its substrate specificity [38]. Recent research has also revealed that HDAC6 interacts with the transcription factor STAT3 [39], a crucial regulator of immune responses in the tumor microenvironment, and impacts the expression of the immunosuppressive molecule PD-L1 [40]. These effects are responsible for the immunomodulatory properties of some HDAC6 inhibitors (HDAC6is) [41-43]. Most selective HDAC6is exhibit weak/modest cytotoxicity in cancer cells while displaying significant antitumor effects as potential immunomodulatory agents in vivo through either single therapy or in combination with anti-PD-1 immune checkpoint inhibitors [44-46]. Thus, in light of the unique structural and functional properties of HDAC6, selective HDAC6is are being widely investigated as a potential treatment for various cancers, autoimmune disorders, and neurological disorders [38,47-49]. At present, five HDAC6is including ricolinostat (ACY-1215, 6), citarinostat (ACY-241, 7), KA2507 (structure not disclosed), CKD-504 (structure not disclosed), and CKD-506 (structure not disclosed) have been advanced into clinical trials primarily with a focus on the treatment of various cancers [37]. Moreover, it has recently been reported that the brain-penetrant selective HDAC6i EKZ-001 (bavarostat), containing 18F radioisotope, was used to investigate its HDAC6 binding in the brains of nonhuman primates and human through positron emission tomography (PET) [50,51]. Ricolinostat (Fig. 2A) is the first HDAC6 inhibitor to reach phase II clinical trials for cancers as a single agent and in combination with other chemotherapies such as lenalidomide, dexamethasone, bortezomib, and (nab)paclitaxel, and its investigation has been extended to non-cancer indications, such as diabetic neuropathic pain [52]. Citarinostat (Fig. 2A) is the second HDAC6 inhibitor to be investigated in phase I trials for melanoma and non-small cell lung cancer (NSCLC), and it is also being evaluated in combination with the immune checkpoint blockers nivolumab and ipilimumab [53]. These compounds show high structural similarity and are only considered partially-selective HDAC6is with about 10- to 15-fold selectivity over HDAC1-3 (Fig. 2A) [54,55]. Based on the short-comings of these clinical-stage HDAC6is, a lot of potent and selective HDAC6is with improved selectivity and better pharmacokinetic profiles has been identified in the last few years [37,56,57].
Fig. 2.

(A) Structures and in vitro HDAC activities of ricolinostat (6) and citarinostat (7). (B) Mechanism of the zinc-assisted Lossen rearrangement for hydroxamic acids, occurring inside the catalytic cavity of HDACs.
A typical pharmacophore of HDACis usually consists of four structural features (Fig. 2A): a zinc-binding group (ZBG) that interacts with the zinc ion (Zn2+) at the bottom of the catalytic site; a linker that interacts with the narrow hydrophobic channel; a capping group (Cap) that can explore additional interactions over the rim region of the catalytic pocket to strengthen the target engagement further; and a connecting unit (CU) area between the linker and the Cap. The Cap can adopt various structural features from simple alkyl chains to bulky polycyclic aromatic rings. At the same time, the linker region can be selected from a long aliphatic chain, a short phenyl/benzyl group, as well as a heterocyclic moiety. Among different types of ZBGs, the hydroxamic acid function is predominant in the discovery of HDAC6is, and only a limited number of non-hydroxamate ZBG has been identified with comparable HDAC6 potency and selectivity over other HDAC isoforms [37,56]. Moreover, the argument is still ongoing about the drug-like properties of hydroxamic acids as this functional group is responsible for the insufficient pharmacokinetics parameters of pan-HDACis’ and inadequate drug exposure. Moreover, hydroxamate may also have mutagenic potential that is related to the Lossen rearrangement occurred in vivo (Fig. 2B), affording mutagenic isocyanate products, hence impairing applications of hydroxamate-based HDAC6is where long-term treatments are required. Although some hydroxamate-based HDAC6is were found to be negative in the Ames test, which is an initial mutagenicity assessment [43,58,59], the identification of novel non-hydroxamate ZBGs is still the preferred approach to fully avoid the unfavorable properties of hydroxamates. During the past 15 years, our continuous efforts towards the discovery of HDAC6is bearing the mercaptoacetamide (MCA) moiety as an alternative ZBG has led to the development of ligands with nanomolar HDAC6 potency and thousand-fold selectivity over HDAC1. In this review, we summarize for the first time the structure-activity relationships (SARs) of MCA-based HDAC6is, analyze the molecular aspects of their selective HDAC6 inhibition on the basis of available inhibitor-HDAC co-crystal structures, and provide our perspective on their pharmacokinetics and pharmacological properties.
2. The development of MCA-based HDAC6is
2.1. Phenyl-derived caps
Romidepsin (8, Fig. 3A) is the only FDA-approved non-hydroxamate HDACi, which disulfide moiety can be converted into the active metabolite 4 in vivo through reduction catalyzed by glutathione reductase [60]. This finding has inspired the use of the disulfide group as well as a variety of other sulfur-bearing groups in the design of other novel HDACis. These include inter alia thiol [61-63], sulfamide [64], N-hydroxypyridine-2-thione [65], trithiocarbonate [66], thiazolidinedione [67], 3-hydroxypyridine-2-thione [68,69], sulfonamide [70], sulfone [70], and mercaptoacetamide [71] groups (Fig. 3B). Unlike others, the mercaptoacetamide (MCA) moiety has been demonstrated to represent a useful non-hydroxamate ZBG for the discovery of selective HDAC6is. In 2005, Suzuki and coworkers [70] were the first to incorporate the MCA moiety into HDACis. However, only the inhibition of total HDAC was reported for the vorinostat-like MCA analogs 10–12 (Fig. 3C and Table 1) in which 11 (n = 3), containing a five methylene alkyl chain as the linker, showed the most potent activity (total HDAC inhibition, IC50 = 0.39 μM) [70]. In 2006, Silverman and coworkers [72] further reported HDAC inhibitory activities of 11 and 12 against total HDACs (IC50 = 2.44 and 0.15 μM) and HDAC8 isoform (IC50 = 3.89 and 0.69 μM). The Kozikowski group resynthesized 11 and further tested it against Class I and IIb HDACs (unpublished), which yielded IC50 values of 0.33 μM and 3.76 μM potency against HDAC6 and HDAC1 (Table 1), respectively, while exhibiting modest activity (IC50 > 10 μM) against HDAC2, 8, and 10.
Fig. 3.

(A) Structure of romidepsin (8) and its active metabolite (4). (B) Sulfur-derived chelating moieties reported as HDAC ZBGs. (C) Structures of trichostatin A (9, TSA), vorinostat (1), and MCA-derived analogs (10–12).
Table 1.
HDAC inhibitory activity of phenyl- and benzyl-capped MCAs 10–26.
| Compound | Total HDAC (IC50, μM) | Class I (IC50, μM) |
Class IIb (IC50, μM) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC6 | HDAC10 | SIb | Ref | Original No. | ||
| SAHA (1)a | 0.28 (0.08) | 0.03 | 0.94 | 0.25 | 0.64 | 0.033 | 0.30 | 1 | [70, 71] | – |
| TSA (9)a | 0.004 | 0.004 | 0.014 | 0.002 | 1.38 | 0.001 | 0.005 | 4 | [71] | – |
| 10 | 11.00 | n.a.c | n.a. | n.a. | n.a. | n.a. | n.a. | n.d.d | [70] | 5c |
| 11a | 0.39 (0.30, 2.44) | 3.76 | >30 | n.a. | 14.1 (200, 3.89) | 0.34 | >30 | 11.1 | [70-72, 74] | 5b, 10e, 4, 1a |
| 12 | 3.00 (0.15) | n.a. | n.a. | n.a. | 0.69 | n.a. | n.a. | n.d. | [70] | 5a, 7 |
| 13a | 1.10 | 3.36 | 18.8 | n.a. | 7.53 | 0.19 | 8.71 | 17.6 | [71] | 4h |
| 14a | 0.90 | 2.42 | 10.4 | n.a. | 8.75 | 0.54 | 5.24 | 4.5 | [71] | 4i |
| 15a | 0.75 | 2.60 | 15.6 | n.a. | 11.4 | 0.52 | 4.34 | 5 | [71] | 5c |
| 16 | 0.63 | 10.9 | >30 | n.a. | 13.9 | 2.01 | >30 | 5.4 | [71, 73] | 5d, 6 |
| 17a | 1.00 | 5.18 | >30 | n.a. | 11.0 | 1.16 | 9.26 | 4.5 | [71] | 5e |
| 18 | 0.80 | 5.26 | >30 | n.a. | 12.3 | 0.22 | 8.78 | 23.9 | [71, 73] | 4a, 13 |
| 19 | 4.70 | 11.9 | >30 | n.a. | >30 | 0.62 | 29.7 | 19.2 | [71, 73] | 4b, 12 |
| 20 | 0.20 | 1.08 | 6.46 | n.a. | 10.2 | 0.11 | 1.54 | 9.8 | [71, 73] | 4c, 11 |
| 21 | 0.45 | 1.43 | 4.64 | n.a. | 19.1 | 0.48 | 3.43 | 3 | [71, 73] | 4d, 7 |
| 22a | 0.60 | 5.38 | 17.0 | n.a. | 15.6 | 0.32 | 17.7 | 16.8 | [71] | 10a |
| 23a | 1.70e | 2.82 | >30 | n.a. | n.a. | 0.68 | 4.61 | 4.1 | [75] | BC-4-93 |
| 24 | 1.70 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.d. | [71] | 5a |
| 25 | 5.00 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.d. | [71] | 5b |
| 26a | 0.20 | 14.7 | >30 | n.a. | 16.7 | 1.54 | >30 | 9.5 | [71] | 4k |
Unpublished HDAC isoform enzymatic data. IC50 values are the mean of two experiments obtained from curve-fitting of a 10-point enzymatic assay starting from 30 μM with 3-fold serial dilution against HDACs (Reaction Biology Corp, Malvern, PA).
SI, HDAC1/6 selectivity index.
n.a., not available.
n.d., not determined.
IC50 value was calculated from pIC50 value.
Based on these findings, the Kozikowski group further prepared the related analogs 13 and 14 (Fig. 4) by reversing the direction of amide moiety [71]. Compound 13, which has the same alkyl linker as 11, exhibited a slight improvement in HDAC6 potency (IC50 = 0.19 μM) and selectivity (SI = 17-folds). Further replacement of the amide CU by a urea group afforded phenyl- or benzyl-capped analogs 15–17 [71], which resulted in weaker HDAC6 potency and only about 5-fold selectivity over HDAC1. Additionally, MCAs 18–26 (Fig. 4), inspired by the N,N-dimethylaminophenyl Cap of trichostatin A (TSA, 9, Fig. 3), were first disclosed in 2005. In these compounds, modifications were made to both linkers and CUs [71], and some of their HDAC isoform inhibition results were reported in 2007 [73]. In the case of inhibitors 18–21, containing the same amide CU as in 13–14, compounds 18 (n = 1) and 20 (n = 3) displayed comparable submicromolar HDAC6 potency (IC50 = 0.22 and 0.11 μM), while the latter compound exhibits lower selectivity over HDAC1 (SI = 24.5- and 9.5-folds). Notably, compound 22, deviating from 20 by a retro-amide moiety, was almost two times more selective than 20 (SI = 17- and 9.5-folds), opposing the results of the pair of 11 and 13 [73]. The replacement of the alkyl linker with a para-xylylene moiety (26) caused a 10-fold loss of activity relative to 20 [71]. In addition, several MCAs together with several hydroxamate-based pan-HDACis were selected to assess their possible neuroprotective effects in primary cortical neurons using the homocysteic acid (HCA) model of oxidative stress [73]. Unlike the hydroxamates that were found to exhibit neuronal toxicity at high concentrations, the MCAs (e.g., 16 and 18) protected the neurons from HCA toxicity in a concentration-dependent manner without obvious toxicity, reaching full protection beginning at 10 μM. Moreover, non-toxic MCAs did not increase the levels of acetylated histone H4 (Ac-H4) in rat primary cortical neurons up to a concentration of 50 μM, while the toxic hydroxamates markedly enhanced those levels, indicating that the neuronal toxicity is likely correlated with their ability to inhibit HDAC1 and HDAC2 [73].
Fig. 4.

Phenyl- and benzyl-capped MCA-based HDAC6is 13–26.
2.2. Biaryl-derived caps
Among the biphenyl-capped analogs 27–29 (Fig. 5), only 29 (n = 3) showed submicromolar HDAC6 potency (IC50 = 0.78 μM, Table 2) and modest selectivity (SI = 6-folds) compared with the corresponding phenyl-capped analog 13 (IC50 = 0.19 μM; SI = 17.5-folds, Table 1). Further optimization with the aim of improving their selectivity by incorporating amino acid fragments into the Caps to enhance the interaction with the rim of the target surface was attempted [76]. The retro-amide analogs 30–33 bearing glycine, l-phenylalanine, l-proline, and l-tryptophan, respectively, exhibited two to three times improved HDAC6 inhibition compared to the parent compound 29; however, their selectivities over HDAC1 are still not adequate (SI = 6- to 10-folds) [76].
Fig. 5.

Biaryl-capped MCA-based HDAC6is 27–55.
Table 2.
HDAC inhibitory activity of biaryl-derived MCAs 27–55.
| Compound | Total HDAC (IC50, μM) | Class I (IC50, μM) |
Class IIb (IC50, μM) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC6 | HDAC10 | SIb | Ref | Original No. | ||
| SAHA (1)a | 0.28 (0.08) | 0.03 | 0.94 | 0.25 | 0.64 | 0.033 | 0.30 | 1 | [70, 71] | – |
| TSA (9)a | 0.004 | 0.004 | 0.014 | 0.002 | 1.38 | 0.001 | 0.005 | 4 | [71] | – |
| 27a | 0.8 | 14.8 | >30 | n.a.c | 11.0 | 2.05 | >30 | 7.2 | [71] | 4e |
| 28a | 13.0 | 14.0 | >30 | n.a. | >30 | 8.66 | 28.8 | 1.6 | [71] | 4f |
| 29a | 0.55 | 4.72 | 25.9 | n.a. | n.a. | 0.78 | 10.8 | 6 | [71] | 4g |
| 30 | n.a. | 3.96 | 16.0 | n.a. | 6.03 | 0.39 | 7.43 | 10.1 | [76] | 10a |
| 31 | n.a. | 2.76 | >30 | n.a. | 5.19 | 0.45 | 7.22 | 6.1 | [76] | 10b |
| 32 | n.a. | 1.95 | 12.5 | n.a. | 4.23 | 0.21 | 6.07 | 9.3 | [76] | 10c |
| 33 | n.a. | 7.09 | >30 | 4.33 | 11.3 | 0.66 | >30 | 10.7 | [76] | 10d |
| 34 | n.a. | 5.7 | 28.0 | 14.0 | n.a. | 0.26 | n.a. | 22 | [77] | 7a |
| 35 | n.a. | >30 | >30 | >30 | n.a. | 1.1 | n.a. | >27 | [77] | 7b |
| 36 | n.a. | >30 | >30 | 15.0 | n.a. | 0.28 | n.a. | >107 | [77] | (R)-7b |
| 37 | n.a. | >30 | >30 | >30 | n.a. | >30 | n.a. | n.d.d | [77] | (S)-7b |
| 38 | n.a. | >30 | >30 | >30 | n.a. | >30 | n.a. | n.d. | [77] | 7c |
| 39 | n.a. | 9.6 | 14.0 | 5.5 | n.a. | 0.85 | n.a. | 11.3 | [77] | 16 |
| 40 | n.a. | 1.5 | n.a. | 3.4 | n.a. | 0.83 | n.a. | 1.8 | [77] | (R)-16 |
| 41 | n.a. | >30 | n.a. | >30 | n.a. | >30 | n.a. | n.d. | [77] | (S)-16 |
| 42 | n.a. | >30 | >30 | >30 | n.a. | 2.7 | n.a. | >11 | [77] | 13a |
| 43 | n.a. | >30 | n.a. | n.a. | n.a. | 4.8 | n.a. | >6.2 | [77] | 13a-cis |
| 44 | n.a. | 28 | n.a. | n.a. | n.a. | 3.5 | n.a. | 8 | [77] | 13a-trans |
| 45 | n.a. | >30 | >30 | >30 | n.a. | 9.8 | n.a. | >3 | [77] | 13b |
| 46 | n.a. | >30 | >30 | >30 | n.a. | >30 | n.a. | n.d. | [77] | 13c |
| 47 | n.a. | >30 | >30 | >30 | n.a. | >30 | n.a. | n.d. | [77] | 20a |
| 48 | n.a. | >30 | >30 | >30 | n.a. | >30 | n.a. | n.d. | [77] | 20b |
| 49 | n.a. | 2.7 | 7.5 | 1.4 | n.a. | 2.0 | n.a. | 1.4 | [77] | 18a |
| 50 | n.a. | 3.7 | 9.4 | 2.1 | n.a. | 2.6 | n.a. | 1.4 | [77] | 18b |
| 51 | n.a. | >30 | >30 | >30 | n.a. | >30 | n.a. | n.d. | [77] | 18c |
| 52 | n.a. | >30 | >30 | >30 | n.a. | >30 | n.a. | n.d. | [77] | 18d |
| 53 | 2.2 | 2.3 | n.a. | n.a. | n.a. | 1.8 | n.a. | 1.3 | [79] | 12a |
| 54 | 1.5 | 1.4 | n.a. | n.a. | n.a. | 0.88 | n.a. | 1.6 | [79] | 12b |
| 55 | 8.6 | >10 | n.a. | n.a. | n.a. | 2.1 | n.a. | >4.8 | [79] | 12c |
Unpublished HDAC isoform enzymatic data. IC50 values are the mean of two experiments obtained from curve-fitting of a 10-point enzymatic assay starting from 30 μM with 3-fold serial dilution against HDACs (Reaction Biology Corp, Malvern, PA).
SI, HDAC1/6 selectivity index.
n.a., not available.
n.d., not determined.
Phenylisoxazole Caps were also applied in the development of MCA-based HDAC6is (Fig. 5 and Table 2) [77], inspired by the corresponding hydroxamate-based compounds [78]. Compounds 34–38 present the same linker as 13 (IC50 = 0.19 μM; SI = 17.5-folds, Table 1), while containing a 5-methyl-3-phenylisoxazole moiety as the Cap in which extra α-methyl groups were introduced into the MCA ZBG to investigate their effects. The compound 34 without any methyl substituents (IC50 = 0.26 μM) maintained submicromolar HDAC6 activity comparable with that of 13. In contrast, the racemic compound 35 containing one methyl group showed a significant drop in potency (IC50 = 1.1 μM) and a complete loss of activity against HDAC1-3. Intriguingly, the evaluation of enantiomers 36 (R configuration) and 37 (S configuration) revealed that the R enantiomer retained full HDAC6 potency (IC50 = 0.28 μM) while the S enantiomer was inactive (IC50 > 30 μM). Molecular docking studies conducted by Kalin and coworkers using a HDAC2 crystal structure (PDB code 3MAX) elucidated differences in the binding mode of each enantiomer [77]. The in silico results suggested that the R α-methyl-MCA moiety of 36 better explored the biding cavity, engaging in coordination with the Zn2+, and additional hydrogen bonding interactions at the bottom of the cavity. Moreover, the R enantiomer was able to assume a more folded geometry, thus exploring additional hydrogen bonding interactions between the Cap and the rim of the catalytic cavity [77]. It was therefore not surprising that the dimethyl-substituted MCA 38 exhibited a complete loss of activity since steric hindrance impairs its interaction with the enzyme [77]. In the cases of compounds 39–41 containing an ether as the CU, both racemic 39 and the R enantiomer 40 showed almost identical potency against HDAC6 (IC50 = 0.85 and 0.83 μM), while the S enantiomer 41 was inactive against all isozymes, thus corroborating the findings for 37 regarding the loss of activity for S α-methyl-MCAs [77].
The replacement of the amide CU of 34 with an alkenyl moiety afforded 42 (Fig. 5 and Table 2), which showed micromolar HDAC6 activity (IC50 = 2.7 μM) but slightly improved selectivity over HDAC1 (SI = 30-folds) [77]. It is noted that the individual cis and trans isomers, 43 and 44, displayed modest activity against HDAC1 and similar micromolar activities against HDAC6, thus suggesting that their linker configurations do not significantly impact their potencies. Further evaluation of α-methyl-substituted MCAs bearing the alkene CU indicates a significant reduction in potency of the monomethylated compound 45. At the same time, the dimethylated compound 46 has no activity against any HDAC isozyme up to the concentration of 30 μM [77]. Moreover, the MCA analogs 47 and 48, combining the 3-methyl-5-phenylisoxazole Cap and a cinnamoyl CU-linker, were inactive against all tested isozymes [77]. Additional phenylisoxazolyl-derived compounds, 49–52, containing alkyne CU-linkers and an α-methyl MCA ZBG were also prepared. The racemic mixtures of monomethyl MCAs 49 and 50 exhibited micromolar activity against HDAC6 (IC50 = 2.0 and 2.6 μM) while losing isozyme selectivity. As observed for all previous compounds bearing dimethyl MCAs, neither of the corresponding compounds 51 and 52 bearing an alkyne CU exhibited any activity [77].
Molecular docking studies and HDAC enzymatic inhibitory data suggested that the R configuration of the monomethyl MCAs is preferred, which seemed to exhibit comparable HDAC6 potency with the parent MCA. However, this conclusion cannot be fully addressed since the corresponding derivatives bearing the parent MCA were not prepared to compare with the methyl MCA analogs 39–52 in the original article [77]. Furthermore, it was found that the added methyl group significantly reduced the oxidative dimerization tendency of thiols [77]. Additionally, the neuroprotective effects of these MCAs were investigated in a primary cortical neuron model of oxidative stress-induced neurodegeneration. MCA 34 displayed full neuroprotection at a concentration of 10 μM. The corresponding hydroxamate showed modest neuroprotective activity but also neuronal toxicity at the same concentration [77]. Intriguingly, the inactive S-enantiomers (e.g., 37 and 41) showed the ability to prevent glutathione depletion, and other monomethyl MCAs containing alkene- or alkyne-CUs (e.g., 45, 49, and 50) also demonstrated neuroprotective effects at 10 μM. These results may suggest that there is an additional neuroprotection mechanism independent from their HDAC6 inhibitory activity. However, the underlying mechanism was not further investigated and discussed in the original reference. [77].
In 2016, Zhao and coworkers reported a series of MCAs (53–55) bearing a 3-phenyl-1H-pyrazole moiety as the Cap and an amide group as the CU (Fig. 5 and Table 2) [79]. All three compounds exhibited micromolar inhibitory activities against total HDACs, HDAC1, and HDAC6. Although none of them showed good selectivity over HDAC1, the length of the alkyl chain linker seemed to influence their selectivity slightly. It should be noted that, based on the structures of 52–55, three corresponding mercaptopropanamides were prepared and evaluated against HDACs in the original article [79]. None of them display inhibitory activity against HDACs up to 10 μM, underscoring the uniqueness of MCA for maintaining HDAC potency.
2.3. Fused aryl-derived caps
Exploration was also carried out on fused aryl Caps that led to the discovery of quinoline- and tetrahydroquinoline-based MCAs (56–75) (Fig. 6 and Table 3) [71,73,75,80-82]. In comparison with the phenyl- or biphenyl-capped analogs described above, compounds bearing 3-quinolyl (56 and 60), 6-quinolyl (61), and 8-quinolyl (62 and 63) Caps exhibited much better HDAC6 potency (IC50 < 0.1 μM) and enhanced selectivity over HDAC1 ranging from 15-folds to 44-folds (Table 1) [71,73]. The inhibitory activity of 57–60 (Table 3) against total HDACs further suggest that the length of alkyl linker is also critical for maintaining potent HDAC inhibitory activity [82]. Notably, compound 63 was renamed W2 and exhibited the ability to improve learning and memory in a mouse model of Alzheimer’s disease while showing low nanomolar HDAC6 potency and hundred-fold selectivity when retested several years after its first publication (IC50 = 0.02 μM; SI = 153-folds) [83]. Replacement of the alkyl linker in 63 by a para-tolyl moiety afforded compound 64 with a 23-fold lower HDAC6 activity (IC50 = 2.21 μM) [80]. The para-tolyl moiety is considered as a useful linker in the hydroxamate-based inhibitors to generate good selectivity over class I HDAC isoforms that data was not determined for 64. Therefore, although the HDAC6 potency of 64 was indeed decreased, this compound might remain the ability to maintain good selectivity over HDAC1. Similarly, the incorporation of a valine moiety between the 3-quinolyl Cap and the alkyl amide linker of 60, so as to generate structures 65 and 66, resulted in modest HDAC total inhibition for the racemic compounds [75]. It is noted that the conversion of the amide CU of the 8-quinolyl-capped compound 63 into amine (67–71) and ether (72) generated highly selective and potent HDAC6is [80,81]. Among them, compound 68 bearing five methylene units as the linker is the most potent and selective HDAC6i containing the MCA moiety as ZBG (IC50 = 0.0013 μM; SI = 4700-folds) [80]. Shorter or longer alkyl linkers, as in compounds 67 and 69, caused a 6- and 100-fold decrease in HDAC6 activity (IC50 = 0.0078 and 0.178 μM), respectively, thus validating the optimal length (n = 5) for the linker in this series [80]. Subsequent evaluation of α-tubulin acetylation, a typical approach to assess HDAC6 inhibition in cell lines, showed that the treatment of rat primary cortical neurons with 68 led to a 10-fold increase in the level of Ac-Tub, but no significant upregulation of the levels of acetylated histone H3 (Ac-H3) at the concentration of 1 μM, corroborating its HDAC6 potency and selectivity. Moreover, 68 also displayed an ability to increase T-regulatory (Treg) suppressive function in a dose-dependent manner [80]. The introduction of two chlorine substituents at both the C5- and C7-positions of the 8-quinolyl Cap only modestly influences activity or selectivity of the resulting compounds 70 and 71, while significantly improving their permeability and metabolic stability in liver microsomes [81]. Moreover, the replacement of the amino CU of 71 with an ether resulted in a two-fold decrease in HDAC6 potency in the case of 72 [58]. Tetrahydroquinolyl-capped MCAs (73–75) were also prepared. Compounds 73 and 74 demonstrated a micromolar range of inhibition on HDAC6. In contrast, 75, containing an amide CU instead of an amine moiety, exhibited a promising selectivity (SI > 1110-folds) and nanomolar HDAC6 potency (IC50 = 0.027 μM) [80], indicating further optimization of this tetrahydroquinoline class may lead to the identification of more potent and selective HDAC6is. However, the related SAR studies on this series have not been reported.
Fig. 6.

Fused aryl-capped MCA-based HDAC6is 56–81.
Table 3.
HDAC inhibitory activity of fused aryl-capped MCAs 56–81.
| Compound | Total HDAC (IC50, μM) | Class I (IC50, μM) |
Class IIb (IC50, μM) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC6 | HDAC10 | SIb | Ref | Original No. | ||
| SAHA (1)a | 0.28 (0.08) | 0.03 | 0.94 | 0.25 | 0.64 | 0.033 | 0.30 | 1 | [70, 71] | – |
| TSA (9)a | 0.004 | 0.004 | 0.014 | 0.002 | 1.38 | 0.001 | 0.005 | 4 | [71] | – |
| 56a | 0.40 | 3.12 | 6.81 | n.a.c | 4.93 | 0.09 | >30 | 34.7 | [71] | 4m |
| 57 | 39.5 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | 82 | 3a |
| 58 | 0.66 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | [82] | 3b |
| 59 | 2.80 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | [82] | 3c |
| 60a | 1.10 (0.05) | 4.11 | 10.8 | n.a. | 5.97 | 0.09 | 16.6 | 45.6 | [71, 82] | 3d, 10c |
| 61a | 0.90 | 2.41 | 16.3 | n.a. | 7.00 | 0.11 | 11.7 | 21.9 | [71] | 10d |
| 62a | 0.25 | 2.85 | 15.1 | n.a. | 7.57 | 0.19 | 7.93 | 15 | [71] | 4l |
| 63 | 0.04 | 3.22 | 7.38 (1.25) | 1.32 | 6.12 | 0.09 (0.02) | 10.7 (10.7) | 36 (161) | [71, 73, 83] | 10b, 2, W2 |
| 64 | n.a. | n.a. | n.a. | n.a. | n.a. | 2.21 | n.a. | n.d.d | [80] | 4 |
| 65 | 25.1e | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.d. | [75] | BC-6-34 |
| 66 | 3.02e | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | n.d. | [75] | BC-6-30 |
| 67 | n.a. | 13.8 | n.a. | n.a. | n.a. | 0.008 | n.a. | 1730 | [80] | 2a |
| 68 | n.a. | 4.7 | n.a. | n.a. | n.a. | 0.001 | n.a. | 4700 | [80] | 2b |
| 69 | n.a. | 24.9 | n.a. | n.a. | n.a. | 0.18 | n.a. | 138 | [80] | 2c |
| 70 | n.a. | 6.88 | n.a. | n.a. | 3.45f | 0.003 | n.a. | 2290 | [81] | 13a |
| 71 | n.a. | 6.57 | n.a. | n.a. | n.a. | 0.015 | n.a. | 438 | [81] | 13b |
| 72 | n.a. | >30 | n.a. | n.a. | n.a. | 0.033 | n.a. | >909 | [81] | 13c |
| 73 | n.a. | n.a. | n.a. | n.a. | n.a. | 0.22 | n.a. | n.d. | [80] | 3a |
| 74a | n.a. | >30 | n.a. | n.a. | n.a. | 2.33 | n.a. | >12.8 | [80] | 3c |
| 75a | n.a. | >30 | n.a. | n.a. | n.a. | 0.027 | n.a. | >1110 | [80] | 3b |
| 76 | n.a. | >30 | n.a. | n.a. | n.a. | 0.064 | n.a. | >468 | [81] | 7a |
| 77 | n.a. | 28.7 | n.a. | n.a. | n.a. | 1.57 | n.a. | 18.3 | [81] | 7b |
| 78 | n.a. | >30 | n.a. | n.a. | n.a. | 0.24 | n.a. | >125 | [81] | 7c |
| 79 | n.a. | 29.3 | n.a. | n.a. | n.a. | 0.065 | n.a. | 451 | [81] | 7d |
| 80 | n.a. | 7.49 | n.a. | n.a. | 11.7f | 0.011 | n.a. | 681 | [81] | 7e |
| 81 | n.a. | n.a. | n.a. | n.a. | n.a. | 0.53 | n.a. | n.d. | [81] | 24 |
Unpublished HDAC isoform enzymatic data. IC50 values are the mean of two experiments obtained from curve-fitting of a 10-point enzymatic assay starting from 30 μM with 3-fold serial dilution against HDACs (Reaction Biology Corp, Malvern, PA).
SI, HDAC1/6 selectivity index.
n.a.: not available.
n.d.: not determined.
IC50 value was calculated from pIC50 value.
Table S2, Supporting Information of Ref[81].
The Kozikowski group also developed some MCAs capped with indoles without a CU group. A combination of a 5,6-dichloroindole cap and six methylene units afforded 76, which showed nanomolar potency against HDAC6 and excellent selectivity over HDAC1 (IC50 = 0.064 μM; SI = 468-folds). Interestingly, the incorporation of additional substituents at the C3-position of the indole ring reduced the activity for both 77 and 78 (a 24-fold and 3.7-fold decrease, respectively), indicating that the C3-position is less able to tolerate bulky rigid groups. The 6-chloro-5-fluoroindolyl-capped compound 79 exhibits slightly weaker HDAC6 activity and selectivity relative to 76 (IC50 = 0.065 μM; SI = 451-folds). Additionally, when the 5,6-dichloroindole cap was attached to the MCA moiety through a shorter alkyl linker (pentamethylene chain), the resulting compound 80 exhibited a 5.8-folds improvement in HDAC6 potency (IC50 = 0.011 μM) and improved selectivity over HDAC1 compared to 76 (SI = 681- and 468-folds). The introduction of a secondary thiol function into the linker (81) resulted in a significant loss of activity, thus highlighting the conservative feature of alkyl linkers of MCA-based HDAC6is [81].
2.4. MCA-based prodrugs
It is well known that a free thiol group, as observed in MCA and other thiol-derived HDACis, may be subject to oxidation reactions in vitro and in vivo, which could impair their inhibitory activity [63]. According to the literature, several prodrug strategies have been implemented to avoid off-target reactions of thiol-derived HDACis [61-63,84]. Thioesters and disulfides represent the majority of the designed MCA-based prodrugs. The thioester linkage is susceptible to hydrolysis reaction inside cells, thus releasing the active thiol group in the cytosol [85]. The thioacetate 82 (Fig. 7) converted from 11 facilitated the antiparasitic evaluation against Schistosoma mansoni, which was able to release the active derivate 11 inside the cultured schistosomula, leading to the reduction of viability and apoptosis induction at 20 μM [74]. Moreover, in two independent studies using HeLa nuclear extracts containing a mixture of HDACs, 82 remained modest HDAC inhibitory activities (IC50 = 20.1 and 22.0 μM, respectively) relative to 11 (IC50 = 2.4 and 0.4 μM, respectively) [72,84]. Improved membrane permeability is an additional benefit that can be obtained by thiol-derived prodrugs. Interestingly, the S-isobutyryl prodrug 83 exhibited higher levels of Ac-Tub in HEK-293 cells compared to its active form 80 at the concentration of 10 μM, which may be attributed to the increased cellular permeability of 83 [81].
Fig. 7.

MCA-based HDACi prodrugs 82–85.
Disulfide prodrugs can be converted into their active thiol compounds through reduction of the disulfide linkage [86]. Noteworthy, HDAC6 is mainly located in the cell cytosol, which is known to be highly reducing, thus favoring the conversion of disulfide prodrugs into their free MCA derivatives [87]. It was found that the disulfide prodrugs 84 and 85 (Fig. 7) released the corresponding MCAs 70 and 80 efficiently in human and mouse liver microsomes while also exhibiting comparable abilities to enhance the level of Ac-Tub in HEK-293 cells relative to their active forms [81]. Moreover, it was also observed that both disulfides were able to increase the Ac-Tub levels in mouse cortex, indicating that they are capable of crossing the blood-brain barrier (BBB) and have the potential to be further investigated in animal models of CNS disorders [81].
3. Unique molecular basis for the selectivity of MCA-based HDAC6is
Crystallographic studies using Danio rerio HDAC6 (drHDAC6) in complex with different HDACis were first reported in 2016, which initiated the elucidation of the structural features of HDAC6 [21,88]. Presently, more than 50 X-ray crystal structures of HDAC6-CD2 complexes with various HDAC6is have been published, thus providing a better understanding of the molecular basis for the potency and selectivity of HDAC6is [89]. However, the majority of the published structures are for the hydroxamate-based inhibitors, and only one HDAC6 crystal structure has been reported for HDAC6 in complex with an MCA-based analog (Fig. 8) [87].
Fig. 8.

Crystal structures of drHDAC6 and smHDAC8 complexes with HDACis. (A) drHDAC6/vorinostat (1, yellow, PDB code 5EEI); (B) smHDAC8/vorinostat (1, yellow, PDB code 4BZ6); (C) drHDAC6/ricolinostat (6, purple, PDB code 5WGL); (D) drHDAC6/panobinostat (2, green, PDB code 5EF8); (E) smHDAC8/compound 86 (rose red, PDB code 6GXA); (F) drHDAC6/tubastatin A (87, brown, PDB code 6THV); (G) drHDAC6/compound 80 (white, PDB code 6MR5); (H) smHDAC8/compound 11 (orange, PDB code 4CQF) (I) drHDAC6/bavarostat (88, cyan, PBD code 6DVO). Selected drHDAC6/smHDAC8 residues are shown in stick representation with atoms colored pink/cyan (carbon), red (oxygen), blue (nitrogen), and yellow (sulfur). The active-site Zn2+ and water molecules are shown as grey and red spheres, respectively. The structures and HDAC inhibition data are shown accordingly.
In comparison with other isoforms, HDAC6 has a relatively large and wide solvent-exposed surface (~14 Å). Therefore, bulky and rigid polycyclic Cap groups, as observed for 6 and 7, lead to favorable binding interactions in many selective HDAC6is [21], particularly strong hydrophobic interactions with the “L1-loop pocket”, the selectivity-determining area defined by the key residues H463, P464, F583, and L712 [43,90,91]. The hydrophobic tunnel formed by F583 and F643 is able to establish interactions with the linker of HDAC6is (e.g., a benzyl group) to stabilize the ligand in the catalytic pocket. Besides the van der Waals contacts and hydrophobic interactions, there are also specific hydrogen-bonding interactions observed between the Cap, linker, or CU area of HDAC6is and key amino acid residues (e.g., S531) that play critical roles in the substrate recognition process of HDAC6 [89,92]. Intriguingly, each ZBG demonstrate distinct coordination geometry with the Zn2+ at the bottom of the cavity, which may be essential for determining the HDAC6 isoform selectivity of the MCA-based ligands.
In the negatively charged alkylhydroxamate moiety of the pan-HDACi vorinostat (1) (Fig. 8A, PDB code 5EEI) and in the cinnamoylhydroxamate moiety of the pan-HDACi panobinostat (2) (Fig. 8D, PDB code 5EF8) [88], the carbonyl group (C═O) accepts a hydrogen bond from Y745. Moreover, the NH group donates a hydrogen bond to H574 (neutral state), while the deprotonated OH group accepts a hydrogen bond from H573 (positively charged). The carbonyl and oxyanion groups engage in bidentate hydroxamate-Zn2+ coordination, while the Zn2+ also coordinates with D612, H614, and D705, forming a tetrahedral coordination geometry. In the Schistosoma mansoni HDAC8 (smHDAC8) crystal in complex with vorinostat (1, Fig. 8B, PDB code 4BZ6) and another cinnamoylhydroxamate-based analog 86 (Fig. 8E, PDB code 6GXA) [74,93], the deprotonated hydroxamate moiety establishes hydrogen bonding interactions with the tandem histidine residues H141 and H142, as well as Y341, forming an identical bidentate hydroxamate-Zn2+ coordination as observed in the drHDAC6/vorinostat complex. These observations may be related to the broad-spectrum HDAC inhibition of hydroxamate-based HDACis. On the other hand, the drHDAC6/ricolinostat co-crystal (6, Fig. 8C, PDB code 5WGL) exhibits the same Zn2+ coordination mode as observed for vorinostat (1), as the compound contains a vorinostat-like long-chain aliphatic linker. However, the large and rigid N,N-diphenyl-2-aminopyrimidine Cap of ricolinostat (6) can occupy the surface, while the amide group of the CU and the nitrogen of the amino-pyrimidinyl Cap form two water-mediated hydrogen bonds with the key residues S531 and D460, respectively. These additional hydrogen bond interactions are likely responsible for the HDAC6 potency and selectivity presented by ricolinostat (6) [92].
The capless phenylhydroxamate itself is a selective HDAC6i (HDAC6 IC50 = 115 nM; HDAC8, IC50 = 1900 nM), which coordinates to Zn2+ with typical bidentate geometry [94]. Moreover, phenylhydroxamate has also served as a useful linker and ZBG for the discovery of selective HDAC6is [95]. The drHDAC6 crystallographic studies provided critical insights that phenylhydroxamate-based selective HDAC6is capped with a rigid polycyclic ring or bulky bifurcated groups exhibit unusual monodentate hydroxamate-Zn2+ coordination geometries in the active site [90,92,96]. Moreover, these phenyl-based linkers are able to stabilize the engagement of the inhibitor through strong π-π stacking interactions with the key phenylalanine residues F583 and F643 in the narrow hydrophobic tunnel. As illustrated by the crystal complex of drHDAC6 with tubastatin A (87), a widely used selective HDAC6i (Fig. 8F, PDB code 6THV) [97], the deprotonated OH group of the phenylhydroxamate group accepts a hydrogen bond from Y745, in contrast to the hydroxamates linked to an alkyl or cinnamoyl group where it is the C═O group that accepts a hydrogen bond from Y745. At the same time, its C═O group accepts a hydrogen bond from a water molecule. The tandem histidine residues H573 and H574 interact with the water molecule rather than binding with the NH-O− moiety of the hydroxamate. Additionally, the oxyanion group coordinates with zinc directly, while the carbonyl group engages with the Zn2+ through water-mediated coordination. As the monodentate coordination mode is slightly less stable (0.5 kcal/mol) than the typical bidentate coordination, the interactions between the surface and the large Caps are important to stabilize this binding mode. On the other hand, it has been reported that in some arylhydroxamate-based HDAC6is in which CUs (such as amine or amide) are monosubstituted by a capping group, the hydroxamate reaches deeper into the cavity and engages in a bidentate coordination with Zn2+ (illustrated for the case of bavarostat (88) in Fig. 8I, PBD code 6DVO) [43,91,96]. Moreover, the CU/linker of these monosubstituted HDAC6is present crucial direct (or indirect) hydrogen bonding interactions with S531, which is an essential residue responsible for the substrate recognition of HDAC6, thus enhancing the selectivity of these ligands.
In 2005, a molecular docking study was performed for MCA-based analog 11 using the crystal structures of HDAC8 (PDB code 1T64, 1T67, 1T69, and 1VKG), which suggested bidentate coordination with the Zn2+ through its sulfur (HS…Zn2+) and carbonyl oxygen (CO…Zn2+) of the MCA moiety, with no involvement of water molecules [70]. Moreover, the hypothesis of bidentate coordination related to MCA-based HDACis was corroborated by docking simulations of 63 using an HDAC1 (PDB code 1C3S) homology 3D model [71]. However, the only available HDAC6 crystal structure in complex with the selective MCA-based HDAC6i 80 (IC50 = 0.011 μM; SI = 681-folds; Table 3) presents a different MCA-zinc coordination geometry compared to that shown by the hydroxamate-based inhibitors and docking studies of MCAs (Fig. 8G, PDB code 6MR5) [87]. The carbonyl group (C═O) of the MCA accepts a hydrogen bond from Y745 in the same manner as observed for the alkylhydroxamates. The NH group donates a hydrogen bond to H574 (neutral state), while the deprotonated thiolate (S−) group accepts a hydrogen bond from the positively charged H573. Moreover, the C-S−-Zn2+ angles (114° and 120°) and S−-Zn2+ distance (2.3 Å) indicate an ideal thiolate-metal coordination interaction [98]. In comparison with the important contribution of the C═O group to hydroxamate-zinc bidentate or water-mediated monodentate coordination, the C═O group in the MCA moiety is too far removed to be able to properly engage in coordination with Zn2+. Moreover, the dichloroindole Cap of 80 is located over the L1 pocket area allowing it to form optimal van der Waals contacts with H463, P464, and L712, which is also important for its excellent HDAC6 selectivity. The MCA version of vonoristat (1), compound 11, exhibits improved selectivity over Class I HDACs relative to vonoristat (Table 1). The smHDAC8 crystal in complex with 11 (Fig. 8H, PDB code 4CQF) demonstrates that its MCA moiety exhibits a similar thiolate-zinc coordination geometry as observed for 80 [74]. However, while vonoristat shows dual hydrogen bonding interactions with the tandem H141 and H142 residues (Fig. 8B), only one hydrogen bond is established between the S− group and H141, while the NH group is too far away to interact with H142. It is worthwhile to mention that the empirical monodentate thiolate-Zn2+ coordination of 11 and 80 contradicts the original bidentate hypothesis proposed in the previous in silico models. The distinct monodentate zinc coordination of the MCAs on both HDAC6 and HDAC8 may explain their selectivity, further underscoring MCA’s unique properties as a useful ZBG for the design of selective and non-genotoxic HDAC6is.
4. DMPK and pharmacological studies of MCA
Compared to the extensive biological evaluation of hydroxamate-based HDACis, only limited work has been conducted at present on the pharmacokinetics (PK), pharmacodynamics (PD), and pharmacological effects of MCA-based HDAC6is in models of cancer and Alzheimer’s disease. These contributions have mainly focused on the characterization of the N,N-dimethylaminophenyl-capped compound 20 and 8-quinolyl-capped compound 63. In 2008 and 2009, Jung and coworkers determined the ADME properties in vitro and PK/PD correlations in vivo for these two compounds [99-101].
Experimental LogD values shown in Table 4 indicate that replacement of hydroxamate moiety with MCA decreases the molecular polarity, while the dimethylamino group of 20 and the quinoline ring of 63, respectively, maintain their lipophilicity in a reasonable range [99]. Moreover, their Caps containing a basic nitrogen atom allow the molecules to be protonated in the acidic environment of the stomach (pH = 1.2), which leads to a significant improvement in their solubilities [99]. Generally, compounds with permeability coefficients (Papp (A–B)) higher than 3 × 10−6 cm/s and efflux ratios lower than 2.5 show the ability to cross the BBB [102]. The permeability assays in Caco-2 cells (Table 4) demonstrate that both 20 and 63, as well as vorinostat (1) can be classified as moderately permeable (2 × 10−6 cm/s < Papp(A–B) < 20 × 10−6 cm/s) and exhibit favorable efflux ratios for brain penetration [99]. It is noted that 63 shows higher permeability coefficients in both directions and a more favorable efflux ratio compared to 20, indicating a slightly superior CNS access. Further investigation demonstrated that P-glycoprotein (P-gp) and multidrug resistance-associated protein 1 (MRP1) transporters are responsible for the efflux of 63 [100]. Plasma and liver microsomal stability assays performed in different species (Table 4) indicate that the half-lives (t1/2) of 20 and 63 are comparable, ranging from 43 min to 173 min [99,101].
Table 4.
ADME/PK profiles of the MCA-based HDAC6is 20 and 63.a
| Compound Structure |
20 | 63 | SAHA | |
|---|---|---|---|---|
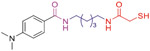 |
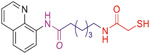 |
|||
| HDAC isoform (IC50, μM) | HDAC6 | 0.11 | 0.02 | 0.03 |
| HDAC1 | 1.08 | 3.22 | 0.03 | |
| SI | 9.8 | 161 | 1 | |
| LogD at 37°C | 2.19 ± 0.1 | 2.64 ± 0.1 | 1.46 ± 0.2 | |
| Solubility (μg/mL) | PBS (pH 7.4, 37°C, 24 h) | ~120 | ~150 | ~400 |
| HCl (pH 1.2, 37°C, 24 h) | ~1000 | ~1000 | ~100 | |
| Permeability in Caco-2 cells | Papp (A-B) (10−6, cm/s) | 7.33 ± 0.3 | 15.0 ± 0.4 | 3.00 ± 0.2 |
| Papp (B-A) (10−6, cm/s) | 10.0 ± 0.4 | 24.7 ± 0.12 | 6.33 ± 0.3 | |
| Efflux ratio | 1.36 | 1.64 | 2.11 | |
| Plasma stability (t1/2, min) | Human | 77 | 79 | 75 |
| Mouse | 173 | 58 | 115 | |
| Porcine | 89 | 69 | 87 | |
| Rat | 99 | 92 | 86 | |
| Liver microsomal stability (t1/2, min) | Human | 68 | 83 | n/a |
| Dog | 43 | 72 | n/a | |
| Rat | 70 | 66 | n/a | |
| PK parameters in mice (400 mg/kg, IP) | Cmax (μmol/L) | 1.54 ± 0.26 | 1.81 ± 0.34 | n/a |
| tmax (h) | 0.5 | 0.5 | n/a | |
| AUC (μmol/L × h) | 4.23 ± 0.43 | 4.97 ± 0.60 | n/a | |
| t1/2 (h) | 1.98 ± 0.21 | 2.20 ± 0.33 | n/a | |
| CL (L/h) | 4.87 ± 0.2 | 4.05 ± 0.15 | n/a |
Subsequently, compounds 20 and 63 were dosed in mice in a range from 0.5 to 400 mg/kg through intraperitoneal (IP) injection to determine the maximum tolerated dose (MTD) [101]. The behavior of the mice was reported to remain normal after treating with 20 over the entire dose range. On the other hand, the mice appear to become incapacitated 15 min after treating with 400 mg/kg of 63, but they fully recovered within 1 h. Even though 400 mg/kg is relatively high for PK studies, Jung and coworkers further determined PK parameters in plasma for both compounds at this dose, through IP administration [101]. The maximum peaks were observed at 0.5 h after the treatment with either compound. Moreover, 63 exhibited slightly higher maximum concentration (Cmax) and area under the curve (AUC), longer half-life (t1/2), and lower clearance rate (CL) compared to 20 (Table 4), thus suggesting that 63 has a better plasma PK profile. The level of Ac-H4 was used as a biomarker to investigate HDAC target engagement of 20 and 63 in vivo. In the dose range from 0.5 to 400 mg/kg, Ac-H4 levels were significantly increased in the spleen after 4 h treatment and were induced in a dose-dependent manner [101]. Furthermore, brain and liver tissues were collected at 0, 0.5, 1, 2, 4, and 6 h posttreatment (400 mg/kg) [101]. The maximum increase of Ac-H4 levels appeared in both tissues at 0.5 h after treatment, which was well correlated with the drug exposure determined in the plasma PK studies. In addition, the enhanced histone acetylation observed in the brain supports their ability to readily cross the BBB, in line with their favorable permeability coefficients, as mentioned above. It was reported that compound 20 displayed greater hyperacetylation effects than 63 in both dose-dependent and time-dependent PD assessments, which is relatively consistent with its lower HDAC6 selectivity compared to 63. Histone acetylation is, of course, more suitable to use in assessing the ability of a compound to inhibit HDAC class I isoforms. It should be noted that 63 (SI = 161-folds) has a much better selectivity over HDAC1 relative to 20 (SI = 9.8-folds), thus suggesting that the level of Ac-Tub was supposed to a more appropriate biomarker for 63 in the PD assessment.
As several HDACis have been approved by the FDA as effective therapeutic agents for cancer, the above PK/PD correlation results encouraged Jung and coworkers to investigate the antitumor effects of 20 and 63 in prostate cancer models [101]. Noteworthy, both compounds exhibited low-micromolar levels of anti-proliferative effects on prostate cancer cells (PC-3 and LNCap) while having no apparent inhibitory effects on nonmalignant cells (267-B1 and RWPE-1). Moreover, in vivo studies using mice xenografted with PC-3 tumors revealed that IP administration of 20 and 63 for 28 days at the dose of 0.5 mg/kg, which was determined by the minimum dose to increase Ac-H4 level according to the PD assessment abovementioned, led to significant inhibition on the tumor growth [101]. Notably, the antitumor effects were sustained until the termination of these studies (60 days), and no mortality or body-weight loss was observed during the treatment. However, it is hard to conclude that these antitumor effects were associated with their HDAC6 inhibitory activities, as histone acetylation rather than α-tubulin acetylation was utilized as a biomarker to perform their PD assessments as described above [101]. Another study was reported in 2017 using monomorphic malignant human glioma cells (A172, U373MG, and T98G) to further explore the antitumor effects of 63 [103]. Unlike moderate anti-proliferative effects on prostate cancer cells in the previous study, compound 63 did not show cytotoxicity at concentrations up to 50 μM in glioma cells, while the levels of Ac-Tub were significantly enhanced without influence on the levels of Ac-H3 in A172 and U373MG cells. Moreover, the studies also revealed that 63 has the ability to suppress the phosphorylation of FAK/STAT3, which in turn downregulates the activity of MMP-2, eventually leading to reduced cell migration and invasion in A172 cells.
HDAC6 has also emerged as a potential therapeutic target for neurodegenerative disorders (e.g., Alzheimer’s disease, Charcot-Marie-Tooth disease, and amyotrophic lateral sclerosis), mainly associated with its capability to modulate the levels of Ac-Tub, HSP90, and peroxiredoxin [37,49]. To examine the effects of 63 on Alzheimer’s disease models, N2a cells overexpressing hAPP were initially treated with 63 (1 or 5 μM) [83]. It was found that 63 led to a 20% decrease in human Aβ40 and Aβ42 levels. Furthermore, 63 also significantly reduced rodent Aβ40 by 20% in primary cortical neurons. After 24 h of treatment with 63 (5 μM), it was observed that cell surface APP increased by two-fold in N2a cells and in primary neurons, indicating that 63 influences Aβ by regulating APP trafficking [83]. To further explore the molecular mechanisms of the decrease in Aβ levels caused by 63, the expression of genes involved in Aβ synthesis and degradation was evaluated. After treating primary cortical neurons with 63 (5 μM) for 24 h, most genes involved in Aβ synthesis (e.g., Psen1, Psen2, and Ncstn) were downregulated. In contrast, genes related to Aβ degradation (e.g., Ece1, Mmp2, and Ctsd) were upregulated. These results suggest that compound 63 impacts Aβ levels by down-regulating Aβ production pathways while upregulating Aβ clearance pathways. Finally, animal efficacy studies were conducted by treating hAPP 3 × Tg mice with 63 at a dose of 50 mg/kg (IP injection) for four weeks [83]. The results demonstrated that 63 decreased Aβ levels and phosphorylated tau (Thr181) levels and alleviated the learning and memory deficits exhibited in aged hAPP 3 × Tg mice (9-10-month-old). It has been demonstrated that deficits in learning and memory are correlated with dendritic spine density [104,105]. Moreover, Aβ accumulation reduces dendritic spine density and impairs cognitive performance [106]. Subsequent molecular mechanism studies [107] revealed that 63 regulates dendritic spine formation through the RasGRF1 and ERK signaling pathways, influences dendritic spine number by changing excitatory synapses in primary hippocampal neurons, and eventually promotes dendritic spine density in mice (50 mg/kg, IP). Both efficacy studies described above provide evidence that the CNS penetrant MCA compound 63 has the therapeutic potential to be the first non-hydroxamate selective HDAC6i for the treatment of Alzheimer’s disease.
5. Conclusion
Given the unique structural and functional features of HDAC6, selective HDAC6is have become an attractive research area for the discovery of therapeutic treatments for cancers and neurodegenerative disorders in the last decade. It is noted that most HDAC6is with in vivo efficacy belong to the hydroxamate class, and their potential mutagenicity is likely to limit their application in long-term treatments. On the other hand, the mercaptoacetamide moiety has proven to be an effective ZBG to replace the hydroxamate group while, in some cases, retaining good HDAC6 potency and selectivity. In this review, we discuss for the first time the evolution of these MCA-based inhibitors, compare their distinct coordination modes with the catalytic zinc ion in different HDAC isoforms using the structures of inhibitor-enzyme co-crystals of hydroxamate- and MCA-based HDACis, and summarize the DMPK characteristics and pharmacological properties of this series. Overall, this information may provide new insights into the potential of the mercaptoacetamide moiety as an alternative ZBG for the discovery of HDAC6 selective inhibitors while avoiding the potential mutagenicity of the hydroxamate class.
Acknowledgments
We thank Dr. Werner Tueckmantel and Dr. Guiping Zhang for proofreading the article and providing comments.
Funding
Funded by NIH R01NS079183, R43HD093464, and R41AG058283 (A.P.K.).
Abbreviations:
- ADME
absorption-distribution-metabolism-excretion properties
- Aβ40
plasma amyloid β-peptide 40
- Aβ42
plasma amyloid β-peptide 42
- ERK
extra-cellular-signal-regulated kinase
- FAK
focal adhesion kinase
- FDA
Food and Drug Administration
- hAPP
human amyloid precursor protein
- LogD
the logarithm of partition coefficient for ionizable compounds
- MMP-2
matrix metalloproteinase-2
- PDB
protein data bank
- PD-1
programmed cell death protein
- PD-L1
programmed death-ligand 1
- Ras
reticular activating system
- RasGRF1
Ras-protein specific guanine nucleotide releasing factor 1
- SAHA
suberoylanilide hydroxamic acid
- STAT3
signal transducer and activator of transcription 3
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Carew JS, Giles FJ, Nawrocki ST, Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy, Canc. Lett 269 (2008) 7–17. [DOI] [PubMed] [Google Scholar]
- [2].Liu B, Lin Y, Darwanto A, Song X, Xu G, Zhang K, Identification and characterization of propionylation at histone H3 lysine 23 in mammalian cells, J. Biol. Chem 284 (2009) 32288–32295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bao X, Wang Y, Li X, Li XM, Liu Z, Yang T, Wong CF, Zhang J, Hao Q, Li XD, Identification of ‘erasers’ for lysine crotonylated histone marks using a chemical proteomics approach, Elife 3 (2014), e02999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Seidel C, Schnekenburger M, Dicato M, Diederich M, Histone deacetylase 6 in health and disease, Epigenomics 7 (2015) 103–118. [DOI] [PubMed] [Google Scholar]
- [5].Shen S, Kozikowski AP, Why hydroxamates may not be the best histone deacetylase inhibitors-What some may have forgotten or would rather forget? ChemMedChem 11 (2016) 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pharmacology Review for Belinostat. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/206256Orig1s000PharmR.pdf.
- [7].Ning ZQ, Li ZB, Newman MJ, Shan S, Wang XH, Pan DS, Zhang J, Dong M, Du X, Lu XP, Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity, Canc. Chemother. Pharmacol 69 (2012) 901–909. [DOI] [PubMed] [Google Scholar]
- [8].Yao Y, Tu Z, Liao C, Wang Z, Li S, Yao H, Li Z, Jiang S, Discovery of novel class I histone deacetylase inhibitors with promising in vitro and in vivo antitumor activities, J. Med. Chem 58 (2015) 7672–7680. [DOI] [PubMed] [Google Scholar]
- [9].Savelieva M, Woo MM, Schran H, Mu S, Nedelman J, Capdeville R, Population pharmacokinetics of intravenous and oral panobinostat in patients with hematologic and solid tumors, Eur. J. Clin. Pharmacol 71 (2015) 663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Xu S, De Veirman K, Vanderkerken K, Van Riet I, Vorinostat-induced bone loss might be related to drug toxicity, Bone 57 (2013) 384–385. [DOI] [PubMed] [Google Scholar]
- [11].Warren KE, McCully C, Dvinge H, Tjornelund J, Sehested M, Lichenstein HS, Balis FM, Plasma and cerebrospinal fluid pharmacokinetics of the histone deacetylase inhibitor, belinostat (PXD101), in non-human primates, Canc. Chemother. Pharmacol 62 (2008) 433–437. [DOI] [PubMed] [Google Scholar]
- [12].Doi T, Hamaguchi T, Shirao K, Chin K, Hatake K, Noguchi K, Otsuki T, Mehta A, Ohtsu A, Evaluation of safety, pharmacokinetics, and efficacy of vorinostat, a histone deacetylase inhibitor, in the treatment of gastrointestinal (GI) cancer in a phase I clinical trial, Int. J. Clin. Oncol 18 (2013) 87–95. [DOI] [PubMed] [Google Scholar]
- [13].Zhao C, Dong H, Xu Q, Zhang Y, Histone deacetylase (HDAC) inhibitors in cancer: a patent review (2017-present), Expert Opin. Ther. Pat 30 (2020) 263–274. [DOI] [PubMed] [Google Scholar]
- [14].Zhang Y, Gilquin B, Khochbin S, Matthias P, Two catalytic domains are required for protein deacetylation, J. Biol. Chem 281 (2006) 2401–2404. [DOI] [PubMed] [Google Scholar]
- [15].Zou H, Wu Y, Navre M, Sang BC, Characterization of the two catalytic domains in histone deacetylase 6, Biochem. Biophys. Res. Commun 341 (2006) 45–50. [DOI] [PubMed] [Google Scholar]
- [16].Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL, Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation, Proc. Natl. Acad. Sci. U.S.A 100 (2003) 4389–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Noack M, Leyk J, Richter-Landsberg C, HDAC6 inhibition results in tau acetylation and modulates tau phosphorylation and degradation in oligodendrocytes, Glia 62 (2014) 535–547. [DOI] [PubMed] [Google Scholar]
- [18].Zhang M, Xiang S, Joo HY, Wang L, Williams KA, Liu W, Hu C, Tong D, Haakenson J, Wang C, Zhang S, Pavlovicz RE, Jones A, Schmidt KH, Tang J, Dong H, Shan B, Fang B, Radhakrishnan R, Glazer PM, Matthias P, Koomen J, Seto E, Bepler G, Nicosia SV, Chen J, Li C, Gu L, Li GM, Bai W, Wang H, Zhang X, HDAC6 deacetylates and ubiquitinates MSH2 to maintain proper levels of MutSalpha, Mol. Cell 55 (2014) 31–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kutil Z, Skultetyova L, Rauh D, Meleshin M, Snajdr I, Novakova Z, Mikesova J, Pavlicek J, Hadzima M, Baranova P, Havlinova B, Majer P, Schutkowski M, Barinka C, The unraveling of substrate specificity of histone deacetylase 6 domains using acetylome peptide microarrays and peptide libraries, Faseb. J 33 (2019) 4035–4045. [DOI] [PubMed] [Google Scholar]
- [20].Osko JD, Christianson DW, Structural basis of catalysis and inhibition of HDAC6 CD1, the enigmatic catalytic domain of histone deacetylase 6, Biochemistry 58 (2019) 4912–4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Miyake Y, Keusch JJ, Wang L, Saito M, Hess D, Wang X, Melancon BJ, Helquist P, Gut H, Matthias P, Structural insights into HDAC6 tubulin deacetylation and its selective inhibition, Nat. Chem. Biol 12 (2016) 748–754. [DOI] [PubMed] [Google Scholar]
- [22].Saito M, Hess D, Eglinger J, Fritsch AW, Kreysing M, Weinert BT, Choudhary C, Matthias P, Acetylation of intrinsically disordered regions regulates phase separation, Nat. Chem. Biol 15 (2019) 51–61. [DOI] [PubMed] [Google Scholar]
- [23].Osko JD, Christianson DW, Binding of inhibitors to active-site mutants of CD1, the enigmatic catalytic domain of histone deacetylase 6, Acta Crystallogr. F Struct. Biol. Commun 76 (2020) 428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR, Yao TP, Lane WS, Seto E, HDAC6 modulates cell motility by altering the acetylation level of cortactin, Mol. Cell 27 (2007) 197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP, HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor, Mol. Cell 18 (2005) 601–607. [DOI] [PubMed] [Google Scholar]
- [26].Mortenson JB, Heppler LN, Banks CJ, Weerasekara VK, Whited MD, Piccolo SR, Johnson WE, Thompson JW, Andersen JL, Histone deacetylase 6 (HDAC6) promotes the pro-survival activity of 14-3-3zeta via deacetylation of lysines within the 14-3-3zeta binding pocket, J. Biol. Chem 290 (2015) 12487–12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Parmigiani RB, Xu WS, Venta-Perez G, Erdjument-Bromage H, Yaneva M, Tempst P, Marks PA, HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation, Proc. Natl. Acad. Sci. U.S.A 105 (2008) 9633–9638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Majid T, Griffin D, Criss Z 2nd , Jarpe M, Pautler RG, Pharmocologic treatment with histone deacetylase 6 inhibitor (ACY-738) recovers Alzheimer’s disease phenotype in amyloid precursor protein/presenilin 1 (APP/PS1) mice, Alzheimers Dement 1 (2015) 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].d’Ydewalle C, Krishnan J, Chiheb DM, Van Damme P, Irobi J, Kozikowski AP, Vanden Berghe P, Timmerman V, Robberecht W, Van Den Bosch L, HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease, Nat. Med 17 (2011) 968–974. [DOI] [PubMed] [Google Scholar]
- [30].Mo ZY, Zhao XB, Liu HQ, Hu QH, Chen XQ, Pham J, Wei N, Liu Z, Zhou JD, Burgess RW, Pfaff SL, Caskey CT, Wu CB, Bai G, Yang XL, Aberrant GlyRS-HDAC6 interaction linked to axonal transport deficits in Charcot-Marie-Tooth neuropathy, Nat. Commun 9 (2018) 1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Benoy V, Van Helleputte L, Prior R, d’Ydewalle C, Haeck W, Geens N, Scheveneels W, Schevenels B, Cader MZ, Talbot K, Kozikowski AP, Vanden Berghe P, Van Damme P, Robberecht W, Van Den Bosch L, HDAC6 is a therapeutic target in mutant GARS-induced Charcot-Marie-Tooth disease, Brain 141 (2018) 673–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Picci C, Wong VSC, Costa CJ, McKinnon MC, Goldberg DC, Swift M, Alam NM, Prusky GT, Shen S, Kozikowski AP, Willis DE, Langley B, HDAC6 inhibition promotes alpha-tubulin acetylation and ameliorates CMT2A peripheral neuropathy in mice, Exp. Neurol 328 (2020) 113281. [DOI] [PubMed] [Google Scholar]
- [33].Guo W, Naujock M, Fumagalli L, Vandoorne T, Baatsen P, Boon R, Ordovas L, Patel A, Welters M, Vanwelden T, Geens N, Tricot T, Benoy V, Steyaert J, Lefebvre-Omar C, Boesmans W, Jarpe M, Sterneckert J, Wegner F, Petri S, Bohl D, Vanden Berghe P, Robberecht W, Van Damme P, Verfaillie C, Van Den Bosch L, HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients, Nat. Commun 8 (2017) 861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Rossaert E, Pollari E, Jaspers T, Van Helleputte L, Jarpe M, Van Damme P, De Bock K, Moisse M, Van Den Bosch L, Restoration of histone acetylation ameliorates disease and metabolic abnormalities in a FUS mouse model, Acta Neuropathol. Commun 7 (2019) 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Xu X, Kozikowski AP, Pozzo-Miller L, A selective histone deacetylase-6 inhibitor improves BDNF trafficking in hippocampal neurons from Mecp2 knockout mice: implications for Rett syndrome, Front. Cell. Neurosci 8 (2014) 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gold WA, Lacina TA, Cantrill LC, Christodoulou J, MeCP2 deficiency is associated with reduced levels of tubulin acetylation and can be restored using HDAC6 inhibitors, J. Mol. Med 93 (2015) 63–72. [DOI] [PubMed] [Google Scholar]
- [37].Shen S, Kozikowski AP, A patent review of histone deacetylase 6 inhibitors in neurodegenerative diseases (2014-2019), Expert Opin. Ther. Pat 30 (2020) 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Brindisi M, Saraswati AP, Brogi S, Gemma S, Butini S, Campiani G, Old but gold: tracking the new guise of histone deacetylase 6 (HDAC6) enzyme as a biomarker and therapeutic target in rare diseases, J. Med. Chem 63 (2020) 23–39. [DOI] [PubMed] [Google Scholar]
- [39].Keremu A, Aimaiti A, Liang Z, Zou X, Role of the HDAC6/STAT3 pathway in regulating PD-L1 expression in osteosarcoma cell lines, Canc. Chemother. Pharmacol 83 (2019) 255–264. [DOI] [PubMed] [Google Scholar]
- [40].Lienlaf M, Perez-Villarroel P, Knox T, Pabon M, Sahakian E, Powers J, Woan KV, Lee C, Cheng F, Deng S, Smalley KSM, Montecino M, Kozikowski AP, Pinilla-Ibarz J, Sarnaik A, Seto E, Weber J, Sotomayor EM, Villagra A, Essential role of HDAC6 in the regulation of PD-L1 in melanoma, Mol. Oncol 10 (2016) 735–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Woan KV, Lienlaf M, Perez-Villaroel P, Lee C, Cheng F, Knox T, Woods DM, Barrios K, Powers J, Sahakian E, Wang HW, Canales J, Marante D, Smalley KSM, Bergman J, Seto E, Kozikowski A, Pinilla-Ibarz J, Sarnaik A, Celis E, Weber J, Sotomayor EM, Villagra A, Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: enhanced antitumor immunity and impaired cell proliferation, Mol. Oncol 9 (2015) 1447–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Tavares MT, Shen S, Knox T, Hadley M, Kutil Z, Barinka C, Villagra A, Kozikowski AP, Synthesis and pharmacological evaluation of selective histone deacetylase 6 inhibitors in melanoma models, ACS Med. Chem. Lett 8 (2017) 1031–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Shen S, Hadley M, Ustinova K, Pavlicek J, Knox T, Noonepalle S, Tavares MT, Zimprich CA, Zhang G, Robers MB, Barinka C, Kozikowski AP, Villagra A, Discovery of a new isoxazole-3-hydroxamate-based histone deacetylase 6 inhibitor SS-208 with antitumor activity in syngeneic melanoma mouse models, J. Med. Chem 62 (2019) 8557–8577. [DOI] [PubMed] [Google Scholar]
- [44].Tsuji G, Okiyama N, Villarroel VA, Katz SI, Histone deacetylase 6 inhibition impairs effector CD8 T-cell functions during skin inflammation, J. Allergy Clin. Immunol 135 (2015) 1228–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ryu HW, Shin DH, Lee DH, Won HR, Kwon SH, A potent hydroxamic acid-based, small-molecule inhibitor A452 preferentially inhibits HDAC6 activity and induces cytotoxicity toward cancer cells irrespective of p53 status, Carcinogenesis 39 (2018) 72–83. [DOI] [PubMed] [Google Scholar]
- [46].Knox T, Sahakian E, Banik D, Hadley M, Palmer E, Noonepalle S, Kim J, Powers J, Gracia-Hernandez M, Oliveira V, Cheng F, Chen J, Barinka C, Pinilla-Ibarz J, Lee NH, Kozikowski A, Villagra A, Selective HDAC6 inhibitors improve anti-PD-1 immune checkpoint blockade therapy by decreasing the anti-inflammatory phenotype of macrophages and down-regulation of immunosuppressive proteins in tumor cells, Sci. Rep 9 (2019) 6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Greer JM, McCombe PA, The role of epigenetic mechanisms and processes in autoimmune disorders, Biologics 6 (2012) 307–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Li T, Zhang C, Hassan S, Liu X, Song F, Chen K, Zhang W, Yang J, Histone deacetylase 6 in cancer, J. Hematol. Oncol 11 (2018) 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Prior R, Van Helleputte L, Klingl YE, Van Den Bosch L, HDAC6 as a potential therapeutic target for peripheral nerve disorders, Expert Opin. Ther. Targets 22 (2018) 993–1007. [DOI] [PubMed] [Google Scholar]
- [50].Celen S, Rokka J, Gilbert TM, Koole M, Vermeulen I, Serdons K, Schroeder FA, Wagner FF, Bleeser T, Hightower BG, Hu J, Rahal D, Beyzavi MH, Vanduffel W, Van Laere K, Kranz JE, Hooker JM, Bormans G, Cawthorne CJ, Translation of HDAC6 PET imaging using [(18)F]EKZ-001-cGMP production and measurement of HDAC6 target occupancy in nonhuman primates, ACS Chem. Neurosci 11 (2020) 1093–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Koole M, Van Weehaeghe D, Serdons K, Herbots M, Cawthorne C, Celen S, Schroeder FA, Hooker JM, Bormans G, de Hoon J, Kranz JE, Van Laere K, Gilbert TM, Clinical validation of the novel HDAC6 radiotracer [(18)F]EKZ-001 in the human brain, Eur. J. Nucl. Med. Mol. Imag (2020), 10.1007/s00259-020-04891-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Clinical Trials for Ricolinostat. https://clinicaltrials.gov/ct2/results?term=ACY-1215&Search=Search (accessed Mar 27, 2020).
- [53].Clinical Trials for Citarinostat. https://clinicaltrials.gov/ct2/results?term=ACY-241&Search=Search. (Accessed 27 March 2020).
- [54].Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M, Jarpe M, van Duzer JH, Mazitschek R, Ogier WC, Cirstea D, Rodig S, Eda H, Scullen T, Canavese M, Bradner J, Anderson KC, Jones SS, Raje N, Pre-clinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma, Blood 119 (2012) 2579–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Huang P, Almeciga-Pinto I, Jarpe M, van Duzer JH, Mazitschek R, Yang M, Jones SS, Quayle SN, Selective HDAC inhibition by ACY-241 enhances the activity of paclitaxel in solid tumor models, Oncotarget 8 (2017) 2694–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wang XX, Wan RZ, Liu ZP, Recent advances in the discovery of potent and selective HDAC6 inhibitors, Eur. J. Med. Chem 143 (2018) 1406–1418. [DOI] [PubMed] [Google Scholar]
- [57].Faria Freitas M, Cuendet M, Bertrand P, HDAC inhibitors: a 2013-2017 patent survey, Expert Opin. Ther. Pat 28 (2018) 365–381. [DOI] [PubMed] [Google Scholar]
- [58].De Vreese R, Van Steen N, Verhaeghe T, Desmet T, Bougarne N, De Bosscher K, Benoy V, Haeck W, Van Den Bosch L, D’Hooghe M, Synthesis of benzothiophene-based hydroxamic acids as potent and selective HDAC6 inhibitors, Chem. Commun 51 (2015) 9868–9871. [DOI] [PubMed] [Google Scholar]
- [59].Kozikowski AP, Shen S, Pardo M, Tavares MT, Szarics D, Benoy V, Zimprich CA, Kutil Z, Zhang G, Barinka C, Robers MB, Van Den Bosch L, Eubanks JH, Jope RS, Brain penetrable histone deacetylase 6 inhibitor SW-100 ameliorates memory and learning impairments in a mouse model of Fragile X syndrome, ACS Chem. Neurosci 10 (2019) 1679–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].VanderMolen KM, McCulloch W, Pearce CJ, Oberlies NH, Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): a natural product recently approved for cutaneous T-cell lymphoma, J. Antibiot 64 (2011) 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Itoh Y, Suzuki T, Kouketsu A, Suzuki N, Maeda S, Yoshida M, Nakagawa H, Miyata N, Design, synthesis, structure-selectivity relationship, and effect on human cancer cells of a novel series of histone deacetylase 6-selective inhibitors, J. Med. Chem 50 (2007) 5425–5438. [DOI] [PubMed] [Google Scholar]
- [62].Suzuki T, Kouketsu A, Itoh Y, Hisakawa S, Maeda S, Yoshida M, Nakagawa H, Miyata N, Highly potent and selective histone deacetylase 6 inhibitors designed based on a small-molecular substrate, J. Med. Chem 49 (2006) 4809–4812. [DOI] [PubMed] [Google Scholar]
- [63].Giannini G, Vesci L, Battistuzzi G, Vignola D, Milazzo FM, Guglielmi MB, Barbarino M, Santaniello M, Fanto N, Mor M, Rivara S, Pala D, Taddei M, Pisano C, Cabri W, ST7612AA1, a thioacetate-omega(gamma-lactam carboxamide) derivative selected from a novel generation of oral HDAC inhibitors, J. Med. Chem 57 (2014) 8358–8377. [DOI] [PubMed] [Google Scholar]
- [64].Wahhab A, Smil D, Ajamian A, Allan M, Chantigny Y, Therrien E, Nguyen N, Manku S, Leit S, Rahil J, Petschner AJ, Lu AH, Nicolescu A, Lefebvre S, Montcalm S, Fournel M, Yan TP, Li Z, Besterman JM, Deziel R, Sulfamides as novel histone deacetylase inhibitors, Bioorg. Med. Chem. Lett 19 (2009) 336–340. [DOI] [PubMed] [Google Scholar]
- [65].Muthyala R, Shin WS, Xie J, Sham YY, Discovery of 1-hydroxypyridine-2-thiones as selective histone deacetylase inhibitors and their potential application for treating leukemia, Bioorg. Med. Chem. Lett 25 (2015) 4320–4324. [DOI] [PubMed] [Google Scholar]
- [66].Dehmel F, Weinbrenner S, Julius H, Ciossek T, Maier T, Stengel T, Fettis K, Burkhardt C, Wieland H, Beckers T, Trithiocarbonates as a novel class of HDAC inhibitors: SAR studies, isoenzyme selectivity, and pharmacological profiles, J. Med. Chem 51 (2008) 3985–4001. [DOI] [PubMed] [Google Scholar]
- [67].Mohan R, Sharma AK, Gupta S, Ramaa CS, Design, synthesis, and biological evaluation of novel 2,4-thiazolidinedione derivatives as histone deacetylase inhibitors targeting liver cancer cell line, Med. Chem. Res 21 (2012) 1156–1165. [Google Scholar]
- [68].Patil V, Sodji QH, Kornacki JR, Mrksich M, Oyelere AK, 3-Hydroxypyridin-2-thione as novel zinc binding group for selective histone deacetylase inhibition, J. Med. Chem 56 (2013) 3492–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Sodji QH, Patil V, Kornacki JR, Mrksich M, Oyelere AK, Synthesis and structure-activity relationship of 3-hydroxypyridine-2-thione-based histone deacetylase inhibitors, J. Med. Chem 56 (2013) 9969–9981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Suzuki T, Matsuura A, Kouketsu A, Nakagawa H, Miyata N, Identification of a potent non-hydroxamate histone deacetylase inhibitor by mechanism-based drug design, Bioorg. Med. Chem. Lett 15 (2005) 331–335. [DOI] [PubMed] [Google Scholar]
- [71].Chen B, Petukhov PA, Jung M, Velena A, Eliseeva E, Dritschilo A, Kozikowski AP, Chemistry and biology of mercaptoacetamides as novel histone deacetylase inhibitors, Bioorg. Med. Chem. Lett 15 (2005) 1389–1392. [DOI] [PubMed] [Google Scholar]
- [72].Gu W, Nusinzon I, Smith RD Jr., Horvath CM, Silverman RB, Carbonyl- and sulfur-containing analogs of suberoylanilide hydroxamic acid: potent inhibition of histone deacetylases, Bioorg. Med. Chem 14 (2006) 3320–3329. [DOI] [PubMed] [Google Scholar]
- [73].Kozikowski AP, Chen Y, Gaysin A, Chen B, D’Annibale MA, Suto CM, Langley BC, Functional differences in epigenetic modulators-superiority of mercaptoacetamide-based histone deacetylase inhibitors relative to hydroxamates in cortical neuron neuroprotection studies, J. Med. Chem 50 (2007) 3054–3061. [DOI] [PubMed] [Google Scholar]
- [74].Stolfa DA, Marek M, Lancelot J, Hauser AT, Walter A, Leproult E, Melesina J, Rumpf T, Wurtz JM, Cavarelli J, Sippl W, Pierce RJ, Romier C, Jung M, Molecular basis for the antiparasitic activity of a mercaptoacetamide derivative that inhibits histone deacetylase 8 (HDAC8) from the human pathogen schistosoma mansoni, J. Mol. Biol 426 (2014) 3442–3453. [DOI] [PubMed] [Google Scholar]
- [75].Tang H, Wang XS, Huang XP, Roth BL, Butler KV, Kozikowski AP, Jung M, Tropsha A, Novel inhibitors of human histone deacetylase (HDAC) identified by QSAR modeling of known inhibitors, virtual screening, and experimental validation, J. Chem. Inf. Model 49 (2009) 461–476. [DOI] [PubMed] [Google Scholar]
- [76].Kozikowski AP, Chen Y, Gaysin AM, Savoy DN, Billadeau DD, Kim KH, Chemistry, biology, and QSAR studies of substituted biaryl hydroxamates and mercaptoacetamides as HDAC inhibitors-nanomolar-potency inhibitors of pancreatic cancer cell growth, ChemMedChem 3 (2008) 487–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Kalin JH, Zhang H, Gaudrel-Grosay S, Vistoli G, Kozikowski AP, Chiral mercaptoacetamides display enantioselective inhibition of histone deacetylase 6 and exhibit neuroprotection in cortical neuron models of oxidative stress, ChemMedChem 7 (2012) 425–439. [DOI] [PubMed] [Google Scholar]
- [78].Kozikowski AP, Tapadar S, Luchini DN, Kim KH, Billadeau DD, Use of the nitrile oxide cycloaddition (NOC) reaction for molecular probe generation: a new class of enzyme selective histone deacetylase inhibitors (HDACIs) showing picomolar activity at HDAC6, J. Med. Chem 51 (2008) 4370–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Wen J, Niu Q, Liu J, Bao Y, Yang J, Luan S, Fan Y, Liu D, Zhao L, Novel thiol-based histone deacetylase inhibitors bearing 3-phenyl-1H-pyrazole-5-carboxamide scaffold as surface recognition motif: design, synthesis and SAR study, Bioorg. Med. Chem. Lett 26 (2016) 375–379. [DOI] [PubMed] [Google Scholar]
- [80].Segretti MC, Vallerini GP, Brochier C, Langley B, Wang L, Hancock WW, Kozikowski AP, Thiol-based potent and selective HDAC6 inhibitors promote tubulin acetylation and T-regulatory cell suppressive function, ACS Med. Chem. Lett 6 (2015) 1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lv W, Zhang G, Barinka C, Eubanks JH, Kozikowski AP, Design and synthesis of mercaptoacetamides as potent, selective, and brain permeable histone deacetylase 6 inhibitors, ACS Med. Chem. Lett 8 (2017) 510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Anandan SK, Ward JS, Brokx RD, Bray MR, Patel DV, Xiao XX, Mercaptoamide-based non-hydroxamic acid type histone deacetylase inhibitors, Bioorg. Med. Chem. Lett 15 (2005) 1969–1972. [DOI] [PubMed] [Google Scholar]
- [83].Sung YM, Lee T, Yoon H, DiBattista AM, Song JM, Sohn Y, Moffat EI, Turner RS, Jung M, Kim J, Hoe HS, Mercaptoacetamide-based class II HDAC inhibitor lowers Abeta levels and improves learning and memory in a mouse model of Alzheimer’s disease, Exp. Neurol 239 (2013) 192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Suzuki T, Nagano Y, Kouketsu A, Matsuura A, Maruyama S, Kurotaki M, Nakagawa H, Miyata N, Novel inhibitors of human histone deacetylases: design, synthesis, enzyme inhibition, and cancer cell growth inhibition of SAHA-based non-hydroxamates, J. Med. Chem 48 (2005) 1019–1032. [DOI] [PubMed] [Google Scholar]
- [85].Lombardi PM, Cole KE, Dowling DP, Christianson DW, Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes, Curr. Opin. Struct. Biol 21 (2011) 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Furumai R, Matsuyama A, Kobashi N, Lee KH, Nishiyama M, Nakajima H, Tanaka A, Komatsu Y, Nishino N, Yoshida M, Horinouchi S, FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases, Canc. Res 62 (2002) 4916–4921. [PubMed] [Google Scholar]
- [87].Porter NJ, Shen S, Barinka C, Kozikowski AP, Christianson DW, Molecular basis for the selective inhibition of histone deacetylase 6 by a mercaptoacetamide inhibitor, ACS Med. Chem. Lett 9 (2018) 1301–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Hai Y, Christianson DW, Histone deacetylase 6 structure and molecular basis of catalysis and inhibition, Nat. Chem. Biol. 12 (2016) 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Osko JD, Christianson DW, Structural determinants of affinity and selectivity in the binding of inhibitors to histone deacetylase 6, Bioorg. Med. Chem. Lett 30 (2020) 127023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Vogerl K, Ong N, Senger J, Herp D, Schmidtkunz K, Marek M, Muller M, Bartel K, Shaik TB, Porter NJ, Robaa D, Christianson DW, Romier C, Sippl W, Jung M, Bracher F, Synthesis and biological investigation of phenothiazine-based benzhydroxamic acids as selective histone deacetylase 6 inhibitors, J. Med. Chem 62 (2019) 1138–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Porter NJ, Osko JD, Diedrich D, Kurz T, Hooker JM, Hansen FK, Christianson DW, Histone deacetylase 6-selective inhibitors and the influence of capping groups on hydroxamate-zinc denticity, J. Med. Chem 61 (2018) 8054–8060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Porter NJ, Mahendran A, Breslow R, Christianson DW, Unusual zinc-binding mode of HDAC6-selective hydroxamate inhibitors, Proc. Natl. Acad. Sci. U.S.A 114 (2017) 13459–13464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Bayer T, Chakrabarti A, Lancelot J, Shaik TB, Hausmann K, Melesina J, Schmidtkunz K, Marek M, Erdmann F, Schmidt M, Robaa D, Romier C, Pierce RJ, Jung M, Sippl W, Synthesis, crystallization studies, and in vitro characterization of cinnamic acid derivatives as smHDAC8 inhibitors for the treatment of schistosomiasis, ChemMedChem 13 (2018) 1517–1529. [DOI] [PubMed] [Google Scholar]
- [94].Porter NJ, Wagner FF, Christianson DW, Entropy as a driver of selectivity for inhibitor binding to histone deacetylase 6, Biochemistry 57 (2018) 3916–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].De Vreese R, D’Hooghe M, Synthesis and applications of benzohydroxamic acid-based histone deacetylase inhibitors, Eur. J. Med. Chem 135 (2017) 174–195. [DOI] [PubMed] [Google Scholar]
- [96].Osko JD, Porter NJ, Narayana Reddy PA, Xiao YC, Rokka J, Jung M, Hooker JM, Salvino JM, Christianson DW, Exploring structural determinants of inhibitor affinity and selectivity in complexes with histone deacetylase 6, J. Med. Chem 63 (2020) 295–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Shen S, Svoboda M, Zhang G, Cavasin MA, Motlova L, McKinsey TA, Eubanks JH, Barinka C, Kozikowski AP, Structural and in vivo characterization of Tubastatin A, a widely used histone deacetylase 6 inhibitor, ACS Med. Chem. Lett 11 (2020) 706–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Chakrabarti P, Geometry of interaction of metal ions with sulfur-containing ligands in protein structures, Biochemistry 28 (1989) 6081–6085. [DOI] [PubMed] [Google Scholar]
- [99].Konsoula R, Jung M, In vitro plasma stability, permeability and solubility of mercaptoacetamide histone deacetylase inhibitors, Int. J. Pharm 361 (2008) 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Konsoula Z, Jung M, Involvement of P-glycoprotein and multidrug resistance associated protein 1 on the transepithelial transport of a mercaptoacetamide-based histone-deacetylase inhibitor in Caco-2 cells, Biol. Pharm. Bull 32 (2009) 74–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Konsoula Z, Cao H, Velena A, Jung M, Pharmacokinetics-pharmacodynamics and antitumor activity of mercaptoacetamide-based histone deacetylase inhibitors, Mol. Canc. Therapeut 8 (2009) 2844–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Ghose AK, Herbertz T, Hudkins RL, Dorsey BD, Mallamo JP, Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery, ACS Chem. Neurosci 3 (2012) 50–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Nam JH, Cho HJ, Kang H, Lee JY, Jung M, Chang YC, Kim K, Hoe HS, A Mercaptoacetamide-based class II histone deacetylase inhibitor suppresses cell migration and invasion in monomorphic malignant human glioma cells by inhibiting FAK/STAT3 signaling, J. Cell. Biochem 118 (2017) 4672–4685. [DOI] [PubMed] [Google Scholar]
- [104].Vivar C, Potter MC, van Praag H, All about running: synaptic plasticity, growth factors and adult hippocampal neurogenesis, Curr. Top Behav. Neurosci 15 (2013) 189–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Brewster AL, Lugo JN, Patil VV, Lee WL, Qian Y, Vanegas F, Anderson AE, Rapamycin reverses status epilepticus-induced memory deficits and dendritic damage, PLoS One 8 (2013), e57808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Ripoli C, Piacentini R, Riccardi E, Leone L, Li Puma DD, Bitan G, Grassi C, Effects of different amyloid beta-protein analogues on synaptic function, Neurobiol. Aging 34 (2013) 1032–1044. [DOI] [PubMed] [Google Scholar]
- [107].Song JM, Sung YM, Nam JH, Yoon H, Chung A, Moffat E, Jung M, Pak DT, Kim J, Hoe HS, A Mercaptoacetamide-based class II histone deacetylase inhibitor increases dendritic spine density via RasGRF1/ERK pathway, J. Alzheimers Dis 51 (2016) 591–604. [DOI] [PubMed] [Google Scholar]